
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The aromatic nature of auracycles and diauracycles based on calculated ring-current strengths†
Daniel
Blasco
 *a and
Dage
Sundholm
*b
*a and
Dage
Sundholm
*b
aDepartamento de Química, Instituto de Investigación en Química (IQUR), Universidad de La Rioja, Madre de Dios 53, 26006, Logroño, Spain. E-mail: daniel.blascos@unirioja.es
bDepartment of Chemistry, Faculty of Science, University of Helsinki, P. O. Box 55 (A. I. Virtasen aukio 1), FIN-00014, Helsinki, Finland. E-mail: dage.sundholm@helsinki.fi
First published on 31st May 2024
Abstract
We have calculated the magnetically induced current density susceptibility for gold-containing organometallic molecular rings using the gauge-including magnetically induced currents (GIMIC) method. The aromatic nature has been determined by calculating the strength of the magnetically induced ring current susceptibility, which is often called ring current. To our knowledge, we show here for the first time that gold-containing organometallic rings may be aromatic or antiaromatic sustaining ring currents in the presence of an external magnetic field. The calculated aromatic character of the rings agrees with the aromatic nature one expects when using Hückel's aromaticity rules. The studied auracycles and diauracycles with 4n electrons in the conjugated orbitals generally sustain a weak paratropic ring current, whereas those having 4n + 2 electrons in the conjugated orbitals sustain a diatropic ring current that is almost as strong as that of benzene. The number of electrons are obtained by assuming that each C, N and Au atom of the ring contribute one electron, and a H atom connected to a N atom in the ring increases the number of electrons by one. An electron-attracting ligand at Au removes one electron from the ring. Formation of a short Au–Au bonding diauracycles reduces the number of electrons in the ring by two.
1 Introduction
The metalloaromaticity concept refers to organometallic molecules that exhibit chemical and physical properties similar to those of classical organic aromatic molecules such as benzene.1,2 These properties include planar molecular rings with bond-length equalization, distinctive 1H NMR chemical shifts of exo and endocyclic protons due to the magnetically induced ring current, and an enhanced thermodynamical stability relative to non-aromatic analogues. Examples of such metal-containing molecules include metallabenzenes, metallabenzenoids, metallabenzynes, (spiro)metalloles, metallapentalenes, and metallapentalynes, which are obtained by the formal replacement of a CH group (or C atom) of the parental molecule by an isolobal metallic fragment. In particular, gold-containing molecular rings can also be constructed by replacing C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C moieties of organic rings by C–Au–N.3
C–C moieties of organic rings by C–Au–N.3
Aromaticity itself is an already fuzzy concept,4,5 but it becomes even fuzzier for this kind of molecules.6 The main reason is that there is no consensus on the number of occupied metal d orbitals participating in the π binding with the conjugated bonds of the organic fragment, leading to an unclear number of electrons in the conjugated orbitals. Möbius and Craig-Möbius aromaticity models,7–10 for which 4n π electrons lead to aromaticity, are invoked for explaining disagreements between the classical Hückel's 4n + 2 π-electron rule and the experimental properties of metalloaromatic molecules.
The first reported stable metallabenzene was osmabenzene [OsC5H4(SMe)(CO)(I)(PPh3)2], which was synthesized by Roper et al.11 The synthesis was followed by other examples where the ring contains osmium, iridium, platinum, ruthenium, or rhenium.1,12–24 Surprisingly, no aurabenzene, i.e. a metallabenzene containing gold, has yet been reported, despite the fact that platinum(II) and iridium(I) are isoelectronic with gold(III). Representative examples of aromatic platinum and iridium containing molecular rings are platinabenzenes,25,26 iridafurans,27 spiroaromatic platinacycles,28 and the antiaromatic platinacorrole.29 Many reviews have been written about metallabenzenes.1,13–16,19,30–33
There are few examples of gold complexes such as gold(III) auracyclopentadienes that may exhibit metalloaromaticity. Usón et al. prepared the first auracyclopentadiene complex [AuCl(C4Ph4)(tht)] (tht = tetrahydrothiophene) and its dimer [Au(μ2-Cl)(C4Ph4)]2 by the transmetallation reaction of [Sn(C4Ph4)Me2] and [AuCl3(tht)].34 These complexes further react with neutral and anionic ligands with release of the labile tht ligand or leading to chloride bridge cleavage, respectively.34,35 Structural evidence was provided for [AuCl(C4Ph4)(phen)] (phen = phenanthroline), which features a planar five-membered AuC4 ring shown in Fig. 1.35 This AuC4 core has also been structurally characterized as an intermediate in the gold(III)-catalyzed conversion of enyne-amines into cyclopentadienes,36 and as a precursor of a (η1-cyclobutadiene)gold(I) complex, which is obtained by reductive elimination.37 Other types of gold-containing aromatic rings may remain to be discovered, as the aromatic nature of gold complexes has not yet been studied in depth.
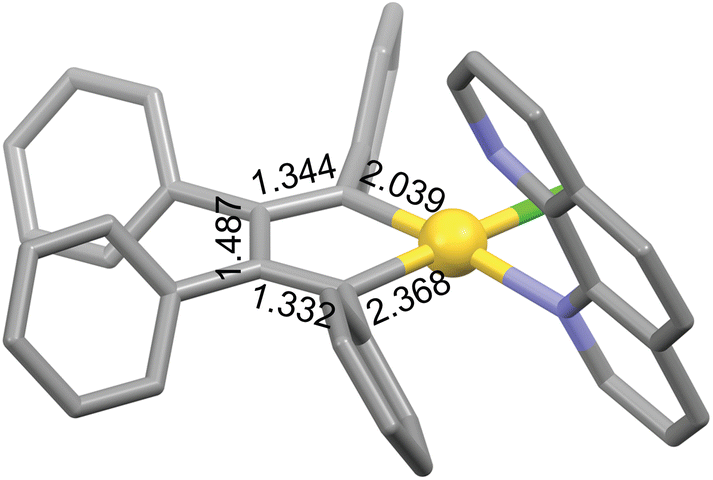 | ||
| Fig. 1 The AuC4 core of [AuCl(C4Ph4)(phen)] (Cambridge Structural Database refcode: BULPED).35 Bond distances are given in Å. Hydrogen atoms are omitted for clarity. Color code: C, grey; Au, yellow; Cl, green; N, blue. | ||
Here, we examine the magnetically induced current (MIC) density susceptibility and the aromatic behaviour of model auracycles and diauracycles by performing calculations with the gauge-including magnetically induced currents (GIMIC) method. A globally aromatic (antiaromatic) molecule is able to sustain a strong net diatropic (paratropic) MIC density around the whole molecular ring, where a diatropic ring current means that the current-density flux is in the classical circulation direction and paratropic represents a current-density flux in the opposite direction. The employed computational methods are presented in section 2. The results obtained in the computational studies on auracycles and diauracycles are discussed in section 3. The study is summarized and conclusions are drawn in section 4.
