
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-reducing precursors for aluminium metal thin films: evaluation of stable aluminium hydrides for vapor phase aluminium deposition†
Niklas
Huster
 a,
Rita
Mullins
b,
Michael
Nolan
b and
Anjana
Devi
*acde
a,
Rita
Mullins
b,
Michael
Nolan
b and
Anjana
Devi
*acde
aInorganic Materials Chemistry, Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: anjana.devi@ruhr-uni-bochum.de
bTyndall National Institute, Lee Maltings, University College Cork, Cork T12 R5CP, Ireland
cLeibniz Institute for Solid State and Materials Research, Helmholtzstr. 20, 01069 Dresden, Germany
dFraunhofer Institute for Microelectronic Circuits and Systems (IMS), Finkenstr. 61, 47057, Duisburg, Germany
eChair of Materials Chemistry, TU Dresden, Bergstr. 66, 01069, Dresden, Germany
First published on 11th April 2024
Abstract
Thin films of Al as interconnect materials and those of AlN as wide bandgap semiconductor and piezoelectric material are of great interest for microelectronic applications. For the fabrication of these thin films via chemical vapor deposition (CVD) based routes, the available precursor library is rather limited, mostly comprising aluminium alkyls, chlorides, and few small amine-stabilized aluminium hydrides. Herein, we focused on rational precursor development for Al, their characterization and comparison to existing precursors comprising stabilized aluminium hydrides. We present and compare a series of potentially new and reported aluminium hydride precursors divided into three main groups with respect to their stabilization motive, and their systematic structural variation to evaluate the physicochemical properties. All compounds were comprehensively characterized by means of nuclear magnetic resonance spectroscopy (NMR), Fourier-transform infrared spectroscopy (FTIR), elemental analysis (EA), electron-impact ionization mass spectrometry (EI-MS) and thermogravimetric analysis (TGA). Promising representatives were further evaluated as potential single source precursors for aluminium metal formation in proof-of-concept experiments. Structure and reaction enthalpies with NH3 or H2 as co-reactants were calculated via first principles density functional theory simulations and show the great potential as atomic layer deposition (ALD) precursors for Al and AlN thin films.
Introduction
Aluminium (Al) is one of the more earth abundant elements, with huge variety of applications, ranging from simple applications like packaging material to light weight material for aerospace applications, while being comparably inexpensive in contrast to many transition metals or rare-earth metals.1–5 For the microelectronic industry Al and especially Al-composite films have become indispensable,6–9 with trimethylaluminium (TMA) as one of the prominent precursors for vapor phase deposition and the TMA/H2O as one of the most studied atomic layer deposition (ALD) processes.10 Although the majority of Al-related chemical vapor deposition-based processes are targeted towards the deposition of Al2O3 thin films,11 AlN and Al thin films recently have drawn increased interest for applications in microelectronics as insulator and wide bandgap semiconductor (AlN),12–14 gate electrode material (Al)9 or for hydrogen storage applications.15–17For the fabrication of such thin films chemical vapor deposition (CVD) and its above-mentioned variant ALD are the methods of choice. Whereas Al2O3 thin films and the respective processes for their fabrication are well investigated and established, processes for the deposition of AlN and especially Al-metal thin films are less explored and usually require strongly reducing process conditions like NH3, N2H4, or H2 co-reactants or plasmas,18,19 owing to the highly negative electrochemical potential (Al3+ ↔ Al0 = E° = −1.66 V).20 A strategy, providing those reducing conditions directly from the precursor and thus avoiding rather harsh (plasma) or hazardous (NH3, N2H4) process conditions, is to employ Al-hydride (or alane) complexes as precursor. For CVD and ALD of AlN, the utilization of such complexes, in the form of simple amine stabilized aluminium hydrides (such as dimethylethylamine alane or dimethylamido alane), has been reported before.21–25 For Al thin films, the reports for depositions employing aluminium hydrides are mostly limited to CVD.26–29 Ternary material systems like AlTiN have been fabricated using alane precursors such as dimethylethylamine alane in combination with titanium tetrachloride and ammonia.30
A concept which should be mentioned here is the exploitation of a rationally designed balance between stabilization and directed decomposition pathways for a targeted (thermal) decomposition during a process. Such balanced precursors could in principle function as single source precursors (SSP) in CVD. First reports for vapor phase depositions of Al-metal thin films can be dated back to the mid 90′s, employing dimethylethylamine alane in CVD and laser assisted CVD as reported by Karpov et al.26,27 and Han et al.28 More recent publications by Blakeney et al.31,32 investigating alanes as precursors in ALD are describing the deposition of Al metal thin films in a thermal ALD process employing the diamine stabilized alane Al(Me2NCH2CH2NtBu)H2 with AlCl3 as co-reactant. The growth of Al thin films could be shown in a low temperature range of 100–140 °C, with calculated bulk resistivities of 3.03 μΩ cm for a 77 nm film deposited at 120 °C. The deposition of AlN using [AlH2NMe2]3 and NH3 plasma as the co-reactant was reported by Buttera et al. in a temperature range of 100–250 °C.21
Following these concepts, we focused on the chemistry of aluminium hydride complexes targeting application as precursors for vapor phase deposition processes, with respect to influence of precursor ligand and coordinationsphere on the physicochemical properties, especially thermal properties, combining thermal stability and high volatility, and long-term stability for high shelf-life times.
When designing precursors for the material systems Al and AlN, certain parameters must be considered. As Al is prone to oxidation, oxygen must be avoided in the precursor's ligand and especially in the coordinationsphere and thus, inhibiting potential oxide formation during the process by incorporation of oxygen atoms originating from the precursor itself in CVD process. Hence, the focus of this comparative study lies on C and N coordinating ligands. A further issue which must be considered is the rather low stability of Al–H bonds, which can be addressed by stabilization through dative bonds, through introduction of multidentate ligands or a combination of both. Structural variations of sidechains and substituents can furthermore be exploited to fine-tune the thermal properties and reactivity of a potential precursor and tailor them for a specific process.
Herein, we present the synthesis and characterization of nine different mono- and bi-dentate nitrogen coordinating and nitrogen dative bond stabilized alanes (Scheme 1). All complexes are characterized by means of nuclear magnetic resonance (NMR) spectroscopy, elemental analysis (EA), fourier transform infra red spectroscopy (FTIR) and in case of 1 and 2 by electron-impact ionization mass spectrometry (EI-MS). In the case of compound 1, the solid-state structure was analyzed by means of single crystal X-ray diffraction (SCXRD). Thermal properties were investigated via thermogravimetric analysis (TGA) as well as thermal stability tests. We employed computational methods namely density functional theory (DFT) studies to elucidate the structure of selected representatives and their reactivity towards NH3 and H2 was investigated to gain insights for their potential applications in ALD.
 | ||
| Scheme 1 Overview of investigated Al compounds categorized into groups by the stabilizing ligands. Structural modifications undertaken in the ligand surroundings are highlighted in blue. | ||
Results and discussion
Precursor synthesis
To provide a strong stabilization motif for the relatively unstable alane scaffold, the concept of this work was to introduce and investigate different ligand systems and coordination motifs and evaluate the impact on the physicochemical properties of the resulting compounds and their applicability as potential precursors for vapor phase deposition processes.Herein, three different dimethylaminopropyl (DMP) based compounds, namely the previously reported dihydride bis(dimethylaminopropylalane) (1, [Al(DMP)H2]2),33 and two new monohydride compounds dimethylaminopropylchloridoalane (2, Al(DMP)HCl), and dimethylaminopropylmethylalane (3, Al(DMP)HMe) were synthesized. The DMP ligand features an excellent balance of reactivity and stabilization through dative bonding of the amine group, which could be shown previously for the potential TMA substitutes reported by Mai et al.34,35 Furthermore, this ligand features decomposition pathways for clean, targeted decomposition as previously shown by Zanders et al.,36 where DMP was employed as ligand for a zinc alkyl reducing agent for ALD of cobalt films.
