
A ratiometric substrate for rapid evaluation of transfer hydrogenation efficiency in solution†
Yen-An
Young
,
Huong T. H.
Nguyen
,
Hieu D.
Nguyen
 ,
Tuhin
Ganguly
,
Yennie H.
Nguyen
and
Loi H.
Do
*
,
Tuhin
Ganguly
,
Yennie H.
Nguyen
and
Loi H.
Do
*
Department of Chemistry, University of Houston, 4800 Calhoun Road, Houston, TX 77204, USA. E-mail: loido@uh.edu
First published on 10th May 2024
Abstract
A cyclometalated iridium(III) complex bearing a self-immolative quinolinium moiety was developed as a ratiometric substrate for transfer hydrogenation studies. This photoluminescent probe allowed the rapid screening of a variety of Ir catalysts using a microplate reader, offering a convenient method to assess activity using a minimum amount of catalyst sample.
Transition metal catalysts capable of promoting transfer hydrogenation (TH), which are reactions involving the exchange of H− equivalents between hydride donors and acceptors, have numerous applications.1–3 For example, they are used in the stereoselective reduction of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN groups in chemical synthesis4,5 and the conversion of dioxygen to hydrogen peroxide in small-molecule activation6 (Scheme 1A). Researchers have shown that TH catalysts can also be applied in living systems, including as redox modulators for anti-cancer therapy,7,8 activity-based platforms for sensing endogenous formate,9 detoxification agents against harmful aldehydes,10 and cofactors in artificial metalloenzymes.11,12 Although intracellular catalysts13–15 are commonly derivatized with various ligand substituents or targeting groups,11,16,17 such modifications risk negatively impacting their catalytic performance. Thus, having a fast and reliable method to evaluate TH catalysts could accelerate their discovery and development.
O and CN groups in chemical synthesis4,5 and the conversion of dioxygen to hydrogen peroxide in small-molecule activation6 (Scheme 1A). Researchers have shown that TH catalysts can also be applied in living systems, including as redox modulators for anti-cancer therapy,7,8 activity-based platforms for sensing endogenous formate,9 detoxification agents against harmful aldehydes,10 and cofactors in artificial metalloenzymes.11,12 Although intracellular catalysts13–15 are commonly derivatized with various ligand substituents or targeting groups,11,16,17 such modifications risk negatively impacting their catalytic performance. Thus, having a fast and reliable method to evaluate TH catalysts could accelerate their discovery and development.
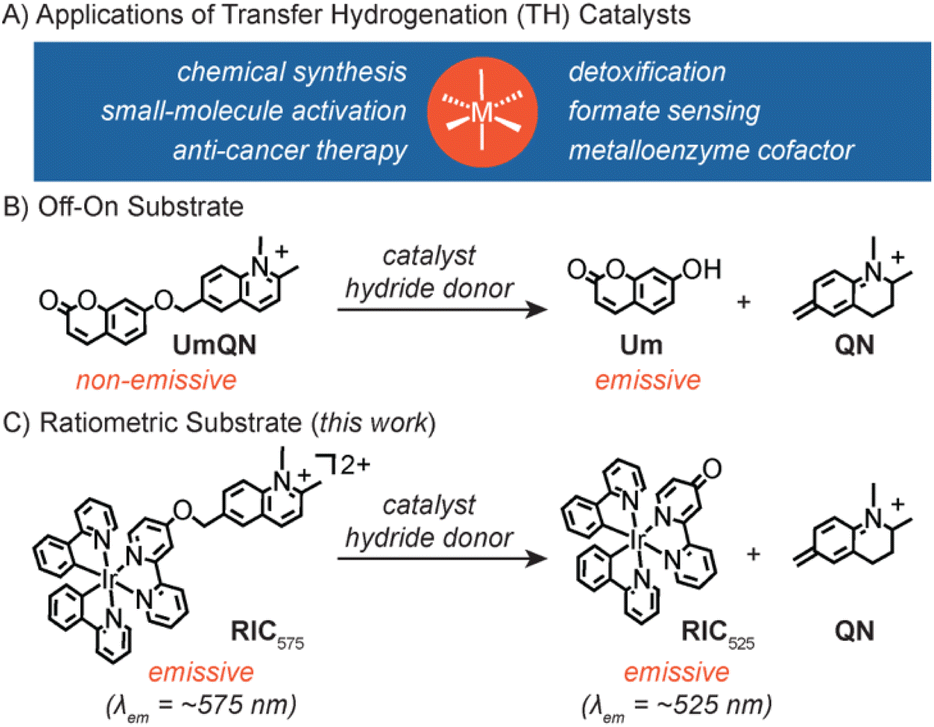 | ||
| Scheme 1 Various applications of transfer hydrogenation (TH) catalysts (A) and off–on (B) and ratiometric (C) substrates used to study their activity. | ||
Herein, we introduce a user-friendly photoluminescence-based method to screen for TH catalyst activity using a microplate reader. Our strategy relies on the use of a ratiometric substrate that allows in situ quantification of the reaction progress.18,19 This approach enables simultaneous monitoring of multiple reactions using minimum amounts of sample, which is particularly attractive for studies of catalysts that are costly (e.g., those containing Ir,20 Ru,21 or Os22) or difficult to obtain on large scales.
