
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new biphenol-dipicolylamine based ligand and its dinuclear Zn2+ complex as fluorescent sensors for ibuprofen and ketoprofen in aqueous solution†
Daniele
Paderni‡
 a,
Eleonora
Macedi‡
a,
Gina Elena
Giacomazzo
b,
Mauro
Formica
a,
Luca
Giorgi
a,
Barbara
Valtancoli
b,
Patrizia
Rossi
c,
Paola
Paoli
c,
Luca
Conti
*b,
Vieri
Fusi
*a and
Claudia
Giorgi
b
a,
Eleonora
Macedi‡
a,
Gina Elena
Giacomazzo
b,
Mauro
Formica
a,
Luca
Giorgi
a,
Barbara
Valtancoli
b,
Patrizia
Rossi
c,
Paola
Paoli
c,
Luca
Conti
*b,
Vieri
Fusi
*a and
Claudia
Giorgi
b
aDepartment of Pure and Applied Sciences, University of Urbino, via Ca’ le Suore, 2-4, 61029 Urbino, Italy. E-mail: vieri.fusi@uniurb.it
bDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, FI, Italy. E-mail: luca.conti@unifi.it
cDepartment of Industrial Engineering, University of Florence, via S. Marta 3, 50139 Florence, Italy
First published on 15th May 2024
Abstract
In this work, the study of the new ligand 3,3′-bis[N,N-bis(pyridine-2-ylmethyl)aminomethyl]-2,2′-dihydroxybiphenyl (L) is reported, where a central 2,2′-biphenol (BPH) fluorophore was functionalized at 3,3′-positions with two dipicolylamine (DPA) side arms as receptor units. Following the synthesis and full chemical–physical characterization, the acid–base and Zn2+-coordination abilities of L were investigated through a combination of potentiometric, UV-Vis, fluorescence, NMR, XRD and DFT measurements. The optical properties of the ligand turned out to be strongly dependent on the pH, being straightforwardly associated with the protonation state of the BPH moiety, whereas its peculiar design allowed to form stable mono and dinuclear Zn2+ complexes. In the latter species, the presence of two Zn2+ ions coordinatively unsaturated and placed at close distance to each other, prompted us to test their usefulness as metallo-receptors for two environmental pollutants of great relevance, ibuprofen and ketoprofen. Potentiometric and fluorescence investigations evidenced that these important non-steroidal anti-inflammatory drugs (NSAIDs) are effectively coordinated by the metallo-receptors and, of relevance, both the stability and the fluorescence properties of the resulting ternary adducts are markedly affected by the different chemical architectures of the two substrates. This study aims at highlighting the promising perspectives arising from the use of polyamino phenolic ligands as chemosensors for H+/Zn2+ and other additional anionic targets in their metal-complexed forms.
Introduction
Emerging pollutants (EPs) are defined as any synthetic or naturally occurring chemical or any microorganism that is not commonly monitored in the environment but has the potential to enter the environment and cause known or suspected adverse ecological and/or human health effects. More than 700 EPs and their metabolites can be found in the European aquatic environment (https://www.norman-network.net). They can either be released from pollution sources, such as water treatment plants, or come from the atmosphere or plantations or breeding farms. Their identification can be due either to the availability of new detection methods, able to unveil the presence of long-term substances, or to the synthesis of new chemicals or changes in use of existing chemicals. Among the classes of EPs, pharmaceuticals, pesticides, disinfection by-products and wood preservation and industrial chemicals are the main ones.1Non-steroidal anti-inflammatory drugs (NSAIDs) are listed among the EPs.2–5 Indeed, the consumption of drugs by the population is continuously growing, and NSAIDs represent the most commonly prescribed classes of medications for pain and inflammation. They can enter the environment through industrial, municipal, pharmaceutical and hospital wastewater,6 which raises concern about the toxicity towards aquatic life and, finally, human health.2
Being EPs, NSAIDs are not routinely monitored, for this reason no easy-to-use analysis for their detection is available. Their determination instead relies on complex and expensive techniques such as high-performance liquid chromatography (HPLC) or gas-chromatography (GC). The use of fluorescent chemosensors could be an alternative and more advantageous strategy, in terms of sensitivity, response time and cost, for the detection, monitoring and sequestering of NSAIDs in solution.7–14
NSAIDs are weak carboxylic acids and can be easily deprotonated to give the anionic form in aqueous solution. The recognition of anions in solution is a challenging task, due to crucial properties such as multiple protonation equilibria, diverse geometries, high solvation energies and size of the anion. In water, anion recognition is an even more tough goal, due the hydration of both receptor and substrate, that hampers their mutual interaction.15–18 Both free ligands19–32 and metal complexes33–41 can be employed as receptors for anions. The use of free ligands implies the formation of non-covalent interactions, such as hydrogen bonds or π-stacking, whereas the metal centers of complexes can act as binding sites for the carboxylate moieties of anionic guests,42 also offering the receptor a structural organization (preorganization). Moreover, polynuclear metal complexes are an advantageous choice, in that they allow for the cooperation of multiple metal ions in the formation of the binding site, offering higher stability and selectivity to the formed adduct.13,33,34,38,43–50
In the present paper the synthesis of the new ligand 3,3′-bis[N,N-bis(pyridine-2-ylmethyl)aminomethyl]-2,2′-dihydroxybiphenyl (L, Scheme 1) is reported, featuring two dipicolylamine fragments (DPA) appended to the 3,3′-positions of a central 2,2′-biphenol (BPH) fluorophore. The polyamine sites gathered on the DPA units favour the solubility of L in aqueous medium and can be exploited, along with the involvement of the oxygen donors of the BPH unit, to form stable metal complexes in solution, that can act, in turn, as suitable metallo-receptors for anion binding.51,52 Moreover, the insertion of the BPH fluorophore confers on the ligand peculiar photochemical properties and the ability to signal via fluorescence emission the occurred interaction with the guest.33,38,45,53–56
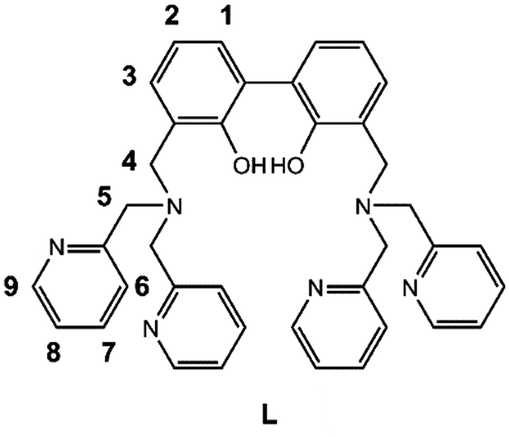 | ||
| Scheme 1 The ligand L together with labels for NMR resonances. | ||
Herein, besides the synthesis of the novel receptor, the study of its acid–base and binding properties towards Zn2+ in aqueous medium (water/acetonitrile 80/20, v/v) through potentiometry, UV-Vis absorption, fluorescence emission and NMR spectroscopies is presented. The solid-state structure of L was also analysed by single crystal X-ray diffraction. Lastly, the ability of the dinuclear Zn2+ complexes of L to behave as metallo-receptors for NSAIDs, in particular ibuprofen and ketoprofen, was investigated by potentiometry and fluorescence emission spectroscopy, revealing the ability of this system to detect the two guests in aqueous solution via an ON–OFF fluorescence mechanism.
Results and discussion
Synthesis and X-ray crystal structure of L
The synthetic pathway to obtain L is depicted in Scheme 2.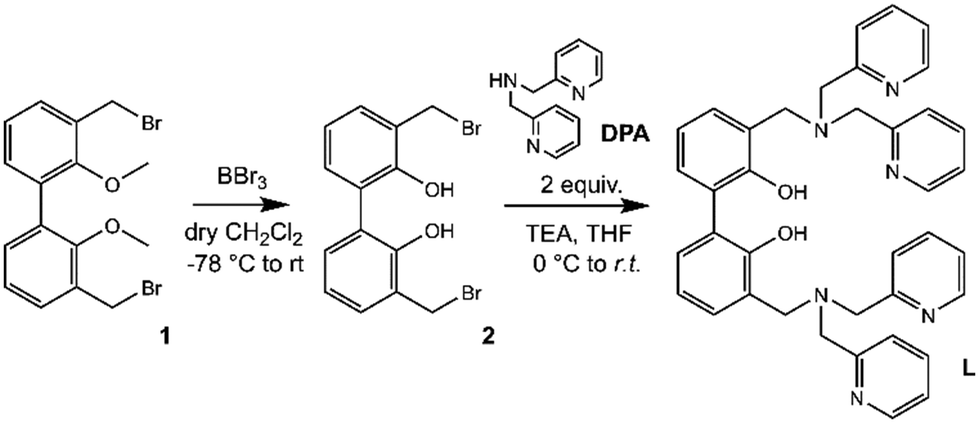 | ||
| Scheme 2 Synthetic procedure followed for the preparation of receptor L. | ||
Compound 1 (3,3′-dibromomethyl-2,2′-dimethoxybiphenyl) was synthesized as previously described,33,54–56 starting from 2,2′-biphenol, whose hydroxy functions were protected with methyl groups57 prior to activate the 3,3′ positions with butyllithium and add further methyl groups by using dimethyl sulphate in diethyl ether.58
Finally, the inserted methyl groups were brominated by using N-bromosuccinimide to give compound 1.59 Following the demethylation of phenolic oxygen atoms by using boron tribromide in dry DCM and, successively, H2O to quench the reaction (2, Fig. S1†), two equivalents of commercial di-(2-picolyl)amine (DPA) were reacted with the obtained compound 2 in dry THF in the presence of a base to give L (Fig. S2 and S3, ESI†).
Suitable crystals for single crystal X-ray diffraction analysis were obtained for the free ligand. As shown in Fig. 1, in the asymmetric unit of L two independent molecules (A and B in the following) of the ligand are present. Crystallographic data and refinement parameters of L are reported in Table S1.†
 | ||
| Fig. 1 ORTEP view (30% ellipsoid probability) of the asymmetric unit of L. For sake of clarity, only the most populated model of the disordered moiety (molecule labelled with “A”, see experimental details) and only the hydrogen bonded to oxygen atoms were shown. | ||
The two molecules are quite well superimposable (Fig. S4, ESI†), showing just a little difference in the disposition of the pyridine rings (Fig. S4 and Table S2, ESI†). More in particular, the 2,2′-biphenol fragment assumes the same conformation in A and B, being the inter-annular C2–C1–C7–C12 dihedral angle −63.0(2)°and −65.6(2)° in A and B, respectively. In addition, the O1⋯O2 distance is comparable in A and B (3.013(2) vs. 3.058(2) Å, in A and B, respectively).
As already observed in phenol derivatives bearing a DPA moiety in ortho position,60 each O–H group of the A and B molecules is involved in an intramolecular bifurcated hydrogen bond61 with the nitrogen atoms of the closest tertiary amine and pyridine (N1 and N2 for O1–H1o and N4 and N6 for O2–H2O, Table 1 and Fig. S5 and S6, ESI†). Finally, the two nitrogen atoms not involved in intermolecular H-bonds (N3 and N5) point outside the ligand. No significant intermolecular H-bonds contacts are present in the crystal packing.
| X–H⋯Y | X⋯Y (Å) | H⋯Y (Å) | X–H⋯Y (°) | |
|---|---|---|---|---|
| O1–H1o⋯N1 | A | 2.722(2) | 1.93(2) | 150(2) |
| B | 2.745(2) | 1.97(2) | 152(2) | |
| O1–H1o⋯N2 | A | 3.60(1)/3.33(2) | 2.92(2)/2.73(2) | 136(2)/126(2) |
| B | 3.187(2) | 2.56(2) | 147(2) | |
| O2–H2O⋯N4 | A | 2.844(2) | 2.14(2) | 140(2) |
| B | 2.777(2) | 2.03(2) | 147(2) | |
| O2–H2O⋯N6 | A | 3.092(2) | 2.38(2) | 141(2) |
| B | 3.187(2) | 2.56(2) | 132(2) |
Acid–base properties of L
Prior to evaluate the coordination ability of L towards Zn2+, its acid–base properties were investigated through a combination of potentiometric, UV-Vis and fluorescence titrations. Due to the scarce solubility of the ligand in pure water, measurements were carried out in mixed H2O/CH3CN 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 (v/v) solutions at 298 ± 0.1 K.
:20 (v/v) solutions at 298 ± 0.1 K.
The stepwise protonation constants (logK) of L obtained via potentiometric titrations are reported in Table 2, whereas the corresponding distribution diagram of the species present in solutions is reported in ESI (Fig. S7†).
K) of L potentiometrically determined in H2O/CH3CN 80:20 (v/v) NMe4Cl 0.1 M at 298 ± 0.1 K
As shown in Table 2, the neutral species L behaves as a tetraprotic base and as a monoprotic acid, with the ligand being present in solution in its monoanionic H−1L− form at alkaline pH values. Analogously to similar BPH-containing systems previously described54 and considering that the full deprotonation of the BPH unit typically takes place only in strong alkaline conditions (pKa > 14),62 one acid hydrogen atom of the BPH unit is retained in the overall pH range investigated by potentiometric measurements (2 ≤ pH ≤ 12).
The analysis of protonation constants indicates that H−1L− behaves as a rather strong base in the first protonation equilibrium, being associated with a logK of 10.47. This value closely resembles those reported in the literature for the protonation of phenolate groups (pKa = 10);62 in this case it must be taken into account that the presence of the tertiary amine functions close to the BPH fragment may allow for the stabilization of the BPH proton via H-bonding (as also evidenced by the single crystal X-ray diffraction analysis), increasing the basicity of the monoanionic species and, therefore, the pKa value.52 The involvement of a phenolate function in this first protonation step is thus strongly suggested, together with a probable sharing of this first proton between the BPH and the close tertiary amine function.
A net drop in basicity is then observed with further protons additions, as evidenced by a gap of approximately 5.7 log units moving from the first to the second protonation step. More likely, the second and third protonation steps involve the tertiary amines. Their constant values (4.72, 4.65) are very similar, due to their mutual distance that make the two sites behave independently from each other. Finally, the last protonation steps involve two pyridine units of two different DPA moieties. This behaviour agrees well with that of a similar ligand containing a 2,5-diphenyl[1,3,4]oxadiazole (PPD) fluorophore connected to two DPA units,63 where the low basicity of the tertiary amines is ascribed to the attached strong electron-withdrawing groups.
The role played by the DPA and BPH units in the acid–base behaviour of the ligand has been further investigated by UV-Vis and fluorescence spectroscopy. As shown in Fig. 2a, where the electronic absorption spectra of L as a function of pH are reported, the ligand displays at acidic pH an intense and broad absorption centered at ∼261 nm with a minor shoulder at 283 nm, whereas a new intense band, with a maximum at 313 nm, strongly appears in the spectra by increasing the pH above 8.5. Since the absorption of pyridines typically falls within the 240–280 nm range,64–66 and taking into account previous studies on the UV-Vis behaviour of the free BPH unit,62 this new band can be easily attributed to the deprotonation of a phenolic function of the central BPH moiety. In fact, the absorption properties of 2,2′-biphenyldiols are markedly affected by the protonation state of their hydroxyl functions, and their absorption bands corresponding to the neutral and monoanionic forms, respectively centered at around 278 and 307 nm, can be taken as diagnostic of the different BPH protonation degrees; the band of the dianionic form is not generally observed, unless in very strong alkaline conditions.
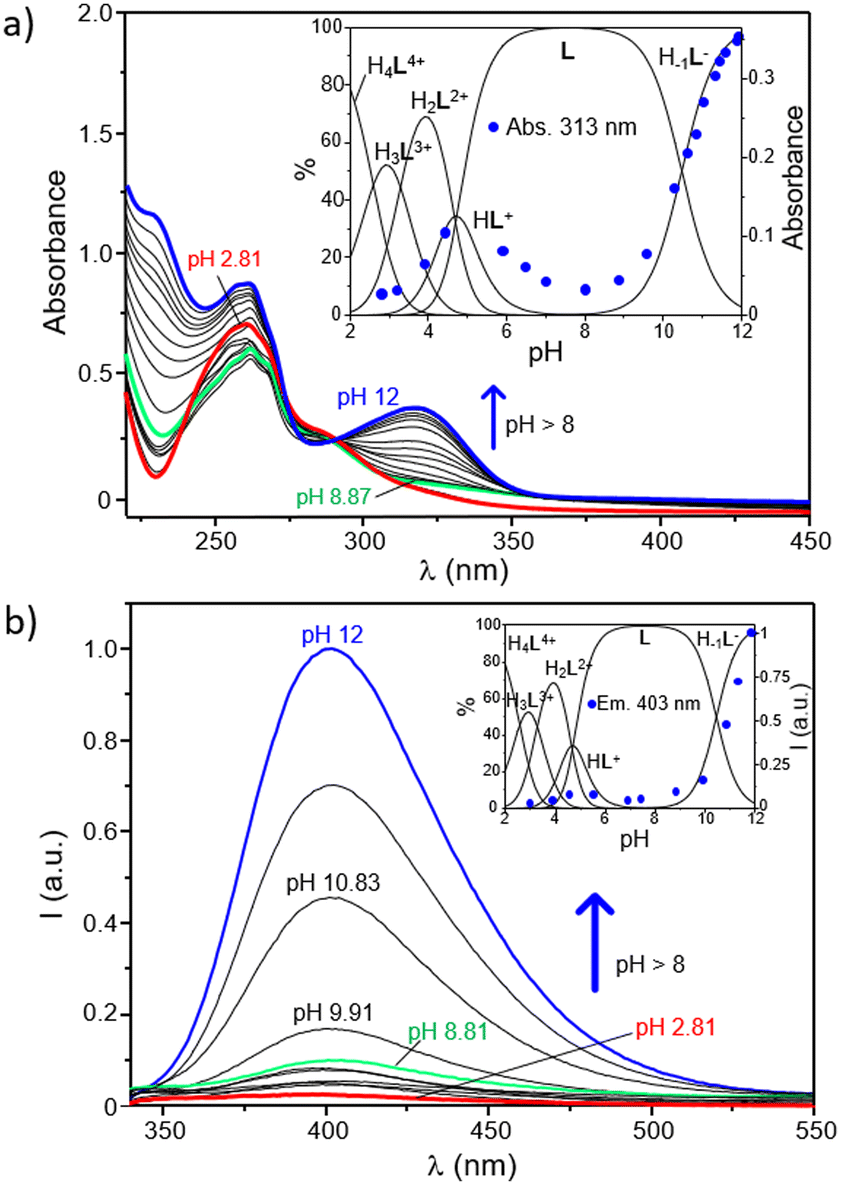 | ||
| Fig. 2 Absorption spectra (a) and fluorescence emission (b) of an aqueous solution of L at different pH values. In the insets are respectively shown the absorbance at 314 nm and the intensity of emission at 402 nm as a function of pH and overlapped with the distribution diagram of the protonated species present in solution. ([L] = 3 × 10−5 M, H2O/CH3CN 80:20 (v/v), λexc = 288 nm, λem = 403 nm). | ||
The pH-dependence of the absorption at 313 nm is better highlighted by the inset of Fig. 2a, where it is reported overlapped with the distribution diagram of the protonated species in solution. From this trend it is clear that such absorption is readily enhanced as soon as the H−1L− is formed in solution. This is in agreement with potentiometric data, confirming that the deprotonation of a phenol function of BPH takes place only at pH ≥ 8.5, along with the formation in solution of H−1L−. In other words, the monoanionic motif of BPH would be present only in marked alkaline conditions (pH ≥ 8.5), when H−1L− is present in solution, whereas both the acidic protons of the BPH moiety are retained in all the other species of the ligand. However, the absorption at 313 nm ascribed to the monoanionic form of BPH shows a little increase also along with the formation of the HL+ species (see inset of Fig. 2a), suggesting that in all protonation steps the proton distribution can be affected by the formation of different H-bonds depending on the species formed.
The analogous experiments conducted via fluorescence measurements are reported in Fig. 2b. As shown, the ligand displays a sharp OFF–ON switching of fluorescence emission, centered at around 403 nm, moving from acidic to alkaline conditions. This is denoted from the poor emission registered in a wide range of pH (up to ∼8.5) and the progressive enhancement observed at pH ≥8.5 that accompanies the formation of the H−1L− species (Fig. 2b, inset). This behaviour is probably ascribable to the changes in the protonation state of the BPH unit of L. In fact, just like the absorption properties, also the fluorescence emission of such fluorophore is highly influenced by its protonation degree.62,67,68 Generally, the formation of an intramolecular H-bond between the two oxygen functions of BPH in its monoanionic form induces a fluorescence emission gain, due to the increased co-planarity and rigidity of BPH; on the other hand, the possible formation of H-bonds between the oxygen functions of BPH in its neutral form and the close amine groups decreases the emission intensity, due to the loss of co-planarity and consequent nonradiative relaxation processes of the excited state.
Also in the present case, the most emissive species is indeed the monoanionic H−1L− species, which is characterized by an emission centered at 403 nm. Therefore, it is reasonable to assume that the low emission observed at 2 ≤ pH < 8.5 can be associated with the presence of the fully protonated BPH unit in L, whereas the burst of emission observed at pH ≥ 8.5 should be related to the deprotonation of BPH to give the monoanionic form in H−1L−, in well agreement with potentiometric and UV-Vis data. Once again, a little increase of the emission can be observed during the formation of the HL+ species (4 < pH < 6), suggesting either a partial deprotonation or the achievement of a partial coplanarity of BPH aromatic rings.
On the other side, a potential contribution deriving from the different protonation states of pyridines of DPA units should be also considered, as their protonation degree could efficiently quench the emission of close aromatic fluorophores.69–71 However, in the present case the restoring of the ligand emission is generated in a range of pH between 8.5 and 12, in which, according to potentiometric data, no changes in the protonation states of pyridines are involved. This would confirm a predominant role played by the BPH fluorophore in the emission mechanism. Accordingly, and in agreement with potentiometric data, a tentative distribution of the acidic hydrogen atoms in the different protonated species of the ligand was provided in Fig. S8 (ESI†).
Coordination of Zn2+
The stability constants for the complexation of L with Zn2+ potentiometrically determined are reported in Table 3, whereas the distribution diagrams of the complexed species present in solution for the L/Zn2+ and L/2Zn2+ systems are reported in Fig. S9 (ESI†). As shown, the peculiar topology of L imparts to the ligand the capability to form stable mono and dinuclear complexes with Zn2+, with the latter being the only ones present in solution in the L/2Zn2+ system, whereas they coexist with the mononuclear species when the ligand to metal molar ratio is equal to 1:1.
:20 (v/v) NMe4Cl 0.1 M at 298 ± 0.1 K
| Reaction | LogKa |
|---|---|
|
a Values in parenthesis are standard deviations on the last significant figures.
b Logβ values.
|
|
| H−1L− + Zn2+ = Zn(H−1L)+ | 13.30 (9) |
| Zn(H−1L)+ + Zn2+ = Zn2(H−1L)3+ | 8.18 (8) |
| H−1L− + 2Zn2+ = Zn2(H−1L)3+ | 21.48 (9)b |
| Zn2(H−1L)3+ = Zn2(H−2L)2+ + H+ | −7.25 (9) |
| H−1L− + 2Zn2+ = Zn2(H−2L)2+ + H+ | 14.24 (9)b |
| Zn2(H−2L)2+ + OH− = Zn2(H−2L)(OH)+ | 3.50 (9) |
| Zn2(H−2L)(OH)+ + OH− = Zn2(H−2L)(OH)2 | 3.39 (9) |
The fact that the logK value for the addition of Zn2+ to the H−1L− species (logK = 13.30) is only slightly lower compared to the corresponding one previously found for a BPH-containing analogue featuring two tri-amine dien side arms in place of DPA units (logK = 14.79, 0.15 M NaCl),54 leads us to hypothesize a similar coordination environment within the two ligands, namely with the metal ion being mainly stabilized by the nitrogen atoms of a DPA unit plus one or both oxygen atoms of BPH.
Interestingly, contrary to what previously found for the open chain dien analogue54 and other BPH-containing polyamine macrocyclic receptors,33 no evidence for the binding of Zn2+ by the neutral form L of the ligand was found in this case: this, which can be ascribed to the lower coordination properties of the DPA with respect to the aliphatic diethylentriamine unit, further supports the necessary involvement of the BPH moiety in the Zn2+-coordination (vide infra, Fig. 3d).
 | ||
| Fig. 3 Absorption (a) and fluorescence emission spectra (b) of an aqueous solution of L in the absence and presence of increasing Zn2+ concentrations at pH 9. In the inset of (a) are reported the absorptions at 313 and 283 nm (respectively blue and grey diamonds), together with the variation of the absorption wavelength (red dash-dotted line) as a function of the different [Zn2+]/[L] molar ratios ([L] = 3 × 10−5 M (a and b), H2O/CH3CN 80:20 (v/v), λexc = 288 nm). (c) 1H NMR spectra of L (bottom) and Zn2(H−2L)2+ complex (top) in DMSO-d6 along with signal assignments; for the NMR labelling see Scheme 1. (d) Proposed modes of coordination for the Zn(H−1L)+ and Zn2(H−2L)3+ complexes. | ||
The presence of a second equivalent of the metal gives rise to the formation of dinuclear species, prevailing in solution even from strong acidic conditions (pH > 2) (Fig. S9b, ESI†). In a wide range of pH, from 2 to values around neutrality, the predominant species turns out to be Zn2(H−1L)3+, for which the addition of the second Zn2+ to Zn(H−1L)+ occurs with a lower constant (logK = 8.18) compared to the addition of the metal to H−1L− (logK = 13.30), as naturally expected due to the electrostatic repulsion between the two metal ions. It can be also noted that this complex represents the predominant species in acidic conditions (pH ≤ 6) even in the L/Zn2+ system with 1 to 1 molar ratio (Fig. S9a, ESI†), unveiling a high stability of both metal ions in this species. This suggests a similar coordination pattern for both Zn2+ ions in which one DPA unit and one oxygen of BPH coordinate one metal ion. The absence of any proton addition supports the involvement of all protonable sites in the metal coordination.
Around neutrality the most abundant species is the Zn2(H−2L)2+ one, which prevails in a wide range of pH, from ca. 7 to ca. 10.5 (Fig. S9b, ESI†). The formation of hydroxyl species in more alkaline conditions unveils the capability of the dinuclear complexes to bind additional external groups, which likely contribute to saturate the coordination requirements of the two Zn2+ ions. More in detail, the addition of OH− to Zn2(H−2L)2+ and Zn2(H−2L)(OH)+ occurs with similar constant values (3.50 and 3.39 log units, respectively), thus hinting that, contrary to the free ligand, metal coordination can promote the full deprotonation of BPH in the Zn2(H−2L)2+ species. Interestingly enough, no evidence for the formation of hydroxylate species was found in case of the mononuclear Zn(H−1L)+ complex, leading us to speculate that in this species the coordination requirements of Zn2+ are fully saturated (vide infra).
Given the central role of the dinuclear complexes as metallo-receptors for additional anionic guests, and to get more insights into the stoichiometry of these species, which clearly depends on the degree of protonation of the BPH and DPA groups, a combination of UV-Vis, fluorescence and NMR studies was carried out.
In Fig. 3a and b are respectively reported the UV-Vis and fluorescence spectra of aqueous solutions of the ligand registered at fixed pH in the absence and presence of increasing concentrations of Zn2+. Measurements were performed at pH 9 to allow for the formation of the neutral form L and then monitor the switch first to the mononuclear Zn(H−1L)+ and then to the dinuclear Zn2(H−2L)2+ species with increasing [Zn2+]/[L] molar ratios.
As shown in Fig. 3a, in the absence of Zn2+ the ligand displays its characteristic uncoordinated absorption profile, with an intense absorption at 261 nm and a shoulder at ca. 283 nm. Then, by increasing the Zn2+ concentration, this latter band undergoes a progressive decrease along with the simultaneous appearance of a new red-shifted absorption, centered at around 313 nm. This trend is better evidenced by the inset of Fig. 3a, where the variation of the absorbance at 313 nm (blue diamonds) as a function of the different [Zn2+]/[L] molar ratios, remarks a net change which goes along with the formation in solution of the 1:1 species, followed by a less pronounced effect accompanying the transition to the 2:1 one. The relative maximum of absorption (λmax) is influenced by metal coordination, too, as it moves from 283 nm, in the absence of Zn2+, to 304 and 313 nm, respectively, for [Zn2+]/[L] molar ratios of 1:1 and 2:1 (dashed red line, inset of Fig. 3a). No significant variations were instead observed for [Zn2+]/[L] ≥ 2, confirming the formation of the dinuclear complexes for the higher metal-to-ligand molar ratios tested.
As the absorption at 313 nm is ascribable to the phenolate group, these data would confirm that the coordination of Zn2+ proceeds along with the deprotonation of the central BPH unit. This was also indicated by the sharp increase in intensity of this band accompanying the formation in solution of the Zn2(H−1L)3+ species with increasing the pH of a L/Zn2+ system in 1:2 molar ratio (Fig. S10, ESI†). In addition, the fact that the greatest variations in terms of both changes in intensity and maximum of absorbance are observed in correspondence of the formation of the 1:1 species suggests a somewhat mediated situation in the mononuclear Zn(H−1L)+ form. It is indeed reasonable to assume that both the phenolate and phenolic groups of the monoanionic BPH unit are simultaneously involved in the coordination to the metal, justifying the net changes underwent by absorption profiles in correspondence of the formation of this species. Further stabilization via OH⋯NH-bonds belonging to the phenol group and the closest nitrogen atom of a DPA unit can be also envisaged (Fig. 3d, left). Moreover, such a disposition would allow to completely satisfy the coordination requirements of Zn2+, and it is therefore supported by the lack of potentiometric evidence for the formation of hydroxylate species of Zn(H−1L)+, as well as of any proton addition to the mononuclear species.
On the other side, the absorbance changes observed for [Zn2+]/[L] ratios ≥1 would hint at a coordination rearrangement promoted by further metal addition, resulting in the two cationic guests being each one coordinated by a phenolate group of a fully deprotonated BPH moiety (Fig. 3d, right), in agreement with the crystal structure already described of the Zn2+-dinuclear complex of the dien-analogue.54 These proposed modes of coordination, which are sketched in Fig. 3d, were further supported by fluorescence, NMR and DTF studies (vide infra).
Interestingly, by looking at the analogous fluorescence experiment shown in Fig. 3b, it is clear that, contrary to what previously observed for other BPH-polyamine-Zn2+ complexes45,54 in this case, the metal coordination promotes a sharp quenching of the ligand emission, initially centered at ca. 403 nm. The strong CHEQ (chelating enhancement of the quenching) effect observed in our case seems to be ascribable to the presence in L of two DPA side arms instead of the open-chain or macrocyclic polyamine fragments of previously studied compounds and can be assumed to be dependent on the presence of both the BPH unit and the DPA side arms. On one side, the Zn2+-coordination promotes the deprotonation of phenolic functions of BPH providing a potential OFF–ON fluorescence switch. However, on the other side the coordination of DPA-pyridines to Zn2+ would play an opposite ON–OFF fluorescence switch, as expected considering the known capability of pyridines to efficiently quench the emission of nearby aromatic fluorophores in their complexed forms.69,72,73 Coordination of pyridine moieties, indeed, increases their oxidizing potential, making the LUMO accessible to a oxidative PET process from the excited BPH fluorophore to the electron-poor coordinated pyridines (Scheme 3).
 | ||
| Scheme 3 Schematic representation of the oxidative PET process suggested upon Zn2+ coordination of the pyridine moieties of the DPA unit of L. | ||
Therefore, in this view the sharp CHEQ effect observed for the Zn2+-coordination could be explained as the result of a negative balance between these opposite mechanisms.
From Fig. 3b it can be also noted that a marked quenching of the fluorescence emission at 403 nm is achieved yet in the presence of one Zn2+ ion; similar to the observed variations in the UV-Vis spectra, this would further support the hypothesis that both the oxygen donors of BPH are involved in the coordination to the metal in the Zn(H−1L)+ species, as sketched in Fig. 3d (left).
Moreover, a closer look at the fluorescence spectra of dinuclear complexes (Fig. 3b) unveils significant differences between the maximum of emission of their spectra and those of mononuclear species at pH 9, as testified by the blue-shift of 2597 cm−1 displayed by the emission wavelength of Zn2(H−2L)2+ (364 nm) compared to that of Zn(H−1L)+ (403 nm). Such differences, which can be better appreciated in the study of ternary systems (Fig. 7, vide infra), would account for the different protonation degrees adopted by BPH in these species.
Lastly, the dinuclear complex Zn2(H−2L)2+ was further analysed through NMR spectroscopy. The complex was obtained as described in the Experimental section and measurements were performed in DMSO-d6 to allow for the complete dissolution of the complex at the higher concentrations needed for NMR experiments.
In Fig. 3c are reported the 1H NMR proton spectra of the free ligand and Zn2(H−2L)2+ complex. As shown, in both spectra the number of signals (9) suggests a C2v symmetry on the NMR time-scale, as also confirmed by the 13 resonances observed in the 13C NMR spectra. The symmetry of the molecule is then maintained also upon the coordination of the metal ions. Upon addition of Zn2+, the aromatic resonances belonging to the phenol unit (H1, H2, H3) shifted up-field, while those of the pyridine rings (H6, H7, H8, H9) moved down-field. The two aliphatic resonances showed a different behaviour: H4 shifted up-field whereas H5 moved down-field and split into two signals, that partially overlap in DMSO-d6. All these observations would confirm that the whole molecule is involved in the coordination of the two Zn2+ ions, justifying the de-shielding of the pyridine rings and shielding of the phenol fragments. This last result is ascribable to the deprotonation of the phenol functions upon the coordination of metal ions, in agreement with what discussed above (see Fig. 3d, right).
The deprotonation of the –OH group is also confirmed by the fact that a signal (broad singlet) at 10.95 ppm is present in the spectrum of the free ligand while it is absent in that of the complex (data not shown). The splitting of H5 in an AB system also suggests the stiffening of the structure upon Zn2+ binding.
DFT calculations
A theoretical study at the DFT level has been performed in the present study to gain more insight into the structural features of the Zn2+-complexes. To this aim, the B3LYP functional74–77 along with the TZV (for C, H, O, N)78,79 and SDD80 (for Zn) basis sets were used in Gaussian16.81The neutral ligand obtained by X-ray diffraction analysis (vide supra) has been optimized (Fig. 4, left; the non-disordered independent molecule was chosen to this purpose) and its geometry compared with the obtained structure, revealing a very good superimposition (RMSD: 0.1863 Å). The deprotonated species H−1L− was also calculated (Fig. 4, right), revealing, as expected, the ability to form an intra-BPH hydrogen bond (see Table S4†), that necessarily increases the coplanarity of the BPH system (34.29° vs. 62.68° between the two mean planes containing the aromatic rings of BPH, see Table S4, ESI†). This possibly confirms and explains the higher emission of the H−1L− species compared to the neutral L.
 | ||
| Fig. 4 Molecular drawings and atom labelling schemes for neutral L and H−1L− species at the DFT-optimized geometries. Non-H,C atoms are depicted in ball and stick style. H-bonds are depicted as blue dotted lines (for L only the strongest ones were shown, see “Synthesis and X-ray crystal structure of L” paragraph). | ||
The optimized geometries of the mono- and dinuclear complexes (Fig. 5a, b, c–f, and Tables S3, S4, ESI†) are in good agreement with the structural hypotheses drawn by potentiometric and spectroscopic measurements. In mononuclear cases, a N3O2 penta-coordinated (square pyramid (sp) for Zn(H−1L)+(1) (τ82 = 0.2), between sp and trigonal bipyramid (tbp) for Zn(H−1L)+(2) (τ = 0.49)) environment around Zn2+ is observed, contrarily to the N3O tetracoordinated (trigonal pyramid (tp) for Zn2(H−2L)2+ (τ483 = 0.81), tp for Zn2(H−1L)3+(1) (τ4 = 0.81/0.82), seesaw for Zn2(H−1L)3+(2) (τ4 = 0.48)) environment found in dinuclear cases.
 | ||
| Fig. 5 (a and b) Molecular drawings and atom labelling schemes for the different species (1, 2) of the mononuclear complex Zn(H−1L)+ and (c–f) of the dinuclear complex Zn2(H−2L)2+ (c and d: two different views are shown of Zn2(H−2L)2+) at the DFT-optimized geometries. The different species (1, 2) of the monoprotonated dinuclear complex Zn2(H−1L)3+ are shown in (e and f) Non-H,C atoms are depicted in ball and stick style. H-bonds are depicted as blue dotted lines. | ||
In the mononuclear complex Zn(H−1L)+(1), featuring a mono-deprotonated BPH fragment, only one DPA moiety is involved in the coordination of the zinc cation, together with both BPH oxygen atoms, that converge towards the cation. In addition, a H-bond between the –OH group and the closest pyridine of the uncoordinated DPA unit is observed (Fig. 5a). An additional optimization on this complex was performed in order to confirm that the two BPH-oxygen atoms are able to chelate the metal cation. The resulting optimization led to a similar Zn(H−1L)+(2) complex (Fig. 5b), where a 4 + 1 coordination around Zn2+ can be described, due to a Zn1–N1 bond distance (2.352 Å, see Table S3, ESI†) a little bit longer than those found in the Cambridge Structural Database (CSD)84 for tetra- and penta-coordinated Zn2+ complexes. Moreover, the Zn(H−1L)+(2) species differs for the intramolecular H-bond interaction, indeed the tertiary amine function instead of the pyridine nitrogen atom of the uncoordinated DPA moiety is involved. However, the difference in the Gibbs free energy (ΔG) of the two species was calculated to be negligible (2.5 kcal mol−1), suggesting that in solution both species of Zn(H−1L)+ could exist. Anyway, in both cases the zinc cation is shared between the two oxygen atoms of the BPH moiety.
In the dinuclear complex Zn2(H−2L)2+, the fully deprotonated BPH almost flipped over, with the two –O− phenolate functions lying on opposite sides (Fig. 5c and d). This gives rise to a butterfly-like structure where each DPA unit along with the closest BPH-oxygen atom coordinate a Zn2+ cation in a tetracoordinated N3O environment.
The protonated complex Zn2(H−1L)3+(1) was also optimized, and it well superimposes to Zn2(H−2L)2+ (Fig. 5e). An additional optimization was performed on the protonated complex to assess its ability to form an intra-BPH hydrogen bond. The results confirmed the possible formation of such interaction, producing the Zn2(H−1L)3+(2) species (Fig. 5f) where, however, the Zn2–O2 bond distance (2.651 Å) is significantly longer than those found in the CSD database. Anyway, such Zn2(H−1L)3+(2) complex features the same Gibbs free energy (ΔG = of 0.9 kcal mol−1) of the butterfly-like structure (Zn2(H−1L)3+(1)). Therefore, also in this case, it could tentatively be inferred that in solution both butterfly-like (1) and the H-bonded (2) species of Zn2(H−1L)3+ could exist.
Ketoprofen and ibuprofen binding by the Zn2+-dinuclear complexes of L
In the dinuclear complexes formed by L, the tendency to easily form hydroxylate species unveils the presence of unsaturated Zn2+ ions that are prone to add further anionic guests, such as hydroxyl ions as well as external anionic substrates. Moreover, in these species two cations are nicely accommodated at a short distance between each other, thus providing two distinct anchoring sites that can cooperate in the binding of a given anionic guest.Altogether, these considerations prompted us to study the abilities of the dinuclear Zn2+ complexes of L, hereinafter referred to as R systems, to act as metallo-receptors towards the two environmentally relevant NSAIDs ibuprofen (HIbu) and ketoprofen (HKT). Of note, while the formation of classical “binary” L–A systems (L = fluorescent receptor, A = HIbu or HKT) has been only recently considered as a potential approach for the fluorescence detection of such important analytes,14,85,86 their binding/recognition by optimally designed metallo-receptors (R) in so-called “ternary” R-A systems has been much less explored.13
Similar to protonation and Zn2+ coordination studies, the capacity of R to effectively bind the two emerging pollutants was investigated in aqueous solution through potentiometric titrations in H2O/CH3CN 80:20 (v/v) NMe4Cl 0.1 M at 298 ± 0.1 K, whereas fluorescence measurements were carried out to test the capability of metallo-receptors to detect their presence via fluorescence signalling.
Preliminary to the study of the ternary R-A systems, the acid–base behaviour of HIbu and HKT was potentiometrically investigated, which resulted in pKa values respectively of 5.28 and 4.80 (Table 4). These values closely resemble those already reported for these drugs in similar mixed acetonitrile/water media,87,88 but it should be pointed out that they are significantly greater compared to those derived in pure aqueous solutions;89 this is in line with a generally observed trend that accounts for the increased amount of the unionized form of the drugs with increasing contents of the organic counterpart.42,90,91
K values) potentiometrically determined in H2O/CH3CN 80:20 (v/v) NMe4Cl 0.1 M at 298 ± 0.1 K
| Reaction | LogKa |
|---|---|
| a Values in parenthesis are standard deviations on the last significant figures. | |
| Ibu− + H+ = HIbu | 5.28 (3) |
| KT− + H+ = HKT | 4.80 (4) |
| Zn2(H−1L)3+ + Ibu− = [Zn2(H−1L)(Ibu)]2+ | 5.92 (7) |
| Zn2(H−2L)2+ + Ibu− = [Zn2(H−2L)(Ibu)]+ | 3.66 (8) |
| Zn2(H−1L)3+ + KT− = [Zn2(H−1L)(KT)]2+ | 6.72 (7) |
| Zn2(H−2L)2+ + KT− = [Zn2(H−2L)(KT)]+ | 4.29 (9) |
The study of the interaction between the metallo-receptors R and HIbu or HKT through potentiometric technique was accomplished by employing R:A molar ratios varying from 0.2:1 to 2:1, to ascertain the stoichiometry of all the adducts formed in solution. The resulting stability constants are summarized in Table 4, while the corresponding distribution diagrams of the species present in solution are reported in Fig. 6.
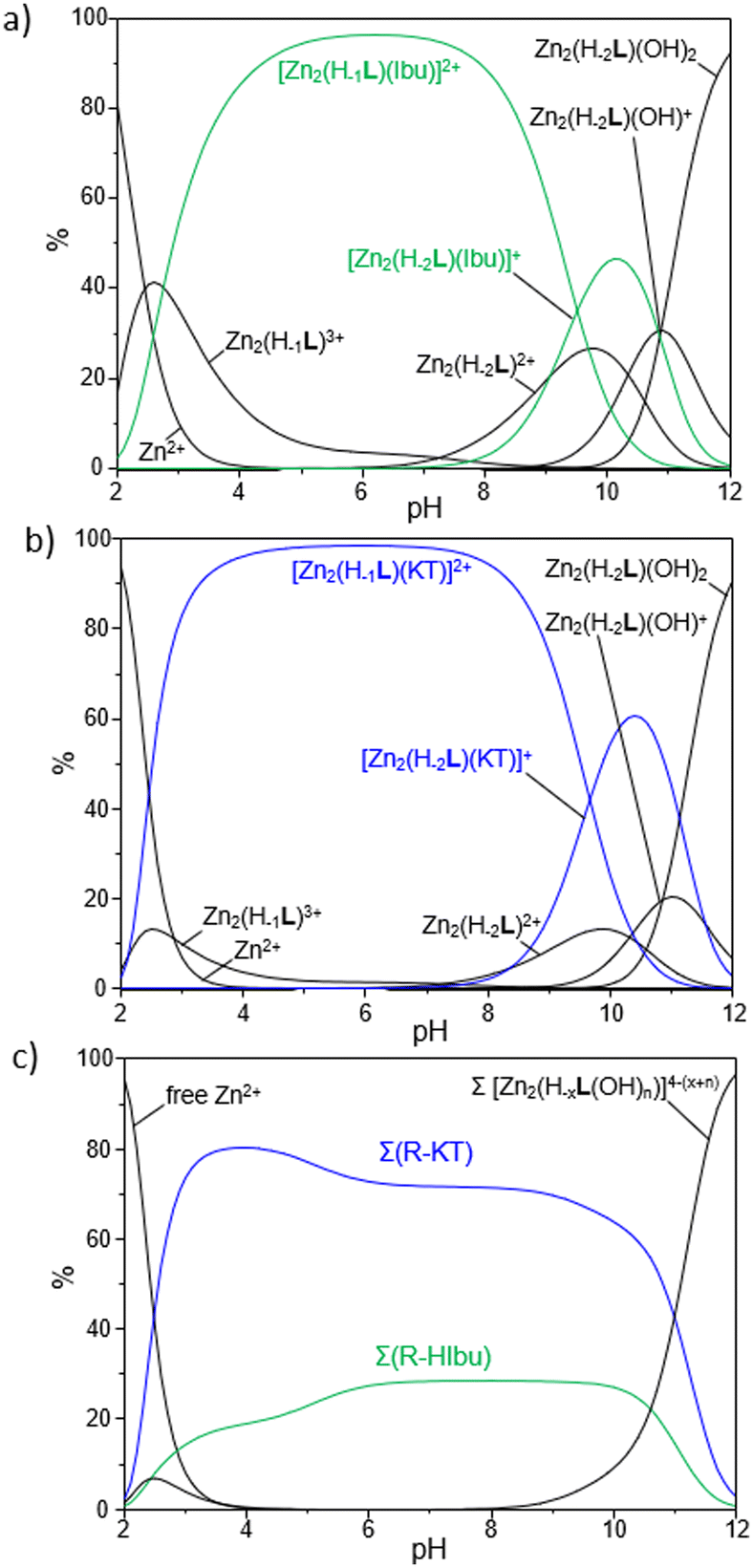 | ||
| Fig. 6 Binding of NSAIDS by the metallo-receptor R as evaluated through potentiometric technique: distribution diagrams of the species formed in solution for a system R/A in 1:1 molar ratio with A = HIbu (a) and HKT (b). In (c) is reported a selectivity diagram showing the percentage of the two anionic guests bound to the metallo-receptor as a function of pH. ([R] = [A] = 1 × 10−3 M, H2O/CH3CN 80:20 (v/v) NMe4Cl 0.1 M at 298 ± 0.1 K). | ||
First, it should be noted that only R-A adducts with 1:1 stoichiometry were found in our experimental conditions, thus ruling out the possibility to have multiple anionic guests coordinated to a single R unit but rather hinting at the simultaneous binding of a given substrate by the two Zn2+-based anchoring sites, likely via a bridge disposition of the carboxylate functions of NSAIDs (vide infra).
The analysis of equilibrium constants indicates that R forms stable adducts with both HIbu and HKT, with logK values for the addition of Ibu− or KT− to the different protonated forms of R (Zn2(H−1L)3+ and Zn2(H−2L)2+) ranging from 3.66 to 6.72. These values turn out to be comparable, or up to 2.9 log units higher, relative to those reported in the literature for the coordination of monocarboxylic acids (benzoate) by Zn2+-based polyamine receptors;92,93 no potentiometric data are instead available in the literature for the HIbu or HKT binding by such a class of metallo-receptors.
Interestingly, among the two NSAIDs, ketoprofen forms the most stable adducts with R, as highlighted, for instance, by a logK value of 4.29 for the addition of KT− to Zn2(H−2L)2+; the corresponding equilibria for the addition of Ibu− to the same metallo-receptor results in a logK of 3.66. From data reported in Table 4 it is also possible to highlight a common trend, by which the binding of Ibu− or KT− to R is markedly affected by the charges gathered on the interacting species. This can be appreciated by the increase of 2.43 log units for the addition of KT− passing from Zn2(H−2L)2+ to Zn2(H−1L)3+; a similar effect (2.26 log units) was also given by ibuprofen.
As reported in Fig. 6a and b, which show the distribution diagrams of the species present in solution for the ternary R-A systems, the adducts formed by R with ibuprofen (green curves in Fig. 6a) and ketoprofen (blue curves in Fig. 6b) are easily formed in solution, with the [Zn2(H−1L)A]2+ species (A = Ibu−, KT−) being the predominant ones in a wide range of pH, from pH 3 to ca. pH 9. The [Zn2(H−2L)A]+ species are instead formed in more alkaline conditions and prevail in a narrower range of pH (9 < pH < 11). As a general observation, the formation of ternary adducts in this range of pH (3 < pH < 11) is favoured by the simultaneous presence in solution of highly charged forms of both the metallo-receptor and the anionic substrates; the protonation of drugs to give their neutral forms or the formation of hydroxyl species of R, respectively in more acidic and basic conditions, would indeed disfavour the formation of such adducts.
The higher stability of adducts formed by Zn2(H−1L)3+ and Zn2(H−2L)2+ with ketoprofen compared to ibuprofen represents an interesting finding, which could be tentatively rationalized on the basis of the different structural characteristics of the two NSAIDs. Indeed, beyond the central role played by the electrostatic interactions between the two cationic anchoring sites of R and the negatively charged carboxylate function, common to both substrates, it is reasonable to assume that a further contribution to the stability of adducts may arise from π-stacking interactions between the aromatic moieties of NSAIDs and those of R, with both the central BPH and DPA-based groups potentially involved. In this view, the presence of the more extended 3-benzoylphenyl group in KT− would impart a greater π-stacking stabilization compared to the phenyl group in Ibu−, thus justifying the higher equilibrium constants found for the ternary adducts with ketoprofen.
The higher affinity of R towards ketoprofen can be even better appreciated by the selectivity diagrams for the systems R-HIbu-HKT (Fig. 6c), obtained by calculating the relative distribution diagrams and plotting the overall percentages of R-A adducts formed in solution as a function of pH. These diagrams clearly highlight the preferential binding of ketoprofen in a wide range of pH, from pH ∼ 3 to 11.
Lastly, the chance to detect a fluorescence variation of R as a result of the coordination to the two NSAIDs was evaluated. To this end, aqueous solutions of R were added with increasing amounts of anionic substrates (Ibu− and KT−) and the resulting spectra were collected. Due to the scarce emission of R at neutral pH, measurements were performed at more alkaline conditions (pH 10), where the residual emission of the uncoordinated R system allowed to follow the variations on the emission properties induced by the presence of targeted anions. As shown in Fig. 7a and as previously mentioned, the emission of dinuclear complexes is significantly blue-shifted if compared to the free ligand, being characterized by a maximum centered at ca. 364 nm plus a broad shoulder at lower frequencies. No shifts in the wavelength of the maximum of emission were observed upon increasing the concentration of both the anions, which instead led to a strong decrease of the intensity of emission, at least in the case of ketoprofen. In particular, a strong switch ON–OFF effect, as high as the 65% of the starting R emission, was induced by the presence of a relatively small amount, 5 equivalents, of drug; the same concentration of ibuprofen had instead only a small influence on the receptor emission (Fig. 7a, inset). Therefore, the coordinative selectivity pointed out by potentiometric data, which indicated ketoprofen as the substrate forming the most stable adducts with R, was also paralleled by the same optical trend.
 | ||
| Fig. 7 (a) Fluorescence emission of the metallo-receptor R in the absence and presence of increasing amounts of ketoprofen at pH 10; in the inset are shown the variation of the intensity of emission at 364 nm as a function of the different concentrations of ketoprofen and ibuprofen ([R] = 1 × 10−4 M, H2O/CH3CN 80:20 (v/v)). (b) Model proposed for the coordination of ketoprofen and ibuprofen by the metallo-receptor [Zn2(H−2L)]2+, charges are omitted for clarity. Green and blue colours are respectively used to indicate ibuprofen and ketoprofen. | ||
This would indicate that, beyond the negatively charged carboxylate groups, that likely drive the binding via electrostatic interaction with the two Zn2+ centers of R, the different aromatic extensions gathered on the two NSAIDs play a role in stabilizing the resulting ternary adducts as well as in modulating their fluorescence properties. It is reasonable to envisage that the greater aromatic portion of HKT imparted by the 3-benzoylphenyl moiety, would not only reinforce its interaction with the metallo receptor, but also affects more its emission through a higher π–π stabilization if compared to ibuprofen, bearing the less extended phenyl group.
To sum up, taken together potentiometric and fluorescence data hint at a coordination mode in the R-A adducts where the two Zn2+ ions of the metallo-receptor can cooperate in the binding of NSAIDs, which are in turn likely coordinated to the two metal centers through their carboxylate functions, in a bridge disposition with a Zn–O–C–O–Zn arrangement (Fig. 7b).
Further contributions may also arise by π-stacking interactions between their aromatic functions and those of the metallo-receptor, with the differences among the chemical architectures of the two substrates playing a central role in ruling both stability and emissive properties of the resulting ternary adducts. Such a proposed mode of coordination, schematically sketched in Fig. 7b, would be in line with what previously suggested for the carboxylate (but also phosphate) binding by analogous dinuclear Zn2+ polyamine complexes94–97 and with the crystal structure reported by Fenton and coworkers for the acetate complex of their alkoxide-bridged dinuclear Zn2+ receptor.98
Conclusions
Herein we report on the synthesis, chemical–physical characterization and binding/recognition properties of the dinuclear Zn2+-complexes towards ibuprofen and ketoprofen of the new biphenol-dipicolylamine based ligand 3,3′-bis[N,N-bis(pyridine-2-ylmethyl)aminomethyl]-2,2′-dihydroxybiphenyl (L). Following the synthesis, the novel ligand was fully characterized through NMR, UV-Vis, CHN, DFT and X-Ray diffraction analyses.The acid–base behaviour of L in aqueous solution was investigated through a combination of potentiometric, UV-Vis and fluorescence techniques. These indicated that L behaves as a monoprotic acid and a tetraprotic base in the 2–12 pH range investigated and that its optical properties are strictly related to the protonation state of its central BPH fluorophore, with the H−1L− species being the most emissive one. Thanks to its peculiar topology L easily hosts Zn2+ ions in aqueous solution, leading to the formation of stable mono- and dinuclear complexes in a wide range of pH. In this respect, the combination of potentiometric, UV-Vis, fluorescence, NMR and DFT studies permitted to gain important information on the coordination modes adopted by the mono- and dinuclear complexed species formed by the ligand. In particular, these data suggested that both the phenolate and phenolic groups of the monoanionic BPH unit are simultaneously involved in the coordination to the metal in the mononuclear Zn(H−1L)+ form, with the Zn2+ ion displaying a N3O2 pentacoordinated environment. On the contrary, the two cationic guests in the dinuclear complexed species are likely coordinated by the fully deprotonated BPH unit in a coordinatively unsaturated N3O tetracoordinated environment. This latter feature, along with the close distance between the two metals in the receptor cavity, makes such systems (herein referred to as R systems) attractive as metallo-receptors for external anionic guests. For that reason, their coordination and sensing abilities towards two emerging pollutants belonging to the NSAIDs family, ibuprofen and ketoprofen, were investigated.
Potentiometric measurements revealed that R strongly bind both these substrates, leading to the formation of stable ternary adducts in a wide range of pH. Of worth noting, all of these species feature a 1:1 stoichiometry, suggesting the coordination of analytes, likely through their carboxylate functions, in a bridge disposition between the two metal centers, with a Zn–O–C–O–Zn arrangement. Comparative studies pointed out a different binding selectivity among the two NSAIDs, with ketoprofen leading to the formation of the most stable adducts with R. Such coordinative selectivity was also paralleled by the same optical trend, as witnessed by the stronger quenching of fluorescence emission induced by the coordination of R to ketoprofen, if compared to ibuprofen. This suggests that, beyond the electrostatic interactions driven by their carboxylate groups, the different aromatic extensions gathered on the two NSAIDs play a role, too, in governing the stability of the resulting ternary adducts, as well as in modulating their fluorescence properties.
In conclusion, this work highlights the promising perspectives arising from the use of polyamino polyphenolic ligands, not only as potential chemosensors for H+/Zn2+, but even as suitable metallo-receptors for the recognition and sensing of elusive environmental pollutants.
Experimental section
General methods
All chemicals were purchased in the highest quality commercially available and used without further purification. Solvents were RP grade, unless otherwise indicated, and were dried prior to use.Synthesis
Ligand L was obtained following the synthetic procedure reported in Scheme 1; the synthesis of the intermediate 1 was accomplished as previously described.33,54–59Potentiometric measurements
The equilibrium constants for the ligand protonation, Zn2+-complex stability and formation of the ternary adducts with ketoprofen/ibuprofen were determined through potentiometric titrations in degassed H2O/CH3CN 80:20 (v/v) NMe4Cl 0.1 M at 298 ± 0.1 K, by employing procedures and equipment already described.99–101
Briefly, a combined glass electrode was calibrated as a hydrogen-ion concentration probe by titrating known amounts of HCl with CO2-free NaOH solutions, while using an Ag/AgCl electrode in saturated KCl as reference electrode. The standard potential Eo and the ionic product of water (pKw = 14.99 (1)),102,103 were determined by the Gran's method.104
All the employed solutions were prepared by using freshly boiled, doubly deionized water, saturated with anhydrous nitrogen prior to uses; NaOH solutions were standardized against carbonate free potassium hydrogen phthalate and stored under nitrogen atmosphere.
Measurements were carried out by using a ligand concentration of 1 × 10−3 M, in a range of pH within 2–11. In the complexation experiments the Zn2+ concentration was varied from 0.9[L] to 1.8[L] whereas in the study of ternary systems the concentration of the anionic substrates was varied from 0.5[L] to 5[L], to establish all the different stoichiometries of the species formed in solution. From the EMF data, the equilibrium constants were determined by using the program HYPERQUAD105 and the distribution diagrams of the species present in solution for each system were calculated by the Hyss program.106
Spectrophotometric and fluorescence measurements
Electronic UV-Vis absorption and fluorescence emission spectra were respectively collected on a PerkinElmer Lambda 6 spectrophotometer and on a PerkinElmer LS55 spectrofluorimeter. Fluorescence spectra were collected by employing a 288 nm excitation wavelength, in H2O/CH3CN 80:20 (v/v) mixture. All measurements were performed at 298.0 ± 0.1 K.
NMR measurements
NMR spectra were recorded on a Bruker Avance 200 spectrometer (Bruker Italia, Milano, Italy) operating at 200.13 and 50.32 MHz for 1H and 13C, respectively, equipped with a PABBO Z-gradient direct probe and a variable temperature unit. 1H and 13C NMR spectra were referenced to residual solvent signals.The complex Zn2(H−2L)2+ was synthesized by dissolving the ligand in a D2O/CD3CN 80/20 (v/v) mixture at a concentration of 5 × 10−3 mol dm−3 at acidic pH (about 3), followed by the addition of two equivalents of Zn2+ as perchlorate salt and adjustment of pH to about 9, to obtain the target complexed species. Due to the limited solubility in such experimental conditions, the complex precipitated as a white solid, that was isolated by filtration, washed with methanol and then dissolved in DMSO-d6 affording its full dissolution. The NMR tube was kept for 5 min at a temperature of 298.1 K after the dissolution of the complex before starting the acquisition of the spectrum.
Single Crystal X-ray diffraction (SCXRD) data collection and structure solution
Single crystal X-ray diffraction data of L were collected on a Bruker Apex-II diffractometer equipped with a CCD detector (T = 100 K), Cu-Kα radiation (λ = 1.54184 Å), several crystals were tested, and the best one was chosen for the data collection. Data were collected with the APEX2107 software, while data integration and reduction were performed with the Bruker SAINT software.108 The crystal structure was solved using the SIR-2014109 package and refined by full-matrix least squares against F2 using all data (SHELXL-2018/3).110 All the non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms linked to the oxygen atoms, O1 and O2, in both the independent molecules of L were found in the Fourier Density Maps, their coordinates were freely refined while their thermal parameter was set in accordance with that of the atoms to which they are bonded. All the other hydrogen atoms were set in calculated position. One of the pyridine rings in one of the two independent molecules (the one labelled with “A”, see Fig. Pat1 and Pat-S1†) is affected by disorder. Such disorder was modelled by using two positions for the atoms N2, C15, C18 and C19 (see Fig. Pat-S2†), as well as for the hydrogen atoms bonded to such atoms. The occupancy factors were freely refined (final values 0.42127 and 0.57873 for each model).Geometrical calculations, for both structures, were performed by PARST97111 and molecular plots were produced by the program ORTEP-3.112
ORTEP view of the asymmetric unit of L is reported in Fig. 2, whereas in Table S1 (ESI†) crystallographic data and refinement parameters of L are reported.
Theoretical calculations
Theoretical calculations were performed at the DFT level with the Gaussian 16 suite of programs (rev. C0.1).81 The B3LYP hybrid functional74–77 was adopted, along with Ahlrichs’ TZV78,79 basis set for all atomic species and SDD pseudopotential80 within Gen basis input for Zn atom. The reliability of the stationary points was assessed by the evaluation of the vibrational frequencies. The molecular geometry optimizations on L and H−1L− were started from the structural data of the ligand, while the molecular geometry optimizations on mono- and dinuclear zinc complexes were started from conveniently modified related structural data. The alternative binding modes of complexes Zn(H−1L)+ and Zn2(H−1L)2+ were generated from the initially optimized structures by appropriately modifications and performing subsequent re-optimization. The program GaussView 6.1.113 was used to investigate the optimized structures.Author contributions
D. P.: investigation, G. E. G.: investigation, M. F.: funding acquisition, supervision, L. G.: supervision, B. V.: supervision, P. R.: investigation, writing – original draft, P. P.: supervision, E. M.: investigation, formal analysis, writing – original draft, writing – review & editing, L. C.: conceptualization, investigation, writing – original draft, writing – review & editing, V. F.: conceptualization, supervision, writing – review & editing, funding, C. G.: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially funded by the European Union – NextGenerationEU within the framework of PNRR Mission 4 – Component 2 – Investment 1.1 under the Italian Ministry of University and Research (MUR) programme “PRIN 2022” – grant number 2022HYH95P – Wastezilla – CUP: H53D23003860006. This work was also supported by the European Union – NextGenerationEU – under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY – CUP [H33C22000430006]. This work was also partially funded by the University of Urbino (Grant DISPEA_ASSEGNAZIONE_ATENEO_SICUREZZA_ALIMENTARE).The publication was made by a researcher (LC) with a research contract co-funded by the European Union – PON Research and Innovation 2014-2020 in accordance with Article 24, paragraph 3a, of Law No. 240 of December 30, 2010, as amended, and Ministerial Decree No. 1062 of August 10, 2021.
Ms Anna Rita Pierleoni and Dr Gianluca Ambrosi are acknowledged for their help with NMR measurements. Mr Lorenzo Mari is gratefully acknowledged for the help with optical investigation.
G.E.A. Green Economy and Agriculture Centro per la Ricerca s.r.l. (via Ciliegiole 99, 51100 Pistoia, Italy) is gratefully acknowledged for supporting this project.
CRIST (Centro di Servizi di Cristallografia Strutturale – University of Firenze), where the X-ray diffraction data were collected, is greatly acknowledged.
References
- V. Geissen, H. Mol, E. Klumpp, G. Umlauf, M. Nadal, M. Van Der Ploeg, E. A. T. M. Van De Zee and J. Ritsema, Int. Soil Water Conserv. Res., 2015, 3, 57–65 CrossRef.
- S. K. Khetan and T. J. Collins, Chem. Rev., 2007, 107, 2319–2364 CrossRef CAS PubMed.
- C. G. Daughton and T. A. Ternes, Environ. Health Perspect., 1999, 107, 907 CrossRef CAS PubMed.
- M. Godoy and J. Sánchez, in Antibiotic Materials in Healthcare, Elsevier, 2020, pp. 221–230 Search PubMed.
- A. Rastogi, M. K. Tiwari and M. M. Ghangrekar, J. Environ. Manage., 2021, 300, 113694 CrossRef CAS PubMed.
- N. Vieno and M. Sillanpää, Environ. Int., 2014, 69, 28–39 CrossRef CAS PubMed.
- S. Shakya and I. M. Khan, J. Hazard. Mater., 2021, 403, 123537 CrossRef CAS PubMed.
- N. Dey and C. J. E. Haynes, ChemPlusChem, 2021, 86, 418–433 CrossRef CAS PubMed.
- A. Gogoi, S. Mukherjee, A. Ramesh and G. Das, Anal. Chem., 2015, 87, 6974–6979 CrossRef CAS PubMed.
- A. K. Mahapatra, R. Maji, K. Maiti, S. S. Adhikari, C. Das Mukhopadhyay and D. Mandal, Analyst, 2014, 139, 309–317 RSC.
- S. J. Dickson, E. V. B. Wallace, A. N. Swinburne, M. J. Paterson, G. O. Lloyd, A. Beeby, W. J. Belcher and J. W. Steed, New J. Chem., 2008, 32, 786–789 RSC.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed.
- E. Macedi, L. Giorgi, M. Formica, P. Rossi, D. Paderni, P. Paoli and V. Fusi, ChemPlusChem, 2023, 88, e202200364 CrossRef CAS PubMed.
- G. Picci, M. C. Aragoni, M. Arca, C. Caltagirone, M. Formica, V. Fusi, L. Giorgi, F. Ingargiola, V. Lippolis, E. Macedi, L. Mancini, L. Mummolo and L. Prodi, Org. Biomol. Chem., 2023, 21, 2968–2975 RSC.
- G. W. Bates and P. A. Gale, Recognition of Anions, Springer, Heidelberg, 2008 Search PubMed.
- A. Bianchi, K. Bowman-james and E. Garcia-Espana, Supramolecular chemistry of anions, Wiley-VCH, Weinheim, 1997 Search PubMed.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- N. Alashkar, M. Arca, H. Alnasr, M. Lutter, V. Lippolis and K. Jurkschat, Eur. J. Inorg. Chem., 2020, 2020, 3925–3936 CrossRef CAS.
- A. Andrés, C. Bazzicalupi, A. Bencini, A. Bianchi, V. Fusi, E. Garcia-España, C. Giorgi, N. Nardi, P. Paoletti, J. A. Ramirez and B. Valtancoli, J. Chem. Soc., Perkin Trans. 2, 1994, 2367–2373 RSC.
- J. Zhao, D. Yang, X. J. Yang and B. Wu, Coord. Chem. Rev., 2019, 378, 415–444 CrossRef CAS.
- M. Formica, V. Fusi, E. Macedi, P. Paoli, G. Piersanti, P. Rossi, G. Zappia and P. Orlando, New J. Chem., 2008, 32, 1204–1214 RSC.
- F. Bartoli, A. Bencini, L. Conti, C. Giorgi, B. Valtancoli, P. Paoli, P. Rossi, N. Le Bris and R. Tripier, Org. Biomol. Chem., 2016, 14, 8309–8321 RSC.
- Y. Hu, S. Long, H. Fu, Y. She, Z. Xu and J. Yoon, Chem. Soc. Rev., 2021, 50, 589–618 RSC.
- S. A. Boer, E. M. Foyle, C. M. Thomas and N. G. White, Chem. Soc. Rev., 2019, 48, 2596–2614 RSC.
- L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, Chem. Commun., 2019, 55, 5195 RSC.
- M. Formica, V. Fusi, L. Giorgi, E. Macedi, G. Piersanti, M. A. Varrese and G. Zappia, Supramol. Chem., 2010, 22, 365–379 CrossRef CAS.
- S. Amatori, G. Ambrosi, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini, P. Rossi and M. A. Varrese, Chem. – Eur. J., 2012, 18, 4274–4284 CrossRef CAS PubMed.
- S. Amatori, G. Ambrosi, E. Borgogelli, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli, P. Rossi and A. Tassoni, Inorg. Chem., 2014, 53, 4560–4569 CrossRef CAS PubMed.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, E. MacEdi, G. Piersanti, M. Retini, M. A. Varrese and G. Zappia, Tetrahedron, 2012, 68, 3768–3775 CrossRef CAS.
- F. Bettazzi, D. Voccia, A. Bencini, C. Giorgi, I. Palchetti, B. Valtancoli and L. Conti, Eur. J. Inorg. Chem., 2018, 2018, 2675–2679 CrossRef CAS.
- C. Becker, P. C. Trapp, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2022, 51, 6565–6575 RSC.
- T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42(2), 388–397 CrossRef CAS PubMed.
- G. E. Giacomazzo, D. Paderni, L. Giorgi, M. Formica, L. Mari, R. Montis, L. Conti, E. Macedi, B. Valtancoli, C. Giorgi and V. Fusi, Molecules, 2023, 28, 2031 CrossRef CAS PubMed.
- P. Rossi, E. Macedi, M. Formica, L. Giorgi, P. Paoli and V. Fusi, ChemPlusChem, 2020, 85, 1179–1189 CrossRef CAS PubMed.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Chem. – Eur. J., 2011, 17, 1670–1682 CrossRef CAS PubMed.
- F. M. Dolgushin and I. L. Eremenko, Russ. Chem. Rev., 2021, 90, 1493–1519 CrossRef.
- M. M. Naseer and K. Jurkschat, Chem. Commun., 2017, 53, 8122 RSC.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, A. Guerri, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2009, 48, 5901–5912 CrossRef CAS PubMed.
- M. Formica, V. Fusi, D. Paderni, G. Ambrosi, M. Inclan, M. P. Clares, B. Verdejo and E. Garcia-España, Molecules, 2021, 26, 2352 CrossRef CAS PubMed.
- O. Francesconi, M. Gentili, F. Bartoli, A. Bencini, L. Conti, C. Giorgi and S. Roelens, Org. Biomol. Chem., 2015, 13, 1860–1868 RSC.
- C. Bazzicalupi, A. Bencini, E. Berni, A. Bianchi, P. Fornasari, C. Giorgi and B. Valtancoli, Eur. J. Inorg. Chem., 2003, 2003, 1974–1983 CrossRef.
- J. Morais Missina, L. Conti, P. Rossi, A. Ienco, G. Gioppo Nunes, B. Valtancoli, L. Chelazzi and P. Paoli, Inorg. Chim. Acta, 2021, 523, 120319 CrossRef CAS.
- G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Chem. Soc. Rev., 2020, 49, 765–838 RSC.
- C. Liu, M. Wang, T. Zhang and H. Sun, Coord. Chem. Rev., 2004, 248, 147–168 CrossRef CAS.
- G. Ambrosi, C. Battelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, R. Pontellini and L. Prodi, New J. Chem., 2009, 33, 171–180 RSC.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli and P. Rossi, Inorg. Chem., 2009, 48, 10424–10434 CrossRef CAS PubMed.
- L. Conti, N. Flore, M. Formica, L. Giorgi, M. Pagliai, L. Mancini, V. Fusi, B. Valtancoli and C. Giorgi, Inorg. Chim. Acta, 2021, 519, 120261 CrossRef CAS.
- L. Conti, L. Mummolo, G. M. Romano, C. Giorgi, G. E. Giacomazzo, L. Prodi and A. Bencini, Molecules, 2021, 26, 527 CrossRef CAS PubMed.
- A. Erxleben, Front. Chem., 2019, 7, 82 CrossRef CAS PubMed.
- D. Montagner, V. Gandin, C. Marzano and A. Erxleben, Eur. J. Inorg. Chem., 2014, 25, 4084–4092 CrossRef.
- H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928 RSC.
- D. R. Rice, K. J. Clear and B. D. Smith, Chem. Commun., 2016, 52, 8787–8801 RSC.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni and R. Pontellini, Inorg. Chim. Acta, 2009, 362, 2667–2677 CrossRef CAS.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, A. Guerri, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2007, 46, 309–320 CrossRef CAS PubMed.
- G. Ambrosi, P. Dapporto, M. Formica, V. Fusi, L. Giorgi, A. Guerri, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Chem. – Eur. J., 2003, 9, 800–810 CrossRef CAS PubMed.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli and P. Rossi, Inorg. Chem., 2016, 55, 7676–7687 CrossRef CAS PubMed.
- R. A. Fernandes and S. V. Mulay, J. Org. Chem., 2010, 75, 7029–7032 CrossRef CAS PubMed.
- H. Gilman, J. Swiss and L. C. Cheney, J. Am. Chem. Soc., 1940, 62, 1963–1967 CrossRef CAS.
- T. Kaneda, S. Umeda, H. Tanigawa, S. Misumi, Y. Kai, H. Morii, K. Miki and N. Kasai, J. Am. Chem. Soc., 1985, 107, 4802–4803 CrossRef CAS.
- J. K. Bjernemose and C. J. McKenzie, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o1275–o1276 CrossRef CAS.
- C. Aakeröy, Cryst. Growth Des., 2001, 1, 255–255 CrossRef.
- J. Mohanty, H. Pal and A. V. Sapre, Bull. Chem. Soc. Jpn., 1999, 72, 2193–2202 CrossRef CAS.
- M. Formica, G. Ambrosi, V. Fusi, L. Giorgi, M. Arca, A. Garau, A. Pintus and V. Lippolis, New J. Chem., 2018, 42, 7869–7883 RSC.
- R. J. L. Andon, J. D. Cox and E. F. G. Herington, Trans. Faraday Soc., 1954, 50, 918 RSC.
- F. A. Mautner, R. C. Fischer, N. M. H. Salem, A. J. Darbonne, S. L. Silhan, Z. Haghighijoo, S. P. Sahu, F. R. Louka and S. S. Massoud, Inorg. Chim. Acta, 2022, 535, 120871 CrossRef CAS.
- P. Chaibuth, N. Chuaytanee, J. Hojitsiriyanont, K. Chainok, S. Wacharasindhu, O. Reiser and M. Sukwattanasinitt, New J. Chem., 2022, 46, 12158–12168 RSC.
- M. Jonsson, J. Lind and G. Merényi, J. Phys. Chem. A, 2002, 106, 4758–4762 CrossRef CAS.
- J. W. J. Bridges, P. J. P. Creaven and R. T. R. Williams, Biochem. J., 1965, 96, 872–878 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. N. Gunaratne and C. P. McCoy, Chem. Commun., 1996, 2399–2400 RSC.
- A. J. Parola, J. C. Lima, F. Pina, J. Pina, J. S. de Melo, C. Soriano, E. García-España, R. Aucejo and J. Alarcón, Inorg. Chim. Acta, 2007, 360, 1200–1208 CrossRef CAS.
- S. A. de Silva, A. Zavaleta, D. E. Baron, O. Allam, E. V. Isidor, N. Kashimura and J. M. Percarpio, Tetrahedron Lett., 1997, 38, 2237–2240 CrossRef CAS.
- L. Fabbrizzi, F. Gatti, P. Pallavicini and L. Parodi, New J. Chem., 1998, 22, 1403–1407 RSC.
- R. Aucejo, J. Alarcón, E. García-España, J. M. Llinares, K. L. Marchin, C. Soriano, C. Lodeiro, M. A. Bernardo, F. Pina, J. Pina and J. Seixas de Melo, Eur. J. Inorg. Chem., 2005, 2005, 4301–4308 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 59, 1200 CrossRef.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revis. C.01, Inc., Wallingford CT, 2016 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- L. Yang, D. R. Powell and R. P. Houser, J. Chem. Soc., Dalton Trans., 2007, 955–964 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- G. M. Romano, L. Mummolo, M. Savastano, P. Paoli, P. Rossi, L. Prodi and A. Bencini, Chem. Commun., 2022, 58, 7022–7025 RSC.
- L. Duan and Y. Zhao, React. Funct. Polym., 2021, 158, 104759 CrossRef CAS PubMed.
- M. Herrador, Talanta, 2002, 56, 769–775 CrossRef CAS PubMed.
- A. Avdeef, K. Box, J. E. Comer, M. Gilges, M. Hadley, C. Hibbert, W. Patterson and K. Tam, J. Pharm. Biomed. Anal., 1999, 20, 631–641 CrossRef CAS PubMed.
- M. Meloun, S. Bordovská and L. Galla, J. Pharm. Biomed. Anal., 2007, 45, 552–564 CrossRef CAS PubMed.
- R. M. Watkinson, C. Herkenne, R. H. Guy, J. Hadgraft, G. Oliveira and M. E. Lane, Skin Pharmacol. Physiol., 2009, 22, 15–21 CrossRef CAS PubMed.
- V. K. Mourya and T. R. Saini, Indian Res. J. Pharm. Sci., 1997, 59, 200–202 CAS.
- M. Savastano, M. Fiaschi, G. Ferraro, P. Gratteri, P. Mariani, A. Bianchi and C. Bazzicalupi, Molecules, 2020, 25, 1355 CrossRef CAS PubMed.
- M. Kruppa and B. König, Chem. Rev., 2006, 106, 3520–3560 CrossRef CAS PubMed.
- J. Chen, S. Cao, D. Wang, S. Wu and X. Wang, J. Braz. Chem. Soc., 2009, 20, 13–18 CrossRef CAS.
- E. Kinoshita, M. Takahashi, H. Takeda, M. Shiro and T. Koike, Dalton Trans., 2004, 1189 RSC.
- J. Chen, X. Wang, Y. Zhu, J. Lin, X. Yang, Y. Li, Y. Lu and Z. Guo, Inorg. Chem., 2005, 44, 3422–3430 CrossRef CAS PubMed.
- D. H. Lee, S. Y. Kim and J. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777–4780 CrossRef CAS PubMed.
- H. Adams, D. Bradshaw and D. E. Fenton, J. Chem. Soc., Dalton Trans., 2002, 925–930 RSC.
- M. Becatti, A. Bencini, S. Nistri, L. Conti, M. G. Fabbrini, L. Lucarini, V. Ghini, M. Severi, C. Fiorillo, C. Giorgi, L. Sorace, B. Valtancoli and D. Bani, Sci. Rep., 2019, 9, 10320 CrossRef PubMed.
- L. Conti, C. Giorgi, B. Valtancoli, P. Paoli, P. Rossi, A. Marchionni, E. Faggi and A. Bencini, ChemPlusChem, 2020, 85, 659–671 CrossRef CAS PubMed.
- M. C. Aragoni, M. Arca, A. Bencini, C. Caltagirone, L. Conti, A. Garau, B. Valtancoli, F. Isaia, V. Lippolis, F. Palomba, L. Prodi and N. Zaccheroni, Supramol. Chem., 2017, 29, 912–921 CrossRef CAS.
- A. Garau, A. Bencini, A. J. Blake, C. Caltagirone, L. Conti, F. Isaia, V. Lippolis, R. Montis, P. Mariani and M. A. Scorciapino, Dalton Trans., 2019, 48, 4949–4960 RSC.
- A. Garau, G. Picci, A. Bencini, C. Caltagirone, L. Conti, V. Lippolis, P. Paoli, G. M. Romano, P. Rossi and M. A. Scorciapino, Dalton Trans., 2022, 51, 8733–8742 RSC.
- G. Gran, Analyst, 1952, 77, 661–671 RSC.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- Bruker APEX2, Bruker AXS Inc., 2012 Search PubMed.
- Bruker SAINT, Bruker AXS Inc., 2012 Search PubMed.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- M. Nardelli and IUCr, J. Appl. Crystallogr., 1995, 28, 659–659 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 6.1.1, Semichem Inc., Shawnee Mission, KS, 2019 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR characterization, solid-state details, acid–base properties of L, Zn2+ binding properties, DFT calculations details. CCDC 2344129. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00935e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |