
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Twinned versus linked organometallics - bimetallic “half-baguette” pentalenide complexes of Rh(I)†
Hugh J.
Sanderson
 a,
Gabriele
Kociok-Köhn
b,
Claire L.
McMullin
a and
Ulrich
Hintermair
*ac
a,
Gabriele
Kociok-Köhn
b,
Claire L.
McMullin
a and
Ulrich
Hintermair
*ac
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: u.hintermair@bath.ac.uk
bMaterial and Chemical Characterisation Facility, University of Bath, Claverton Down, Bath BA2 7AY, UK
cInstitute for Sustainability, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 28th February 2024
Abstract
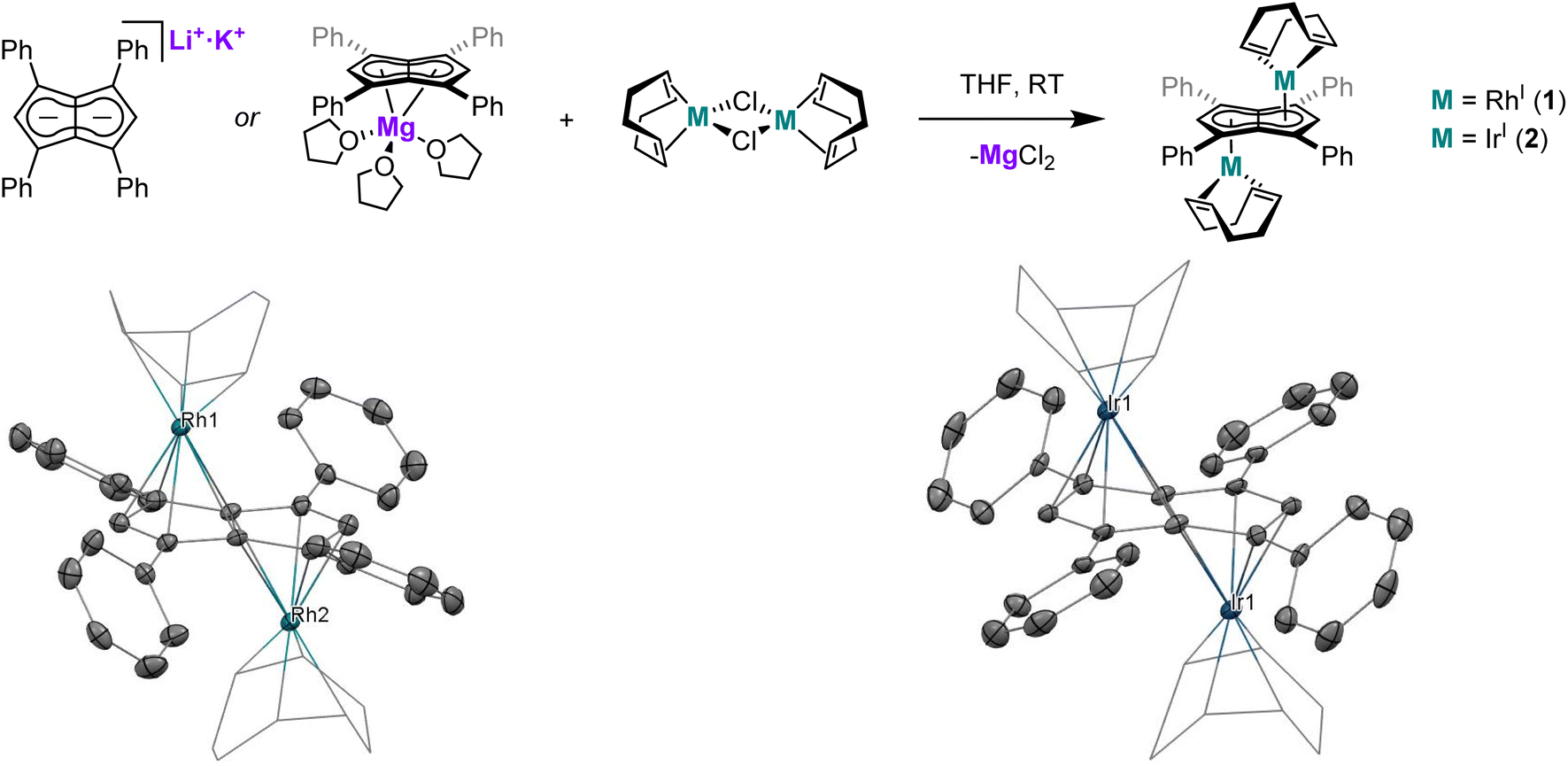
The application of Mg[Ph4Pn] and Li·K[Ph4Pn] in transmetalation reactions to a range of Rh(I) precursors led to the formation of “half-baguette” anti-[RhI(L)n]2[μ:η5:η5Ph4Pn] (L = 1,5-cyclooctadiene, norbornadiene, ethylene; n = 1, 2) and syn-[RhI(CO)2]2[μ:η5:η5Ph4Pn] complexes as well as the related iridium complex anti-[IrI(COD)]2[μ:η5:η5Ph4Pn]. With CO exclusive syn metalation was obtained even when using mono-nuclear Rh(I) precursors, indicating an electronic preference for syn metalation. DFT analysis showed this to be the result of π overlap between the adjacent M(CO)2 units which overcompensates for dz2 repulsion of the metals, an effect which can be overridden by steric clash of the auxiliary ligands to yield anti-configuration as seen in the larger olefin complexes. syn-[RhI(CO)2]2[μ:η5:η5Ph4Pn] is a rare example of a twinned organometallic where the two metals are held flexibly in close proximity, but the two d8 Rh(I) centres did not show signs of M–M bonding interactions or exhibit Lewis basic behaviour as in some related mono-nuclear Cp complexes due to the acceptor properties of the ligands. The ligand substitution chemistry of syn-[RhI(CO)2]2[μ:η5:η5Ph4Pn] was investigated with a series of electronically and sterically diverse donor ligands (P(OPh)3, P(OMe)3, PPh3, PMe3, dppe) yielding new mono- and bis-substituted complexes, with E-syn-[RhI(CO)(P{OR})3]2[μ:η5:η5Ph4Pn] (R = Me, Ph) characterised by XRD.
Introduction
The past decade has witnessed renewed interest in bimetallic complexes and the opportunities they offer in terms cooperative effects that derive from attractive or repulsive metal–metal interactions1 within molecularly defined homometallic and heterobimetallic complexes of the s-, d- and f-block.2–4 Bimetallics featuring Rh(I) are a particularly useful test for intermetallic effects due to its diamagnetic nature, useful 103Rh coupling information, wide range of known mono-nuclear complexes and rich reactivity (stoichiometric as well as catalytic). The majority of known bimetallic Rh(I) complexes consist of a ligand with two distinct binding sites connected by a linker moiety, where the size and nature of the linker determines the proximity and the extent of electronic communication between the two Rh(I) centres.5–19 With a view on cooperative effects it is thus useful to distinguish between linked bimetallics (two monometallic complexes tethered together) and twinned bimetallics where the two metals are held closely enough for direct metal–metal interactions to exist.20,21 This distinction is not purely a function of atomic distances but depends on orbital overlap and is thus a sliding scale akin to the Robin–Day classification,22,23 where the extremes of a truly twinned bimetallic would be class III and a distantly linked bimetallic class I. A particularly interesting ligand platform to explore bimetallic effects is pentalenide (Pn2−, C8H62−), the bicyclic analogue of cyclopentadienide (Cp−, C5H5−), itself one of the most widely used ligands in organometallic chemistry.24 The bicyclic nature of Pn2− offers unique advantages over its monocyclic relative cyclooctatetraenide (COT2−, C8H82−), including the formation of mono- and di-nuclear complexes paired with the ability to engage in a wide range of hapticities that allow for flexibility in the metal binding.25 Monometallic Pn2− complexes often show folding around the central bridgehead carbon–carbon bond to engage in η8 bonding, a mode that thus far is exclusive to early d- and f-block metals with a d0 electron count.26,27 Bimetallic Pn2− complexes can be classified as either syn- or anti-based on the relative orientation of the two metals, and can exhibit a range of coordination modes to Pn2− with η5/η5, η5/η3, η3/η3 and η5/η1 known to exist.27 The η1/η1 mode is also known for Si and Sn.28–30Syn-Pn2− complexes are most common for the homobimetallic double-sandwich (or perhaps more apposite, baguette) complexes of the type M2Pn2 which are known for Ti–Ni31–35 as well as Rh and Pd.36 Although intriguing compounds with regards to their structure, bonding and (photo)electrochemistry, in terms of chemical reactivity and potential use in bimetallic catalysis1–4 the corresponding syn-bimetallic “half-baguette” complexes (analogous to the widely used mononuclear “half-sandwich” Cp complexes) would be very interesting to access and study. Several examples of such homobimetallic half-baguette complexes (M2LxPn) of Mn,37,38 Fe,39–43 Co,43 Ni,44 Ru,45,46 Re37,38 Rh and Ir47 have been reported bearing a range of differently substituted pentalenide frameworks, including PnR (R = Ph, Me, NMe2, H), 1,3-(SiMe3)2Pn2−, 1,3,5-(SiMe3)3Pn2− and 1,2,3,4,5,6-Me6Pn2− (Pn*). However, the factors that lead to syn/anti selectivity are not yet clear, nor have any of these complexes been used in ligand exchange reactions or catalysis. Intriguingly, the close proximity of two metals in syn bimetallic Pn2− complexes can result in various and flexible degrees of metal–metal bonding. This can range from a double bond as in the homobimetallic baguette complex [Ti(Pn†)]2 (Pn† = 1,4-(iPr3Si)2Pn) which can undergo a wide range of reactivity including CO2,48,49 H2 and CH activation,50,51 to cases where the metal centres repel each other, such as syn-[M(CO)2]2[Pn*] (M = Co, Rh, Ir) which were found to have M–M bond orders of −0.11, −0.06 and 0.16 for Co, Rh and Ir, respectively.27,47 Further modulation of the reactivity within pentalenide complexes can be achieved by ligand exchange reactions. O'Hare and co-workers have derivatised their monometallic group 4 Pn*2− dichloride complexes to introduce a variety of alkyl (Me, allyl, CH2Ph), phenyl and amide (NMe2, NPh2) ligands to study their effects on stoichiometric CO2 activation,52–54 and similar modulation has allowed for the synthesis of group 4 mixed sandwich complexes.55–57 However, auxiliary ligand exchange reactions of late d-block pentalenide complexes are unknown save for Manriquez and co-workers who reported that [Cp*RuII(μ:η5:η3Pn)RhI(COD)] will liberate COD to form [Cp*RuII(μ:η5:η3Pn)RhI(CO)2] when exposed to a CO atmosphere.58We have recently described the facile and modular syntheses of tetra-substituted dihydropentalenes59 and introduced the first tetra-arylated pentalenide, M2[Ph4Pn] (M = Li, Na, K)60 and the first alkaline earth pentalenide, Mg[Ph4Pn].61,62 The increased solubility and ease of separation of MgX2 transmetalation by-products (through induced precipitation by addition of 1,4-dioxane63,64) are key advantages of Mg[Ph4Pn] over M2[Ph4Pn] which make it a prime candidate to explore the coordination chemistry of Ph4Pn2− by way of salt metathesis. The majority of known pentalenide complexes contain pentalenide scaffolds containing electron-donating groups (such as alkyl and trialkylsilyl substituents), and the electron-withdrawing nature of the phenyl substituents should make Ph4Pn2− significantly less reducing and thus more suited to bind to electron-rich, late d-block metals. Indeed, recent DFT calculations have shown that the presence of the four phenyl groups reduces the HOMO–LUMO gap of Ph4Pn2− by 0.26 eV relative to Pn2− alongside a lowering in the absolute energy levels of both the HOMO and the LUMO (by 0.20 eV and 0.46 eV respectively).65 The phenyl groups of Ph4Pn2− may allow for additional metal-arene interactions66–68 that have not been seen with known (non-arylated) pentalenide complexes. In this work, we report the synthesis and characterisation of a series of bimetallic Rh(I) half-baguette pentalenide complexes and compare the outcome of transmetalation with Mg[Ph4Pn] to that of Li·K[Ph4Pn]. Furthermore, we explore the reactivity of a dirhodium tetracarbonyl complex (syn-[RhI(CO)2]2[μ:η5:η5Ph4Pn]) in ligand substitution reactions to evaluate the tolerance of the Ph4Pn2− scaffold to a range of donor and acceptor ligands.
Results and discussion
Synthesis and structure of syn- and anti-bimetallic Rh(I) and Ir(I) complexes
XRD analysis of single crystals of 1 grown from standing of a THF solution at −35 °C revealed the two rhodium(I) centres to be bound to Ph4Pn2− in an anti-geometry, with rhodium–C5–centroid (Rh–Ct) distances of 1.9391(11) Å and 1.9502(11) Å (Fig. 1). The Cw–Ct–Rh angles of 86.4° and 87.4° suggested the Rh(I) centres to sit closer to the wingtip carbons than the bridgehead carbons (CB), as also reflected in the corresponding Rh–C distances where the Rh–Cw distances were on average 0.15 Å shorter than the Rh–CB distances. The heterobimetallic Pn2− complex anti-[Cp*RuII(μ:η5:η3Pn)RhI(COD)] is the only other known pentalenide complex to contain a RhI(COD) fragment, exhibiting Rh–Ct distances (1.944 Å) and Rh–Ct–Cw angles (86.6°) similar to that of 1 in the solid state but without any reported Rh–Cw coupling in solution.58
 | ||
| Fig. 1 Synthesis of anti-[RhI(COD)]2[μ:η5:η5Ph4Pn] (1) and anti-[IrI(COD)]2[μ:η5:η5Ph4Pn] (2) (top) and corresponding X-ray crystal structures with thermal ellipsoids at the 50% probability level (bottom; hydrogen atoms omitted for clarity). | ||
Using the same procedure as described for 1, the analogous reaction of [IrI(COD)(μ-Cl)]2 with Mg[Ph4Pn] resulted in the formation of the isostructural and isoelectronic complex anti-[IrI(COD)]2[μ:η5:η5Ph4Pn] (2). The two equivalent Hw protons of 2 were found at the same chemical shift as in 1 (6.10 ppm in C6D6) but a 7 ppm upfield shift of Cw was noticed in the Ir complex (92.3 ppm vs. 99.1 ppm for Rh). XRD analysis of single crystals of 2 confirmed the anti-geometry of the two iridium(I) centres as in 1. The M–Ct distances in 2 (1.9241(19) Å, 1.9290(17) Å) were up to 0.026 Å shorter than in the rhodium complex 1 but otherwise the two compounds were iso-structural. The only previously reported iridium Pn2− complex syn-[IrI(CO)2]2[μ:η5:η5Pn*]47 had an Ir–Ct distance of 2.167 Å, significantly longer than in 2. However, this difference is due to a combination of differences in the auxiliary ligands, Pn2− frameworks, and geometry. 2 also exhibited some deviation towards Cw, as reflected in the shorter Ir–Cw distances than the Ir–CB distances (by 0.17 Å) and the more acute Cw–Ct–Ir angles (86.3° and 86.9°), greater than what was noted for 1.
Rh(I) and Ir(I) d8 complexes typically prefer 16 e− configuration so a slight deviation from η5 to η3 may be expected, as η5 coordination would yield a formal 18 e− count for each metal. The hapticity of the L3X2-type pentalenide ligand can be quantified with the displacement parameter Δ defined as the difference in the mean M–CB and M–CNB distances (CNB = non-bridgehead carbons).47,69,70 Relatively small ring slippage values of Δ = 0.13 and 0.15 can thus be calculated for 1 and 2, respectively, which suggest a formal η5 coordination of both Rh(I) and Ir(I) to Ph4Pn2−. The slightly larger value for 2 compared to 1 may be attributed to increased atomic size of Ir versus Rh.47 In contrast, the Rh centre in anti-[Cp*RuII(μ:η5:η3Pn)RhI(COD)] was reported as bound η3 to Pn2−, suggesting a formal 16 e− count. However, based on the XRD data reported for this complex, a value of Δ = 0.20 can be calculated which is markedly less than what was reported for syn-[RhI(CO)2]2[Pn*] and that found for complex 5 (vide infra) which are both considered η5 and thus 18 e− per Rh(I). Complexes 1 and 2 also displayed close structural similarities to their mononuclear analogues [CpMI(COD)] (M = Rh,71,72 Ir73), with the COD ligand adopting the expected distorted boat conformation in all cases.
