
Ladder-like heteropolynuclear assemblies via cyanido bridges and platinum(II)–thallium(I) bonds: structural and photophysical properties†
Mina
Sadeghian‡
ab,
David
Gómez de Segura‡
b,
Mohsen
Golbon Haghighi
 *a,
Nasser
Safari
a,
Elena
Lalinde
*b and
M. Teresa
Moreno
*b
*a,
Nasser
Safari
a,
Elena
Lalinde
*b and
M. Teresa
Moreno
*b
aDepartment of Chemistry, Shahid Beheshti University, Evin, Tehran 19839-69411, Iran. E-mail: m_golbon@sbu.ac.ir
bDepartamento de Química, Instituto de Investigación en Química (IQUR), Complejo Científico Tecnológico, Universidad de La Rioja, 26006 Logroño, Spain. E-mail: elena.lalinde@unirioja.es; teresa.moreno@unirioja.es
First published on 10th April 2024
Abstract
We describe the mononuclear anionic cyanido-pentafluorophenyl complexes, (NBu4)[Pt(C^N)(C6F5)(CN)] [C^N = 7,8-benzoquinolate (bzq) 1, 2-(2,4-difluorophenyl)pyridinate (dfppy) 2] and the heteropolynuclear derivatives [{Pt(C^N)(C6F5)(CN)}Tl] (C^N = bzq 3, dfppy 4). These complexes were synthesized via a two-step modular synthesis by reaction of the corresponding potassium salts K[Pt(C^N)(C6F5)(CN)], prepared in situ from [Pt(C^N)(C6F5)(DMSO)] and KCN in acetone/H2O, with TlPF6. The structures of {[Pt(bzq)(C6F5)(CN)Tl]·THF}n (3·THF)n and [{Pt(dfppy)(C6F5)(CN)}Tl]4·dioxane [4]4·dioxane, determined by X-ray crystallography, confirm the presence of Pt(II)–Tl(I) bonds [2.9795(6)–3.0736(3) Å], but in the dfppy complex, the incorporation of dioxane, causes a significant structural change. Thus, whereas [3·THF]n achieves a bent-ladder shape extended double chain Tl⋯[Pt⋯Tl]n⋯Pt supported by lateral bridging [Pt](μ-CN)[Tl] ligands, [4]4·dioxane is formed by discrete Pt4Tl4 rectangular aggregates stabilized by [Pt](μ-CN)[Tl] and Pt⋯Tl bonds, which are connected by dioxane bridging molecules through Tl⋯O(dioxane) additional contacts. Solid state emissions are redshifted compared with the mononuclear derivatives 1 and 2 and have been assigned, with the support of theoretical calculations on Pt4Tl4 models, to metal–metal′-to-ligand charge transfer (3MM′LCT [d/s σ*(Pt, Tl) → π*(C^N)]) for 3 and mixed 3MM′LCT/3IL for 4. In fluid THF solution, the complexes are not emissive. At 77 K, 3 and 4 exhibit bright emissions attributed to the formation of bimetallic [{Pt(C^N)(C6F5)(CN)}Tl(THF)x], and anionic [Pt(C^N)(C6F5)(CN)]− fragments. Furthermore, both 3 and 4 exhibit a reversible mechanochromism with a red shift of the emissions upon crushing, suggesting some degree of shortening of metal–metal separation. Finally, complex 3 shows solvatochromic behavior with color/luminescence changes by treatment with a drop of MeOH, CH2Cl2, THF or Et2O, with shifts from 583 in 3-MeOH to 639 nm in 3-THF. However, 4 only demonstrates a bathochromic response to MeOH.
Introduction
The study of metal–metal interactions between closed (d10, d10s2) and pseudoclosed (d8) shell metal ions has been of significant research interest in the design of a wide array of polymetallic discrete compounds or supramolecular networks.1 Many of these metallophilic interactions involve dative bonds between a Lewis base (donor) and a Lewis acid (acceptor) to give the denominated Metal Only Lewis Pairs (MOLP) complexes.2 In many cases, the presence of metal–metal interactions is associated with a luminescent behavior and this can be influenced by the metal–metal distances, the number and type of metals or ligands, the geometry, the orientation between the units and even the secondary contacts in the solid state.1e,f,3 Furthermore, some of these linkages suffer modifications in the color and/or in the emission as a result of external stimuli, being perfect candidates for the design of smart materials.4Since the earliest pioneering luminescent complex trans-[Tl2Pt(CN)4],5 heterometallic systems containing Pt–Tl bonds between Pt(II) with a high-lying filled 5dz2 donor orbital and Tl(I) as acceptor (6pz, π*) are often associated with intense photoluminescence. In this regard, numerous Pt(II)–Tl(I) backbones have been documented with various structural configurations, supported and unsupported by bridging ligands, comprising binuclear (PtTl),6 trinuclear (Pt2Tl,4b,7 PtTl25,8), tetranuclear [paired (PtTl),6a,8c,9 and trigonal (Pt3Tl)10], hexanuclear clusters (Pt2Tl4,11 Pt3Tl310b) or infinite networks.4a,b,6a,7,8b,c,11a,c,12
Among them, some luminescent cyanido heterometallic assemblies based on Pt(II)–Tl(I) backbones have been reported.13 Our group described cyanido-pentafluorophenyl extended complexes [trans,trans,trans-Tl2{Pt(C6F5)2(CN)2}·(CH3COCH3)2]n (two-dimensional, 2D), {Tl[Tl{cis-Pt(C6F5)2(CN)2}]·(H2O)}n (one-dimensional, 1D) and a discrete dimer [Tl{cis-Pt(C6F5)2(CN)(PPh2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)}]2,8c and the cyanido-alkynyl {trans-[PtTl2(CCTol)2(CN)2]·2MeOH}n 1D-chain.8b Within this area, it has been found that the use of the strong field cyclometalated ligands coordinated to Pt favors the formation of strong Pt → Tl bonds by raising the energy of the 5dz2 orbitals on the Pt center.4a,b,6a,7b,c,10,12c With cyclometalated-cyanido platinum motifs, extended 2D networks of composition [PtTl(C^N)(CN)2] (C^N = bzq, ppy)6a and [PtTl(Naph∧E)(CN)2] [Naph∧N = 1-(naphthalene-2-yl)-1H-pyrazolyl; Naph∧C* = 3-methyl-1-(naphthalene-2-yl)-1H-imidazolyl-2-ylidene]12c have been published. In all of them, the formation of additional secondary inter or intramolecular Tl⋯NC contacts typically determines the final arrangement of the units. Our group has also reported the utilization of cyclometalated-pentafluorophenyl derivatives, (NBu4)[Pt(C^N)(C6F5)2] (C^N = bzq)4b and the solvate [Pt(C^N)(C6F5)S] (C^N = bzq, S = acetone; C^N = ppy, S = DMSO)4a to form extended Pt–Tl chains, in which the occurrence of intramolecular Tl⋯Fortho contacts play a significant role in stabilizing the Pt–Tl bonds.
CPh)}]2,8c and the cyanido-alkynyl {trans-[PtTl2(CCTol)2(CN)2]·2MeOH}n 1D-chain.8b Within this area, it has been found that the use of the strong field cyclometalated ligands coordinated to Pt favors the formation of strong Pt → Tl bonds by raising the energy of the 5dz2 orbitals on the Pt center.4a,b,6a,7b,c,10,12c With cyclometalated-cyanido platinum motifs, extended 2D networks of composition [PtTl(C^N)(CN)2] (C^N = bzq, ppy)6a and [PtTl(Naph∧E)(CN)2] [Naph∧N = 1-(naphthalene-2-yl)-1H-pyrazolyl; Naph∧C* = 3-methyl-1-(naphthalene-2-yl)-1H-imidazolyl-2-ylidene]12c have been published. In all of them, the formation of additional secondary inter or intramolecular Tl⋯NC contacts typically determines the final arrangement of the units. Our group has also reported the utilization of cyclometalated-pentafluorophenyl derivatives, (NBu4)[Pt(C^N)(C6F5)2] (C^N = bzq)4b and the solvate [Pt(C^N)(C6F5)S] (C^N = bzq, S = acetone; C^N = ppy, S = DMSO)4a to form extended Pt–Tl chains, in which the occurrence of intramolecular Tl⋯Fortho contacts play a significant role in stabilizing the Pt–Tl bonds.
