
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing the electron donation in a dinucleating ligand family: molecular and electronic structures in a series of CoIICoII complexes†
Felix
Depenbrock
,
Thomas
Limpke
,
Anja
Stammler
,
Jan
Oldengott
,
Hartmut
Bögge
and
Thorsten
Glaser
 *
*
Lehrstuhl für Anorganische Chemie I, Fakultät für Chemie, Universität Bielefeld, Universitätsstrasse 25, D-33615 Bielefeld, Germany. E-mail: thorsten.glaser@uni-bielefeld.de
First published on 16th May 2024
Abstract
We have developed a family of dinucleating ligands with varying terminal donors to generate dinuclear peroxo and high-valent complexes and to correlate their stabilities and reactivities with their molecular and electronic structures as a function of the terminal donors. It appears that the electron-donating ability of the terminal donors is an important handle for controlling these stabilities and reactivities. Here, we present the synthesis of a new dinucleating ligand with potentially strong donating terminal imidazole donors. As CoII ions are sensitive to variations in donor strength in terms of coordination number, magnetism, UV-Vis-NIR spectra, redox potentials, we probe the electron donation ability of this new ligand in CoIICoII complexes in comparison to the parent CoIICoII complexes with terminal pyridine donors and we synthesize the analogous CoIICoII complexes with terminal 6-methylpyridines and methoxy-substituted pyridines. The molecular structures show indeed strong variations in coordination numbers and bond lengths. These differences in the molecular structures are reflected in the magnetic properties and in the d–d transitions demonstrating that the molecular structures remain intact upon dissolution. The redox potentials are analyzed with respect to the electron donation ability and are the only handle to observe an effect of the methoxy-substituted pyridines. All data taken together show the following order of electron donating ability for the terminal donors: 6-methylpyridines ≪ pyridines < methoxy-substituted pyridines ≪ imidazoles.
Introduction
Nature frequently employs metalloproteins with a dinuclear active site and regulates their molecular and electronic structures and hence reactivity for function not only by the choice of the metal ions but also of the ligands.1 A deeper understanding of the influence of the ligands on molecular and electronic structures and hence reactivity would be of benefit to use these biological catalysts as a blueprint for the development and optimization of molecular catalysts.2 In this respect, we have developed a dinucleating ligand system3 comprised of two tetradentate coordination compartments that vary in the terminal donors including carboxylates,4 phenolates, and pyridines (Scheme 1).5 | ||
| Scheme 1 The bis(tetradentate) dinucleating N8 ligands used in this study. | ||
For example, we were able to model the frequently observed μ-1,2-peroxo FeIIIFeIII intermediate in dinuclear non-heme diiron proteins (NHFe2) and to vary its reactivity: (i) using susan6-Me, the stable complex [(susan6-Me){FeIII(μ-1,2-peroxo)(μ-O)FeIII}]2+ was crystallized and protonated in solution to an unprecedented μ-1,2-hydroperoxo FeIIIFeIII species,6 while (ii) using susan, [(susan){FeIII(μ-1,2-peroxo)(μ-O)FeIII}]2+ is a reactive intermediate.7 Similarly, [(susan6-Me){CuICuI}]2+ reacts with O2 to a reactive high-valent {CuIII(μ-O)2CuIII} species, while [(susan){CuICuI}]2+ reacts with O2 to an also reactive {CuII-μ-1,2-peroxo-CuII} intermediate.8 In dicobalt chemistry, the μ-1,2-peroxo complex [(susan){CoIII(μ-1,2-peroxo)(μ-OH)CoIII}]3+ is stable and can only be reversibly deprotonated with a strong base in non-aqueous solution at low temperatures. In contrast, the oxidized [(susan){CoIII(μ-1,2-superoxo)(μ-OH)CoIII}]4+ becomes deprotonated in aqueous solution above pH 2 and releases O2 modelling the last step in water oxidation catalysis.9 Independently of our work, interest in the reactivity of cobalt complexes has recently increased again.10
The detailed characterization and analysis of the molecular and electronic structures of our aforementioned complexes established that the reactivity of the dinuclear peroxo and high-valent complexes is controlled by the electron donation ability of the dinucleating ligands. Generally, the electron donation ability of a ligand can be tuned by the introduction of electron donating and electron withdrawing substituents quantified for aromatic substituents by the Hammett parameters.11 A higher electron density in the ligand can on the one hand increase the electron donation of the ligand to the metal ion by σ- and π-donor interactions and on the other hand decrease the back-donation of electron density from the metal ion to the ligand by π-acceptor interactions. All three interactions (σ- and π-donor, π-acceptor) increase the strength and hence shorten the metal–ligand bond. However, they do not correlate with the spectrochemical series. The latter classifies the ligands in their ability to induce a ligand-field splitting ranging from “weak ligands” to “strong ligands”. Although both π-donor and π-acceptor ligands increase the strength of the metal–ligand bond, π-donors are “weak ligands” and π-acceptors are “strong ligands”.
The ligand susan6-Me is less electron donating than the ligand susan due to the longer M–N6-Me-py bonds enforced by the sterical demand of the 6-methyl substituents. In order to further tune the reactivity of peroxo intermediates and to stabilize high-valent intermediates, we intend to increase the electron donation capabilities of our dinucleating ligand system and initially used phenolate donors that are both strong σ- and π-donors. However, although those complexes are oxidized at relatively low potentials, the oxidation is not metal-centered but ligand-centered resulting in coordinated phenoxyl radicals.5,12 More recently, we reported the synthesis of the ligand susanOMe,13 a substitution pattern that was already successfully employed with tris(2-pyridylmethyl)amine (tpa) ligands.14 This substitution increases the electron density in the pyridine π-system reducing the π-acceptor character.
Considering that nature uses imidazole ligands in form of the amino acid histidine and not pyridine donors, it is interesting to consider the substitution of the pyridine donors in susan by imidazole donors. Here, we report the synthesis of the ligand cool that employs methylimidazole as terminal donors (Scheme 1). Imidazoles are considered as π-donor ligands, whereas pyridines are considered to be π-acceptor ligands.15 A chemical intuitive interpretation is that in imidazoles the six π electrons are shared only over five atoms instead of six atoms in pyridines so that the higher electron density per atom facilitates π-donor interaction and increases the energy of the π-donor orbitals. Therefore, the electron donation capability of the ligand cool should be stronger than of the ligands susan and susanOMe. With the ligand susan, we already synthesized a series of dinuclear CoIICoII complexes, namely [(susan){CoII(CH3CN)2}2]4+, [(susan){CoIICl}2]2+, [(susan){CoIIBr}2]2+, and [(susan){CoII(μ-OH)CoII}]3+.16 Here we employ the new ligand cool as well as the ligands susanOMe and susan6-Me to extend this series of dinuclear CoIICoII complexes and to evaluate the influence of the terminal nitrogen donors and the exogenous ligands on the molecular structures evaluated by single-crystal X-ray diffraction and on the electronic structures evaluated by magnetism, UV-Vis-NIR spectroscopy, and electrochemistry. These investigations result in the following sequence of electron donation ability: susan6-Me ≪ susan < susanOMe ≪ cool.
Experimental section
Synthesis of compounds
Solvents and starting materials were of the highest commercially available purity and used as received except for CH3CN, which was dried according to standard procedures.17 Tetramine 1 (= 4,7-dimethyl-1,4,7,10-tetrazadecane) was synthesized by a modified literature procedure.18 The ligands susan6-Me (ref. 19) and susanOMe (ref. 13) were synthesized according to the procedures reported previously. Although we experienced no difficulties, perchlorate salts are potentially hazardous and should only be handled in small quantities and with adequate precautions. The assignments of the NMR resonances in all products were supported by 2D COSY, HMBC, and HMQC spectroscopy and the numbering was done according to the numbering scheme in Fig. S2 + S3.†Crystal structure determination
Single-crystals were removed from the mother liquor, coated with oil, and measured at 100(2) K. For crystals of [(susan6-Me){CoII(μ-OH)2CoII}](ClO4)2·MeOH a Bruker X8 prospector ultra three-circle diffractometer with 4K CCD detector, CuKα radiation, and Quazar™ Montel multilayer optics was used. Other crystals were measured on a Bruker KAPPA APEX II four-circle diffractometer equipped with 4K CCD detector. On this device, CuKα radiation with Quazar™ Montel multilayer optics was used to measure crystals of [(susan6-Me){CoII(CH3CN)2}2](ClO4)4 and MoKα radiation with a focusing graphite monochromator for all other compounds. Empirical absorption corrections using equivalent reflections were performed with the programs SADABS 2012/1 for crystals of [(cool){CoII(CH3CN)}2](ClO4)4·2CH3CN and SADABS-2016/2 for all other data sets.20 The structures were solved and refined vs. F2 with the programs SHELXS/T/L21,22 using OLEX2.22 Crystal data and details concerning data collections and structure refinements are given in Table S1.†Hydrogen atoms were found and refined for all bridging hydroxo-groups, all other hydrogen atoms were generated. All crystal structures contain counter ions of which several showed disorder to various extend. This disorder was resolved when possible and refined using the appropriate constraints. Crystal structures of [(susanOMe){CoIICl}2](ClO4)2·3MeOH and [(susanOMe){CoIIBr}2](ClO4)2·3MeOH are isostructural. Both contain solvent accessible voids, a channel along the 21 screw axis and a smaller void (Fig. S5†). The scattering contribution of the disordered MeOH molecules inside the voids was masked using OLEX2.23,24 Two CH3CN solvent molecules in [(susanOMe)CoII(μ-OH)CoII](ClO4)3·3CH3CN suffered from substantial disorder and were also masked using the OLEX2 routine.23,24
[(susan6-Me){CoII(CH3CN)2}2](ClO4)4 and [(cool){CoII(CH3CN)}2](ClO4)4·2CH3CN both crystallize with one half molecule in the asymmetric unit with the second half generated by a center of inversion. [(susan6-Me){CoII(μ-OH)2CoII}](ClO4)2·MeOH, [(susanOMe){CoIICl}2](ClO4)2·3MeOH, [(susanOMe){CoIIBr}2](ClO4)2·3MeOH [(susanOMe)CoII(μ-OH)CoII](ClO4)3·3CH3CN, [(cool){CoIICl}2](ClO4)2, [(cool){CoIIBr}2](ClO4)2, and [(cool)CoII(μ-OH)CoII](BPh4)2(CF3SO3)·2CH3CN all crystallize with a whole molecule in the asymmetric unit.