When the transmetalations of [RhI(COD)(μ-Cl)]2 and [IrI(COD)(μ-Cl)]2 were performed using Li·K[Ph4Pn]60 instead of Mg[Ph4Pn], the same colour changes (dark red for Rh, dark yellow for Ir) were observed and analysis by 1H and 13C{1H} NMR spectroscopy confirmed the formation of 1 and 2, respectively (Fig. S4, S5, S9 and S10†). This observation showed that the nature of the counter cation(s) of Ph4Pn2− does not influence the outcome of transmetalation reactions in donor solvents such as THF where these salts exist as solvent-separated ion pairs, despite the formal presence of a LiCp-like unit60 which was found to be too reducing in the case of reacting Li2[Pn*] with Rh(I) and Ir(I) precursors.47 This difference in reactivity likely reflects the electronic nature of the pentalenide used, in line with recent calculations showing Ph4Pn2− to have significantly lower frontier orbital energies than Pn2− and its alkylated derivatives,65 making it a less reducing π base (donor) and a better π acid (acceptor).
 | ||
| Fig. 2 Synthesis of anti-[RhI(NBD)]2[μ:η5:η5Ph4Pn] (3) (top) and its X-ray crystal structure with thermal ellipsoids at the 50% probability level (bottom; hydrogen atoms omitted for clarity). | ||
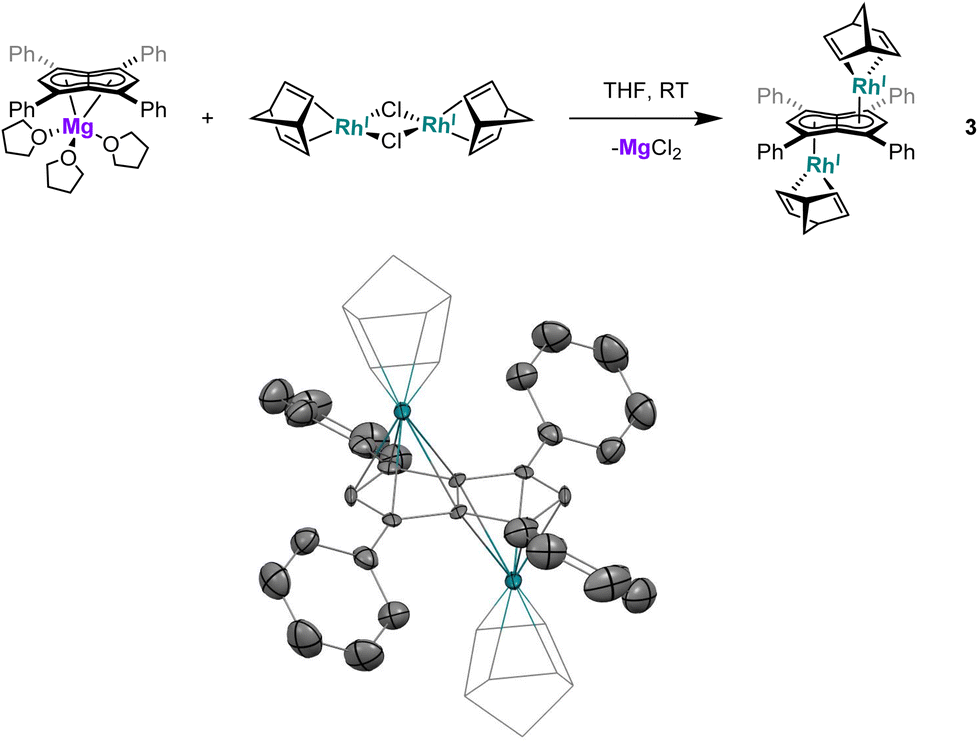
Utilising the ethylene complex [RhI(C2H4)2(μ-Cl)]2 featuring a minimally substituted, monodentate olefin ligand in a transmetalation with Mg[Ph4Pn] in THF resulted in an immediate colour change from orange to dark yellow. The 1H NMR spectrum of the mixture revealed the appearance of a new Hw signal at 6.21 ppm, indicating successful transmetalation. After 10 minutes a black solid started to form, presumed to be elemental rhodium from decomposition, indicating the product to be unstable at room temperature. Lower temperatures and lower polarity solvents did not fully prevent this decomposition but slowed it sufficiently for analysis. In C6D6Hw of the product appeared as a doublet at 5.98 ppm (2JRhH = 0.7 Hz), 0.1–0.2 ppm upfield relative to the COD and NBD analogues. In the 13C{1H} NMR spectrum Cw was resolved as a doublet at 98.6 ppm (1JRhC = 5.2 Hz), suggesting the electronics of the COD, NBD and ethylene complexes to be very similar. Crystals of 4 could be grown from the reaction mixture at −35 °C after MgCl2 had been removed via addition of dioxane and filtration. Complex 4 was found to be isostructural to 1 and 3 by XRD (Fig. 3), with an anti-configuration and Rh–Ct distances of 1.9408(9) Å and 1.9383(9) Å, and Rh–CtCw angles of 86.6° and 87.8°.
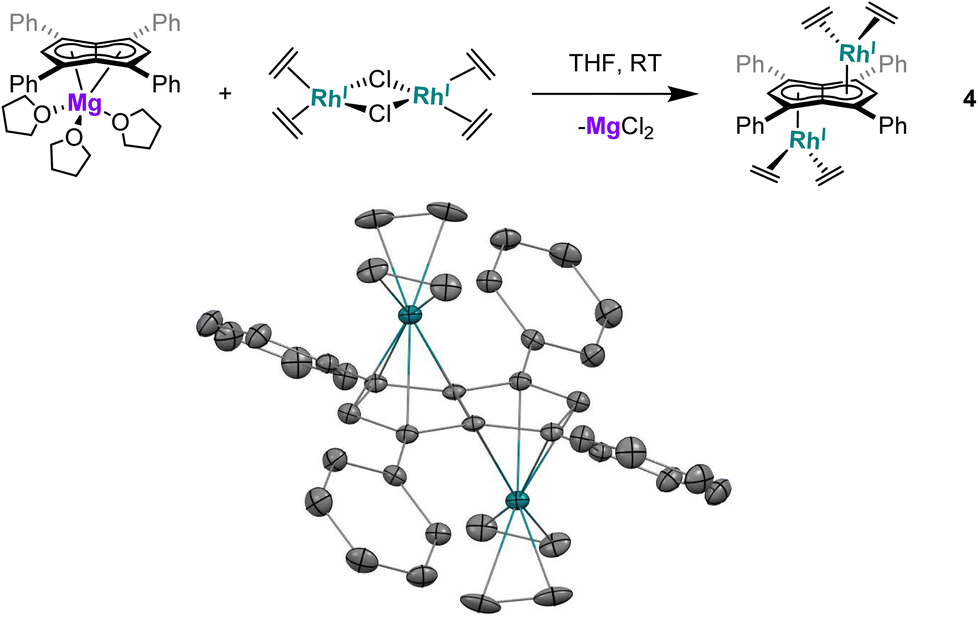 | ||
| Fig. 3 Synthesis of anti-[RhI(C2H4)2]2[μ:η5:η5Ph4Pn] (4) (top) and its X-ray crystal structure with thermal ellipsoids at the 50% probability level (bottom; hydrogen atoms omitted for clarity). | ||
 | ||
| Fig. 4 Synthesis of syn-[RhI(CO)2]2[μ:η5:η5Ph4Pn] (5, top) and its X-ray crystal structure with thermal ellipsoids at the 50% probability level (bottom; hydrogen atoms omitted for clarity). | ||
| δ Hw/ppm | δ Cw/ppm | δ CO/ppm | ν CO /cm−1 | |
|---|---|---|---|---|
| a Calculated from reported NMR data. b IR data recorded using a KBr pellet. | ||||
| [Rh(CO) 2 ] 2 [Ph 4 Pn] (5) | 6.91 (2JRhH = 0.45 Hz) | 90.1 (1JRhC = 6.96 Hz) | 194.5 (1JRhC = 83.8 Hz) | 2064, 2046, 2025, |
| [Rh(CO) 2 (Ph 2 Cp)] 74 | 5.99 (2JRhH = 0.45 Hz) | 83.7 (1JRhC = 3.17 Hz) and 85.4 (1JRhC = 3.64 Hz) | 191.3 (1JRhC = 84.0 Hz) | 2040, 1962 |
| [Rh(CO) 2 ] 2 [Pn*] 47 | n/a | 71.0 | 196.3 (1JRhC = 49.1 Hz) | 2034, 1999, 1965, 1946b |
| [Rh(CO) 2 ][Cp*] 75 | n/a | n/a | 195.1 (1JRhC = 75.6 Hz)a | 2016, 1948 |
The X-ray crystal structure of 5, depicted in Fig. 4, confirmed a syn-dirhodium arrangement. A comparison of the key structural features with syn-[RhI(CO)2]2[Pn*] as well as complexes 1–4 can be found in Table 2. As in the Pn*2− analogue, the two RhI(CO)2 fragments were located next to each other on the Ph4Pn2− with an intermetallic distance of 2.9130(4) Å, too long to ascribe any direct Rh–Rh interaction36,47 as also reflected in a formal shortness ratio (FSR) of 1.16 using a R1 value of 1.247.76,77 O'Hare related this increased distance to the C5 rings of Pn*2− being partially bent away at a hinge angle of 11.4° (defined as the angle between the plane of the Cw and non-wingtip carbons (CNW) and the plane of CNW and CB),47 with DFT analysis showing the HOMO of [RhI(CO)2]2[Pn*] to contain a Rh–Pn* antibonding component that is offset by the Rh–CO π-backdonation.27,475 exhibited a smaller hinge angle of 10.2° due to the increased π-accepting ability of the phenyl substituents in Ph4Pn2−. However, despite the changes in electronics of the pentalenide ligand an identical ring slippage value of Δ = 0.27 was found. This is noticeably larger than for the anti-complexes 1–4 signifying a greater deviation towards η3 in the syn-systems, further evidenced by the more acute Rh–Ct–Cw angles of 82.4° and 82.6°, suggesting some electronic repulsion between the RhI(CO)2 fragments. While complex 4 had a L–Rh–L versus pentalenide dihedral angle of 86.1° (close to the ideal 90° geometry, Fig. S61†), in 5 the two L–Rh–L units were angled at 77.8° relative to the pentalenide. This outward bending together with the greater Δ value towards η3 and 10.2° pentalenide hinging in 5 all contribute to an enlarged Rh–Rh distance signifying intermetallic repulsion. Previous DFT analysis of [RhI(CO)2]2[Pn*] had revealed an out of phase interaction of the metal dz2 orbitals in the HOMO−1 leading to a formal negative bond order of −0.06.27,47
| δ Hw/ppm | δ Cw/ppm | M-M/Å | M-Ct/Å | M-Ct-Cw/ ° | Ring slippage Δ | |
|---|---|---|---|---|---|---|
| anti-[Rh I (COD)] 2 [Ph 4 Pn] (1) | 6.17 | 99.3 (1JRhC = 5 Hz) | 4.5305(5) | 1.9391(11), 1.9502(11) | 86.4, 87.4 | 0.11, 0.15 |
| anti-[Ir I (COD)] 2 [Ph 4 Pn] (2) | 6.28 | 93.6 | 4.5411(5) | 1.9241(19), 1.9290(17) | 86.3, 86.9 | 0.15 |
| anti-[Rh I (NBD)] 2 [Ph 4 Pn] (3) | 6.22 (2JRhH = 0.8 Hz) | 97.4 (1JRhC = 6 Hz) | 4.5462(9) | 1.938(3) | 86.1 | 0.15 |
| anti-[Rh I (C 2 H 4 ) 2 ] 2 [Ph 4 Pn] (4) | 5.98 (2JRhH = 0.7 Hz) | 98.6 (1JRhC = 5 Hz) | 4.4862(3) | 1.9383(9), 1.9408(9) | 86.6, 87.8 | 0.14, 0.10 |
| anti-[Cp*Ru II (μ:η 5 :η 3 Pn)Rh I (COD)] 58 | 5.84 | n/a | 4.4166(4) | 1.944 | 86.6 | 0.20 |
| syn-[Rh I (CO) 2 ] 2 [Ph 4 Pn] (5) | 6.50 (2JRhH = 1.7 Hz) | 89.8 (1JRhC = 7 Hz) | 2.913(6) | 1.9917(9), 1.9940(9) | 82.4, 82.6 | 0.27 |
| syn-[Rh I (CO) 2 ] 2 [Pn*] 47 | n/a | n/a | 2.9130(4) | 1.976 | 83.0, 83.1 | 0.27 |
The average Rh–CO bond length found in 5 was 1.876(1) Å, 0.01 Å longer than in syn-[RhI(CO)2]2[Pn*] suggesting less Rh–CO backbonding in 5. This was further supported by IR spectroscopy where 5 exhibited four carbonyl bands at 1992, 2025, 2046 and 2064 cm−1, similar to what was reported for [RhI(CO)2(Ph2Cp)] (1962 and 2040 cm−1),74 and shifted to higher wavenumbers compared to syn-[RhI(CO)2]2[Pn*]47 indicative of a stronger CO bond and weaker Rh–CO backbonding as a reflection of the increased π-accepting ability of Ph4Pn2− over Pn*2−.
Interested in whether the formation of the syn-isomer was partially influenced by the use of a dimeric rhodium precursor, the reaction was repeated with the monomeric [RhI(acac)(CO)2], with the formation of 5 confirmed by NMR and IR spectroscopy. Moreover, the influence of the cation was found to be negligible as the reaction of either [RhI(acac)(CO)2] or [RhI(CO)2(μ-Cl)]2 with Li·K[Ph4Pn] also resulted in clean formation of 5 (Scheme 1).