In this context, we decided to expand the investigation of this type of system to neutralization reactions of new heteroleptic anionic metalloligands [Pt(C^N)(C6F5)(CN)]− (C^N = bzq, dfppy) with Tl+. We report the synthesis, structures and optical properties of new neutral heterometallic systems based on Pt(II)–Tl(I) bonded [{Pt(C^N)(C6F5)(CN)}Tl]n units, distinctly connected by Pt(μ-CN)Tl bonds. We have found that the C^N moiety and the crystallization solvent have a remarkable effect on the final structures and photoluminescent behaviour, which has been analyzed by computational studies.
Results and discussion
Synthesis and characterization
The mononuclear complexes (NBu4)[Pt(C^N)(C6F5)(CN)] (C^N = bzq 1, dfppy 2) were prepared, as yellow solids in high yield, by displacement of dimethyl sulfoxide by cyanido in the corresponding precursors [Pt(C^N)(C6F5)(DMSO)] using one equivalent of (NBu4)CN in acetone at room temperature (Scheme 1i). The products were fully characterized by means of multinuclear [1H, 19F{1H}, 13C{1H}] NMR and IR spectroscopy, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF), and elemental analysis (CHN). 1H and 13C{1H} NMR signals were assigned based on 2D experiments, including 1H–1H (COSY) and 1H–13C (HSQC and HMBC) correlations. | ||
| Scheme 1 Synthesis of 1–4, (i) 1 equiv. of (NBu4)CN, acetone, (ii) 1 equiv. of KCN, acetone/H2O (3/1), 298 K, (iii) 1 equiv. of TlPF6. | ||
Their IR spectra displayed the characteristic ν(CN) stretching vibration at 2102 (1) and 2110 cm−1 (2), consistent with a terminal CN− bond trans to CC^N.14 Their MALDI-TOF(−) MS spectra display in both complexes the molecular peak anion [M]− as the parent peak together with the peak corresponding to the diplatinum anion [Pt2(C^N)2(C6F5)2(CN)]− in less abundance, which suggests the propensity of these cyanido mononuclear complexes to form binuclear complexes through a cyanido bridge under MALDI-TOF conditions.14 The 1H NMR of 1 and 2 in CDCl3 show the signals for the cyclometalated and NBu4 groups with their expected integration (Fig. S1–S3†). The 19F{1H} NMR spectra display the three typical signals (Fo, Fp and Fm, AA′MXX′ spin system) of the C6F5 ligand, and in the case of 2, additionally, the two expected fluorine resonances of the cyclometalated dfppy ligand (F10 and F8) with long-range Pt satellites (4JPt–F = 50 and 40 Hz). The coordination of the cyanido takes place with retention of the geometry, as is reflected in the high values of 3Jo–F–Pt (530 1, 515 Hz 2),4g,15 in coherence with the relatively low trans influence of the N pyridinic atom. The 13C{1H} NMR spectra show the resonance of the cyanido at δ 147.7 1, 146.3 2, further supported by the 13C{1H} NMR spectrum of the in situ prepared mononuclear complex K[Pt(bzq)(C6F5)(13CN)] (1′) in CDCl3 (Fig. S2†), which displays a sharp resonance at δ 146.5 with 195Pt satellites (1JPt–C = 881 Hz). These values compare well with those recently reported for K[Pt(ppy)(p-MeC6H4)(13CN)] (δ 157.4, 1JPt–C = 882 Hz).14
With the aim of investigating the metallophilic affinity of the anionic platinum substrates toward Tl(I), we decided to examine the reactivity of the synthons “[Pt(C^N)(C6F5)(CN)]−” toward TlPF6. Thus, the heteropolynuclear complexes, 3 and 4 were synthesized via a two-step modular synthesis (Scheme 1ii, iii). The reactions of [Pt(C^N)(C6F5)(DMSO)] with KCN in a mixture acetone/H2O render “in situ” the anionic complexes K[Pt(C^N)(C6F5)(CN)] and their treatment with one equivalent of TlPF6 gives rise to the final compounds of stoichiometry [{Pt(C^N)(C6F5)(CN)}Tl] (C^N = bzq 3, dfppy 4) as orange (3) or yellow (4) solids in high yields. This procedure avoids the presence of (NBu4)PF6, difficult to separate from the reaction media. Attempts to obtain Pt2Tl complexes by treatment of the anionic Pt complexes (NBu4)[Pt(C^N)(C6F5)(CN)] with TlPF6 in a molar relation 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 results in the formation of solids of identical stoichiometry, [{Pt(C^N)(C6F5)(CN)}Tl]. Their molecular structures were determined by single-crystal X-ray diffraction (see below). In their IR spectra, the ν(CN) absorption appear slightly shifted to higher frequencies (2108 3, 2120 cm−14) in relation to the precursors, confirming the electronic perturbation of the platinum fragment and/or the coordination of the CN− to the acidic Tl center. The MALDI-TOF(+) mass spectra in THF solution show the peak corresponding to the binuclear [Pt(C^N)(C6F5)(CN)Tl]+ fragment, although in low abundance, suggesting the partial retention of these Pt–Tl fragments in solution.
:1 results in the formation of solids of identical stoichiometry, [{Pt(C^N)(C6F5)(CN)}Tl]. Their molecular structures were determined by single-crystal X-ray diffraction (see below). In their IR spectra, the ν(CN) absorption appear slightly shifted to higher frequencies (2108 3, 2120 cm−14) in relation to the precursors, confirming the electronic perturbation of the platinum fragment and/or the coordination of the CN− to the acidic Tl center. The MALDI-TOF(+) mass spectra in THF solution show the peak corresponding to the binuclear [Pt(C^N)(C6F5)(CN)Tl]+ fragment, although in low abundance, suggesting the partial retention of these Pt–Tl fragments in solution.
Both complexes are only soluble in donor solvents, in which this type of donor acceptor Pt → M bonds are usually broken or involved in dynamic processes. Conductivity measurements in THF solution are close to 1:1 electrolytes (suggesting a remarkable dissociation process into the anion [Pt(C^N)(C6F5)(CN)]− and [Tl(THF)x]+). Not surprisingly, the corresponding 1H and 13C{1H} NMR spectra of 3 and 4 in THF-d8 show chemical shifts and coupling constants very close to those of precursors (Fig. S4 and S5†). In the 13C{1H} NMR spectra, the cyanido signal was assigned at δ 145.2 (3) and 145.5 (4), confirmed by the 13C{1H} NMR spectrum of the in situ prepared [{Pt(dfppy)(C6F5)(13CN)}Tl] (4′) in THF-d8, which displays a sharp resonance at δ 144.5 flanked by 195Pt satellites (1JPt–C = 887 Hz) (Fig. S6†). The most significant change was observed in the 3Jo–F–Pt coupling constants of the ortho-fluorine resonances of the C6F5 group in the 19F{1H} NMR spectra. Thus, in both complexes (3 and 4) the value of the 3Jo–F–Pt coupling is notably smaller than in the corresponding precursors (435 3/530 Hz 1, Δ = 77; 445 4/515 Hz 2, Δ = 70 Hz). This fact is coherent with the retention to some extend of the Pt–Tl bond in solution, which reduces the electronic density at the Pt center. Notwithstanding, no F–Tl coupling is observed at room temperature, indicating that the Pt–Tl bond is involved in a fast dynamic process on the NMR time scale. In agreement with this behavior, no signal could be detected in the 195Pt NMR spectra.
