
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplified photomodulation of a bis(dithienylethene)-substituted phosphine†
Anastasiia
Sherstiuk
 ab,
Marc
Villabona
b,
Agustí
Lledós
b,
Jordi
Hernando
*b,
Rosa María
Sebastián
*bc and
Evamarie
Hey-Hawkins
*a
ab,
Marc
Villabona
b,
Agustí
Lledós
b,
Jordi
Hernando
*b,
Rosa María
Sebastián
*bc and
Evamarie
Hey-Hawkins
*a
aFaculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Leipzig University, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bDepartment of Chemistry, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, Bellaterra, 08193 Barcelona, Spain. E-mail: jordi.hernando@uab.cat; rosamaria.sebastian@uab.cat
cCentro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, Cerdanyola del Vallès, Bellaterra, 08193 Barcelona, Spain
First published on 27th February 2024
Abstract
Phosphine ligands play a crucial role in homogeneous catalysis, allowing fine-tuning of the catalytic activity of various metals by modifying their structure. An ultimate challenge in this field is to reach controlled modulation of catalysis in situ, for which the development of phosphines capable of photoswitching between states with differential electronic properties has been proposed. To magnify this light-induced behavior, in this work we describe a novel phosphine ligand incorporating two dithienylethene photoswitchable moieties tethered to the same phosphorus atom. Double photoisomerization was observed for this ligand, which remains unhindered upon gold(I) complexation. As a result, the preparation of a fully ring-closed phosphine isomer was accomplished, for which amplified variation of phosphorus electron density was verified both experimentally and by computational calculations. Accordingly, the presented molecular design based on multiphotochromic phosphines could open new ways for preparing enhanced photoswitchable catalytic systems.
Introduction
Diarylethenes (DAEs) are amongst the principal molecular photoswitches used for the development of light-responsive compounds, materials and processes.1–7 In part, this is due to the excellent photochemical properties that most DAEs exhibit, namely reversible photoisomerization between ring-open (o) and ring-closed states (c) with high thermal stability, conversion efficiency and fatigue resistance.1,2 In addition, DAEs undergo large geometrical and electronic changes upon o–c photoswitching, a feature that is exploited to accomplish light-control in a variety of applications (e.g., information storage and processing,1,2,8 chemical reactivity and catalysis,5,9,10 soft matter actuators4,6 and (bio)imaging3,11). In several of these cases, the photoinduced performance of the final system would benefit from the incorporation of multiple DAE units in a single molecular construct,12–14 for instance, to obtain multistate behavior in molecular switches for larger information storage density, or to amplify the electronic modulation between the two states of a photoswitchable catalyst or reagent.However, the development of high-performance multiphotochromic molecules based on DAEs is not straightforward. In many of these systems, complete photoisomerization of their DAE units is inhibited by excited state energy transfer between neighboring ring-open and ring-closed units. As a result, only partial photoswitching can be reached where one (or more) of the DAE moieties in the construct remain in their initial open state.12–14 This detrimental effect is very sensitive to the distance between neighboring DAE photochromes as well as the electronic properties of the linkers through which they are tethered.12–14 In particular, only very few examples have been reported where full photocyclization was observed for two very close DAE groups separated by short spacers such as silylene,15 phenylene16,17 and divinylene bridges.18 By contrast, the use of much longer linkers favors multiple DAE photoswitching, though at the cost of preventing through-bond electronic communication between nearby photochromic units.14,19,20
In this work, we tackled this challenge by directly connecting two dithienylethene (DTE) photochromic units, the most common type of DAE, through a phosphine bridge. Dithienylethene–phosphine tethers have been proposed for the light-control of coordination compounds and catalytic reactions, as the electronic changes occurring upon DTE photoisomerization can modulate the electron-donating ability of ligands.5,21 To our knowledge, this goal has only been explored by attaching one DTE unit to phosphines, either through the central22 or the lateral thiophene23–26 rings of the photochromic moiety. Consequently, only limited photomodulation of phosphine properties can be accomplished in this way. Herein, we hypothesized that this light-promoted effect could be further amplified by introducing additional DTE units to the same ligand, for which we developed bisDTE-substituted phosphine 1 that can present three different isomer states: fully ring-open (oo), fully ring-closed (cc) and an intermediate form with one ring-open and one ring-closed unit (oc) (Scheme 1). The structure of compound 1 was designed on the basis of two main principles: (i) two DTE units were connected to the same phosphorus atom, which should be double affected by photochrome isomerization, and (ii) a strong electron-withdrawing group (EWG) was installed in the external thiophene ring of both DTE units, which only communicates with the phosphorus atom on the other thiophene moiety upon light-induced ring-closure. As a result, the effect of DTE photocyclization on the electronic features of the phosphorus atom in 1 should be maximized if full photoisomerization from the initial oo isomer to the final cc state is accomplished. Trifluoromethyl ketone was selected as EWG in this design, as it has been reported to benefit the photoswitching properties of DTEs (e.g., higher photocyclization quantum yields and conversions as well as photostability).26,27
