
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of the heteroatom on the magnetic and luminescence properties of hybrid lanthanide-substituted Keggin-type polyoxometalates†
Janire
Bustamante-Fernández
 ab,
Estibaliz
Ruiz-Bilbao
b,
Corina
Rodríguez-Esteban
b,
Mathieu
Gonidec
c,
José A.
García
d,
Luis
Lezama
a,
Juan M.
Gutiérrez-Zorrilla
ab,
Itziar
Oyarzabal
*be and
Beñat
Artetxe
*a
ab,
Estibaliz
Ruiz-Bilbao
b,
Corina
Rodríguez-Esteban
b,
Mathieu
Gonidec
c,
José A.
García
d,
Luis
Lezama
a,
Juan M.
Gutiérrez-Zorrilla
ab,
Itziar
Oyarzabal
*be and
Beñat
Artetxe
*a
aDepartamento de Química Orgánica e Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco UPV/EHU, P.O. Box 644, 48080 Bilbao, Spain. E-mail: benat.artetxe@ehu.eus
bBCMaterials, Edificio Martina Casiano, 3rd Floor, UPV/EHU Science Park, Barrio Sarriena s/n, 48940 Leioa, Spain. E-mail: itziar.oyarzabal@bcmaterials.net
cUniv. Bordeaux, CNRS, Bordeaux INP, ICMCB, UMR 5026, F-33600 Pessac, France
dDepartamento de Física Aplicada II, Facultad de Ciencia y Tecnología. Universidad del País Vasco UPV/EHU, P.O. Box 644, 48080 Bilbao, Spain
eIKERBASQUE, Basque Foundation for Science, 48009 Bilbao, Spain
First published on 25th July 2024
Abstract
Replacement of the heteroatom from Si to Ge has a strong influence on the luminescence properties of a series of hybrid, sandwich-type K5[Ln(α-GeW11O39)(C20H22Br2N2O4)]·14H2O (1Ge-Ln, Ln = Sm to Lu) anions. Interestingly, the Gd and Yb derivatives retain their ability to display slow relaxation of magnetisation.
The combination of lacunary polyoxometalates (POMs) that act as inorganic multidentate O donor ligands with lanthanide(III) ions1,2 (Ln) constitutes a suitable approach for the preparation of systems exhibiting interesting catalytic,3,4 optical5,6 and magnetic properties.7,8 In fact, the high oxophilicity and coordination numbers of 4f metal ions allow the generation of some of the largest and most outstanding POMs known to date.9–11 In spite of their structural simplicity, small sandwich-type species that enclose a single LnIII ion between two monolacunary fragments are some of the most explored families. This interest mainly originates from their potential applications in molecular magnetism, including fields like molecular spintronics,12 data-storage13 and quantum computing.14 In close analogy to double-decker type coordination complexes,15 the strong magnetic anisotropy together with the large ground-state magnetic moments of Ln ions has allowed Peacock–Weakley type anions to show single-molecule magnet (SMM) behaviour. Examples of these sandwich-type species with monolacunary polyoxotungstates enclosing a central 4f metal in a square antiprismatic D4d geometry include [Ln(W5O18)2]9− (Ln = Tb, Dy, Ho and Er) and [Ln(β2-SiW11O39)2]13− (Ln = Dy, Ho, Er and Yb) anions.16,17 These Lindqvist- or Keggin-type fragments display some advantages in comparison with classical organic ligands used in coordination complexes: (i) POMs show higher thermal and chemical stability both in solution and the solid state; (ii) the rigidity of the ligand can lead to highly symmetric environments for Ln centres, and (iii) their large size and diamagnetism ensure magnetic isolation over the neighbouring species. Furthermore, 4f metal ions can be sensitised via photoexcitation of the O → W ligand-to-metal charge-transfer states to result in intense light emission.18,19
Despite their potential, there are only a few reports on families of mononuclear complexes with the Ln ion displaying simultaneous coordination with lacunary polyoxotungstates and organic ligands, to the best of our knowledge. Monolacunary Keggin-type anions were reacted with different Ln/organic ligand combinations such as Dy/phthalocyanine and Sm/benzoate leading to systems exhibiting slow relaxation of magnetisation and tuneable luminescence, respectively.20,21 It is also noteworthy that the [n-NBu4]3[LnH(PW11O39)(phen)2]·H2O (phen = phenanthroline) family exhibits interesting magneto-luminescence properties.22,23
Very recently, some of us published a series of hybrid anions formed by Ln-containing Keggin-type anions and the compartmental organic ligand N,N′-dimethyl-N,N′-bis(2-hydroxy-3-formyl-5-bromobenzyl)-ethylenediamine (H2L, Fig. 1).24 This molecule displays an external O4 site which can accommodate large oxophilic 4f centres and an inner N2O2 pocket available for the coordination of smaller 3d metal ions. This combination can result in heterometallic 3d–4f complexes, which represents a suitable way to improve the SMM behaviour of a given system.25 In a first approach, we selected the monolacunary Keggin type tungstosilicate and reacted it with mid-to-late Ln cations, leading to a family of ten isostructural compounds, namely K5[Ln(α-SiW11O39)(H2L)]·14H2O (1Si-Ln, Ln = Sm to Lu). Encouraged by the fact that the Gd and Yb derivatives showed slow relaxation of magnetisation and that efficient emission in the visible and NIR region was reported for different members of the family, we decided to evaluate the influence of the modification of its constituents on the final magneto-luminescence properties.
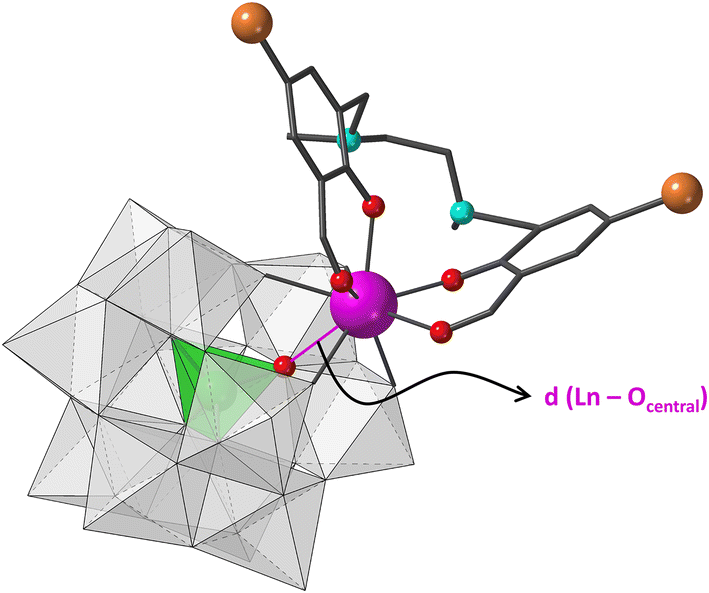 | ||
| Fig. 1 Molecular structure of hybrid [Ln(H2L)(α-GeW11O39)]5− anions in 1Ge-Ln. The Ln⋯Ocentral bond is represented as a purple line and discussed in the text. Colour code: WO6, grey; GeO4, green; Ln, purple; C, black, O, red; N, blue; Br, orange. H atoms have been omitted for clarity. | ||
First, different first row transition metal salts were included in the original reaction, but no incorporation of 3d metal was observed regardless of the synthetic conditions (nature of the 3d metal and the counterion, reaction temperature, aqueous reaction media). Then, we tried to evaluate the effect of the heteroatom without modifying the total charge of the anion by replacing monolacunary Keggin type tungstosilicates with tungstogermanates. Following the same synthetic procedure, nine isostructural compounds namely K5[Ln(α-GeW11O39)(H2L)]·14H2O (1Ge-Ln, Ln = Sm to Yb) were isolated (Fig. S1–S7 and Tables S1–S6†). Single crystal X-ray diffraction studies revealed that the molecular structure of each hybrid polyanion is similar to those of the 1Si-Ln derivatives (Fig. 1). These are composed of a trivalent Ln cation simultaneously coordinated by (i) the Keggin-type monolacunary fragment through the four O atoms delimiting the vacant site and (ii) the outer O4 site from the organic ligand formed by two aldehyde and two phenoxy groups. Regarding the crystal packing, the weak supramolecular interactions including π–π stacking between aromatic rings and Br⋯Br bonds involving H2L ligands give rise to hexameric assemblies formed by different hybrid units (Fig. S7†).
The geometry of the LnO8 coordination polyhedra was studied through continuous shape measures (CShM),26 which show that the lowest distortion values were obtained using the biaugmented trigonal prism (BTPR, C2v, CShM: 0.556–0.707) and square antiprism (SAPR, D4d, CShM: 0.852–1.061) as reference shapes (Table S7†). The BTPR vs. SAPR shape map (Fig. S8†) displays low deviation values for all the 4f ions from the minimal distortion pathway between both reference shapes (<0.3). This fact confirms that their geometry can be best described as BTPR distorted towards SAPR. The scatter of LnO8 polyhedra together with those from compounds 1Si-Ln in the shape map evidences that they can be easily classified into two different groups, with those reported in this work being closer to ideal BTPR. This difference arises from the out-of-pocket coordination mode of Ln ions toward the Keggin skeleton. The larger size of the heteroatom in 1Ge-Ln allows 4f ions to be better incorporated into the lacunary site, in such a way that the distances to central O atoms are shortened in ca. 0.2 Å when moving from Si to Ge (Ln⋯OGe = 3–3.1 Å, Fig. 1). This fact decreases the average folding angle between the aromatic rings of the organic ligand in ca. 2°.
In order to explore if the changes in the local geometry of the Ln impact the magnetic properties, direct-current (dc) magnetic susceptibility measurements were carried out by using single crystals of 1Ge-Ln (Fig. S9 and S10, Table S8†). Starting with 1Ge-Gd, the nearly constant value of the χMT product in the studied temperature range implies the absence of significant interactions between Gd ions, which is in agreement with the large distances between the Ln ions (>8 Å) within the crystal packing. The fitting of the Q-band EPR spectra at room temperature (Fig. S11†) yields D = 0.0831 cm−1, E = 0.0244 cm−1, gx = 1.993, gy = 1.992 and gz = 1.983, which effectively reproduce the susceptibility and magnetisation data when g = 2. Compared to 1Si-Gd, the modifications in the Ln coordination environment lead to negligible differences in the zero field splitting parameters (D and E) and energy levels (Fig. S12, Table S9†), in agreement with the isotropic nature of gadolinium ions. As in the case of 1Si-Gd, 1Ge-Gd displays slow relaxation of magnetisation at low temperatures (Fig. 2 and S13–S15†). The Arrhenius fit of the relaxation times at higher temperatures leads to an energy barrier of 8.1 cm−1 with τ0 = 6.5 × 10−6 s, which is significantly larger than the separation between the ground and first excited states (0.40 cm−1). This, together with the deviation of the relaxation times from linearity and the relatively large α values obtained in the Cole–Cole plots (0.17 (2 K)–0.02 (8 K)), suggests the presence of multiple relaxation processes different from Orbach (i.e., Raman).27
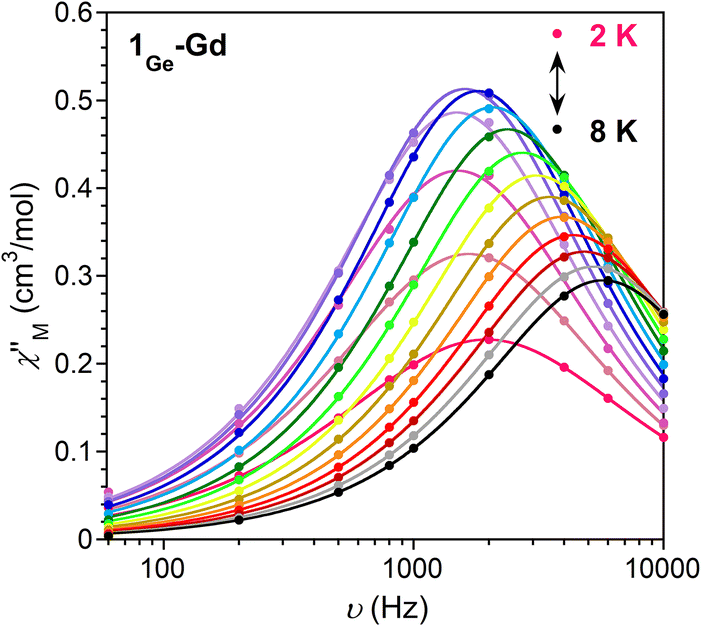 | ||
| Fig. 2 Frequency dependence of the out-of-phase component of the ac susceptibility at Hdc = 2500 Oe for 1Ge-Gd. The experimental data are denoted by circles; solid lines represent the best fit to the data. | ||
In the case of 1Ge-Sm and 1Ge-Eu, the χMT products at room temperature (0.35 and 1.39 cm3 K mol−1, respectively) are higher than expected (0.09 and 0 cm3 K mol−1, respectively), which implies the existence of thermally populated excited states (Fig. S16†). Upon cooling, the χMT values decrease until reaching 0.022 and 0.019 cm3 K mol−1 at 2 K, respectively. The fitting of the magnetic susceptibility of 1Ge-Eu to the equation proposed by Kahn28 yields λ = 332 cm−1 and δ = 0.13%, with λ being the spin–orbit coupling parameter and δ the percentage of EuII paramagnetic impurities. The value of λ is close to that extracted from luminescence measurements (λ = 314 cm−1, see below) and suggests a slightly larger separation between the ground and first excited states than in 1Si-Eu (332 cm−1 for 1Ge-Euvs. 321 cm−1 for 1Si-Eu, Fig. S18†). The same behaviour is observed for 1Ge-Sm, in which the separation between the ground and first excited states is increased from 125 cm−1 (1Si-Sm) to 140 cm−1 (1Ge-Sm) according to the photoluminescence measurements (see below).
The room temperature χMT products of the remaining compounds are close to the expected values (Fig. S9, Table S8†). As the temperature decreases, the χMT products exhibit relative stability and, in most cases, they experience a sudden drop below ∼50 K due to the depopulation of the MJ sublevels of the Ln ions. The presence of populated low-lying states prevents these compounds from achieving the expected saturation values at 2 K and 7 T (Fig. S10, Table S8†).
Regarding the ac susceptibility, 1Ge-Dy exhibits a modest frequency dependence with the maxima of the χ′′M signals below ∼2.7 K (Fig. S19–S21†), while 1Ge-Yb displays clear slow relaxation of magnetisation below ∼9 K (Fig. S22–S24†). Compared to the silicon-based counterparts, the maxima of χ′′M signals are shifted towards higher temperatures in both compounds, enabling the extraction of the energy barrier for 1Ge-Dy (Ueff = 14.3 cm−1, τ0 = 6.3 × 10−9 s). Even though the observed improvements could be associated with an increase in the energy gap between the ground and first excited states of Dy and Yb, the large α values (0.26 (2 K)–0.40 (2.7 K) for 1Ge-Dy; 0.12 (5 K)–0.05 (9 K) for 1Ge-Yb) and the deviation of the relaxation times from linearity indicate that the magnetic relaxation does not occur exclusively via the Orbach mechanism. In fact, photoluminescence studies of 1Ge-Yb reveal a larger separation between the ground and first excited states (343 cm−1 for 1Ge-Ybvs. 261 cm−1 for 1Si-Yb), but the energy barriers obtained by the Arrhenius fitting (26.4 cm−1 for 1Ge-Yb and 14.8 cm−1 for 1Si-Yb) are notably lower. Thus, the relaxation of magnetisation in 1Ge-Yb occurs most likely through a Raman mechanism, as observed in related YbIII-based compounds.29
When it comes to photophysical properties, solid state photoluminescence was studied for 1Ge-Sm, 1Ge-Eu and 1Ge-Tb in the visible region and for 1Ge-Er and 1Ge-Yb in the near infrared (NIR) region. First, the excitation of 1Ge-Sm and 1Ge-Eu (Fig. S25†) was monitored around their most intense bands at 614 nm and 599 nm, respectively. The resulting spectra display broad bands in the 300–450 nm range, which imply that the antenna effect is operative in these compounds.
Emission spectra were recorded at different temperatures (from 15 K to room temperature) after irradiation at 375 and 430 nm. Excitation wavelengths correspond to the maxima of either the UV-Vis spectrum of the H2L ligand (Fig. S26†) or the excitation spectrum of 1Ge-Eu, respectively. In both cases, when the temperature increases, not only the intensity of the signals decreases, but the distinction between sublevels also appears less differentiated. The emission spectra of 1Ge-Eu (Fig. S27 and S28†) exhibit five bands centred at 580, 596, 614, 653 and 701 nm, which are attributed to the 5D0 → 7FJ (J = 0, 1, 2, 3 and 4) manifolds. The relative intensity of the emission bands is virtually identical to that of 1Si-Eu with the strongest transition corresponding to the hypersensitive 5D0 → 7F2, which provides intense reddish luminescence (Fig. 3, S29 and S30†). The additional splitting of this specific transition when going from 1Ge-Eu (five signals) to 1Si-Eu (four signals) is in line with the lower symmetry of {EuO8} centres in the former compound. Observation of the forbidden 5D0 → 7F0 and 5D0 → 7F3 transitions indicates that EuIII ions occupy a low-symmetry site,30 as expected from the biaugmented trigonal prismatic coordination geometry. The average λ parameter was extracted from the positions of the emission bands and it is similar to that found in 1Si-Eu (314 cm−1 for 1Ge-Euvs. 310 cm−1 for 1Si-Eu).
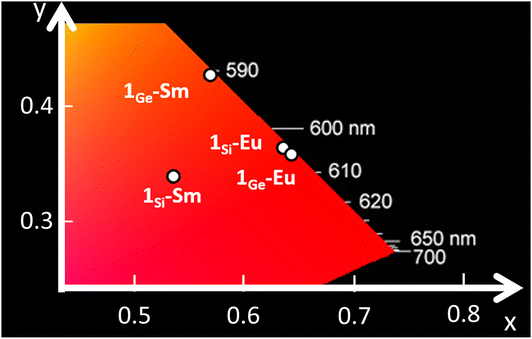 | ||
| Fig. 3 CIE 1931 x, y chromaticity coordinates as a function of the emission wavelengths for 1Si-Sm, 1Ge-Sm, 1Si-Eu and 1Ge-Eu. | ||
In the case of 1Ge-Sm and 1Ge-Tb, spectra were collected under excitation with a continuous HeCd laser at 325 nm due to the lower intensity of the emission. Analogous behaviour to that shown by 1Si-Tb was observed for 1Ge-Tb, which exhibits a very weak metal-centred emission (Fig. S31†) due to the out-of-pocket coordination of the TbIII ion. Even though the energy transfer from the ligand to the Ln is not very efficient in 1Ge-Tb, the spectrum of 1Ge-Sm (Fig. S32†) shows three emission bands that are associated with the transitions 4G5/2 → 6HJ (J = 5/2, 7/2 and 9/2) located at 563, 599 and 646 nm, respectively.31 The positions of the bands indicate a slightly higher λ parameter in 1Ge-Sm (293 cm−1) than that in 1Si-Sm (280 cm−1). The ratio of the intensities for the first (J = 5/2) and the most intense bands (J = 7/2) increases from 1Si-Sm (ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to 1Ge-Sm (ca. 1:5), in such a way that a clear colour shift to orange-yellowish is observed with the replacement of the heteroatom (Fig. 3, S29 and S30†). These emission bands almost vanished at room temperature, in line with the low absolute quantum yield (<0.01%) measured for 1Ge-Sm, which is similar to that for 1Si-Sm.
:3) to 1Ge-Sm (ca. 1:5), in such a way that a clear colour shift to orange-yellowish is observed with the replacement of the heteroatom (Fig. 3, S29 and S30†). These emission bands almost vanished at room temperature, in line with the low absolute quantum yield (<0.01%) measured for 1Ge-Sm, which is similar to that for 1Si-Sm.
The luminescence decay curve of the most intense line was measured for 1Ge-Eu upon excitation at 375 nm and 430 nm (Fig. S33†). Data were fitted to single and double exponential functions: I = A0 + A1exp(−t/τ1) (15–150 K) and I = A0 + A1exp(−t/τ1) + A2exp(−t/τ2) (298 K), with τn being the luminescence lifetime, A0 background and An weighting parameters (Table S10†). At lower temperatures, values of τ1 = ca.800 μs are achieved at both wavelengths, whereas a double exponential function was needed to fit decay curves at room temperature, leading to a significant decrease in the lifetime with τ1 ∼ 100 μs and τ2 = 600 μs, which suggests that there is a thermally activated nonradiative deactivation process of the excited states. Although the obtained lifetimes are similar to those obtained for 1Si-Eu, the emission decay curves obtained for 1Ge-Eu at low temperatures are better fitted to a single exponential function in contrast to that for 1Si-Eu, for which two decay components were required. Interestingly, the absolute quantum yields at room temperature are three times higher for 1Ge-Eu than those for 1Si-Eu, with an average value of 1.18% in contrast to the 0.36% found for the silicon-based counterpart.
Additionally, the emission in the near infrared region was studied for 1Ge-Er and 1Ge-Yb upon excitation at 325 nm (Fig. S34†). The emission spectrum of 1Ge-Er shows the most intense line at 1535 nm, which can be assigned to the 4I13/2 → 4I15/2 transition. In this case, the intensity of the emission was very weak even at low temperatures, so the measurements were performed only at 15 K. However, the high intensity of the quadruplet arising from the 2F5/2 → 2F7/2 transition in 1Ge-Yb and expanding between 970 and 1050 nm allows the collection of the spectrum from 15 K up to room temperature. The high intensity of the emission allowed the decay curves to be measured (Fig. S35†). The calculated values are on the order of a few μs, ranging from 5302(9) ns at 20 K to 2865(12) ns at room temperature. As in the case of 1Ge-Eu, the room temperature lifetime is lower because of the presence of more than one thermally activated non-radiative deactivation mechanism. Nevertheless, in contrast to 1Si-Yb, 1Ge-Yb emits at room temperature, constituting the first POM-based system with this behaviour and one of the few YbIII-based coordination complexes in the literature.32–34 It is noteworthy that these systems are of high interest for bioimaging applications (human tissues are relatively transparent to NIR light),35 as well as for telecommunication devices and lasers.
In summary, our study shows how the replacement of the heteroatom of a POM provokes a significant change in the photoluminescence properties of several lanthanide-based hybrid organic–inorganic compounds. The slight modifications in the bond lengths of the Ln ions lead to a clear colour shift to orange in 1Ge-Sm upon photoirradiation, whereas 1Ge-Yb is one of the few Yb-based compounds exhibiting slow relaxation of magnetisation (below ∼9 K) and NIR emission at room temperature. Regarding the magnetic properties, the frequency dependent ac susceptibility signals of the Gd, Dy and Yb derivatives are slightly shifted towards higher temperatures. This trend implies that the in-pocket coordination mode could be favourable for Ln ions to exhibit more efficient slow relaxation of magnetisation and luminescent emission. Thus, we plan to extend these systematic studies to different lacunary POM fragments, including monolacunary Keggin anions with larger heteroatoms.
Author contributions
J. B. F., E. R. B., I. O., and B. A.: formal analysis, writing – original draft and review and editing. J. B. F., C. R. E., J. A. G., M. G. and L. L.: investigation and data curation. I. O., B. A., and J. M. G. Z.: supervision, conceptualisation and funding acquisition.Data availability
Data available on request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. B. F. thanks Eusko Jaurlaritza/Gobierno Vasco (EJ/GV) for her predoctoral fellowship (PRE_2023_1_0230). This study forms part of the Quantum Communication Program and was supported by MCIN with funding from NextGenerationEU (PRTR-C17.I1), as well as by IKUR Strategy under the collaboration agreement between Ikerbasque Foundation and Fundación BCMaterials on behalf of the Department of Education of EJ/GV. The authors thank UPV/EHU and EJ/GV (Projects: EHU-N23/03 and IT1722-22) for funding, the Magnetic Measurement service at the ICMCB for the SQUID measurements, and SGIker for the technical and human support (UPV/EHU, ERDF, EU).References
- C. Boskovic, Acc. Chem. Res., 2017, 50, 2205–2214 CrossRef CAS PubMed.
- C. M. Granadeiro, D. Juliao, S. O. Ribeiro, L. Cunha-Silva and S. S. Balula, Coord. Chem. Rev., 2023, 476, 214914 CrossRef CAS.
- G. Trautwein, B. El Bakkali, J. Alcañiz-Monge, B. Artetxe, S. Reinoso and J. M. Gutiérrez-Zorrilla, J. Catal., 2015, 331, 110–117 CrossRef CAS.
- G.-P. Yang, S.-X. Shang, B. Yu and C.-H. Hu, Inorg. Chem. Front., 2018, 5, 2472–2477 RSC.
- K. Zheng and P. Ma, Dalton Trans., 2024, 53, 3949–3958 RSC.
- C. M. Granadeiro, R. A. S. Ferreira, P. C. R. Soares-Santos, L. D. Carlos, T. Trindade and H. I. S. Nogueira, J. Mater. Chem., 2010, 20, 3313–3318 RSC.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464–7478 RSC.
- Z. X. Yang, F. Gong, D. Lin and Y. Huo, Coord. Chem. Rev., 2023, 492, 215205 CrossRef CAS.
- B. S. Bassil, M. H. Dickman, I. Römer, B. von der Kammer and U. Kortz, Angew. Chem., Int. Ed., 2007, 46, 6192–6195 CrossRef CAS PubMed.
- S. Reinoso, M. Giménez-Marqués, J. R. Galán-Mascarós, P. Vitoria and J. M. Gutiérrez-Zorrilla, Angew. Chem., Int. Ed., 2010, 49, 8384–8388 CrossRef CAS PubMed.
- F. Hussain, F. Conrad and G. R. Patzke, Angew. Chem., Int. Ed., 2009, 48, 9088–9091 CrossRef CAS PubMed.
- J. J. Baldoví, S. Cardona-Serra, A. Gaita-Ariño and E. Coronado, Adv. Inorg. Chem., 2017, 69, 213–249 CrossRef.
- Z.-X. Yang, F. Gong, D. Lin and Y. Huo, Coord. Chem. Rev., 2023, 492, 215205 CrossRef CAS.
- S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874–8875 CrossRef CAS PubMed.
- M. A. AlDamen, S. Cardona-Serra, J. M. Clemente Juan, E. Coronado, A. Gaita-Ariño, C. Martí-Gastaldo, F. Luis and O. Montero, Inorg. Chem., 2009, 48, 3467–3479 CrossRef CAS PubMed.
- Z. Li, Z.-H. Lv, Y.-Q. Sun, X.-X. Li and S.-T. Zheng, CCS Chem., 2022, 4, 2938–2945 CrossRef CAS.
- B. Artetxe, S. Reinoso, L. San Felices, J. M. Gutiérrez-Zorrilla, J. A. García, F. Haso, T. Liu and C. Vicent, Chem. – Eur. J., 2015, 21, 7736–7745 CrossRef CAS PubMed.
- H. Wu, B. Yan, R. Liang, V. Singh, P. Ma, J. Wang and J. Niu, Dalton Trans., 2019, 49, 388–394 RSC.
- S. Sarwar, S. Sanz, J. van Leusen, G. S. Nichol, E. K. Brechin and P. Kögerler, Dalton Trans., 2020, 49, 16638–16642 RSC.
- W. A. Cañón-Mancisidor, M. Zapata-Lizama, P. Hermosilla-Ibáñez, C. Cruz, D. Venegas-Yazigi and G. Mínguez Espallargas, Chem. Commun., 2019, 55, 14992–14995 RSC.
- M. Zapata-Lizama, P. Hermosilla-Ibáñez, D. Venegas-Yazigi, G. Mínguez Espallargas, L. J. Queiroz Maia, G. Gasparotto, R. C. De Santana and W. A. Cañón-Mancisidor, Inorg. Chem. Front., 2020, 17, 3049–3062 RSC.
- E. Ruiz-Bilbao, M. Pardo-Almanza, I. Oyarzabal, B. Artetxe, L. San Felices, J. A. García, J. M. Seco, E. Colacio, L. Lezama and J. M. Gutiérrez-Zorrilla, Inorg. Chem., 2022, 61, 2428–2443 CrossRef CAS PubMed.
- A. Zabala-Lekuona, J. M. Seco and E. Colacio, Coord. Chem. Rev., 2021, 441, 213984 CrossRef CAS.
- D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. – Eur. J., 2005, 11, 1479–1494 CrossRef CAS PubMed.
- G. Handzlik, M. Magott, M. Arczyński, A. M. Sheveleva, F. Tuna, M. Sarewicz, A. Osyczka, M. Rams, V. Vieru, L. F. Chibotaru and D. Pinkowicz, J. Phys. Chem. Lett., 2020, 11, 1508–1515 CrossRef CAS PubMed.
- O. Kahn, Molecular Magnetism, Dover Publications, New York, 2021 Search PubMed.
- I. Oyarzabal, B. Artetxe, A. Rodríguez-Diéguez, J. Á. García, J. M. Seco and E. Colacio, Dalton Trans., 2016, 45, 9712–9726 RSC.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- Y. Zheng, J. Lin and Q. Wang, Photochem. Photobiol. Sci., 2012, 11, 1567–1574 CrossRef CAS PubMed.
- R. Jankowski, J. J. Zakrzewski, O. Surma, S. Ohkoshi, S. Chorazy and B. Sieklucka, Inorg. Chem. Front., 2019, 6, 2423–2434 RSC.
- P. F. Muldoon, G. Collet, S. V. Eliseeva, T. L. Luo, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2020, 142, 8776–8781 CrossRef PubMed.
- C. Liu, S. V. Eliseeva, T. L. Luo, P. F. Muldoon, S. Petoud and N. L. Rosi, Chem. Sci., 2018, 9, 8099–8102 RSC.
- I. Martinić, S. V. Eliseeva and S. Petoud, J. Lumin., 2017, 189, 19–43 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Thermal and spectroscopic data, crystallographic data, SHAPE measurement results, and additional magnetic and luminescence data. CCDC 2351436–2351445. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01379d |
| This journal is © The Royal Society of Chemistry 2024 |