Deep red suitable crystals for X-ray diffraction studies of 3·THF were grown by slow evaporation of a concentrated THF–d8 solution of 3. For the dfppy complex 4, orange crystals of stoichiometry [4]4·dioxane were obtained by slow diffusion of n-hexane into a saturated THF solution of 4 with one drop of 1,4-dioxane at room temperature. The crystallographic parameters and selected bond lengths and angles are summarized in Table 1 and Tables S1 and S2.† Unfortunately, due to the limitation of their solubility, it was not possible to obtain crystals in other solvents. Both compounds (3 and 4) crystallize in the same space group (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) of the triclinic crystal system. Crystallographic characterization of 3·THF shows the formation of a double chain framework of stoichiometry {[Pt(bzq)(C6F5)(CN)Tl]·THF}n (3·THF)n, in which each Pt is coordinated to two Tl centers and each Tl to two Pt centers and the chains are connected by bridging [Pt](μ-CN)[Tl] ligands. The asymmetric unit contains two [{Pt(bzq)(C6F5)(CN)}Tl] fragments and two tetrahydrofuran molecules (see Fig. 1a), one coordinated to Tl(2) and the other as crystallization solvent. The structure reveals a pseudooctahedral environment around the platinum center, formed by the C and N of the cyclometalated bzq group, the C6F5 ligand, a cyanido group (trans to Cbzq) and two trans donor–acceptor Pt–Tl bonds, with a non-linear sequence [Tl(1)–Pt(1)–Tl(2) 156.90(9), Pt(1)–Tl(2)–Pt(2) 156.74(9), Tl(2)–Pt(2)–Tl(1) 166.81(6), Pt(2)–Tl(1)–Pt(1) 166.98(6)°]. The Pt–Tl distances [Pt(1)–Tl(1) 3.0279(7), Pt(1)–Tl(2) 3.0402(7), Pt(2)–Tl(2) 3.0140(7), Pt(2)–Tl(1) 2.9795(6) Å] are in the usual range for Pt(II)–Tl(I) separations found in related compounds with unsupported Pt–Tl bonds between the metal centers.4a,b,6a,7a,7c,8c,12c,16 The cyanido coordinated to Pt(2) contacts through the N to the Tl(2) of the adjacent chain, whereas the cyanido coordinated to Pt(1) is simultaneously interacting with Tl(1), thus generating a bent ladder-like assembly (Fig. 1b). The angles CN⋯Tl deviated notably from linearity [C(34)–N(2)–Tl(2) 135.4(2)° and C(14)–N(4)–Tl(1) 141.4(2)°], indicating a notable electrostatic interaction between the Tl centers and the cyanido groups. Both Tl centers show different environments. The Tl(1) is coordinated to both trans platinum centers, Pt(1) and Pt(2), and to the nitrogen of a cyanido bridging group of the neighboring chain [Tl(1)–N(4) 2.669(3) Å], whereas Tl(2) is coordinated to both Pt centers and stablishes a longer bond with the N of a cyanido group [Tl(2)–N(2) 2.7223(3) Å], due to an additional bonding interaction to the oxygen of one THF molecule [Tl(2)–O(1) 2.667(2) Å]. Additionally, both Tl atoms interact with two ortho-fluorine atoms of the C6F5 ring of the two adjacent units with relatively short Tl⋯F intramolecular contacts [2.929–3.203 Å],7a,8c giving rise to a final five (Tl(1)) and six (Tl(2)) coordination, respectively (Fig. 1c). These oF⋯Tl interactions contribute to the stability, but are likely responsible for the deviation from linearity of the chain. The resulting ladder-like assembly is extended along the “a” axis defined by the two chains with repeated Pt(1)/Tl(1)–Pt(1)/Tl(1)–Pt(2)/Tl(2)–Pt(2)/Tl(2) fragments. These double chains are separated by THF molecules (Fig. S7†).
) of the triclinic crystal system. Crystallographic characterization of 3·THF shows the formation of a double chain framework of stoichiometry {[Pt(bzq)(C6F5)(CN)Tl]·THF}n (3·THF)n, in which each Pt is coordinated to two Tl centers and each Tl to two Pt centers and the chains are connected by bridging [Pt](μ-CN)[Tl] ligands. The asymmetric unit contains two [{Pt(bzq)(C6F5)(CN)}Tl] fragments and two tetrahydrofuran molecules (see Fig. 1a), one coordinated to Tl(2) and the other as crystallization solvent. The structure reveals a pseudooctahedral environment around the platinum center, formed by the C and N of the cyclometalated bzq group, the C6F5 ligand, a cyanido group (trans to Cbzq) and two trans donor–acceptor Pt–Tl bonds, with a non-linear sequence [Tl(1)–Pt(1)–Tl(2) 156.90(9), Pt(1)–Tl(2)–Pt(2) 156.74(9), Tl(2)–Pt(2)–Tl(1) 166.81(6), Pt(2)–Tl(1)–Pt(1) 166.98(6)°]. The Pt–Tl distances [Pt(1)–Tl(1) 3.0279(7), Pt(1)–Tl(2) 3.0402(7), Pt(2)–Tl(2) 3.0140(7), Pt(2)–Tl(1) 2.9795(6) Å] are in the usual range for Pt(II)–Tl(I) separations found in related compounds with unsupported Pt–Tl bonds between the metal centers.4a,b,6a,7a,7c,8c,12c,16 The cyanido coordinated to Pt(2) contacts through the N to the Tl(2) of the adjacent chain, whereas the cyanido coordinated to Pt(1) is simultaneously interacting with Tl(1), thus generating a bent ladder-like assembly (Fig. 1b). The angles CN⋯Tl deviated notably from linearity [C(34)–N(2)–Tl(2) 135.4(2)° and C(14)–N(4)–Tl(1) 141.4(2)°], indicating a notable electrostatic interaction between the Tl centers and the cyanido groups. Both Tl centers show different environments. The Tl(1) is coordinated to both trans platinum centers, Pt(1) and Pt(2), and to the nitrogen of a cyanido bridging group of the neighboring chain [Tl(1)–N(4) 2.669(3) Å], whereas Tl(2) is coordinated to both Pt centers and stablishes a longer bond with the N of a cyanido group [Tl(2)–N(2) 2.7223(3) Å], due to an additional bonding interaction to the oxygen of one THF molecule [Tl(2)–O(1) 2.667(2) Å]. Additionally, both Tl atoms interact with two ortho-fluorine atoms of the C6F5 ring of the two adjacent units with relatively short Tl⋯F intramolecular contacts [2.929–3.203 Å],7a,8c giving rise to a final five (Tl(1)) and six (Tl(2)) coordination, respectively (Fig. 1c). These oF⋯Tl interactions contribute to the stability, but are likely responsible for the deviation from linearity of the chain. The resulting ladder-like assembly is extended along the “a” axis defined by the two chains with repeated Pt(1)/Tl(1)–Pt(1)/Tl(1)–Pt(2)/Tl(2)–Pt(2)/Tl(2) fragments. These double chains are separated by THF molecules (Fig. S7†).