The design of our ratiometric probe was inspired by a self-immolative umbelliferone-quinolinium substrate (UmQN) developed by Ward and coworkers (Scheme 1B).11,12 It was shown that when this compound was reduced via TH catalysis, release of umbelliferone (Um) led to an emission turn-on. Although this off–on substrate was used to evaluate TH reactions in the periplasm of E. coli, the total amount of starting substrates inside the cells could not be quantified by fluorescence microscopy because they are non-emissive. To further complicate matters, small molecules such as substrates and molecular catalysts can diffuse in and out of cells so obtaining accurate yields in living systems is highly challenging. To overcome these limitations, we sought to develop a fluorophore-quinolinium construct that would exhibit different emission properties in the starting and product forms so that the reaction yields could be determined by monitoring the emission intensity ratio at two different wavelengths. Such ratiometric substrates may allow non-invasive studies of intracellular TH reactions in real time without having to lyse the cells and analyse their contents by methods such as high performance liquid chromatography. After considering a variety of fluorophore candidates, we selected to use a cyclometalated iridium(III) complex as the reporter due to its tunable emission, chemical stability, and ease of preparation (Scheme 1C).23–28 In addition, its phosphorescence may be more easily distinguished from biological self-fluorescence.29
The synthesis of our ratiometric iridium complex (RIC575, where the subscript denotes its emission maximum) proceeded starting from 2,6-dimethylquinoline (Scheme S1†). Monobromination using N-bromosuccinimide (NBS) afforded compound 1 (68% yield), which was then subjected to nucleophilic attack by 2-bromo-4-hydroxypyridine to furnish 2 in 73% yield. Combining 2 in the presence of 2-(tributylstannyl)pyridine under Stille cross-coupling conditions gave the bipyridine species 3 (62% yield). Metalation of 3 was accomplished by treatment with [Ir(ppy)2Cl]2 (ppy = 2-phenylpyridine), anion exchange using AgOTf (OTf− = triflate anion), and then alkylation using methyl triflate to afford RIC575 in 38% yield (based on 3, Scheme 2A).
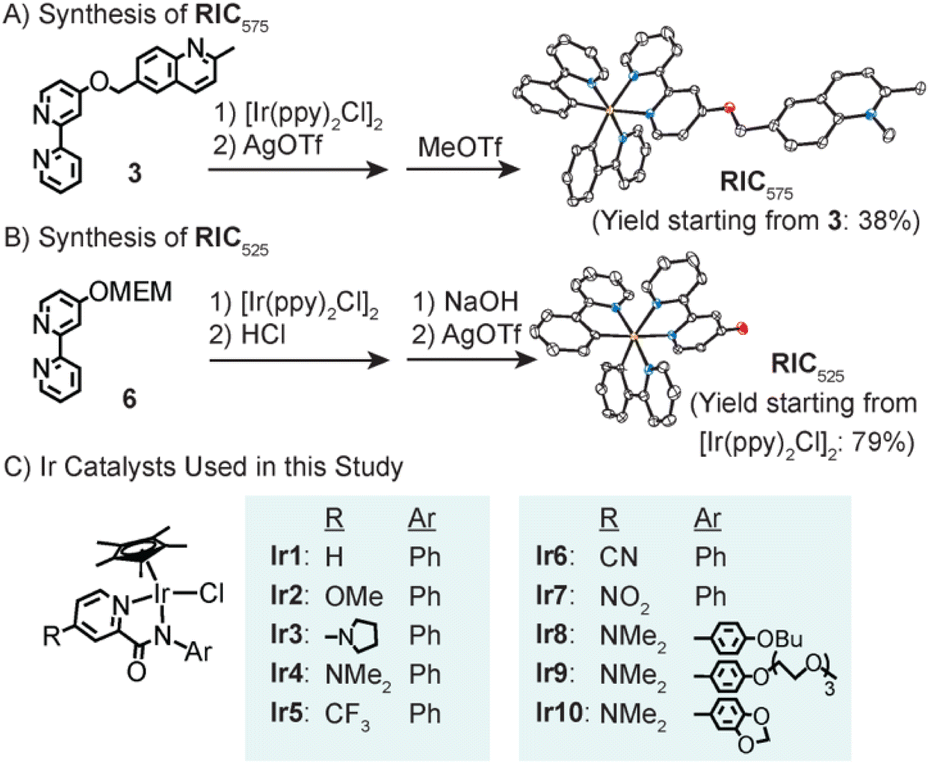 | ||
| Scheme 2 Synthesis of RIC575 (A) and RIC525 (B) starting from their corresponding bipyridine ligands. The molecular structures of RIC575 and RIC525 are depicted with 50% thermal ellipsoids based on their respective X-ray crystallographic data (orange = iridium, blue = nitrogen, red = oxygen atoms). The two OTf− anions in RIC575 were omitted for clarity. In part C, the Ir catalysts used for TH studies are shown. | ||