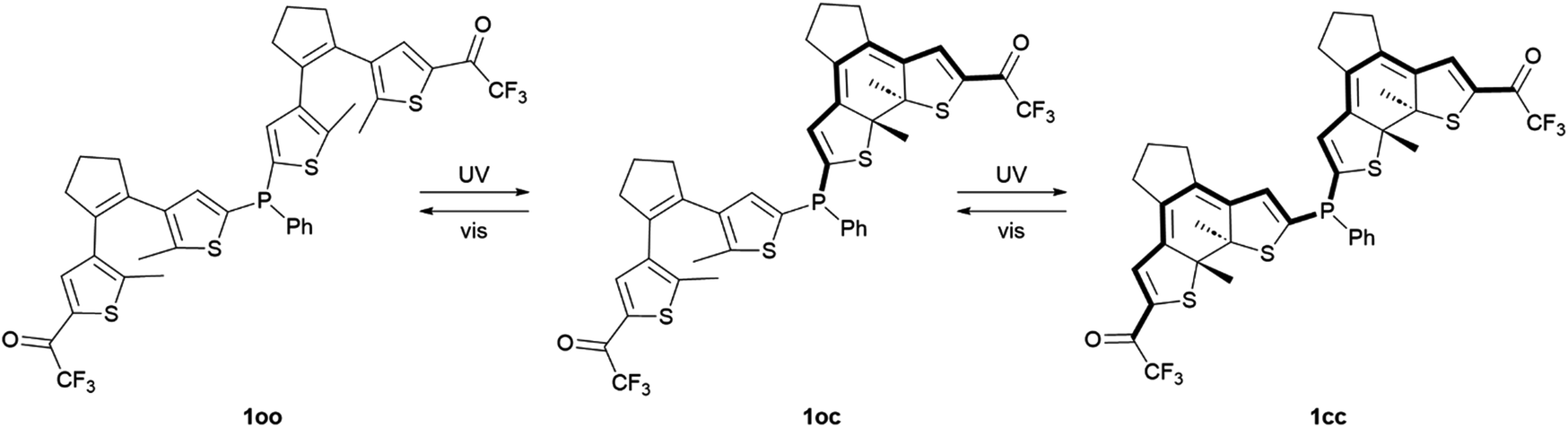 | ||
| Scheme 1 Two-step reversible photoisomerization of the phosphine ligand 1 studied in this work. | ||
Results and discussion
Synthesis
Bis(dithienylethenyl)phosphine 1 was synthesized through sequential lithiation-mediated reactions from 1,2-bis(2-chloro-5-methylthien-4-yl)cyclopentene (2), a common intermediate for the preparation of DTE derivatives (Scheme 2a).28 At first, the phosphanyl group was introduced by performing chlorine–lithium exchange with t-butyllithium (tBuLi) and reacting 2 equivalents of the resulting monolithiated substrate with 1 equivalent of PhPCl2 to yield compound 3 (52% yield). In a second step, the remaining chlorine atoms in 3 were substituted for the trifluoroacetyl function through a similar lithiation protocol using ethyl trifluoroacetate as an electrophile source, which finally led to target ligand 1 in its fully ring-open state oo (49% yield). In the 31P{1H} NMR spectrum this compound presents a singlet at δ = −33.4 ppm that very much resembles the reported value for phenyldi-2-thienylphosphine (δ = −33.6 ppm),29 thereby corroborating the introduction of two DTE units tethered to the central phosphanyl group in 1. The introduction of the external trifluoromethyl ketones in the final ligand was confirmed by a singlet registered at δ = −71.9 ppm in the 19F NMR spectrum, which is in agreement with other trifluoroacetyl-functionalized DTEs (δ ca. −72 ppm).26,27 One of these described compounds, monoDTE-substituted phosphine 5o (Scheme 2b),26 was used herein as a reference in the photochemical and electronic characterization of 1. | ||
| Scheme 2 (a) Synthetic route to bisDTE-substituted phosphine 1 and its gold(I) complex 4. (i) tBuLi/THF, 0.5 eq. PPhCl2, (ii) tBuLi/THF, CF3COOEt, (iii) [AuCl(SMe2)]/CH2Cl2. (b) Structure of the monoDTE-substituted phosphine 5 used as a reference in this work.26 | ||
To further investigate the properties of the obtained ligand 1 upon metal binding, the monophosphine gold(I) complex 4 in its ring-open state oo (89% yield, Scheme 2a) was prepared by reaction with (dimethylsulfide)gold(I) chloride. For this compound, complexation was corroborated through the downfield shift of the singlet in the 31P{1H} NMR spectrum to δ = 6.1 ppm.
Photochemical characterization
As depicted in Scheme 1, DTE-based 1 is expected to undergo photoinduced isomerization between three distinctive states: oo, oc and cc. This photochemical behavior was investigated both experimentally by UV-vis absorption and NMR measurements in organic solvents as well as through TD-DFT calculations at the CAM-B3LYP-D3/6,31-G(d,p) level.First, the UV-vis absorption spectrum of the synthesized 1oo isomer was recorded in cyclohexane (Fig. 1a). Similar to other DTE derivatives,2 the open-state absorption spectrum of 1 resembles that of substituted thiophenes and features a distinct absorption band with λabs,max = 273 nm corresponding to a dithienylethene π → π* electronic transition (Table 1, Fig. S12a and Tables S1 and S2 in the ESI†). Notably, the presence of the electron-withdrawing trifluoroacetyl group results in the observation of an absorption shoulder that extends up to λabs ∼ 400 nm, which should allow irradiation of 1oo with less energetic UV-A light to promote photoisomerization. Indeed, when a cyclohexane solution of 1oo was illuminated at λexc = 355 nm, the emergence of a broad, red-shifted peak at λabs,max = 599 nm was registered, which is characteristic of the lowest-energy π → π* electronic transition of closed-state DTEs exhibiting higher conjugation pathways (Fig. S12b and Tables S1 and S2 in the ESI†).2 For 1, this spectral change made the initial colorless solution turn deep blue, a behavior already reported for other trifluoroacetyl-functionalized DTEs upon photocyclization.26,27 UV-induced photoisomerization of 1 was further confirmed by subsequent irradiation of the sample with visible light (λexc = 532 nm). The absorption band at λabs,max = 599 nm disappeared entirely while the original spectrum of the initial 1oo isomer was recovered, i.e., quantitative ring-opening of the previously formed photocyclized species occurred (Fig. 1b). The reversible open-close photoisomerization of 1 could be repeated for ten consecutive cycles of illumination with UV (365 nm) and visible (520 nm) radiation without observing any spectral sign of photodegradation, thus proving the high fatigue resistance of the bisDTE-substituted phosphine 1 (Fig. 1c).
 | ||
| Fig. 1 Photochemical properties of 1 in cyclohexane solution. (a) Variation of the UV-vis absorption spectrum of 1oo (c = 3.12 × 10−5 M) upon irradiation at λexc = 355 nm until a photostationary state is obtained (PSS@355 nm). (b) Variation of the UV-vis absorption spectrum of PSS@355 nm for 1 (c = 1.83 × 10−5 M) upon irradiation at λexc = 532 nm until the initial spectrum of 1oo is recovered. (c) Variation of the ring-closed absorbance of 1 (c = 2.39 × 10−5 M) in the visible region (λexc = 599 nm) upon 10 consecutive photoswitching cycles with UV and visible light (λexc = 365 and 520 nm). | ||
| λ abs o [nm] (ε [M−1 cm−1])a | λ abs c [nm] (ε [M−1 cm−1])b | PSSo–c compositionc [%] | Φ oo–oc | Φ oc–cc | Φ cc–oc | Φ oc–oo | |
|---|---|---|---|---|---|---|---|
a Wavelength and molar absorptivity coefficient of the absorption band maxima of the open isomer (for 1, 4 and 6, the oo state) in cyclohexane.
b Wavelength and molar absorptivity coefficient of the maximum of the visible absorption band of the closed isomer (for 1, 4 and 6, the cc state) in cyclohexane.
c Composition of the PSS reached for the photocyclization process in toluene-d8 upon irradiation at λexc = 365 nm. DTEoo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DTEoc:DTEcc (for 1, 4 and 6) and DTEo:DTEc (for 5) concentration ratios are given.
d Photocyclization quantum yields measured in cyclohexane at λexc = 355 nm. For ligand 5, a single Φo–c value is given for its o → c ring-closing process.