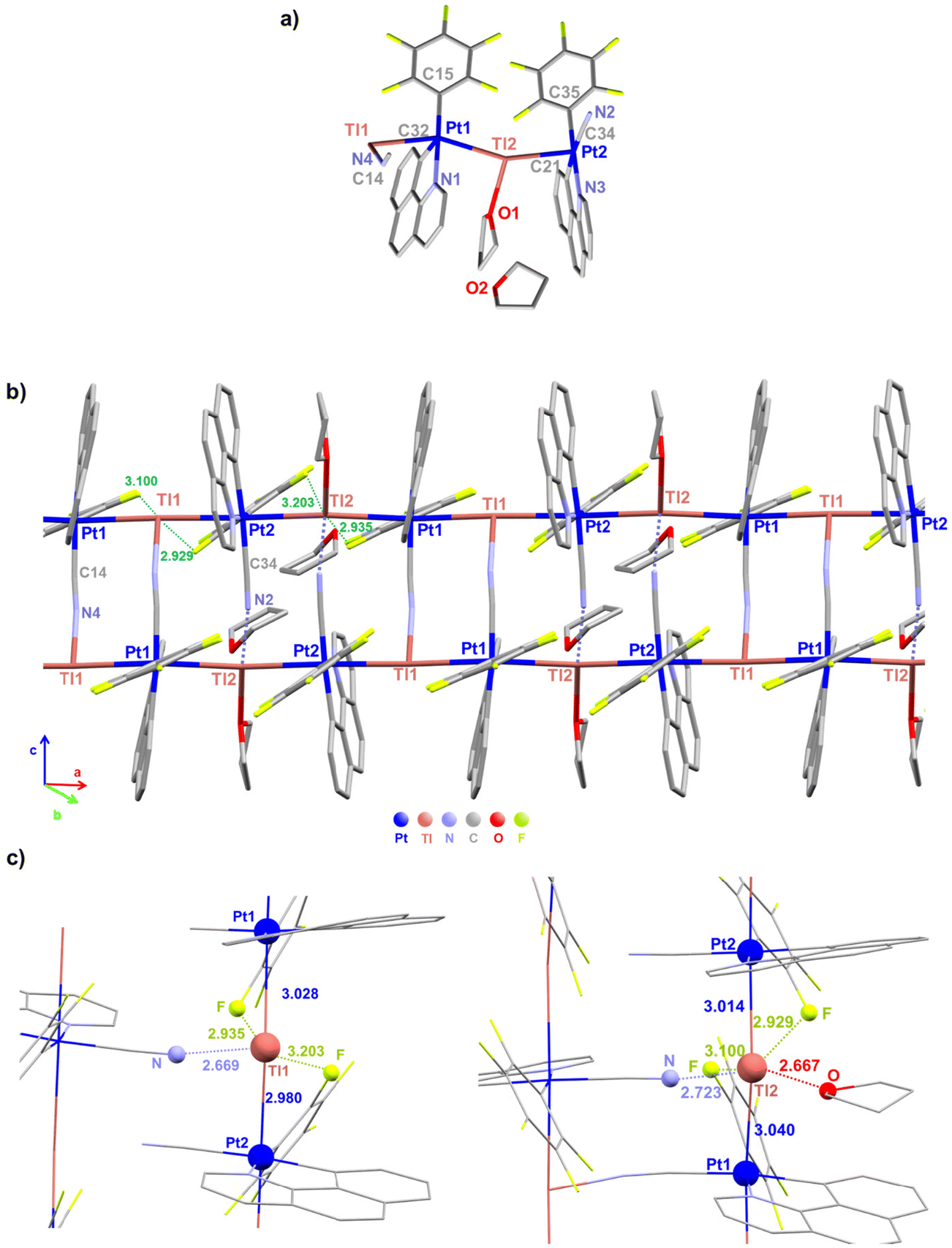 | ||
| Fig. 1 View of the molecular structure of {[Pt(bzq)(C6F5)(CN)Tl]·THF}n (3·THF)n. (a) Asymmetric unit of the unit cell, (b) supramolecular structure showing the ladder shape assembly and (c) views of the coordination environments of both Tl centers. | ||
| {[Pt(bzq)(C6F5)(CN)Tl]·THF}n (3·THF)n | |||
|---|---|---|---|
| Pt(1)–Tl(1) | 3.0279(7) | Pt(1)–N(1) | 2.097(2) |
| Pt(1)–Tl(2) | 3.0402(7) | Pt(1)–C(15) | 2.030(3) |
| Pt(2)–Tl(2) | 3.0140(7) | Pt(1)–C(14) | 2.024(3) |
| Pt(2)–Tl(1)#1 | 2.9795(6) | Pt(2)–C(35) | 2.024(3) |
| Tl(1)–N(4) | 2.669(3) | Pt(2)–C(34) | 2.027(3) |
| Tl(2)–N(2) | 2.7223(3) | Pt(2)–C(21) | 2.052(3) |
| Tl(2)–O(1) | 2.667(2) | Pt(2)–N(3) | 2.097(2) |
| Pt(1)–C(1) | 2.051(3) | ||
| Tl(1)–Pt(1)–Tl(2) | 156.90(9) | Pt(1)–Tl(2)–Pt(2) | 156.74(9) |
| Tl(2)–Pt(2)–Tl(1) | 166.81(6) | Pt(2)–Tl(1)–Pt(1) | 166.98(6) |
| C(34)–N(2)–Tl(2) | 135.4(2) | C(14)–N(4)–Tl(1) | 141.4(2) |
| [{Pt(dfppy)(C6F5)(CN)}Tl]4·C4H8O2[4]4·dioxane | |||
|---|---|---|---|
| Pt(1)–Tl(1) | 3.0736(3) | Pt(1)–C(12)#1 | 2.021(5) |
| Pt(1)–Tl(2) | 3.0518(3) | Pt(1)–C(13)#1 | 2.016(4) |
| Pt(2)–Tl(2) | 2.9258(3) | Pt(2)–C(31) | 2.014(4) |
| Tl(1)–N(4) | 2.596(4) | Pt(2)–C(30) | 2.007(4) |
| Tl(2)–N(2) | 2.626(4) | Pt(2)–C(19) | 2.025(4) |
| Pt(1)–C(1)#1 | 2.039(4) | Pt(2)–N(3) | 2.075(4) |
| Pt(1)–N(1)#1 | 2.082(4) | ||
| Tl(1)–Pt(1)–Tl(2) | 162.723(8) | Pt(1)–Tl(2)–Pt(2) | 160.299(8) |
| C(12)–N(2)–Tl(2) | 142.8(4) | C(30)–N(4)–Tl(1) | 140.6(4) |
In the case of the 2-(2,4-difluorophenyl)pyridinate complex 4, the presence of dioxane originates that the one-dimensional polymeric structure breaks down into discrete heteroocta-metallic Pt4Tl4 rectangular aggregates (ca. 4.6 × 9.56 Å), which are also stabilized by Pt(II)–Tl(I) bonds and bridging cyanido ligands, as is shown in Fig. 2a. The structure contains two different Pt and Tl centers and one molecule of dioxane (C4H8O2) per tetramer as [4]4·dioxane. Thus, the two Pt(1) atoms, located on the long edges of the rectangle, exhibit a similar pseudooctahedral environment formed by the C and N of the dfppy ligand, the Cipso of the C6F5 ring, a cyanido ligand and two trans thallium centers [Tl(1) and Tl(2)]. The Pt(1)–Tl(1,2) bonds are slightly longer than those found in 3·THF [Pt(1)–Tl(1) 3.0736(3), Pt(1)–Tl(2) 3.0518(3) Å; Tl(1)–Pt(1)–Tl(2) 162.723(8)°], which is in accordance with its blue shifted color in relation to the bzq complex, either in crystals (orange [4]4·dioxanevs. red 3·THF) or as pristine solids (yellow 4, orange 3). However, the Pt(2) centers, located at the corner of the rectangle, exhibit a distorted square pyramid environment due to the formation of only one Pt–Tl bond [C,Ndfppy, CC6F5, CCN and Tl(2)]. Due to the lower coordination index, the Pt(2)–Tl distance [Pt(2)–Tl(2) 2.9258(3) Å] is the shortest one. In the same line, as shown in Fig. 2b, the Tl(2) centers, localized along the edges, achieve a distorted octahedral environment with the Pt centers [Pt(1), Pt(2)] located in the axial position and the equatorial plane formed by the N of a cyanido, N(2) bonded to Pt(1), two o-F of adjacent C6F5 groups [Tl(2)⋯F 2.894 and 3.103 Å] and one fluorine atom of a dfppy ligand [Tl(2)⋯F 3.022 Å] of a close octanuclear unit. The Tl(1) at the corners adopts a very distorted hexacoordination formed by the Tl(1)–Pt(1) bond, the bridging NNCPt2, three fluorine contacts [two o-FC6F5 (intra-unit 3.016 Å) (inter-unit 3.197 Å) and one Fdfppy of an adjacent unit (3.188 Å)] and the oxygen of the dioxane molecule [Tl(1)–O(1) 2.945 Å], which acts as a bridge between the Pt4Tl4 aggregates. Within the aggregates, the Tl(1)–N distance is slightly shorter than Tl(2)–N [Tl(1)⋯N(4) 2.596(4) vs. Tl(2)⋯N(2) 2.626(4) Å], whereas the CN⋯Tl angles are similar [C(12)–N(2)–Tl(2) 142.8(4) vs. C(30)–N(4)–Tl(1) 140.6(4)°]. The crystal packing reveals the presence of π⋯π interactions between dfppy ligands of neighboring Pt4Tl4 units (3.269–3.315 Å) (Fig. S8†), presumably contributing to the stability of the packing.
 | ||
| Fig. 2 (a) View of the molecular structure of [{Pt(dfppy)(C6F5)(CN)}Tl]4·C4H8O2[4]4·dioxane showing the Tl⋯F and Tl⋯O(dioxane) contacts, (b) view of the coordination environment of both Tl centers. | ||
In the case of the dfppy complex (4), although the disruption of the ladder-chain observed for the bzq system (3) and formation of the octanuclear aggregates could be attributed to the coordination of the dioxane, it cannot be ruled out that the presence of the electron-withdrawing fluorine atoms on the cyclometalated ligand weakens the Pt–Tl bonds due to the lower electron density on the Pt centers.
Photophysical properties
 | ||
| Fig. 3 Top: UV-vis absorption spectra of complexes 1–4 in THF (5 × 10−5 M) at 298 K. Down: schematic representation of the frontiers orbitals of 1, 2 and models 3·(THF)3 and 4·(THF)3 with selected excitations. | ||
The solid-state diffuse reflectance spectra of all complexes are shown in Fig. S10 and Table S3.† Pristine solids 3 (orange) and 4 (yellow) display additional red-shifted absorptions (440–575 nm 3; 410–525 nm 4) in relation to their respective solution UV-vis spectra, in coherence with their color. These low-energy bands reflect the effect of the platinum-thallium bonds in the molecular transitions in the solid state and according to calculations, can be mainly ascribed to 1MM'LCT transitions due to the strong contribution of the metals in the high occupied molecular orbitals and the involvement of the cyclometalated groups in the target low-lying orbitals.