2 Computational details
The electronic structure calculations were carried out using TURBOMOLE versions 7.7.1 and 7.8-β.38,39 The starting structures were built from scratch and optimized at the density functional theory (DFT) level using the ωB97X-D functional,40 the def2-TZVP basis sets on all atoms41 and the 60-electron def2-ecp effective core potential (ECP) for gold.42,43 The molecular structures were verified to be minima by calculating vibrational frequencies using the aoforce module of TURBOMOLE.44 The molecular structures were reoptimized at the second-order Møller–Plesset (MP2) level of theory with def2-TZVP basis sets using the ricc2 module of TURBOMOLE.41,45,46 The structures optimized at the MP2 level were used in the calculations of the magnetically induced current (MIC) densities using the gauge-including magnetically induced currents (GIMIC) method.47–49 The current densities, which are also called ring-current strengths, were calculated at the DFT level using the BHandHLYP functional (LIBXC ID 436) and def2-TZVP basis sets.41,50,51 In the MIC calculations, the core electrons of Au were replaced by the def2-ecp relativistic ECP.42We also performed magnetic shielding calculations at the all-electron scalar-relativistic exact two-component (1C-X2C) level, and at the two-component (2C-X2C) level including spin–orbit effects.52,53 Triple-ζ polarization basis sets developed for one- and two-component all-electron relativistic electronic structure calculations were used.54
The aromatic nature of the gold-containing rings was determined by integrating the current density passing through a plane, which is perpendicular to the ring and cuts half of it.47,48 The ring-current strengths were also calculated by numerical line integration of Ampère-Maxwell's law.55 The geometric center of the ring and the vortex center of the current density were assumed to coincide. At the X2C levels, the ring-current strengths were only calculated using Ampère-Maxwell's law because the picture change effect is considered in the magnetic shielding calculations but not in the current-density calculations. The obtained ring-current strength yields the aromatic nature that can be compared to the one estimated from the number of electrons in the conjugated bonds of the ring.
The MIC density pathways were plotted with ParaView version 5.11.0.56 The strength of the MIC density is represented with a black-red-orange-yellow-white color scale.
3 Results and discussion
3.1 Auracycles
We have designed gold-containing analogues of regular Hückel aromatic benzene, pyridine and the cycloheptatrienyl cation by replacing a CH unit with a gold(I) atom (see Fig. 2). The negative charge of the carbon bridge is neutralized with the positive one of gold(I). The positive charge of [Au(C6H6)]+ is further neutralized with a chloride ligand attached to the metal. Our attempts to obtain an optimized structure for gold(I) auracyclopentadienide [Au(C4H4)]− were unsuccessful due to convergence problems in the self-consistent field optimization of the orbitals. However, we designed the gold(III) analogue [AuCl(C4H4)(NH3)] (4) as a model of the reported gold(III) auracyclopentadienes.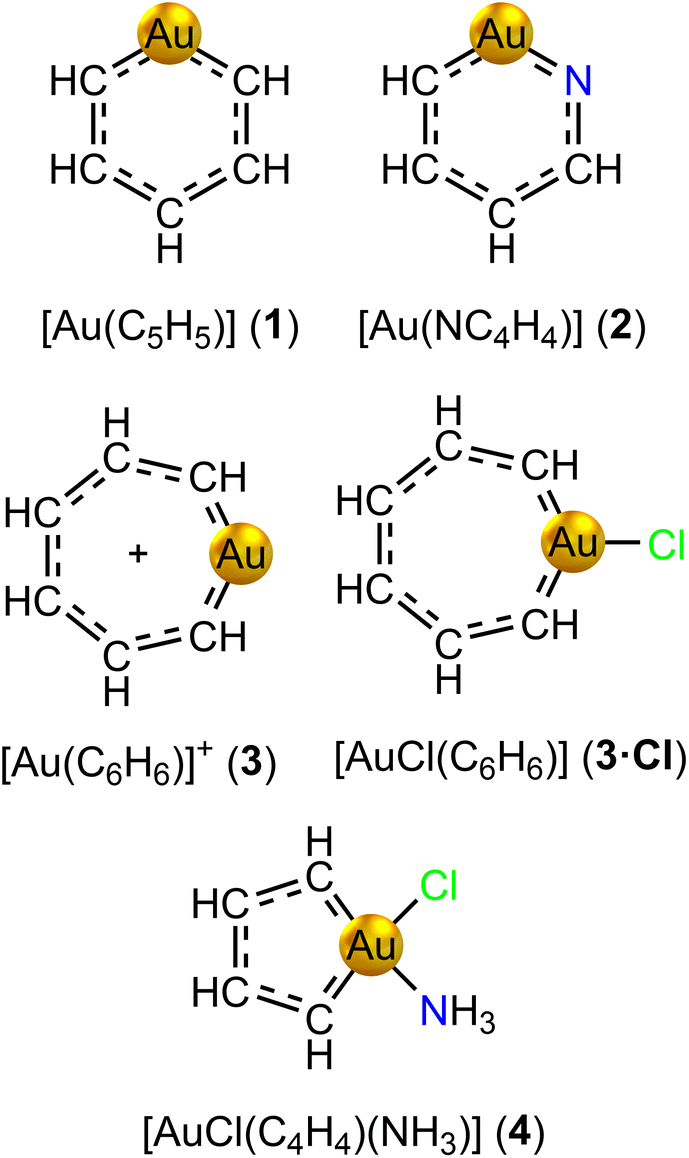 | ||
| Fig. 2 Molecular structures of aurabenzene (1), aurapyridine (2), auratropylium cation (3) and its chloride complex (3·Cl), and auracyclopentadiene (4) models. | ||
The number of electrons in the conjugated ring was estimated by assuming that each C, N and Au atom contribute one electron and an H atom connected to a N atom increases the number of electrons in the ring by one. Substituting Cl or other electron attracting moieties to Au removes an electron from the ring.
The optimized molecular structure of aurabenzene [Au(C5H5)] (1) belongs to the C2 point group. It deviates from the regular structure of benzene due to the C–Au–C angle of 103.1° imposed both by the larger size of Au with respect to the replaced CH group, and by the tendency of Au to linear dicoordination in the formal +1 oxidation state. While the AuC5 core is planar, the hydrogen atoms are bent out of the plane. A similar bent structure is found for ortho-aurapyridine [Au(NC4H4)] (2), where the substitution of an ortho-CH group with a N atom breaks the symmetry to the C1 point group. The auratropylium cation [Au(C6H6)]+ (3) features a boat-shaped structure with the gold atom pointing out of the mean plane, and belonging to the Cs point group. Conversely, the structure of the auratropylium chloride complex [AuCl(C6H6)] (3·Cl) is perfectly planar belonging to the C2v point group. Molecule [AuCl(C4H4)(NH3)] (4) is also planar, belonging to the Cs point group. The analysis of the frontier molecular orbitals collected in Fig. S1† reveals that Au 5d orbitals do not participate much in the π-conjugation of molecules 1–4.
