
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to ligand-stabilized PH-containing phosphenium complexes†
David
Biskup
 a,
Gregor
Schnakenburg
a,
Arturo
Espinosa Ferao
*b and
Rainer
Streubel
*a
a,
Gregor
Schnakenburg
a,
Arturo
Espinosa Ferao
*b and
Rainer
Streubel
*a
aInstitut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany. E-mail: r.streubel@uni-bonn.de
bDepartamento de Química Orgánica, Facultad de Química, Campus de Espinardo, Universidad de Murcia, 30100 Murcia, Spain. E-mail: artuesp@um.es
First published on 5th January 2024
Abstract
While the chemistry of phosphenium compounds, including metal complexes thereof, is very well established, few derivatives having a P–H bond have been described, yet. This work describes rational access to donor-stabilised phosphenium metal complexes possessing a P–H bond using protonation reactions of stable phosphinidene complex adducts. While most Brønsted–Lowry acids yield formal 1,1-addition products at the phosphorus centre under the loss of the donor, super-strong acids having weakly coordinating anions enable access to donor-stabilised P–H phosphenium complex salts. The latter possess N-methylimidazole as a donor (to phosphorus) and the N–P interaction has been studied theoretically.
Introduction
The first cationic dicoordinate phosphorus compounds, namely, phosphamethinecyanines, were described by Dimroth and Hoffmann in 1964, being stabilised by π-donation of the electron-rich neighbouring atoms and charge delocalization.1 Afterwards, a broad range of compounds containing phosphenium cations stabilised by π-donor-substituents were described, e.g. amino(aryl)phosphenium salts,2 diamino phosphenium salts,2 1,3,2-diazaphospholidinium salts,3 1,3,2-diazaphosphinanium salts,4 or 1H-1,2,3-diazaphospholium salts.5 The latter, analogues of N-heterocyclic carbenes, were first prepared by Pudovik6 and Denk,7 and further intensely investigated by Gudat.8 Niecke and Kröher reported a zwitterionic metallaphosphenium salt in 1976.9 Recently, the synthesis of a kinetically stabilised donor-free phosphenium salt I was reported by Olaru, Mebs and Beckmann (Fig. 1).10 Phosphenium salts are usually prepared via halide abstraction of halophosphanes.11 A different access was described by Niecke via the protonation of a phosphaalkene12 or an imino-phosphane13 and later by Grützmacher through methylation of a diphosphene derivative.14 The chemistry of donor-to-nonmetal element adducts started with a report of Burford on cationic low-coordinate phosphorus species such as phosphadiazonium and phosphenium adducts.15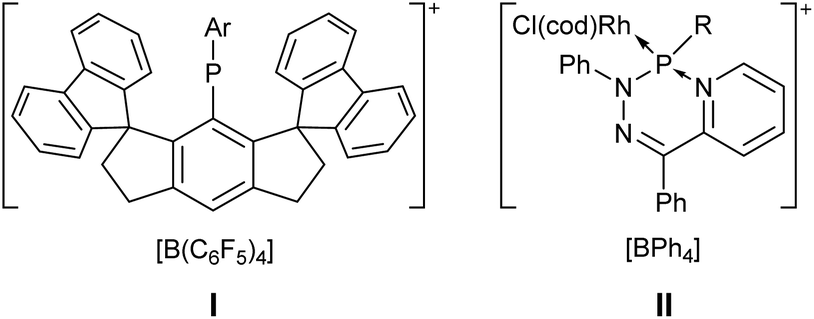 | ||
| Fig. 1 The first stable donor-free phosphenium salt I and the first N-donor-to-phosphenium complex II.10,18 | ||
Similar to carbene metal complexes, PR2 metal complexes can show an electrophilic or nucleophilic character dependent on the nature of the metal, its co-ligands and the PR2 fragment itself.16 Hence, the P-centre can behave as either a phosphenium or a phosphido ligand. The majority of phosphenium complexes are cationic but neutral complexes have also been described in literature.16 However, cationic phosphido complexes are exceptional and were only obtained by using electron-rich metal centres without π-acidic co-ligands.16
Several pathways to phosphenium metal complexes were described including ligand substitution, salt metathesis, alkoxy abstraction, halide abstraction and elimination reactions.16,17 Reactions of phosphenium complexes with neutral nucleophiles were only investigated sparsely, predominantly by Nakazawa and Gudat. In many cases, reactions were observed at the metal instead of the P centre and thus, to date, the class of donor-to-phosphenium complex adducts have been only rarely reported.17–19 Very recently, Ragogna and Gilroy described the first N-donor-to-phosphenium complex adducts II where the N-donor centre was tethered as P-substituent (Fig. 1).18 The first non-stabilised P–H functional phosphenium complex was reported by Schrock and Churchill in 1982.20 Afterwards, only a few further examples were described and sparsely investigated.16,21 In 2007 Delpech reported on the protonation and alkylation of a terminal phosphinidene ruthenium complex to yield respective phosphenium complexes.22 Reactions of a terminal phosphinidene complex with dihydrogen yielded only primary phosphane complexes or respective diphosphane complexes.23 The first transition-metal-free N-heterocyclic carbene-to-phosphenium adduct bearing a P–H unit was reported by Bertrand, but this inversely polarised P–H phosphaalkene24 should be better named as a zwitterion, i.e., imidazolium phosphanide.
The question came up if neutral electrophilic, terminal phosphinidene complexes25 bearing only π-acidic co-ligands might be used, somehow, to access P–H substituted phosphenium complexes. Such phosphinidene complexes were intensely studied via thermal chelotropic elimination or extrusion-type chemistry using 7-phosphanorbornadiene,26 phosphirane,27 2H-azaphosphirene28 or phosphepine complexes.29 An alternative access to such species, usually generated at elevated temperatures, can be achieved via M/X phosphinidenoid complexes at low temperatures. So far, a broad scope of nucleo- and electrophilic chloride substitution reactions, ring expansion reactions, 1,2-addition to polar π-systems, oxidative single electron transfer reactions and the formal insertion reactions into OH- or NH-bonds were identified during the past years.30 A first attempt to synthesise an imidazole-stabilised transient electrophilic terminal phosphinidene complex was undertaken by Mathey in 2006. He studied the reaction of a 7-phosphanorbornadiene complex with N-methylimidazole at elevated temperatures, but only cyclic polyphosphanes were obtained as final products when no trapping reagents were present; therefore, the adduct was only proposed as an intermediate.31 Very recently, we reported on the synthesis and isolation of the first N-methylimidazole-to-phosphinidene complex adduct which could release an electrophilic, terminal phosphinidene complex under mild conditions. But we also observed a stunning thermal decomposition to yield white phosphorus as a final product when no trapping reagents were present.32 Donor-to-phosphinidene complex adducts are expected to exhibit an increased negative charge at phosphorus compared to the genuine phosphinidene complexes showing a less pronounced electrophilic behaviour, i.e. reactions with stronger donors occurred while no reactivity towards alkenes could be observed at ambient temperature.32,33 Therefore, an ambiguous reactivity of the donor-to-phosphinidene complex adducts is expected also enabling P-nucleophilic reactions.
Herein, we report on the reactivity of an N-methylimidazole-to-phosphinidene complex adduct towards a wide variety of Brønsted–Lowry acids intending to gain access to isolable donor-to-phosphenium complex adducts as potential precursors for genuine P–H substituted phosphenium complexes. The reaction pathway of the 1,1-addition of methanol has been computationally studied as a case in point. Furthermore, the varying donor-to-phosphorus interactions were inspected.