Starting from Al(DMP)Cl2, which was synthesized according to the procedure described by Mai et al.,34,351 and 2 were prepared via salt-metathesis reaction with one or two equivalents of LiH, respectively. While the reactants for 1 were employed as solids and dissolved in Et2O at −50 °C, for the synthesis of 2 a slurry of LiH in Et2O was added to a solution of Al(DMP)Cl2 in Et2O via cannula. Work-up of the crude products was done via sublimation for the solid compound 1, and via distillation in case of 2. Crystals of 1 suitable for SCXRD could be picked directly from the sublimation finger after work-up, confirming and refining the previously reported structure of Dümichen et al.33 Crystallographic data are given in the ESI (Tables SI 1, SI 2 and Fig. SI 1†). Compound 2 is being isolated for the first time and it is a highly reactive liquid at room temperature, slightly fuming at O2 concentrations as low as 15 ppm, presumably under HCl elimination, and solidifies amorphous at −30 °C. Thus, no suitable crystals of 2 could be isolated for SCXRD. However, the liquid aggregation is favored for the application as precursor, ensuring constant surface for evaporation. It is also worth mentioning, that this is the only liquid compound among the series of starting reagent Al(DMP)Cl2, and products [Al(DMP)H2]2 and Al(DMP)HCl.
Synthesis of 3 was done by treating 2 with a stoichiometric amount of methyl lithium (MeLi) at −80 °C. It should be noted that the stability of 3 is significantly lower than for the analogues 1 and 2, resulting in rather limited proof for distinct compound characterization. Work-up via distillation of the liquid crude product of 3 resulted in decomposition, which also takes place upon dissolution in dried and degassed NMR solvents (C6D6, CDCl3). Thus, compound formation can only be assumed and supported by FTIR (ν(Al–H) = 1690 cm−1) as well as 1H NMR spectroscopy showing a mixture of 3 and its decomposition product, matching to those of the free ligand (dimethylaminopropane) and its fragments; however, a distinct signal assignment or integration was not possible due to strong signal overlap and would have limited significance. Considering the targeted application and the prerequisite in terms of thermal stability, further attempts to isolate, and characterize this compound were not undertaken. Attempts to synthesize the amide analogue Al(DMP)(NMe2)H by treatment of 2 with one equivalent of lithium dimethyl amide were not successful.
As a comparative standard, the simple amine stabilized alane trimethylamine alane (4, AlH3:NMe3) and the amides tris(dimethylamido alane) (5, [AlH2NMe2]3) and diethylamido alane (6, AlH2NEt2) were synthesized in a conventional salt metathesis reaction of LiAlH4 and the hydrochloride of the respective amines in a one-pot synthesis and could be isolated in yields of 90–95% via crystallization. Procedures were adapted from Ruff et al.37 Those compounds are fairly facile to synthesize, but lack shelf-life and are highly reactive upon contact to the ambient. Even though no pyrophoricity as strong as for TMA is observed, contaminated paper tissue starts fuming and even burning with higher amounts of compound upon air contact.
For further comparison of stabilizing coordination motifs, the all nitrogen coordinating 2-dimethylaminoethane-1-(tert-butylamido)alane (7, Al(NtBu)CH2CH2(NMe2)H2), previously reported by Blakeney et al.,31 was synthesized alongside the dimeric methyl substituted analogue bis(2-dimethylaminoethane-1-methylamidoalane) (8, [Al(NMe)CH2CH2(NMe2)H2]2) reintroducing and allowing β-H elimination, which was aggravated in the previous precursor design of 7 by Blakeney et al. By introduction of a tert-butyl substituent, the β-H elimination decomposition pathway was blocked for increased stability. Pushing the reintroduction of facilitated decomposition further, the amine derivative 2-dimethylaminoethane-1-amidoalane (9, Al(NH)CH2CH2(NMe2)H2) was synthesized. However, this compound readily decomposes, resulting in significantly lowered yields and degradation of the complex over time, forming a brittle foam under H2 release (Fig. SI 2†). Compounds 7–9, were synthesized reacting the respective diamine with in situ prepared AlH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 as described by Blakeney et al.31 While 7 could be isolated spectroscopically pure in high yields of 92%, 8 was isolated in a significantly lower yield of 41%, while yields of 9 are unreliable due to limited stability and mixture of product and decomposition by-product.
38 as described by Blakeney et al.31 While 7 could be isolated spectroscopically pure in high yields of 92%, 8 was isolated in a significantly lower yield of 41%, while yields of 9 are unreliable due to limited stability and mixture of product and decomposition by-product.
Depending on the solubility, all compounds were characterized via1H and 13C NMR spectroscopy. Additionally, elemental analysis (CHN-EA) was performed, confirming the expected composition. For the less stable compounds 8 and 9 deviations of ∼2% can be seen for the C and N content, which can be attributed to self-decomposition as well as decomposition upon air and moisture contact, as a short exposure to the ambient is inevitable during sample submission.