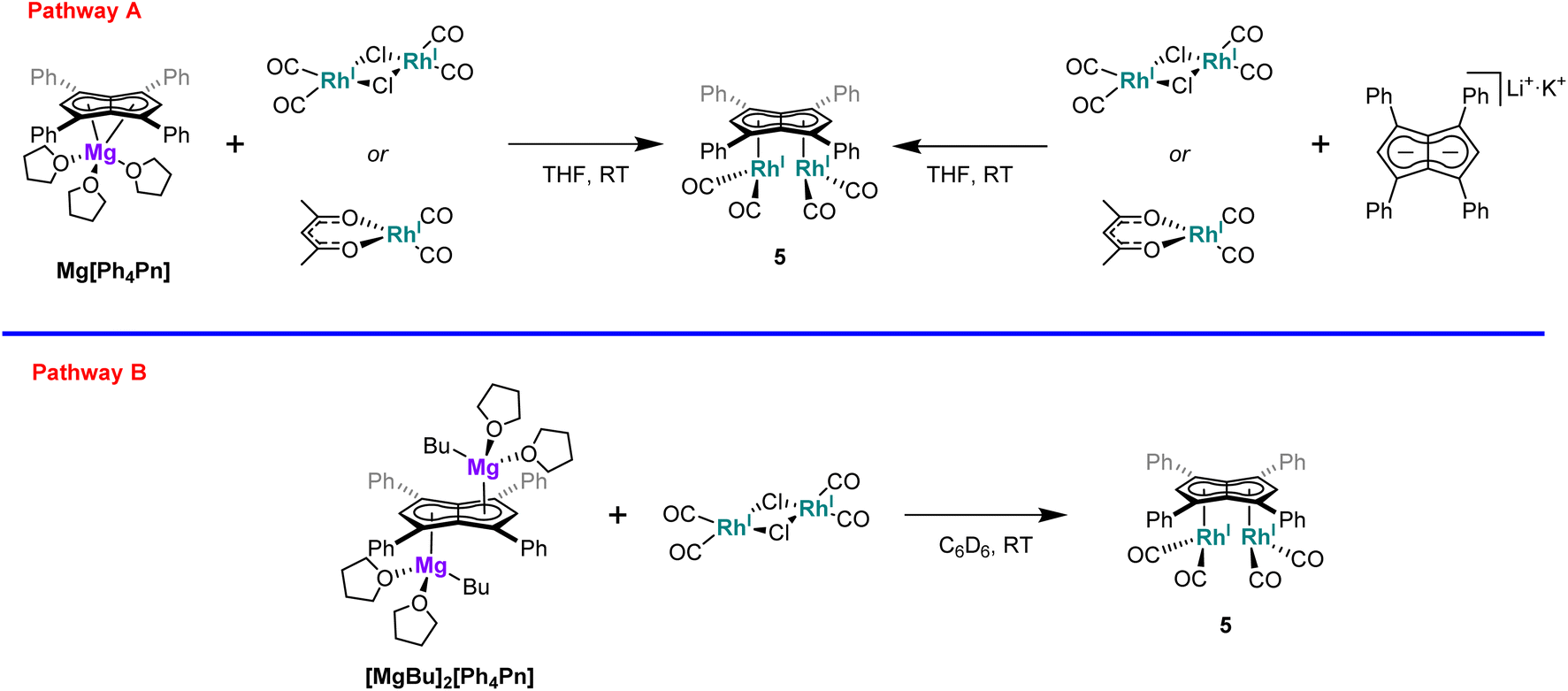 | ||
| Scheme 1 Synthetic pathways to [RhI(CO)2]2[Ph4Pn]. | ||
Electronic structure and reactivity
To gain deeper insight into the origin of syn versus anti formation on 4 and 5, comparative Density Functional Theory (DFT) calculations were carried out on syn-4, anti-4, syn-5 and anti-5 (see ESI† for details). The optimised geometries of anti-4 and syn-5 showed good agreement with the crystallographic data, and anti-4 was found to be 1.2 kcal mol−1 lower in energy than syn-4 whereas syn-5 was 4.6 kcal mol−1 more stable than anti-5. These results are fully in line with our experimental findings and show syn/anti selectivity in the transmetalation of solvent-separated group 1 and group 2 pentalenide salts with Rh(I) and Ir(I) precursors to be thermodynamically controlled. NBO calculations on syn-4 and syn-5 returned Wiberg Bond Index (WBI) values of 0.06 and 0.05 between the two Rh(I) centres, respectively, indicating no significant M–M interactions. As reported for the Pn*2− system,47 the HOMO−1 of syn-5 contained an out-of-phase (repulsive) interaction between the Rh dz2 orbitals, whilst an in-phase (attractive) interaction between the two co-planar Rh(CO)2 moieties was found in the HOMO of syn-5 (Fig. 5). Interestingly, the HOMO of syn-4 contained a similar positive interaction between the two adjacent Rh(C2H4)2 moieties (Fig. 6), suggesting that there is a general energetic preference for syn-metallation in pentalenide complexes of Rh(I) and Ir(I) with π acceptor ligands despite a repulsive M–M interaction. As the switch in isomer preference from syn to anti when going from CO to C2H4 shows, steric clashing between the ancillary ligands easily overrides the net electronic preference for syn to yield anti-geometry in a bimetallic more akin to two linked (or fused) Cp complexes.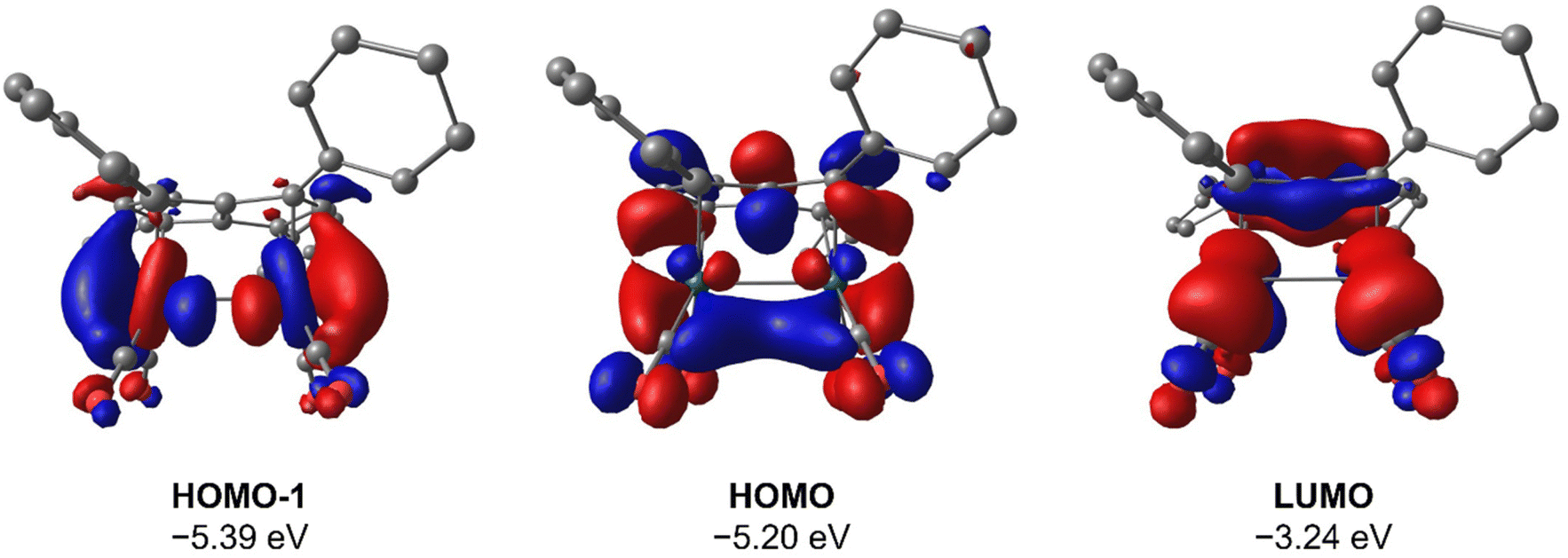 | ||
| Fig. 5 HOMO−1, HOMO and LUMO of syn-[Rh(CO)2]2[Ph4Pn] (syn-5) at the BP86/BS2//BP86/BS1 level of theory. Hydrogen atoms omitted for clarity. | ||
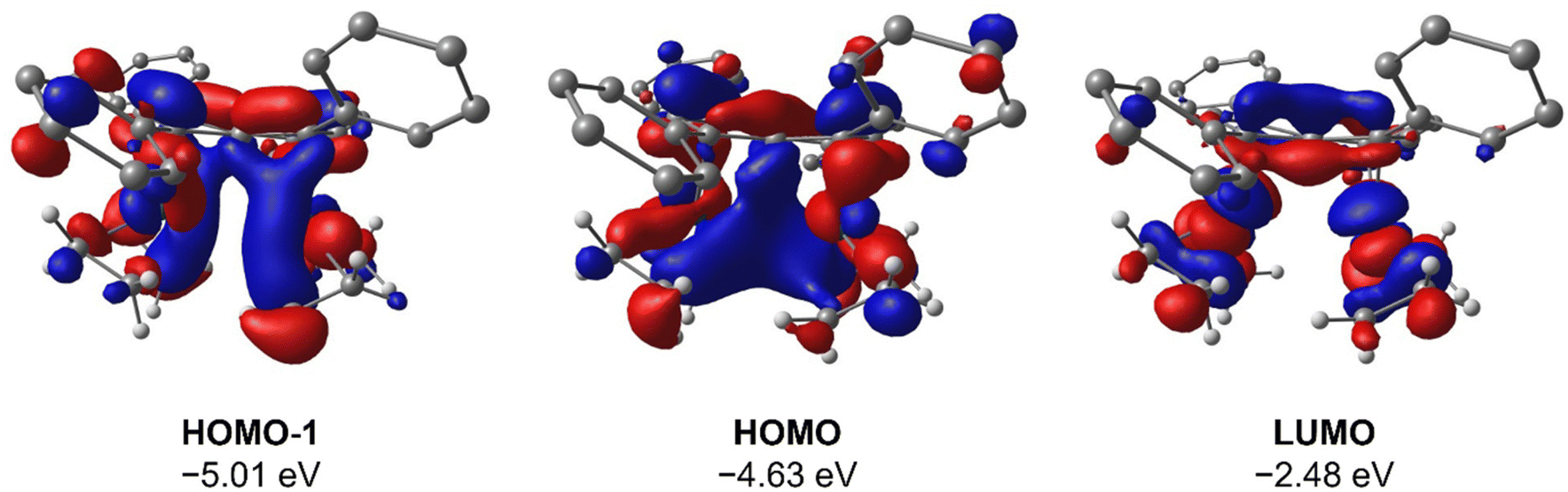 | ||
| Fig. 6 HOMO−1, HOMO and LUMO of syn-[Rh(C2H4)2]2[Ph4Pn] (syn-4) at the BP86/BS2//BP86/BS1 level of theory. Hydrogen atoms of the pentalenide omitted for clarity. | ||
To investigate whether the orientation of the metals resulted in any significant changes in their electronic structure the UV-vis spectra of 1, 5 and the monomeric derivative of 5, [RhI(CO)2(Ph2Cp)] were recorded in THF (Fig. S25†). All three displayed peaks attributable to π–π* transitions of the π-ligands (320 nm for 1 and 5, 350 nm for [Rh(CO)2(Ph2Cp)]), with a slight red shift for the monometallic complex also observed in the group 1 salts of Ph4Pn2− and Ph2Cp−,60 suggesting the orientation of the metals to have negligible influence on the main electronic transitions.
Given the repulsive dz2 interactions found in the HOMO−1 of the twinned syn bimetallics, we tested the influence of the metal orientation on the Lewis basicity of anti-3 and syn-5. Related mononuclear four-coordinate d8 complexes are known to display nucleophilic behaviour by virtue of their filled, non-bonding dz2 orbital which may act as a σ-donor to a range of Lewis acids.78 This effect has even been seen (and exploited for intermetallic reactivity in metal-based Lewis pairs79–82) in formally five-coordinate 18 electron η5-CpRh(I) complexes that have an empty, non-bonding dz2 orbital,79 where their nucleophilic behaviour may either arise from an electron-rich Rh–L bond or a temporary η5–η3 slip of the Cp ligand. However, addition of AlCl3 to anti-3 and syn-5 in THF showed no changes in their 1H NMR spectra even after one week at room temperature. Control reactions with 1 and [RhI(CO)2(Ph2Cp)] also showed no reactivity with AlCl3, highlighting the low nucleophilicity of these complexes which may be ascribed to the phenyl groups lowering the HOMO and LUMO of the Pn2− and increase the π-acceptor character of the ligand.65 This is in contrast to the electron-rich Rh(I) complexes furnished with strongly σ-donating phosphines (e.g. PMe3) and electron rich π-ligands (e.g.Cp*) which serve to raise the energy of the HOMO and thus increase their nucleophilicity.83–85
Ligand substitution chemistry of [RhI(CO)2]2[Ph4Pn] (5)
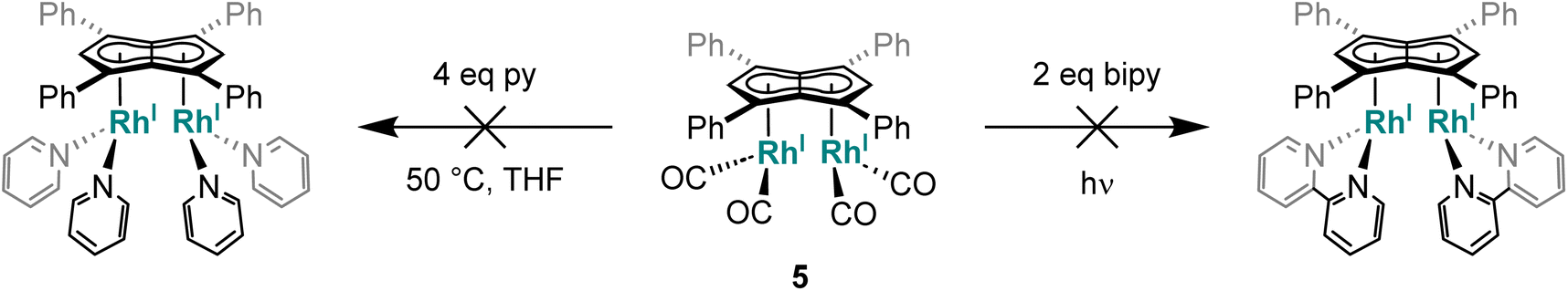 | ||
| Scheme 2 Attempted introduction of pyridyl ligands to syn-[RhI(CO)2]2[Ph4Pn]. | ||
This surprising inertness of 5 either reflects either a kinetic barrier due to steric shielding by the phenyl substituents of the pentalenide or a thermodynamic preference of the complex for strong acceptor ligands like CO. To probe this question further we tried to introduce a range of P-donor ligands with varying degrees of donor/acceptor abilities.
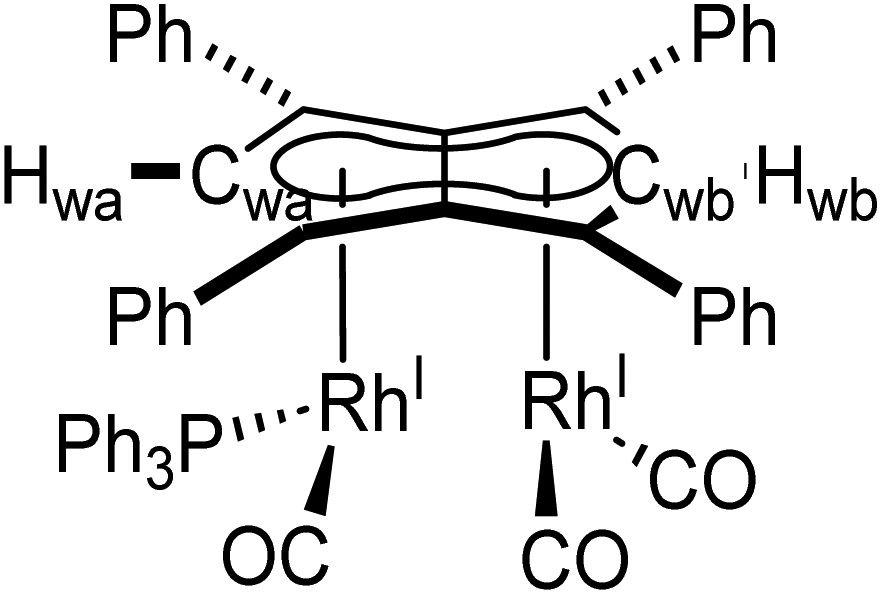
Phosphines. The addition of one equivalent of PPh3 to 5 in THF led to no discernible colour change over several days at room temperature, however, the 1H NMR spectrum showed a desymmetrisation of the pentalenide ligand consistent with monosubstitution to form [RhI(CO)2;RhI(CO)(PPh3)][μ:η5:η5Ph4Pn] (6) (Scheme 3). Two singlets at 6.23 ppm and 6.71 ppm in the 1H NMR spectrum could be assigned to two new wingtips Hwa and Hwb respectively (as confirmed by 1H-31P HMBC), indicating an electronically asymmetric Ph4Pn2− environment with one C5-ring bound to a more electron-rich RhI(CO)(PPh3) fragment and one C5-ring bound to a more electron-deficient RhI(CO)2 fragment. This was reflected with the chemical shift of Hwb being closer to that of Hw in 5, with the slight upfield shift of 0.2 ppm reflective of the delocalised electronic nature of the Ph4Pn2− anion. The 31P{1H} NMR spectrum of 6 revealed a doublet centered at 48.1 ppm (1JRhP = 202 Hz), similar to that reported for [CpRhI(CO)(PPh3)] (52.7 ppm, 1JRhP = 199.5 Hz).94 The UV-vis spectrum of 6 displayed similar π–π* transitions of the Ph4Pn2− in the UV region around 300 nm to that of 5, however, with an additional broad band centred at 838 nm (ε = 7383 M−1 cm−1) which was absent for the symmetrical complexes 5 and 11 (Fig. S56 and 57†) and indicated intermetallic charge transfer between the electronically inequivalent Rh(I) centres in 6.22,23
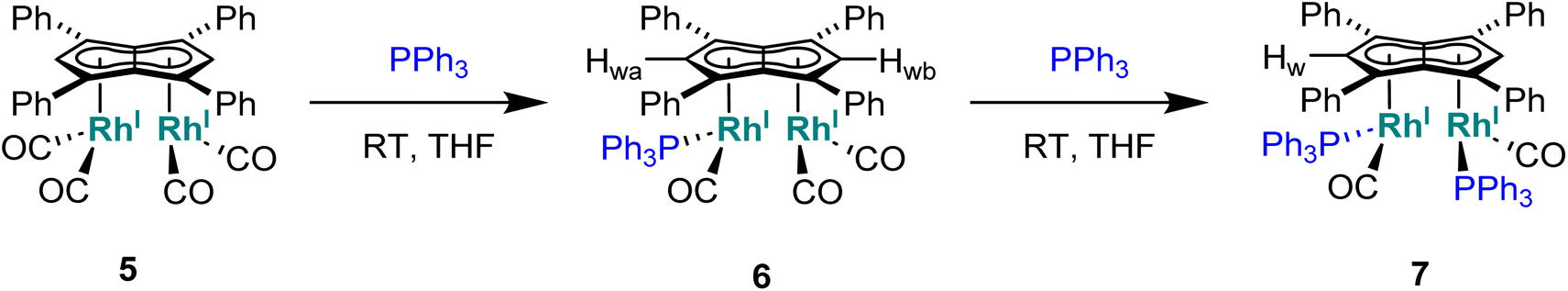 | ||
| Scheme 3 Synthesis of mono and bis-triphenylphosphine derivatives of syn-[RhI(CO)2]2[Ph4Pn] (5). | ||
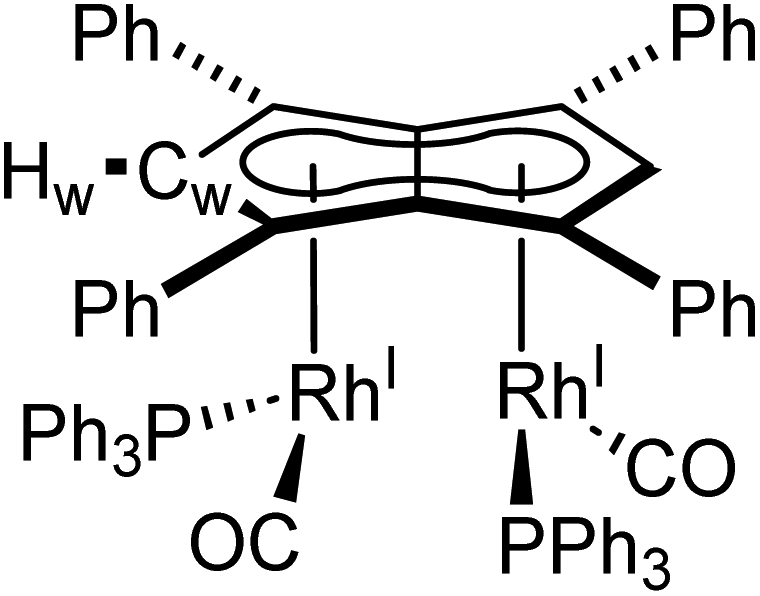
When two equivalents of PPh3 were added to 5, initially only the monosubstituted complex 6 was observed in the 1H and 31P{1H} NMR spectra of the solution (Fig. S31†), but over the course of 12 hours the emergence of a new doublet at 51.4 ppm (1JRhP = 198 Hz) along with the diminishing of the signals assigned to 6 was observed in the 31P{1H} NMR spectrum. The new signal is ascribed to the symmetrically disubstituted product 7, but full characterisation was impeded by the transformation not going to completion and all attempts at isolating 7 from the dynamic mixture being unsuccessful. The electronic similarity of 7 to 6 and to [CpRhI(CO)(PPh3)]94 reflected in their NMR data strongly suggested each RhI(CO)2 centre underwent mono-substitution instead of one RhI(CO)2 centre undergoing double substitution to yield a species akin to [CpRhI(PPh3)2] (31P = 57.3 ppm, 1JRhP = 222 Hz).95 Using more than two equivalents of PPh3 per 5 led to complex mixtures that contained 6 and 7 but also a multitude of other products (Fig. S31†), showing the introduction of phosphines to be more easily achievable than pyridines albeit with some barrier likely arising from steric hindrance (PPh3 being much larger than CO).