The ring-current strengths of auracycles 1–4 calculated at the DFT/BHandHLYP/def2-TZVP level of theory are collected in Table 1, together with those of benzene, pyridine, and the cycloheptatrienyl cation, for comparative purposes. Calculation of the MIC density shows that 1 sustains a strong net diatropic ring current of 10.51 nA T−1, which is almost as strong as the one for benzene of 12.44 nA T−1.47 Visualization of the MIC density pathways of 1 as 3D streamlines (see the ESI†) shows that the Au atom acts as a vortex centre connecting the diatropic ring current on both sides of the molecule, probably because the ring is not completely planar. A paratropic ring current flows along the inner rim of the molecular ring as also in benzene. The ring-current strength of 2 is 7.41 nA T−1, revealing that it is less aromatic than 1 and pyridine. The MIC density pathways of 2 are similar to those of 1, with an additional coiling of the MIC around the C–N bond. The auratropylium models 3 and 3·Cl corresponding to (C7H7)+ are weakly aromatic sustaining a ring current of 3.82 and 4.21 nA T−1, respectively. 3 and 3·Cl have a diatropic MIC density pathway around the Au atom that extends to the adjacent C atoms. They have a diatropic ring current along the outer edge and a paratropic ring current inside the ring. Molecule 4 is weakly antiaromatic sustaining a net paratropic ring current of −4.41 nA T−1 inside the ring and a diatropic atomic current around the Au atom.
| Molecule | Net | Diatropic | Paratropic |
|---|---|---|---|
| [Au(C5H5)] (1) | 10.51 | 15.63 | −5.11 |
| [Au(NC4H4)] (2) | 7.41 | 13.06 | −5.65 |
| [Au(C6H6)]+ (3) | 3.82 | 11.92 | −8.10 |
| [AuCl(C6H6)] (3·Cl) | 4.21 | 12.08 | −7.87 |
| [AuCl(C4H4)(NH3)] (4) | −4.41 | 6.20 | −10.61 |
| C6H6 | 12.44 | 17.40 | −4.96 |
| C5H5N | 12.01 | 17.08 | −5.07 |
| (C7H7)+ | 12.43 | 17.70 | −5.27 |
We have also calculated the ring-current strength of molecules 1–4 at all-electron relativistic levels of theory, which are reported in the ESI.† The ring-current strength of 1 is 14.32 nA T−1 at the fully relativistic 2C-X2C level. Spin–orbit effects increase the ring-current strength by 3.81 nA T−1 or 36% as compared to the one calculated at the quasi-relativistic level using ECPs. The ring-current strength calculated at the scalar X2C level is 10.43 nA T−1, which agrees well with the strength calculated using the ECP.