Infrared spectroscopy
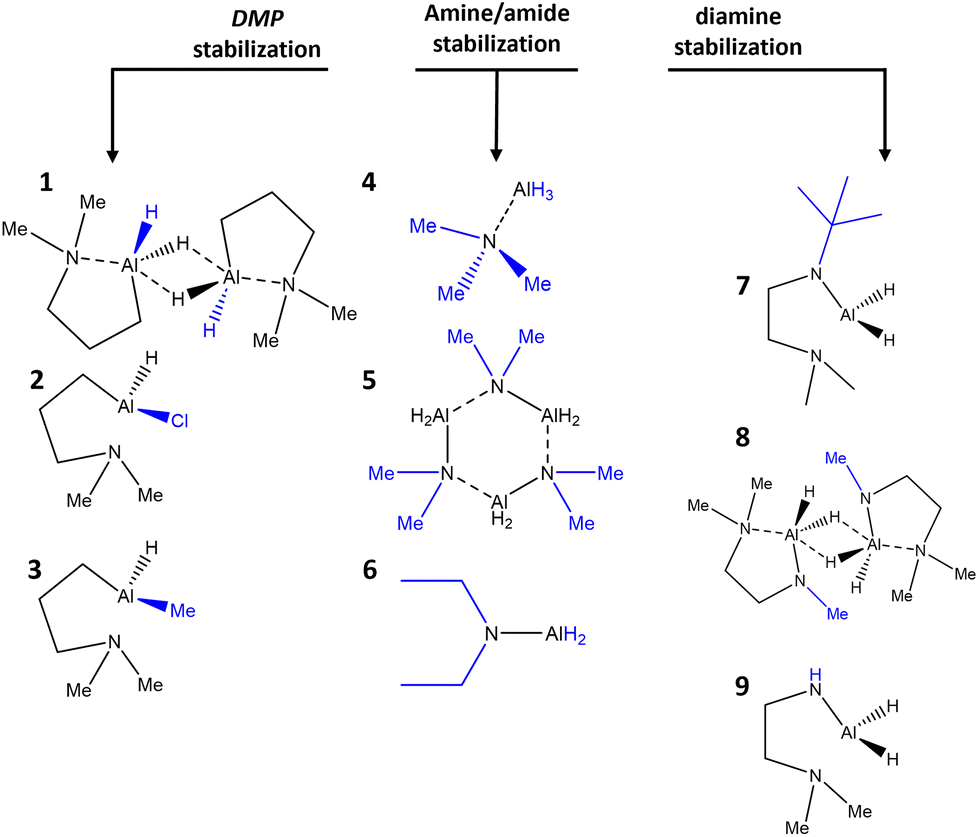
As a complementary analysis tool to EA and NMR spectroscopy, FTIR was performed, as shown for three representative cases (2, 4 and 7) of each group of stabilized alanes (Fig. 1). As the signal of aluminium bond hydrides in 1H NMR often is significantly broadened due to coupling with the 27Al nucleus39 and thus may differ from expected values for the integration, or are not visible at all, FTIR can supplement compound characterization, as the Al–H bond gives a vibrational frequency signal in range of 1750–1900 cm−1.21,39,40 Same applies for the less stable compounds decomposing in solution (3, 8 and 9). The respective ν(Al–H) signals can be seen for all investigated compounds. The vibrational modes of the atomic structures of complexes 2, 4 and 7 can also be calculated using the aoforce program in TURBOMOLE. The Al–H vibrational modes are at 1788.93 cm−1, 1830.32 cm−1 and 1770.01 cm−1 for AlH3:NMe3, Al(DMP)HCl and Al(Me2NCH2CH2NtBu)H2 respectively which are all comparable to the experimental vibrational modes in Fig. 1.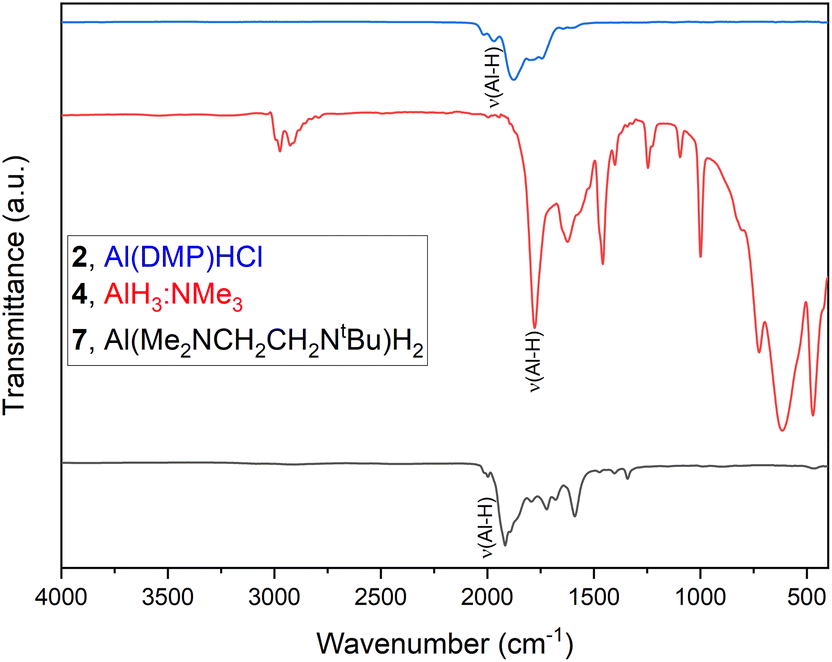 | ||
| Fig. 1 FTIR spectroscopy of compounds 2, 4 and 7. | ||
Mass spectrometry
Apart from NMR, EA and FTIR measurements, compounds 1 and 2 were investigated by means of EI-MS (Fig. 2 and Table 1). Besides confirming the formation of the targeted compounds, this method provides possible decomposition pathways under harsh EI-MS conditions, although the fragmentation cannot be directly compared to the decomposition that occurs during a CVD process. Especially for alanes, the tendency of dimer formation for intermolecular stabilization can be traced, which has been observed before for complex 1.33 For the dihydride 1 the expected dimer [M2]+ signal at m/z 299.22 can be seen with a relative abundance (rel. abund.) of 3.52%, whereas the monomer peak [M]+ at m/z 114.11 gives the strongest signal with 100%. For 2, no dimer formation can be seen and only the monomer [M]+ signal is found at m/z 148.12, 5.67%. For both compounds a peak at m/z 199.24 is found, most likely corresponding to a [Al(DMP)2]+ species, indicating in situ ligand exchange reaction/rearrangement, as such signals are not found in NMR, which would show characteristic coupling effects in the respective spectrum. This shows the versatile nature of the DMP ligand and Al(DMP)-complexes, being able to rearrange and coordinate in multiple ways, as well as form volatile decomposition products.33,36 For dimer 1, a peak ([M2 − DME]+) at m/z 158.18 (3.37%) is found corresponding to fragmentation of the dimer with the cleavage of a dimethyl ethylamine fragment (DME, NMe2Et). Additional peaks below m/z 100 can be assigned to the cleaved DMP ligand and its fragmentation, as well as the Al metal center of the complexes. | ||
| Fig. 2 EI-MS spectra of compounds 1 (top) and 2 (bottom). | ||
| Compound | 1 | 2 | ||
|---|---|---|---|---|
| Fragment | m/z | Rel. abund. (%) | m/z | Rel. abund. (%) |
| [M2]+ | 299.22 | 3.52 | — | — |
| [Al(DMP)2]+ | 199.24 | 8.68 | 199.24 | 6.91 |
| [M2 − DME]+ | 158.18 | 3.37 | — | — |
| [M]+/[Al(DMP)Cl]+ | — | — | 148.12 | 5.67 |
| [M]+/[Al(DMP)H]+ | 114.11 | 100 | 114.15 | 3.48 |
| [DMP]+ | 86.11 | 16.23 | 87.12 | 2.73 |
| [M − NMe2]+ | 72.05 | 30.50 | — | — |
| [NMe3]+ | 58.08 | 86.22 | 58.08 | 100 |
| [Pr]+/[NMe2]+ | 42.05 | 18.15 | 42.04 | 10.67 |
| [Al]+ | 26.99 | 28.78 | — | — |
Thermal analysis