Results and discussion
Insertion reactions with water and alcohols
While (almost) no reactivity towards water has been observed for P-CPh3 substituted Li/Cl phosphinidenoid group 6 metal complexes, surprisingly, the N-methylimidazole-to-phosphinidene complex adduct 1 exhibited high reactivity towards water. The formation of hydroxy(triphenylmethyl)phosphane complex 2 strongly bound to N-methylimidazole (N-MeIm) via O–H⋯N hydrogen bonding was observed (Scheme 1).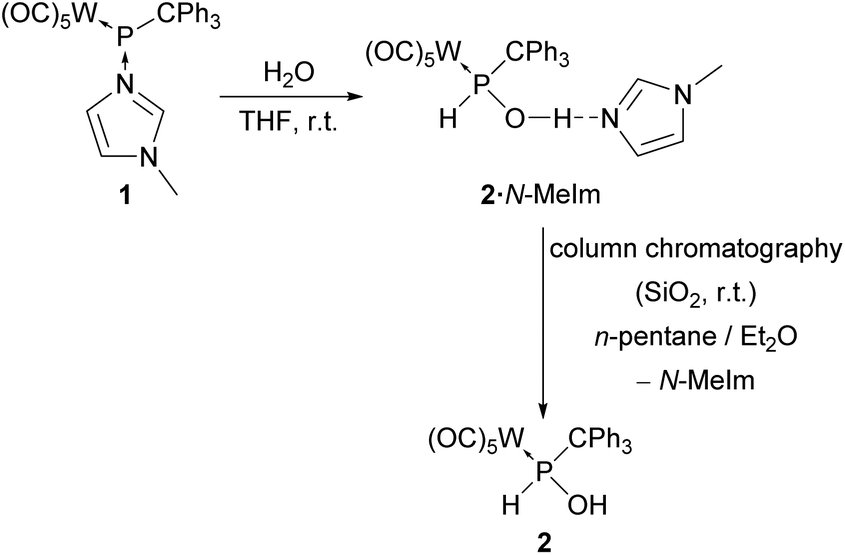 | ||
| Scheme 1 Hydrolysis of complex 1. | ||
The 31P NMR spectrum of 2·N-MeIm in THF-d8 displays a single resonance signal at 94.7 ppm (1JP,H = 337 Hz, 1JW,P = 267 Hz), and the presence of one equivalent of N-methylimidazole was identified in the 1H NMR spectrum. Additionally, the OH proton resonance is strongly deshielded showing a slightly broadened signal at 12.94 ppm, indicating a rather strong hydrogen bonding to the N3 atom of the N-MeIm unit. The N-MeIm was not separable from complex 2via evaporation in vacuo (<0.02 mbar) or via recrystallization. However, a clean separation was achieved via column chromatography at ambient temperature using an n-pentane/diethyl ether mixture as the eluent under argon atmosphere (Scheme 1).
The 31P NMR signal of 2 appears slightly downfield-shifted compared to 2·N-MeIm at 99.2 ppm (1JP,H = 341 Hz, 1JW,P = 270 Hz). In the 1H NMR spectrum of 2, the resonance of the OH group shifted upfield by 4.55 ppm to 8.39 ppm, revealing the loss of the hydrogen bonding to N-MeIm. This tendency was even stronger if a weaker coordinating solvent such as dichloromethane-d2 was used; here, the resonance appeared at 3.79 ppm (2JP,H = 5 Hz). The other proton resonances changed only marginally when transitioning from 2·N-MeIm to pure 2 or by changing the solvent. The solvent change has almost no effect on the 31P resonance, as the signal shifted from 99.2 to 100.7 ppm. The molecular structure of complex 2 was confirmed by single-crystal X-ray diffraction analysis (Fig. 2).
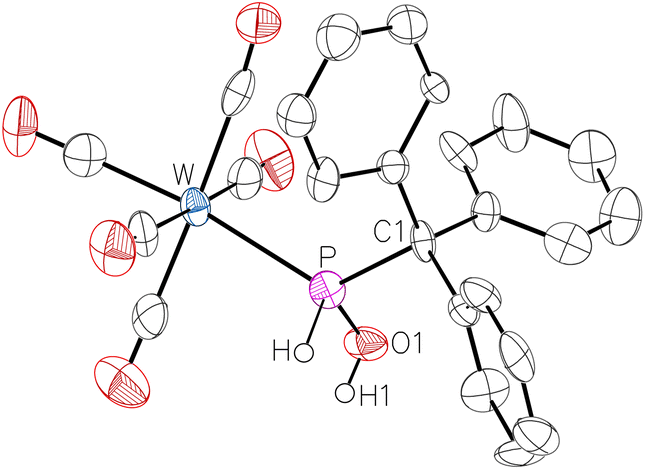 | ||
| Fig. 2 Molecular structure of 2. Thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity except for those bound to phosphorus or oxygen atoms. Selected bond lengths/Å and bond angles/°: W–P 2.527(3), P–O1 1.575(8), P–C1 1.919(10), O1–P–W 113.8(3), O1–P–C1 105.8(4), C1–P–W 124.1(3). | ||
The reaction of complex 1 with methanol and tert-butanol gave the OH insertion products 3a (δ(31P) = 127.2 ppm, 1JP,H = 343 Hz, 3JP,H = 13 Hz, 1JW,P = 274 Hz) and 3b (δ(31P) = 97.4 ppm, 1JP,H = 322 Hz, 1JW,P = 283 Hz) (Scheme 2). Previously, we reported on these products using a different approach.34 In this study, the reaction progress decreased strongly with the increasing steric demand of the organic substituent of the alcohol. For example, the reaction of 1 with methanol was complete within one day, yielding 3, while the reaction with tert-butanol took 14 days to reach completion.
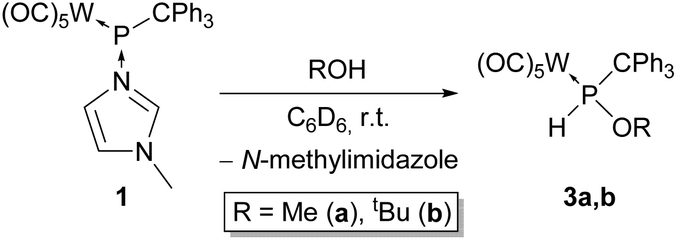 | ||
| Scheme 2 Synthesis of alkoxy(triphenylmethyl)phosphane complexes 3a,b. | ||
Previous studies on ligand substitution in model complex adduct 1′ (tert-butyl instead of trityl group at phosphorus) revealed that both SN1 (elimination–addition) and SN2 (addition–elimination) mechanisms could take place, depending on the incoming ligand.32 To unveil the exact mechanism involved in the addition of ROH, quantum chemical (QC) calculations were performed for the model reaction of 1′ with MeOH (see the Computational Details section in the ESI†). These calculations suggest some interesting aspects described below. First, the associative (addition of the incoming ligand as the first step) SN2-type mechanism turns out not to be a simple displacement of the nucleofugic N-MeIm ligand at P by the incoming MeOH nucleophile but rather seems to start by P-protonation with concomitant deprotonation of the acidic H2 at the imidazole ligand affording 4′·MeOH (Scheme 3). The reprotonation of the imidazole C2 should then occur via the addition of the methoxy unit to a carbonyl ligand at the metal fragment (5′), which allows intramolecular MeO group transfer to the P atom with the elimination of the N-MeIm ligand, affording the final (model) product 3a′ (Scheme 3). The methoxy group transfer can occur from different conformers (5′ax/5′eq) that enable either the energetically favoured (axial) approach to phosphorus, with the nucleofuge occupying an axial position, or the less favoured equatorial position, the latter requiring subsequent pseudorotation at the P centre (Fig. 3). The suggested alternative SN1-like mechanism (Scheme 3) involves the initial rather endergonic dissociation of the N-MeIm ligand, affording the terminal phosphinidene intermediate 6′, followed by thermoneutral addition of MeOH giving rise to the zwitterionic species 7′ (Fig. 3).
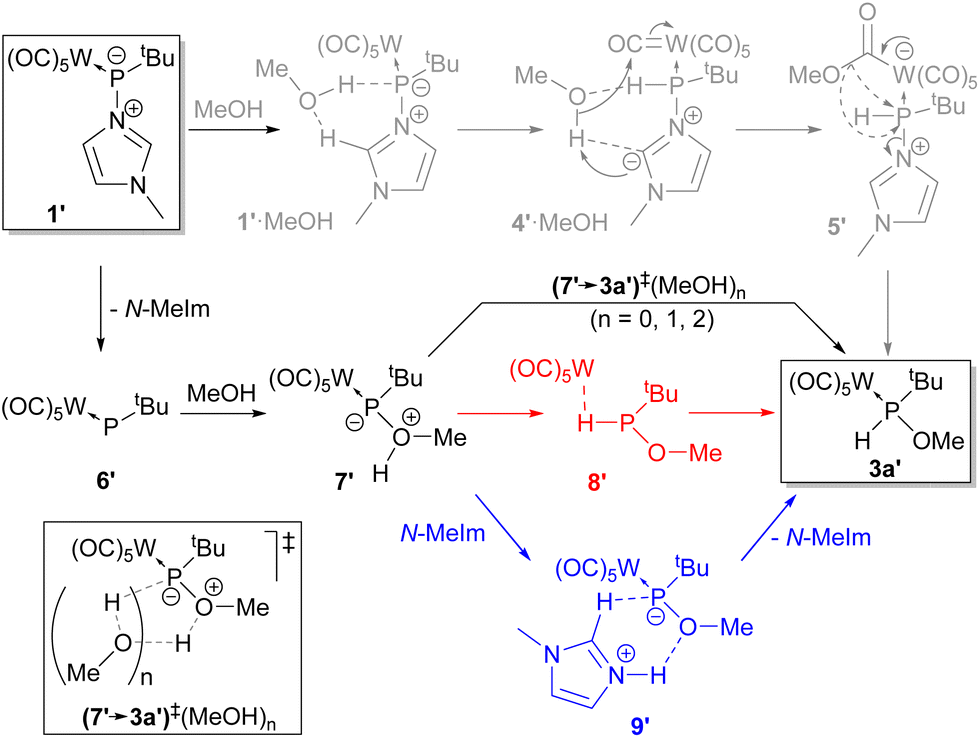 | ||
| Scheme 3 Proposed mechanism for the addition of MeOH to model complex adduct 1′. | ||
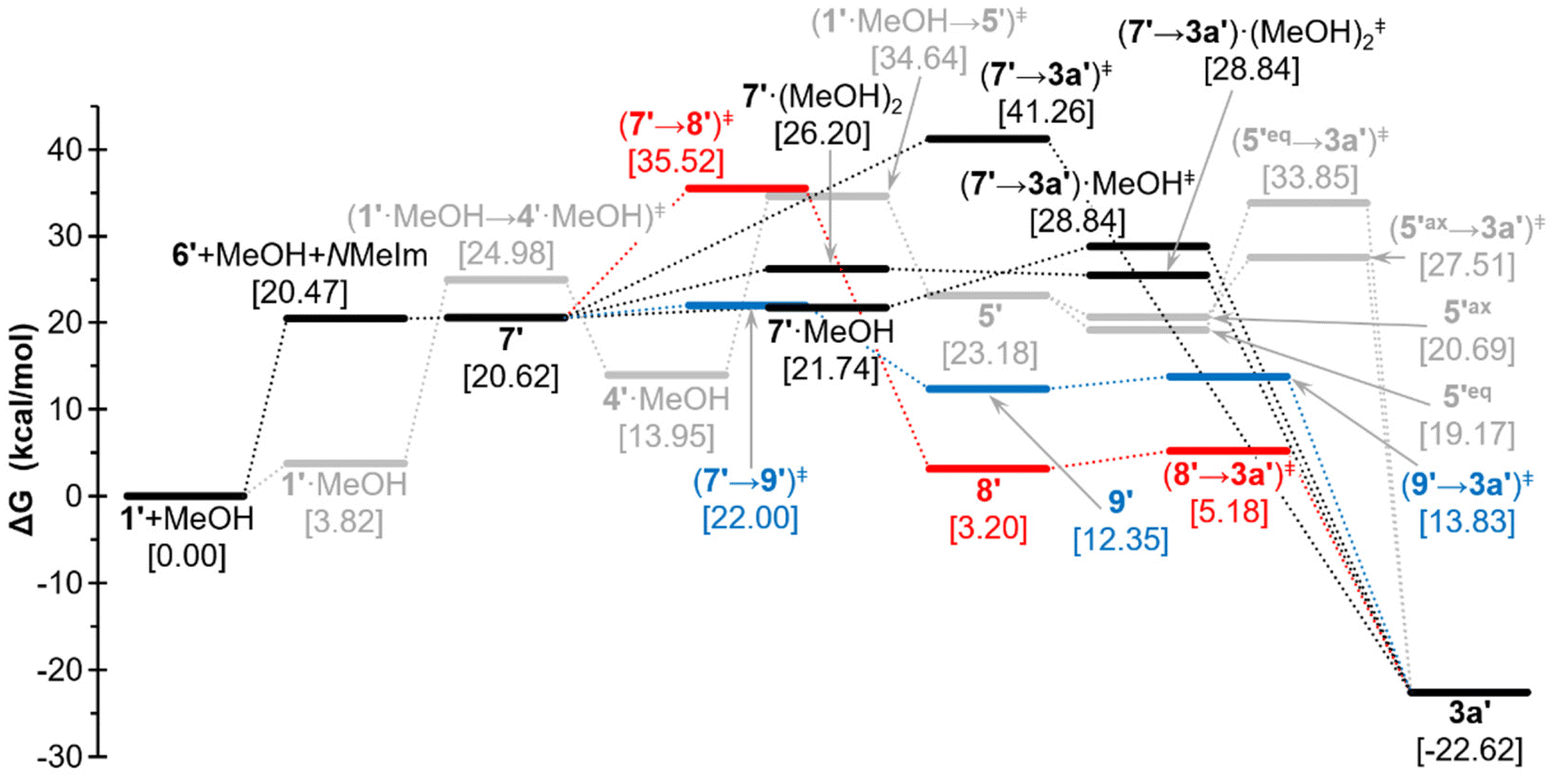 | ||
| Fig. 3 Computed [CPCM(tol)/PWPB95-D3/def2-QZVPP(ecp)//CPCM(tol)/PBEh-3c] Gibbs energy profile for the addition of methanol to model complex adduct 1′. Colour code used for SN2- (grey) and SN1-type mechanism with direct or MeOH-assisted H-transfer (black), N-MeIm-assisted H-transfer (blue) or involving decomplexation/recomplexation (red). | ||
The O-to-P proton shift can occur intramolecularly (7′→3a′) over a moderately high barrier, or across a lower energy pathway involving simultaneous P-decomplexation (8′, the electron rich-metal centre forming a sort of hydrogen bond: dW⋯H = 1.949 Å, MBOW⋯H = 0.213; (OC)ax–W⋯H angle 164.9°) and followed by small barrier P-complexation.
On the other hand, the O-to-P hydrogen shift can occur intermolecularly by the action of one or two MeOH molecules, (7′→3a′)‡(MeOH)n, enabling a proton transfer chain with an increasingly lower energy barrier. The intramolecular transfer can also take place with the lowest computed barrier in two steps involving N-MeIm, via the imidazolium salt 9′, constituting the lowest energy pathway for the 7′→3a′ transformation (Scheme 3, Fig. 3).
Therefore, according to these calculations (Fig. 3), the overall minimum energy pathway for the addition of MeOH to 1′ shows some slight preference for the (dissociative) SN1 mechanism (1′→6′→7′) with N-MeIm-mediated proton migration (7′→9′→3a′), although the SN2 pathway is also possible due to the small energy difference. The insertion of water on 1′ is expected to occur by a similar mechanism.
Reactions with strong acids
When strong(er) Brønsted–Lowry acids were added to complex 1 an additional reaction was observed, i.e., formation of N-methylimidazolium salts. Therefore, the addition of two equivalents of the acids was essential to reach completion. The reaction of complex 1 with trifluoroacetic acid or hydrogen chloride in dichloromethane resulted in the formation of the P-trifluoroacetoxy or P-chloride phosphane complexes 10a,b (Scheme 4). Compound 10b was already reported and characterised in a prior study by us via a different route using hydrogen chloride to substitute a P-bound tert-butylamino group.35 The products were extracted with n-pentane (10a) or diethyl ether (10b) from the residue containing N-methylimidazolium trifluoroacetate or chloride, respectively, thus obtaining both products in good yields.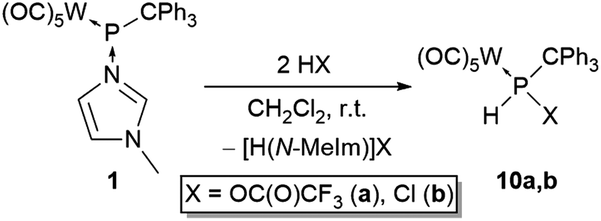 | ||
| Scheme 4 Synthesis of complexes 10a,b. | ||
The 31P NMR spectrum of 10a showed a downfield-shifted resonance signal at 112.1 ppm compared to the hydroxyphosphane (phosphinous acid) 2 and alkoxyphosphane (phosphinous ester) complexes 3a,b whereas 10b appeared at higher field (71.2 ppm). The large, pure negative inductive effect of the perfluorinated acetoxy group, exerted via the P–C σ bond, seems to be the origin of this deshielding effect. In the same vein, the 1JP,H coupling constant of 10a was significantly larger compared to complexes 2, 3 and 10b and, accordingly, also the P–H proton (8.42 ppm, 2JW,H = 7 Hz).
Single crystals suitable for X-ray diffraction analysis of 10a were obtained; the results confirmed the proposed molecular structure clearly showing the P–O connectivity (Fig. 4). It should be noted that the new protocol has furnished 10b with significantly improved yields (>99%) compared to 53% via the old route.35
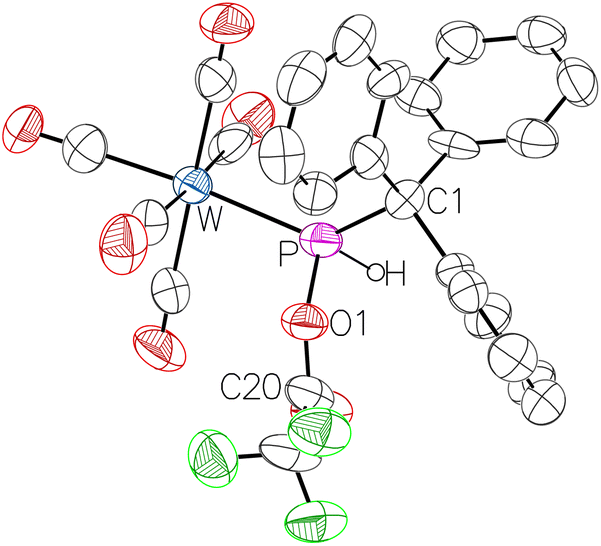 | ||
| Fig. 4 Molecular structure of 10a. Thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity except for the one bound to phosphorus. Selected bond lengths/Å and bond angles/°: W–P 2.452(3), P–O1 1.695(9), P–C1 1.915(12), O1–P–W 106.6(3), O1–P–C1 97.5(5), C1–P–W 130.6(4), C20–O1–P 124.0(9). | ||
Reactions with acids possessing weakly coordinating anions
To avoid the substitution of the P-bound N-methylimidazole of complex 1 by the acid counteranions, the reactivity of 1 towards three different super-strong acids with weakly coordinating anions (WCAs)36 was examined. The reaction of 1 with one equivalent of trifluoromethanesulfonic acid led to the selective formation of the N-MeIm-stabilised phosphenium complex 11a bearing trifluoromethanesulfonate as counteranion (Scheme 5).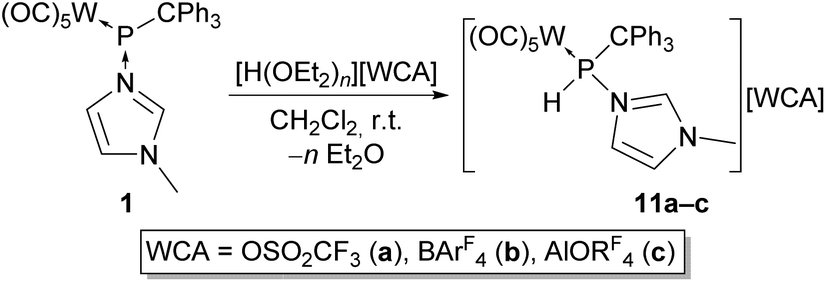 | ||
| Scheme 5 Synthesis of phosphanylimidazolium complex salts 11a–c (BArF4 = tetrakis{3,5-bis(trifluoromethyl)phenyl}borate, AlORF4 = tetrakis(nonafluoro-tert-butoxy)aluminate). | ||
The 31P NMR spectrum of 11a displayed a resonance at 65.5 ppm (1JP,H = 373 Hz, 1JW,P = 278 Hz). The highfield shift compared to complexes 10a,b suggests the presence of the P-bound donor. Interestingly, we noted that the PH and C2H protons exhibited similar deshielded resonance signals in the 1H NMR spectrum (Table 1). The assumption that the deshielding of the C2H proton stems from unexpected hydrogen bonding with the trifluoromethanesulfonate anion was finally confirmed by X-ray diffraction analysis, revealing an O6–H1 distance of 2.405 Å (Fig. 5; please note that here C1 equals C2 in the NMR experiments). Seemingly the P-protonation leads to an enhanced partial positive charge at the phosphorus and the N-MeIm unit, thus creating a significant increase in the acidic character of the PH and C2H hydrogen atoms.
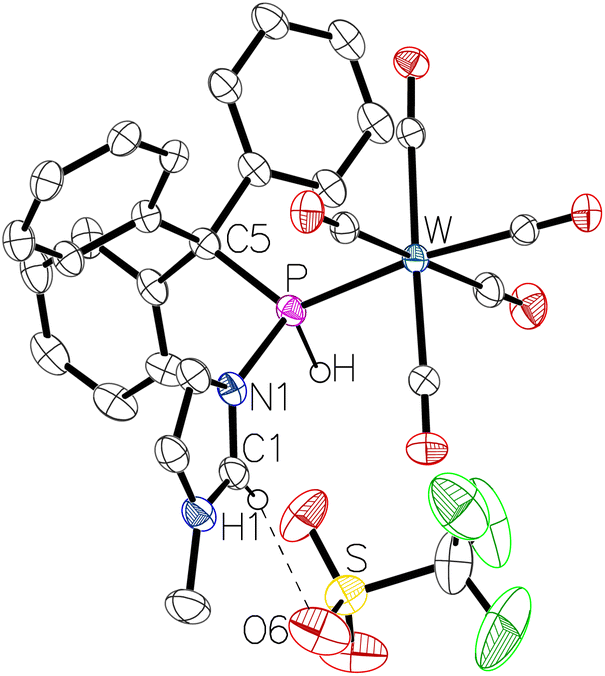 | ||
| Fig. 5 Molecular structure of 11a. Thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity except for those bound to phosphorus or involved in hydrogen bonding. Selected bond lengths/Å and bond angles/°: W–P 2.4815(12), P–C5 1.894(5), P–N1 1.776(4), O6–H1 2.40499(10), N1–P–W 110.94(14), N1–P–C5 103.78(19), C5–P–W 127.57(15). | ||
| Entry | δ(1H)/ppm | δ(15N)/ppm | |Δδ(15N)|/ppm | ||
|---|---|---|---|---|---|
| C2–H | P–H | N–CH3 | N–P | ||
| a “–Cr” denotes the respective pentacarbonylchromium(0) complexes, obtained similarly, as explained in the ESI.† | |||||
| 1 32 | 6.41 | — | −215.2 | −183.9 | 31.3 |
| 11a | 9.12 | 9.41 | −205.0 | −200.3 | 4.7 |
| 11b | 7.26 | 8.84 | −203.1 | −194.6 | 8.5 |
| 11c | 7.27 | 8.84 | −204.2 | −195.7 | 8.5 |
| 12 | 8.71 | — | −202.4 | −191.4 | 11.0 |
| 1-Cr 32 , | 6.45 | — | −215.2 | −177.7 | 37.5 |
| 11a-Cr | 9.11 | 8.94 | −205.8 | −200.3 | 5.5 |
| 11c-Cr | 7.20 | 8.34 | −204.2 | −195.7 | 8.5 |
| 12-Cr | 8.66 | — | −203.1 | −191.4 | 11.7 |
When the reaction of complex 1 with Brookhart's acid37 [H(OEt2)2][BArF4] was performed (Scheme 5), the resulting complex cation was even more separated from the anion, namely tetrakis[3,5-bis(trifluoromethyl)phenyl]borate. In this case, the phosphorus resonance of 11b was at 77.7 ppm (1JP,H = 353 Hz, 1JW,P = 284 Hz), further downfield-shifted compared to 11a. The distinct solvent separation became clear from the 1H NMR spectrum as a strong highfield-shifted C2H resonance was detected thus providing strong evidence for the loss of the hydrogen bond of the anion to the C2H proton. The molecular structure of 11b was confirmed by single-crystal X-ray diffraction analysis (Fig. 6).
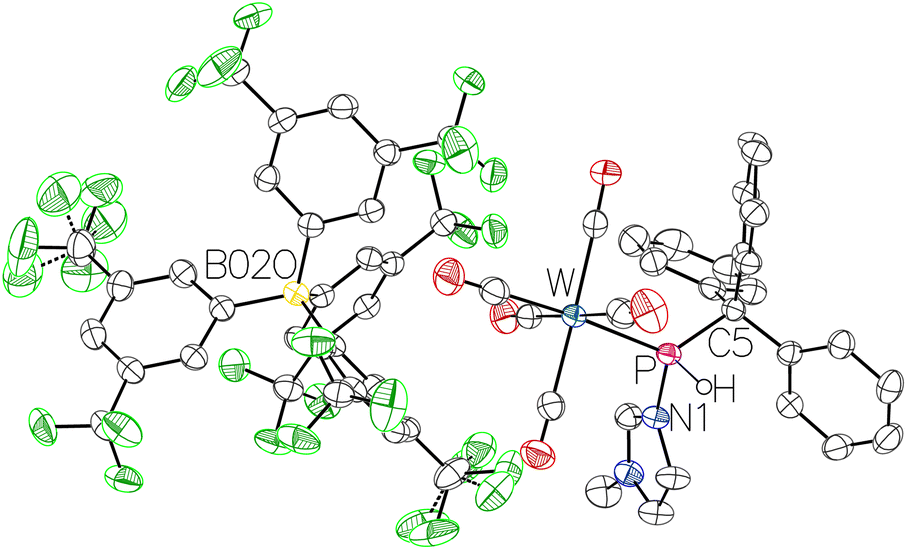 | ||
| Fig. 6 Molecular structure of 11b. Thermal ellipsoids are set at 50% probability and solvent molecules and hydrogen atoms are omitted for clarity except for the one bound to phosphorus. Selected bond lengths/Å and bond angles/°: W–P 2.4611(4), P–N1 1.7773(14), P–C5 1.9239(16), N1–P–W 113.30(5), N1–P–C5 102.46(7), C5–P–W 130.29(5). | ||
To garner further support for the influence of the anion on the cationic part and, hence, the deshielding effects, complex 1 was treated with Krossing's acid [H(OEt2)2][Al{OC(CF3)3}4],38 yielding the corresponding aluminate salt 11c (Scheme 5). As anticipated, the tetrakis(nonafluoro-tert-butoxy)aluminate ([AlORF4]−) did not cause a significant change in the 31P or 1H NMR parameters of 11c (77.8 ppm, 1JP,H = 354 Hz, 1JW,P = 284 Hz) compared to 11b. To experimentally address the bonding situation within the N-methylimidazole donor, gradient-enhanced (ge) 2D NMR 1H,15N heteronuclear multiple bond correlation (HMBC) experiments were conducted.
The difference of the 15N NMR chemical shifts of N1 and N3 in complexes 11a–c is small compared to complex 1 (Table 1), suggesting a significant delocalisation of the positive charge within the imidazolium ring of complexes 11a–c as expected. Interestingly, the 15N NMR chemical shifts of the nitrogen nuclei of 11a were most similar, supposing a stronger stabilisation of the positive charge in the imidazolium ring due to the coordination of the trifluoromethanesulfonate to the C2H proton. The molecular structure of complex 11c was also confirmed by single crystal X-ray diffraction analysis (see ESI Fig. S108†), but the data quality does not justify any further discussion of structural parameters.
As phosphanylimidazolium complexes 11a–c can also be formally described as N-methylimidazole-to-phosphenium complex adducts 11data–c (Fig. 7), we became interested in a more in-depth theoretical P–N bonding analysis. Experimentally, the description of 11data–c, implying a weak P–N bond interaction, was supported by the finding that the mass spectrum of complex 11b showed the P–H substituted phosphenium complex (m/z 599.0) as the base peak, providing first evidence for the ease of P–N bond cleavage under mass spectrometry conditions.
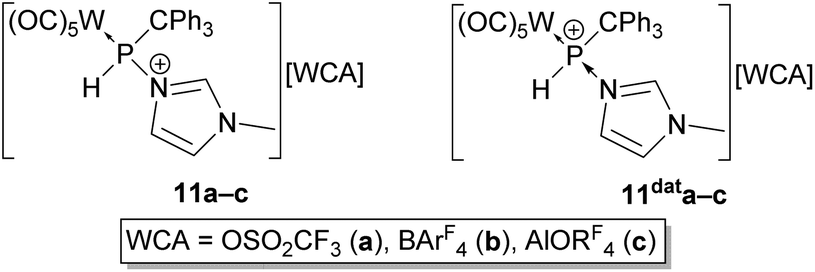 | ||
| Fig. 7 Canonical structures of complexes 11a–c, including the formal representation as N-methylimidazole-to-phosphenium complex adducts 11data–c. | ||
Two model P–H substituted phosphenium complex adducts [tBu-PH(L)–W(CO)5]+ were analysed by quantum chemical calculations. The profile of the Laplacian of the electron density along the central part of the L–P bond path has been shown to have diagnostic character to typify its dative or covalent bonding nature.32,39 In the case of neutral 3a′ (L: MeO−) two valence–shell charge concentration (VSCC) bands (Fig. 8), corresponding to the two atoms engaged in the bond, appear within the basin of the donor atom (O) delimited by the bond critical point (BCP). Within the framework of the quantum theory of atoms-in-molecules (AIM),40 a BCP is a point between two atoms where the gradient of the electron density with respect to the spatial coordinates is zero, indicating a minimum in electron density along the bond path. The positive and relatively large value of the Laplacian at the BCP (∇2ρ = 13.43 e Å−5) points to an essentially covalent nature of the P–OMe bond in 3a′.
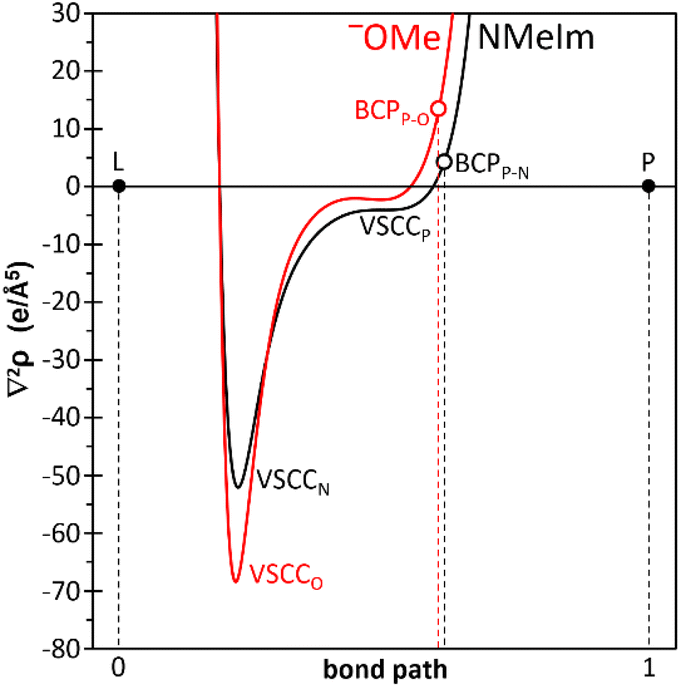 | ||
| Fig. 8 Computed [B3LYP-D3/def2-TZVPP(ecp)//B3LYP-D3/def2-TZVP(ecp)] variation of the Laplacian of electron density ∇2ρ for 3a′ (red) and 11′+ (black) along the L–P bond path. | ||
Similarly, in case of model cation 11′+ (L: N-MeIm), the two VSCC bands are located at the basin of the N donor atom (τVSCC = 0.0423), but the very decreased positive value of the Laplacian of the electron density at the BCP (∇2ρ = 4.21 e Å−5) is indicative of the mostly dative character of the N → P bond, thus paralleling the reported behaviour for phosphinidene complex adduct 1,32 and confirming the best description as 11data–c (Fig. 8) for products arising from protonation with super-strong acids having WCAs.
Facile P–H deprotonation
The P-bound proton in complexes 11a–c is expected to have a highly acidic character due to the proposed significant phosphenium character. This should be very pronounced for complexes 11b,c since the anions display “innocent” behaviour and no additional interaction with the C2–H proton was detected spectroscopically by NMR. Interestingly, when complex 11c was dissolved in tetrahydrofuran or tetrahydrofuran-d8 an immediate colour change from colourless to yellow was observed, and the 1H and 31P NMR spectra showed the reformation of the N-methylimidazole-to-phosphinidene complex adduct 1, thus revealing a surprisingly facile deprotonation at the P-centre by the very weak base THF (Scheme 6).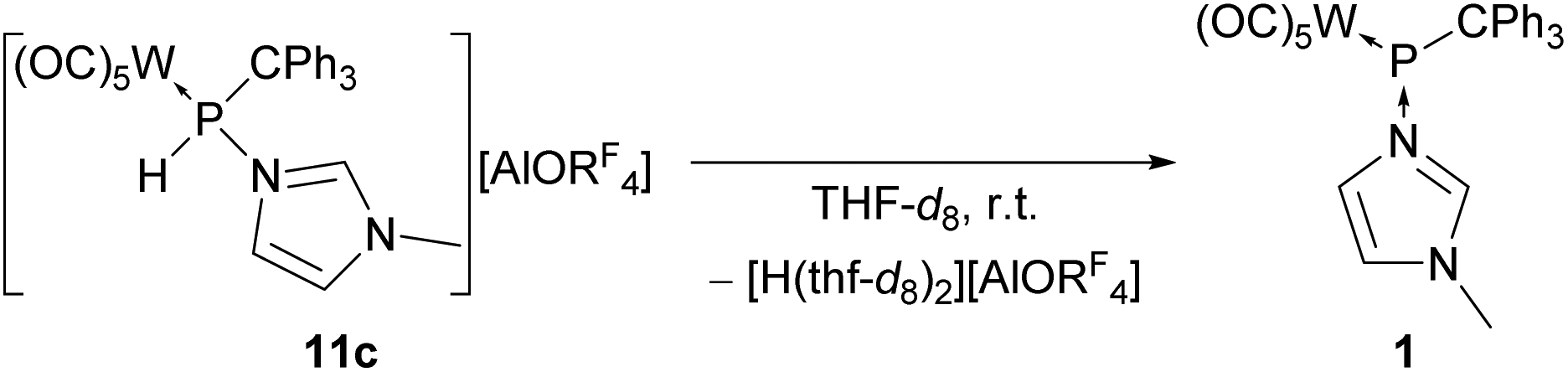 | ||
| Scheme 6 Deprotonation of the phosphanylimidazolium complex 11c in THF-d8 to complex 1 and the proposed formation of [H(thf-d8)2][AlORF4]. | ||
Since no polymerization of the solvent was observed, and a slightly broadened signal at 7.62 ppm with an integral of 1 proton appeared in the 1H NMR spectrum (see ESI Fig. S74†) when dissolved in THF-d8, we assume that [H(thf-d8)2][AlORF4] was formed, especially as the 1H NMR data are in good accordance with those reported for the respective non-deuterated acid by Krossing when dichloromethane-d2 was used as solvent.38
The gas-phase acidity, in terms of Gibbs free energy ΔGacid (kcal mol−1), was computed as reported elsewhere,41 uncovering a remarkable value for 11′+ (244.4), slightly larger than that of Me2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OH+ (188.9) and Me3PH+ (222.8), but significantly lower than that of CF3SO3H (293.6; 299.542), differently substituted imidazoles (299.4–343.9)43 or CH3COOH (341.5; 341.144).
OH+ (188.9) and Me3PH+ (222.8), but significantly lower than that of CF3SO3H (293.6; 299.542), differently substituted imidazoles (299.4–343.9)43 or CH3COOH (341.5; 341.144).
P-Alkylation of the N-MeIm-to-phosphinidene complex adduct
To get access to more inert formal N-methylimidazole-to-phosphenium complex adducts, the P-alkylation was examined to avoid unwanted acid–base reactions. Complex 1 reacted with methyl trifluoromethanesulfonate in a clean fashion (Scheme 7), and the 31P NMR resonance signal of the selectively formed N-methylimidazole-to-methyl(triphenylmethyl)phosphenium complex adduct 12 was observed at 116.4 ppm (1JW,P = 274 Hz). Complex 12 was easily isolated via crystallization from a diethylether/THF mixture at −40 °C.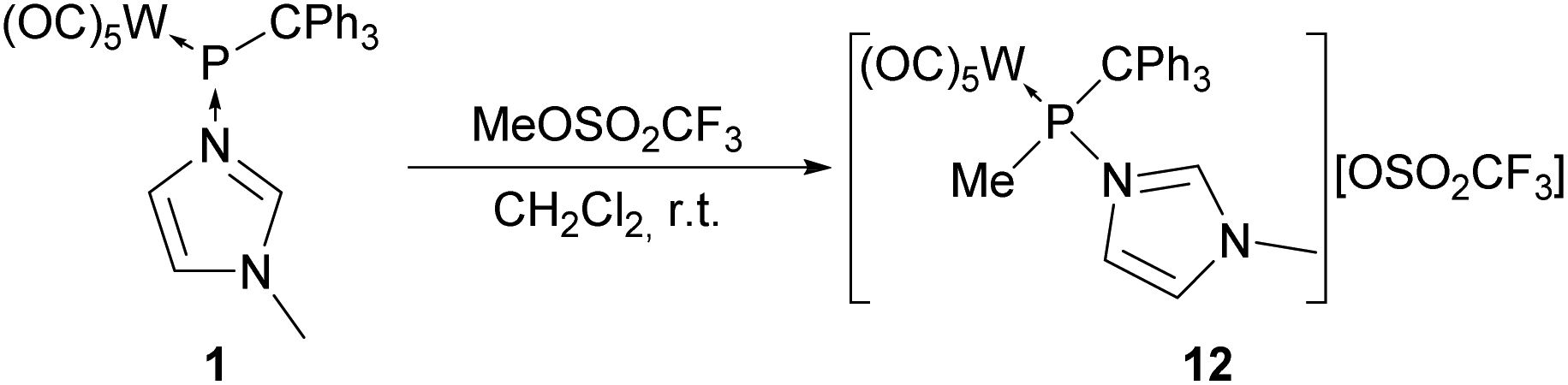 | ||
| Scheme 7 Synthesis of methylphosphanylimidazolium complex 12. | ||
The differences in the 15N NMR chemical shifts of the two nitrogen nuclei in complex 12 are more than doubled (Δδ(15N) = 11.0 ppm) compared to those of complexes 11a–c again indicating weaker P–N interactions resulting in an increased phosphenium character. Nevertheless, the Δδ(15N) value is still relatively small compared to the one of complex 1, suggesting that the positive charge still is significantly located within the N-methylimidazole moiety. These observations are in good accordance with the Laplacian of the electron density ∇2ρP–L (11′+ (4.21 e Å−5) > 12′+ (3.74 e Å−5) > 1′ (3.15 e Å−5)32) and with the (natural) charge transfer qN(L) from the N-MeIm ligand (11′+ (0.409 e) > 12′+ (0.386 e) > 1′ (0.320 e)). Noteworthy, this trend is not in line with the differences in natural charges at the N-MeIm ligand qN(N3)–qN(N1) (1′ (0.207 e) ≪ 11′+ (0.280 e) < 12′+ (0.293 e)).
Unexpectedly, the 1H and 13C{1H} NMR spectra of complex 12 revealed a more complicated pattern of resonance signals for the triphenylmethyl group in which the nuclei of every phenyl group were magnetically different, thus indicating a hindered rotation at the P–C bond. Particularly, the ortho-CH protons differed significantly for each phenyl group with broad resonance signals at 7.64, 7.30 and 6.61 ppm. This indicates the increased steric hindrance at phosphorus due to the P-bound methyl group. Again a weak P–N interaction was indicated by the mass spectrum of 12 as the phosphenium complex (m/z 613.0) and the free phosphenium cation [P(CPh3)Me]+ (m/z 289.1) were detected.
The greater stability of the P-Me derivative 12 enabled to grow single crystals suitable for X-ray diffraction analysis (Fig. 9).
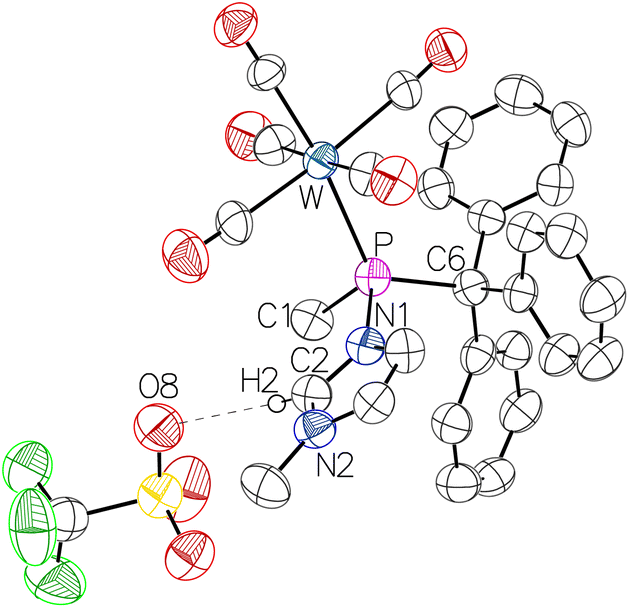 | ||
| Fig. 9 Molecular structure of 12. Thermal ellipsoids are set at 50% probability and solvent molecules and hydrogen atoms are omitted for clarity except for the one involved in hydrogen bonding. Selected bond lengths/Å and bond angles/°: W–P 2.4996(19), P–N1 1.801(6), P–C1 1.837(7), P–C6 1.947(7), O8–H2 2.32189(10), N1–C2 1.335(9), N2–C2 1.321(9), N1–P–W 109.4(2), N1–P–C1 97.1(3), N1–P–C6 104.6(3), C1–P–W 111.5(3), C1–P–C6 106.3(3), C6–P–W 124.3(2). | ||
As expected, the molecular structure of 12 revealed the hydrogen bond interaction between the anion and the C2-bound hydrogen (O8–H2 distance: 2.322 Å). The P1–N1 distance of 1.801(6) Å is slightly shorter than the one in complex 1 (1.813 Å)32 indicating a slightly stronger P–N bond and, hence, a less dative (more covalent) character. However, the N1–C2 and N2–C2 bond lengths are similar, indicating a considerably stronger covalency (higher ∇2ρP–N) of the P–N bond together with a pronounced contribution of a delocalised positive charge within the imidazole ring compared to complex 1.
Conclusions
We have demonstrated that the N-methylimidazole-to-phosphinidene complex adduct 1 undergoes 1,1-addition reactions with weak Brønsted–Lowry acids, such as water and alcohols, by displacing the donor entity. When complex 1 was treated with stronger acids HX, such as trifluoroacetic acid and hydrogen chloride, two equivalents of the acid were needed to complete the 1,1-addition reaction, as the released N-methylimidazole formed rapidly the respective N-methylimidazolium salts. To avoid substitution and achieve exclusively the P-protonation of complex 1, reactions with super-strong acids were performed, having weakly coordinating anions (WCA), i.e., trifluoromethanesulfonic acid, Brookhart's acid [H(OEt2)2][BArF4] and Krossing's acid [H(OEt2)2][AlORF4]. In all cases, P-protonation and salt formation were achieved, but the anion–cation interaction was different. Remarkably, an increased acidity of the P-bound proton was disclosed in the case of the aluminate salt which was analysed by theory. In total, a novel access to donor-stabilised phosphenium complexes bearing a P–H bond has been developed, including an in-depth analysis of the P–N bonding.Data availability
Synthetic details and analytical data, including depictions of all spectra and detailed accounts of the methods applied are documented in the ESI.†Author contributions
DB planned and carried out the experiments and collected and evaluated analytical data. GS measured and evaluated the crystallographic data. AEF carried out the theoretical study and the discussion. RS provided the concept and the supervision of the investigation. DB, AEF and RS wrote the manuscript. The ESI† was prepared by DB, AEF and RS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (STR 411/45-1) and the University of Bonn. We are grateful to Prof. Dr A. C. Filippou and Prof. Dr S. Höger for the use of X-ray facilities. A. E. F. wishes to acknowledge the computational resources used at the computation centre at Servicio de Cálculo Científico (SCC – University of Murcia).References
-
(a) R. Allmann, Chem. Ber., 1966, 99, 1332 CrossRef CAS
; (b) R. Allmann, Angew. Chem., Int. Ed. Engl., 1965, 4, 150 CrossRef
- M. G. Thomas, C. W. Schultz and R. W. Parry, Inorg. Chem., 1977, 16, 994 CrossRef CAS
- S. Fleming, M. K. Lupton and K. Jekot, Inorg. Chem., 1972, 11, 2534 CrossRef CAS
- B. E. Maryanoff and R. O. Hutchins, J. Org. Chem., 1972, 37, 3475 CrossRef CAS
-
(a) P. Friedrich, G. Huttner, J. Luber and A. Schmidpeter, Chem. Ber., 1978, 111, 1558 CrossRef CAS
- I. A. Litvinov, V. A. Naumov, T. V. Griaznova, A. N. Pudovik and A. M. Kibardin, Dokl. Akad. Nauk SSSR, 1990, 312, 3159 Search PubMed
- M. K. Denk, S. Gupta and A. J. Lough, Eur. J. Inorg. Chem., 1999, 1999, 41 CrossRef
- D. Gudat, A. Haghverdi, H. Hupfer and M. Nieger, Chem. – Eur. J., 2000, 6, 3414 CrossRef CAS
-
(a) S. Pohl, Chem. Ber., 1979, 112, 3159 CrossRef CAS
- M. Olaru, S. Mebs and J. Beckmann, Angew. Chem., Int. Ed., 2021, 60, 19133 CrossRef CAS PubMed
-
(a) M. Yoshifuji, Sci. Synth., 2009, 42, 63 Search PubMed
- D. Gudat, E. Niecke, B. Krebs and M. Dartmann, Chimia, 1985, 39, 277 CAS
- D. Gudat, M. Nieger and E. Niecke, J. Chem. Soc., Dalton Trans., 1989, 693 RSC
- S. Loss, C. Widauer and H. Grützmacher, Angew. Chem., Int. Ed., 1999, 38, 3329 CrossRef CAS PubMed
-
(a) S. S. Chitnis and N. Burford, Dalton Trans., 2015, 44, 17 RSC
- L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606 CrossRef CAS
- D. Gudat, A. Haghverdi and M. Nieger, J. Organomet. Chem., 2001, 617–618, 383 CrossRef CAS
- A. E. R. Watson, M. J. Grant, P. D. Boyle, P. J. Ragogna and J. B. Gilroy, Inorg. Chem., 2022, 61, 18719 CrossRef CAS PubMed
-
(a) P. Ai, C. Gourlaouen, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2016, 55, 1219 CrossRef CAS PubMed
- S. M. Rocklage, R. R. Schrock, M. R. Churchill and H. J. Wasserman, Organometallics, 1982, 1, 1332 CrossRef CAS
-
(a)
R. Schmitt, PhD thesis, University of Würzburg, 2005
- R. Menye-Biyogo, F. Delpech, A. Castel, V. Pimienta, H. Gornitzka and P. Rivière, Organometallics, 2007, 26, 5091 CrossRef CAS
- M. P. Duffy, L. Y. Ting, L. Nicholls, Y. Li, R. Ganguly and F. Mathey, Organometallics, 2012, 31, 2936 CrossRef CAS
- L. L. Liu, D. A. Ruiz, F. Dahcheh and G. Bertrand, Chem. Commun., 2015, 51, 12732 RSC
-
(a) K. Lammertsma, Top. Curr. Chem., 2003, 229, 95 CrossRef CAS
- A. Marinetti, F. Mathey, J. Fischer and A. Mitschler, J. Am. Chem. Soc., 1982, 104, 4484 CrossRef CAS
- F. Mercier, B. Deschamps and F. Mathey, J. Am. Chem. Soc., 1989, 111, 9098 CrossRef CAS
-
(a) R. Streubel, Top. Curr. Chem., 2002, 223, 91 CrossRef
- M. L. G. Borst, R. E. Bulo, C. W. Winkel, D. J. Gibney, A. W. Ehlers, M. Schakel, M. Lutz, A. L. Spek and K. Lammertsma, J. Am. Chem. Soc., 2005, 127, 5800 CrossRef CAS PubMed
-
(a) A. Schmer, P. Junker, A. Espinosa Ferao and R. Streubel, Acc. Chem. Res., 2021, 54, 1754 CrossRef CAS PubMed
- I. Kalinina and F. Mathey, Organometallics, 2006, 25, 5031 CrossRef CAS
- D. Biskup, G. Schnakenburg, R. T. Boeré, A. Espinosa Ferao and R. K. Streubel, Nat. Commun., 2023, 14, 6456 CrossRef CAS PubMed
- D. Biskup, G. Schnakenburg, R. T. Boeré, A. Espinosa Ferao and R. Streubel, Dalton Trans., 2023, 52, 13781 RSC
- R. Streubel, A. W. Kyri, L. Duan and G. Schnakenburg, Dalton Trans., 2014, 43, 2088 RSC
- R. Streubel, A. Schmer, A. W. Kyri and G. Schnakenburg, Organometallics, 2017, 36, 1488 CrossRef CAS
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982 CrossRef CAS PubMed
- M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 1992, 11, 3920 CrossRef CAS
- I. Krossing and A. Reisinger, Eur. J. Inorg. Chem., 2005, 2005, 1979 CrossRef
-
(a) A. Espinosa Ferao, A. García Alcaraz, S. Zaragoza Noguera and R. Streubel, Inorg. Chem., 2020, 59, 12829 CrossRef CAS PubMed
-
(a)
R. F. W. Bader, Atoms in molecules. A quantum theory, Oxford University Press, Oxford, 1990 CrossRef
-
(a) I. A. Topol, G. J. Tawa, R. A. Caldwell, M. A. Eissenstat and S. K. Burt, J. Phys. Chem. A, 2000, 104, 9619 CrossRef CAS
- I. A. Koppel, R. W. Taft, F. Anvia, S.-Z. Zhu, L.-Q. Hu, K.-S. Sung, D. D. DesMarteau, L. M. Yagupolskii and Y. L. Yagupolskii, J. Am. Chem. Soc., 1994, 116, 3047 CrossRef CAS
- N. Wang and J. K. Lee, J. Org. Chem., 2019, 84, 14593 CrossRef CAS PubMed
- S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin and W. G. Mallard, J. Phys. Chem. Ref. Data, 1988, 17(Suppl. 1) CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2302946–2302953. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03869f |
| This journal is © The Royal Society of Chemistry 2024 |