The appearance of a strong ring current in the presence of a magnetic field is only one of the several ways in which aromaticity manifests itself. Thus, the proposed aromatic behaviour of auracycles 1–4 has been further studied by calculating the associated thermodynamic stabilization using the isomerization stabilization energy (ISE) method.57 The ISE is calculated as the total energy difference between a methyl derivative of the aromatic molecule and its non-aromatic exocyclic methylene isomer. This method has been successfully applied to assess the aromaticity of several related metallabenzenes for which experimental evidence is available.20 The considered tautomerization processes and the corresponding ISE values calculated at the MP2/def2-TZVP level of theory are shown in Fig. 3. The obtained ISE values confirm the aromatic character suggested by the calculated ring-current strengths, and decrease in the series 2 > 1 > 3 > 4. The trend agrees well with that obtained in the calculations of the ring-current strengths, although the correlation is not perfect. In particular, the ISE of molecule 2 of 29.4 kcal mol−1 is ca. 10 kcal mol−1 larger than the one of molecule 1, whereas 1 exhibits the strongest ring current in the series. This could be explained by the presence of the N atom.
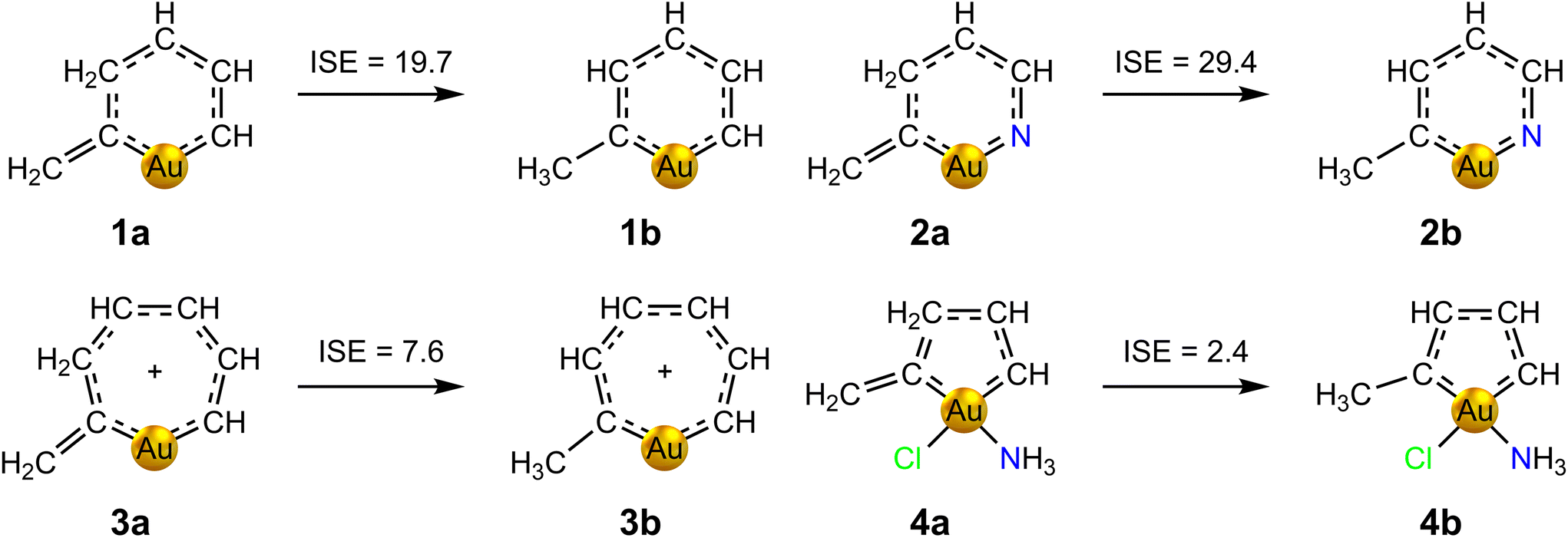 | ||
| Fig. 3 Isomerization stabilization energies (in kcal mol−1) of auracycles 1–4, calculated at the MP2/def2-TZVP level of theory. | ||
3.2 Diauracycles
We have designed three series of model diauracycles of increasing size from six (5–7, Fig. 4) to eight (8–14, Fig. 5) to ten (15–17, Fig. 6) members, and studied their aromatic behaviour. The contribution of each atom to the total number of electrons has been elucidated by analyzing the effect on the calculated MIC strength of binding Cl and H atoms to Au and N, respectively. Adding Cl is expected to oxidize gold(I) ([AuI]: [Xe] 4f14 5d10) to gold(II) ([AuII]: [Xe] 4f14 5d9), whereas binding H to N increases the number of electrons in the bond conjugation by one. The oxidized Au atoms can form a short Au–Au bond that also removes two electrons from the ring. The expected aromatic nature according to Hückel's aromaticity rules can then be determined.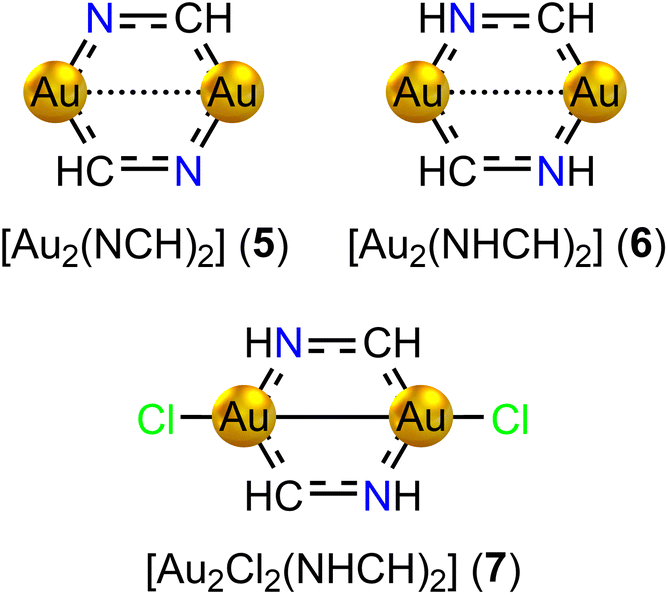 | ||
| Fig. 4 The molecular structures of the six-membered diauracycle models 5–7. | ||
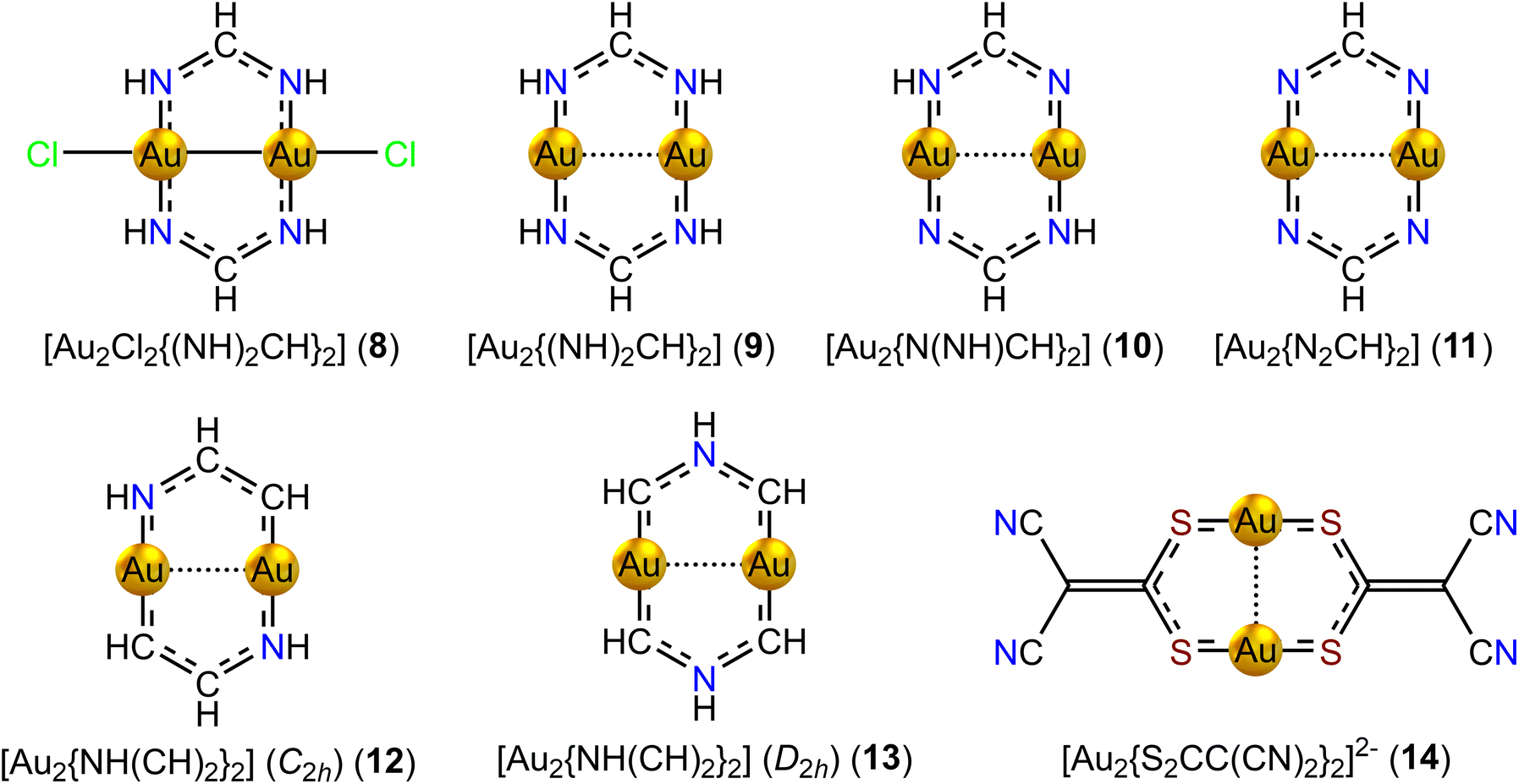 | ||
| Fig. 5 The molecular structures of the eight-membered diauracycle models 8–14. | ||
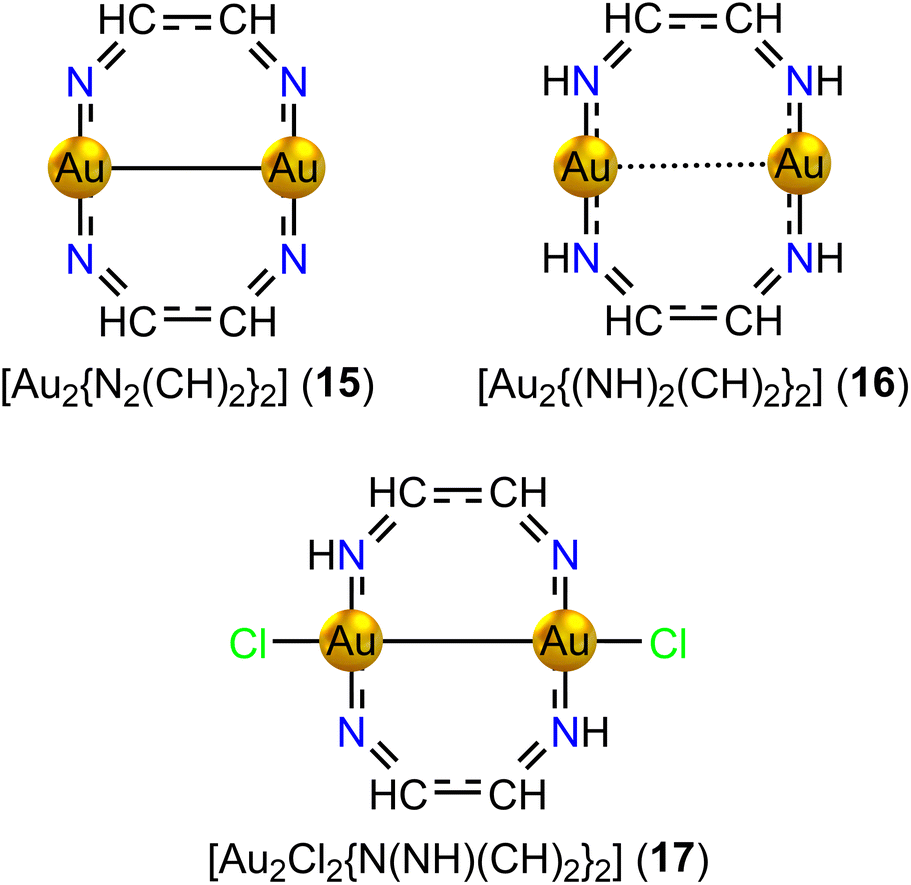 | ||
| Fig. 6 The molecular structures of the ten-membered diauracycle models 14–17. | ||
Adding two H atoms to the N atoms of [Au2(NCH)2] yields [Au2(NHCH)2] (6, C2h) with 8 electrons in the bond conjugation of the six-membered ring. The Au–Au distance is 264.0 pm. The strength of the ring-current inside the ring is −2.96 nA T−1. The ring is weakly antiaromatic as expected from Hückel's aromaticity rule. It has diatropic atomic currents around the Au atoms. The Au–Au bond of 6 sustains a diatropic ring current of 3.98 nA T−1, which is more than twice the strength of the Au–Au bond current of 5 with a longer Au–Au distance.