To evaluate the thermal properties of the synthesized compounds, aiming for the potential application as precursors in CVD and ALD, TG is an important method to determine the thermal characteristics of a precursor. Thus, TG was conducted within a temperature range from 35 to 450 °C. Fig. 3 shows the respective TG graphs divided into three groups, (a) DMP-stabilized alanes, (b) amine stabilized and (c) diamine stabilized alanes. Onsets of volatilization (Tvol.) were determined via 1% mass loss and are summarized in the ESI, in Table SI 3.† All compounds show single step evaporation, except 9, which shows decomposition at temperatures below 100 °C. This is in good accordance with decomposition over longer storage times (∼1 week) even at −30 °C, where the compound forms a “solid foam” (Fig. SI 2†), most likely attributable to hydride elimination of the adjacent Al-hydride and amine-hydrogen. While such decomposition of compound 9 at room temperature and slightly above does not favor it as a CVD/ALD precursor, the intended concept of targeted decomposition via hydrogen elimination can be validated.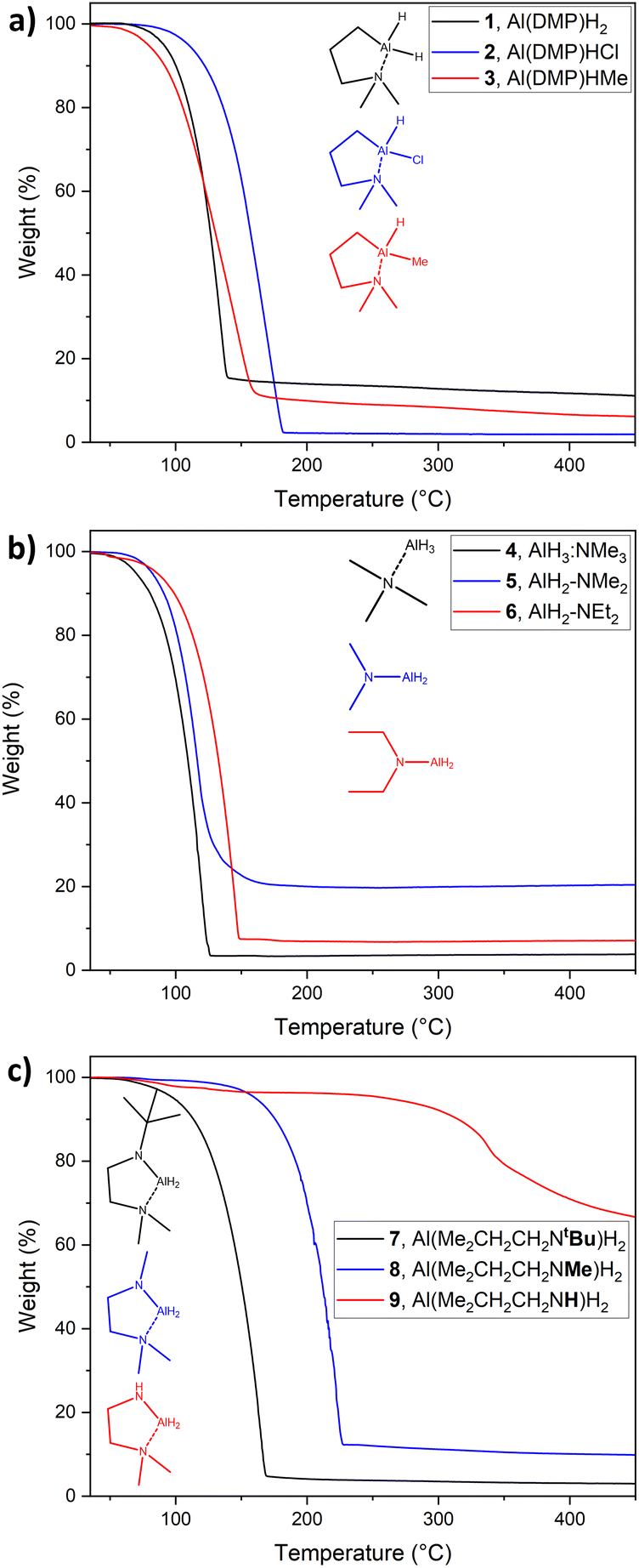 | ||
| Fig. 3 TG of compounds (a) 1–3, (b) 4–6 and (c) 7–9 in a temperature range from 30 °C to 450 °C. Stuctures and names given as monomers for clarity. | ||
The DMP-stabilized compounds 1–3 (Fig. 3(a)) exhibit Tvol. ranging from 56.8 °C for 3 to 87.5 °C for the chloride derivate 2, with the dimeric alane 1 in-between those temperatures with a Tvol. of 74.1 °C. The residual masses (r.m.) are found to be within a range of 1.9 wt% for 2 to 13.1 wt% for 1, indicating a higher thermal stability for 2, than for the dihydride 1 and methyl-hydride 3. As an overall trend, the DMP-stabilized alanes prove to be less volatile in direct comparison to the amine stabilized alanes 4–6, which is expected considering the higher molecular mass.
Among the diamine stabilized compounds (Fig. 3(c)), the tert-butyl derivate 7 is with an onset of evaporation of 67.2 °C and a r.m. of 2.8 wt% the most volatile and most stable representative in this group. The methyl analogue 8 shows a higher residual mass of 9.2 wt%, as the potential decomposition pathway via β-hydrogen elimination at the MeN group is possible in contrast to 7, where no β-hydrogen is present at the tBuN group. The rapid decomposition of 9, already observed during synthesis and storage, is also observed in TGA resulting in a high r.m. of 61.9 wt%, while the respective Tvol. must be looked at critically due to the high tendency for decomposition, which affects the mass loss and thus the Tvol. derived by 1% mass loss.
Residues inside the TG crucibles, as far as visible, have a grey metallic appearance, hinting for the formation of Al, thus having the potential to be used as a single source precursor (SSP) for metallic Al thin films. To further investigate this observation, SSP tests were conducted as discussed in the following section.
Single source precursor tests
As a preliminary test to estimate the potential use as SSP, with respect to the intrinsic reducing capability of the Al hydrides upon exposure to thermal stress (elevated temperatures), the compounds were heated in a Schlenk tube under vacuum (∼1 × 10−2 mbar). These conditions were chosen to mimic the conditions in vapor phase deposition processes, where the precursor is decomposed thermally, either via substrate heating (CVD) or from energy assisted deposition techniques such as plasma enhanced (PE-) or photo assisted ALD.41Fig. 4 shows the representative compounds amongst each group (compounds 2, 4 and 7), as synthesized and post-heating, as well as a powder X-ray diffractogram (XRD) of the metallic deposits collected as powder from the flask wall.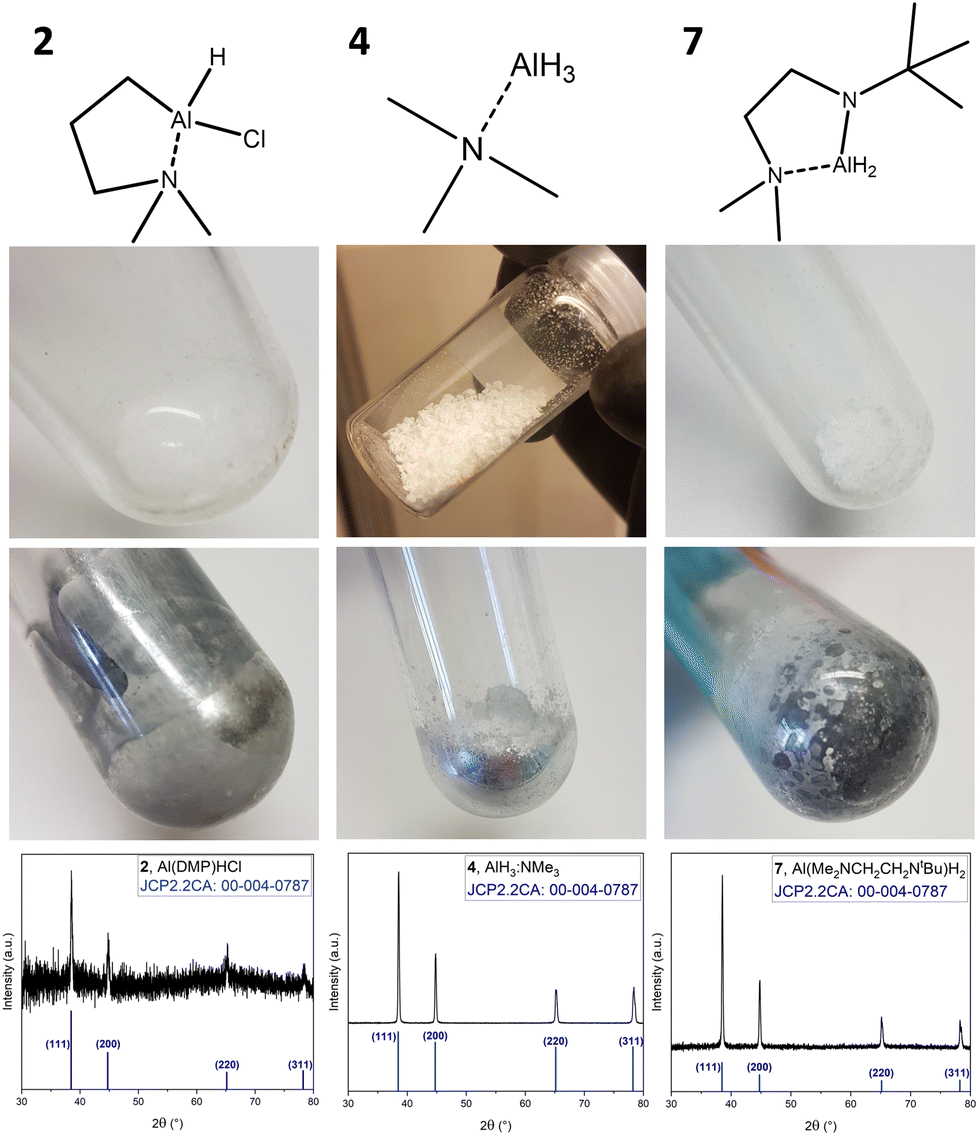 | ||
| Fig. 4 SSP testing of compounds 2, 4 and 7, with images of the respective compound before and after heat treatment and powder XRD patterns of the metallic residue. | ||
The recorded powder XRD diffractograms match the reference for Al metal (reference pattern: JCP2.2CA:00-004-0787), confirming decomposition to form Al.