To rationalize the experimental observations, DFT and TD-DFT calculations in THF solution and in solid state have been carried out for 1–4 (Tables S4–S9 and Fig. S11–S16†). For the mononuclear precursors 1 and 2, which are optimized based on the reported crystallographic structures,14 the calculated S0 → S1 transition in THF agrees with the experimentally observed transition, and arises from HOMO → LUMO (Fig. 3). The HOMOs are mainly centered on the Pt(C^N) units (Pt/bzq 30/63% 1, 41/50% 2) with small contribution of the CN− (6% 1, 9% 2), whereas the LUMOs are located on the cyclometalated ligand (96% 1, 92% 2) (Tables S4, S5 and Fig. S11†). Therefore, the LE absorption band of mononuclear complexes can be assigned to a mixed 1MLCT/1IL transition, localized on the Pt(C^N) fragment, with a minor contribution of the cyanido. Similar results were obtained in solid state (Tables S8, S9 and Fig. S14†).
For the absorption of complexes 3 and 4 in THF solution, calculations for model systems of solvated bimetallic [Pt(C^N)(C6F5)(CN)Tl(S)3] (S = THF) were performed. In these units, the Tl ion is coordinated to the Pt fragment through metal and one ortho-fluorine atom and to three oxygens atoms from solvent molecules, achieving a five coordination. Calculations reveal that, upon formation of the Pt–Tl bond, the contribution of the platinum to the HOMO decreases notably, with a minor Tl contribution, increasing the weight of the cyclometalated group [Pt/Tl/C^N 21/1/73% 3·(THF)3; 30/1/63% 4·(THF)3] and, as a consequence, the HOMO is stabilized in both complexes in relation to the precursor anionic fragments (Fig. 3-down and Fig. S13†). As shown in Fig. 3, which shows the frontiers orbitals, in both models, the Pt–Tl bonding is reflected in the H-1 with a remarkable contribution of the C6F5 ring in the dfppy complex [Pt/Tl/C6F5 65/17/6% 3·(THF)3; 46/8/35% 4·(THF)3]. In both models, the target LUMO (and also L + 1) are mainly located on the C^N ligands with minor metal contribution [LUMO, Pt/Tl/C^N 4/2/93% 3·(THF)3; 8/5/85% 4·(THF)3]. The LUMOs are also stabilized in relation to the anionic precursors, but to a lesser extent than the HOMOs and, as a consequence, the calculated more intense low lying S1 excitation, which arises from HOMO → LUMO, is blue shifted in relation to the precursors (ca. 10 nm) and has mainly mixed 1IL/1MLCT character. The close S2 transition [363 3·(THF)3; 344 nm 4·(THF)3] has 1IL/1MM′CT character in the bzq compound, 3·(THF)3, and mixed 1IL/1MM′CT/1L′LCT (L′ = C6F5) contribution in 4·(THF)3. Considering these results and the notable dynamic dissociation, suggested by the NMR data for the heterometallic complexes, the blue shifted low-lying electronic absorption in THF solution could be attributed to mixed 1IL/1MLCT with some 1MM′CT and 1L′LCT contribution in 3 and 4, respectively.
In the solid state, the presence of extended Pt–Tl–Pt–Tl bonds connected by CN− bridges modifies the electronic structures. For complex 3, a short double chain Pt4Tl4 similar to that seen for 4 has been computed based on the X-ray (Tables S8, S9 and Fig. S15†). For model 3, the HOMOs (HOMO, HOMO−1), with antibonding character, are mainly located at the metal centers (HOMO/H-1 Pt 46/47%; Tl 32/30%) with low contribution of the ligands (bzq 12/12%; C6F5 5/7%), whereas the low lying LUMO and L + 1 are placed at the adjacent benzoquinolate ligands (Fig. 4a) with minor contribution of metals (Pt/Tl LUMO 9/12%; L + 1 8/10). In the case of 4, the contribution of metals in the HOMOs decreases (Pt 34/32%; Tl 9/10%), whereas increases the weight of the dfppy ligand (50/53%) (Fig. 4b). For 3, the computed low-lying more intense excitations (S1 443 and S3 431 nm) involve transitions from HOMO and H-1 to LUMO and L + 1, having strong metal–metal′-to-ligand charge transfer (1MM′LCT). For 4, the computed most intense low-lying absorption, S2 (HOMO → LUMO, 384 nm), is blue shifted in relation to 3 and has mixed 1MM′LCT/1IL character.
 | ||
| Fig. 4 Selected frontier MOs and vertical excitations in the solid state for 3 (optimized model Pt4Tl4 based on the reported crystallographic structure) and 4 (optimized model Pt4Tl4). | ||
 | ||
| Fig. 5 Normalized excitation and emission spectra of (a) 3, (b) 4 in the solid state at 298 and 77 K. | ||
| Solid 298 K | Solid 77 K | THF 77 K | PSa | |||||
|---|---|---|---|---|---|---|---|---|
| λ em/nm (λex/nm) | Φ PL | τ/μs | λ em/nm (λex/nm) | τ/μs | λ em/nm (λex/nm) | λ em/nm (λex/nm) | τ/μs | |
| a 10% wt. b with additional minor peak at 458 nm. | ||||||||
| 1 | 500, 566 (415) | 0.05 | 3.1 (49%), 11.8 (51%) | 496, 532, 570 (415) | 186.6 (67%), 21.4 (33%) | 480, 515, 555, 600 (415) | 523, 568 (415) | 41.5 (21.6%), 12.2 (78.4%) |
| 2 | 472, 497, 530, 575 (405) | 0.02 | 2.8 (46%), 13.7 (54%) | 470, 497, 530, 575 (385) | 15.1 | 457, 490, 520 (385) | 472, 496, 526 (410) | 11.7 |
| 3 | 595 (460) | 0.13 | 14.2 | 658 (525) | 480, 520, 560 (415) | 546 (385) | 21.9 | |
| 4 | 546 (460) | 0.17 | 11.5 | 563 (450) | 559 (415)b | 523 (385) | 12.7 | |
Both precursors, 1 and 2, display in rigid media (solid, glassy) a typical vibronic-structured emission originating from 3IL/3MLCT emissive state with predominantly 3IL character, which is typical of cyclometalated complexes14,17 and supported by TD-DFT calculations. In the case of the bzq derivative 1, the emission profile in the solid state at room temperature presents an additional excimeric low energy feature at 566 nm. In agreement with this assignment, the excitation spectra monitored at the lower and higher energy emission bands are similar (Fig. S17a†). The energy peak maximum is blue shifted in 2 compared to 1, in line with the presence electron-acceptor fluorine atoms in the phenyl ring of 2 (λmax 457–472 2vs. 480–523 nm 1, Fig. S17†) and the measured lifetimes are significantly longer in the bzq complex. This fact, which has been previously observed in other Pt(bzq) compounds,14,18 is attributed to a lower platinum contribution into the excited state and to the stronger structural rigidity of the bzq ligand compared to the dfppy group. As shown in Fig. S16,† the SOMO and SOMO−1 and the spin density for both anion complexes support a higher Pt and MLCT contribution into the excited state for complex 2 (SOMO/SOMO−1 5/24% 2vs. 1/16% 1).
The formation of Pt⋯Tl bonding interactions in both heterometallic complexes 3 and 4 are reflected in their optical properties. Both pristine solids are brightly emissive in solid state. The dfppy hetero-octanuclear aggregate 4 exhibits in solid state at room temperature an unstructured yellow emission centered at 546 nm (ϕ = 17%), which is slightly red shifted at low temperature (563 nm). For the double extended chain bzq derivative, 3, as shown in Fig. 5, the emission is somewhat broader and red shifted (595 nm) at room temperature compared to 4, which agrees with the presence of a shorter Pt⋯Tl separation (see Table 1) and a more delocalized low-lying cyclometalated ligand (bzq vs. dfppy). Furthermore, upon cooling at 77 K, the emission is significantly more red-shifted (658 nm) than in the case of 4 (595 → 658 nm 3; 546 → 563 nm 4), a behavior which is generally attributed to some degree of thermal contraction of the Pt⋯Tl separation at low temperature and has many precedents in metal⋯metal chain systems.4b,19 In both complexes, the excitation profile resembles the absorption spectra in the solid state with a maximum notable red shifted in 3 in relation to 4 (520 3, 450 nm 4), indicating that the absorption in these low energy regions is responsible for the corresponding emission. The large Stokes shifts and the lifetimes measured at room and at low temperature in the range of microseconds (Table 2) confirm the occurrence of phosphorescence processes. For the bzq derivative 3, the computed first vertical excitation at the ground state geometry (T1, 537 nm) for the short double chain Pt4Tl4, in the gas phase, corresponds to the HOMO–LUMO transition having 3MM′LCT character. In 4, the calculated lowest T1 is blue shifted (439 nm) and involves a combination of several electronic transitions from HOMO and H-1 to LUMO-L+3, which are mainly generated from the metals and the cyclometalated group (HOMO, H-1 Pt 32–34% Tl 9–10%, dfppy 50–53%; LUMO–LUMO+3 Pt 15–7%, Tl 25–3%, dfppy 53–88%). The emission is therefore ascribed to phosphorescence with mixed 3MM′LCT/3IL character.