e Photocycloreversion quantum yields measured in cyclohexane at λexc = 532 nm. For ligand 5, only Φc–o value is given for its c → o ring-opening process. :DTEoc:DTEcc (for 1, 4 and 6) and DTEo:DTEc (for 5) concentration ratios are given.
d Photocyclization quantum yields measured in cyclohexane at λexc = 355 nm. For ligand 5, a single Φo–c value is given for its o → c ring-closing process.
e Photocycloreversion quantum yields measured in cyclohexane at λexc = 532 nm. For ligand 5, only Φc–o value is given for its c → o ring-opening process.
|
|||||||
| 1 | 273 (32423), 336 (5820) |
599 (22000) |
7:84:9 |
0.435 | 0.020 | 0.032 | 0.022 |
| 4 | 262 (47527), 336 (7727) |
589 (26542) |
10:77:13 |
0.246 | 0.007 | 0.026 | 0.023 |
| 5 26 | 268 (35673), 339 (6431) |
598 (12261) |
9:91 |
0.480 | 0.012 | ||
| 6 | 272 (42900), 330 (9582) |
593 (28500) |
10:71:19 |
0.402 | 0.022 | 0.019 | 0.018 |
As previously described for other systems bearing multiple DTE units,12–14 a critical parameter of the photoswitching performance of 1 is the extent of its UV-induced photocyclization process, i.e., whether its fully ring-closed isomer 1cc can be produced. This issue could not be investigated by UV-vis absorption spectroscopy, as no clear spectral shift with the irradiation time was observed for the absorption band in the visible region characteristic of ring-closed DTE species. According to the TD-DFT calculations, this is to be expected during the formation of 1oc and 1cc, because both compounds must present similar spectral maxima for their lowest-energy electronic transitions (Fig. S12b and Tables S1 and S2 in the ESI†). For this reason, we analyzed the ring-closing reaction of 1 upon UV irradiation by NMR spectroscopy in toluene-d8 (Fig. 2 and Fig. S1 and S2 in the ESI†). For this study, we had to consider the particular stereochemistry of DTE photocyclization, which produces a racemic mixture of two ring-closed enantiomers due to its conrotatory nature.2 As a result, UV-induced photoisomerization of bisDTE-functionalized phosphine 1oo should generate a diastereomeric mixture of two pairs of enantiomers for the state oc, which could give rise to two distinctive sets of NMR signals, and a diastereomeric mixture of two meso forms and one pair of enantiomers for the state cc, which could produce three separate sets of NMR signals (Scheme S1 in the ESI†). This behavior was indeed experimentally observed by 31P NMR spectroscopy, where one, two and three different resonances lying at significantly different spectral regions could be identified for 1oo, 1oc and 1cc after UV irradiation, respectively (Fig. 2 and Fig. S1 in the ESI†). Therefore, this result demonstrates the capacity of 1 to undergo full DTE ring-closing, a quite remarkable feature that has been seldom reported for compounds bearing multiple dithienylethene units separated at short distances.15–18
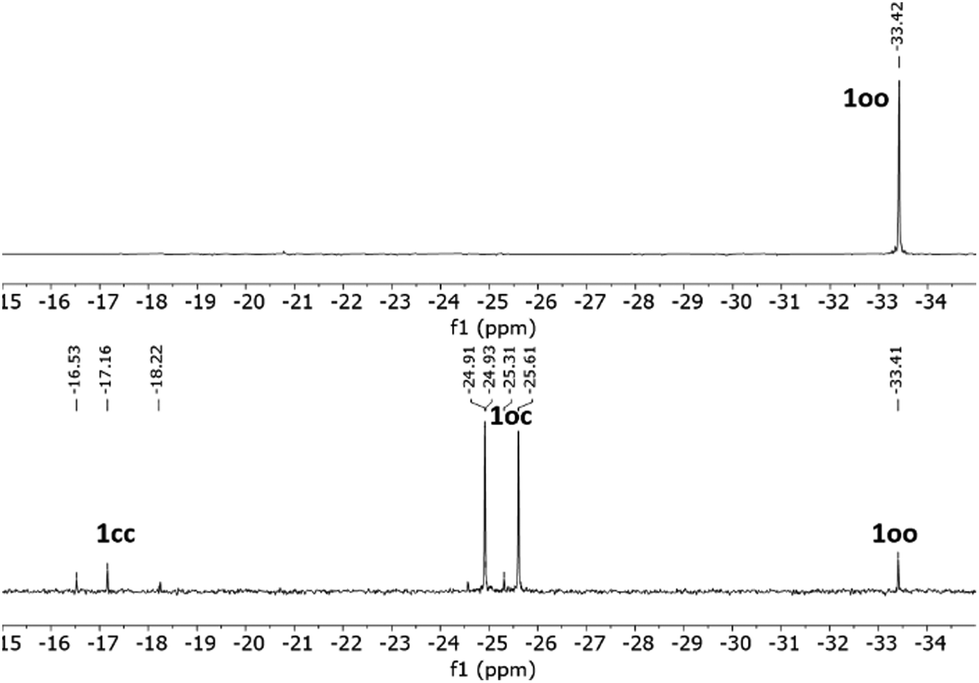 | ||
| Fig. 2 31P{1H} NMR spectra (121 MHz, toluene-d8) of 1oo (top) and the PSS@365 nm (bottom), where partial DTE photocyclization produces a mixture of oo, oc and cc isomers. | ||
However, the efficiency of complete DTE photocyclization was found to be moderate for 1. Instead, a photostationary state (PSS) mainly enriched in the intermediate oc isomer was observed to form upon irradiation at λexc = 365 nm in toluene-d8. In particular, the oo:oc:cc molar ratio determined for the PSS mixture produced was 7:84:9, which corresponds to a 52% efficacy in DTE ring-closing that could not be incremented by using other more polar solvents (Table 1 and Fig. S3 in the ESI†). This is clearly lower than for the reference, monoDTE phosphine 5, which generates 91% of the fully closed isomer under the same irradiation conditions.26 To rationalize this behavior, the separate oo → oc and oc → cc photocyclization quantum yields were determined for 1 (Φoo–oc = 0.435 and Φoc–cc = 0.020, Table 1). On the one hand, Φoo–oc closely resembles the photocyclization quantum yield of reference 5 (Φo–c = 0.480),26 which indicates that the first ring-closing step in 1 is not significantly affected by the presence of a nearby ring-open DTE unit. By contrast, a 20-fold reduction in Φ was measured for the second ring-closing reaction of 1, which proceeds notably less effectively and more slowly when a close-by DTE unit is already in the closed state (Fig. S4 in the ESI†). As reported for many other multiphotochromic systems,12–14 through-bond and through-space intramolecular energy transfer from the photoexcited open DTE group to the closed DTE unit in 1oc must account for this situation, which eventually leads to photoconversion back to 1oo instead of full photocyclization to 1cc. The intramolecular nature of this behavior was confirmed by investigating the photocyclization of 1 at increasing concentrations, which did not affect the ring-closing efficiency and, therefore, excluded the occurrence of intermolecular effects (Fig. S5 in the ESI†). This conclusion was further supported by the TD-DFT calculations, where electronic transitions that transfer electron density from the ring-open to the ring-closed units of 1oc contribute to the UV absorption of this compound (Tables S1 and S2 in the ESI†). By contrast, intramolecular DTE interactions did not seem to affect the efficacy of the ring-opening reactions of 1, as similar Φcc–oc and Φoc–oo values were obtained that are of the same order of magnitude as the photocycloreversion quantum yield of reference 5.26
Metal complexation often leads to a change in the photochemical behavior of DTE-based ligands.20,25,30 For this reason, we evaluated both experimentally and computationally the photochemical properties of gold(I) complex 4 (Scheme 2), which can also present three different oo, oc and cc isomers. Resembling free ligand 1, a solution of the open-state complex 4oo in cyclohexane exhibited strong absorption in the UV region with an intense peak at λabs,max = 262 nm and an additional shoulder at λabs,max = 336 nm (Table 1, Fig. S6a and S13a in the ESI†). In addition, the expected spectral changes accounting for DTE photoisomerization were observed upon UV irradiation of 4oo cyclohexane solutions. A new band in the visible part of the spectrum appeared, which was accompanied by a color change of the sample to deep blue, i.e., gold(I) complex 4 also undergoes photocyclization (Table 1 and Fig. S6a in the ESI†). Although a 10 nm hypsochromic shift was observed for this band relative to free ligand 1 (λabs,max = 589 nm), the TD-DFT calculations proved that it could also be attributed to π → π* transitions associated with the closed-state DTE units of the complex, and these transitions present similar spectral maxima for both the oc and cc isomers of 4 (Fig. S13b and Tables S1 and S2 in the ESI†).