These observations clearly indicate and confirm the SSP concept reported in previous publications26–28 for amine stabilized alanes transferable to the comparable and more stabilized compounds 1–3, 7 and 8 presented in this study. These findings also show a high reactivity in general and more specific towards relevant co-reactants such as NH3 and H2 that could enable thermal ALD for AlN and Al thin film deposition, respectively. Thus, to substantiate this hypothesis, in terms of structure and reactivity of these precursors, we conducted a detailed computational study of three representative precursors, namely compounds 2, 4 and 7.
Density functional theory simulations of Al hydride complexes
The structure of the three aluminium hydride complexes AlH3:NMe3 (4), Al(DMP)HCl (2) and Al(Me2NCH2CH2NtBu)H2 (7) were investigated using first principles density functional theory (DFT).Bond dissociation energies EBD for the loss of ligand, L, were calculated using:
| EBD = (EL + EP–L) − EP |
The computed total energy of the precursor molecule and a free ligand are denoted by EP and EL respectively, while the computed total energy of the precursor after loss of a ligand, L, is EP–L.
The relaxed geometries of the aluminium complexes are shown in Fig. 5. The bond lengths and bond angles for all three complexes are shown in Tables 2 and 3. All Al–H bond lengths in AlH3:NMe3 are the same and the dative Al–N bond is consistent with the value of 2.06 Å from Gundersen et al. 1972.42
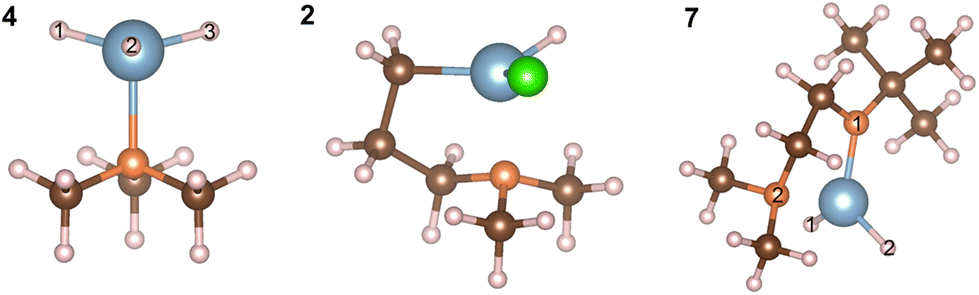 | ||
| Fig. 5 Atomistic structures of relaxed Al-hydride complexes from DFT: AlH3:NMe3 (4), Al(DMP)HCl (2) and Al(Me2NCH2CH2NtBu)H2 (7). Al, N, C, H and Cl are blue, orange, brown, pink and green respectively. | ||
| AlH3:NMe3 | Al(DMP)HCl | Al(Me2NCH2CH2NtBu)H2 | |||
|---|---|---|---|---|---|
| H1–Al–H2 | 117.28° | H–Al–Cl | 113.37° | H1–Al–H2 | 115.70° |
| H2–Al–H3 | 117.25° | C–Al–H | 121.37° | H1–Al–N1 | 103.88° |
| H3–Al–H1 | 117.26° | C–Al–N | 89.93° | H1–Al–N2 | 119.70° |
| H1–Al–N | 99.64° | N–Al–Cl | 105.03° | H2–Al–N1 | 103.60° |
| H2–Al–N | 99.58° | H–Al–N | 102.69° | H2–Al–N2 | 118.81° |
| H3–Al–N | 99.65° | Cl–Al–C | 117.93° | N1–Al–N2 | 86.82° |
To understand the experimental results regarding reactivity, we consider the energetics of ligand elimination from each precursor. The computed energies for elimination of H by breaking the Al–H bond are 137.42 kJ mol−1, 136.37 kJ mol−1 and 132.58 kJ mol−1 for AlH3:NMe3, Al(DMP)HCl and Al(Me2NCH2CH2NtBu)H2 respectively. These are relatively low energies, essentially identical across all Al-hydrides, suggesting facile loss of H from each precursor.
Considering AlH3:NMe3 (4), the energy required to eliminate NMe3 through breaking the Al–N bond is 125.92 kJ mol−1 showing, as expected from the structure of the complex, a weaker bond is present between Al and N, which is more favourable to dissociate compared to the Al–H bond. This is consistent with the abundance of [NMe3]+ found in the mass spectrometry data (Table 1).
In compound 2, Al bonds to the DMP ligand through an Al–C bond and the energy needed to eliminate the DMP ligand is 418.94 kJ mol−1, significantly higher than the corresponding Al–H energy and suggesting a high stability of this metal–ligand coordination. There is also an Al–Cl bond in Al(DMP)HCl with bond dissociation energy 378.35 kJ mol−1 which is more stable than Al–H but less stable than Al–C. In 7, Al(Me2NCH2CH2NtBu)H2, Al bonds to the Me2NCH2CH2NtBu ligand through N labelled 1 in Fig. 5 with a computed bond dissociation energy 456.82 kJ mol−1.
To provide an initial assessment of the chemical reactivity of these precursors for vapor phase deposition, we consider the interaction of the three hydride complexes with H2 to assess H2 as a co-reactant. The interaction energy is computed as:
| Eint = ∑EP − ∑ER |
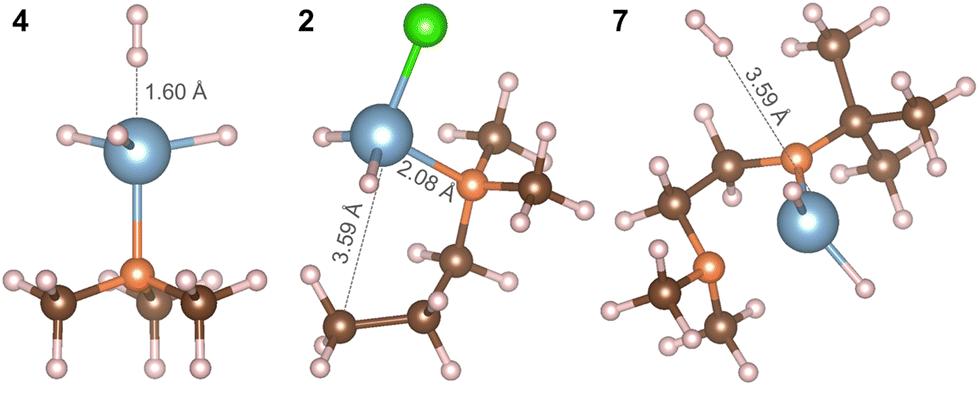 | ||
| Fig. 6 Atomistic structures of complexes AlH3:NMe3 (4), Al(DMP)HCl (2) and Al(Me2NCH2CH2NtBu)H2 (7) after the interaction with H2. Colour scheme is the same as in Fig. 5. | ||
As a consequence, the Al–N dative bond in Al(DMP)HCl is now shorter, changing from 2.11 Å in the precursor to 2.08 Å after interaction with H2. Therefore, with H2 present, complex 2 can decompose through a favorable insertion of H into the original Al–C bond to DMP.
In Fig. 7 NH3 can interact with all three precursors with favorable interaction energies, −36.00 kJ mol−1 for AlH3:NMe3, −76.95 kJ mol−1 for Al(DMP)HCl and −19.51 kJ mol−1 for Al(Me2NCH2CH2NtBu)H2. The Al–N dative bond in AlH3:NMe3 breaks and Al forms a new Al–N bond with NH3 making mono(ammonia)alane.43 In Al(DMP)HCl, NH3 donates a H to form a C–H bond resulting in the formation of dimethylproplylamine. Al forms a new Al–N bond with NH2 with a bond length of 1.82 Å. Similarly for Al(Me2NCH2CH2NtBu)H2, N1 forms a new N–H bond and Al forms a new Al–N bond with NH2 of 1.85 Å. This results in the formation of N,N-dimethyl-N′-(2-methyl-2-propanyl)-1,2-ethanediamine. Compared to H2, NH3 is shown to be a more reactive reducing agent with these Al complexes, which therefore shows some promise for AlN deposition.
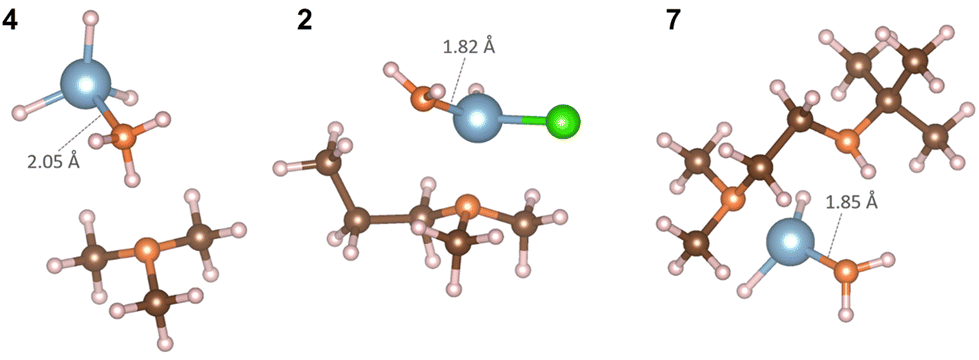 | ||
| Fig. 7 Atomistic structures of complexes AlH3:NMe3 (4), Al(DMP)HCl (2) and Al(Me2NCH2CH2NtBu)H2 (7) after the interaction with NH3. Colour scheme is the same as in Fig. 5. | ||
Conclusion
In this study we targeted aluminium hydrides as potential single source precursors for chemical vapor phase deposition processes of Al metal, with the scope of improving the lack of thermal stability in existing Al complexes, through ligand stabilization. Three different ligand classes were chosen, namely small alkyl amines, dimethylaminopropyl and diamines. The significant impact of coordination sphere and ligand design on the complexes stability and intermolecular coordination behavior is demonstrated from comparative FTIR and NMR analyses. Thermal decomposition and residual mass analysis yielded metallic aluminium as the main decomposition product, indicating the potential for the targeted application as CVD precursor. This was further confirmed by DFT simulations, where in addition to the relaxed geometries of the complexes, bond dissociation energies were computed, giving insight into favored decomposition pathways. To assess the potential as a possible ALD precursor, reaction enthalpies for reactions with H2 and NH3 were calculated, revealing NH3 as the more reactive potential co-reactant for all three investigated compounds. With this study we aim to highlight the great potential of stabilized aluminium hydrides as a currently underestimated precursor class for chemical vapor phase deposition processes, especially for the more sensitive material systems Al and AlN. Future work will focus on validating these exciting results in ALD process development for Al and AlN thin film deposition.Experimental
Precursor synthesis and characterization
All reactions and handling of air- and moisture sensitive compounds were carried out under dried argon atmosphere (Air Liquide, 99.995%) using conventional Schlenk techniques. Glassware were baked out and silanized prior to use. Sample preparation for further analysis was carried out in an Argon filled glovebox (Mbraun). All commercially available chemicals were used without further purification. Solvents were dried by a solvent purification system (MBraun) and stored over molecular sieves (4 Å) under an argon atmosphere. NMR-solvents were degassed and dried over activated molecular sieves (4 Å).Starting reagents were prepared as described in literature. Al(DMP)Cl2 was synthesized following a route described by Mai et al.34 Literature known compounds, either for comparison or for first time thermal evaluation were synthesized according to the respective reported procedures: compound 7 was synthesized as described by Blakeney et al.,31 and 4–6 following the procedure reported by Ruff et al.37
NMR-spectra were recorded on Bruker Avance III 300 and referenced to the internal solvent signal (C6D6). Spectra analysis was done with the software MestReNova v10.0.2-15465 from Mestrelab Research S.L. Elemental analysis (EA) was performed on an Elementar Vario Micro Cube. Electron-impact ionization mass spectra (EI-MS) were recorded at the RubioSpec Service Center of the Ruhr-University Bochum with a Varian MAT spectrometer at an ionizing energy of 70 eV. IR spectra were recorded on a FTIR spectrometer Spectrum Two by PerkinElmer placed in an argon filled glove box, utilizing an UATR Two ATR-unit by PerkinElmer. Thermogravimetric analysis (TGA) was performed on a Netzsch STA 409 PC at ambient pressure (sample size ≈ 10 mg), with a heating rate of 5 K min−1 (N2 flow rate = 90 sccm), placed in an argon (Air Liquide, 99.995%) filled glovebox (SylaTech). In the DFT calculations, all structures were fully optimized with no symmetry constraints using the TURBOMOLE suite44,45 of quantum chemistry programs. All calculations were performed with the PBE exchange–correlation functional46 and a polarized triple ζ basis set (def-TZVPP).47,48 The SCF convergence criterion was set to 10−6 Ha and a medium (m3) grid was used for the integrations.