Adding two Cl atoms to the Au atoms of 6 yields [Au2Cl2(NHCH)2] (7, C2h). 7 has a short Au–Au distance of 244.8 pm. A very strong diatropic bond current of 9.73 nA T−1 circulates around the short Au–Au bond. The short Au–Au distance and population analysis indeed suggest that gold is in the +2 oxidation state. Since the Cl atoms remove two electrons from the ring and a Au–Au bond is formed, the ring fulfills Hückel's 4n rule for antiaromaticity. However, the ring of 7 is nonaromatic sustaining a net global ring current of −1.18 nA T−1. The ring-current strengths of the six-membered diauracycles 5–7 calculated at the DFT/BHandHLYP/def2-TZVP level of theory are given in Table 2.
| Molecule | Net | Diatropic | Paratropic |
|---|---|---|---|
| [Au2(NCH)2] (5) | 12.43 | 18.13 | −5.70 |
| [Au2(NHCH)2] (6) | −2.96 | 8.67 | −11.63 |
| [Au2Cl2(NHCH)2] (7) | −1.18 | 9.40 | −10.58 |
The corresponding model compound without the Cl atoms 9 has 12 electrons in the conjugated bonds. 9 is weakly antiaromatic sustaining a paratropic ring current of −1.88 nA T−1. The Au–Au distance of 273.6 pm is much longer than for 8. 9 also sustains a weak diatropic bond current of 2.69 nA T−1 in the vortex of the Au–Au bond. The calculated Au–Au distances of the model compounds 8 and 9 agree qualitatively with the experimental Au–Au distances for [Au2Cl2(2,6-Me2PhN2CH)2] and [Au2(2,6-Me2PhN2CH)2].
Removing two of the four H atoms of the NH moieties from 9 yields [Au2N(NH)CH2] (10, Ci). The s electrons of the Au atoms contribute to the 10 electrons in the conjugated bonds. The Au–Au distance is 279.5 pm. The eight-membered ring is weakly aromatic sustaining a ring current of 5.78 nA T−1. There is a weak diatropic ring current of 1.97 nA T−1 between the two Au atoms.
Removing all four H atoms from the NH moieties of 9 leads to [Au2(N2CH)2] (11, C2h), whose ground state is an aromatic triplet state sustaining a ring current of 11.12 nA T−1. The Au–Au distance is 263.3 pm and a bond current of 3.65 nA T−1 circles around the vortex of the Au–Au bond. The ring has formally 8 electrons in the conjugated bonds fulfilling Hückel's 4n aromaticity rule for triplet states. Structure optimization at the RI-MP2/def2-TZVP level failed for this molecule.
Replacing two of the NH moieties of 9 by CH yields [{Au2NH(CH)2}2] (12, C2h), with 10 electrons in the conjugated bonds. 12 has a Au–Au distance of 250.0 pm suggesting that the oxidation state is AuII, which also agrees with the population analysis. The formation of the Au–Au bond reduces the total number of conjugated electrons to 8, leading to antiaromaticity. 12 is nonaromatic sustaining a very weak paratropic ring current of −1.69 nA T−1, whereas a strong diatropic bond current of 9.88 nA T−1 circulates around the vortex of the short Au–Au bond.