As noted before, complexes 3 and 4 are soluble in THF and in this solvent the emission is lost at room temperature, a behavior that can be attributed to partial and reversible rupture of the Pt–Tl bonds. Upon cooling the solutions (5 × 10−5 M) to 77 K (Fig. S18†), bright emission is developed mainly formed by an unstructured low energy band (560 3, 559 nm 4), which is attributed to the formation of bimetallic fragments [{Pt(C^N)(C6F5)(CN)}Tl(THF)x] in the frozen solution, together with an overlapped minor high energy structured band (λmax 480 nm 3, 458 nm 4), associated with the presence of anionic [Pt(C^N)(C6F5)(CN)]− fragments. Similar behavior has been previously reported in related heteropolynuclear complexes featuring unsupported Pt–Tl bonds.4b,8c
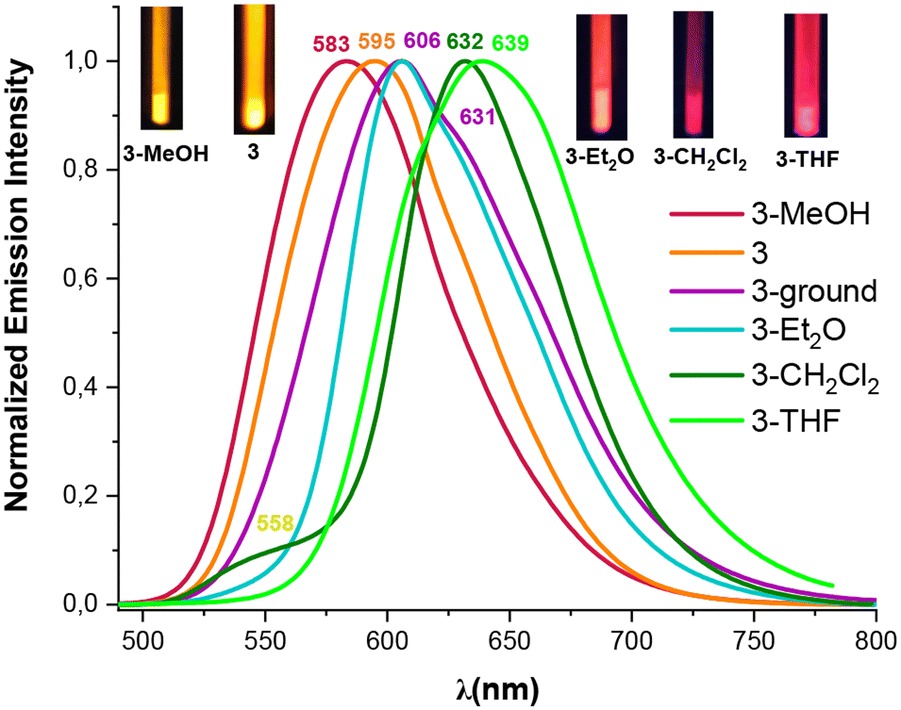 | ||
| Fig. 6 Normalized emission spectra at 298 K of the orange solids 3, 3-ground and the solvates (3-solvent) (λex 405–415 nm). | ||
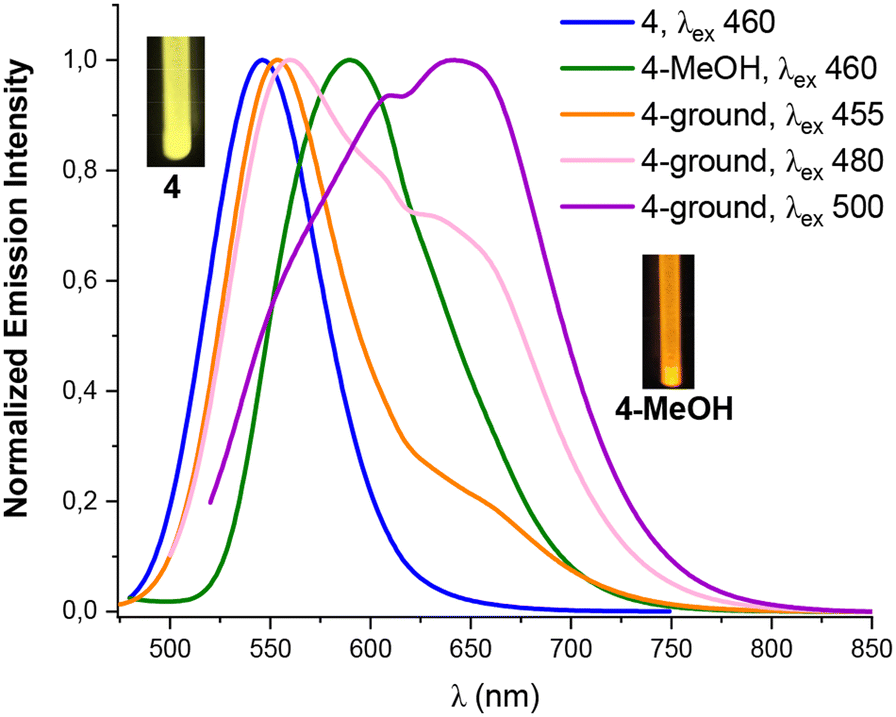 | ||
| Fig. 7 Normalized emission spectra at 298 K of the orange solid 4, 4-MeOH and 4-ground (with different λexc). | ||
Interestingly, we observed that the treatment of powder 3 with a drop of MeOH, CH2Cl2, THF or Et2O results in color and luminescence changes, thus exhibiting solvatochromic behavior (Table S10 and Fig. S20†). As shown in Fig. 6, when solid 3 was treated with a drop of Et2O, CH2Cl2 and THF a red shift in its color and emission band takes place from orange to pink-red (630 nm), to crimson (632 nm) and to deep pink color (639 nm), respectively. This bathochromic shift from 595 nm in solvent-free solid 3 to 639 nm in 3-THF suggests that the solvent molecules allow the platinum–thallium bond to be shortened and to form a strong interaction, decreasing the gap associated with the metal–metal-to ligand charge transition 3MM′LCT, leading to change in the color of the compound. Curiously, treatment of pristine solid 3 with a drop of MeOH causes a color change from deep orange to bright yellow-orange under UV light (reflected in its absorption spectrum, Fig. S10†), and shows a blue shift in the emission band both, at 298 K (595 → 583 nm) and at 77 K (658 → 650 nm). However, in contrast to complex 3, complex 4 demonstrates a bathochromic response to MeOH. Thus, upon addition of a drop of MeOH solvent to the yellow solid 4, the color of the complex changes from yellow to orange and a notable red shift appears in the emission spectrum, both at room and at low temperature (298/77 K 590/662 nm Fig. 7 and Fig. S21†). This bathochromic response suggests that the incorporation of MeOH likely provokes a partial or total shortening of the Pt–Tl bonds. Surprisingly, no similar response was observed with other solvents (Et2O, CH2Cl2 or THF).