Bis(3-dimethylaminopropylalane), {[Al(DMP)H2]2}, (1)
The procedure from Dümichen et al.33 was adapted and slightly modified 1.0 g of Al(DMP)Cl2 (5.43 mmol, 1 eq.) and 86 mg of LiH (10.87 mmol, 2 eq.) were filled in a Schlenk flask, cooled to −50 °C and cold diethyl ether (Et2O) was slowly added under stirring. The reaction mixture was warmed to room temperature (RT), stirred for 20 h and subsequently refluxed at 45 °C. Precipitated solids were filtered off and the solvent was evaporated in vacuum. The remaining colorless solid was purified via sublimation at 45 °C, yielding 375 mg (60%) of spectroscopically pure product. Crystals for SCXRD were collected directly from the sublimation finger.1H NMR (250 MHz, C6D6): δ (ppm) = 4.21 (s, 2H), 1.90 (t, 2H), 1.83 (s, 6H), 1.57 (p, 2H), 0.50 (t, 2H). 13C NMR (75 MHz, C6D6): δ (ppm) = 63.5, 45.5, 22.5. EA: calc. (%): C: 52.15, H: 12.25, N: 12.16 found: C: 51.97, H: 12.24, N: 13.68. m.p. = 55 °C.
Chloro[3-(dimethylamino)propyl]alane, Al(DMP)HCl, (2)
The synthesis of compound 5 was performed following the synthesis route adapted for compound 4. To a solution of 3.0 g of Al(DMP)Cl2 (16.3 mmol, 1 eq.) in Et2O, a slurry of 285 mg LiH (35.86 mmol, 2.2 eq.) in Et2O was added at −50 °C. The solution was warmed to RT and stirred for 20 h followed by one hour reflux at 45 °C. Precipitated solids were filtered off and the solvent was evaporated in vacuum, leaving a colorless, oily residue. The product was isolated via distillation in vacuum (∼10−2 mbar) at 45 °C, yielding 2.05 g (82%) of a spectroscopically pure colorless oil.1H NMR (250 MHz, C6D6): δ (ppm) = 4.35 (s, 1H), 1.89 (br, 2H), 1.82 (br, 6H), 1.37 (br, 2H), 0.25 (t, 2H). 13C NMR (75 MHz, C6D6): δ (ppm) = 62.9, 44.8, 21.2, 20.5. EA: calc. (%): C: 40.14, H: 8.76, N: 9.36 found: C: 40.22, H: 8.44, N: 9.75. IR (cm−1): ν(C–H) = 2937, ν(Al–H) = 1822, δ(C–H) = 1467, ν(C–N) = 1015.
Stability for the following compounds 3, 8 and 9 is very limited, especially in solution, resulting in ambiguous NMR data, and slightly higher deviations in EA.
Methyl[3-(dimethylamino)propyl]alane, Al(DMP)HMe, (3)
To a solution of 1 g of compound 2 (6.68 mmol, 1 eq.) in 20 mL of Et2O, 4.39 mL of 1.6 M MeLi in Et2O (7.02 mmol, 1.05 eq.) was added dropwise at −80 °C. The solution was warmed to RT and stirred for 20 h. Precipitated solids were filtered off and the solvent was evaporated in vacuum leaving 458 mg (53%) of a colorless liquid. The product was analyzed as obtained. Attempts for further purification via vacuum distillation resulted in decomposition of the product, leaving a viscous colorless oil. The fast decomposition can also be seen in NMR, as the compound decomposes in solution over time. Thus, NMR signals given below are accompanied by signals close by corresponding to by-products resulting from decomposition.1H NMR (250 MHz, C6D6): δ (ppm) = 3.81 (br, 1H), 2.29 (qq, 1H), 2.00 (br, 5H), 1.92 (t, 2H), 1.69 (s, 4H), 1.57 (p, 2H), 1.23 (br, 2H), 0.89 (t, 2H), 0.22 (br, 2H), 0.04 (br, 1H), −0.52 (s, 3H). 13C NMR (75 MHz, C6D6): δ (ppm) = 63.0, 45.5, 22.9. IR (cm−1): ν(C–H) = 2910, ν(Al–H) = 1690, δ(C–H) = 1460, ν(C–N) = 1184, ν(C–H) = 1028.
1-(Methylamido)-2-(dimethylaminoethyl)alane, [Al(NMe)CH2CH2(NMe2)H2]2, (8)
To a slurry of 619 mg LiAlH4 in 60 mL Et2O (16.31 mmol, 3 eq.), a solution of 725 mg AlCl3 (5.44 mmol, 1 eq.) in 20 mL Et2O was added dropwise at −50 °C and stirred for 30 min while warming to RT. Subsequently the mixture was cooled down to −50 °C and a solution of 2.0 g of N,N,N′-trimethylethylendiamine (19.57 mmol, 3.6 eq.) in 20 mL of Et2O was added dropwise. The reaction mixture was stirred overnight, and the precipitated solids were removed via filtration followed by solvent extraction in vacuum. The product was isolated without further purification yielding 1.06 g (41%) of a white solid.1H NMR (300 MHz, C6D6) δ (ppm) = 2.69 (s, 3H), 2.08 (s, 6H). 13C NMR (75 MHz, C6D6) δ (ppm) = 57.3, 51.1, 48.1, 41.6. EA: calc. (%): C: 46.14, H: 11.62, N: 21.52 found: C: 44.10, H: 10.91, N: 20.92. IR (cm−1): ν(C–H) = 2966, ν(C–H) = 2867, ν(Al–H) = 1744, δ(C–H) = 1457, ν(C–N) = 1280, ν(C–N) = 1148, ν(C–N) = 1086, ν(C–N) = 1020.
1-(Amido)-2-(dimethylaminoethyl)alane, Al(NH)CH2CH2(NMe2)H2, (9)
Compound 9 was prepared in an analogous procedure like for compound 8 using 718 mg LiAlH4 (18.91 mmol, 3 eq.), 840 mg AlCl3 (6.30 mmol, 1 eq.) and 2.0 g of N,N-dimethylethylendiamine (22.69 mmol, 3.6 eq.). Precipitated solids were removed via filtration and the solvent was evaporated in vacuum. The product was isolated without further purification yielding 2.17 g (83%) of a white solid. It should be stated that the stability of 9 is rather limited and it slowly decomposes at RT, forming a brittle foam under H2 release. Hence, yield is referring to collected white solid and not necessarily the desired product. Further analysis via NMR showed only decomposed residues of the product. 1H NMR and 13C NMR measurements resulted in decomposition of 9 in solution and thus no reliable NMR data could be obtained.EA: calc. (%): C: 41.37, H: 11.28, N: 24.12 found: C: 38.91, H: 9.45, N: 22.73. IR (cm−1): ν(C–H) = 2835, ν(Al–H) = 1768, ν(Al–H) = 1660, δ(C–H) = 1459, ν(C–N) = 1272, ν(C–N) = 1064, ν(C–N) = 1022.
Author contributions
All authors have given approval to the final version of the manuscript. N. H. was involved in project management, ideation, synthesis, analysis, and data interpretation; R. M., DFT simulations and interpretation; N. H. and R. M., manuscript writing; A. D. and M. N., ideation, manuscript review and project supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the DFG-ANR project REACTIVE (project number 490773082). M. N. and R. M. acknowledge financial support from Science Foundation Ireland-NSF China Partnership Program, NITRALD Grant number: 17/NSFC/5279. A. D. acknowledges Fraunhofer Attract Programme for the support. The authors thank Martin Wilken and Dr. Dominik Naglav-Hansen for measuring and refining the crystal structure.References
- G. K. Deshwal and N. R. Panjagari, J. Food Sci. Technol., 2020, 57, 2377–2392 CrossRef PubMed.
- M. Lamberti and F. Escher, Food Rev. Int., 2007, 23, 407–433 CrossRef CAS.
- Fundamentals of Aluminium Metallurgy, ed. R. Lumley, Woodhead, Oxford, 2011 Search PubMed.