The NH moiety can also be placed in the middle between the two CH groups. The symmetric [{Au2NH(CH)2}2] (13, D2h) has a long Au–Au distance of 276.5 pm. 13 has 10 electrons in the conjugated bonds and is aromatic sustaining a diatropic ring current whose strength is 12.32 nA T−1. A weak diatropic bond current of 2.50 nA T−1 circulates around the vortex of the long Au–Au bond. Thus, the electronic structures of the symmetric 13 and non-symmetric 12 [{Au2NH(CH)2}2] isomers are different. The charge of the Au atoms of 13 is 0.46e as compared to 0.85e for 12.
Other kinds of diauracycles like [{Au2S2CC(CN)2}2]2− (14, D2) have also been synthesized.5914 has a Au–Au distance of 275.8 pm. Since the ring has several C–S and S–Au single bonds, it is expected to sustain a weak ring current, if any. The calculated ring-current strength of 14 is −1.97 nA T−1 and the Au–Au bond sustains a diatropic bond current of 2.65 nA T−1. The ring-current strengths of the eight-membered diauracycles 8–14 calculated at the DFT/BHandHLYP/def2-TZVP level of theory are given in Table 3.
| Molecule | Net | Diatropic | Paratropic |
|---|---|---|---|
| [Au2Cl2{(NH)2CH}2] (8) | −3.02 | 7.17 | −10.19 |
| [Au2{(NH)2CH}2] (9) | −1.88 | 8.30 | −10.18 |
| [{Au2N(NH)CH}2] (10) | 5.78 | 12.14 | −6.36 |
| [Au2(N2CH)2] (11) | 11.12 | 17.06 | −5.94 |
| [{Au2NH(CH)2}2] (12) | −1.69 | 7.58 | −9.27 |
| [{Au2NH(CH)2}2] (13) | 12.32 | 17.65 | −5.33 |
| [{Au2S2CC(CN)2}2]2− (14) | −1.97 | 8.40 | −10.37 |
Adding four H atoms to the N atoms of 15 yielding [Au2{(NH)2(CH)2}2] (16, D2h) increases the Au–Au distance to 284.8 pm, which suggests that the s electrons of Au contribute to the conjugated bonds of the ring. 16 with 14 electrons in the ring is strongly aromatic. It sustains a strong global diatropic ring current and two weaker paratropic ring currents inside the Au2N2C2 rings that touch each other in the center of the Au–Au interaction forming a sandglass-shaped ring current. The strength of the ring current along the organic fragment is 13.83 nA T−1 and the net ring current passing between the gold atoms is 3.33 nA T−1.
Removing H atoms from two of the N atoms of 16 and adding Cl atoms to the Au atoms yield [{Au2Cl2N(NH)(CH)2}2] (17, C2). The molecular structure of 17 has a short Au–Au distance of 250.1 pm. Population analysis and the short Au–Au distance suggest that the s electrons are transferred to Cl and the Au atoms are in oxidation state AuII forming a Au–Au bond. Thus, only 8 electrons contribute to the conjugated bonds. However, 17 is nonaromatic instead of antiaromatic sustaining a weak ring current of −0.44 nA T−1. A strong diatropic bond current of 10.56 nA T−1 circulates around the vortex of the short Au–Au bond. The ring-current strengths of the eight-membered diauracycles 15–17 calculated at the DFT/BHandHLYP/def2-TZVP level of theory are given in Table 4.
| Molecule | Net | Diatropic | Paratropic |
|---|---|---|---|
| [Au2{(N2(CH)2)}2] (15) | −22.01 | 23.89 | −1.88 |
| [Au2{(NH2)(CH2)}2] (16) | 13.83 | 17.78 | −3.95 |
| [Au2Cl2{(NH2)(CH2)}2] (17) | −0.44 | 4.61 | −5.05 |
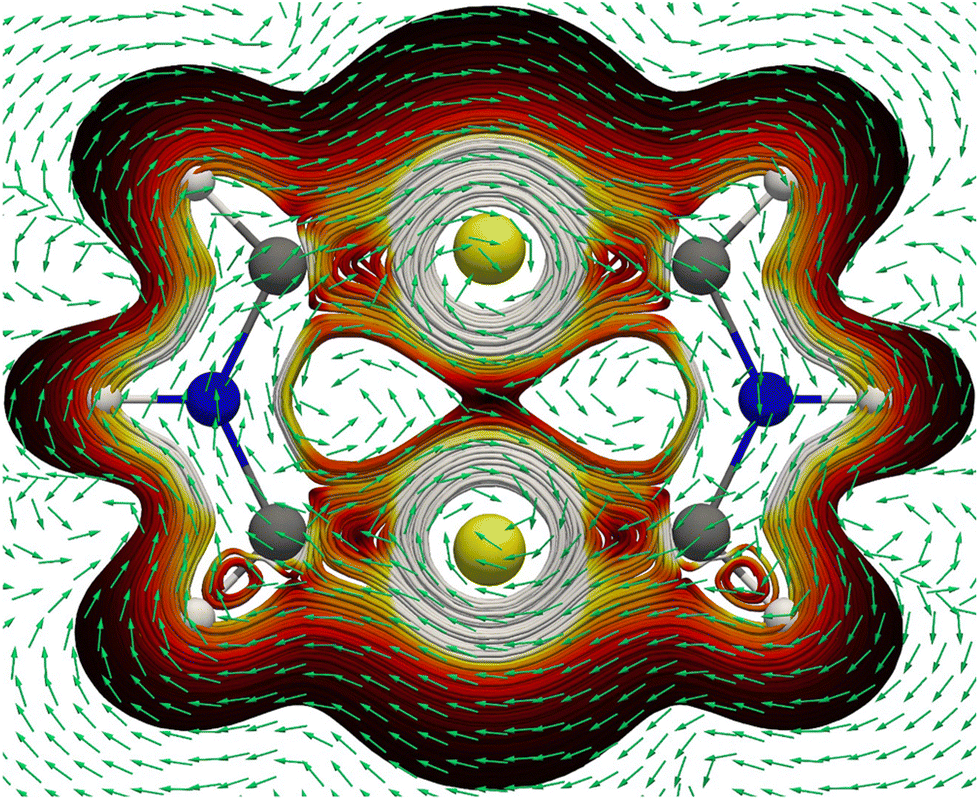 | ||
| Fig. 7 The 3D streamline plot of the MIC density pathways of the aromatic [Au2{(CH)2NH}2] (13). The flux direction of the MIC density is indicated by the green arrows that point in the clockwise (anticlockwise) direction when the MIC density is diatropic (paratropic). Color code: C, grey; H, white; Au, yellow; N, blue. | ||
 | ||
| Fig. 8 The strength of the current-density vortex (I(x) in nA T−1) of the Au–Au bond as a function of the Au–Au distance (x in pm). The ring-current strengths were calculated at the DFT/ωB97X-D level using the Ampère-Maxwell approach. The a, b and c parameters of I(x) = a + b(240 − x) + c/(180 − x) are optimized to fit I(x) to the numerical values in the ESI.† | ||
4 Conclusions
The magnetically induced current (MIC) density susceptibility has been calculated for gold-containing organometallic molecular rings of different size by using the gauge-including magnetically induced currents (GIMIC) method. The aromatic nature of the rings has been investigated by calculating the strength of the magnetically induced ring current. The obtained aromatic nature was compared to the one judged from the number of electrons in the conjugated bonds according to Hückel's aromaticity rules. The number of electrons in the conjugated bonds have been obtained by assuming that each conjugated C, N and Au atom contribute one electron. A H atom connected to a N atom in the ring increases the number of electrons in the bond conjugation by one. Substituting a chloride ligand or another electron attracting moiety to Au removes one electron from the ring. The formation of a short Au–Au bond in diauracycles also consumes two electrons from the ring.When the ring has 4n + 2 electrons in the conjugated orbitals it is aromatic sustaining a strong diatropic ring current, whereas auracycles and diauracycles with 4n electrons generally sustain a weak paratropic ring current. The studied molecules with 4n electrons in the conjugated ring are with one exception weakly antiaromatic or non-aromatic. The calculations show that gold-containing organometallic rings follow Hückel's aromaticity rules and can indeed be aromatic or antiaromatic depending on the number of electrons in the conjugated bonds. More generally, one can state that the studied molecules with odd number of occupied conjugated orbitals are aromatic or weakly aromatic, whereas the studied molecules with even number of occupied conjugated orbitals are antiaromatic or nonaromatic. The generalized rule holds for both open-shell and closed-shell molecules.60 Note that antiaromaticity is actually a stabilization with respect to nonaromaticity, otherwise it would not exist. The ring-current strengths, the number of electrons in the conjugated ring, and the aromatic nature of the studied molecules are summarized in Table 5.