Conclusions
The new mononuclear anionic (NBu4)[Pt(C^N)(C6F5)(CN)] (C^N = bzq 1, dfppy 2) and heterometallic [{Pt(C^N)(C6F5)(CN)}Tl] (C^N = bzq 3, dfppy 4) compounds have been prepared and their photophysical properties investigated with the support of theoretical calculations. Structures of (3·THF)n and [4]4·dioxane contain Pt(II)–Tl(I) bonds, but they show different arrangements. In the bzq complex, (3·THF)n, each Pt interacts with two Tl(I) ions and each Tl(I) with two Pt centers, generating a bent ladder-like extended assembly where two chains are connected by bridging cyanido ligands [Pt](μ-CN)[Tl]. To the best of our knowledge, this double chain network is unknown in PtTl systems. In the case of the dfppy complex, [4]4·dioxane, the presence of dioxane breaks the polymeric structure into discrete heterooctametallic Pt4Tl4 rectangular aggregates, connected by one molecule of dioxane, also stabilized by Pt–Tl bonds and bridging cyanido ligands. Notwithstanding, the presence of electron-withdrawing fluorine atoms on the dfppy ligand produces lower electron density on the Pt centers, also contributing to the weakening of the Pt–Tl bonds. In both structures, the secondary interactions of the Tl(I) centers with the ortho-fluorine atoms of the C6F5 rings of adjacent units and with oxygen atoms of THF (3) or dioxane (4) also contributes to the stability.In the solid-state, the PtTl complexes 3 and 4 exhibit unstructured emissions (595 3, 546 nm 4) red shifted in relation to the mononuclear derivatives (500 1, 472 nm 2), mainly attributed to 3MM′LCT [d/s σ*(Pt, Tl) → π*(C^N)] in 3, with mixed 3MM′LCT/3IL character in 4. However, in glassy THF solution, both derivatives exhibit a dual emission, corresponding the unstructured low energy band to the formation of bimetallic [{Pt(C^N)(C6F5)(CN)}Tl(THF)x] fragments and the minor structured high energy band to the anionic [Pt(C^N)(C6F5)(CN)]− units. Interestingly, complexes 3 and 4 show a reversible mechanochromic behavior with red shifted color and luminescence changes, likely due to some degree of shortening of Pt–Tl separations, when pressure is applied. In addition, 3 displays strong solvatochromism in the solid state with MeOH, CH2Cl2, THF or Et2O, showing shifts from 583 in 3-MeOH to 639 nm in 3-THF, which can be attributed to modulation of the Pt–Tl separations resulting from the effect of the solvent molecules. However, 4 only demonstrates response to MeOH.
Experimental section
General comments
The neutral precursor complexes, [Pt(C^N)(C6F5)(DMSO)] [C^N = 7,8-benzoquinolate (bzq), (2-(2,4-difluorophenyl)pyridinate) (dfppy)] were prepared according to published procedure.17b Other reagents were used as received. The reactions with Tl(I) salts were performed in the absence of light. The microanalyses were carried out with a CA FLASH 2000 (Thermo Fisher Scientific) microanalyzer. Mass spectra were recorded on a Microflex MALDI-TOF Bruker spectrometer operating in the linear and reflector modes using THF solutions of the complexes and dithranol as the matrix. NMR spectra were recorded on a Bruker AVANCE ARX 400 MHz spectrometer at 298 K. IR spectra were obtained on a Fourier Transform PerkinElmer Spectrum UATR Two spectrophotometer, with the diamond crystal ATR attachment, which covers the region between 4000 and 450 cm−1. The chemical shifts (δ) are reported in units of parts per million (ppm) relative to external standards (TMS for 1H and 13C{1H} and CFCl3 for 19F{1H}) and coupling constant were given in hertz (Hz). The UV-vis absorption spectra were measured with a Hewlett-Packard 8453 spectrophotometer. Diffuse Reflectance UV-vis (DRUV) spectra were carried out in SiO2 pellets, using a Shimazdu UV-3600 spectrophotometer with a Harrick Praying Mantis accessory, and recalculated following the Kubelka Munk function. Excitation and emission spectra were obtained in a Shimazdu RF-60000. The measurements in solid state and PS films were carried out on air and in solutions under N2 atmosphere. The lifetime measurements up to 10 μs at 298 K at all samples at 77 K were performed with a Jobin Yvon Horiba Fluorolog operating in the phosphorimeter mode (with an F1–1029 lifetime emission PMT assembly, using a 450 W Xe lamp) and the Jobin Yvon software packing, that works with Origin 6.0. The decay data were analysed by tail fitting to the functions “one-phase exponential decay function with time constant parameter” (ExpDec1) and “two-phase exponential decay function with time constant parameters (ExpDec 2)”. The lifetimes below 10 μs at 298 K were measured with a Datastation HUB-B with a nanoLED controller, using the technique “Time Correlated Single Photon Counting” (TCSPC). Quantum yields were measured using a Hamamatsu Absolute PL Quantum Yield Measurement System C11347-11.:20 mL), removing the aqueous layer. It was repeated for three times. Treatment of the organic layer with MgSO4, filtration through Celite, evaporation to dryness and treatment with n-hexane (5 mL) allowed to obtain 1 as a yellow solid (yield: 437 mg, 68%). Elem. Anal. Calcd for C36H44F5N3Pt (808.85): C, 53.46; H, 5.48; N, 5.20. Found: C, 53.80; H, 5.48; N, 5.10%; IR (cm−1): 2102 (s) ν(CN); MALDI-TOF (−) m/z (%): 565.97 [Pt(bzq)(C6F5)(CN)]− (100), 1105.98 [Pt2(bzq)2(C6F5)2(CN)]− (54). 1H NMR (400 MHz, CDCl3, 298 K, δ): 9.80 (dd, 3JH–H = 5.2, 4JH–H = 1.0, 3JPt–H = 28.6, H2), 8.33 (dd, 3JH–H = 8.0, 4JH–H = 1.0, H4), 7.78 (d, 3JH–H = 8.8, H5), 7.55–7.38 (m, 4H, H3,6–8), 7.20 (d, 3JH–H = 7.0, 3JPt–H = 46.2, H9), 3.03 (m, 8H, N-CH2–), 1.45 (m, 8H, N-CH2-CH2–, NBu4), 1.26 (sx, 8H, 3JH–H = 8.0, –CH2-CH3, NBu4), 0.88 (t, 12H, 3JH–H = 8.0, –CH3, NBu4). 13C{1H} NMR (100.6 MHz, CDCl3, 298 K, δ): 158.3 (s, C13), 157.1 (s, C10), 150.8 (s, C2), 147.7 (s, CN), 144.3 (s, C14), 136.6 (s, C4), 134.2 (s, C9), 133.7 (s, C11), 129.7 (s, C5), 129.5 (s, C8), 126.7 (s, C12), 122.6 (s, C6), 121.7 (s, C3), 121.4 (s, C7), 58.6 (s, C1′), 23.7 (s, C2′), 19.5 (s, C3′), 13.5 (s, C4′). 19F{1H} NMR (376.5 MHz, CDCl3, 298 K, δ): −115.4 (dm, 3JPt–Fo = 530, 2Fo), −165.8 (m, 2Fm, Fp).
X-ray diffraction
X-ray intensity data were collected using Molybdenum graphite monochromatic (Mo-Kα) radiation with Bruker APEX-II diffractometer at a temperature of 100 K using APEX-II programs for all complexes. Structures were solved by Intrinsic Phasing using SHELXT20 with the WinGX graphical user interface.21 Multi-scan absorption corrections were applied to all the data sets and refined by full-matrix least squares on F2 with SHELXL.22 Hydrogen atoms were positioned geometrically, with isotropic parameters Uiso = 1.2Ueq (parent atom) for aromatic hydrogens and CH2 and Uiso = 1.5Ueq (parent atom) for methyl groups. Finally, the structures show some residual peaks in the vicinity of the platinum atoms but with no chemical meaning.Computational details
Calculations were performed with the program suite Gaussian 1623 on complexes with Beckés three-parameter functional combined with Lee–Yang–Parŕs correlation functional (B3LYP).24 Optimizations on the singlet state (S0) in THF and in gas phase of the mononuclear models 1 and 2 were performed based on the reported crystallographic structures.14 For 3 and 4, calculations in THF solutions were performed for model systems of solvated bimetallic species [Pt(C^N)(C6F5)(CN)Tl(S)3] (S = THF) [3·(THF)3, 4·(THF)3], using as starting point the molecular geometry obtained by X-ray diffraction analysis. Calculations in gas phase on heteropolynuclear structures of 3 and 4 (considered as Pt4Tl4) were made starting from the X-Ray data. No imaginary frequency was found in the vibrational frequency analysis of the final equilibrium geometries. The LANL2DZ basis set was used for the platinum and thallium centers and all-electron 6-31G (d, p) basis set was applied for other atoms.25 Solvent effects were taken into consideration by the Polarizable Continuum Model (PCM)26 implemented in the Gaussian 16 software, in the presence of THF. The TD-DFT method was employed for calculations of the electronic absorption spectra. The predicted emission wavelengths were calculated by the energy difference between the triplet state at its optimized geometry and the singlet state at the triplet geometry. The results were visualized with GaussView 6. Overlap populations between molecular fragments were calculated using the GaussSum 3.0 program.27Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministerio de Ciencia e Innovación (Project PID2019-109742GB-I00) funded by MCIN/AIE/10.13039/501100011033. D. G. S. is grateful to UR for a PhD grant.References
- (a) M. E. Moret, Top. Organomet. Chem., 2011, 35, 157–184 CrossRef CAS PubMed; (b) S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC; (c) M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC; (d) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC; (e) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (f) Z. N. Chen, N. Zhao, Y. Fan and J. Ni, Coord. Chem. Rev., 2009, 253, 1–20 CrossRef CAS; (g) M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2022, 144, 22805–22825 CrossRef CAS PubMed.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329–4346 CrossRef CAS PubMed.