The cleaved cyclometalated iridium species RIC525 (where the subscript denotes its emission maximum) was also prepared (Scheme S2†). The starting 2-bromo-4-hydroxypyridine was protected using 2-methoxyethoxymethyl (MEM) chloride to give 5, followed by Stille cross-coupling with 2-(tributylstannyl)pyridine to furnish bipyridine 6. Combining 6 with [Ir(ppy)2Cl]2 and then treatment with HCl gave the corresponding anionic iridium complex (RIC525′). To obtain the neutral form, it was stirred in the presence of NaOH, followed by the addition of AgOTf to afford RIC525 with a yield of 79% based on the amount of [Ir(ppy)2Cl]2 used in the previous step.
To obtain structural characterization, X-ray crystallography was used to analyse single crystals of RIC575 and RIC525. The structure of RIC575 showed the expected octahedral iridium center with a quinolinium moiety attached to the bipyridine ligand (Fig. S62†). The presence of two triflate anions supports the dicationic nature of the complex. The structure of RIC525 also reveals an octahedral Ir geometry but the absence of any counterions in the crystallographic unit cell indicates that this species is charge neutral overall (Fig. S63†). Comparison of the bond metrics in RIC575vs.RIC525 suggests that the former contains a 4-alkoxypyridyl donor whereas the latter contains a 4-pyridonate donor coordinated to iridium (Fig. 1A).30–32 For example, in RIC575, the C–C and C–N bond distances of ∼1.33–1.39 Å in the functionalised pyridine ring are typical of an aromatic structure (Fig. 1A).33 Additionally, its C(3)–O(1) bond length of 1.35 Å is expected for a carbon–oxygen single bond (average = ∼1.37 Å). In contrast, the corresponding six-membered ring in RIC525 exhibits contracted or elongated C–C/C–N bonds relative to those in RIC575, suggesting that it is non-aromatic. The only exception is that the C(4)–C(5) bond distance in RIC525 is similar to that in RIC575 (∼1.37 Å), which may be due to the conjugation of its C4 and C5 p-orbitals with the π-system of the adjacent pyridine ring. Lastly, the C(3)–O(1) bond distance of 1.28 Å in RIC525 is consistent with a CO carbonyl group.33
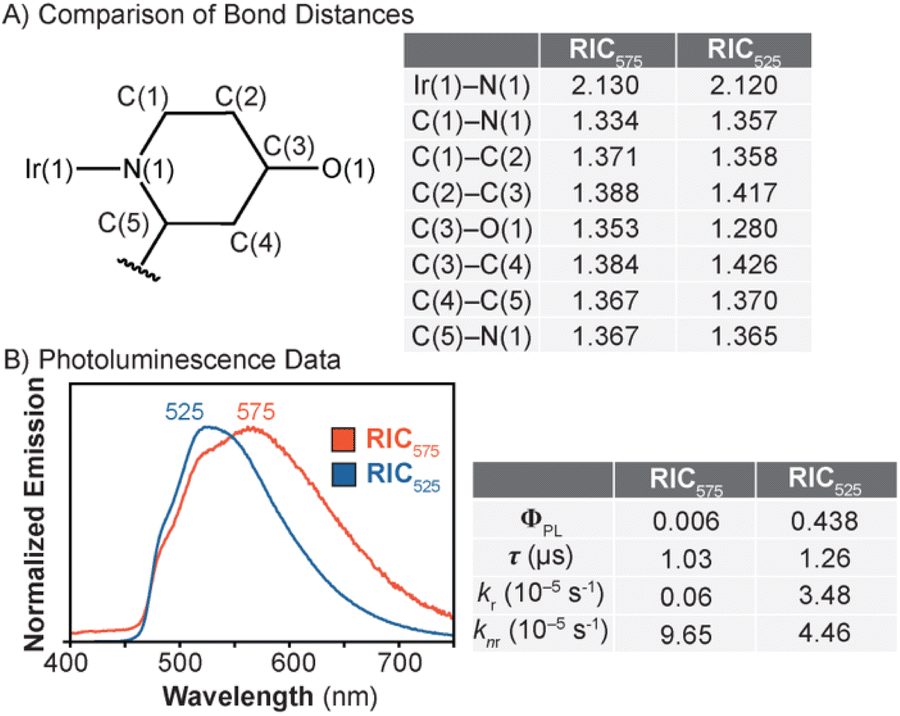 | ||
| Fig. 1 Comparison of key bond distances (Å) between RIC575 and RIC525 (A) and their corresponding photophysical properties (B). The emission spectra were recorded in anhydrous MeOH at 20 °C under N2 with excitation at 350 nm. | ||
Due to the electronic differences in their ancillary ligands, RIC575 and RIC525 were expected to display different photophysical properties.23 As shown in Fig. 1B, when RIC575 was excited with 350 nm light in MeOH under N2, a broad emission peak was observed with a maximum at 575 nm and shoulders at 490 and 520 nm. Our data indicate that the non-radiative decay rate (knr) was about 160-fold faster than the radiative decay rate (kr), presumably due to the emission quenching effects of its quinolinium moiety.34 For comparison, the iridium