As in the case of the free ligand, partial photoconversion of 4oo under UV irradiation was revealed by NMR spectroscopic analysis. Thus, an oo:oc:cc molar ratio of 10:77:13 with an overall 52% content in closed DTE units was determined by 31P NMR spectroscopy for the PSS accomplished at λexc = 365 nm (Table 1 and Fig. S7–S9 in the ESI†). This result could also be attributed to the decrement in ring-closing quantum yield upon partial photocyclization due to intramolecular excited state energy transfer between neighboring open and closed DTE units in the complex. Thus, a 35-fold decrease in Φoc–cc was measured relative to Φoo–oc for 4, in a similar fashion to free ligand 1 (Table 1). Therefore, metal complexation did not cause notable effects on the capacity of the bisDTE-functionalized phosphine to undergo full photocyclization, which allows preparing the double ring-closed isomer 4cc though with minor efficiency. However, it must be noted that lower Φoo–co and Φoc–cc values were determined for complex 4 in comparison to 1, which we tentatively attributed to two main factors. First, TD-DFT computations showed that UV irradiation of 4 does not only lead to photoexcitation of the open DTE units, but also to metal-to-ligand and ligand-to-metal charge transfer transitions that should not lead to photoisomerization (Fig. S13a and Tables S1 and S2 in the ESI†). As quantum yield analysis does not account for this effect, the apparent Φoo–oc and Φoc–cc values obtained from the overall complex absorption should decrease relative to the free ligand. Second, the 10 nm hypsochromic absorption shift measured for the closed DTE units in 4 relative to 1 could favor intramolecular energy transfer from open DTE groups and further hamper 4oc–4cc photocyclization. Photoinduced ring-opening of the bisDTE ligand was found to be less affected in the gold(I) complex, and quantitative photocycloreversion could also be promoted for 4 through irradiation at λexc = 532 nm with similar Φcc–oc and Φoc–oo values (Table 1 and Fig. S6b in the ESI†). As a result, complex 4 demonstrated good fatigue resistance, enduring 10 reversible photoswitching cycles without significant degradation (Fig. S6c in the ESI†).
Photomodulation of the electronic properties of phosphine 1
The introduction of DTE photochromes into phosphine ligands pursues modulating their metal-binding properties upon photoisomerization, eventually aiming to light-control the catalytic activity in situ of the resulting complexes.5,10 For this goal to be accomplished, the phosphorus atom of these compounds should undergo a sufficient change in its electronic properties upon photoswitching of the DTE units to which it is connected. Due to the nature of the phosphorus–selenium bond,29,31 a common method to assess the modulation of the phosphorus σ-donating ability upon DTE photoisomerization is through the coupling of 77Se to the 31P nuclei in the corresponding phosphine selenides.23,25,26 Generally, the rearrangement of the electron density caused by DTE photocyclization results in the retraction of electron density from the substituents at the 5-position in thiophene, thus leading to a decrease in phosphorus σ-donating properties for the closed state of the DTE-based phosphines that can be evidenced by an increase in 1JP,Se for the selenide derivative (Δ(1JP,Se) > 0 upon ring-closing).23,25,26 Herein we envisioned to amplify this effect through a two-fold strategy. First, by introduction of the strong electron-withdrawing trifluoroacetyl group at position 5 of the other thiophene ring of the system, which should only become electronically connected to the phosphanyl moiety upon DTE ring-closing. As we have established before for monoDTE-based phosphine 5,26 this design leads to a larger decrease in 1JP,Se upon photocyclization compared to other DTE-functionalized phosphines (Δ(1JP,Se) = 14 Hz, Table 2). In the bisDTE-based phosphine 1, a second factor should allow further increasing the photomodulation of electronic properties of the phosphine, namely the additive effect arising from the photoisomerization of the two DTE units linked to the phosphorus atom.| Δ(1JP,Se) [Hz]a | Δ(qMullikenP)b | Δ(%sP)c | Δ(BEP–Au)d [kcal mol−1] | |||||
|---|---|---|---|---|---|---|---|---|
| a Difference in 1JP,Se for the corresponding selenides measured in CDCl3. b Difference in Mulliken charges in electronic units on the phosphorus atom. c Difference in percentage of s character of the lone pair of electrons at phosphorus. d Difference in phosphine–Au bond energy. | ||||||||
| oo → oc | oo → cc | oo → oc | oo → cc | oo → oc | oo → cc | oo → oc | oo → cc | |
| 1 | 11 | 19 | 0.011 | 0.036 | 0.52 | 1.58 | 1.52 | 2.13 |
| o → c | o → c | o → c | o → c | |||||
| 5 | 1426 | 0.01426 | 0.7926 | 1.23 | ||||
This hypothesis was successfully validated by evaluating the variation in 31P–77Se coupling constant for the different isomers of the selenide derivative 6 (Scheme 3). By reacting 1oo with grey selenium powder in CDCl3, phosphine selenide 6oo was first prepared, which exhibited a very similar photoswitching behavior to the free phosphine and the gold(I) complex. Thus, it partially photoconverted to compounds 6oc and 6cc upon irradiation at 365 nm with 57% photocyclization efficiency, a process that could be fully reverted by excitation with visible light (Table 1 and Fig. S10 and S11 in the ESI†). When analyzing the sequential photoisomerization process of 6 by 31P NMR spectroscopy, a total change in 1JP,Se of 19 Hz was determined for the fully closed state 6cc in comparison to 6oo (Table 2). To our knowledge, this is the highest modulation measured to date for DTE-based phosphines,23,25,26 and it indeed exceeds the variation reported from 2-thienyldiphenylphosphine (1JP,Se = 743 Hz) to tri-2-thienylphosphine (1JP,Se = 757 Hz).29
 | ||
| Scheme 3 Photoisomerization-induced variation of 1JP,Se in the oo, oc and cc isomers of phosphine selenide 6. | ||
To corroborate this experimental result, several descriptors commonly used to assess the electronic properties of ground state phosphines were computed using DFT calculations at the B3LYP-D3/6,31G(d,p) level. On the one hand, clear decrements in the Mulliken charge on the phosphorus atom of 1 were predicted upon DTE ring-closing (Δ(qMullikenP), Table 2). While the first DTE photocyclization process in 1 should result in a similar Δ(qMullikenP) value as for monoDTE-based phosphine 5, the second ring-closing reaction should lead to further lowering of the electron density on phosphorus. This effect is typically accompanied by changes in the composition of the lone pair of electrons at the phosphorus atom with an increased participation of the s orbital. Indeed, according to natural bond orbital (NBO) analysis,32 the photoconversion from the 1oo to the 1cc state must lead to a progressive increment of the contribution of the s orbital to the lone pair of electrons at phosphorus, eventually reaching twice the variation computed for phosphine 5 (Table 2).
Finally, we computationally evaluated how the electron density change on the phosphorus atom in compound 1 and reference 5 upon photoisomerization would affect the ligand–metal bond energies of their respective gold(I) complexes (Table 2). As expected, weaker phosphorus–gold(I) bonds were predicted after DTE ring-closing in both ligands. More importantly, double DTE photocyclization in 4 should result in almost a two-fold variation in ligand–metal binding energies compared to the complex with monoDTE-based phosphine 5. In combination with the experimental and theoretical data disclosed in this section, this result confirms the capacity to accomplish large photomodulation in the electronic properties of phosphines by tethering two photoswitchable DTE units to a central phosphorus atom.
Conclusions
In this work we reported the synthesis and characterization of a bis(dithienylethenyl)phosphine bearing two photoswitchable moieties connected to the same phosphorus atom. Upon light irradiation, reversible transformation between open–open, open–closed and closed–closed states was accomplished for this compound due to sequential DTE photoisomerization, in contrast to many other multichromophoric systems where several DTE units are arranged at short distances. More importantly, photoconversion allowed switching the communication between the phosphorus atom and the electron-withdrawing trifluoromethyl ketone substituent of the nearby DTE units on and off. In combination with the additive effect caused by double DTE photoisomerization, this resulted in an unprecedented light-modulation of the electronic properties of the phosphine, as proven experimentally and through computational calculations. As the phosphine's ability to undergo photocyclization remained unhindered upon complexation with gold(I), this study provides valuable insights for the design of novel photoswitchable phosphines with amplified activity for light-controlled catalysis.Experimental
General procedures
All reactions were carried out under nitrogen atmosphere in the absence of air and water using standard Schlenk line techniques. All solvents (hexanes, CH2Cl2, cyclohexane) were dried and degassed prior to use. THF was distilled over sodium/benzophenone and stored over 4 Å activated molecular sieves. CDCl3 was degassed by freeze–pump–thaw cycling. Toluene-d8 was degassed with nitrogen. All starting materials and reagents were commercially purchased and used without further purification. Flash column chromatography was done using silica gel (230–400 mesh) using a stream of nitrogen.NMR spectra were recorded on a BRUKER Avance III HD 400 MHz, BRUKER Ascend 300 MHz and BRUKER Ascend 400 MHz at 25 °C. Tetramethylsilane (TMS) was used as an internal reference in 1H and 13C NMR spectra; all other nuclei were referenced to TMS using Ξ scale.33 Chemical shifts are reported in parts per million (ppm). Assignment of 1H and 13C NMR signals was carried out using 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC NMR experiments. IR spectra were recorded on FT-IR spectrometers Thermo Scientific Nicolet iS5 and BRUKER Alpha II. Electrospray ionization mass spectrometry was carried out with BRUKER Impact II, BRUKER Esquire 3000+ and micrOTOF-Q II BRUKER spectrometers in positive ion mode. UV-vis absorption spectra were recorded on an Agilent HP 8453 spectrophotometer using HPLC quality solvents and 1 cm quartz cuvettes. Photoisomerization studies were carried out using different irradiation sources (365 nm and 520 nm LEDs (Chanzon) and a Nd:YAG pulsed laser (Brilliant, Quantel, λexc = 355 or 532 nm)).
Photochemical characterization
DTE photoswitching in 1 and 4 was monitored by UV-vis and NMR spectroscopy. PSS composition was determined via31P or 19F NMR spectroscopy from a PSS state produced by irradiating a toluene-d8 solution in an NMR tube with the appropriate wavelength. UV-vis absorption spectra of the closed-state isomers shown in Fig. S12 and S13 in the ESI† were estimated from the PSS@365 nm and the corresponding open-state UV-vis spectra. Photoisomerization quantum yields were determined by monitoring the variation of the UV-vis absorption spectra of 1 and 4 in cyclohexane upon irradiation with UV (for photocyclization, λexc = 355 nm) or visible light (for photocycloreversion, λexc = 532 nm). A kinetic model previously reported that accounts for the sequential photoisomerization of DTE units in 1 and 4 was used to separately determine the photoisomerization quantum yields of their oo, oc and cc isomers.20 To apply this model, we assumed the UV-vis absorption spectrum of each DTE unit to be independent of the isomerization state of the other, i.e., the extinction coefficients of open DTE units are the same in the oo and oc states, while those of closed DTE groups are equal in the oc and cc states, as suggested by the TD-DFT calculations and observed in previous work on DTE dimers.13 In all the cases, the irradiation intensities used in our photoisomerization quantum yield experiments were determined by monitoring the photocyclization and photocycloreversion processes of 1,2-bis(2-methyl-5-phenyl-3-thienyl)perfluorocyclopentene in n-hexane as a reference (Φoc = 0.59 and Φco = 0.013).34Computational methods
DFT calculations were carried out using the Gaussian16 program package.35 Geometry optimizations were conducted without any constraints using the B3LYP functional36 with Grimme's D3 correction to account for dispersion effects.37 Geometry optimizations were performed in THF using the solvation model density (SMD) continuum model38 with basis set 1 (BS1). BS1 included the 6-31G(d,p) basis set for the main group atoms39 (H, C, O, F, P, S) and the Stuttgart-Dresden SDD effective core potential (ECP) and its corresponding double-ζ basis set,40 with a set of f polarization functions41 for Au. Frequency calculations were performed for all the optimized geometries to determine the stationary points as either minima or transition states. Energies in THF were refined through single-point calculations of the optimized BS1 geometries with an extended basis set (BS2). BS2 consisted of the def2-TZVP for main group atoms, and the quadruple-ζ def2-QZVP basis set for Au, together with the def2 ECP.42 Frontier molecular orbitals and natural bond orbital (NBO)43 analysis were calculated at the B3LYP-D3/BS1 level in THF using the SMD continuum model. TD-DFT calculations were carried out using the CAM-B3LYP functional44 with Grimme's D3 correction to account for dispersion effects.37 The first 15 electronic transitions were calculated in cyclohexane using the SMD continuum model with the BS1 described above.Synthetic procedures
45 in 20 mL THF was cooled to −78 °C (hexanes/N2(l)), and then 0.69 mL (1.6 mol L−1, 1.10 mmol, 1.1 eq.) tBuLi in n-pentane were added dropwise. The resultant bright yellow mixture was kept stirring for 65 min at −78 °C, followed by the addition of 0.07 mL (0.50 mmol, 0.5 eq.) of dichlorophenylphosphine in one swift motion. The reaction mixture was left overnight to warm up to room temperature, and then quenched with a degassed brine solution. Under nitrogen atmosphere, the phases were separated, the aqueous phase was extracted with THF (2 × 5 mL), and the combined organic phases were dried over degassed anhydrous Na2SO4. After canula filtration, the product mixture was absorbed on silica gel and purified through flash column chromatography (hexanes). After solvent removal in vacuo, a white oil was obtained (0.182 g, 52% yield).
R f 0.29 (hexanes).
1 H NMR (300 MHz, CDCl3, δ): 7.35–7.29 (m, 5H), 6.98 (d, J = 6.6 Hz, 2H), 6.57 (s, 2H), 2.80–2.68 (m, 8H), 2.09–1.97 (m, 10H), 1.87 (s, 6H) ppm.
13 C{ 1 H} NMR (75 MHz, CDCl3, δ): 142.0, 139.0 (d, 1JC,P = 6.3 Hz), 137.4 (d, 2JC,P = 27.8 Hz), 136.8 (d, 3JC,P = 8.3 Hz), 135.2, 135.2, 134.2, 133.9 (d, 1JC,P = 23.3 Hz), 133.3, 131.9 (d, 2JC,P = 19.1 Hz), 128.7, 128.4 (d, 3JC,P = 6.7 Hz), 127.0, 125.1, 38.4, 23.1, 14.8, 14.3 ppm.
31 P{ 1 H} NMR (162 MHz, CDCl3, δ): −33.4 (s) ppm.
IR (ATR, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ): 3068 (w), 3050 (w), 2948 (s), 2914 (s), 2841 (s), 2730 (w), 1547 (w), 1456 (m), 1434 (s), 1376 (w), 1307 (w), 1288 (w), 1202 (m), 1162 (m), 1026 (w), 1010 (s), 990 (m), 964 (w), 829 (m), 742 (m), 696 (m), 652 (w), 530 (m), 519 (m), 482 (m), 430 (w) cm−1.
): 3068 (w), 3050 (w), 2948 (s), 2914 (s), 2841 (s), 2730 (w), 1547 (w), 1456 (m), 1434 (s), 1376 (w), 1307 (w), 1288 (w), 1202 (m), 1162 (m), 1026 (w), 1010 (s), 990 (m), 964 (w), 829 (m), 742 (m), 696 (m), 652 (w), 530 (m), 519 (m), 482 (m), 430 (w) cm−1.
HRMS (ESI-TOF, m/z): calculated for [M + Na]+ 717.0472; found 717.0479.
:20). After solvent removal in vacuo, a yellow oil was obtained (0.100 g, 49% yield). Caution: the product is very sensitive to oxygen nucleophiles (water, alcohols); special care should be taken during the work up to avoid formation of the corresponding hydrate.
R
f
0.18 (hexanes/dichloromethane 80:20).
1 H NMR (300 MHz, CDCl3, δ): 7.57 (s, 2H), 7.32–7.21 (m, 5H), 6.89 (d, J = 6.6 Hz, 2H), 2.87–2.71 (m, 8H), 2.16–2.03 (m, 10H), 1.99 (s, 6H) ppm.
13 C{ 1 H} NMR (101 MHz, CDCl3, δ): 173.0 (q, 2JC,F = 36.7 Hz), 150.1, 142.2, 138.9, 138.4 (d, 1JC,P = 6.3 Hz), 138.1 (q, 4JC,F = 3.2 Hz), 137.3, 137.0 (d, 2JC,P = 27.7 Hz), 136.3 (d, 3JC,P = 8.3 Hz), 134.6 (d, 1JC,P = 23.9 Hz), 133.0, 132.2, 131.8 (d, 2JC,P = 19.3 Hz), 128.9, 128.5 (d, 3JC,P = 6.9 Hz), 116.6 (q, 1JC,F = 290.7 Hz), 38.4, 38.3, 23.1, 15.6, 14.7 ppm.
19 F NMR (376 MHz, CDCl3, δ): −71.9 (s) ppm.
31 P{ 1 H} NMR (162 MHz, CDCl3, δ): −33.3 (s) ppm.
IR (ATR, ): 2952 (w, νC–H), 2917 (w, νC–H), 2843 (w, νC–H), 1680 (s, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1527 (w), 1425 (m), 1331 (w), 1246 (w), 1220 (m), 1194 (m), 1137 (s), 1025 (m), 999 (m), 970 (m), 923 (m), 867 (m), 801 (s), 753 (m), 739 (m), 716 (m), 694 (m), 684 (m), 660 (m), 580 (m), 528 (m), 490 (m), 426 (m) cm−1.
O), 1527 (w), 1425 (m), 1331 (w), 1246 (w), 1220 (m), 1194 (m), 1137 (s), 1025 (m), 999 (m), 970 (m), 923 (m), 867 (m), 801 (s), 753 (m), 739 (m), 716 (m), 694 (m), 684 (m), 660 (m), 580 (m), 528 (m), 490 (m), 426 (m) cm−1.
HRMS (ESI-TOF, m/z): calculated for [M + H]+ 819.1078; found 819.1095.
UV-vis (cyclohexane, λmax (ε)): 273 (32423), 336 (5820) nm (M−1 cm−1).
1 H NMR (400 MHz, CDCl3, δ): 7.52–7.43 (m, 5H), 7.43–7.37 (m, 2H), 7.11 (d, J = 10.0 Hz, 2H), 2.83–2.76 (m, 8H), 2.15–2.03 (m, 16H).
13 C{ 1 H} NMR (75 MHz, CDCl3, δ): 172.9 (q, 2JC,F = 36.5 Hz), 150.0, 145.5 (d, 4JC,P = 3.1 Hz), 139.6 (d, 2JC,P = 15.3 Hz), 138.4, 137.9 (q, 4JC,F = 3.2 Hz), 137.3 (d, 3JC,P = 12.6 Hz), 136.3, 134.5, 132.6 (d, 2JC,P = 15.3 Hz), 132.3, 132.0, 130.9 (d, 1JC,P = 63.7 Hz), 129.2 (d, 3JC,P = 12.4 Hz), 126.5 (d, 1JC,P = 64.3 Hz), 116.5 (q, 1JC,F = 290.8 Hz), 38.3, 38.1, 23.1, 15.5, 14.8 ppm.
19 F NMR (376 MHz, CDCl3, δ): −71.9 (s) ppm.
31 P{ 1 H} NMR (162 MHz, CDCl3, δ): 6.1 (s) ppm.
IR (ATR, ): 2955 (m, νC–H), 2922 (m, νC–H), 2850 (m, νC–H), 1680 (s, νCO), 1527 (w), 1434 (s), 1424 (s), 1332 (w), 1246 (w), 1221 (m), 1194 (s), 1139 (s), 1100 (s), 1061 (m), 1016 (s), 911 (m), 868 (s), 751 (s), 739 (s), 716 (s), 687 (s), 659 (m), 581 (w), 525 (s), 507 (s), 453 (m) cm−1.
HRMS (ESI-TOF, m/z): calculated for [M + Na]+ 1073.0252; found 1073.0243.
UV-vis (cyclohexane, λmax (ε)): 262 (47527), 336 (7727) nm (M−1 cm−1).
Preparation of phosphine selenides
Phosphine selenides were prepared by the addition of grey selenium powder to an NMR tube containing 1 in CDCl3 and leaving the tube at 30 °C for 30 min. The reaction proceeded with 100% yield. The obtained selenide (6oo) was isolated by filtration and then characterized photochemically.1 H NMR (400 MHz, CDCl3, δ): δ 7.70–7.62 (m, 2H), 7.54–7.51 (m, 2H), 7.48–7.42 (m, 1H), 7.40–7.34 (m, 2H), 7.00 (d, J = 9.4 Hz, 2H), 2.83–2.73 (m, 8H), 2.12–2.03 (m, 16H) ppm.
13 C{ 1 H} NMR (151 MHz, CDCl3, δ): 173.0 (q, 2JC,F = 36.4 Hz), 150.1, 145.2 (d, 3JC,P = 3.4 Hz), 138.5, 138.2 (d, 2JC,P = 9.8 Hz), 137.9 (q, 4JC,F = 3.1 Hz), 136.8 (d, 3JC,P = 13.3 Hz), 136.6, 134.1, 132.8 (d, 1JC,P = 84.6 Hz), 132.3, 132.1 (d, 3JC,P = 3.3 Hz), 131.5 (d, 2JC,P = 12.3 Hz), 130.8 (d, 1JC,P = 89.9 Hz), 128.6 (d, 2JC,P = 13.4 Hz), 116.6 (q, 1JC,F = 290.7 Hz), 38.2, 38.2, 23.1, 15.5, 14.9 ppm.
19 F NMR (376 MHz, CDCl3, δ): −71.9 (s) ppm.
31 P{ 1 H} NMR (162 MHz, CDCl3) δ 8.3 (s) ppm.
IR (ATR, ): 2958 (w, νC–H), 2925 (w, νC–H), 2850 (w, νC–H), 1724 (w), 1681 (s, νCO), 1591 (w), 1518 (w), 1436 (m), 1376 (w), 1333 (w), 1260 (w), 1221 (w), 1195 (m), 1143 (s), 1098 (m), 1014 (m), 909 (w), 868 (m), 799 (w), 731 (s), 689 (w), 658 (w), 548 (w), 526 (w), 455 (w) cm−1.
HRMS (ESI-TOF, m/z): calculated for [M + Na]+ 921.0062; found 921.0055.
UV-vis (cyclohexane, λmax (ε)): 272 (42900), 330 (9582) nm (M−1 cm−1).
Author contributions
A. S. – performed most of the experimental work, DFT calculations, data analysis and prepared the original draft; M. V. – performed part of the photochemical experiments; J. H. – performed data analysis, supervised the project and prepared the original draft; A. L. – supervised the DFT calculations and revised the draft; R. M. S. and E. H.-H. – supervised the project, revised the draft and acquired funding. All authors have read and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 860322. M. V., J. H. and R. M. S. also acknowledge support from Generalitat de Catalunya through grant 2021 SGR 00064. A. S. is thankful for receiving doctoral funding from the Marie Skłodowska-Curie grant agreement no. 860322 and additional financial support from the Graduate School Building with Molecules and Nano-objects (BuildMoNa).References
- M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174 CrossRef CAS PubMed.
- T. Fukaminato, S. Ishida and R. Métivier, NPG Asia Mater., 2018, 10, 859 CrossRef CAS.
- J. Boelke and S. Hecht, Adv. Opt. Mater., 2019, 7, 1900404 CrossRef.
- D. Majee and S. Presolski, ACS Catal., 2021, 11, 2244 CrossRef CAS.
- H.-B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, 2108289 CrossRef CAS.
- Z. Li, X. Zeng, C. Gao, J. Song, F. He, T. He, H. Guo and J. Yin, Coord. Chem. Rev., 2023, 497, 215451 CrossRef CAS.
- J. Andréasson and U. Pischel, Chem. Soc. Rev., 2015, 44, 1053 RSC.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982 RSC.
- G. C. Thaggard, J. Haimerl, R. A. Fischer, K. C. Park and N. B. Shustova, Angew. Chem., Int. Ed., 2023, 62, e202302859 CrossRef CAS.
- O. Nevskyi, D. Sysoiev, J. Dreier, S. C. Stein, A. Oppermann, F. Lemken, T. Janke, J. Enderlein, I. Testa, T. Huhn and D. Wöll, Small, 2018, 14, 1703333 CrossRef.
- A. Perrier, F. Maurel and D. Jacquemin, Acc. Chem. Res., 2012, 45, 1173 CrossRef CAS.
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719 RSC.
- N. M.-W. Wu, M. Ng and V. W.-W. Yam, Nat. Commun., 2022, 13, 33 CrossRef CAS.
- J. Areephong, W. R. Browne and B. L. Feringa, Org. Biomol. Chem., 2007, 5, 1170 RSC.
- K. Matsuda and M. Irie, J. Am. Chem. Soc., 2001, 123, 9896 CrossRef CAS.
- S. Kobatake and M. Irie, Tetrahedron, 2003, 59, 8359 CrossRef CAS.
- D. Zhang, C. Fan, C. Zheng and S. Pu, Dyes Pigm., 2017, 136, 669 CrossRef.
- (a) S. Kobatake, S. Kuma and M. Irie, Bull. Chem. Soc. Jpn., 2004, 77, 945 CrossRef CAS; (b) R. Arai, S. Uemura, M. Irie and K. Matsuda, J. Am. Chem. Soc., 2008, 130, 9371 CrossRef CAS; (c) J. Areephong, H. Logtenberg, W. R. Browne and B. L. Feringa, Org. Lett., 2010, 12, 2132 CrossRef CAS; (d) B. Li, J.-Y. Wang, H.-M. Wen, L.-X. Shi and Z.-N. Chen, J. Am. Chem. Soc., 2012, 134, 16059 CrossRef CAS; (e) S. Wei, X. Li, C. Fan, G. Liu and S. Pu, Tetrahedron, 2017, 73, 6164 CrossRef CAS; (f) T. Biellmann, A. Galanti, J. Boixel, J. A. Wytko, V. Guerchais, P. Samorì and J. Weiss, Chem. – Eur. J., 2018, 24, 1631 CrossRef CAS; (g) Y. Wang, Q. Zhou, X. He, Y. Zhang, H. Tan, J. Xu, C. Wang, W. Wang, X. Luo, J. Chen and L. Xu, Chin. Chem. Lett., 2022, 33, 1613 CrossRef CAS.
- L. Ordronneau, V. Aubert, R. Métivier, E. Ishow, J. Boixel, K. Nakatani, F. Ibersiene, D. Hammoutène, A. Boucekkine, H. Le Bozec and V. Guerchais, Phys. Chem. Chem. Phys., 2012, 14, 2599 RSC.
- F. Medici, N. Goual, V. Delattre, A. Voituriez and A. Marinetti, ChemCatChem, 2020, 12, 5573 CrossRef CAS.
- (a) Z. Xu, Y. Cao, B. O. Patrick and M. O. Wolf, Chem. – Eur. J., 2018, 24, 10315 CrossRef CAS; (b) F. Buß, M. Das, D. Janssen-Müller, A. Sietmann, A. Das, L. F. B. Wilm, M. Freitag, M. Seidl, F. Glorius and F. Dielmann, Chem. Commun., 2023, 59, 12019 RSC.
- D. Sud, R. McDonald and N. R. Branda, Inorg. Chem., 2005, 44, 5960 CrossRef CAS.
- (a) F.-R. Dai, B. Li, L.-X. Shi, L.-Y. Zhang and Z.-N. Chen, Dalton Trans., 2009, 10244 RSC; (b) J. Yin, Y. Lin, X. Cao, G.-A. Yu, H. Tu and S. H. Liu, Dyes Pigm., 2009, 81, 152 CrossRef CAS; (c) J. Liang, J. Yin, Z. Li, C. Zhang, Di Wu and S. H. Liu, Dyes Pigm., 2011, 91, 364 CrossRef CAS; (d) H.-M. Wen, J.-Y. Wang, M.-Q. Hu, B. Li, Z.-N. Chen and C.-N. Chen, Dalton Trans., 2012, 41, 11813 RSC; (e) B. Li, Y.-H. Wu, H.-M. Wen, L.-X. Shi and Z.-N. Chen, Inorg. Chem., 2012, 51, 1933 CrossRef CAS.
- G. Bianchini, G. Strukul, D. F. Wass and A. Scarso, RSC Adv., 2015, 5, 10795 RSC.
- A. Sherstiuk, A. Lledós, P. Lönnecke, J. Hernando, R. M. Sebastián and E. Hey-Hawkins, ChemRxiv DOI:10.26434/chemrxiv-2023-vw3zz.
- M. Villabona, S. Wiedbrauk, F. Feist, G. Guirado, J. Hernando and C. Barner-Kowollik, Org. Lett., 2021, 23, 2405 CrossRef CAS.
- A. D. Sponza, Di Liu, E. P. Chen, A. Shaw, L. Diawara and M. Chiu, Org. Biomol. Chem., 2020, 18, 7238 RSC.
- D. W. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51 RSC.
- (a) M. T. Indelli, S. Carli, M. Ghirotti, C. Chiorboli, M. Ravaglia, M. Garavelli and F. Scandola, J. Am. Chem. Soc., 2008, 130, 7286 CrossRef CAS; (b) V. Aubert, V. Guerchais, E. Ishow, K. Hoang-Thi, I. Ledoux, K. Nakatani and H. Le Bozec, Angew. Chem., Int. Ed., 2008, 47, 577 CrossRef CAS; (c) M. N. Roberts, C.-J. Carling, J. K. Nagle, N. R. Branda and M. O. Wolf, J. Am. Chem. Soc., 2009, 131, 16644 CrossRef CAS PubMed; (d) M. N. Roberts, J. K. Nagle, M. B. Majewski, J. G. Finden, N. R. Branda and M. O. Wolf, Inorg. Chem., 2011, 50, 4956 CrossRef CAS; (e) J. C.-H. Chan, W. H. Lam, H.-L. Wong, N. Zhu, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12690 CrossRef CAS; (f) L. Ordronneau, V. Aubert, R. Métivier, E. Ishow, J. Boixel, K. Nakatani, F. Ibersiene, D. Hammoutène, A. Boucekkine, H. Le Bozec and V. Guerchais, Phys. Chem. Chem. Phys., 2012, 14, 2599 RSC.
- (a) H. A. Bent, Chem. Rev., 1961, 61, 275 CrossRef CAS; (b) C. C. Levin, J. Am. Chem. Soc., 1975, 97, 5649–5655 CrossRef CAS; (c) A. Muller, S. Otto and A. Roodt, Dalton Trans., 2008, 650 RSC.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1 CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458 CrossRef CAS.
- M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, (Revision B.01), Gaussian Inc., Pittsburgh, PA, 2016 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 2003, 56, 2257 CrossRef; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chem. Acc., 1990, 77, 123 Search PubMed.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- R. S. Sánchez, R. Gras-Charles, J. L. Bourdelande, G. Guirado and J. Hernando, J. Phys. Chem. C, 2012, 116, 7164 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt04246d |
| This journal is © The Royal Society of Chemistry 2024 |