CCDC numbers (Table S1†) contain the supplementary crystallographic data for this paper.
Other physical measurements
Infrared spectra (400–4000 cm−1) of solid samples were recorded on a Bruker Vertex 70 as KBr disks. ESI mass spectra were recorded on a Bruker Esquire 3000 ion trap mass spectrometer equipped with a standard ESI source. 1H and 13C{1H} NMR spectra were measured on a Bruker Avance III 500 HD or a Bruker Avance III 300 spectrometer using the solvent as an internal standard. UV-Vis-NIR absorption spectra were measured on a JASCO V770 spectrophotometer at 20 °C. Cyclic and square-wave voltammograms (CVs and SWs) were measured by use of an EG&G potentiostat/galvanostat 273A on N2-flushed solutions containing 0.1 M TBAPF6 as supporting electrolyte in a conventional electrochemical cell. The working electrode was a GC electrode, the counter electrode was a platinum wire, and the reference electrode was Ag/0.01 M AgNO3/CH3CN. The potentials are referenced versus the ferrocenium/ferrocene (Fc+/Fc) couple used as an internal standard. SWs have been recorded with typical frequency 60 Hz. CVs were routinely measured with scan rates of 200 mV s−1. Magnetic susceptibility data were measured on powdered samples in the temperature range 2–300 K by using a SQUID magnetometer (Quantum Design MPMS XL-7 EC) with a field of 1.0 T. Variable-temperature variable-field (VTVH) measurements were performed in various static fields (1–7 T) in the range 2–10 K with the magnetization equidistantly sampled on a 1/T temperature scale. For calculations of the molar magnetic susceptibilities, χm, the measured susceptibilities were corrected for the underlying diamagnetism of the sample holder and the sample by using tabulated Pascal's constants.Results and discussion
Synthesis
In analogy to the other N8 ligands of our dinucleating ligand system,5,13,19 the ligand cool was synthesized by a reductive amination25 of tetramine 1 with 1-methyl-1H-imidazole-carbaldehyde (Scheme 2). Reacting cool with Co(ClO4)2·6H2O in CH3CN resulted in [(cool){CoII(CH3CN)}2](ClO4)4. The reaction of cool with Co(ClO4)2·6H2O and NEt3 in different solvents resulted in the formation of crystalline [(cool){CoII(μ-OH)CoII}](ClO4)3. Single-crystals provided single-crystal X-ray diffraction data, that were not of sufficient quality to allow a satisfactory refinement. Using Co(OTf)2·7H2O and subsequent addition of NaBPh4 provided single-crystals of sufficient quality for single-crystal X-ray diffraction and analyzed as [(cool){CoII(μ-OH)CoII}](BPh4)2(OTf)·2CH3CN. For the synthesis of complexes of cool with exogenous halide ligands, the synthetic strategy of the analogue susan complexes was adapted.16 The reaction of cool with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CoCl2·6H2O/Co(ClO4)2·6H2O in MeOH/EtOH provided the complex [(cool){CoIICl}2](ClO4)2, using CoBr2 instead of CoCl2·6H2O provided [(cool){CoIIBr}2](ClO4)2.
:1 mixture of CoCl2·6H2O/Co(ClO4)2·6H2O in MeOH/EtOH provided the complex [(cool){CoIICl}2](ClO4)2, using CoBr2 instead of CoCl2·6H2O provided [(cool){CoIIBr}2](ClO4)2.
 | ||
| Scheme 2 Synthesis of the ligand cool. | ||
In analogy, the reaction of susanOMe with a 1:1 mixture of CoCl2·6H2O/Co(ClO4)2·6H2O or CoBr2/Co(ClO4)2·6H2O in MeOH/EtOH provided [(susanOMe){CoIICl}2](ClO4)2 or [(susanOMe){CoIIBr}2](ClO4)2, respectively. The reaction of susanOMe with Co(ClO4)2·6H2O and NEt3 under a N2-blanketing atmosphere provided the μ-hydroxo bridged complex [(susanOMe){CoII(μ-OH)CoII}](ClO4)3. Attempts to obtain a CH3CN complex with the ligand susanOMe provided only oils. The reaction of susan6-Me and Co(ClO4)2·6H2O in CH3CN provided the complex [(susan6-Me){CoII(CH3CN)2}2](ClO4)4. The bis-μ-hydroxo bridged complex [(susan6-Me){CoII(μ-OH)2CoII}](ClO4)2 was obtained from the reaction of susan6-Me with Co(ClO4)2·6H2O and NEt3 in MeOH.
Structural characterization
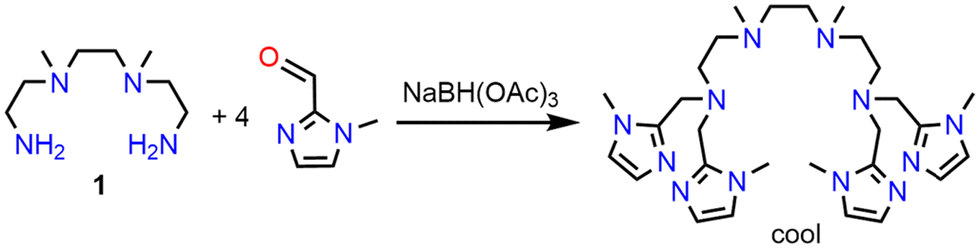
All new complexes were characterized by single-crystal X-ray diffraction. Details of the crystal structures are provided in the Experimental section. Instead of describing the individual molecular structures, the differences between the molecular structures of the nine complexes described herein and of the already published susan complexes will be analyzed to determine the influence of the dinucleating and the exogenous ligands. The molecular structures of the complex cations are shown in Fig. 1, thermal ellipsoid plots in Fig. S4,† and selected interatomic distances are given in Table 1. Mean bond lengths with two decimal places are provided in Table 2 to avoid on the one hand chemically not-relevant differences and on the other hand the inclusion of standard deviations for the comparison.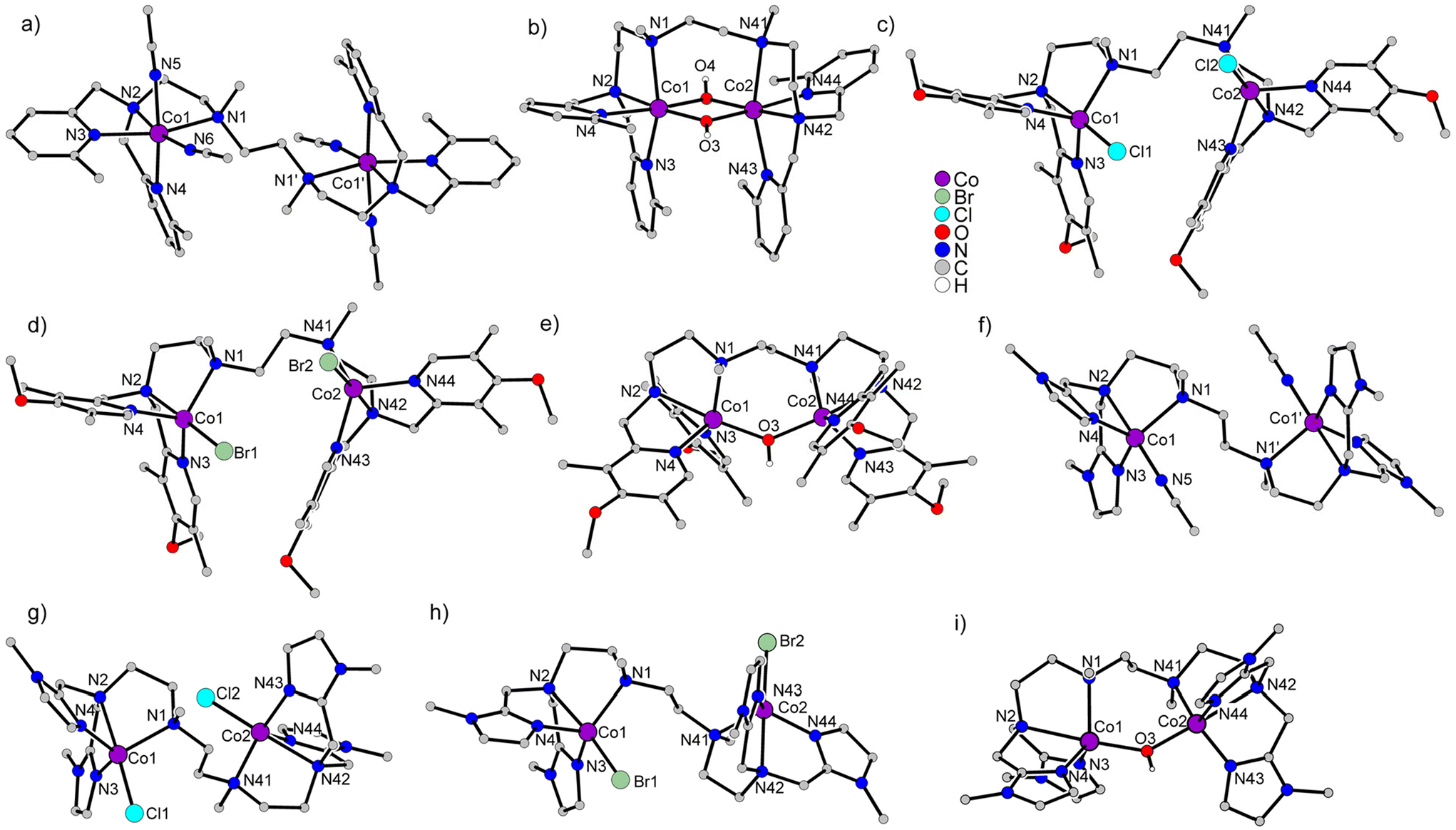 | ||
| Fig. 1 Molecular structures of (a) [(susan6-Me){CoII(CH3CN)2}2]4+ in single-crystals of [(susan6-Me){CoII(CH3CN)2}2](ClO4)4, (b) [(susan6-Me){CoII(μ-OH)2CoII}]2+ in single-crystals of [(susan6-Me){CoII(μ-OH)2CoII}](ClO4)2·MeOH, (c) [(susanOMe){CoIICl}2]2+ in single-crystals of [(susanOMe){CoIICl}2](ClO4)2·3MeOH, (d) [(susanOMe){CoIIBr}2]2+ in single-crystals of [(susanOMe){CoIIBr}2](ClO4)2·3MeOH, (e) [(susanOMe)CoII(μ-OH)CoII]3+ in single-crystals of [(susanOMe)CoII(μ-OH)CoII](ClO4)3·3CH3CN, (f) [(cool){CoII(CH3CN)}2]4+ in single-crystals of [(cool){CoII(CH3CN)}2](ClO4)4·2CH3CN, (g) [(cool){CoIICl}2]2+ in single-crystals of [(cool){CoIICl}2](ClO4)2, (h) [(cool){CoIIBr}2]2+ in single-crystals of [(cool){CoIIBr}2](ClO4)2, and (i) [(cool)CoII(μ-OH)CoII]3+ in single-crystals of [(cool)CoII(μ-OH)CoII](BPh4)2(CF3SO3)·2CH3CN. Hydrogen atoms, counter ions and solvent molecules are omitted for clarity. | ||
| [(susan6-Me){Co (CH3CN)2}2](ClO4)4 | [(susan6-Me){Co(μ-OH)2Co}](ClO4)2·MeOH | [(susanOMe){CoCl}2](ClO4)2·3MeOH | [(susanOMe){CoBr}2](ClO4)2·3MeOH | [(susanOMe)Co(μ-OH)Co] (ClO4)3·3CH3CN | |||||
|---|---|---|---|---|---|---|---|---|---|
| Co1-side | Co2-sidea | Co1-side | Co2-sidea | Co1-side | Co2-sidea | Co1-side | Co2-sidea | ||
| Co1–N1 | 2.3175(14) | 2.327(2) | 2.403(2) | 2.135(2) | 2.112(2) | 2.1381(18) | 2.1265(19) | 2.1398(14) | 2.1456(15) |
| Co1–N2 | 2.1028(14) | 2.148(2) | 2.141(2) | 2.172(2) | 2.1736(19) | 2.177(2) | 2.1688(17) | 2.1952(15) | 2.1998(16) |
| Co1–N3 | 2.1950(14) | 2.220(2) | 2.225(2) | 2.070(2) | 2.051(2) | 2.068(2) | 2.0580(17) | 2.0739(14) | 2.0595(15) |
| Co1–N4 | 2.1918(14) | 2.261(2) | 2.236(2) | 2.064(2) | 2.046(2) | 2.069(2) | 2.0488(19) | 2.0419(15) | 2.0731(15) |
| Co1–Xb | 2.1984(15) | 1.9557(19) | 2.105(2) | 2.2763(7) | 2.3050(7) | 2.4186(4) | 2.4486(3) | 1.9756(13) | 1.9694(13) |
| Co1–Yb | 2.0718(15) | 2.1530(19) | 1.9540(19) | ||||||
| Co1⋯Co2c | 7.8707(6) | 3.1763(6) | 5.7559(5) | 5.7568(4) | 3.6616(5) | ||||
| N1–Co1–N2 | 81.56(5) | 80.83(8) | 79.47(9) | 82.28(8) | 81.55(8) | 82.37(7) | 81.55(7) | 81.11(5) | 81.70(6) |
| N1–Co1–N3 | 159.65(5) | 156.58(8) | 155.98(8) | 124.38(8) | 125.93(8) | 123.82(7) | 127.23(7) | 130.46(6) | 103.60(6) |
| N1–Co1–N4 | 87.42(5) | 83.23(8) | 81.94(8) | 108.59(8) | 111.80(8) | 110.15(7) | 109.97(7) | 106.20(6) | 131.58(6) |
| N1–Co1–Xb | 93.27(5) | 99.86(8) | 93.13(8) | 100.25(6) | 97.61(6) | 99.23(5) | 97.94(5) | 97.33(6) | 97.79(6) |
| N1–Co1–Y | 91.15(6) | 94.60(7) | 95.62(8) | ||||||
| N2–Co1–N3 | 78.58(6) | 75.76(8) | 76.61(9) | 77.82(9) | 77.54(7) | 77.71(9) | 77.42(6) | 76.59(6) | 78.32(6) |
| N2–Co1–N4 | 82.72(5) | 79.31(8) | 80.80(9) | 77.99(8) | 78.32(8) | 78.26(8) | 78.11(7) | 78.82(6) | 76.08(6) |
| N2–Co1–Xb | 90.25(6) | 167.69(8) | 93.58(8) | 177.36(6) | 178.09(6) | 178.05(5) | 177.45(5) | 172.89(5) | 173.37(6) |
| N2–Co1–Yb | 169.97(6) | 90.37(8) | 170.53(9) | ||||||
| N3–Co1–N4 | 94.37(5) | 91.96(8) | 92.07(8) | 116.91(8) | 111.80(8) | 116.10(8) | 112.07(7) | 111.89(6) | 112.99(6) |
| N3–Co1–Xb | 82.49(5) | 103.10(8) | 90.52(8) | 101.19(7) | 101.67(6) | 102.20(6) | 101.02(5) | 99.53(6) | 108.16(6) |
| N3–Co1–Yb | 107.99(6) | 86.04(7) | 108.37(8) | ||||||
| N4–Co1–Xb | 172.76(5) | 112.99(8) | 173.08(8) | 100.41(6) | 103.59(6) | 100.10(5) | 104.39(5) | 108.24(6) | 99.66(6) |
| N4–Co1–Yb | 103.93(6) | 169.67(8) | 106.69(8) | ||||||
| Xb–Co1–Yb | 83.27(6) | 77.32(8) | 78.52(8) | ||||||
| Co1–O3–Co2 | 102.87(9) | 136.30(7) | |||||||
| Co1–O4–Co2 | 101.21(8) | ||||||||
| [(cool){Co(CH3CN)}2](ClO4)4·2CH3CN | [(cool){CoCl}2](ClO4)2 | [(cool){CoBr}2](ClO4)2 | [(cool)Co(μ-OH)Co](BPh4)2(CF3SO3)·2CH3CN | ||||
|---|---|---|---|---|---|---|---|
| Co1-side | Co2-sidea | Co1-side | Co2-sidea | Co1-side | Co2-sidea | ||
| a The numbering scheme of the Co2 side or molecule has been adapted according to the Co1 side. b X = Br, Cl, CH3CN, or μ-OH; Y = CH3CN, or μ-OH. c Intramolecular Co⋯Co distance and angle, which is Co1⋯Co1′ with −x, 1 − y, 2 − z for [(susan6-Me){CoII(CH3CN)2}2](ClO4)4. d X = Br, Cl, MeCN, or μ-OH. e Intramolecular Co⋯Co distance and angle, which is Co1⋯Co1′ with 2 − X, 1 − Y, 1 − Z for [(cool)CoII(μ-OH)CoII](BPh4)2(CF3SO3)·2CH3CN. | |||||||
| Co1–N1 | 2.093(2) | 2.116(3) | 2.107(3) | 2.1097(16) | 2.1145(16) | 2.1488(11) | 2.1411(11) |
| Co1–N2 | 2.273(2) | 2.320(3) | 2.342(2) | 2.3327(16) | 2.3069(17) | 2.3407(11) | 2.3820(11) |
| Co1–N3 | 1.998(2) | 2.016(3) | 2.013(3) | 2.0080(17) | 2.0107(16) | 2.0263(11) | 1.9924(12) |
| Co1–N4 | 2.001(2) | 2.024(3) | 2.000(3) | 2.0015(17) | 2.0198(17) | 2.0210(12) | 2.0148(11) |
| Co1–Xd | 2.052(2) | 2.3015(9) | 2.2898(9) | 2.4421(3) | 2.4471(3) | 1.9586(10) | 1.9769(10) |
| Co1⋯Co2e | 6.1573(19) | 5.8531(9) | 5.9247(4) | 3.6323(2) | |||
| N1–Co1–N2 | 81.89(8) | 80.43(10) | 80.06(9) | 80.50(6) | 80.95(6) | 79.67(4) | 80.08(4) |
| N1–Co1–N3 | 121.59(9) | 119.72(10) | 122.02(10) | 122.00(6) | 119.66(7) | 122.73(4) | 110.24(5) |
| N1–Co1–N4 | 110.51(9) | 107.58(11) | 116.48(11) | 115.00(7) | 108.75(7) | 109.04(5) | 118.01(5) |
| N1–Co1–Xd | 98.87(9) | 101.76(8) | 102.76(8) | 104.83(5) | 101.69(5) | 98.32(4) | 101.42(4) |
| N1–Co1–Y | |||||||
| N2–Co1–N3 | 78.53(8) | 75.60(10) | 76.83(9) | 76.82(6) | 75.87(6) | 76.11(4) | 77.18(4) |
| N2–Co1–N4 | 78.88(8) | 76.61(11) | 77.11(10) | 77.03(6) | 76.84(7) | 77.75(4) | 76.08(4) |
| N2–Co1–Xd | 177.82(9) | 176.78(7) | 176.09(7) | 173.93(4) | 176.76(4) | 175.98(4) | 175.43(4) |
| N2–Co1–Yd | |||||||
| N3–Co1–N4 | 118.54(9) | 118.86(11) | 108.91(11) | 110.66(7) | 118.50(7) | 115.10(4) | 118.51(5) |
| N3–Co1–Xd | 102.71(9) | 101.22(8) | 103.63(8) | 102.29(5) | 101.11(5) | 102.31(4) | 106.13(4) |
| N3–Co1–Yd | |||||||
| N4–Co1–Xd | 98.94(9) | 104.79(8) | 99.13(8) | 97.85(5) | 103.89(5) | 106.24(4) | 99.46(4) |
| N4–Co1–Yd | |||||||
| Xd–Co1–Yd | |||||||
| Co1–O3–Co2 | 134.73(6) | ||||||
| Co1–O4–Co2 | |||||||
| Co–N1 | Co–N2 | Co–N3 | Co–N4 | Co–X | Co–Y | τ | J/cm−1 | g | D/cm−1 | E p/V vs. Fc+/Fca | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Values in italics are E°′ for reversible redox waves. | ||||||||||||
X(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) Y)CH
3
CN Y)CH
3
CN
|
||||||||||||
| susan | 2.22 | 2.15 | 2.09 | 2.10 | 2.24 | 2.08 | −0.41 | 2.43 | 40.9 | 0.91, 0.46 | −1.53, −1.9 | |
| susan6-Me | 2.32 | 2.10 | 2.20 | 2.19 | 2.20 | 2.07 | −0.33 | 2.55 | 68.5 | 1.49 | −1.41, −1.9 | |
| cool | 2.09 | 2.27 | 1.99 | 2.00 | 2.05 | 0.94 | −1.2 | 2.19 | 10 | 1.47 | −1.76, −2.4 | |
| X = Cl | ||||||||||||
| susan | 2.12 | 2.23 | 2.05 | 2.07 | 2.29 | 0.88/0.91 | −0.68 | 2.25 | 6.9 | 1.08 | −1.91, −2.1 | |
| susanOMe | 2.12 | 2.17 | 2.06 | 2.05 | 2.29 | 0.88/0.87 | −0.01 | 2.29 | 6.1 | 1.10 | −2.1 | |
| cool | 2.11 | 2.33 | 2.01 | 2.01 | 2.30 | 0.95/0.90 | −0.24 | 2.24 | 3.2 | 1.02 | −2.4 | |
| X = Br | ||||||||||||
| susan | 2.13 | 2.19 | 2.06 | 2.06 | 2.44 | 0.91/0.86 | −0.12 | 2.25 | 5.8 | 0.75 | −1.83, −1.9 | |
| susanOMe | 2.13 | 2.17 | 2.06 | 2.06 | 2.43 | 0.90/0.87 | −0.10 | 2.33 | 6.0 | 0.79 | −2.03, −2.1 | |
| cool | 2.11 | 2.32 | 2.01 | 2.01 | 2.44 | 0.87/0.95 | −0.29 | 2.25 | 4.2 | 0.77 | −2.4 | |
|
X(Y)OH
|
||||||||||||
| susan | 2.17 | 2.20 | 2.08 | 2.08 | 1.96 | 0.74/0.75 | −18.8 | 2.29 | D 1 = 65/D2 = 17 | 1.31 | −1.9 | |
| susan6-Me | 2.37 | 2.14 | 2.22 | 2.25 | 1.96 | 2.13 | +2.4 | 2.46/2.50 | D 1 = −56/D2 = −16 | 1.26, 0.72 | ||
| susanOMe | 2.14 | 2.20 | 2.07 | 2.06 | 1.97 | 0.71/0.70 | −20.8 | 2.29 | D 1 = 56/D2 = 8 | 1.24 | −2.1 | |
| cool | 2.15 | 2.36 | 2.01 | 2.02 | 1.97 | 0.89/0.95 | −14.5 | 2.18 | 86.4 | 1.18 | −2.4 | |
One obvious difference is the coordination number of the CH3CN complexes. Six-coordinate CoII ions were obtained with the ligands susan and susan6-Me having two CH3CN ligands per CoII, while the ligand cool provides five-coordinate CoII ions with one CH3CN ligand. This difference cannot be attributed to sterical effects as the imidazole donors of cool are sterically the least demanding. Hence, this five-coordination demonstrates the higher electron donation capability of cool than of susan and susan6-Me.
The two closely related complexes [(susan){CoII(CH3CN)2}2]4+ and [(susan6-Me){CoII(CH3CN)2}2]4+ nicely demonstrate the effect of the introduction of the 6-Me substituents on the pyridine donors. The N3-subunit diphenylamine (DPA) of the ligand susan coordinates meridionally but that of the ligand susan6-Me coordinates facially. If the ligand susan6-Me coordinated also meridionally, both 6-Me groups would directly point to one CH3CN. This is avoided by the facial coordination of susan6-Me preventing this steric hinderance with one CH3CN ligand. However, the sterical repulsion of the two 6-Me group with the CH3CN ligand (N6) is still effective resulting in longer mean Co–N6-Me-py bonds (2.20 Å) than the mean Co–Npy bonds (2.10 Å). This different coordination of the ligands susan and susan6-Me also influences the Co–Nam distances, which are Co1–N1 = 2.22 Å and Co1–N2 = 2.15 Å with susan, where both amine donors coordinate trans to a CH3CN donor. In contrast, with susan6-Me, Co1–N1 trans to a pyridine elongates to 2.32 Å, while Co1–N2 trans to a CH3CN decreases to 2.10 Å.
In [(cool){CoII(CH3CN)}2]4+, the mean Co–Nim bond lenghts are shorter at 2.00 Å indicating a stronger electron donation ability of the imidazoles. However, a direct comparison between five- and six-coordinate complexes can be misleading as a lower-coordinate metal ion requires more electron donation and therefore shorter bond lengths for similar donor types.
In the chloride and bromide complexes of cool, susanOMe, and susan, the CoII ions are all five-coordinate close to trigonal–bipyramidal (τ ∼ 0.9) allowing a direct comparison of the aromatic nitrogen donors. In [(cool){CoIICl}2]2+ and [(cool){CoIIBr}2]2+, the Co–Nim bonds are 2.01 Å, while the Co–Npy bonds are 2.06 Å in [(susanOMe){CoIICl}2]2+, [(susanOMe){CoIIBr}2]2+, [(susan){CoIICl}2]2+, and [(susan){CoIIBr}2]2+ demonstrating stronger bonds with an imidazole donor compared to a pyridine donor. This indicates besides minor π-bonding effects mainly a stronger σ-donor interaction of the imidazole donors. The shorter Co–Nim bonds result in longer Co–N2 bonds of the amine in cis position (2.32–2.33 Å) compared to 2.17–2.23 Å with the pyridine donors.
For all four ligands, μ-hydroxo-bridged complexes were obtained under basic conditions. While the CoII ions in the complexes with the ligands susan, susanOMe, and cool are five-coordinate with only one μ-hydroxo-bridge, the CoII ions in the complex with the ligand susan6-Me are six-coordinate with two μ-hydroxo-bridges. This difference nicely demonstrates the lower electron donation of the ligand susan6-Me due to the longer CoII–N6-Me-py bonds leading to a coordinatively unsaturated situation for only one μ-hydroxo-bridge. The resulting “diamond-core” in [(susan6-Me){CoII(μ-OH)2CoII}]2+ shows alternating short 1.96 and long 2.13 Å Co–O(H) bonds, which was also observed for corresponding diferrous complexes.26
In the five-coordinate μ-hydroxy-bridged complexes the same trend as in the five-coordinate halide complexes is observed. The Co–Nim bond lengths are 2.02 Å while the Co–Npy are longer in [(susanOMe)CoII(μ-OH)CoII]3+ (2.06/2.07 Å) and in [(susan)CoII(μ-OH)CoII]3+ (2.08 Å). Interestingly, this difference has no significant influence on the Co-μ-OH bond lengths, which are all in the range 1.96–1.97 Å. However, the other apical donor of the trigonal bipyramide (N2) is significantly elongated 2.30 Å with cool compared to 2.20 Å with susan and susanOMe as already observed in the halide complexes.
In summary, the differences in the structural parameters allow to conclude, that the electron-density donation ability is in the order susan6-Me < susan/susanOMe < cool. No significant structural differences are observed for complexes of the ligands susanOMe and susan allowing no differentiation of their electron-donation ability.
Magnetic properties
To investigate the magnetic properties of the dinuclear CoII complexes, the temperature-dependence of the effective magnetic moments, μeff, (Fig. 2) and the variable-temperature variable-field VTVH magnetization (Fig. 3) were measured. These magnetic data were analyzed on the basis of the usual spin-Hamiltonian description for the electronic ground state for exchange coupled systems.27 The spin-Hamiltonian employed was: | (1) |
 | ||
| Fig. 2 Temperature dependence of the effective magnetic moment, μeff, for the dinuclear CoIICoII complexes of the ligands (a) susan6-Me, (b) susanOMe, and (c) cool. Open circles correspond to the experimental data and solid lines correspond to simulations using the spin-Hamiltonian provided in eqn (1) and parameters provided in Table 2. The sample of [(susan6-Me){CoII(μ-OH)2CoII}](ClO4)2 was measured in eicosane. | ||
 | ||
| Fig. 3 Variable-temperature variable-field magnetization data of (a) [(susan6-Me){CoII(CH3CN)2}2](ClO4)4·H2O, (b) [(susan6-Me){CoII(μ-OH)2CoII}](ClO4)2·1.5H2O (measured in eicosane), (c) [(susanOMe){CoIICl}2](ClO4)2·3MeOH, (d) [(susanOMe){CoIIBr}2](ClO4)2·3MeOH, (e) [(susanOMe)CoII(μ-OH)CoII](ClO4)3·1.5H2O, (f) [(cool){CoII(CH3CN)}2](ClO4)4·CH3CN, (g) [(cool){CoIICl}2](ClO4)2·MeOH, and (h) [(cool){CoIIBr}2](ClO4)2·MeOH. Open circles correspond to the experimental data and solid lines correspond to simulations using the spin-Hamiltonian provided in eqn (1) and parameters provided in Table 2. | ||
The complex [(susan6-Me){CoII(CH3CN)2}2](ClO4)4 exhibits μeff = 6.92μB at 300 K that decreases by decreasing temperature with a stronger decrease below 100 K reaching 4.02μB at 2 K (Fig. 2a). The VTVH data exhibit a slight nesting behavior of the iso-field lines (Fig. 3a). Fitting these data provided a small coupling constant J = −0.33 cm−1, a large deviation from ge = 2.00 with g = 2.55, and a large value of D = 68.5 cm−1. The μeff of 6.87μB at 300 K of [(susan6-Me){CoII(μ-OH)2CoII}](ClO4)2 increases by decreasing the temperature, reaches a maximum of 7.45μB at 11 K, and drops to a value of 5.48μB at 2 K (Fig. 2a). This behavior indicates a ferromagnetic coupling between the CoII ions. This sample was measured in eicosane to prevent potential torquing effects in high magnetic fields. The VTVH data (Fig. 3b) show a slight nesting behavior of the iso-field lines. Fitting these data provided the ferromagnetic coupling constant J = 2.4 cm−1, g values of g1 = 2.46 and g2 = 2.50, and zero-field splittings of D1 = −56 cm−1 and D2 = −16 cm−1. Corresponding differences for the two CoII ions have already been observed for [(susan){CoII(μ-OH)CoII}](ClO4)3 (Table 2).16
The halide complexes [(susanOMe){CoIICl}2](ClO4)2·3MeOH and [(susanOMe){CoIIBr}2](ClO4)2 exhibit almost the same temperature-dependence of μeff (Fig. 2b). The value of μeff for [(susanOMe){CoIICl}2](ClO4)2·3MeOH is 6.29μB at 300 K and shows a decrease below 30 K to a value of 4.85μB at 2 K. For [(susanOMe){CoIIBr}2](ClO4)2·3MeOH, μeff is 6.38μB at 300 K and drops to 4.79μB at 2 K. In combination with the VTVH (Fig. 3c + d), the following parameter sets were obtained: X = Cl: g = 2.29, J = −0.01 cm−1, D = 6.1 cm−1; X = Br: g = 2.33, J = −0.10 cm−1 and D = 6.0 cm−1.
The complex [(susanOMe){CoII(μ-OH)CoII}](ClO4)3 exhibits a similar coordination environment for the two CoII ions as in the halide complexes but μeff = 5.47μB at 300 K steadily decreases with decreasing temperature to 0.42μB at 2 K (Fig. 2b). This comparison clearly demonstrates that besides the local magnetic anisotropies, a considerable antiferromagnetic exchange is mediated by the bridging hydroxo ligand. This behavior closely resembles that of the complex [(susan){CoII(μ-OH)CoII}](ClO4)3 (Table 2)16 and fitting provided J = −20.8 cm−1, g = 2.29, D1 = 56 cm−1, and D2 = 8 cm−1.
The three non-bridged complexes of the ligand cool [(cool){CoII(CH3CN)}2](ClO4)4, [(cool){CoIICl}2](ClO4)2, and [(cool){CoIIBr}2](ClO4)2 show μeff of 5.96, 6.04, and 6.15μB at 300 K, respectively (Fig. 2c), that decrease below 50 K to 4.19μB for [(cool){CoIICl}2](ClO4)2 and 3.87μB for [(cool){CoIIBr}2](ClO4)2 at 2 K. The complex [(cool){CoII(CH3CN)}2](ClO4)4 exhibits a slightly stronger decrease of μeff below ∼100 K reaching a lower value of 1.42μB at 2 K. This can be rationalized by π–π-interactions between two neighboring molecules observed in the crystal-structure (Fig. S6†), that lead to a stronger antiferromagnetic exchange. Fitting these data provided the following parameter sets: X = CH3CN: J = −1.2 cm−1, g = 2.19, D = 10.0 cm−1; X = Cl: J = −0.24 cm−1, g = 2.21, D = 3.2 cm−1; X = Br: J = −0.29 cm−1, g = 2.25, D = 4.2 cm−1. The occurrence of the bridging hydroxo ligand in [(cool){CoII(μ-OH)CoII}](BPh4)2(OTf) coincides again with a stronger temperature-dependence from 5.36μB at 300 K to 0.58μB at 2 K (Fig. 2c). The fitting provided J = −15.5 cm−1, g = 2.18, and D = 86.4. The VTVH data were not considered as they provided no contribution of the complex due to the almost not existing magnetization at low temperature.
The magnetic properties reflect the electronic structures of the complexes that are governed for these CoIICoII complexes by their molecular structures. An important aspect is the deviation of the coordination polyhedra from an ideal octahedron. In the latter, the 4T1g ground state has a first-order orbital angular momentum. The larger the distortion from octahedral symmetry, the stronger is the splitting of the 4T1g reducing the orbital angular momentum contribution. When the splitting is large enough, the remaining orbital angular momentum can be treated by second order spin–orbit coupling and phenomenologically by zero-field splitting in the spin-Hamiltonian description. This is manifested here by zero-field splittings |D| in the order of 40–70 cm−1 for six-coordinate complexes and <10 cm−1 for the five-coordinate complexes with exogenous halide ligands. For the μ-hydroxo-bridged complexes, a smaller and a larger |D| is found also for the five-coordinate complexes; a situation already found for the susan complexes.16 In summary, as the electron-donating abilities of the exogenous and dinucleating ligands determine the coordination number, the magnetic properties reflect these differences and again no differentiation can be made between susan and susanOMe.
Electronic absorption spectroscopy
The electronic absorption spectra of the complexes and ligands in CH3CN are shown in Fig. 4. The ligands susan6-Me (ref. 19) and susanOMe (ref. 13) exhibit intense absorptions at 37600 cm−1 (ε = 16.4 × 103 M−1 cm−1) and 38000 cm−1 (ε = 9.9 × 103 M−1 cm−1), respectively, that can be assigned to π–π* transitions of the pyridines. The dinuclear CoIICoII complexes of these ligands exhibit similar bands that exhibit therefore strong ligand π–π* character. The imidazoles in the ligand cool exhibit no such band in this energy region, so that absorptions of the dinuclear CoIICoII complexes in this energy region can be attributed to have contributions from the CoII ions.
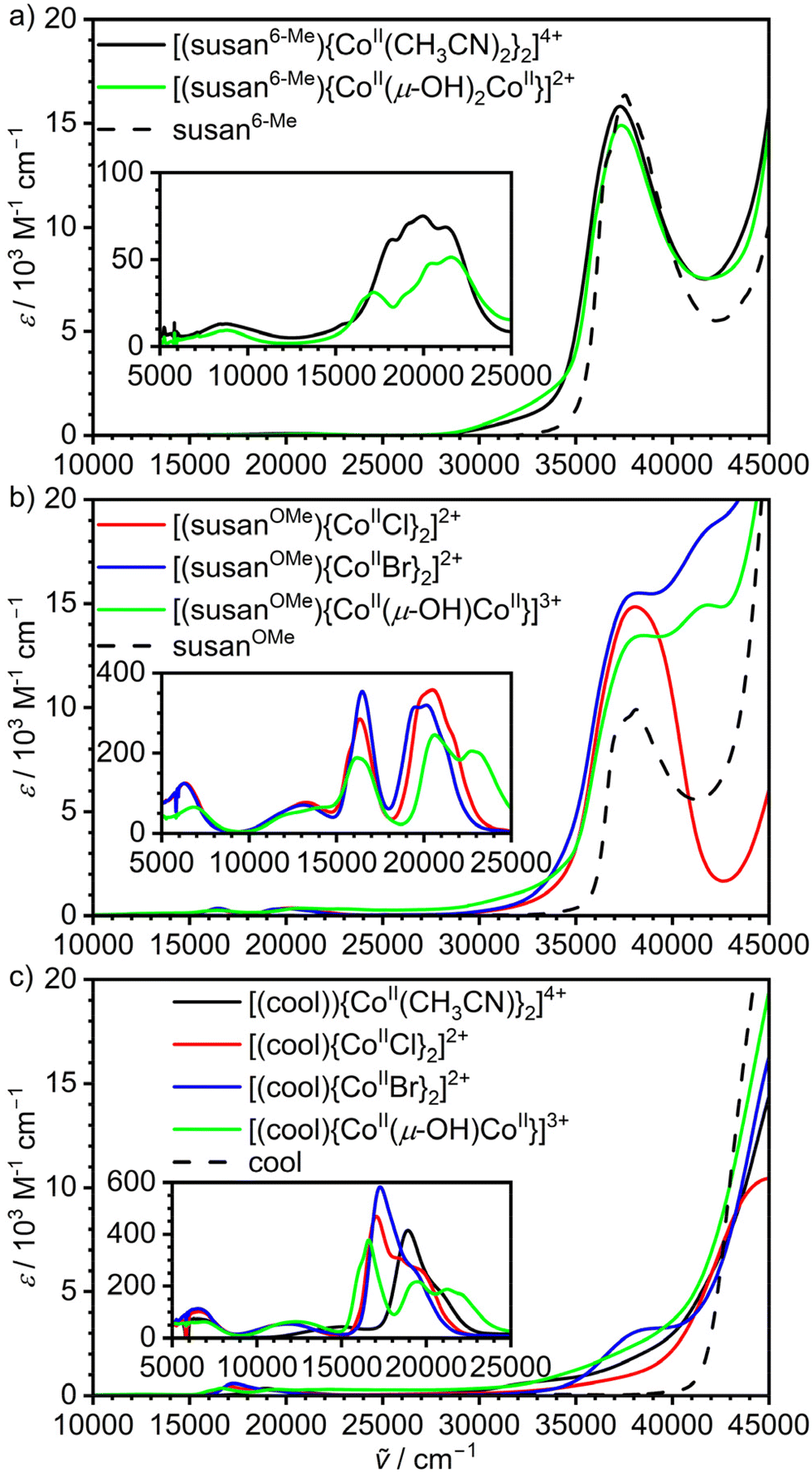 | ||
| Fig. 4 UV-Vis-NIR spectra of the dinuclear CoII2 complexes of the ligands (a) susan6-Me, (b) susanOMe, and (c) cool. and the different ligands dissolved in CH3CN.13,19 | ||
The CoIICoII complexes show multiple absorptions in the range 5000–25000 cm−1 (Fig. 4 insets) that mainly arise from d–d transitions and unspecific more intense absorptions at higher energies that are mainly charge-transfer in nature. The spectra of the CoIICoII complexes of the ligand susan16 closely resemble those of the ligand susanOMe having the same exogenous donors. We presented a detailed assignment and analysis of the d–d transitions of the susan CoIICoII complexes16 and therefore refrain from repeating this here. Instead, we focus on specific signatures that may allow to deduce molecular structures in solution.
Generally, energies and molar extinction coefficients differ significantly for five- and six-coordinated complexes. The six-coordinate complexes show a weak band around 9000 cm−1 ([(susan6-Me){CoII(CH3CN)2}2]4+ 8700 cm−1, ε = 14 M−1 cm−1; [(susan6-Me){CoII(μ-OH)2CoII}]2+ 8800 cm−1, ε = 9 M−1 cm−1) and not well-resolved transitions in the range 17000–23000 cm−1 with ε below 100 M−1 cm−1. The same signature was observed for [(susan){CoII(CH3CN)2}2]4+ while [(susan)CoII(μ-OH)CoII]3+ resembles the spectra of the five-coordinate complexes.16 This shows that not only [(susan6-Me){CoII(CH3CN)2}2]4+ and [(susan){CoII(CH3CN)2}2]4+ but also [(susan6-Me){CoII(μ-OH)2CoII}]2+ remains six-coordinate in solution and that the two hydroxo-bridged complexes [(susan6-Me){CoII(μ-OH)2CoII}]2+ and [(susan)CoII(μ-OH)CoII]3+ can be easily differentiated in solution by their UV-Vis spectra.
The five-coordinate complexes all exhibit a d–d transition below 7000 cm−1 of higher intensity (ε > 50 M−1 cm−1). At higher energies, a well-resolved transition around 16500–17500 cm−1 is characteristic besides a band around 20000 cm−1. The three μ-OH− bridged CoIICoII complexes of susan, susanOMe, and cool exhibit a further distinct band around 22000–23000 cm−1. Hence, this feature is characteristic for the {CoII(μ-OH)CoII} core of the five-coordinate CoIICoII complexes and can – in other words – serve to assign the persistence of the μ-OH− bridge in solution.
The spectra of the CoIICoII complexes of susanOMe (Fig. 4b) show no strong differences with those of susan having the same exogenous donor, but differ significantly from those of cool (Fig. 4c) in the 15000–25000 cm−1 region. The distinct bands below 19000 cm−1 are of lower intensity in the spectra of the susanOMe complexes (ε < 370 M−1 cm−1) than of the cool complexes (ε > 370 M−1 cm−1). Moreover, this band does not significantly shift for the susanOMe complexes (16200–16500 cm−1), while a significant shift is observed for the cool complexes (μ-OH−: 16600 cm−1, Cl−: 17100 cm−1, 17300 cm−1). These absorptions were assigned to 4A′2 → 4A′2 (4P) transitions, which do not involve significant d(z2) character and are therefore not strongly sensitive to the nature of the axial donors in trigonal-bipyramidally coordinated CoII ions.16 Hence, the change of π-acceptor ligands pyridine with π-donor ligands imidazole influence these transitions. A further observation is that the well-resolved transitions around 20000 cm−1 in the susanOMe complexes decrease in intensity and become less well-resolved in the cool complexes.
A special effect is observed for [(cool){CoII(CH3CN)}2]4+ – the only five-coordinate complex with an axial CH3CN donor. The other CoIICoII complexes of cool exhibit one band around 12100 cm−1 and the characteristic absorptions between 16600–17300 cm−1. These bands shift to 15100 and 18900 cm−1, respectively, for [(cool){CoII(CH3CN)}2]4+. This correlates with a change of π-donor ligands (μ-OH−, Cl−, Br−) to a strong π-acceptor ligand (CH3CN) in the axial positions. Hence, these transitions are diagnostic for the nature of the donors in the axial positions.
Electrochemical properties
Cyclic voltammograms were recorded on CH3CN solutions of the dinuclear CoIICoII complexes (Fig. 5). Peak potentials (Table 2) are referenced versus Fc+/Fc used as internal standard. All complexes show irreversible oxidative and reductive processes that are influenced by the dinucleating and the exogenous ligands.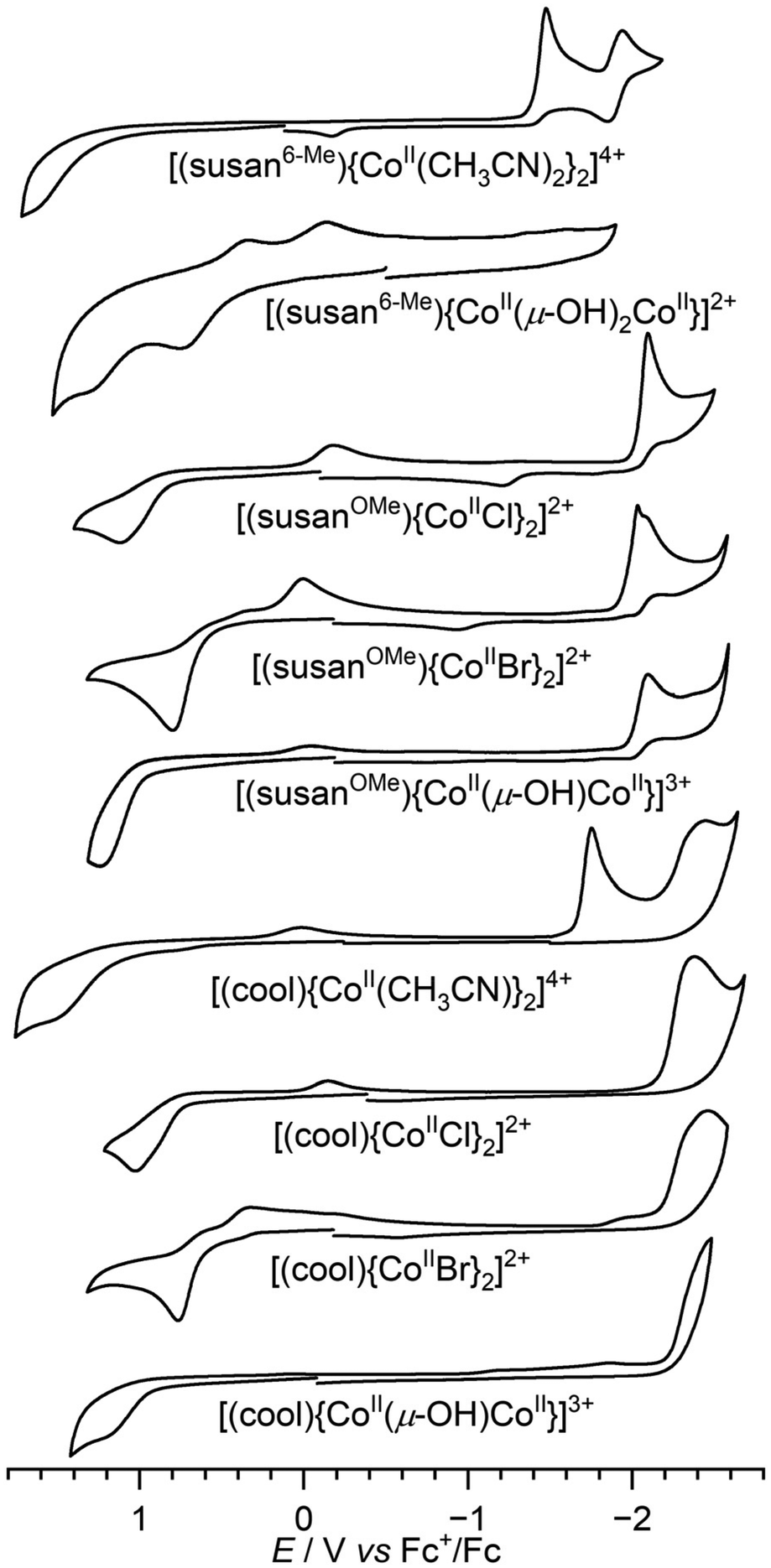 | ||
| Fig. 5 Cyclic voltammograms of the dinuclear complexes in CH3CN solutions containing 0.1 M TBAPF6 as supporting electrolyte. Scan rate is 200 mV s−1. | ||
The effects on the redox processes differ for five- and six-coordinate complexes and will thus be discussed separately. The six-coordinate complex [(susan6-Me){CoII(CH3CN)2}2]4+ shows one oxidative process at 1.49 V that is probably a metal-centered oxidation to a CoIIICoII species, while the first oxidative process in the analogous [(susan){CoII(CH3CN)2}2]4+ is at 0.46 V. This cathodic shift of ∼1 V show that for octahedral coordination the CoIII/CoII redox couple shifts as expected to lower potential with stronger σ-donors (susan vs. susan6-Me). The doubly bridged complex [(susan6-Me){CoII(μ-OH)2CoII}]2+ shows two oxidative processes at 0.72 and 1.26 V, that evoke reductive back currents at 0.30 and −0.18 V, respectively. However, the overall charge differ to that of [(susan6-Me){CoII(CH3CN)2}2]4+ prohibiting a direct comparison of reduction potentials as reduction potentials generally increase by increasing positive charge of the complex according to the Born equation.4,28
In the reductive part, both [(susan6-Me){CoII(CH3CN)2}2]4+ and [(susan){CoII(CH3CN)2}2]4+ show a reversible reduction at −1.9 V. This reduction was assigned to be ligand-centered in the complex with susan, while the higher potential reduction at −1.53 V was assigned to be metal-centered to a CoIICoI species.16 The shift of the latter to higher potential at −1.41 V in the complex with susan6-Me corroborates on the one hand the lower electron donation ability of susan6-Me than of susan as already observed in the oxidative processes and on the other hand the metal-centered assignment.
The five-coordinate complexes exhibit in the reductive part mostly the ligand-centered reduction that is not affected by the exogenous ligands. The trend −1.9 V (susan) > −2.1 V (susanOMe) > −2.4 V (cool) follows the expected trend of increasing electron density in the aromatic systems of susan < susanOMe < cool. Furthermore, reductive processes to metal-centered CoIICoI species at higher potentials are observed besides for [(cool){CoII(CH3CN)}2]4+ (−1.76 V) also for [(susan){CoIIBr}2]2+ (−1.83 V) and [(susanOMe){CoIIBr}2]2+ (−2.03 V). The lower potential of [(susanOMe){CoIIBr}2]2+ than of [(susan){CoIIBr}2]2+ demonstrates the higher electron donation of susanOMe than of susan.
In the oxidative part, the nine five-coordinate complexes varying the ligand (susan, susanOMe, cool) and the exogenous donors (Br−, Cl−, μ-OH−) allow to study the individual contributions to the potential. Considering the variation of the exogenous donors, the potential generally increases in the order Br− (0.75–0.79 V) < Cl− (1.02–1.10 V) < μ-OH− (1.18–1.31 V), which is counter-intuitive to the increasing electron donation ability of the donors in this series. This was rationalized in our study on the susan complexes16 that the oxidation is from five-coordinate CoII high-spin to six-coordinate CoIII low-spin. The latter has antibonding interaction with π-donor ligands so that the increasing π-donor ability Br− < Cl− < μ-OH− destabilizes CoIII low-spin and hence increases the potential.
For the halide complexes, the variation of the potential by changing the dinucleating ligand is relatively small (Cl−: 80 mV; Br−: 40 mV). The strongest variation occurs in the {CoII(μ-OH)CoII} complexes with susan (1.31 V) > susanOMe (1.24) > cool (1.18 V) in-line with the increasing electron donation ability susan < susanOMe < cool.
Conclusions
The differences in the molecular structures of the dinuclear CoIICoII complexes already allow conclusions to be drawn about the electron-donating ability of the ligands cool, susan, susanOMe, and susan6-Me. All complexes of the ligand cool are only five-coordinate including that with exogenous π-accepting CH3CN demonstrating its strongest electron donation capability correlated with shorter Co–Nim than Co–Npy bond lengths in related trigonal-bipyramidally coordinated complexes with Cl−, Br−, and μ-OH−. For the μ-OH− complexes, only the ligand susan6-Me requires two μ-OH− bridges resulting in six-coordinate CoII ions, while the ligands cool, susanOMe, and susan only include one μ-OH− bridge and therefore five-coordinate CoII ions. This shows that the ligand susan6-Me is less electron-donating due to the longer Co–N6-Me-py bonds than the Co–Npy bonds caused by the steric interference of the 6-Me groups oriented towards cis-coordinated ligands. Despite the longer Co–N6-Me-py bonds, the steric demand of the 6-Me groups enforces a facial coordination of the DPA-subunits in [(susan6-Me){CoII(CH3CN)2}2]4+, whereas in the otherwise analogous [(susan){CoII(CH3CN)2}2]4+ the DPA-subunits are meridionally coordinated. Overall, the structural analysis provides the following sequence of electron donation capability: susan6-Me < susanOMe/susan < cool without the possibility for a differentiation between susanOMe and susan.The magnetic data and the extracted spin-Hamiltonian parameters also show characteristic differences by varying the dinucleating and the exogenous ligands. For the complexes without a bridging hydroxo ligand, the zero-field splitting is much stronger for the six-coordinate complexes than for the five-coordinate complexes attributed to the stronger splitting of the octahedral 4T1g ground state by the trigonal bipyramidal coordination environment. There is no significant difference observed in the zero-field splittings for pyridine of imidazole donors. An important difference is the exchange coupling in the μ-hydroxo-bridged complexes, which is slightly ferromagnetic in the bis-μ-hydroxo bridged complex [(susan6-Me){CoII(μ-OH)2CoII}]2+, but antiferromagnetic and stronger in the μ-hydroxo-bridged five-coordinate complexes.
The UV-Vis-NIR spectra exhibit specific trends in the d–d transitions. The complexes that are six-coordinate in the solid-state exhibit generally lower intensities in the solution d–d spectra than the complexes that are five-coordinate in the solid-state (a well-known observation due to lifting of the Laporte rule) confirming that their coordination number is retained upon dissolution. This is supported by a weak band around 9000 cm−1 in the six-coordinate complexes, while the five-coordinate complexes show a more intense band below 7000 cm−1. The μ-hydroxo-bridged complexes show a unique band around 22000–23000 cm−1 confirming their bridged nature upon dissolution. Overall, all significant changes observed in the UV-Vis-NIR spectra can be interpreted consistently with changes in the solid-state structure providing strong evidence that the solid-state structures retain in solution. This will be a valuable tool for the study of the reactivity of these CoIICoII complexes. Moreover, the spectral features in the range 15000–22000 cm−1 vary specifically for the complexes with cool compared to structural analogous complexes of susan and susanOMe demonstrating the differing donor-strength of imidazole, while again no differentiation can be made between susan and susanOMe.
The electrochemical characterization provides a large set of potentials that are dependent on the coordination number, the dinucleating ligand, and the exogenous donor. For the six-coordinate complexes, the oxidation is facilitated by stronger σ-donor and less π-acceptor character and demonstrates a much stronger electron-donation of susan than of susan6-Me. In the five-coordinate complexes, the oxidation is counter-intuitively facilitated by less π-donation assigned to antibonding interactions of π-donors with CoIII low-spin. Irrespective of this, potentials for oxidations and reduction are shifted to lower potentials for susanOMe than for susan (e.g. 200 mV cathodic shift for the reduction of [(susanOMe){CoIIBr}2]2+vs. [(susan){CoIIBr}2]2+) providing an experimental handle for its stronger electron donation ability.
Taking together the information obtained from all measurements, the overall electron donation ability has the order susan6-Me ≪ susan < susanOMe ≪ cool. The +I effect of the 6-Me substituents is minor as the ligand-centered reduction is not affected. The strongly reduced electron donation of susan6-Me originates from the longer Co–N6-Me-py bond due to the sterical demand of the 6-Me groups. The stronger electron donation of susanOMe than of susan originates from the higher electron density in the pyridine rings that reduces the π-acceptor character of the pyridine. This effect is not strong enough to influence bond lengths significantly but strong enough to influence redox potentials. On the other hand, the effect is strong going from susan/susanOMe to cool with significantly shorter Co–Nim bonds demonstrate stronger Co–Nim bonds. This cannot be attributed to the change from a π-acceptor to a π-donor ligand15 but indicates an increasing main σ-donor character of the imidazole than the pyridine donors resulting in an overall stronger electron donation. In summary, the new ligand cool is a significantly stronger electron donating member in our family of dinucleating ligands and the influence on stability and reactivity in peroxo and high-valent complexes is currently under study in our lab.