precursor 4 bearing an unmethylated quinoline ring exhibited a knr that is only 1.9-fold faster than kr (Table S4†). When RIC525 was irradiated with 350 nm light, its emission spectrum showed a peak maximum at 525 nm. This hypsochromic shift, relative to the λem of RIC575, is consistent with the more electron-rich nature of the RIC525 bipyridine ligand, which destabilizes the LUMO energy and consequently, increases the Ir complex's HOMO–LUMO gap. Our results also indicate that RIC525 has a higher quantum yield (ΦPL = 0.438 vs. 0.006) and longer luminescence lifetime (τ = 1.26 vs. 1.03 μs) than that of RIC575 (Fig. 1B). A chromaticity analysis shows that the photoluminescence of RIC575 appears yellow whereas that of RIC525 appears green (Fig. S4†). Because RIC575 and RIC525 have poor solubility in water, we were unable to perform photophysical measurements in aqueous solutions such as biological buffers or cell culture media.
Prior to conducting reaction studies with RIC575, we created a calibration curve using standard solutions containing different ratios of RIC575/RIC525 (Table S6†). Sodium bicarbonate was also added to the mixtures to ensure that RIC525 remains in the neutral form. We found that measuring the emission intensities at 550 (I550) and 600 (I600) nm and plotting the I550/I600 ratio gave a linear correlation with the amount of RIC525 present (Fig. S6†).19 Due to the greater than 70-fold difference in quantum yield between RIC525 and RIC575, the choice of 550 and 600 nm for reaction monitoring (rather than at their emission maxima of 525 and 575 nm, respectively) ensures that signals from the product do not overwhelm signals from the starting material for accurate quantification.
Next, we proceeded to study the TH activity of a series of previously reported [Cp*Ir(4-R-picolinamidate)Cl] complexes (where Cp* = pentamethylcyclopentadienyl anion, R = various functional groups; Scheme 2C).11,35–37 To establish that RIC575 is a viable substrate for TH,11,12 a small-scale reaction was performed using a 1-dram vial (Fig. S61A†). When Ir4 (R = NMe2), RIC575, HCOONa, and NaHCO3 were dissolved in 1 mL of MeOH and stirred at RT, the reaction mixture showed increasing luminescence over the course of 1 h with a change in color from yellow to green (Fig. S61B†), suggesting that RIC525 had formed. The presence of the RIC525 product was further confirmed by emission spectroscopy and mass spectrometry. Reduction of the quinolinium moiety was supported by showing that a model substrate, 1,2,6-trimethylquinolinium triflate, was converted to 1,2,3,4-tetrahydro-1,2,6-trimethylquinoline in the presence of Ir4, HCOONa, and NaHCO3 (Fig. S59†). These results indicate that the conversion of RIC575 to RIC525 by the Ir catalysts and HCOONa occurs on a timescale that is suitable for quantification using a microplate reader.