When the smaller and more σ-donating trimethylphosphine was employed in place of PPh3 a colour change from the dark yellow of 5 to red was noticed over the course of five minutes at room temperature. The 31P{1H} NMR spectrum of this solution showed a signal centred at −24.0 ppm with second order coupling, indicative of a rhodium-phosphorous dimer (Fig. S34†).96 Two broad peaks at −16.2 ppm and −34.4 ppm were also observed suggesting some fluxionality within the product(s) formed. Although all attempts at isolating a clean sample lead to decomposition, standing of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF/pentane solution of the crude reaction mixture at −35 °C overnight led to the formation of crystals of the ion pair [RhII(CO)(PMe3)4][Ph4Pn] (8, Fig. 7) where four PMe3 ligands had detached the Rh-carbonyl centre from the pentalenide. To the best of our knowledge this is the first crystallographically characterised uncomplexed pentalenide and exhibits a perfectly co-planar core with the phenyl rings twisted by 21.2°–26.8° to minimise steric repulsion between adjacent rings and allow some conjugation of the charge into the phenyl substituents. This rotation is less than what was found for Li2[Ph4Pn]60 (29.3°–35.8°) but within the range seen for Mg[Ph4Pn] (17.1°–35.2)°.61
:1 THF/pentane solution of the crude reaction mixture at −35 °C overnight led to the formation of crystals of the ion pair [RhII(CO)(PMe3)4][Ph4Pn] (8, Fig. 7) where four PMe3 ligands had detached the Rh-carbonyl centre from the pentalenide. To the best of our knowledge this is the first crystallographically characterised uncomplexed pentalenide and exhibits a perfectly co-planar core with the phenyl rings twisted by 21.2°–26.8° to minimise steric repulsion between adjacent rings and allow some conjugation of the charge into the phenyl substituents. This rotation is less than what was found for Li2[Ph4Pn]60 (29.3°–35.8°) but within the range seen for Mg[Ph4Pn] (17.1°–35.2)°.61
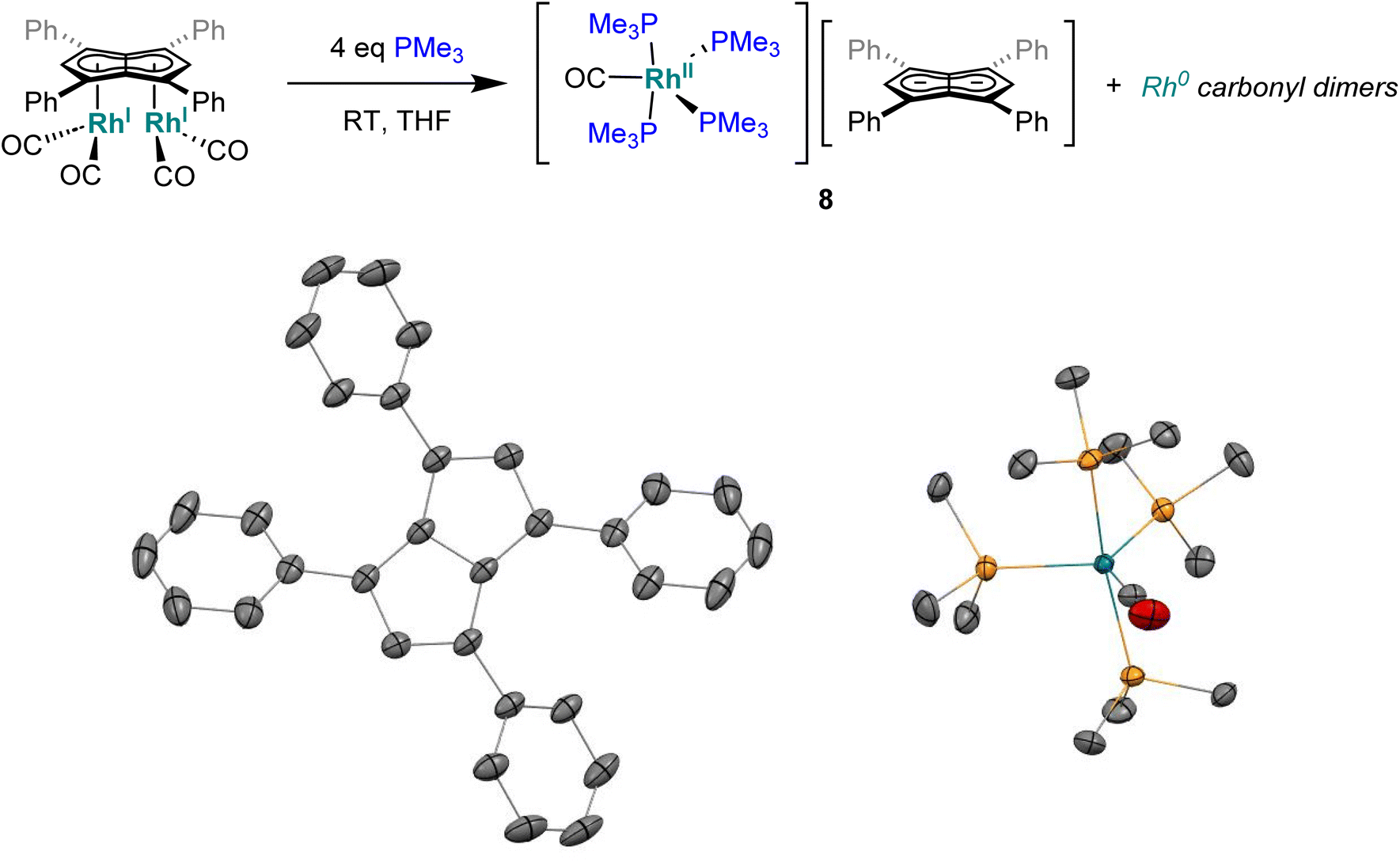 | ||
| Fig. 7 Synthesis of [RhII(PMe3)4(CO)][Ph4Pn] (8) (top) and its X-ray crystal structure with thermal ellipsoids at the 50% probability level (bottom; hydrogen atoms omitted for clarity). | ||
The cationic fragment of 8 consisted of a Rh(II) centre in a distorted trigonal bipyramidal geometry with PMe3 groups occupying both axial and two equatorial positions with one carbonyl group occupying the third equatorial site. Exchange between the equatorial and axial positions is likely responsible for the observation of two inequivalent, broad signals in the 31P{1H} NMR spectra, although variable temperature NMR data were inconclusive (Fig. S35†) due to the paramagnetic nature of Rh(II) which was independently confirmed by the Evans method (Fig. S36†). In the solid state, the Rh–Peq bonds were 0.056 Å longer than Rh–Pax bonds (2.3227(4)–2.3859(7) Å), with the axial PMe3 groups bent away from the equatorial PMe3 to give a Pax–Rh–Pax angle of 165.6° to relive steric clash between the methyl groups. The related cation [RhI(CO)(PMe3)4]+ has previously been reported,97 however, with no spectroscopic or crystallographic data provided. Mononuclear Rh(II) complexes are uncommon and often adopt square planar or octahedral geometries in the presence of bulky chelating ligands with 15 e− or 19 e− counts, respectively,98–104 whereas the cation of 8 is a rare example of a five-coordinate 17 electron complex with monodentate ligands only. The formation of a dicationic Rh(II) complex from the two adjacent Rh(I) centres in 5 is likely the result of a disproportionation reaction induced by the introduction of the strongly σ-donating PMe3 ligand, producing dimeric Rh(0) carbonyl species responsible for the signal at −24.0 ppm in the 31P{1H} NMR spectrum of the product mixture.
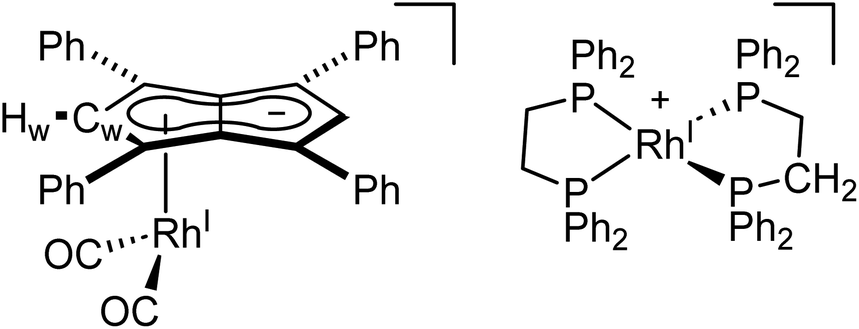
To see if a bidentate ligand would span across the two rhodium centres (μ2) or chelate around a single metal (κ2) in 5 the introduction of bis(diphenylphosphino)ethane (dppe) was tested. Whereas the 1H NMR spectrum showed no significant change in the position of Hw after adding dppe to 5 in THF, the 31P{1H} NMR spectrum showed a new doublet at 57.6 ppm (1JRhP = 128.3 Hz) with Cw still exhibiting rhodium-carbon coupling. Crystals suitable for XRD, grown from standing of the reaction mixture at −35 °C, revealed the formation of the non-contact ion pair [RhI(dppe)2][RhI(CO)2(η5Ph4Pn)] (9, Fig. 8) where one Rh(I) centre was bound to Ph4Pn2− in an η5 coordination fashion with a Rh-Ct distance of 1.959(2) Å, 0.35 Å shorter than the equivalent distance in the di-Rh(I) complex 5. This significant shortening likely is a consequence of the monometallic coordination of RhI(CO)2 to Ph4Pn2− leading to increased interaction of the cationic d8 centre with the dianionic 10π system. There was also less pronounced deviation to η3 in 9 compared to 5, as evidenced by the slight increase in the Rh–Ct–Cw angle (84.1° vs. an average 82.5° in 5) and a decrease in the ring slippage value Δ = 0.23 (cf. 0.27 in 5). Moreover, a smaller hinge angle of 8.4° was found for 9, presumably due to a relaxation of Ph4Pn2 and the loss of steric strain from syn-dimetallation, whilst the uncomplexed C5-ring exhibited a hinge angle of 2°. The Rh–CO distances in 9 were also 0.016 Å shorter than in 5 suggesting a greater degree of back-bonding in the former, which was further supported by the IR spectrum of 9 exhibiting two CO bands at 1980 cm−1 and 1918 cm−1 (Fig. S41†). The [RhI(CO)2(η5Ph4Pn)] anion could also be crystallised with a [Mg2Cl3(THF)6] cation when MgCl2 was not removed by addition of dioxane prior to dppe addition (Fig. S42†). The cationic [RhI(dppe)2]+ resulting from mono-demetallation of 5 by dppe has previously been reported with a 31P NMR signal at 57 ppm (1JRhP = 134.2 Hz),105 consistent with the NMR signature of 9 in the crude reaction mixture. The fact that one signal for Hw and Cw respectively was observed in the NMR spectra of 9 implies rapid ring slippage of the RhI(CO)2 fragment across the pentalenide core in solution.
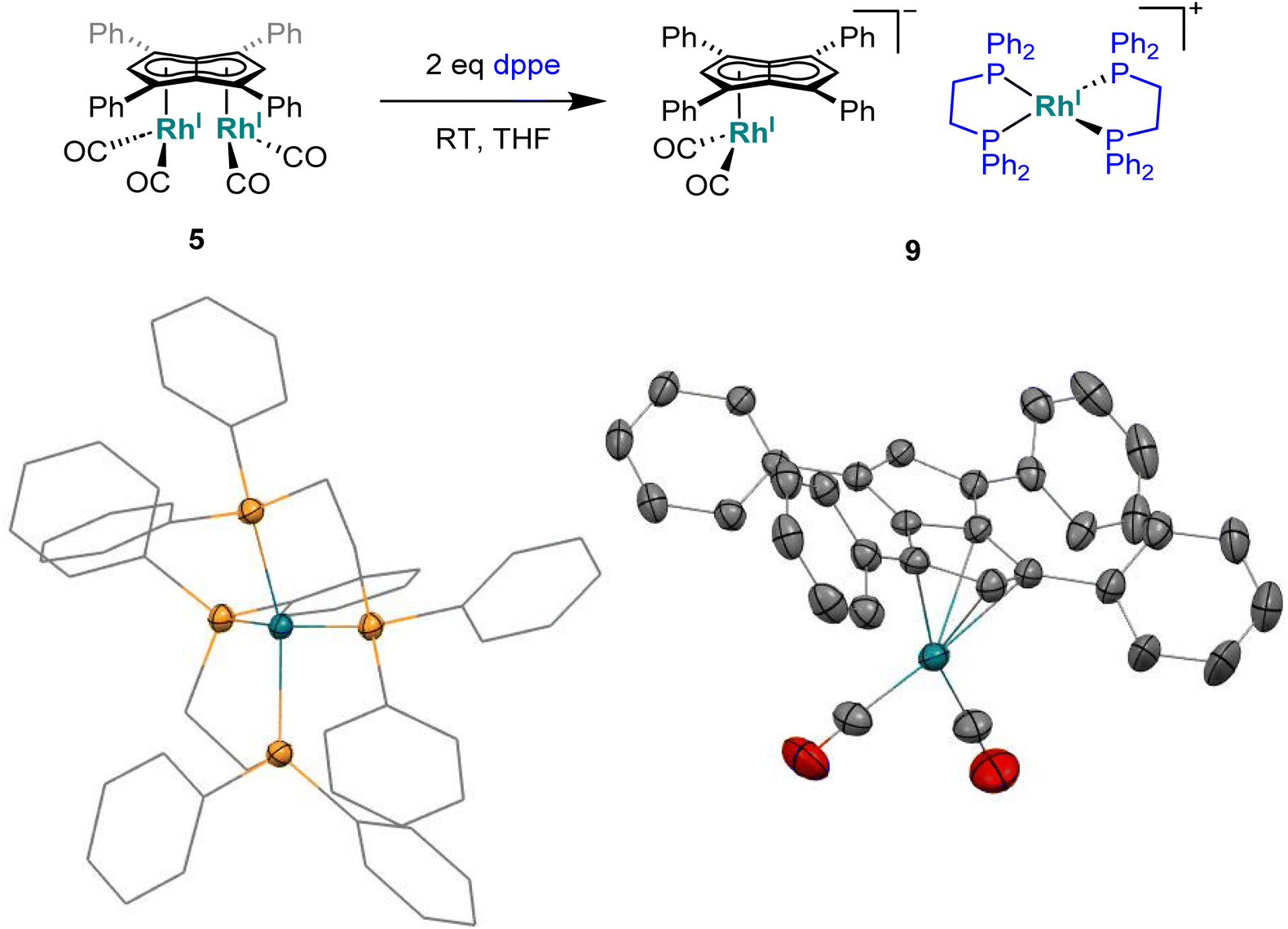 | ||
| Fig. 8 Synthesis of [RhI(dppe)2][RhI(CO)2(η5Ph4Pn)] (9) (top) and its X-ray crystal structure with thermal ellipsoids at the 50% probability level (bottom; hydrogen atoms omitted for clarity). | ||
The above results show that although 5 does not react with mono- or bidentate pyridines, chelating phosphines readily bind to the RhI(CO)2 centre to displace both carbonyls and lead to demetallation of the pentalenide. With monodentate phosphines progressive carbonyl substitution occurs on both RhI(CO)2 centres which in the case of strong σ-donors can lead to a disproportionating demetallation reaction. Thus, ligand exchange reactions using less basic and more π-accepting phosphites106 with lower steric demand in comparison to phosphines were tried next.