| Molecule | # Electrons | I | Arom. |
|---|---|---|---|
| a Triplet state. | |||
| [Au(C5H5)] (1) | 6 | 10.51 | A |
| [Au(NC4H4)] (2) | 6 | 7.41 | A |
| [AuCl(C6H6)] (3) | 6 | 4.21 | WA |
| [AuCl(C4H4)(NH3)] (4) | 4 | −4.41 | WAA |
| [Au2(NCH)2] (5) | 6 | 12.43 | A |
| [Au2(NHCH)2] (6) | 8 | −2.96 | NA |
| [Au2Cl2(NHCH)2] (7) | 4 | −1.18 | NA |
| [Au2Cl2{(NH)2CH}2] (8) | 8 | −3.02 | WAA |
| [Au2{(NH)2CH}2] (9) | 12 | −1.88 | NA |
| [{Au2N(NH)CH}2] (10) | 10 | 5.78 | WA |
| [Au2(N2CH)2] (11)a | 8 | 11.12 | A |
| [{Au2NH(CH)2}2] (12) | 8 | −1.69 | NA |
| [{Au2NH(CH)2}2] (13) | 10 | 12.32 | A |
| [{Au2S2 CC(CN)2}2]2− (14) | 16 | −1.97 | NA |
| [Au2{N2(CH)2}2] (15) | 8 | −22.01 | SAA |
| [Au2{(NH)2(CH)2}2] (16) | 14 | 13.83 | A |
| [{Au2 Cl2N(NH)(CH)2}2] (17) | 8 | −0.44 | NA |
| [AuCl(C4Ph4)(py)] (18) | 4 | −3.33 | WAA |
Even though the number of π electrons determines the global aromatic character, geometrical features such as the planarity and strain of the ring also affect the strength of the ring current. The deviation of the calculated X–Au–X angles from the ideal ones of 180° for linear gold(I) and 90° for square-planar gold(III) is an adequate estimate of the ring strain. The X–Au–X angles and ring-current strengths of the studied molecules are collected in Table S3† for comparative purposes. For instance, the coordination of chloride (3·Cl) to [Au(C6H6)]+ (3) results in a planar ring and releases the ring strain by slightly opening the C–Au–C angle, which consequently increases the ring-current strength and preserves the aromatic character.
Calculation of the ring-current strength of the AuC4 core of [AuCl(C4Ph4)(phen)] (Fig. 1) is complicated because of the presence of the phenyl substituents and of the phenanthroline ligand capping the gold(III)atom. Thus, phenanthroline was replaced with pyridine leading to [AuCl(C4Ph4)(py)] (18), for which experimental evidence also exists.35 The strength of the ring current that was obtained by applying Ampère-Maxwell's law is −3.33 nA T−1 suggesting that it is weakly antiaromatic, which is also suggested by the 4n electrons in the conjugated ring.
To our knowledge, we show here for the first time that gold-containing organometallic rings may be aromatic or antiaromatic sustaining ring currents in the presence of an external magnetic field. Electron attracting ligands such as chloride oxidize AuI to AuII that can form a short Au–Au bond to another gold atom in the ring. There is a current density vortex between the gold atoms, whose strength increases with decreasing Au–Au distance.
Author contributions
DB and DS have performed calculations and written the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research has been supported by The Academy of Finland (project number 340583). DB acknowledges Universidad de La Rioja for the concession of a Margarita Salas postdoc fellowship financed by the Spanish Ministerio de Universidades and the European Union-NextGenerationEU program. We thank Yannick Franzke for a prerelease version of the mpshift program.References
- D. Chen, Y. Hua and H. Xia, Chem. Rev., 2020, 120, 12994–13086 CrossRef CAS PubMed.
- L. J. Wright, Dalton Trans., 2006, 1821–1827 RSC.
- C. A. Celaya, M. Orozco-Ic, M. Dimitrova, L. N. Wirz and D. Sundholm, Chem. Commun., 2020, 56, 5433–5436 RSC.
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Chem. Sci., 2023, 14, 5569–5576 RSC.
- G. Frenking and A. Krapp, J. Comput. Chem., 2007, 28, 15–24 CrossRef CAS PubMed.
- J. Zhu, Commun. Chem., 2020, 3, 161 CrossRef CAS PubMed.
- H. S. Rzepa, Chem. Rev., 2005, 105, 3697–3715 CrossRef CAS PubMed.
- L. N. Wirz, M. Dimitrova, H. Fliegl and D. Sundholm, J. Phys. Chem. Lett., 2018, 9, 1627–1632 CrossRef CAS PubMed.
- D. Craig and N. Paddock, Nature, 1958, 181, 1052–1053 CrossRef CAS.
- K. An, T. Shen and J. Zhu, Organometallics, 2017, 36, 3199–3204 CrossRef CAS.
- G. P. Elliott, W. R. Roper and J. M. Waters, J. Chem. Soc., Chem. Commun., 1982, 14, 811–813 RSC.
- D. L. Thorn and R. Hoffmann, Nouv. J. Chim., 1979, 3, 39–45 CAS.
- J. R. Bleeke, Chem. Rev., 2001, 101, 1205–1228 CrossRef CAS PubMed.
- G. He, H. Xia and G. Jia, Chin. Sci. Bull., 2004, 49, 1543–1553 CrossRef CAS.
- C. W. Landorf and M. M. Haley, Angew. Chem., Int. Ed., 2006, 45, 3914–3936 CrossRef CAS PubMed.
- G. Jia, Organometallics, 2013, 32, 6852–6866 CrossRef CAS.
- C. Zhu, S. Li, M. Luo, X. Zhou, Y. Niu, M. Lin, J. Zhu, Z. Cao, X. Lu, T. Wen, Z. Xie, P. v. R. Schleyer and H. Xia, Nat. Chem., 2013, 5, 698–703 CrossRef CAS PubMed.
- C. Zhu, M. Luo, Q. Zhu, J. Zhu, P. v. R. Schleyer, J. I.-C. Wu, X. Lu and H. Xia, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- B. J. Frogley and L. J. Wright, Coord. Chem. Rev., 2014, 270–271, 151–166 CrossRef CAS.
- I. Fernández, G. Frenking and G. Merino, Chem. Soc. Rev., 2015, 44, 6452–6463 RSC.
- Y. Cai, Y. Hua, Z. Lu, Q. Lan, Z. Lin, J. Fei, Z. Chen, H. Zhang and H. Xia, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2102310118 CrossRef CAS PubMed.
- B. J. R. Cuyacot, Z. Badri, A. Ghosh and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2022, 24, 27957–27963 RSC.
- L. Chen, L. Lin, A. R. Nath, Q. Zhu, Z. Chen, J. Wu, H. Wang, Q. Li, W.-F. Lin, J. Zhu and H. Xia, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2215900120 CrossRef CAS PubMed.
- A. Rabe, Q. Wang and D. Sundholm, Dalton Trans., 2024 Search PubMed , submitted.
- D. Arias-Olivares and D. Páez-Hernández, New J. Chem., 2022, 46, 16708–16716 RSC.
- V. Jacob, T. J. R. Weakley and M. M. Haley, Angew. Chem., Int. Ed., 2002, 41, 3470–3473 CrossRef CAS PubMed.
- M. A. Esteruelas, F. Leon, S. Moreno-Blázquez, M. Oliván and E. Oñate, Inorg. Chem., 2023, 62, 16810–16824 CrossRef CAS PubMed.
- Y. Zhang, J. Wei, Y. Chi, X. Zhang and W.-X. Zhang, J. Am. Chem. Soc., 2017, 39, 5039–5042 CrossRef PubMed.
- K. Miwa, T. Yokota, Q. Wang, T. Sakurai, H. Fliegl, D. Sundholm and H. Shinokubo, J. Am. Chem. Soc., 2024, 146, 1396–1402 CrossRef CAS PubMed.
- Metallabenzenes: An Expert View, ed. L. J. Wright, John Wiley & Sons, Ltd, 2017.
- M. Mauksch and S. Tsogoeva, Chem. – Eur. J., 2010, 16, 7843–7851 CrossRef CAS PubMed.
- I. Fernández and G. Frenking, Chem. – Eur. J., 2007, 13, 5873–5884 CrossRef PubMed.
- M. A. Iron, A. C. B. Lucassen, H. Cohen, M. E. van der Boom and J. M. L. Martin, J. Am. Chem. Soc., 2004, 126, 11699–11710 CrossRef CAS PubMed.
- R. Usón, J. Vicente and M. T. Chicote, J. Organomet. Chem., 1981, 209, 271–279 CrossRef.
- R. Usón, J. Vicente, M. T. Chicote, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1983, 1131–1136 RSC.
- M. Melchionna, M. Nieger and J. Helaja, Chem. – Eur. J., 2010, 16, 8262–8267 CrossRef CAS PubMed.
- Z. R. Wong, T. K. Schramm, M. Loipersberger, M. Head-Gordon and F. D. Toste, Angew. Chem., Int. Ed., 2020, 61, e202202019 CrossRef PubMed.
- TURBOMOLE V7.8-β 2023, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.org.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodynski and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- M. Dolg and X. Cao, Chem. Rev., 2012, 112, 403–480 CrossRef CAS PubMed.
- P. Deglmann, K. May, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2004, 384, 103–107 CrossRef CAS.
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef.
- C. Hättig and K. Hald, Phys. Chem. Chem. Phys., 2002, 4, 2111–2118 RSC.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- GIMIC version 2.0, a current density program; available from https://github.com/qmcurrents/gimic and https://zenodo.org/record/8180435.
- S. Lehtola, C. Steigemann, M. J. Oliveira and M. A. Marques, SoftwareX, 2018, 7, 1–5 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- Y. J. Franzke and F. Weigend, J. Chem. Theory Comput., 2019, 15, 1028–1043 CrossRef CAS PubMed.
- Y. J. Franzke and C. Holzer, J. Chem. Phys., 2023, 159, 184102 CrossRef CAS PubMed.
- Y. J. Franzke, R. Treß, T. M. Pazdera and F. Weigend, Phys. Chem. Chem. Phys., 2019, 21, 16658–16664 RSC.
- R. J. F. Berger, M. Dimitrova, R. T. Nasibullin, R. R. Valiev and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 624–628 RSC.
- J. Ahrens, B. Geveci and C. Law, Visualization Handbook, Elsevier, 2005 Search PubMed.
- P. v. R. Schleyer and F. Pühlhofer, Org. Lett., 2002, 4, 2873–2876 CrossRef CAS PubMed.
- H. E. Abdou, A. A. Mohamed and J. P. Fackler, Inorg. Chem., 2007, 46, 9692–9699 CrossRef CAS PubMed.
- M. C. Gimeno, in The Chemistry of Gold, ed. A. Laguna, John Wiley & Sons, Ltd, Weinheim, 2008, ch. 1, pp. 1–63 Search PubMed.
- R. R. Valiev, T. Kurten, L. I. Valiulina, S. Y. Ketkov, V. N. Cherepanov, M. Dimitrova and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 1666–1674 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of molecules 1–18, X–Au–X angles of molecules 1–18, selected molecular orbitals of molecules 1–5, 13, and 16, MIC strengths of molecules 1–4 at X2C-1C and X2C-2C levels of theory, 3D streamline plots of the MIC density pathways of molecules 1–18. See DOI: https://doi.org/10.1039/d4dt00827h |
| This journal is © The Royal Society of Chemistry 2024 |