Once we identified the optimal reaction conditions for TH, we carried out our catalyst screening experiments in 24-well microplates (Fig. 2A). In these investigations, each well was charged with an Ir catalyst (2.0 μM), HCOONa (0.7 mM), and NaHCO3 (1.25 mM) in MeOH at 28 °C (Fig. 2A). The mixtures were injected with RIC575 (0.2 mM) and then the microplate was inserted into a microplate reader for continuous emission monitoring at 550 and 600 nm. By converting the I550/I600 ratio to the %yield of RIC525 (see Table S7† for example with Ir1), kinetic plots of the reactions were generated (Fig. 2B, left). The kinetic data showed that in all cases, an induction period of ∼10 min was observed, which may be due to catalyst activation (e.g., formation of the initial iridium-hydride species).36 Based on the RIC525 yields after 1 h, the catalyst activity trend follows the order: Ir4 (R = NMe2) > Ir3 (R = pyrrolidinyl) > Ir2 (R = OMe) > Ir1 (R = H) > Ir5 (R = CF3) > Ir6 (R = CN). The poor solubility of Ir7, which bears a nitro group on the pyridyl ring, precluded studies using this complex. The trend observed that electron-rich Ir catalysts are more active than their electron-poor counterparts is fully consistent with previous studies performed in aqueous media.35,36 It has been demonstrated that hydride formation is typically rate-limiting in TH reactions by [Cp*Ir(4-R-picolinamidate)Cl] catalysts and that this step occurs faster with electron-rich than electron-poor catalysts.36 Although a more in-depth analysis of the kinetic data was not pursued in this work, these results validate our ratiometric substrate-based approach for rapid screening of TH catalysts. Because each reaction in our studies requires only 20 nmol (<∼90 μg) of Ir catalyst, a significant amount of material could be saved compared to performing larger scale reactions in standard vials or flasks.
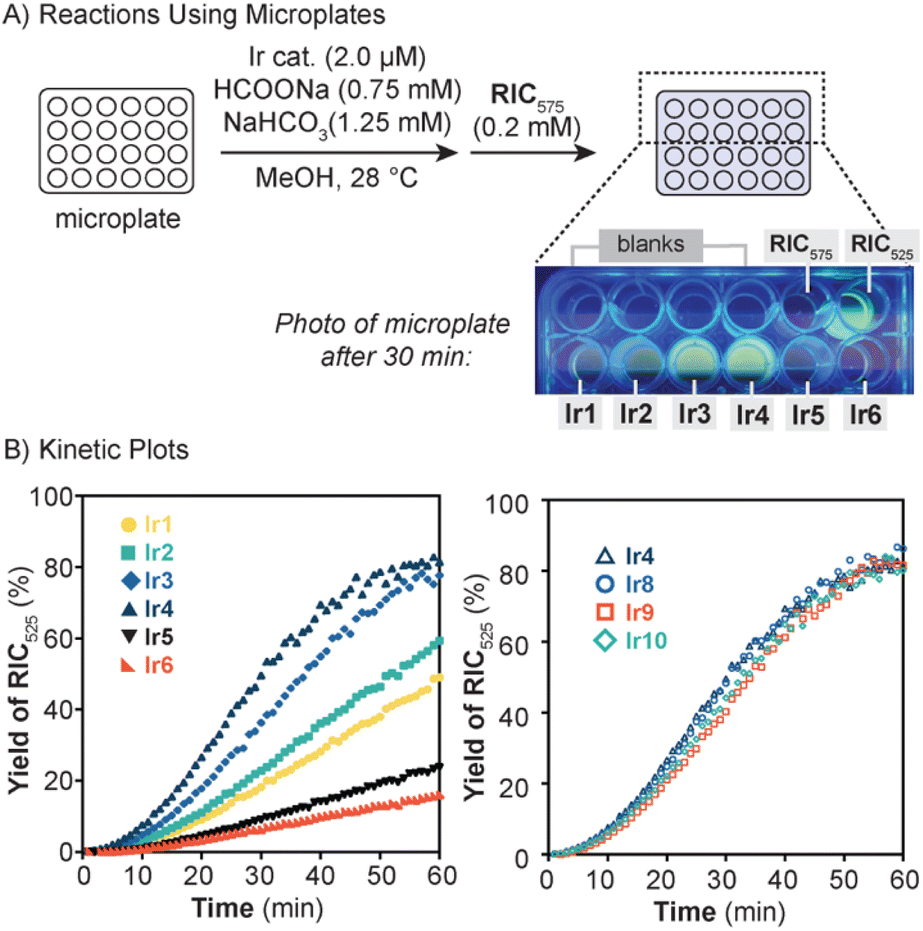 | ||
| Fig. 2 (A) Scheme depicting our TH reaction studies using 24-well microplates. The photo shows a portion of the microplate after the reaction proceeded for 30 min (excitation = 325 nm light). (B) Kinetic plots of TH reactions using RIC575 with catalysts Ir1–Ir6 (left) and Ir4/Ir8–Ir10 (right) acquired using a microplate reader. | ||
To illustrate the utility of our method, we used our protocol to evaluate the performance of several new TH catalysts. When developing a catalyst for in cell or in vivo applications, it is often desirable to customize its biological properties, such as cellular uptake, interactions with biomolecules, subcellular localization, etc., without diminishing its catalytic activity.13 Because Ir4 was the most active in our series (Fig. 2B, left), we chose to derivatize this complex to determine whether the catalyst structure and activity could be tuned independently of each other. By adapting previously reported procedures (see ESI†), we successfully prepared three iridium variants featuring tert-butoxyphenyl (Ir8), triethylene glycol phenyl (Ir9), and 1,3-benzodioxole (Ir10) groups on the picolinamidate ligand (Scheme 2C). These substituents were chosen to represent groups with varying shapes and sizes. The new Ir catalysts were tested in TH reactions by combining them with HCOONa, NaHCO3, RIC575, and MeOH in 24-well microplates. Analysis of the photoluminescence data acquired by the microplate reader produced the kinetic traces shown in Fig. 2B (right). The results revealed that Ir8–Ir10 exhibit nearly identical kinetic profiles as that of their parent catalyst Ir4. This finding corroborates our previous observations that for [Cp*Ir(4-R-picolinamidate)Cl] complexes ligand modifications at the N-phenyl moiety do not impact their catalyst activity.36
In summary, we have developed a self-immolative substrate for photoluminescence monitoring of TH catalysts using a microplate reader. Because the emission maximum of the cleaved product RIC525 is blue-shifted relative to that of the starting RIC575, ratiometric quantification of the reaction yields was possible by measuring the intensity changes at two different wavelengths. Application of our method to studies of several new [Cp*Ir(4-R-picolinamidate)Cl] variants revealed that functionalization of the N-phenyl group does not negatively impact their catalytic activity. Our photoluminescence-based method is user-friendly because it is rapid, requires minimal catalyst sample, and can be readily used by researchers with access to microplate readers, which are available in many synthetic and biochemistry laboratories.
There are numerous non-aqueous applications that may benefit from this new molecular tool, such as in high throughput automation for chemical synthesis or metallodrug discovery.38,39 To enable TH reaction imaging in biological cells,40 the RIC575 probe could be rendered more water soluble by functionalizing it with sulfonate groups; however, how this change may affect its cell permeability remains to be investigated.41 Since different cellular compartments have different pH values, modifying the probe so that its emission properties are not sensitive to pH changes could broaden its biological range. Although in-cell applications were not achieved in this work, it provides proof-of-concept demonstration that cyclometalated Ir-quinolinium substrates are useful for ratiometric quantification of TH reactions in solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Welch Foundation (grant no. E-1894 to L. H. D.) and the National Institutes of Health (grant no. R01GM129276 to L. H. D.) for funding this work. We are grateful to Prof. Tom Teets for helpful suggestions and allowing us to use their fluorimeter.References
- D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- S. Gaillard and J.-L. Renaud, Iron-Catalyzed Hydrogenation, Hydride Transfer, and, Hydrosilylation: An Alternative to Precious-Metal Complexes?, ChemSusChem, 2008, 1, 505–509 CrossRef CAS PubMed.
- A. Matsunami and Y. Kayaki, Upgrading and Expanding the Scope of Homogeneous Transfer Hydrogenation, Tetrahedron Lett., 2018, 59, 504–513 CrossRef CAS.
- P. D. Parker, X. Hou and V. M. Dong, Reducing Challenges in Organic Synthesis with Stereoselective Hydrogenation and Tandem Catalysis, J. Am. Chem. Soc., 2021, 143, 6724–6745 CrossRef CAS PubMed.
- S. Gladiali and E. Alberico, Asymmetric transfer hydrogenation: chiral ligands and applications, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- Z. M. Heiden and T. B. Rauchfuss, Homogeneous Catalytic Reduction of Dioxygen Using Transfer Hydrogenation Catalysts, J. Am. Chem. Soc., 2007, 129, 14303–14310 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Transfer Hydrogenation Catalysis in Cells as a New Approach to Anticancer Drug Design, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- A. C. Carrasco, V. Rodríguez-Fanjul, A. Habtemariam and A. M. Pizarro, Structurally Strained Half-Sandwich Iridium(III) Complexes As Highly Potent Anticancer Agents, J. Med. Chem., 2020, 63, 4005–4021 CrossRef CAS PubMed.
- S. W. M. Crossley, L. Tenney, V. N. Pham, X. Xie, M. W. Zhao and C. J. Chang, A Transfer Hydrogenation Approach to Activity-Based Sensing of Formate in Living Cells, J. Am. Chem. Soc., 2024, 146, 8865–8876 CrossRef CAS PubMed.