- E. A. Starke and J. T. Staley, in Fundamentals of Aluminium Metallurgy, ed. R. Lumley, Woodhead, Oxford, 2011, pp. 747–783 Search PubMed.
- M. Aamir, K. Giasin, M. Tolouei-Rad and A. Vafadar, J. Mater. Res. Technol., 2020, 9, 12484–12500 CrossRef CAS.
- S. P. Douglas and C. E. Knapp, ACS Appl. Mater. Interfaces, 2020, 12, 26193–26199 CrossRef CAS PubMed.
- J. Yang, K. Liu, X. Chen and D. Shen, Prog. Quantum Electron., 2022, 83, 100397 CrossRef.
- C. Fei, X. Liu, B. Zhu, Di Li, X. Yang, Y. Yang and Q. Zhou, Nano Energy, 2018, 51, 146–161 CrossRef CAS.
- L. P. B. Lima, H. F. W. Dekkers, J. G. Lisoni, J. A. Diniz, S. van Elshocht and S. de Gendt, J. Appl. Phys., 2014, 115, 074504 CrossRef.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 121301 CrossRef.
- A. L. Brazeau and S. T. Barry, Chem. Mater., 2008, 20, 7287–7291 CrossRef CAS.
- A. L. Hickman, R. Chaudhuri, S. J. Bader, K. Nomoto, L. Li, J. C. M. Hwang, H. Grace Xing and D. Jena, Semicond. Sci. Technol., 2021, 36, 44001 CrossRef CAS.
- C. Ozgit, I. Donmez, M. Alevli and N. Biyikli, Thin Solid Films, 2012, 520, 2750–2755 CrossRef CAS.
- W. M. Yim, E. J. Stofko, P. J. Zanzucchi, J. I. Pankove, M. Ettenberg and S. L. Gilbert, J. Appl. Phys., 1973, 44, 292–296 CrossRef CAS.
- W. Su, F. Zhao, L. Ma, R. Tang, Y. Dong, G. Kong, Y. Zhang, S. Niu, G. Tang, Y. Wang, A. Pang, W. Li and L. Wei, Materials, 2021, 14, 2898 CrossRef CAS PubMed.
- H. Liu, L. Zhang, H. Ma, C. Lu, H. Luo, X. Wang, X. Huang, Z. Lan and J. Guo, J. Energy Chem., 2021, 52, 428–440 CrossRef CAS.
- R. Zidan, B. L. Garcia-Diaz, C. S. Fewox, A. C. Stowe, J. R. Gray and A. G. Harter, Chem. Commun., 2009, 3717–3719 RSC.
- Y. J. Lee and S.-W. Kang, ECS Solid State Lett., 2002, 5, C91–C93 CrossRef CAS.
- Y. J. Lee and S.-W. Kang, J. Vac. Sci. Technol., A, 2002, 20, 1983 CrossRef CAS.
- D. R. Lide, J. Am. Chem. Soc., 2007, 129(3), 724 Search PubMed.
- S. C. Buttera, P. Rouf, P. Deminskyi, N. J. O'Brien, H. Pedersen and S. T. Barry, Inorg. Chem., 2021, 60, 11025–11031 CrossRef CAS PubMed.
- B. D. Fahlman and A. R. Barron, Adv. Mater. Opt. Electron., 2000, 10, 135–144 CrossRef CAS.
- J. N. Kidder, H. K. Yun, J. W. Rogers and T. P. Pearsall, Chem. Mater., 1998, 10, 777–783 CrossRef CAS.
- J. N. Kidder, J. S. Kuo, A. Ludviksson, T. P. Pearsall, J. W. Rogers, J. M. Grant, L. R. Allen and S. T. Hsu, J. Vac. Sci. Technol., A, 1995, 13, 711–715 CrossRef CAS.
- A. Ludviksson, D. W. Robinson and J. Rogers, Thin Solid Films, 1996, 289, 6–13 CrossRef CAS.
- I. Karpov, G. Bratina, L. Sorba, A. Franciosi, M. G. Simmonds and W. L. Gladfelter, J. Appl. Phys., 1994, 76, 3471–3478 CrossRef CAS.
- I. Karpov, W. Gladfelter and A. Franciosi, Appl. Phys. Lett., 1996, 69, 4191–4193 CrossRef CAS.
- J. Han, K. F. Jensen, Y. Senzaki and W. L. Gladfelter, Appl. Phys. Lett., 1994, 64, 425–427 CrossRef CAS.
- M. Sugiyama, T. Iino, T. Nakajima, T. Tanaka, Y. Egashira, K. Yamashita, H. Komiyama and Y. Shimogaki, Thin Solid Films, 2006, 498, 30–35 CrossRef CAS.
- Y.-H. Shin and Y. Shimogaki, Jpn. J. Appl. Phys., 2004, 43, 8253–8257 CrossRef CAS.
- K. J. Blakeney and C. H. Winter, Chem. Mater., 2018, 30, 1844–1848 CrossRef CAS.
- K. J. Blakeney, P. D. Martin and C. H. Winter, Organometallics, 2020, 39, 1006–1013 CrossRef CAS.
- U. Dümichen, K.-H. Thiele, T. Gelbrich and J. Sieler, J. Organomet. Chem., 1995, 495, 71–75 CrossRef.
- L. Mai, N. Boysen, D. Zanders, T. de los Arcos, F. Mitschker, B. Mallick, G. Grundmeier, P. Awakowicz and A. Devi, Chem. – Eur. J., 2019, 25, 7489–7500 CrossRef CAS PubMed.
- L. Mai, M. Gebhard, T. de los Arcos, I. Giner, F. Mitschker, M. Winter, H. Parala, P. Awakowicz, G. Grundmeier and A. Devi, Chem. – Eur. J., 2017, 23, 10768–10772 CrossRef CAS PubMed.
- D. Zanders, J. Liu, J. Obenlüneschloß, C. Bock, D. Rogalla, L. Mai, M. Nolan, S. T. Barry and A. Devi, Chem. Mater., 2021, 33, 5045–5057 CrossRef CAS.
- J. K. Ruff and M. F. Hawthorne, J. Am. Chem. Soc., 1960, 82, 2141–2144 CrossRef CAS.
- A. E. Finholt, A. C. Bond and H. I. Schlesinger, J. Am. Chem. Soc., 1947, 69, 1199–1203 CrossRef CAS.
- A. J. Downs, D. Duckworth, J. C. Machell and C. R. Pulham, Polyhedron, 1992, 11, 1295–1304 CrossRef CAS.
- S. Chaudhuri, S. Rangan, J.-F. Veyan, J. T. Muckerman and Y. J. Chabal, J. Am. Chem. Soc., 2008, 130, 10576–10587 CrossRef CAS PubMed.
- T. Henke, M. Knaut, C. Hossbach, M. Geidel, L. Rebohle, M. Albert, W. Skorupa and J. W. Bartha, ECS J. Solid State Sci. Technol., 2015, 4, P277–P287 CrossRef CAS.
- A. Almenningen, G. Gundersen, T. Haugen, A. Haaland and Å. Pilotti, Acta Chem. Scand., 1972, 26, 3928–3934 CrossRef CAS.
- M. Czerw, A. S. Goldman and K. Krogh-Jespersen, Inorg. Chem., 2000, 39, 363–369 CrossRef CAS PubMed.
- University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, TURBOMOLE, Karlsruhe, 2010.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: TGA and SCXRD. CCDC 2338876. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00709c |
| This journal is © The Royal Society of Chemistry 2024 |