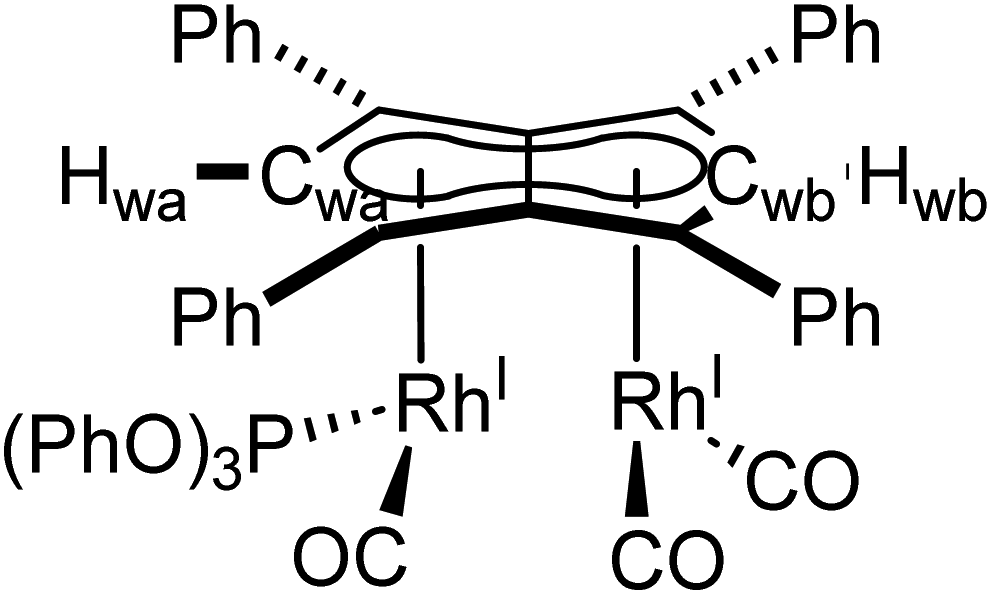
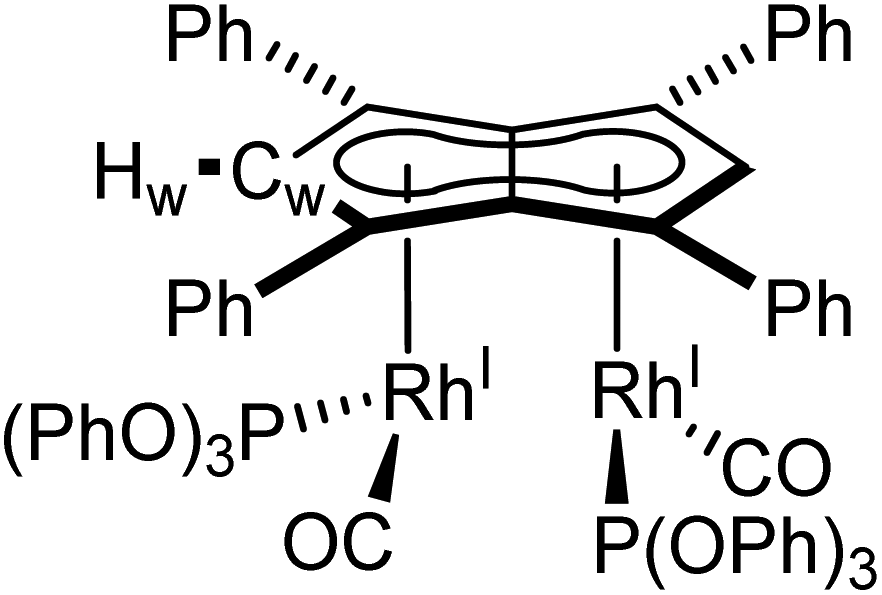
Phosphites. As seen before with PPh3, the addition of one equivalent P(OPh)3 to a solution of 5 in THF resulted in the desymmetrisation of the Ph4Pn2− ligand with two new wingtip signals Hwa and Hwb appearing in the 1H NMR at 6.22 ppm and 6.75 ppm, respectively, suggesting monosubstitution on one Rh centre of 5 to form [RhI(CO)2;RhI(CO)(P{OPh}3)][μ:η5:η5Ph4Pn] (10). The two inequivalent wingtips could be distinguished by 1H–31P-HMBC, with HWa at 6.22 ppm displaying a cross peak with the new 31P signal at 130.6 ppm. The difference in wingtip shift, Δw, of 0.53 ppm is similar to that observed with monosubstitution with PPh3 in 6 (Δw = 0.48 ppm). Interestingly, while the magnitude of the Rh–P coupling in 10 was consistent with that reported for [CpRhI(CO)(P{OPh}3)] (1JRhP = 328.8 Hz), the 31P{H} shift of the asymmetrically twinned bimetallic was found 70 ppm downfield compared to the mononuclear complex (130.6 ppm versus 62.7 ppm).94 When an excess of P(OPh)3 was added to 5, the 1H NMR spectrum showed an increase in the symmetry of the aromatic region again, with a single Hw signal emerging at 5.90 ppm (1 ppm upfield of that of 5) suggesting increased back-bonding from the metals to Ph4Pn2−. These signatures were consistent with each RhI(CO)2 fragment undergoing monosubstitution each to give the symmetrically di-substituted complex [RhI(CO)(P{OPh}3)]2[μ:η5:η5Ph4Pn] (11) instead of forming inequivalent RhI(CO)2 and RhI(P{OPh}3)2 moieties within the same complex. The 31P{1H} NMR spectrum of 11 showed a major peak centred at 129.3 ppm (1JRhP = 342.7 Hz), close to the shift of 10 indicating an electronically similar environment consistent with two RhI(P{OPh}3)(CO) moieties. When two equivalents of P(OPh)3 were used a mixture of the mono- and bis-complexes 10 and 11 was observed, suggesting some (perhaps steric) barrier to forming the bis-product. With four or more equivalents of P(OPh)3 clean formation of 11 was consistently observed, with no signs of demetallation unlike when using phosphines. X-ray crystallographic analysis of 11, the first heteroleptic half-baguette pentalenide complex, confirmed retention of the syn-geometry of metal fragments with the phosphite ligands arranged in an E-geometry (Fig. 9). The Rh–Rh distance of 2.960 Å was slightly longer in 11 than in 5 (2.913 Å), leading to a FSR of 1.19, likely as a consequence of the increased steric crowding around each Rh(I) centre. The Rh–Ct distances were comparable to that of 5 (1.9969(12) Å and 2.0013(11) versus 1.997 Å) and the ring slippage values Δ were marginally lower (0.25 versus 0.27) revealing a slightly smaller degree of deviation towards η3 than in 5. The hinge angle of 11 showed that the C5-rings deviated from planarity by an average of 8.5°, 1.7° less than in 5 whilst the angle between the L–Rh–L plane and Pn plane was 76.7°. The Rh–CO bond lengths were on average 0.03 Å shorter than in 5 suggesting an increase in Rh–CO back-bonding which was further supported by blueshifted CO bands at 1982 and 1961 cm−1 in the IR spectrum of 11 (cf. 1992, 2025, 2046 and 2064 cm−1 in 5). The analogous reaction with P(OMe)3 led to the formation of a variety of products, including a bis-substituted product, E-syn-[RhI(CO)(P{OMe}3)]2[μ:η5:η5Ph4Pn] (12) which was found to be isostructural to 11 by single-crystal XRD exhibiting a ring slippage value of Δ = 0.27, a hinge angle of 9.5° and an angle of 78.2° between the L–Rh–L plane and the plane of Ph4Pn2−.
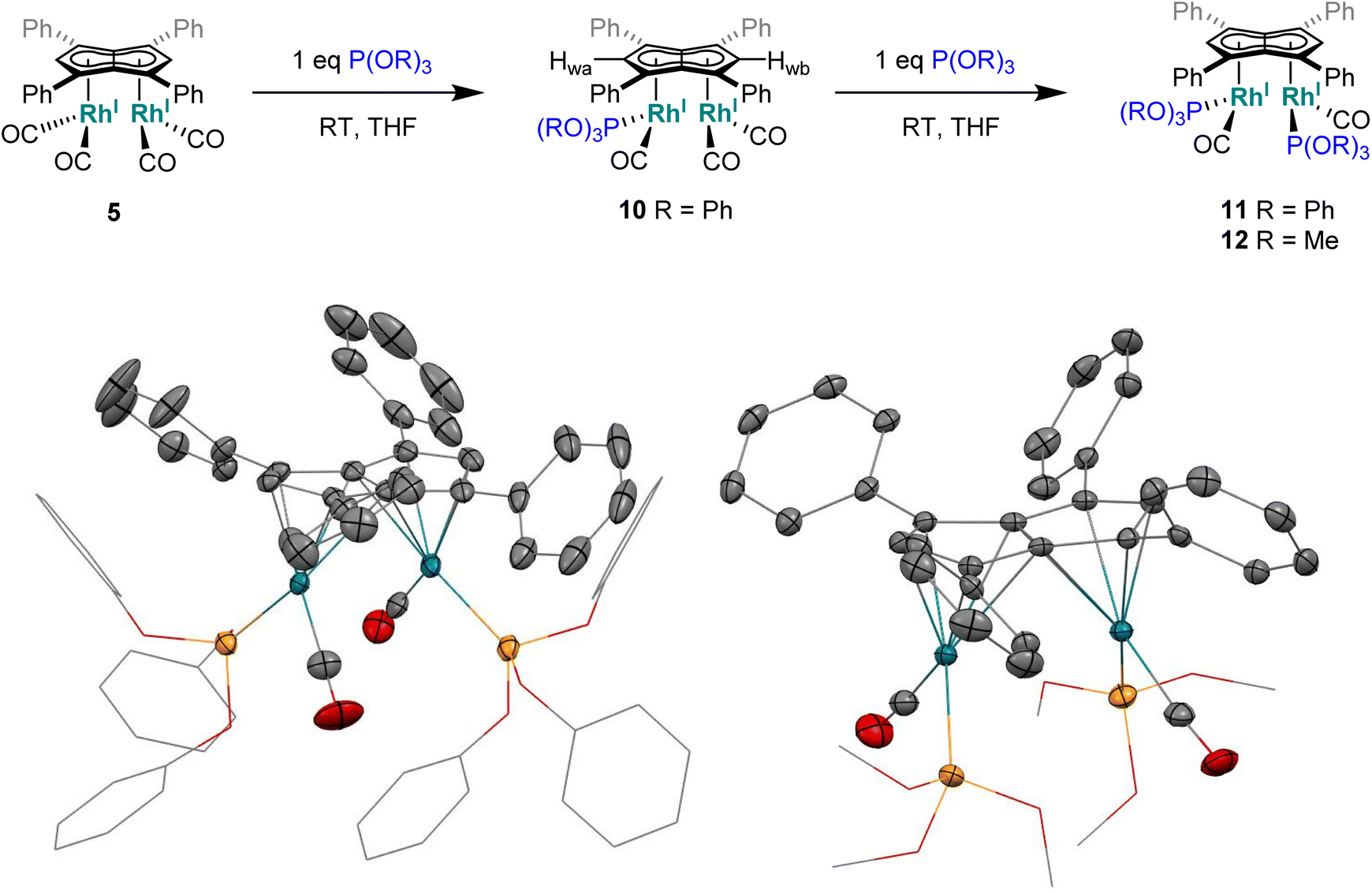 | ||
| Fig. 9 Synthesis of E-syn-[RhI(CO)(P{OPh}3)]2[μ:η5:η5Ph4Pn] (11) and E-syn-[RhI(CO)(P{OMe}3)]2[μ:η5:η5Ph4Pn] (12) (top) and their X-ray crystal structure with thermal ellipsoids at the 50% probability level (bottom; hydrogen atoms omitted for clarity). | ||
Conclusion
We have reported the synthesis of a series of bimetallic Rh(I) half-baguette complexes from transmetalation of Ph4Pn2− salts with [RhIL2X]2 complexes. With either Mg[Ph4Pn] or Li·K[Ph4Pn] the same transmetalation product was obtained, consistent with the notion that in ethereal solution s-block pentalenides exist as solvent-separated ion pairs.60,61 However, Mg[Ph4Pn] provides practical advantages with cleaner formation of the transmetalation product and easy removal of MgCl2 by induced precipitation of [MgCl2(dioxane)]. [RhI(COD)]2[Ph4Pn] (1), [IrI(COD)]2[Ph4Pn] (2), [RhI(NBD)]2[Ph4Pn] (3) and [RhI(C2H4)2]2[Ph4Pn] (4) all formed exclusively as linked anti-bimetallics, whilst the twinned syn-bimetallic [RhI(CO)2]2[Ph4Pn] (5) was obtained regardless whether the monometallic [RhI(acac)(CO)2] or bimetallic [RhI(CO)2(μ-Cl)]2 precursors were used. DFT calculations revealed a mix of repulsive M–M and attractive ML2–ML2 interactions which weakly favour syn arrangement in the absence of steric clash between the auxiliary ligands L. Neither anti-[RhI(NBD)]2[Ph4Pn] nor syn-[RhI(CO)2]2[Ph4Pn] displayed Lewis-basic reactivity towards AlCl3 due to being surrounded by π-acceptor ligands in a distorted trigonal–bipyramidal 18-electron configuration.Attempted ligand exchange reactions of syn-[RhI(CO)2]2[Ph4Pn] with N-donor ligands were unsuccessful, whilst the introduction of strongly σ-donating and/or chelating phosphines led to rhodium abstraction from Ph4Pn2−. With PMe3 this process was accompanied by disproportionation, forming Rh(0) dimers and the ion pair [RhII(PMe3)4(CO)][Ph4Pn] (8). With dppe a single Rh(I) centre was abstracted to yield the salt [RhI(dppe)2][RhI(CO)2(η5Ph4Pn)] (9) featuring an anionic, mono-nuclear pentalenide complex which could serve as a synthon to access heterobimetallic complexes in the future. PPh3 and P(OPh)3 reacted similarly towards 5 to successively yield the monosubstituted complexes [RhI(CO)2;RhI(CO)(PPh3)][Ph4Pn] (6) and [RhI(CO)2;RhI(CO)(P{OPh}3)][Ph4Pn] (10) respectively. The di-substituted complexes E-syn-[RhI(CO)(PPh3)]2[Ph4Pn] (7) and E-syn-[RhI(CO)(P{OR}3)]2[Ph4Pn] (R = Ph 11, R = Me 12) could be accessed using stoichiometric amounts of PPh3 and an excess of P(OR)3 respectively. The latter were characterised by XRD which showed each rhodium centre underwent monosubstitution, with overall retention of the syn-geometry and the phosphite ligands adopting E-conformation due to sterics. These results will be useful in guiding the targeted derivatisation of future transition metal pentalenide complexes to test and exploit intermetallic cooperation in linked and twinned bimetallics. Related monometallic Cp complexes have been employed in a range of chemistries including C–H and Si–H activation107 and cycloadditions108–113 as well as synthons for heterometallic clusters,114,115 offering a wealth of opportunities for bimetallic half-baguette pentalenide complexes.
Experimental
General considerations
All reactions were conducted under argon using standard Schlenk techniques or a MBraun Unilab Plus glovebox unless stated otherwise. All commercially available materials were purchased from Sigma Aldrich, Fisher or Acros.Solvents
Methanol was dried and distilled over magnesium. Toluene was dried and distilled over sodium. THF, hexane and pentane were dried and distilled over potassium. C6D6 was distilled over CaH2 and stored over 4 Å molecular sieves. TMEDA, DME and 1,4-dioxane were distilled over CaH2.Reagents
1,3,4,6-Tetraphenyl-1,2-dihydropentalene (Ph4PnH2),60 Lithium potassium 1,3,4,6-tetraphenylpentalenide (Li·K[Ph4Pn]),60 Magnesium 1,3,4,6-tetraphenylpentalenide (Mg[Ph4Pn]),61 [RhI(COD)(μ-Cl)]2,116 [IrI(COD)(μ-Cl)]2117 and [RhI(NBD)(μ-Cl)]2118 were prepared according to literature procedures. [RhI(CO)2(μ-Cl)]2 and [RhI(C2H4)2(μ-Cl)]2 were used as received.Analysis
NMR spectra were obtained using either a 500 MHz Bruker Avance III, a 500 MHz Bruker Avance III HD with nitrogen cooled CryoProbe Prodigy (BBO) or a 400 MHz Bruker AvanceNeo with room temperature iProbe. Spectra were recorded at 25 °C unless stated otherwise. Chemical shifts (δ) are given in ppm and referenced to residual proton chemical shifts from the NMR solvent for 1H and 13C{1H} spectra. UV-vis, NIR and IR spectroscopy were performed inside a MBraun Unilab Plus glovebox. UV-vis and NIR spectra were obtained using a fibre-optic AvaSpec-2048L photospectrometer (UV-vis) and an AvaSpec-NIR256 (NIR) with an AvaLight-DH-S-BAL light source and 400 μm cables (Avantes), data was collected between 250 nm and 1000 nm with an integration time of 4 ms (UV-vis) and between 910 nm and 1700 nm with an integration time of 0.26 ms (NIR). IR spectra were obtained using a Bruker Alpha II FT-IR (recorded between 4000 and 400 cm−1 with 24 scans at 2 cm−1 resolution).Single X-ray diffraction analysis was carried out at the Material and Chemical Characterisation (MC2) Facility at the University of Bath using a RIGAKU SuperNova Dual EoS2 single crystal diffractometer. Mass spectrometry was carried out at the Material and Chemical Characterisation Facility at the University of Bath using a Bruker MaXis HD ESI-QTOF with direct injection. Samples were prepared in THF inside an argon filled glovebox.
Synthesis of anti-[RhI(COD)]2[μ:η5:η5Ph4Pn] (1)
[Rh(COD)(μ-Cl)]2 (16 mg, 0.03 mmol) was dissolved in 0.5 mL THF and added, by syringe, to a THF solution of Mg[Ph4Pn] (20 mg, 0.03 mmol) in THF (0.5 ml). The reaction was stirred for one hour during which all solid dissolved and a colour change to red was observed. Then dioxane (0.3 mL) was added and the solution stirred for another hour, during which a creamy white precipitate formed. The supernatant was extracted via filtration and concentrated to 0.2 mL volume. To this, pentane (0.5 mL) was added, and the mixture stood overnight at −35 °C. The supernatant was removed, and the resultant red powder dried under vacuum for one hour to give the product (5 mg, 20%). Crystals suitable for XRD could be grown from standing of a THF solution at −35 °C.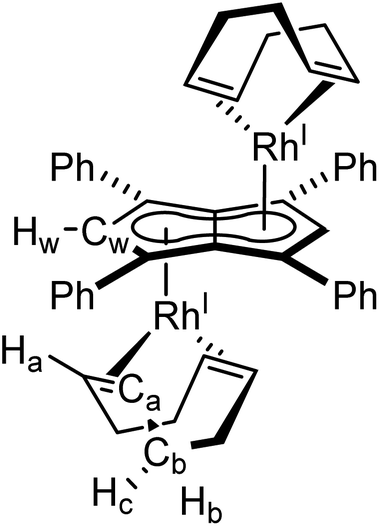
1 H NMR (500 MHz, C6D6) δ: 7.44 (d, 3JHH = 7.2 Hz, 8H, ArH), 7.13–7.09 (m, 11H, ArH), 6.10 (s, 2H, Hw), 3.92 (bs, 8H, Ha), 2.08 (bs, 9H, Hc/Hb), 1.77 (d, 2JHH = 7.9 Hz, 8H, Hc/Hb).
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 136.2, 129.7, 128.1, 126.8, 99.3 (d, 1JRhC = 4.8 Hz, Cw), 91.2 (d, 1JRhC = 4.3 Hz), 89.9, 72.9 (d, 1JRhC = 13.6 Hz, Ca), 32.3 (Cb).
HR APCI-MS (+) m/z expected for [M + H] = 829.1782, found 829.1722
Synthesis of anti-[IrI(COD)]2[μ:η5:η5Ph4Pn] (2)
[Ir(COD)(μ-Cl)]2 (20 mg, 0.03 mmol) was dissolved in 0.5 mL THF and added, by syringe, to a THF solution of Mg[Ph4Pn] (20 mg, 0.03 mmol) in THF (0.5 ml). The reaction was stirred for one hour during which all solid dissolved and a colour change to red was observed. Then dioxane (0.3 mL) was added and the solution stirred for another hour, during which a creamy white precipitate formed. The supernatant was extracted via filtration and concentrated to 0.2 mL volume. To this, pentane (0.5 mL) was added, and the solution stood overnight at −35 °C. The supernatant was removed, and the resultant yellow powder dried under vacuum for one hour to give the product (12 mg, 39%). Crystals suitable for XRD could be grown from standing of a THF solution at −35 °C.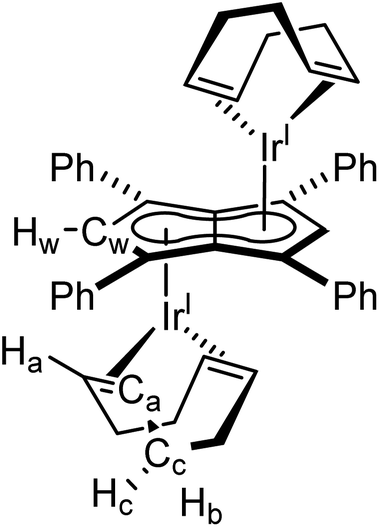
1 H NMR (500 MHz, C6D6) δ: 7.32–7.31 (m, 8H, ArH), 7.10–7.09 (m, 12H, ArH), 6.10 (s, 2H, Hw), 3.98 (bs, 8H, Ha), 2.00–1.98 (m, 9H, Hc/Hb), 1.74–1.72 (m, 8H, Hc/Hb)
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 134.9, 130.2, 128.3, 127.2, 92.3 (Cw), 86.9, 86.3, 56.1 (Ca), 33.4 (Cb)
HR APCI-MS (+) m/z expected for [M + H] = 1009.2931, found 1009.2829
Synthesis of anti-[RhI(NBD)]2[μ:η5:η5Ph4Pn] (3)
Mg[Ph4Pn] (20 mg, 0.03 mmol) was dissolved in THF (0.5 mL) and to this solution [Rh(NBD)(μ-Cl)]2 (14 mg, 0.03 mmol) in THF (0.5 mL) was added dropwise and stirred. Upon addition, complete dissolution of all solid was observed and a colour change to dark yellow was seen. The reaction was stirred for one hour during which a yellow precipitate formed. The precipitate was collected via filtration over a silica frit and washed with hexane (2 × 1 mL) and dried to afford the product as a yellow/orange powder (18 mg, 76%). Crystals suitable for XRD could be grown from standing of a THF solution at −35 °C.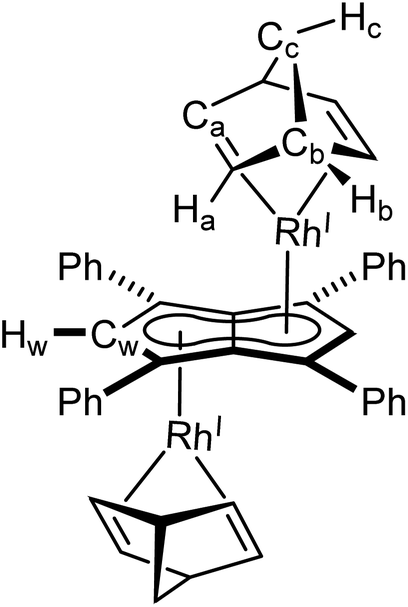
1 H NMR (500 MHz, C6D6) δ: 7.35–7.33 (m, 9H, ArH), 7.07–7.06 (m, 13H ArH), 6.22 (d, 2JRhH = 0.8 Hz, 2H, Hw), 3.4–3.32 (m, 8H, Ha), 3.17 (bs, 5H, Hb), 0.81 (s, 4H, Hc).
13 C{ 1 H} NMR: (126 MHz, C6D6) δ: 136.8, 129.5, 128.4, 126.3, 97.4 (d, 1JRhC = 5.8 Hz, Cw), 88.8 (d, 1JRhC = 4.3 Hz), 88.1, 58.1 (d, 3JRhC = 7 Hz, Cc), 48.5 (Cb), 41.3 (d, 1JRhC = 10.3 Hz, Ca).
HR APCI-MS (+) m/z expected for [M + H] = 797.1156, found 797.1205.
Synthesis of [RhI(C2H4)2]2[μ:η5:η5Ph4Pn] (4)
Mg[Ph4Pn] (20 mg, 0.03 mmol) was dissolved in THF (0.5 mL) and to this solution [Rh(C2H4)2(μ-Cl)]2 (12 mg, 0.03 mmol) in THF (0.5 mL) was added. An immediate colour change from orange to dark green was observed. Full conversion as determined by 1H NMR spectroscopy. Crystals suitable for XRD could be grown from standing of a THF solution at −35 °C, after treatment with dioxane to remove MgCl2 by-product. Due to the instability of this complex, it could not be cleanly isolated to obtain a yield.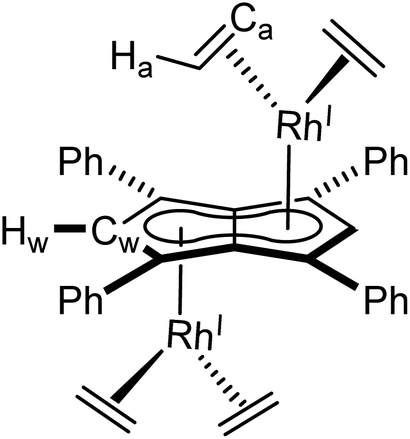
1 H NMR (500 MHz, C6D6) δ: 7.41–7.39 (m, 8H, Ar–H), 7.10–7.09 (m, 12H, Ar–H), 5.98 (d, 2JRhH = 0.7 Hz, 2H, Hw), 2.19 (d, 2JRhH = 1.8 Hz, 16H, Ha).
13 C{ 1 H} NMR: (126 MHz, C6D6) δ: 134.8, 130.8, 129.8, 127.2, 98.6 (d, 1JRhC = 5.2 Hz, Cw), 91.8 (d, 1JRhC = 4.3 Hz), 89.5, 50.4 (d, 1JRhC = 14.2 Hz, Ca).
Synthesis of syn-[RhI(CO)2]2[μ:η5:η5Ph4Pn] (5)
Method A: [Rh(CO)2(μ-Cl)]2 (12 mg, 0.03 mmol) was dissolved in THF (1 mL) and added dropwise to a solution of Mg[Ph4Pn] (20 mg, 0.03 mmol) in THF (0.5 mL) and stirred. A colour change from orange to dark yellow was observed along with complete dissolution of Mg[Ph4Pn]. The reaction was allowed to stir for one hour, after dioxane (0.3 mL) was added and the reaction stirred for a further hour during which a brown precipitate formed. The supernatant was collected via filtration and concentrated to ∼0.3 mL. To this pentane (0.5 mL) was added and the solution stood overnight at −35 °C. The supernatant was decanted off and the powder dried under vacuum to give the product (7 mg, 32%). Crystals suitable for XRD could be grown from standing of a THF/pentane solution (2:1) at −35 °C.
Method B: [Rh(acac)(CO)2] (8 mg, 0.031 mmol) was dissolved in THF (0.5 mL) and added dropwise to a solution of Mg[Ph4Pn] (10 mg, 0.015 mmol) in THF (0.5 mL). A colour change from orange to dark yellow was observed alongside complete dissolution of Mg[Ph4Pn]. Formation of the product was confirmed in situ by 1H NMR.
Method C: Li·K[Ph4Pn] (0.05 mmol) was generated in situ and to this a solution of [Rh(CO)2(μ-Cl)]2 (29 mg, 0.07 mmol) in THF (0.5 mL) was added dropwise. A colour change from red to dark yellow was observed and the product identified in situ by 1H NMR.
Method D: Li·K[Ph4Pn] (0.05 mmol) was generated in situ and to this a solution of [Rh(acac)(CO)2] (51 mg, 0.2 mmol) in THF (0.5 mL) was added dropwise. A colour change from red to dark yellow was observed and the product identified in situ by 1H NMR.
Method E: [MgnBu(THF)2][Ph4Pn] (10 mg, 0.012 mmol) was dissolved in C6D6 (0.5 mL) and to this a solution of [Rh(CO)2(μ-Cl)]2 (5 mg, 0.012 mmol) in C6D6 (0.5 mL) was added slowly. A colour change from bright yellow to dark yellow was observed and the product identified in situ by 1H NMR.
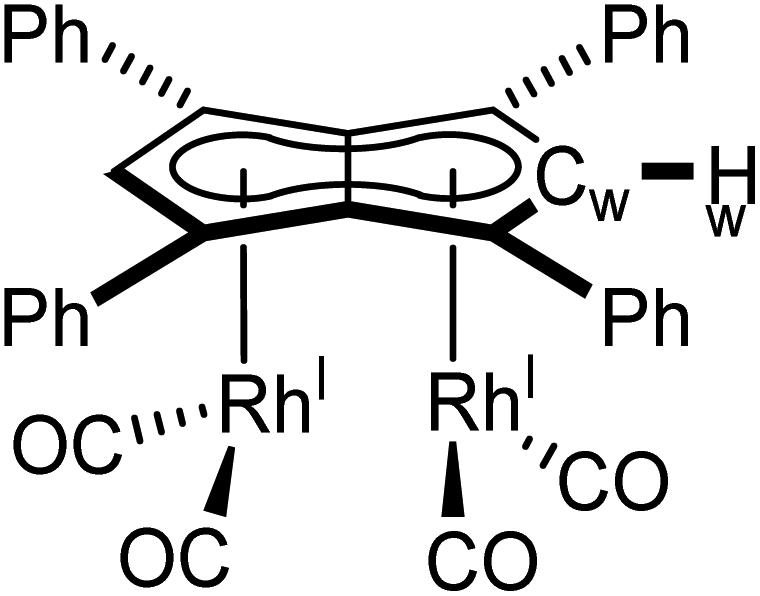
1 H NMR (500 MHz, THF-H8) δ: 7.16 (d, 3JHH = 7.1 Hz, 8H, ArH), 7.12 (d, 3JHH = 7.3 Hz, 3H, ArH), 7.04 (t, 3JHH = 7.7 Hz, 8H, ArH), 6.91 (d, 2JRhH = 1.4 Hz, 2H, Hw).
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 194.5 (d, 1JRhC = 83.8 Hz, CO), 133.7, 130.5, 128.2, 128.2, 112.1, 90.1 (d, 1JRhC = 7.0 Hz, Cw), 81.9 (d, 1JRhC = 4.3 Hz).
HR APCI-MS (+) m/z expected for [M + H] = 724.9706, found = 724.9677.
IR (ν CO , cm−1): 2064, 2046, 2025, 1992.
General procedure for ligand substitution reactions
5 was generated in situ by the addition of [Rh(CO)2(μ-Cl)]2 (12 mg, 0.03 mmol in 0.3 mL THF) to Mg[Ph4Pn] (20 mg, 0.03 mmol in 0.5 mL THF) with formation confirmed by 1H NMR. An appropriate amount of ligand was then added (0.03 mmol, 0.06 mmol, or 0.12 mmol). Solid ligands were added as a THF solution (0.3 mL) and liquids were added directly to the solution of 5, before dioxane (0.2 mL) was added, and the solution left to stand for 10 minutes after which it was syringe filtered to remove the precipitated [MgCl2(dioxane)]∞. In the cases of 6, 7, 8 and 10 the products could not be cleanly isolated and were identified in situ using multi-nuclear 1D and 2D NMR techniques supported by mass spectrometry. The original NMR spectra can be found in Fig. S28–S47.†syn-[RhI(CO)2;RhI(CO)(PPh3)][μ:η5:η5Ph4Pn] (6)
1 H NMR (500 MHz, THF-H8) δ: 6.71 (s, 1H, Hwb), 6.23 (s, 1H, Hwa).
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 195.2 (dd, 1JRhC = 84.3 Hz, 2JCP = 3.3 Hz, CO), 93.4 (Cwa), 88.7 (d, 1JRhC = 6.5 Hz, Cwb).
31 P{ 1 H} NMR (202 MHz, THF-H8) δ: 48.1 ppm (1JRhP = 202 Hz).
HR ESI-MS (+) m/z expected for [M − Rh(CO)2] = 799.1632, found = 799.1622.
syn-[RhI(CO)(PPh3)]2[μ:η5:η5Ph4Pn] (7)
31 P{ 1 H} NMR (202 MHz, THF-H8) δ: 51.4 (1JRhP = 198 Hz).
HR ESI-MS (+) m/z expected for [M] = 1192.1547, found = 1192.1459; m/z expected for [M − Rh(CO)(PPh3)] = 799.1632, found = 799.1590.
[RhII(PMe3)4(CO)][Ph4Pn] (8)
As per the general procedure, PMe3 (0.12 mmol, 1M in THF) was added to a THF solution of 5. A colour change to dark red was observed. To this solution dioxane (0.2 mL) was added and subsequently filtered. Addition of pentane (1 mL, 1:1 ratio with THF) followed by standing of the solution at −35 °C yielded dark red crystals of [RhII(PMe3)4(CO)][Ph4Pn] suitable for X-ray diffraction. Due to the instability of this complex, it could not be cleanly isolated to obtain a yield.
31 P{ 1 H} NMR (202 MHz, THF-H8) δ: −16.2 (bs), −34.4 (bs).
HR APCI-MS (−) m/z expected for [M − Rh(CO)(PMe3)4] = 406.1722, found = 406.1769.
[RhI(dppe)2][RhI(CO)2(η5Ph4Pn)] (9)
The crude reaction mixture was concentrated to circa 0.3 mL, 1 mL hexane added and the solution stood at −35 °C for one hour. The supernatant was removed and the solid dried under vacuum to afford the product (20.3 mg, 45%).
1 H NMR (500 MHz, THF-H8) δ: 7.29 (t, 3JHH = 7 Hz, 6H, ArH), 7.18–7.11 (m, 36H, ArH), 7.04 (t, 3JHH = 7.6 Hz, 8H, ArH), 6.91 (d, 2JRhH = 0.9 Hz, 2H, Hw), 2.11 (bs, CH2).
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 194.4 (d, 1JRhH = 82.8 Hz, CO), 134.0, 133.6, 131.3, 130.5, 129.4, 129.0, 128.9, 128.5, 128.2, 128.1, 121.1, 90.1 (d, 1JRhC = 7.1 Hz, Cw), 81.9 (d, 1JRhC = 4.4 Hz), 42.1 Hz (CH2).
31 P{ 1 H} NMR (202 MHz, THF-H8) δ: 57.6 (d, 1JRhP = 128 Hz).
HR ESI-MS (+) m/z expected for [M − Rh(CO)2(Ph4Pn)] = 899.1756, found = 899.1715.
IR (ν CO , cm−1): 1980, 1918.
syn-[RhI(CO)2;RhI(CO)(P{OPh}3)][μ:η5:η5Ph4Pn] (10)
1 H NMR (500 MHz, THF-H8) δ: 6.75 (s, 1H, Hb), 6.22 (s, 1H, Ha).
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 88.6 (d, 1JRhC = 7.4 Hz, Hwb), 81.9 (d, 1JRhC = 4.5 Hz, Hwa).
31 P{ 1 H} NMR (202 MHz, THF-H8) δ: 130.6 (d, 1JRhP = 328 Hz).
E-syn-[RhI(CO)(P{OPh}3)]2[μ:η5:η5Ph4Pn] (11)
Using 0.12 mmol P(OPh)3. Hexane (1 mL) was added to the crude reaction mixture, and the solution stood overnight at −35 °C. The supernatant was removed, and the resultant brown powder dried under vacuum for 2 hours to give the product (16.4 mg, 41%).
1 H NMR (500 MHz, THF-H8) δ: 7.18–7.15 (m, 13H, ArH), 7.05–6.99 (m, 19H, ArH), 6.91–6.87 (m, 16H, ArH), 5.90 (s, 2H, Hw).
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 152.8 (d, 1JRhC = 10.5 Hz), 135.4, 130.9, 129.7, 127.6, 126.7, 124.7, 122.4 (1JRhC = 3.8 Hz), 109.6, 89.1 (m, Cw), 81.0 [CO not resolved].
31 P{ 1 H} NMR (202 MHz, THF-H8) δ: 128.5 (d, 1JRhP = 344 Hz).
HR ESI-MS (+) m/z expected for [M − Rh(CO)(P{OPh}3)] = 847.1479, found = 847.1561.
IR (ν CO , cm−1): 1982, 1961.
E-syn-[RhI(CO)(P{OMe}3)]2[μ:η5:η5Ph4Pn] (12)
Using 0.06 mmol P(OMe)3. The solvent was removed under vacuum and the resultant brown solid washed with n-hexane (3 × 0.5 mL) to afford the product (21.4 mg, 74%).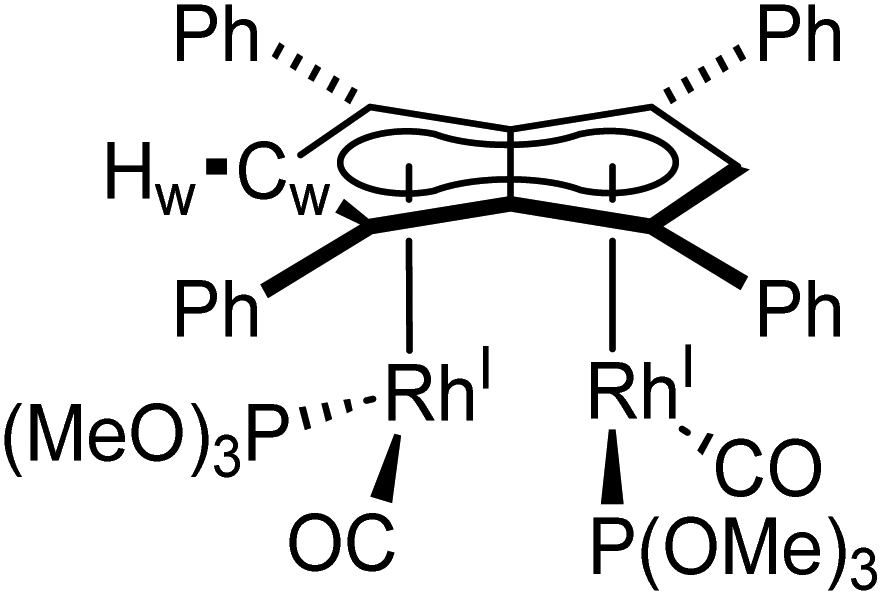
1 H NMR (500 MHz, THF-H8) δ: 7.20 (d, 3JHH = 7 Hz, 8H, ArH), 6.99–6.93 (m, 12H, ArH), 6.64 (s, 2H, Hw) [methyl groups obscured by solvent].
13 C{ 1 H} NMR (126 MHz, THF-H8) δ: 136.5, 130.7, 127.3, 126.2, 110.1, 88.3 (1JRhC = 7 Hz, Cw), 80.1 (dd, 1JRhC = 10 Hz, 2JPC = 4 Hz), 52.2 (d, 2JRhC = 3 Hz, CH3).
31 P{ 1 H} NMR (202 MHz, THF-H8) δ: 146.2 (d, 1JRhP = 309 Hz).
HR ESI-MS (+) m/z expected for [M] = 916.0303, found = 916.0284.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Royal Society (award UF160458) and the University of Bath for funding this work. Dr Mandeep Kaur is acknowledged for help and advice with the synthesis and isolation of several compounds, and Dr Kathryn Proctor and Dr Samantha Lau for assistance with conducting air-sensitive mass spectrometry measurements. This research made use of the Anatra High Performance Computing (HPC) Service at the University of Bath (doi.org/10.15125/b6cd-s854).References
- J. Campos, Bimetallic cooperation across the periodic table, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS PubMed.
- C. M. Farley and C. Uyeda, Organic Reactions Enabled by Catalytically Active Metal–Metal Bonds, Trends Chem., 2019, 1, 497–509 CrossRef CAS.
- J. F. Berry and C. M. Thomas, Multimetallic complexes: synthesis and applications, Dalton Trans., 2017, 46, 5472 RSC.
- J. M. Gil-Negrete and E. Hevia, Main group bimetallic partnerships for cooperative catalysis, Chem. Sci., 2021, 12, 1982–1992 RSC.
- R. H. Lam, S. T. Keaveney, B. A. Messerle and I. Pernik, Bimetallic Rhodium Complexes: Precatalyst Activation-Triggered Bimetallic Enhancement for the Hydrosilylation Transformation, ACS Catal., 2023, 13, 1999–2010 CrossRef CAS.
- M. S. Jeletic, I. Ghiviriga, K. A. Abboud and A. S. Veige, Mono- and bimetallic rhodium(I) complexes supported by new C2-symmetric bis-N-heterocyclic carbene ligands: Metalation via C=C bond cleavage under mild conditions, Organometallics, 2007, 26, 5267–5270 CrossRef CAS.
- F. Min, N. D. Jones, K. Friesen, L. Guanyang, M. J. Ferguson, R. McDonald, R. Lukowski and R. G. Cavell, Bimetallic, spirocyclic, methylene-bridged carbene complexes of rhodium and palladium derived by stepwise metalations of lithiated bis(diphenylphosphoranotrimethylsilylimido)methandiide, Organometallics, 2009, 28, 1652–1665 CrossRef.
- L. A. Oro, D. Carmona and J. Reedijk, Dinuclear rhodium(I) compounds containing diolefins and the 1,6-bis(2′-benzimidazolyl)-2,5-dithiahexane ligand, Inorg. Chim. Acta, 1983, 71, 115–118 CrossRef CAS.
- C. Tan, H. Tinnermann, V. Wee, S. Tan, S. Sung, Q. Wang and R. D. Young, Synthesis of bimetallic rhodium phosphinine complexes with enhanced catalytic activity towards alkyne hydrosilylation, J. Organomet. Chem., 2023, 986, 122617 CrossRef CAS.
- T. A. Koizumi, Y. Ohkura and E. Tsuda, Synthesis, structure, and electrochemical behavior of a new dinuclear Rh(III) complex bridged by a bpp− ligand (bpp− = 3,5-bis(2-pyridyl)pyrazolate), Inorg. Chem. Commun., 2016, 67, 25–28 CrossRef CAS.
- M. Mintert and W. S. Sheldrick, Dinuclear Rh(I,I) and Rh(II,II) complexes containing bridging asymmetrically 2,7-disubstituted naphthyridines, Inorg. Chim. Acta, 1997, 254, 93–98 CrossRef CAS.
- C. M. Thomas, A. Neels, H. Stoeckli-Evans and G. Süss-Fink, Rhodium-Catalysed Carbonylation of Methanol Using a New Multifunctional Ligand Isolation and Structural Characterisation of the Macrocycle [Rh2I6(CO)2(C6H4N3CH2CO2C4H2SCO2CH2C6H4N3)]2, Eur. J. Inorg. Chem., 2001, 3005–3008 CrossRef CAS.
- M. E. Broussard, B. Juma, S. G. Train, W. J. Peng, S. A. Laneman and G. G. Stanley, A Bimetallic Hydroformylation Catalyst: High Regioselectivity and Reactivity Through Homobimetallic Cooperativity, Science, 1993, 260, 1784–1788 CrossRef CAS PubMed.
- J. H. H. Ho, S. W. S. Choy, S. A. MacGregor and B. A. Messerle, Cooperativity in bimetallic dihydroalkoxylation catalysts built on aromatic scaffolds: Significant rate enhancements with a rigid anthracene scaffold, Organometallics, 2011, 30, 5978–5984 CrossRef CAS.
- S. W. S. Choy, M. J. Page, M. Bhadbhade and B. A. Messerle, Cooperative catalysis: Large rate enhancements with bimetallic rhodium complexes, Organometallics, 2013, 32, 4726–4729 CrossRef CAS.
- M. Maekawa, K. Sugimoto, T. Kuroda-Sowa, Y. Suenaga and M. Munakata, Syntheses and structures of dinuclear rhodium(I) complexes and 1-D zigzag-chain rhodium(I) co-ordination polymers bridged by rod-like bidentate nitrogen ligands, J. Chem. Soc., Dalton Trans., 1999, 4357–4362 RSC.
- T. Nakajima, M. Maeda, A. Matsui, M. Nishigaki, M. Kotani and T. Tanase, Unsymmetric Dinuclear RhI2 and RhIRhIII Complexes Supported by Tetraphosphine Ligands and Their Reactivity of Oxidative Protonation and Reductive Dechlorination, Inorg. Chem., 2022, 61, 1102–1117 CrossRef CAS PubMed.
- K. Masada, K. Okabe, S. Kusumoto and K. Nozaki, A dinuclear Rh(−I)/Rh(I) complex bridged by biphilic phosphinine ligands, Chem. Sci., 2023, 14, 8524–8530 RSC.
- L. J. Watson and A. F. Hill, C–H activation in bimetallic rhodium complexes to afford N-heterocyclic carbene pincer complexes, Dalton Trans., 2023, 52, 2164–2174 RSC.
- S. Sun, C. A. Dullaghan, G. B. Carpenter, A. L. Rieger, P. H. Rieger and D. A. Sweigart, Synthesis and Structure of Bimetallic η4,η6-Naphthalene Complexes Containing a Mn-Mn Moiety Bonded in a syn-Facial Manner: A General Route to Homo- and Heteronuclear Bimetallic Complexes of Polyarenes, Angew. Chem., Int. Ed. Engl., 1995, 34, 2540–2542 CrossRef CAS.
- J. A. Reingold, K. L. Virkaitis, G. B. Carpenter, S. Sun, D. A. Sweigart, P. T. Czech and K. R. Overly, Chemical and electrochemical reduction of polyarene manganese tricarbonyl cations: Hapticity changes and generation of Syn- and Anti-facial bimetallic η4,η6-naphthalene complexes, J. Am. Chem. Soc., 2005, 127, 11146–11158 CrossRef CAS PubMed.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, The Localized-to-Delocalized Transition in Mixed-Valence Chemistry, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS PubMed.
- W. Kaim, Concepts for metal complex chromophores absorbing in the near infrared, Coord. Chem. Rev., 2011, 255, 2503–2513 CrossRef CAS.
- P. Jutzi and N. Burford, Structurally Diverse π-Cyclopentadienyl Complexes of the Main Group Elements, Chem. Rev., 1999, 99, 969–990 CrossRef CAS PubMed.
- S. Bendjaballah, S. Kahlal, K. Costuas, E. Bévillon and J. Y. Saillard, The Versatility of Pentalene Coordination to Transition Metals: A Density Functional Theory Investigation, Chem. – Eur. J., 2006, 12, 2048–2065 CrossRef CAS PubMed.
- O. T. Summerscales and F. G. N. Cloke, The organometallic chemistry of pentalene, Coord. Chem. Rev., 2006, 250, 1122–1140 CrossRef CAS.
- F. G. N. Cloke, J. C. Green, A. F. R. Kilpatrick and D. O'Hare, Bonding in pentalene complexes and their recent applications, Coord. Chem. Rev., 2017, 344, 238–262 CrossRef CAS.
- Y. A. Ustynyuk, A. K. Shestakova, V. A. Chertkov, N. N. Zemlyansky, I. V. Borisova, A. I. Gusev, E. B. Tchuklanova and E. A. Chernyshev, Synthesis, structure and fluxional behaviour of isomeric bis(trimethylstannyl)dihydropentalenes, J. Organomet. Chem., 1987, 335, 43–57 CrossRef CAS.
- F. G. N. Cloke, M. C. Kuchta, R. M. Harker, P. B. Hitchcock and J. S. Parry, Trialkylsilyl-substituted pentalene ligands, Organometallics, 2000, 19, 5795–5798 CrossRef CAS.
- R. T. Cooper, F. M. Chadwick, A. E. Ashley and D. O'Hare, Synthesis and Characterization of Group 4 Permethylpentalene Dichloride Complexes, Organometallics, 2013, 32, 2228–2233 CrossRef CAS.
- A. F. R. Kilpatrick, J. C. Green, F. Geoffrey, N. Cloke and N. Tsoureas, Bis(pentalene)di-titanium: a bent double-sandwich complex with a very short Ti-Ti bond, Chem. Commun., 2013, 49, 9434 RSC.
- G. Balazs, F. G. N. Cloke, L. Gagliardi, J. C. Green, A. Harrison, P. B. Hitchcock, A. R. M. Shahi and O. T. Summerscales, A dichromium(II) bis(η8-pentalene) double-sandwich complex with a spin equilibrium: Synthetic, structural, magnetic, and theoretical studies, Organometallics, 2008, 27, 2013–2020 CrossRef CAS.
- A. E. Ashley, R. T. Cooper, G. G. Wildgoose, J. C. Green and D. O'Hare, Homoleptic Permethylpentalene Complexes: ‘Double Metallocenes’ of the First-Row Transition Metals, J. Am. Chem. Soc., 2008, 130, 15662–15677 CrossRef CAS PubMed.
- S. C. Binding, J. C. Green, W. K. Myers and D. O'Hare, Synthesis, Structure, and Bonding for Bis(permethylpentalene)diiron, Inorg. Chem., 2015, 54, 11935–11940 CrossRef CAS PubMed.
- G. Balazs, F. G. N. Cloke, A. Harrison, P. B. Hitchcock, J. Green and O. T. Summerscales, Mn2 bis(pentalene): A mixed-spin bimetallic with two extremes of bonding within the same molecule, Chem. Commun., 2007, 873–875 RSC.
- O. T. Summerscales, C. J. Rivers, M. J. Taylor, P. B. Hitchcock, J. C. Green and F. G. N. Cloke, Double-sandwich pentalene complexes M2(pent†)2 (M = Rh, Pd; Pent† = 1,4-Bis(triisopropylsilyl) pentalene): Synthesis, structure, and bonding, Organometallics, 2012, 31, 8613–8617 CrossRef CAS.
- S. C. Jones, T. Hascall, S. Barlow and D. O'Hare, Pentalene Complexes of Group 7 Metal Carbonyls: An Organometallic Mixed-Valence System with Very Large Metal-Metal Electronic Coupling, J. Am. Chem. Soc., 2002, 124, 11610–11611 CrossRef CAS PubMed.
- S. C. Jones, P. Roussel, T. Hascall and D. O'Hare, Convenient solution route to alkylated pentalene ligands: New metal monoalkylpentalenyl complexes, Organometallics, 2006, 25, 221–229 CrossRef CAS.
- D. F. Hunt and J. W. Russell, Phenylpentalenediiron pentacarbonyl, J. Organomet. Chem., 1972, 46, C22–C24 CrossRef CAS.
- D. F. Hunt and J. W. Russell, A Stable Transition Metal π Complex of Dimethylaminopentalene, J. Am. Chem. Soc., 1972, 94, 7198–7199 CrossRef CAS.
- W. Weidemüller and K. Hafner, Synthesis of Carbonyliron Complexes of Pentalene and Its Derivatives, Angew. Chem., Int. Ed. Engl., 1973, 12, 925–925 CrossRef.
- D. F. Hunt and J. W. Russell, Iron carbonyl complexes of pentalene and dihydropentalene, J. Organomet. Chem., 1976, 104, 373–376 CrossRef CAS.
- A. E. Ashley, G. Balázs, A. R. Cowley, J. C. Green and D. O'Hare, syn-Permethylpentalene Iron and Cobalt Carbonyl Complexes: Proximity Bimetallics Lacking Metal-Metal Bonding, Organometallics, 2007, 26, 5517–5521 CrossRef CAS.
- A. Miyake and A. Kanai, Allyldihydropentalenylene Complexes of Transition Metals, Angew. Chem., Int. Ed. Engl., 1971, 10, 801–802 CrossRef CAS.
- S. A. R. Knox, F. Gordon and A. Stone, Approaches to the Synthesis of Pentalene via Metal Complexes, Acc. Chem. Res., 1974, 7, 321–328 CrossRef CAS.
- J. A. K. Howard, S. A. R. Knox, F. G. A. Stone, A. C. Szary and P. Woodward, Pentalene complexes from cyclo-octatrienes: Crystal structure of η,-(1,5-bistrimethylsilyl)pentalene-1,1,2,2,3,3,3,3-octacarbonyl-triangulo- triruthenium, {Ru3(CO)8[C8H4(SiMe3)2]}, J. Chem. Soc., Chem. Commun., 1974, 788–789 RSC.
- F. M. Chadwick, A. E. Ashley, R. T. Cooper, L. A. Bennett, J. C. Green and D. M. O'Hare, Group 9 bimetallic carbonyl permethylpentalene complexes, Dalton Trans., 2015, 44, 20147–20153 RSC.
- A. F. R. Kilpatrick and F. G. N. Cloke, Reductive deoxygenation of CO2 by a bimetallic titanium bis(pentalene) complex, Chem. Commun., 2014, 50, 2769–2771 RSC.
- A. F. R. Kilpatrick, J. C. Green and F. G. N. Cloke, The Reductive Activation of CO2 Across a Ti=Ti Double Bond: Synthetic, Structural, and Mechanistic Studies, Organometallics, 2015, 34, 4816–4829 CrossRef CAS PubMed.
- N. Tsoureas, J. C. Green and F. G. N. Cloke, C-H and H-H activation at a di-titanium centre, Chem. Commun., 2017, 53, 13117–13120 RSC.
- N. Tsoureas, J. C. Green and F. G. N. Cloke, Bis(pentalene)dititanium chemistry: C-H, C-X and H-H bond activation, Dalton Trans., 2018, 47, 14531–14539 RSC.
- R. T. Cooper, F. M. Chadwick, A. E. Ashley and D. O'Hare, Double CO2 activation by 14-electron η8-permethylpentalene titanium dialkyl complexes, Chem. Commun., 2015, 51, 11856–11859 RSC.
- F. M. Chadwick, R. T. Cooper and D. O'Hare, Zirconium and Hafnium Permethylpentalene Compounds, Organometallics, 2016, 35, 2092–2100 CrossRef CAS.
- E. A. Hamilton, A. F. R. Kilpatrick, Z. R. Turner, D. A. X. Fraser, J. C. Buffet and D. O'Hare, CO2 activation by permethylpentalene amido zirconium complexes, Dalton Trans., 2021, 50, 4494–4498 RSC.
- K. Jonas, P. Kolb, G. Kollbach, B. Gabor, R. Mynott, K. Angermund, O. Heinemann and C. Kruger, Mononuclear Pentalene and Methylpentalene Complexes of Titanium, Zirconium, and Hafnium, Angew. Chem., Int. Ed., 1997, 36, 1714–1718 CrossRef CAS.
- F. M. Chadwick, R. T. Cooper, A. E. Ashley, J. C. Buffet and D. M. O'Hare, Early transition metal permethylpentalene complexes for the polymerization of ethylene, Organometallics, 2014, 33, 3775–3785 CrossRef CAS.
- D. A. X. Fraser, Z. R. Turner, J. C. Buffet and D. O'Hare, Titanium and Zirconium Permethylpentalene Complexes, Pn*MCpRX, as Ethylene Polymerization Catalysts, Organometallics, 2016, 35, 2664–2674 CrossRef CAS.
- F. Burgos, I. Chá Vez, J. M. Manriquez, M. Valderrama, E. Lago, E. Molins, F. Delpech, A. Castel and P. Riviè, A New Heterobimetallic Ru,Rh Complex with a Dianionic Pentalene as Bridging Ligand. Synthesis, Crystal Structure, and Catalytic Activity of [Cp*Ru(μ,-η5,η3-C8H6)Rh(η4-COD)], Organometallics, 2001, 20, 1287–1291 CrossRef CAS.
- N. A. Jenek, M. Balschun, S. M. Boyt and U. Hintermair, Connect Four: Tetraarylated Dihydropentalenes and Triarylated Monocyclic Pentafulvenes from Cyclopentadienes and Enones, J. Org. Chem., 2022, 87, 13790–13802 CrossRef CAS PubMed.
- S. M. Boyt, N. A. Jenek, H. J. Sanderson, G. Kociok-Köhn and U. Hintermair, Synthesis of a Tetraphenyl-Substituted Dihydropentalene and Its Alkali Metal Hydropentalenide and Pentalenide Complexes, Organometallics, 2022, 41, 211–225 CrossRef CAS.
- H. J. Sanderson, G. Kociok-Köhn and U. Hintermair, Synthesis, Structure, and Reactivity of Magnesium Pentalenides, Inorg. Chem., 2023, 62, 15983–15991 CrossRef CAS PubMed.
- F. G. N. Cloke, Pure Appl. Chem., 2001, 73, 233–238 CrossRef CAS.
- W. T. Ford and J. B. Grutzner, 1H and 13C Nuclear Magnetic Resonance Spectra of Cyclopentadienylmagnesium Compounds in Tetrahydrofuran, J. Org. Chem., 1972, 37, 2561–2564 CrossRef CAS.
- P. Sobota and B. Duda, Influence of MgCl2 on Grignard reagent composition in tetrahydrofuran. III, J. Organomet. Chem., 1987, 332, 239–245 CrossRef CAS.
- N. A. Jenek, A. Helbig, S. M. Boyt, S. B. Reeksting, G. Kociok-Köhn, H. Helten and U. Hintermair, The coming of age: Understanding and tuning the electronic structure of pentalenides, ChemRxiv, 2023 Search PubMed.
- P. Zhao, C. D. Incarvito and J. F. Hartwig, Direct observation of β-aryl eliminations from Rh(I) alkoxides, J. Am. Chem. Soc., 2006, 128, 3124–3125 CrossRef CAS PubMed.
- S. Sakamoto, T. Taniguchi, Y. Sakata, S. Akine, T. Nishimura and K. Maeda, Chain: Catalysts for Living Polymerization of Phenylacetylene and Potential Helical Chirality of 1,3,5-Hexatrienes, Helical Struct., 2021, 60, 22201–22206 CAS.
- T. Osswald, I. S. Mikhel, H. Rüegger, P. Butti and A. Mezzetti, Secondary ligand–metal interactions in rhodium(III) and iridium(III) phosphoramidite complexes, Inorg. Chim. Acta, 2010, 363, 474–480 CrossRef CAS.
- J. W. Faller, R. H. Crabtree and A. Habib, Control of Slippage and Conformation in Indenyl Complexes, Organometallics, 1985, 4, 929–935 CrossRef CAS.
- J. L. Atwood, W. E. Hunter, D. C. Hrncir, E. Samuel, H. Alt and M. D. Rausch, Molecular Structures of the Bis(η5-indenyl)dimethyl Derivatives of Titanium, Zirconium, and Hafnium, Inorg. Chem., 1975, 14, 1757–1762 CrossRef CAS.
- H. Adams, N. A. Bailey, B. E. Mann, B. F. Taylor, C. White and P. Yavari, Activation energies for molecular tumbling and cyclopentadienyl rotation in [M(η5-C5H5)(η4-cod)](M = Rh or Ir; cod = cyclo-octa-1,5-diene) and the X-ray crystal structure of and bonding in [Rh(η5-C5H5)(η4-cod)], J. Chem. Soc., Dalton Trans., 1987, 9, 1947–1951 RSC.
- M. Graf, H. C. Böttcher, P. Mayer and M. Scheer, Synthesis and Molecular Structures of [Rh(η5-C5H5)(coe)2] (coe = cis-cyclooctene) and [Rh(η5-C5H5)(η4-cod)] (cod = cis,cis-η,4–1,5-cyclooctadiene), Z. Anorg. Allg. Chem., 2017, 643, 1323–1325 CrossRef CAS.
- I. Y. Ilyin, I. V. Mirzaeva, T. S. Sukhikh, D. V. Bonegardt and T. V. Basova, Study Of The Correlation Between The Structure Of The [Ir(cod)Cp] Complex and its Thermal Properties, J. Struct. Chem., 2021, 62, 1863–1871 CrossRef CAS.
- H. Schumann and S. Stenz, Carbonyl(diphenylcyclopentadienyl) and carbonyl(tetraphenylcyclopentadienyl)rhodium(I) complexes, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 1293–1296 CrossRef CAS.
- T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, Correlating reactivity and selectivity to cyclopentadienyl ligand properties in Rh(III)-catalyzed C-H activation reactions: An experimental and computational study, J. Am. Chem. Soc., 2017, 139, 1296–1310 CrossRef CAS PubMed.
- F. A. Cotton, in Multiple Bonds Between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer-Verlag, 3rd edn, 2006, pp. 35–68 Search PubMed.
- L. Pauling, Atomic Radii and Interatomic Distances in Metals, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- P. Pyykkö, Strong closed-shell interactions in inorganic chemistry, Chem. Rev., 1997, 97, 597–636 CrossRef PubMed.
- S. Bajo, M. G. Alférez, M. M. Alcaide, J. López-Serrano and J. Campos, Metal-only Lewis Pairs of Rhodium with s, p and d-Block Metals, Chem. – Eur. J., 2020, 26, 16833–16845 CrossRef CAS PubMed.
- S. Bajo, M. M. Alcaide, J. López-Serrano and J. Campos, Dehydrogenative Double C−H Bond Activation in a Germylene-Rhodium Complex, Chem. – Eur. J., 2021, 27, 16422–16428 CrossRef CAS PubMed.
- M. G. Alférez, J. J. Moreno, C. Maya and J. Campos, Polarized Au(I)/Rh(I) bimetallic pairs cooperatively trigger ligand non-innocence and bond activation, Dalton Trans., 2023, 52, 3835–3845 RSC.
- M. Navarro, J. J. Moreno, M. Pérez-Jiménez and J. Campos, Small molecule activation with bimetallic systems: a landscape of cooperative reactivity, Chem. Commun., 2022, 58, 11220–11235 RSC.
- B. Klingert and H. Werner, Basische Metalle, XLII. Die Metall-Basizität der Komplexe C5Me5Rh(PMe3)2, C5Me5Rh(C2H4PMe3 und C5Me5Rh(C2H4)P2Me4: Neue Pentamethyl-cyclopentadienylrhodium(I)- und -rhodium(III)-Verbindungen, Chem. Ber., 1983, 116, 1450–1462 CrossRef CAS.
- A. K. Swarnakar, M. J. Ferguson, R. McDonald and E. Rivard, Transition metal-mediated donor–acceptor coordination of low-oxidation state Group 14 element halides, Dalton Trans., 2016, 45, 6071–6078 RSC.
- R. Bissert, H. Braunschweig, R. D. Dewhurst and C. Schneider, Metal-Only Lewis Pairs Based on Zerovalent Osmium, Organometallics, 2016, 35, 2567–2673 CrossRef CAS.
- D. Lionetti, V. W. Day and J. D. Blakemore, Synthesis and Electrochemical Properties of Half-Sandwich Rhodium and Iridium Methyl Complexes, Organometallics, 2017, 36, 1897–1905 CrossRef CAS.
- H. Nakai, K. Jeong, T. Matsumoto and S. Ogo, Catalytic C-F bond hydrogenolysis of fluoroaromatics by [(η5-C5Me5)RhI(2,2′-bipyridine)], Organometallics, 2014, 33, 4349–4352 CrossRef CAS.
- M. Aresta and E. Quaranta, Synthesis, characterization and reactivity of [Rh(bpy)(C2H4)Cl]. A study on the reaction with C1 molecules (CH2O, CO2) and NaBPh4, J. Organomet. Chem., 1993, 463, 215–221 CrossRef CAS.
- J. Lutz, F. Hollmann, T. V. Ho, A. Schnyder, R. H. Fish and A. Schmid, Bioorganometallic chemistry: biocatalytic oxidation reactions with biomimetic NAD+/NADH co-factors and [Cp*Rh(bpy)H]+ for selective organic synthesis, J. Organomet. Chem., 2004, 689, 4783–4790 CrossRef CAS.
- S. I. Johnson, H. B. Gray, J. D. Blakemore and W. A. Goddard, Role of Ligand Protonation in Dihydrogen Evolution from a Pentamethylcyclopentadienyl Rhodium Catalyst, Inorg. Chem., 2017, 56, 11375–11386 CrossRef CAS PubMed.
- V. Ganesan, D. Sivanesan and S. Yoon, Correlation between the Structure and Catalytic Activity of [Cp*Rh(Substituted Bipyridine)] Complexes for NADH Regeneration, Inorg. Chem., 2017, 56, 1366–1374 CrossRef CAS PubMed.
- C. L. Pitman, O. N. L. Finster and A. J. M. Miller, Cyclopentadiene-mediated hydride transfer from rhodium complexes, Chem. Commun., 2016, 52, 9105–9108 RSC.
- M. A. Wright and J. A. Wright, PhotoCORMs: CO release moves into the visible, Dalton Trans., 2016, 45, 6801–6811 RSC.
- T. E. Bitterwolf, Protonation and dimerization reactions of cyclopentadienylrhodiumcarbonylphosphine and phosphite derivatives, Inorg. Chim. Acta, 1986, 122, 175–184 CrossRef CAS.
- D. M. Haddleton and R. N. Perutz, Solution and matrix photochemistry of (η-cyclopentadienyl)bis(ethane)rhodium, J. Chem. Soc., Chem. Commun., 1985, 1372–1374 RSC.
- A. Bara-Estaún, C. L. Lyall, J. P. Lowe, P. G. Pringle, P. C. J. Kamer, R. Franke and U. Hintermair, Understanding Rh-catalysed Hydroformylation with Phosphite Ligands through Catalyst Speciation Analysis by Operando FlowNMR Spectroscopy, ChemCatChem, 2023, 15, e202201204 CrossRef.
- D. Milstein, cis-Hydridoacylrhodium(III) Complexes Not Stabilized by Chelation. Reductive Elimination and Decarbonylation, Organometallics, 1982, 1, 1549–1551 CrossRef CAS.
- D. G. DeWit, Reactivity of mononuclear rhodium(II) compounds, Coord. Chem. Rev., 1996, 147, 209–246 CrossRef.
- P. Paul, B. Tyagi, A. K. Bilakhiya, M. M. Bhadbhade and E. Suresh, Stable mononuclear rhodium(II) polypyridyl complexes:synthesis, spectroscopic and structural characterisation, J. Chem. Soc., Dalton Trans., 1999, 2009–2014 RSC.
- M. P. García, M. V. Jiménez, A. Cuesta, C. Siurana, L. A. Oro, F. J. Lahoz, J. A. López, M. P. Catalán, A. Tiripicchio and M. Lanfranchi, Synthesis and reactivity of mononuclear (pentachlorophenyl)rhodium(II) complexes. Structural relevance of rhodium-o-chlorine secondary bonding, Organometallics, 1997, 16, 1026–1036 CrossRef.
- R. T. Hamazawa, T. Nishioka, I. Kinoshita, T. Takui, R. Santo and A. Ichimura, Thiacalix[3]pyridine produces a stable mononuclear rhodium(II) complex with mutual Jahn–Teller effect, Dalton Trans., 2006, 60, 1374–1376 RSC.
- M. Feller, E. Ben-Ari, T. Gupta, L. J. W. Shimon, G. Leitus, Y. Diskin-Posner, L. Weiner and D. Milstein, Mononuclear Rh(II) PNP-type complexes. Structure and reactivity, Inorg. Chem., 2007, 46, 10479–10490 CrossRef CAS PubMed.
- D. A. Smith, D. E. Herbert, J. R. Walensky and O. V. Ozerov, Monomeric rhodium(II) complexes supported by a diarylamido/bis(phosphine) PNP pincer ligand and their reactivity toward dihydrogen, Organometallics, 2013, 32, 2050–2058 CrossRef CAS.
- K. Fuchigami, N. P. Rath and L. M. Mirica, Mononuclear Rhodium(II) and Iridium(II) Complexes Supported by Tetradentate Pyridinophane Ligands, Inorg. Chem., 2017, 56, 9404–9408 CrossRef CAS PubMed.
- B. R. James and D. Mahajan, Bis(ditertiaryphosphine) complexes of rhodium(I). Synthesis, spectroscopy, and activity for catalytic hydrogenation, Can. J. Chem., 1979, 57, 180–187 CrossRef CAS.
- H. A. Bent, An appraisal of valence-bond structures and hybridization in compounds of the first-row elements, Chem. Rev., 1961, 61, 275–311 CrossRef CAS.
- D. P. Drolet and A. J. Lees, Solution Photochemistry of (η5-C5R5)Rh(CO)2 (R = H, Me) Complexes: Pathways for Potosubstitution and C-H/Si-H Bond Activation Reactions, J. Am. Chem. Soc., 1992, 114, 4186–4194 CrossRef CAS.
- G. M. Reisner, I. Bernal, M. D. Rausch, S. A. Gardner and A. Clearfield, On the cyclodimerization of acetylenes to cyclobutadiene problem: the synthesis, crystal and molecular structure of (η5-cyclopentadienyl)-(η,4-1,2-diphenylcyclobuta[l]phenanthrene)rhodium, J. Organomet. Chem., 1980, 184, 237–254 CrossRef CAS.
- R. S. Dickson and H. P. Kirsch, The effects of bulky substituents in the reactions of alkynes with (η,-C5H5)M(CO)2, M = Co or Rh, Aust. J. Chem., 1981, 34, 1401–1411 CrossRef.
- P. A. Corrigan, R. S. Dickson, S. H. Johnson, G. N. Pain and M. Yeoh, The role of binuclear rhodium complexes as possible intermediates in the formation of cyclopentadienone-rhodium complexes. Reactions between (η,-C5H5),Rh2(CO)(CF3C2CF3) and symmetrically disubstituted alkynes, Aust. J. Chem., 1982, 35, 2203–2214 CrossRef CAS.
- M. Kajitani, T. Suetsugu, R. Wakabayashi, A. Igarashi, T. Akiyama and A. Sugimori, An “adduct” between CpRh(S2C2Z2) and ZC≡CZ (Z = COOCH3) as an intermediate in CpRh(I)-catalyzed synthesis of tetramethyl-2,3,4,5-thiophenetetracarboxylate from elemental sulfur and ZC≡CZ, J. Organomet. Chem., 1985, 293, C15–C18 CrossRef CAS.
- M. Kajitani, R. Ochiai, R. Kikuchi, M. Okubo, T. Akiyama and A. Sugimori, Formation of (η-cyclopentadienyl)(1,2-disubstituted 1,2-ethylenedithiolato)cobalt(III) and -rhodium(III) in reactions of (η-cyclopentadienyl)cobalt(I) or -rhodium(I) species with 1,4-dithiins, Polyhedron, 1990, 9, 1123–1125 CrossRef CAS.
- M. Nomura, H. Hatano, T. Fujita, Y. Eguchi, R. Abe, M. Yokoyama, C. Takayama, T. Akiyama, A. Sugimori and M. Kajitani, Formation, structure, and reactivities of 1:1 adducts between quadricyclane and (η5-cyclopentadienyl)(1,2-diphenyl- or -dimethoxycarbonyl-1,2-ethenedithiolato)rhodium(III), J. Organomet. Chem., 2004, 689, 997–1005 CrossRef CAS.
- D. A. Lesch and T. B. Rauchfuss, Synthesis, Reactivity, and 125Te NMR Studies of (C5H5)RhFe2Te2(CO)x (x = 6, 7), Inorg. Chem., 1983, 22, 1854–1857 CrossRef CAS.
- J. Washington and J. Takats, Alternative Synthesis and Fluxional Behavior of triangulo-Os2Rh(CO)9(C5R5) (R = H, Me), Organometallics, 1990, 9, 925–928 CrossRef CAS.
- J. Tönnemann, J. Risse, Z. Grote, R. Scopelliti and K. Severin, Efficient and Rapid Synthesis of Chlorido-Bridged Half-Sandwich Complexes of Ruthenium, Rhodium, and Iridium by Microwave Heating, Eur. J. Inorg. Chem., 2013, 2013, 4558–4562 CrossRef.
- J. L. Herde, J. C. Lambert, C. V. Senoff and M. A. Cushing, Cyclooctene and 1,5-Cyclooctadiene Complexes of Iridium(I), Inorg. Synth., 2007, 15, 18–20 CrossRef.
- P. A. Boeg, J. Ø. Duus, J. H. Ardenkjær-Larsen, M. Karlsson and S. Mossin, Real-Time Detection of Intermediates in Rhodium-Catalyzed Hydrogenation of Alkynes and Alkenes by Dissolution DNP, J. Phys. Chem. C, 2019, 123, 9949–9956 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional analytical data (NMR, NIR, XRD); Computational methods. CCDC 2309145–2309154. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04325h |
| This journal is © The Royal Society of Chemistry 2024 |