- (a) A. Díez, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2011, 255, 2426–2447 CrossRef; (b) J. R. Berenguer, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2018, 366, 69–90 CrossRef CAS; (c) Q.-C. Zhang, H. Xiao, X. Zhang, L.-J. Xu and Z.-N. Chen, Coord. Chem. Rev., 2019, 378, 121–133 CrossRef CAS; (d) J. R. Berenguer, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2010, 254, 832–875 CrossRef CAS.
- (a) J. R. Berenguer, E. Lalinde, A. Martín, M. T. Moreno, S. Sánchez and H. R. Shahsavari, Inorg. Chem., 2016, 55, 7866–7878 CrossRef CAS PubMed; (b) J. Forniés, N. Giménez, S. Ibáñez, E. Lalinde, A. Martín and M. T. Moreno, Inorg. Chem., 2015, 54, 4351–4363 CrossRef PubMed; (c) Q.-H. Wei, S.-Q. Zhang, X.-J. Ma, Z.-Z. Huang and B. Wang, New J. Chem., 2022, 46, 18990–18995 RSC; (d) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed; (e) A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 4469–4483 CrossRef CAS; (f) X. Zhang, B. Li, Z. H. Chen and Z. N. Chen, J. Mater. Chem., 2012, 22, 11427–11441 RSC; (g) J. R. Berenguer, E. Lalinde, A. Martín, M. T. Moreno, S. Ruiz, S. Sánchez and H. R. Shahsavari, Inorg. Chem., 2014, 53, 8770–8785 CrossRef CAS PubMed.
- J. K. Nagle, A. L. Balch and M. M. Olmstead, J. Am. Chem. Soc., 1988, 110, 319–321 CrossRef CAS.
- (a) J. Forniés, S. Fuertes, A. Martín, V. Sicilia, B. Gil and E. Lalinde, Dalton Trans., 2009, 2224–2234 RSC; (b) R. Usón, J. Forniés, M. Tomás, R. Garde and R. I. Merino, Inorg. Chem., 1997, 36, 1383–1387 CrossRef PubMed; (c) B. R. Barnett, C. E. Moore, P. Chandrasekaran, S. Sproules, A. L. Rheingold, S. DeBeer and J. S. Figueroa, Chem. Sci., 2015, 6, 7169–7178 RSC.
- (a) L. R. Falvello, J. Forniés, R. Garde, A. García, E. Lalinde, M. T. Moreno, A. Steiner, M. Tomás and I. Usón, Inorg. Chem., 2006, 45, 2543–2552 CrossRef CAS PubMed; (b) D. Campillo, Ú. Belío and A. Martín, Dalton Trans., 2019, 48, 3270–3283 RSC; (c) S. Fuertes, A. J. Chueca, A. Martín and V. Sicilia, Cryst. Growth Des., 2017, 17, 4336–4346 CrossRef CAS.
- (a) J. P. H. Charmant, J. Forniés, J. Gómez, E. Lalinde, R. I. Merino, M. T. Moreno and A. G. Orpen, Organometallics, 2003, 22, 652–656 CrossRef CAS; (b) J. R. Berenguer, J. Fernández, E. Lalinde and S. Sánchez, Chem. Commun., 2012, 48, 6384–6386 RSC; (c) J. Forniés, A. García, E. Lalinde and M. T. Moreno, Inorg. Chem., 2008, 47, 3651–3660 CrossRef PubMed.
- A. Díez, J. Forniés, J. Gómez, E. Lalinde, A. Martín, M. T. Moreno and S. Sánchez, Dalton Trans., 2007, 3653–3660 RSC.
- (a) Ú. Belío, S. Fuertes and A. Martín, Inorg. Chem., 2013, 52, 5627–5629 CrossRef PubMed; (b) U. Belío, S. Fuertes and A. Martín, Dalton Trans., 2014, 43, 10828–10843 RSC.
- (a) Á. Díez, J. Fernández, E. Lalinde, M. T. Moreno and S. Sánchez, Inorg. Chem., 2010, 49, 11606–11618 CrossRef PubMed; (b) J. R. Berenguer, J. Forniés, J. Gómez, E. Lalinde and M. T. Moreno, Organometallics, 2001, 20, 4847–4851 CrossRef CAS; (c) J. R. Berenguer, J. Forniés, B. Gil and E. Lalinde, Chem. – Eur. J., 2006, 12, 785–795 CrossRef CAS PubMed.
- (a) W. Chen, F. Liu, D. Xu, K. Matsumoto, S. Kishi and M. Kato, Inorg. Chem., 2006, 45, 5552–5560 CrossRef CAS PubMed; (b) G. Wu and D. Wang, J. Cluster Sci., 2007, 18, 406–413 CrossRef CAS; (c) S. Paziresh, V. Sicilia, I. Ara, A. Martín and S. Fuertes, Organometallics, 2019, 38, 3804–3815 CrossRef CAS; (d) J. R. Stork, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2005, 127, 6512–6513 CrossRef CAS PubMed.
- M. Sadeghian, M. G. Haghighi, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2022, 452, 214310 CrossRef CAS.
- M. Sadeghian, D. Gómez de Segura, M. G. Haghighi, N. Safari, E. Lalinde and M. T. Moreno, Inorg. Chem., 2023, 62, 1513–1529 CrossRef CAS PubMed.
- F. Juliá, D. Bautista and P. González-Herrero, Chem. Commun., 2016, 52, 1657–1660 RSC.
- S. Rajabi, S. Jamali, S. Naseri, A. Jamjah, R. Kia, H. Samouei, P. Mastrorilli, H. R. Shahsavari and P. R. Raithby, Organometallics, 2019, 38, 1709–1720 CrossRef CAS.
- (a) K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC; (b) G. Millán, N. Giménez, R. Lara, J. R. Berenguer, M. T. Moreno, E. Lalinde, E. Alfaro-Arnedo, I. P. López, S. Piñeiro-Hermida and J. G. Pichel, Inorg. Chem., 2019, 58, 1657–1673 CrossRef PubMed; (c) E. Lalinde, M. T. Moreno, R. Lara, I. P. López, E. Alfaro-Arnedo, J. G. Pichel and S. Piñeiro-Hermida, Chem. – Eur. J., 2018, 24, 2440–2456 CrossRef CAS PubMed; (d) N. Giménez, R. Lara, M. T. Moreno and E. Lalinde, Chem. – Eur. J., 2017, 23, 5758–5771 CrossRef PubMed; (e) J. R. Berenguer, J. G. Pichel, N. Giménez, E. Lalinde, M. T. Moreno and S. Piñeiro-Hermida, Dalton Trans., 2015, 44, 18839–18855 RSC; (f) M. S. Sangari, M. G. Haghighi, S. M. Nabavizadeh, A. Pfitzner and M. Rashidi, New J. Chem., 2018, 42, 8661–8671 RSC; (g) S. R. Barzegar-Kiadehi, M. G. Haghighi, M. Jamshidi and B. Notash, Inorg. Chem., 2018, 57, 5060–5073 CrossRef CAS PubMed; (h) M. Jamshidi, S. R. Barzegar-Kiadehi, M. G. Haghighi and B. Notash, Organometallics, 2020, 39, 3099–3111 CrossRef CAS.
- (a) J. R. Berenguer, E. Lalinde, M. T. Moreno, S. Sánchez and J. Torroba, Inorg. Chem., 2012, 51, 11665–11679 CrossRef CAS PubMed; (b) J. Forniés, V. Sicilia, P. Borja, J. M. Casas, A. Díez, E. Lalinde, C. Larraz, A. Martín and M. T. Moreno, Chem. - Asian J., 2012, 7, 2813–2823 CrossRef PubMed.
- G. Y. Zheng and D. P. Rillema, Inorg. Chem., 1998, 37, 1392–1397 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Revision A.03, Inc., Gaussian 16, Wallingford CT, 2016, 2016 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization of complexes (NMR spectra, crystal data), photophysical properties and computational details and photocatalytic studies. CCDC 2337876 and 2337877. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00674g |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |