
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitching of magnetic properties by topotactic reaction in a 1D CN-bridged Ni(II)–Nb(IV) system†
Michał
Heczko
 and
Beata
Nowicka
*
and
Beata
Nowicka
*
Faculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Kraków, Poland. E-mail: beata.nowicka@uj.edu.pl
First published on 8th February 2024
Abstract
Two 1D CN-bridged assemblies: the nearly straight Li2[Ni(cyclam)][Nb(CN)8]·7.5H2O (1) chains and the zigzag-shaped Li2[Ni(cyclam)][Nb(CN)8]·2H2O (2) chains, are obtained in the reaction between [Ni(cyclam)]2+ and [Nb(CN)8]4− in warm concentrated LiCl water solution. Both compounds are composed of alternating bimetallic Ni(II)–Nb(IV) chains and contain incorporated lithium cations, which compensate the negative charge of the coordination skeleton. The straight chain 1 (Ni–Nb–Ni angle = 153.2°) can be reversibly dehydrated under dry nitrogen flow at room temperature to an intermediate dihydrate phase 1d and further transformed to the zigzag-shaped chain 2 (Ni–Nb–Ni angle = 86.6°) by annealing at 150 °C. The process can be reversed by exposure to high humidity at room temperature, upon which 2 is converted back to 1. This water sorption-induced breathing effect is accompanied by changes in magnetic properties, most notably reflected in different values of saturation magnetization and critical field of metamagnetic transition, which indicate that both intra- and inter-chain interactions are affected by the structure reorganization.
Introduction
Cyano-bridged coordination polymers constitute an important group of molecular magnetic materials. The CN ligand mediates relatively strong magnetic interactions and simultaneously allows the formation of versatile structures of different topologies, incorporating organic parts, which may serve as platforms for the introduction of additional properties.1–5 The flexibility of CN-bridged frameworks enables the design of non-rigid structures, in which magnetic properties can be modified by the inclusion of guest molecules or ions. Octacyanoniobate(IV) ion has shown great potential in the construction of multifunctional bimetallic assemblies.6,7 The 3D Nb-based networks with high connectivity show relatively high magnetic ordering temperatures,8,9 with the record value of TC = 210 K among the octacyanometallate-based assemblies for the VII/III–NbIV 3D network.9 Some networks with additional organic ligands were found to undergo structural changes upon reversible dehydration, which enables post-synthetic modifications and tuning of TC.10,11 Several [Nb(CN)8]4− – based compounds show marked magnetocaloric effect,12 including very rare rotating magnetocaloric effect, connected with high magnetic anisotropy.13,14 There are also examples of chiral networks, which exhibit interesting magneto-optical cross effects, including magnetically enhanced second harmonic generation.15 Notably, there is only one report on an octacyanidoniobate(IV)-based 1D coordination polymer.16 It was obtained by the combination of [Nb(CN)8]4− anions with Mn2+ cations coordinated by a flat pentadentate ligand and it shows the topology of a chain with pendant arms. More 1D assemblies are known for analogous tetravalent octacyanidometallates of group 6 metals. They show different topologies, including ladder,17 vertex-sharing squares,18,19 or vertex-sharing trigonal bipyramids.20 However, since [M(CN)8]4− (M = MoIV, WIV) ions are diamagnetic, these chain structures do not show magnetic interactions through CN-bridges, which in many cases lead to interesting phenomena including single chain magnet behaviour.21,22We have shown before the possibility of manipulating the dimensionality and topology of the CN-bridged assemblies based on [Ni(cyclam)]2+ and tetravalent [M(CN)8]4− ions (M = MoIV, WIV) by the addition of LiCl and changing reaction conditions. For the WIV-based precursor we have obtained as many as five coordination polymers of different topology and dimensionality, which include a 3D diamond-like network [Ni(cyclam)]2[W(CN)8]·16H2O, a microporous 2D honeycomb-like network incorporating Li+ ions Li2[Ni(cyclam)]3[W(CN)8]2·24H2O, a 3D two-fold interpenetrating network [Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·11H2O formed as a result of rearrangement of the building blocks, as well as 1D linear and zigzag-shaped chains.23 MoIV-based analogues comprise 3D and 2D structures24 and show interesting photomagnetic properties modified by water sorption.23 By replacing the diamagnetic octacyanidometallates of MoIV and WIV with paramagnetic [Nb(CN)8]4− ions we have constructed a series of 2D honeycomb-like frameworks with tuneable magnetic properties by incorporation of different guest cations.25,26 Here we present the first alternating bimetallic chain composed of octacyanidoniobate(IV) and Ni(II) ions, obtained by the synthetic strategy based on the addition of non-coordinating s-block cations at elevated temperature.23
Experimental section
Materials
The [Ni(cyclam)(NO3)2] and K4[Nb(CN)8]·2H2O precursor complexes were synthesized according to literature methods.27 The lithium chloride salt and all solvents were commercially available and used as supplied.Synthesis of compounds
Structure determination
The single crystal X-ray diffraction measurements for 1 and 2 were performed on a Bruker D8 Quest diffractometer equipped with a Mo Kα radiation source (λ = 0.71073 Å). The structures were solved by direct methods using SHELXT and the refinement was performed using SHELXL.28 All the non-hydrogen atoms were refined anisotropically. The C–H and N–H hydrogen atoms were placed in idealized positions and refined isotropically using the riding model with Uiso(H) equal to 1.2Ueq for the C or N atoms. The H-atoms of crystallization water were found from the difference electron density map. Graphical representations of the structures were prepared with the Mercury CSD 4.3.1 software.29 The analysis of the coordination polyhedra was performed using SHAPE 2.1.30 The crystallographic data are presented in Table 1. CCDC deposition numbers: 2280169 (1), 2280170 (2).†| Compound | 1 | 2 |
|---|---|---|
| Empirical formula | C18H36Li2N12NbNiO6 | C18H28Li2N12NbNiO2 |
| FW | 682.09 | 610.02 |
| T (K) | 100 | 100 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P2/n | C2/c |
| a (Å) | 10.2590(8) | 12.2706(4) |
| b (Å) | 9.6405(7) | 14.4329(5) |
| c (Å) | 15.8896(12) | 14.6110(5) |
| α (°) | 90 | 90 |
| β (°) | 105.385(2) | 94.782(1) |
| γ (°) | 90 | 90 |
| V (Å3) | 1515.2(2) | 2578.61(15) |
| Z | 2 | 4 |
| V/Z (Å3) | 757.6 | 644.7 |
| ρ calc. (Mg m−3) | 1.495 | 1.571 |
| μ (mm−1) | 1.051 | 1.214 |
| F(000) | 702 | 1244 |
| Crystal size (mm) | 0.17 × 0.07 × 0.03 | 0.40 × 0.04 × 0.03 |
| θ range (°) | 2.50–27.5 | 2.50–26.4 |
| Reflections collected | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 012 012 |
15907 |
| Independent | 3483 | 2641 |
| Observed | 3293 | 2455 |
| Parameters | 201 | 171 |
| R int | 0.030 | 0.026 |
| GOF on F2 | 1.206 | 1.186 |
| R 1 [I > 2σ(I)] | 0.0293 | 0.0230 |
| wR2 (all data) | 0.0579 | 0.0476 |
| CCDC deposition number | 2280169 | 2280170 |
Physical measurements
Powder X-ray diffraction data were collected on a Bruker D8 Advance Eco diffractometer equipped with a Cu Kα radiation source (λ = 1.541874 Å), in the Debye–Scherrer geometry. The microcrystalline samples were sealed in glass capillaries and measured in the 5–50° 2θ range at room temperature. The reference powder patterns from SC-XRD measurements were generated using Mercury CSD 4.3.1 software.29 Elemental analyses of CHN were performed on an ELEMENTAR Vario Micro Cube CHNS analyser. IR spectra were collected on a Bruker Alpha II spectrometer with a diamond ATR add-on. The thermogravimetric data were collected using a Netzsch TG 209 F1 Libra apparatus. The water sorption/desorption processes were characterized by the dynamic vapor sorption method using the SMS DVS Resolution apparatus. The isotherm was measured in a 0–90% relative humidity range at a temperature of 25 °C. Every measurement step was performed until a stable mass was achieved. Magnetic susceptibility measurements were performed on a Quantum Design MPMS-3 Evercool magnetometer in magnetic fields up to 70 kOe. The experimental data were corrected for diamagnetism of the sample and the sample holder. The magnetic data were fitted using the PHI programme.31Results and discussion
Synthesis strategy, materials stability, and sorption properties
The combination of the cationic [Ni(cyclam)]2+ building block with polycyanometallate anions led to the formation of a wide group of CN-bridged coordination polymers characterized by diverse network dimensionality and topology.23–26 Moreover, the final product characteristics often depended on the reaction conditions including type of solvent, temperature, presence of additional ions, and building block ratio. We have previously proved that it is possible to obtain as many as six various coordination systems based on [Ni(cyclam)]2+ and [W(CN)8]4− by the manipulation of the above factors.23 The reaction between [Ni(cyclam)]2+ and [Nb(CN)8]4− performed in water solution leads to the formation of a neutral coordination network of the formula [Ni(cyclam)]2[Nb(CN)8]·nH2O,32 which is the most thermodynamically stable phase. When the reaction is carried out in the presence of additional electrolytes at room temperature, the incorporation of appropriate cations into the coordination framework was observed. It resulted in the formation of two-dimensional networks, characterized by the honeycomb-like topology, of the general formula Mx[Ni(cyclam)]3[Nb(CN)8]2·nH2O (x = 2: M = Li+, Na+, NH4+; x = 1: M = Mg2+, Ca2+, Sr2+, Ba2+).25,26 However, these compounds upon contact with water recrystallize to the neutral Ni2Nb network.32In order to construct a 1D NiII–NbIV coordination polymer we used the slightly modified synthetic procedure by which an analogous linear NiII–WIV chain was obtained.23 The reaction temperature was kept below 45 °C in order to avoid the decomposition of the [Nb(CN)8]4− complex, which is less robust than [W(CN)8]4−. The 1420-fold excess of LiCl was used. When the warm solutions of the building blocks were combined and allowed to cool, two types of crystals were formed: plates of the Li2[Ni(cyclam)][Nb(CN)8]·7.5H2O straight chain (1) and needles of the Li2[Ni(cyclam)][Nb(CN)8]·2H2O zigzag chain (2). If the reaction mixture was kept at 35 °C for a week the needle-shaped crystals of 2 recrystallized into well-formed plate-shaped crystals of 1. The purity of the final product was confirmed by powder X-ray diffraction measurement on the sample in mother liquor (Fig. S1†). The recrystallization process suggests that 1 is the thermodynamically favoured phase. The crystals of 1 are stable under mother liquor and in concentrated lithium chloride solution. Upon even short contact with water, the crystals collapse into a fine powder and the compound decomposes. Therefore, in order to remove the remains of highly hygroscopic LiCl, after filtration the crystals of 1 were washed with a small amount of cold (4 °C) tetrahydrofuran, in which LiCl is soluble, and then with cold acetone. The crystals of 1 treated that way are stable under ambient temperature and humidity conditions, as shown by PXRD (Fig. S1†).
The water sorption/desorption measurements, performed using the dynamic vapor sorption gravimetric method (DVS), indicate that the fully hydrated phase containing 7.5H2O per formula unit is stable in the range of 90–2% RH (Fig. 1). Dehydration takes place under dry nitrogen flow at room temperature which leads to phase 1d containing 2H2O per formula unit. The rehydration of 1d occurs in two steps with a short plateau between 20 and 30% RH, which corresponds to the phase containing about 5H2O. At 90% RH the original phase 1 is reproduced, which was confirmed by PXRD (Fig. S1†). Moreover, 1d can also be obtained by keeping 1 under vacuum for about 15 hours, which was confirmed by magnetic measurements. The structure of 1d could not be discerned from the PXRD data (Fig. S1†), however, the shift of most peaks to higher 2θ values suggests decrease in the crystallographic periods, which can be expected upon loss of crystallisation water in the non-rigid coordination polymer structure.33,34 The thermogravimetric analysis of 1 was performed with an initial 6-hour isothermal step, during which the sample was kept under dry nitrogen flow at 25 °C (Fig. S2†). The mass loss observed in this step corroborates with the DVS measurement results. The thermal decomposition marked by a sharp decrease in mass begins at about 200 °C. The slight apparent mass loss at the beginning of the temperature rise step can be attributed to the balance drift.
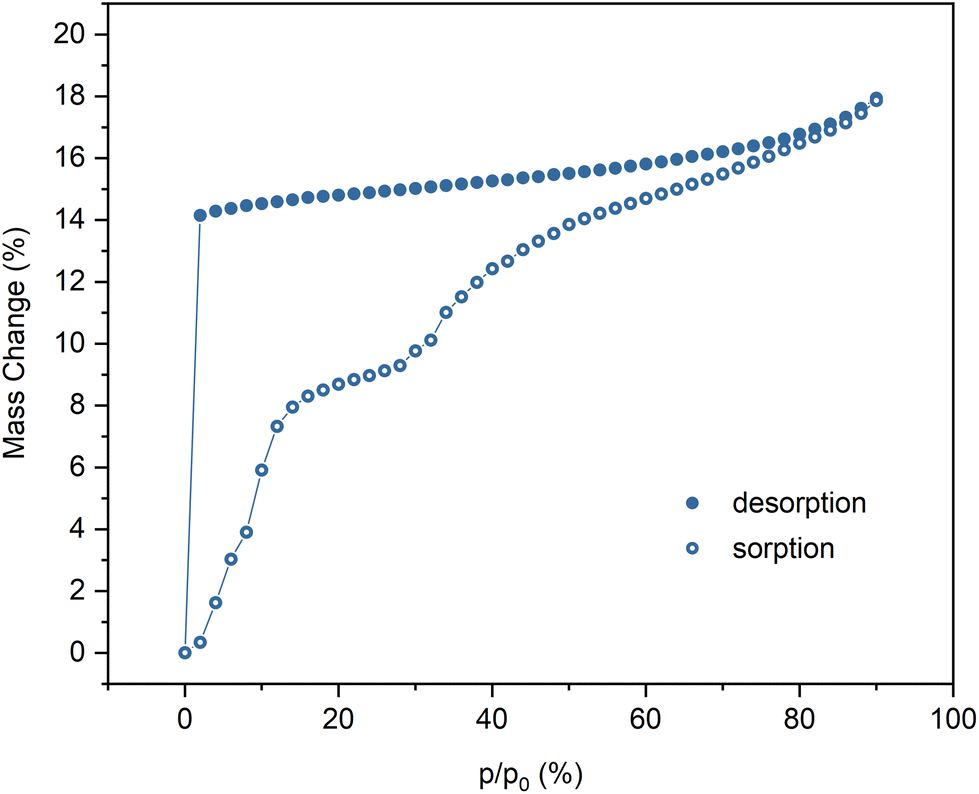 | ||
| Fig. 1 Water sorption isotherm for 1 at 25 °C. | ||
In contrast to the straight chain compound 1, the zigzag-shaped compound 2 could not be obtained in pure form from solution reaction. The synthesis of an isotypic zigzag-shaped chain based on [W(CN)8]4− ions23 was performed at 105 °C. This synthetic procedure could not be adopted, since such high temperature causes decomposition of the more fragile [Nb(CN)8]4− ion. However, pure compound 2 in the powder form can be obtained in a topotactic solid-state reaction. Thermal treatment of 1 or 1d at 150 °C for one hour results in complete conversion to structure 2, which was confirmed by PXRD (Fig. S1†). Compound 2 in both crystalline and powder form is extremely sensitive to contact with liquid water, which causes immediate decomposition. However, it remains unchanged for a few days under ambient temperature and humidity below 50% and can be kept for longer periods of time in a moisture-free atmosphere. Compound 2 can be transformed back to 1 through rehydration by exposure to 90% humidity for about 1 day. However, the transition is accompanied by partial recrystallization to the neutral Ni2Nb polymer.32 The thermogravimetric analysis of 2 (Fig. S3†) reflects the behaviour observed for 1 at higher temperatures, with decomposition beginning at 200 °C.
Structure description
Li2[Ni(cyclam)][Nb(CN)8]·7.5H2O (1) crystallizes in the monoclinic system, space group P2/n (Table 1). The asymmetric unit contains half of the [Ni(cyclam)]2+ cation at the centre of inversion, half of the [Nb(CN)8]4− anion at the 2-fold proper rotation axis as well as one Li+ ion, and three water molecules in general positions (Fig. S4†). The niobium ion is coordinated by eight cyanide ligands with the geometry close to an ideal square antiprism (D4d, CShM = 0.114, Table S1†). The nickel ion is coordinated by four N-atoms of the cyclam ligand in equatorial positions and two N-atoms of the bridging CN− ligands in axial positions, with the geometry close to an ideal octahedron (Oh, CShM = 0.154, Table S2†). The [Ni(cyclam)]2+ and [Nb(CN)8]4− moieties are linked through CN-bridges and form 1D coordination chains along the [−101] crystallographic direction (Fig. 2). The bridging CN− ligands are located on the opposite faces of the square antiprism forming the C1–Nb1–C1′ angle of 144.2° (Table S4†). Additionally, the CN-bridges are slightly bent with the Ni1–N1–C1 angle of 162.3°, which results in the Ni–Nb–Ni angle of 153.2° and Ni–Nb separation of 5.42 Å. The negative charge of the coordination chains, which arises from the charge disparity between the building blocks, is compensated by the presence of Li+. The lithium cation is surrounded by two water molecules and two N-atoms of terminal CN− ligands, resulting in slightly distorted tetrahedral geometry (Td, CShM = 0.611, Table S3†). The [Li(H2O)2]+ units link the neighbouring coordination chains into two-dimensional layers in the (020) crystallographic plane (Fig. S5†). The distance between chains within one layer is 7.46 Å, while the distance between the neighbouring layers is 9.64 Å. The closest contact between the Ni and Nb centres from the adjacent chains is 7.78 Å. These values are comparable to those observed in the isotypic structure based on the [W(CN)8]4− building block.23 The layers are bound by hydrogen bonds (Table S5†) between the terminal CN ligands (N4) and the [Li(H2O)2]+ unit (O2), as well as through the crystallization water molecule (O3) bound to the terminal cyanide (N4) and cyclam ammine group (N12). The structure contains six water molecules per formula unit, which were found from the electron density map. However, there are also well-defined solvent-accessible voids, which presumably contain the additional water found in elemental analysis and sorption measurements.35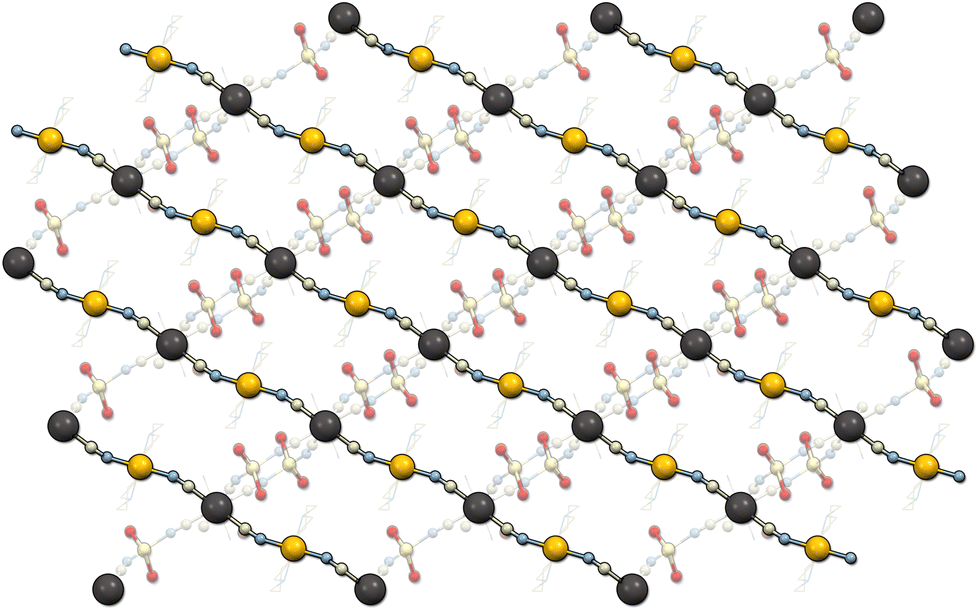 | ||
| Fig. 2 Structure of 1 viewed along the [010] crystallographic direction showing the parallel straight coordination chains (Ni – orange balls, Nb – black balls) linked through the [Li(H2O)2]+ units (shaded out). | ||
Li2[Ni(cyclam)][Nb(CN)8]·2H2O (2) crystallizes in the monoclinic system, space group C2/c (Table 1). The asymmetric unit contains one half of the [Nb(CN)8]4− anion at the 2-fold proper rotation axis, half of the [Ni(cyclam)]2+ cation at the centre of inversion as well as one Li+ ion and one water molecule in general positions (Fig. S6†). In contrast to 1, the geometry of octacyanidoniobate(IV) is closer to an ideal triangular dodecahedron (D2d, CShM = 0.374) rather than square antiprism (Table S1†). The octahedral environment of nickel ion is slightly more distorted than in 1 (Oh, CShM = 0.193). The CN-bridged Ni–Nb coordination chains that extend in the [001] crystallographic direction are distinctly zigzag-shaped (Fig. 3). The bridging CN− ligands are located on the shortest edge of the triangular dodecahedron with the C1–Nb1–C1′ angle of 69.7° (Table S4†). The Ni–Nb–Ni angle within the zigzag chain is larger (86.6°), due to the marked bending of the CN-bridges, in which the Ni1–N1–C1 angle of 156.3° is smaller than in 1. It also results in a slightly shorter Ni–Nb distance of 5.33 Å. Like in 1, two Li+ cations per formula unit are present in the structure to compensate the negative charge of the coordination chain, and each Li+ centre is surrounded by two water molecules and two N atoms of terminal CN ligands. However, since compound 2 contains less crystallization water, the ratio between Li+ and H2O is 1:1. Therefore, two lithium cations share two water molecules and form [Li2(H2O)2]2+ units. As a result, the tetrahedral geometry of Li+ is significantly distorted (Td, CShM = 1.841, Table S3†). Each [Li2(H2O)2]2+ unit is bound to three Nb centres, thus linking the coordination Ni–Nb chains into a 3D network (Fig. S7†). In addition, the water molecules (O1) form hydrogen bonds to the terminal CN ligands (N4) to give connections extending in the [001] direction (Table S6†). The closest distance between the Ni and Nb centres from the neighbouring chains is 7.64 Å, which is shorter by 0.18 Å than in 1.
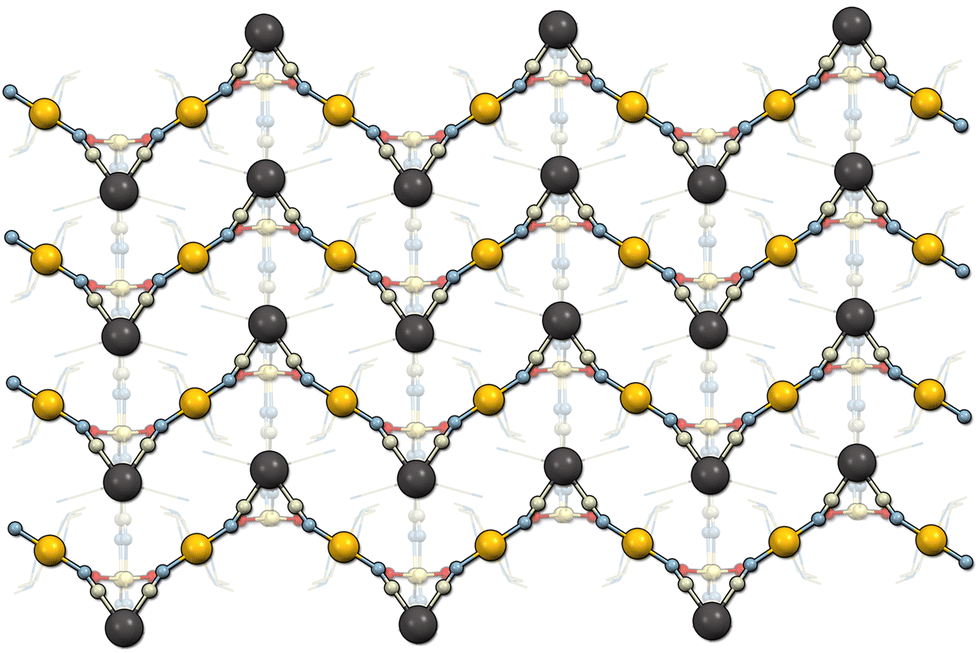 | ||
| Fig. 3 Structure of 2 viewed along the [100] crystallographic direction showing the zigzag-shaped coordination chains. | ||
Magnetic properties
All samples were carefully protected from changing the hydration level during the magnetic measurements. The crystalline sample of 1 was sealed in a glass tube under concentrated lithium chloride solution. Samples of the dehydrated 1d phase were prepared in two ways: sample (i) was dehydrated in the DVS chamber under flow of dry nitrogen until it reached constant mass, then transferred to a glovebox and covered in nujol; sample (ii) was placed in an open glass tube, immobilized with a glass wool pad and dehydrated under vacuum for 1 day then transferred to the magnetometer and kept for additional two hours under vacuum in the magnetometer cavity. The magnetic data collected for both samples of 1d were identical. Sample 2 was prepared by heating sample 1d in an open glass tube at 150 °C for 1 hour. It was then transferred quickly to the magnetometer and kept for half an hour at 100 °C under vacuum in the magnetometer cavity. Detailed magnetic measurements plots are presented in Fig. S8–S10.†The product of magnetic susceptibility and temperature (Fig. 4) in the high-temperature limit reaches 1.49, 1.46, and 1.44 cm3 K mol−1 for 1, 1d, and 2, respectively, which is in good agreement with the value of 1.48 cm3 K mol−1 expected for one NiII (S = 1 and g = 2.1) and one NbIV (S = 1/2 and g = 2.0) per formula unit. The χMT signals for 1 and 2 increase with decreasing temperature reaching the maximum of 2.60 cm3 K mol−1 at 8.2 K (1) and 2.00 cm3 K mol−1 at 6.5 K (2). In the case of 1d, the χMT curve decreases slowly below 75 K and then sharply below 6 K. The magnetization vs. field curves at 1.8 K (Fig. 5) show marked differences between the compounds. For 1 the initial slow linear increase is followed by an inflection point at HC = 15.6 kOe and the fast increase to near saturation value of 3.10Nβ. In the case of the dehydrated phase 1d, there is no discernible inflection point and the maximum magnetization value of 1.44Nβ at 70 kOe is far from the expected saturation. For 2 the inflection point appears at HC = 8.4 kOe and the magnetization reaches 1.93Nβ. The increase in χMT and magnetization reaching saturation indicate that ferromagnetic interactions are present within the straight chains of 1. They arise from the orthogonality of magnetic NiII orbitals and π* orbitals of the CN ligands and were observed in numerous NiII – cyanometallate assemblies with only slightly bent CN bridges. The antiferromagnetic interactions between the chains mediated by hydrogen bonds result in the observed field-induced metamagnetic phase transition between antiferromagnetically and ferromagnetically ordered phases at the critical field HC = 15.6 kOe. This transition is also observed in the FC susceptibility measurements performed at different magnetic fields, where the maximum marking the antiferromagnetic order disappears between 15 and 20 kOe (Fig. S8†). The magnetic susceptibility data for 1 were fitted above 10 K, where the effect of inter-chain antiferromagnetic interactions is negligible (Fig. S8†). The chain structure was approximated by a cyclic fragment composed of 6 ions (3 Ni and 3 Nb) with isotropic interactions using the following exchange Hamiltonian: H = −2J(S1S2 + S2S3 + S3S4 + S4S5 + S5S6 + S6S1). The resulting values of J = 4.8 (±0.2) cm−1, gNi = 2.09, gNb = 2.00 (fixed) are within the range observed for CN-bridged compounds based on NiII and octacyanometallates.7,36,37 For the dehydrated phase 1d the decrease in the χMT product with lowering temperature indicates antiferromagnetic intra-chain interactions. The fitting of the magnetic susceptibility data above 30 K (Fig. S9†) returned the values of J = −1.94 (±0.08) cm−1 and gNi = 2.12. In the magnetization measurements at 1.8 K the maximum value of 1.44Nβ reached at 70 kOe is only slightly higher than expected for the ferrimagnetically ordered chain composed of alternating S = 1 and S = 1/2 centres. The FC susceptibility measurements at the applied field of 20 and 100 Oe show a maximum around 2.2 K, which is not present at the field of 500 Oe. This suggests that the metamagnetic phase transition between antiferromagnetically and ferrimagnetically ordered phases is present but cannot be observed in the magnetization measurements because of the low critical field. Weaker inter-chain antiferromagnetic interactions arise from the lower effective spin value but may also be affected by breaking of the hydrogen bond network upon loss of the crystallization water. Although the structure of 1d could not be established, the negative value of the magnetic exchange constant suggests that the dehydration process causes significant bending of the CN bridges, which results in breaking the orthogonality of NiII dz2 and CN− π* orbitals and switching from ferromagnetic to antiferromagnetic NiII–NbIV interactions within the chain. For the zigzag-shaped chain 2 the slower increase in χMT with decreasing temperature indicates weaker intra-chain interactions than those in the straight chain 1. The fitting of magnetic susceptibility above 10 K (Fig. S10†) resulted in J = 1.88 (±0.03) cm−1, gNi = 2.03. The exchange constant values for 1 and 2, together with the data available for other NiII–NbIV CN-bridged compounds7,36 show a clear trend in the decrease of J with decreasing value of the Ni–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C angle (Table S7†). Similar trend was observed for the polynuclear systems based on NiII and [MV(CN)8]3− ions (M = Mo, W).38 In contrast to 1, for compound 2 the magnetization at 1.8 K and 70 kOe reaches only 1.93Nβ, which is much lower than the expected saturation value. This effect can be attributed to the anisotropy of the NiII ions and their arrangement within the zigzag chain. The octahedral NiII complexes typically show magnetic anisotropy with |D| ranging from 1 to 5 cm−1.39–42 In the straight chain structure of 1, with the CN bridges along the [−101] crystallographic direction, all [Ni(cyclam)]2+ complexes have nearly the same orientation, which ensures that their easy magnetization axes (or planes) coincide. In the zigzag chain structure of 2, the [Ni(cyclam)]2+ complexes have two almost perpendicular orientations, which may result in spin frustration or spin canting effects.43,44 The inter-chain antiferromagnetic interactions between the chains result in the metamagnetic phase transition with HC = 8.4 kOe, which is slightly lower than in the case of 1. The AC susceptibility measurements for 1, 1d, and 2 (Fig. S8–S10†) show no slow relaxation processes. On the basis of the temperature dependence of magnetic susceptibility measured at different fields and magnetization data, the magnetic phase diagrams for 1 and 2 were constructed, which show a rough outline of the magnetic field and temperature regions where antiferromagnetic or ferromagnetic order is present (Fig. S11†).
C angle (Table S7†). Similar trend was observed for the polynuclear systems based on NiII and [MV(CN)8]3− ions (M = Mo, W).38 In contrast to 1, for compound 2 the magnetization at 1.8 K and 70 kOe reaches only 1.93Nβ, which is much lower than the expected saturation value. This effect can be attributed to the anisotropy of the NiII ions and their arrangement within the zigzag chain. The octahedral NiII complexes typically show magnetic anisotropy with |D| ranging from 1 to 5 cm−1.39–42 In the straight chain structure of 1, with the CN bridges along the [−101] crystallographic direction, all [Ni(cyclam)]2+ complexes have nearly the same orientation, which ensures that their easy magnetization axes (or planes) coincide. In the zigzag chain structure of 2, the [Ni(cyclam)]2+ complexes have two almost perpendicular orientations, which may result in spin frustration or spin canting effects.43,44 The inter-chain antiferromagnetic interactions between the chains result in the metamagnetic phase transition with HC = 8.4 kOe, which is slightly lower than in the case of 1. The AC susceptibility measurements for 1, 1d, and 2 (Fig. S8–S10†) show no slow relaxation processes. On the basis of the temperature dependence of magnetic susceptibility measured at different fields and magnetization data, the magnetic phase diagrams for 1 and 2 were constructed, which show a rough outline of the magnetic field and temperature regions where antiferromagnetic or ferromagnetic order is present (Fig. S11†).
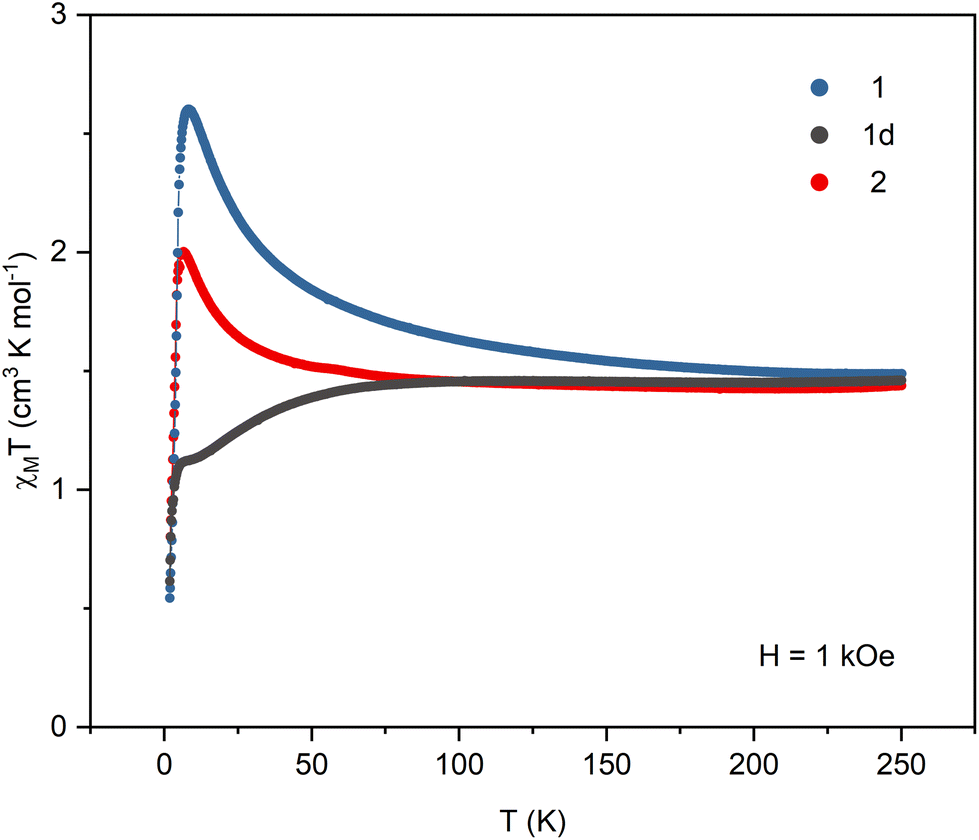 | ||
| Fig. 4 The magnetic susceptibility and temperature product in the function of temperature at the applied field of 1 kOe for 1, 1d, and 2. | ||
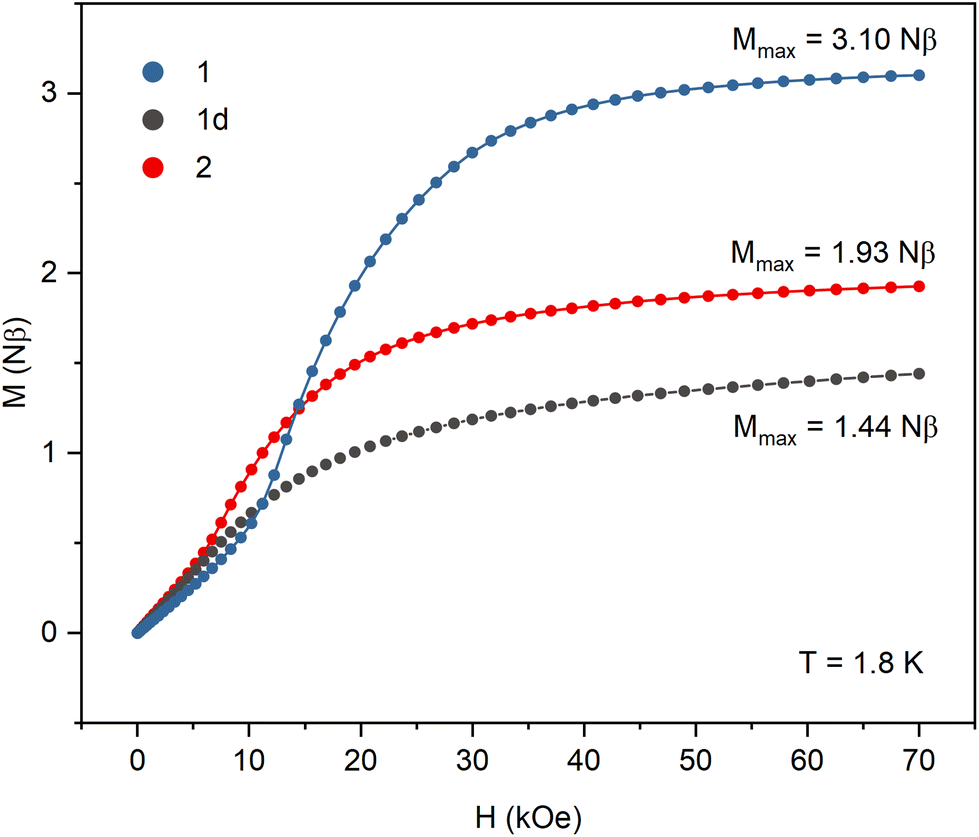 | ||
| Fig. 5 The magnetization vs. magnetic field plot at 1.8 K for 1, 1d, and 2. | ||
Conclusions
Two 1D structures: the straight Li2[Ni(cyclam)][Nb(CN)8]·7.5H2O (1) chain and the zigzag-shaped Li2[Ni(cyclam)][Nb(CN)8]·2H2O (2) chain were obtained by self-assembly process carried out in concentrated LiCl solution. The compounds can be interconverted by a two-step topotactic reaction resulting in ferro- to antiferromagnetic switching of the superexchange interactions through CN-bridges.The formation of alternating bimetallic chains from [Ni(cyclam)]2+ and [W(CN)8]4− is hindered by charge disparity between the building blocks. This problem was overcome by the use of LiCl solution, which enabled incorporation of Li+ cations into the structure to compensate the negative charge of the coordination chain. The solution reaction led to the straight chain 1, with transient formation of the zigzag-shaped chain 2 as a by-product. The latter could be obtained in pure form by dehydration of 1 to the intermediate dihydrate phase 1d followed by thermal treatment at 150°. The structure reorganization during the solid-state conversion from 1 to 2 is quite drastic, as the angle between the bridging cyanides changes from 144.2° to 69.6° and the [Ni(cyclam)]2+ units are rotated. Together with high activation temperature, it indicates the possibility of temporary CN− ligand dissociation, which was found possible for related octacyanidometallates.45–47 Our work shows the utility of the topotactic reactions, which may give access to compounds that are difficult to synthesise in solution reactions. The effects of thermally activated ligand release from the complex in solid state may be different from that in solution, where the vacant coordination site is exposed to solvent. The released ligands trapped in the crystal lattice are more likely to re-form bonds.
The structure reorganization in the topotactic reaction is accompanied by changes in magnetic properties. The intra-chain interactions change from ferro- in 1 to antiferromagnetic in 1d and again to weaker ferromagnetic in 2. The switching is effected by bending of the CN-bridges, which mediate magnetic superexchange, as well as the rearrangement of the relative positions of the anisotropic NiII building blocks. Switching of magnetic properties by dehydration-induced structure changes was discovered over 20 years ago48 and later observed in several CN-bridged networks.49–52 In most cases switching between two structures was discovered, with only a few examples of stepwise dehydration, which allowed characterisation of three phases.10,33,34 For the presented NiII–NbIV chains we have shown that dehydration-induced structural changes can be followed by subsequent structure reorganization activated by thermal treatment without further loss of crystallization water.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the Polish National Science Centre within grant no. 2021/43/B/ST5/02216 (OPUS 22) and the research grant under the “Diamond Grant” program (0193/DIA/2017/46) of the Polish Ministry of Science and Higher Education. The open-access publication of this article has been supported by a grant from the Faculty of Chemistry under the Strategic Programme Excellence Initiative at the Jagiellonian University.References
- B. Sieklucka, R. Podgajny, T. Korzeniak, B. Nowicka, D. Pinkowicz and M. Kozieł, Eur. J. Inorg. Chem., 2011, 305–326 CrossRef CAS.
- S. Chorazy, J. J. Zakrzewski, M. Magott, T. Korzeniak, B. Nowicka, D. Pinkowicz, R. Podgajny and B. Sieklucka, Chem. Soc. Rev., 2020, 49, 5945–6001 RSC.
- B. Nowicka, T. Korzeniak, O. Stefańczyk, D. Pinkowicz, S. Chorąży, R. Podgajny and B. Sieklucka, Coord. Chem. Rev., 2012, 256, 1946–1971 CrossRef CAS.
- M. Magott, B. Gaweł, M. Sarewicz, M. Reczyński, K. Ogorzały, W. Makowski and D. Pinkowicz, Chem. Sci., 2021, 12, 9176–9188 RSC.
- B. Sieklucka and D. Pinkowicz, Molecular Magnetic Materials, Wiley-VCH, Weinheim, Germany, 2017 Search PubMed.
- J. M. Herrera, P. Franz, R. Podgajny, M. Pilkington, M. Biner, S. Decurtins, H. Stoeckli-Evans, A. Neels, R. Garde, Y. Dromzée, M. Julve, B. Sieclucka, K. Hashimoto, S. Okhoshi and M. Verdaguer, C. R. Chim., 2008, 11, 1192–1199 CrossRef CAS.
- D. Pinkowicz, R. Pełka, O. Drath, W. Nitek, M. Bałanda, A. M. Majcher, G. Poneti and B. Sieklucka, Inorg. Chem., 2010, 49, 7565–7576 CrossRef CAS PubMed.
- W. Kosaka, K. Imoto, Y. Tsunobuchi and S. Ohkoshi, Inorg. Chem., 2009, 48, 4604–4606 CrossRef CAS PubMed.
- K. Imoto, M. Takemura, H. Tokoro and S. Ohkoshi, Eur. J. Inorg. Chem., 2012, 2649–2652 CrossRef CAS.
- D. Pinkowicz, R. Podgajny, B. Gaweł, W. Nitek, W. Łasocha, M. Oszajca, M. Czapla, M. Makarewicz, M. Bałanda and B. Sieklucka, Angew. Chem., Int. Ed., 2011, 50, 3973–3977 CrossRef CAS PubMed.
- D. Pinkowicz, R. Podgajny, M. Bałanda, M. Makarewicz, B. Gaweł, W. Łasocha and B. Sieklucka, Inorg. Chem., 2008, 47, 9745–9747 CrossRef CAS PubMed.
- M. Fitta, M. Bałanda, M. Mihalik, R. Pełka, D. Pinkowicz, B. Sieklucka and M. Zentkova, J. Phys.: Condens. Matter, 2012, 24, 506002 CrossRef PubMed.
- P. Konieczny, Ł. Michalski, R. Podgajny, S. Chorazy, R. Pełka, D. Czernia, S. Buda, J. Mlynarski, B. Sieklucka and T. Wasiutyński, Inorg. Chem., 2017, 56, 2777–2783 CrossRef CAS PubMed.
- P. Konieczny, R. Pełka, D. Czernia and R. Podgajny, Inorg. Chem., 2017, 56, 11971–11980 CrossRef CAS PubMed.
- D. Pinkowicz, R. Podgajny, W. Nitek, M. Rams, A. M. Majcher, T. Nuida, S. Ohkoshi and B. Sieklucka, Chem. Mater., 2011, 23, 21–31 CrossRef CAS.
- R. Pradhan, C. Desplanches, P. Guionneau and J. P. Sutter, Inorg. Chem., 2003, 42, 6607–6609 CrossRef CAS PubMed.
- S. L. Ma and S. Ren, J. Inorg. Organomet. Polym., 2009, 19, 382–388 CrossRef CAS.
- S. L. Ma, Y. Ma, S. Ren, S. P. Yan and D. Z. Liao, Struct. Chem., 2008, 19, 329–338 CrossRef CAS.
- N. Ozaki, R. Yamada, K. Nakabayashi and S. Ohkoshi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67, m702–m703 CrossRef PubMed.
- W. Zhang, Z.-Q. Wang, O. Sato and R.-G. Xiong, Cryst. Growth Des., 2009, 9, 2050–2053 CrossRef CAS.
- H.-L. Sun, Z.-M. Wang and S. Gao, Coord. Chem. Rev., 2010, 254, 1081–1100 CrossRef CAS.
- S. Gao, Molecular Nanomagnets and Related Phenomena, Springer Berlin, Heidelberg, 2015 Search PubMed.
- M. Heczko, E. Sumińska, D. Pinkowicz and B. Nowicka, Inorg. Chem., 2022, 61, 13817–13828 CrossRef CAS PubMed.
- M. Heczko, E. Sumińska, B. Sieklucka and B. Nowicka, CrystEngComm, 2019, 21, 5067–5075 RSC.
- M. Heczko, M. Reczyński, C. Näther and B. Nowicka, Dalton Trans., 2021, 50, 7537–7544 RSC.
- M. Reczyński, M. Heczko, M. Kozieł, S. Ohkoshi, B. Sieklucka and B. Nowicka, Inorg. Chem., 2019, 58, 15812–15823 CrossRef PubMed.
- G. Handzlik, M. Magott, B. Sieklucka and D. Pinkowicz, Eur. J. Inorg. Chem., 2016, 4872–4877 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- D. C. M. Llunell, J. Cirera, P. Alemany and S. Alvarez, SHAPE v. 2.1, University of Barcelona, Spain, 2013 Search PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- B. Nowicka, M. Bałanda, M. Reczyński, A. M. Majcher, M. Kozieł, W. Nitek, W. Łasocha and B. Sieklucka, Dalton Trans., 2013, 42, 2616–2621 RSC.
- B. Nowicka, M. Reczyński, M. Bałanda, M. Fitta, B. Gaweł and B. Sieklucka, Cryst. Growth Des., 2016, 16, 4736–4743 CrossRef CAS.
- R. Herchel, J. Tuček, Z. Trávníček, D. Petridis and R. Zbořil, Inorg. Chem., 2011, 50, 9153–9163 CrossRef CAS PubMed.
- L. Glasser, Cryst. Growth Des., 2019, 19, 3397–3401 CrossRef CAS.
- T. Ohno, S. Chorazy, K. Imoto and S. Ohkoshi, Cryst. Growth Des., 2016, 16, 4119–4128 CrossRef CAS.
- D. Visinescu, C. Desplanches, I. Imaz, V. Bahers, R. Pradhan, F. A. Villamena, P. Guionneau and J.-P. Sutter, J. Am. Chem. Soc., 2006, 128, 10202–10212 CrossRef CAS PubMed.
- Y.-Q. Zhang and C.-L. Luo, Dalton Trans., 2008, 4575–4584 RSC.
- A. Wojciechowska, A. Gągor, M. Duczmal, Z. Staszak and A. Ozarowski, Inorg. Chem., 2013, 52, 4360–4371 CrossRef CAS PubMed.
- D. Schweinfurth, J. Krzystek, I. Schapiro, S. Demeshko, J. Klein, J. Telser, A. Ozarowski, C.-Y. Su, F. Meyer, M. Atanasov, F. Neese and B. Sarkar, Inorg. Chem., 2013, 52, 6880–6892 CrossRef CAS PubMed.
- R. Herchel, R. Boča, J. Krzystek, A. Ozarowski, M. Durán and J. Slageren, J. Am. Chem. Soc., 2007, 129, 10306–10307 CrossRef CAS PubMed.
- G. Novitchi, S. Jiang, S. Shova, F. Rida, I. Hlavička, M. Orlita, W. Wernsdorfer, R. Hamze, C. Martins, N. Suaud, N. Guihéry, A.-L. Barra and C. Train, Inorg. Chem., 2017, 56, 14809–14822 CrossRef CAS PubMed.
- D. Shao, S.-L. Zhang, X.-H. Zhao and X.-Y. Wang, Chem. Commun., 2015, 51, 4360–4363 RSC.
- H. Y. Jung, H. L. Jeong, W. C. Seok, C. K. Hyoung and S. H. Chang, Inorg. Chem., 2007, 46, 1529–1531 CrossRef PubMed.
- X. Qi, S. Pillet, C. Graaf, M. Magott, E. E. Bendeif, P. Guionneau, M. Rouzières, V. Marvaud, O. Stefańczyk, D. Pinkowicz and C. Mathonière, Angew. Chem., Int. Ed., 2020, 59, 3117–3121 ( Angew. Chem. , 2020 , 132 , 3141–3145 ) CrossRef CAS PubMed.
- K. Nakabayashi, K. Tomono, Y. Tsunobuchi, W. Kosaka and S. Ohkoshi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, E65, i79–i80 CrossRef PubMed.
- F. J. Birk, D. Pinkowicz and K. R. Dunbar, Angew. Chem., Int. Ed., 2016, 55, 11368–11371 CrossRef CAS PubMed.
- O. Kahn, J. Larionova and J. V. Yakhmi, Chem. – Eur. J., 1999, 5, 3443–3449 CrossRef CAS.
- Y. Sato, S. Ohkoshi, K. Arai, M. Tozawa and K. Hashimoto, J. Am. Chem. Soc., 2003, 125, 14590–14595 CrossRef CAS PubMed.
- M. Ohba, K. Yoneda and S. Kitagawa, CrystEngComm, 2010, 12, 159–165 RSC.
- P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40, 3249–3265 RSC.
- N. Yanai, W. Kaneko, K. Yoneda, M. Ohba and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 3496–3497 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2280169 (1) and 2280170 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03891b |
| This journal is © The Royal Society of Chemistry 2024 |