Data availability
The supplementary crystallographic data for this study have been deposited at the Cambridge Crystallographic Data Centre under accession codes 2341023–2341031.†Author contributions
F. D. conducted the syntheses as well as the spectroscopic, magnetic, and electrochemical characterization. T. L. conducted the initial syntheses of the ligand cool and of some of the complexes. A. S., H. B., and J. O. collected, solved, and refined all the crystallographic data. T. G. designed experiments, assisted with data analysis, and wrote the manuscript with input from all the authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The DFG (Research Unit FOR 5215 “Bioinspired Oxidation Catalysis with Iron Complexes “BioOxCat”, TP1”) and Bielefeld University are gratefully acknowledged for funding.References
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- (a) L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed; (b) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed; (c) S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef CAS PubMed; (d) Y. Liang, J. Wei, X. Qiu and N. Jiao, Chem. Rev., 2018, 118, 4912–4945 CrossRef CAS PubMed; (e) A. J. Jasniewski and L. Que, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed; (f) C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed; (g) E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef CAS PubMed.
- (a) T. Glaser, Coord. Chem. Rev., 2019, 380, 353–377 CrossRef CAS; (b) S. Walleck and T. Glaser, Isr. J. Chem., 2020, 60, 1019–1031 CrossRef CAS.
- J. B. H. Strautmann, S. Walleck, H. Bögge, A. Stammler and T. Glaser, Chem. Commun., 2011, 47, 695–697 RSC.
- J. B. H. Strautmann, S. Dammers, T. Limpke, J. Parthier, T. P. Zimmermann, S. Walleck, G. Heinze-Brückner, A. Stammler, H. Bögge and T. Glaser, Dalton Trans., 2016, 45, 3340–3361 RSC.
- S. Walleck, T. P. Zimmermann, H. Hachmeister, C. Pilger, T. Huser, S. Katz, P. Hildebrandt, A. Stammler, H. Bögge, E. Bill and T. Glaser, Nat. Commun., 2022, 13, 1376 CrossRef CAS PubMed.
- T. P. Zimmermann, N. Orth, S. Finke, T. Limpke, A. Stammler, H. Bögge, S. Walleck, I. Ivanović-Burmazović and T. Glaser, Inorg. Chem., 2020, 59, 15563–15569 CrossRef CAS PubMed.
- L. Siebe, C. Butenuth, A. Stammler, H. Bögge, S. Walleck and T. Glaser, Inorg. Chem., 2024, 63, 2627–2639 CrossRef CAS PubMed.
- F. Depenbrock, T. Limpke, E. Bill, D. J. SantaLucia, M. van Gastel, S. Walleck, J. Oldengott, A. Stammler, H. Bögge and T. Glaser, Inorg. Chem., 2023, 62, 17913–17930 CrossRef CAS PubMed.
- (a) J. B. Gordon, A. C. Vilbert, M. A. Siegler, K. M. Lancaster, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2019, 141, 3641–3653 CrossRef CAS PubMed; (b) A. Chandra, M. Ansari, I. Monte-Pérez, S. Kundu, G. Rajaraman and K. Ray, Angew. Chem., Int. Ed., 2021, 60, 14954–14959 CrossRef CAS PubMed; (c) M. S. Møller, J. Kongsted and C. J. McKenzie, Dalton Trans., 2021, 50, 4819–4829 RSC; (d) Z.-Y. Chen, Z.-H. Long, X.-Z. Wang, J.-Y. Zhou, X.-S. Wang, X.-P. Zhou and D. Li, Inorg. Chem., 2021, 60, 10380–10386 CrossRef CAS PubMed; (e) A. A. DeLucia, K. A. Kelly, K. A. Herrera, D. L. Gray and L. Olshansky, Inorg. Chem., 2021, 60, 15599–15609 CrossRef CAS PubMed; (f) M. K. Goetz, J. E. Schneider, A. S. Filatov, K. A. Jesse and J. S. Anderson, J. Am. Chem. Soc., 2021, 143, 20849–20862 CrossRef CAS PubMed; (g) P. Kumar, L. Devkota, M. C. Casey, A. A. Fischer, S. V. Lindeman and A. T. Fiedler, Inorg. Chem., 2022, 61, 16664–16677 CrossRef CAS PubMed; (h) J. Amtawong, A. I. Nguyen and T. D. Tilley, J. Am. Chem. Soc., 2022, 144, 1475–1492 CrossRef CAS PubMed; (i) W. Mao, D. Fehn, F. W. Heinemann, A. Scheurer, M. van Gastel, S. A. V. Jannuzzi, S. DeBeer, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2022, 61, e202206848 CrossRef CAS PubMed; (j) Y.-F. Su, W.-Z. Luo, W.-Q. Lin, Y.-B. Su, Z.-J. Li, Y.-J. Yuan, J.-F. Li, G.-H. Chen, Z. Li, Z.-T. Yu and Z. Zou, Angew. Chem., Int. Ed., 2022, 61, e202201430 CrossRef CAS PubMed; (k) J. B. Gordon, T. Albert, S. Yadav, J. Thomas, M. A. Siegler, P. Moënne-Loccoz and D. P. Goldberg, Inorg. Chem., 2023, 62, 392–400 CrossRef CAS PubMed; (l) N. Zhao, M. K. Goetz, J. E. Schneider and J. S. Anderson, J. Am. Chem. Soc., 2023, 145, 5664–5673 CrossRef CAS PubMed; (m) A. A. DeLucia and L. Olshansky, Inorg. Chem., 2024, 63, 1109–1118 CrossRef CAS PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- (a) J. B. H. Strautmann, C.-G. Freiherr von Richthofen, G. Heinze-Brückner, S. DeBeer, E. Bothe, E. Bill, T. Weyhermüller, A. Stammler, H. Bögge and T. Glaser, Inorg. Chem., 2011, 50, 155–171 CrossRef CAS PubMed; (b) J. B. H. Strautmann, C.-G. Freiherr von Richthofen, S. DeBeer George, E. Bothe, E. Bill and T. Glaser, Chem. Commun., 2009, 2637–2639 RSC.
- S. Finke, A. Stammler, J. Oldengott, S. Walleck and T. Glaser, Dalton Trans., 2023, 52, 17548–17561 RSC.
- G. Xue, D. Wang, R. de Hont, A. T. Fiedler, X. Shan, E. Münck and L. Que Jr., Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20713–20718 CrossRef CAS PubMed.
- R. J. Sundberg, R. F. Bryan, I. F. Taylor and H. Taube, J. Am. Chem. Soc., 1974, 96, 381–392 CrossRef CAS.
- F. Depenbrock, T. Limpke, A. Stammler, J. Oldengott, H. Bögge and T. Glaser, Eur. J. Inorg. Chem., 2022, e202100992 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of laboratory chemicals, Elsevier/Butterworth-Heinemann, Amsterdam, Boston, 6th edn, 2009 Search PubMed.
- M. F. Braña, J. M. Castellano, D. Perron, C. Maher, D. Conlon, P. F. Bousquet, J. George, X.-D. Qian and S. P. Robinson, J. Med. Chem., 1997, 40, 449–454 CrossRef PubMed.
- S. Dammers, T. P. Zimmermann, S. Walleck, A. Stammler, H. Bögge, E. Bill and T. Glaser, Inorg. Chem., 2017, 56, 1779–1782 CrossRef CAS PubMed.
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- (a) C. He and S. J. Lippard, Inorg. Chem., 2001, 40, 1414–1420 CrossRef CAS PubMed; (b) J. Kuzelka, S. Mukhopadhyay, B. Spingler and S. J. Lippard, Inorg. Chem., 2003, 42, 6447–6457 CrossRef CAS PubMed; (c) S. V. Kryatov, S. Taktak, I. V. Korendovych, E. V. Rybak-Akimova, J. Kaizer, S. Torelli, X. Shan, S. Mandal, V. L. MacMurdo, A. Mairata i Payeras and L. Que, Inorg. Chem., 2005, 44, 85–99 CrossRef CAS PubMed; (d) F. L. B. Röhs, S. Dammers, A. Stammler, J. Oldengott, H. Bögge, E. Bill and T. Glaser, Eur. J. Inorg. Chem., 2022, 2022, e202200177 CrossRef.
- The program package JulX was used for spin-Hamiltonian simulations and fittings of the data by a full-matrix diagonalization approach (E. Bill, unpublished results).
- (a) M. Born, Z. Phys., 1920, 1, 45–48 CrossRef CAS; (b) T. Beissel, F. Birkelbach, E. Bill, T. Glaser, F. Kesting, C. Krebs, T. Weyhermüller, K. Wieghardt, C. Butzlaff and A. X. Trautwein, J. Am. Chem. Soc., 1996, 118, 12376–12390 CrossRef CAS; (c) T. Glaser, M. Heidemeier, J. B. H. Strautmann, H. Bögge, A. Stammler, E. Krickemeyer, R. Huenerbein, S. Grimme, E. Bothe and E. Bill, Chem. – Eur. J., 2007, 13, 9191–9206 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details crystal structure determination, FTIR spectra, NMR spectra, (pdf) X-ray crystallographic data (cif). CCDC 2341023–2341031. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00877d |
| This journal is © The Royal Society of Chemistry 2024 |