- R. D. Jana, A. H. Ngo, S. Bose and L. H. Do, Organoiridium Complexes Enhance Cellular Defense Against Reactive Aldehydes Species, Chem. – Eur. J., 2023, 29, e202300842 CrossRef CAS PubMed.
- J. G. Rebelein, Y. Cotelle, B. Garabedian and T. R. Ward, Chemical Optimization of Whole-Cell Transfer Hydrogenation Using Carbonic Anhydrase as Host Protein, ACS Catal., 2019, 9, 4173–4178 CrossRef CAS PubMed.
- J. Zhao, J. G. Rebelein, H. Mallin, C. Trindler, M. M. Pellizzoni and T. R. Ward, Genetic Engineering of an Artificial Metalloenzyme for Transfer Hydrogenation of a Self-Immolative Substrate in Escherichia coli's Periplasm, J. Am. Chem. Soc., 2018, 140, 13171–13175 CrossRef CAS PubMed.
- A. H. Ngo, S. Bose and L. H. Do, Intracellular Chemistry: Integrating Molecular Inorganic Catalysts with Living Systems, Chem. – Eur. J., 2018, 24, 10584–10594 CrossRef CAS PubMed.
- H. Madec, F. Figueiredo, K. Cariou, S. Roland, M. Sollogoub and G. Gasser, Metal complexes for catalytic and photocatalytic reactions in living cells and organisms, Chem. Sci., 2023, 14, 409–442 RSC.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Intracellular Catalysis with Selected Metal Complexes and Metallic Nanoparticles: Advances toward the Development of Catalytic Metallodrugs, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Transition Metal Catalysis in the Mitochondria of Living Cells, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- J. W. Southwell, R. Herman, D. J. Raines, J. E. Clarke, I. Böswald, T. Dreher, S. M. Gutenthaler, N. Schubert, J. Seefeldt, N. Metzler-Nolte, G. H. Thomas, K. S. Wilson and A.-K. Duhme-Klair, Siderophore-Linked Ruthenium Catalysts for Targeted Allyl Ester Prodrug Activation within Bacterial Cells, Chem. – Eur. J., 2023, 29, e202202536 CrossRef CAS PubMed.
- M. H. Lee, J. S. Kim and J. L. Sessler, Small molecule-based ratiometric fluorescence probes for cations, anions, and biomolecules, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- Z. Song, R. T. K. Kwok, E. Zhao, Z. He, Y. Hong, J. W. Y. Lam, B. Liu and B. Z. Tang, A Ratiometric Fluorescent Probe Based on ESIPT and AIE Processes for Alkaline Phosphatase Activity Assay and Visualization in Living Cells, ACS Appl. Mater. Interfaces, 2014, 6, 17245–17254 CrossRef CAS PubMed.
- Y. Wei, Y. Liang, R. Luo and L. Ouyang, Recent advances of Cp*Ir complexes for transfer hydrogenation: focus on formic acid/formate as hydrogen donors, Org. Biomol. Chem., 2023, 21, 7484–7497 RSC.
- R. Noyori and S. Hashiguchi, Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- J. P. C. Coverdale, C. Sanchez-Cano, G. J. Clarkson, R. Soni, M. Wills and P. J. Sadler, Easy To Synthesize, Robust Organo-osmium Asymmetric Transfer Hydrogenation Catalysts, Chem. – Eur. J., 2015, 21, 8043–8046 CrossRef CAS PubMed.
- M. S. Lowry and S. Bernhard, Synthetically Tailored Excited States: Phosphorescent, Cyclometalated Iridium(III) Complexes and Their Applications, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- D. N. Chirdon, C. E. McCusker, F. N. Castellano and S. Bernhard, Tracking of Tuning Effects in Bis-Cyclometalated Iridium Complexes: A Combined Time Resolved Infrared Spectroscopy, Electrochemical, and Computational Study, Inorg. Chem., 2013, 52, 8795–8804 CrossRef CAS PubMed.
- V. Mdluli, S. Diluzio, J. Lewis, J. F. Kowalewski, T. U. Connell, D. Yaron, T. Kowalewski and S. Bernhard, High-throughput Synthesis and Screening of Iridium(III) Photocatalysts for the Fast and Chemoselective Dehalogenation of Aryl Bromides, ACS Catal., 2020, 10, 6977–6987 CrossRef CAS.
- S. DiLuzio, V. Mdluli, T. U. Connell, J. Lewis, V. VanBenschoten and S. Bernhard, High-Throughput Screening and Automated Data-Driven Analysis of the Triplet Photophysical Properties of Structurally Diverse, Heteroleptic Iridium(III) Complexes, J. Am. Chem. Soc., 2021, 143, 1179–1194 CrossRef CAS PubMed.
- Y. Wu, G. D. Sutton, M. D. S. Halamicek, X. Xing, J. Bao and T. S. Teets, Cyclometalated iridium-coumarin ratiometric oxygen sensors: improved signal resolution and tunable dynamic ranges, Chem. Sci., 2022, 13, 8804–8812 RSC.
- P.-N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, Highly Efficient Red-Emitting Bis-Cyclometalated Iridium Complexes, J. Am. Chem. Soc., 2018, 140, 10198–10207 CrossRef CAS PubMed.
- S. Zhao, L. Chen, Y. Yang and X. Liu, Research progress of phosphorescent probe for biological imaging, J. Mol. Struct., 2022, 1269, 133855 CrossRef CAS.
- P. Beak and F. S. Fry Jr, The Equilibrium between 2-hydroxypyridine and 2-pyridone in the gas phase, J. Am. Chem. Soc., 1973, 95, 1700–1702 CrossRef CAS.
- J. DePasquale, I. Nieto, L. E. Reuther, C. J. Herbst-Gervasoni, J. J. Paul, V. Mochalin, M. Zeller, C. M. Thomas, A. W. Addison and E. T. Papish, Iridium Dihydroxybipyridine Complexes Show That Ligand Deprotonation Dramatically Speeds Rates of Catalytic Water Oxidation, Inorg. Chem., 2013, 52, 9175–9183 CrossRef CAS PubMed.
- O. E. Oladipupo, M. C. Prescott, E. R. Blevins, J. L. Gray, C. G. Cameron, F. Qu, N. A. Ward, A. L. Pierce, E. R. Collinson, J. F. Hall, S. Park, Y. Kim, S. A. McFarland, I. Fedin and E. T. Papish, Ruthenium Complexes with Protic Ligands: Influence of the Position of OH Groups and π Expansion on Luminescence and Photocytotoxicity, Int. J. Mater. Sci., 2023, 24, 5980 CAS.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, Appendix A: Typical Interatomic Distances in Organic Compounds and Organometallic Compounds and Coordination Complexes of the d- and f-block metals, Structure Correlation, 1994, 751–858 CAS.
- K. L. VanDenburgh, Y. Liu, T. Sadhukhan, C. R. Benson, N. M. Cox, S. Erbas-Cakmak, B. Qiao, X. Gao, M. Pink, K. Raghavachari and A. H. Flood, Multi-state amine sensing by electron transfers in a BODIPY probe, Org. Biomol. Chem., 2020, 18, 431–440 RSC.
- A. H. Ngo, M. Ibañez and L. H. Do, Catalytic Hydrogenation of Cytotoxic Aldehydes Using Nicotinamide Adenine Dinucleotide (NADH) in Cell Growth Media, ACS Catal., 2016, 6, 2637–2641 CrossRef CAS.
- A. H. Ngo and L. H. Do, Structure–Activity Relationship Study of Half-Sandwich Metal Complexes in Aqueous Transfer Hydrogenation Catalysis, Inorg. Chem. Front., 2020, 7, 583–591 RSC.
- Z. Almodares, S. J. Lucas, B. D. Crossley, A. M. Basri, C. M. Pask, A. J. Hebden, R. M. Phillips and P. C. McGowan, Rhodium, Iridium, and Ruthenium Half-Sandwich Picolinamide Complexes as Anticancer Agents, Inorg. Chem., 2014, 53, 727–736 CrossRef CAS PubMed.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Autonomous Discovery in the Chemical Sciences Part I: Progress, Angew. Chem., Int. Ed., 2020, 59, 22858–22893 CrossRef CAS PubMed.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Autonomous Discovery in the Chemical Sciences Part II: Outlook, Angew. Chem., Int. Ed., 2020, 59, 23414–23436 CrossRef CAS PubMed.
- J. Cui, S. Zang, H. Nie, T. Shen, S. Su, J. Jing and X. Zhang, An ICT-based fluorescent probe for ratiometric monitoring the fluctuations of peroxynitrite in mitochondria, Sens. Actuators, B, 2021, 328, 129069 CrossRef CAS.
- M. R. Schreier, X. Guo, B. Pfund, Y. Okamoto, T. R. Ward, C. Kerzig and O. S. Wenger, Water-Soluble Tris(cyclometalated) Iridium(III) Complexes for Aqueous Electron and Energy Transfer Photochemistry, Acc. Chem. Res., 2022, 55, 1290–1300 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, photophysical studies, and reaction data. CCDC 2336124 and 2336125. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00891j |
| This journal is © The Royal Society of Chemistry 2024 |