DOI:
10.1039/D4DT01399A
(Paper)
Dalton Trans., 2024,
53, 11876-11883
Electron-rich pyridines with para-N-heterocyclic imine substituents: ligand properties and coordination to CO2, SO2, BCl3 and PdII complexes†
Received
13th May 2024
, Accepted 17th June 2024
First published on 20th June 2024
Abstract
Electron-rich pyridines with π donor groups at the para position play an important role as nucleophiles in organocatalysis, but their ligand properties and utilization in coordination chemistry have received little attention. Herein, we report the synthesis of two electron-rich pyridines 1 and 2 bearing N-heterocyclic imine groups at the para position and explore their coordination chemistry. Experimental and computational methods were used to assess the donor ability of the new pyridines showing that they are stronger donors than aminopyridines and guanidinyl pyridines, and that the nature of the N-heterocyclic backbone has a strong influence on the pyridine donor strength. Coordination compounds with Lewis acids including the CO2, SO2, BCl3 and PdII ions were synthesized and characterized. Despite the ambident character of the new pyridines, coordination preferentially occurs at the pyridine-N atom. Methyl transfer experiments reveal that 1 and 2 can act as demethylation reagents.
Introduction
Pyridines have been proved to be remarkably versatile synthetic building blocks and important tools in coordination chemistry and catalysis. The parent pyridine is a rather weak base and is therefore often incorporated into polydentate chelating ligands such as bipyridines and terpyridines. Donor-substituted, electron-rich pyridines, on the other hand, play an important role as nucleophilic organocatalysts for a number of synthetic transformations such as acylations,1–9 esterifications10,11 and silylations.12–14 Zipse and coworkers calculated the methyl cation affinity (MCA) and proton affinity (PA) of a series of electron-rich pyridines and showed that the MCA is a useful descriptor for catalytic activity.15–18 Pyridines with π donor substituents at the para position proved to be particularly active catalysts,1,4 and 4-dimethylaminopyridine (DMAP) and 4-pyrrolidinopyridine (PPY) have been regarded as benchmarks for decades (Fig. 1).1,2,19,20 Later, annulated aminopyridines such as 9-azajulolidene21(I), 3,4-diaminopyridines15,22,23 and 3,4,5-triaminopyridines,23,24 including 4-guanidyl-substituted pyridines (II, III),3,25 were found to outperform these benchmarks in acylation reactions. Phosphazenyl-based pyridines (IV) are among the most active catalysts, but their use is limited due to the possible cleavage of the PN bond with the irreversible formation of phosphine oxides.26,27 Apart from neutral pyridines, the incorporation of anionic groups leads to negatively charged pyridines (V–VII), which outperform DMAP in terms of their nucleophilicity and catalytic activity in the urethane-forming reaction.28–30
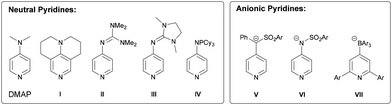 |
| | Fig. 1 Selected examples of neutral and anionic electron-rich pyridines. | |
To date, the development of electron-rich pyridines has mainly been focused on their application in organocatalysis, while their properties as ligands in coordination chemistry have received little attention. The attempt to prepare transition metal complexes with VII was unsuccessful due to single electron transfer processes from the electron-rich pyridine.31,32 2,3,5,6-Tetrakis(tetramethylguanidino)pyridine also readily undergoes electron transfer processes, yet CuI complexes have been prepared. However, the complexation of the CuI ions occurs via the guanidino rather than the pyridine donor atoms.33,34 Terpyridines bearing dialkylamino groups were employed as ligands in homoleptic RuII, CoII, ZnII and FeII complexes.35,36 The latter complex has recently been used as a catalyst in the electroreduction of CO2.37
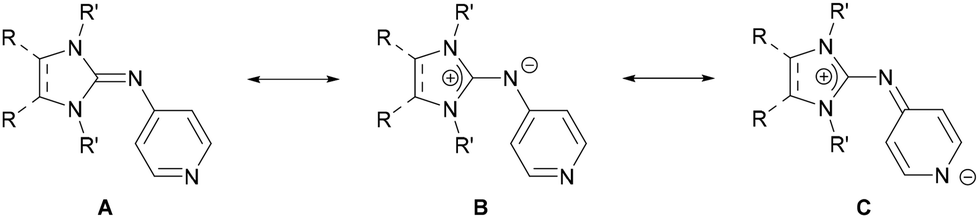
We have shown that N-heterocyclic imines (NHIs)38,39 are very effective substituents for generating highly electron-rich phosphines or porphyrins.40–46 An attractive feature of NHIs is that variations in the backbone strongly affect the ability of the exocyclic N atom to act as a π donor, providing a tool for electronic fine-tuning without compromising steric properties.47 Based on the molecular electrostatic potential (MESP)48–50 topology analysis, Krishnapriya and Suresh recently indicated that NHI substituents are very effective at increasing the electron-density at the pyridine-N atom which leads to strong donor interactions with metal centers.51 Here we report the synthesis of two electron-rich pyridines bearing different NHI groups at the para position (Fig. 2) and investigate their potential to act as strong donor ligands in coordination chemistry.
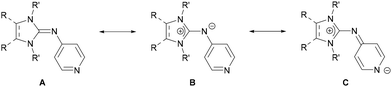 |
| | Fig. 2 Resonance structures of NHI-pyridines. | |
Results and discussion
Pyridines 1 and 2 were synthesized according to the reported procedure for the synthesis of N-heterocyclic imines52,53via coupling of 4-aminopyridine and N,N’-diisopropyl-4,5-dimethyl-2-chloroimidazolium tetrafluoroborate or N,N’-di-tert-butyl-2-chloroimidazolinium tetrafluoroborate, respectively (Fig. 3). The attempted synthesis of 1 in analogy to the preparation of III54 using triethylamine as the base did not result in the selective formation of 1. Instead, the disubstituted dication 3 was observed, as a result of the reaction of the aminopyridine with two equivalents of the imidazolium salt. Using a mixture of triethylamine and KF, 3 was formed selectively, and it was isolated after aqueous workup in 41% yield. To prevent the generation of the stable dication 3, we opted to increase the electrophilicity of the azolium salt via the in situ generation of the corresponding fluoroazolium salts without using NEt3 as an additional base. In fact, heating a suspension of 4-aminopyridine, the chloroazolium salt and KF in acetonitrile in a sealed vessel at 160 °C (1) or 90 °C (2) for three days led to the formation of 1 and 2, which were isolated after extraction with hot n-hexane and recrystallization as white solids in good yield (Fig. 3). The pyridines 1 and 2 are benchtop-stable crystalline solids that are soluble in non-polar and polar organic solvents. No evidence of degradation was detected by 1H NMR analysis after exposure of solid samples of 1 and 2 to atmospheric oxygen for several weeks. The pyridine 1H NMR resonances of 1 (7.84 and 6.17 ppm) are shielded compared to those of 2 (8.49 and 6.50 ppm) indicating that the pyridine ring is more electron-rich in the case of 1 which agrees with the stronger π donor ability of the corresponding NHI substituent.38,42,53 The solid-state structures of 1, 2 and the dication 3 were established from single-crystal X-ray diffraction (SCXRD) studies (Fig. 3). As a common structural feature, the imidazole rings are nearly orthogonal to the pyridine ring (angle between the imidazole plane and pyridine plane for 1: 82.1°, 2: 87.1°, 3: 82.1°) and the C1–N1–C2 bond angles are in a similar range (1: 123.21°, 2: 124.52°, 3: 126.82°). As expected for conjugated π systems, the C–N bond distances (1.317–1.361 Å) are in between those of classical C–N single and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds (cf.Table 1).55,56
N double bonds (cf.Table 1).55,56
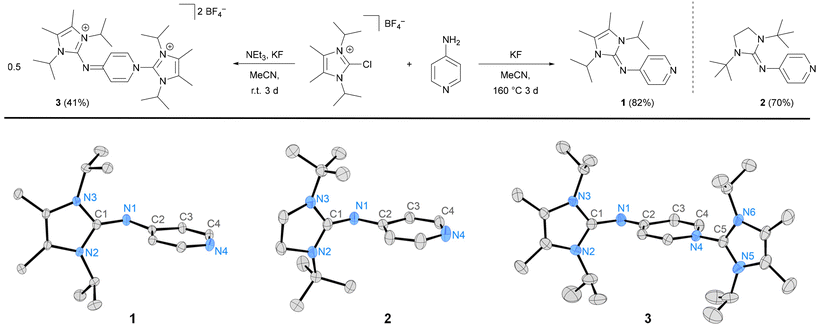 |
| | Fig. 3 Synthesis of pyridines 1, 2 and the dication 3. Molecular structures of 1, 2, and 3 with ellipsoids set at 50% probability. Hydrogen atoms and counter anions are omitted for clarity. Selected structural parameters of 1, 2 and 3 are listed in Table 1. | |
Table 1 Selected bond lengths [Å] and angles [°] of the NHI-pyridine moiety in the solid-state structures of 1, 2, 3, 4, 5, 6, 7 and 8
| |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
| C1–N1 |
1.337(2) |
1.328(2) |
1.377(5) |
1.347(2) |
1.394(2) |
1.339(3) |
1.353(3) |
1.324(4) |
| C1–N2 |
1.367(2) |
1.361(2) |
1.337(5) |
1.355(2) |
1.339(2) |
1.353(3) |
1.353(3) |
1.361(4) |
| C1–N3 |
1.357(2) |
1.359(2) |
1.342(4) |
1.352(2) |
1.339(2) |
1.364(3) |
1.357(3) |
1.354(4) |
| C2–N1 |
1.348(2) |
1.344(2) |
1.317(4) |
1.339(2) |
1.381(3) |
1.349(3) |
1.347(3) |
1.334(4) |
| C2–C3 |
1.416(2) |
1.412(2) |
1.431(5) |
1.426(2) |
1.413(3) |
1.420(3) |
1.416(3) |
1.414(4) |
| C3–C4 |
1.377(2) |
1.374(2) |
1.347(5) |
1.365(2) |
1.367(4) |
1.360(3) |
1.372(3) |
1.372(4) |
| C4–N4 |
1.343(2) |
1.342(2) |
1.378(4) |
1.350(2) |
1.356(3) |
1.353(3) |
1.352(3) |
1.348(4) |
| N1–C1–N2 |
127.6(1) |
124.9(1) |
126.8(3) |
126.5(2) |
125.3(9) |
122.7(2) |
124.7(2) |
125.0(3) |
| C1–N1–C2 |
123.2(2) |
124.5(1) |
119.2(3) |
121.2(2) |
116.4(2) |
122.2(2) |
119.4(2) |
129.5(3) |
| N2–C1–N1–C2 |
77.5(2) |
84.8(2) |
87.9(5) |
76.6(2) |
89.8(2) |
58.2(2) |
62.6(2) |
86.3(4) |
The reactivity of 1 towards the Lewis acids CO2, SO2, BCl3 and PdII complexes was investigated to determine which nitrogen donor site binds preferentially to Lewis acids and to rank the ligand donor strength. The interaction of pyridine with CO2 has been thoroughly studied due to its relevance in the context of CO2 conversion. While unsubstituted pyridine is not basic enough to form isolable adducts with CO2,57,58 spectroscopic evidence exists for the carbamate radical anion, which is the reduced form and has been proposed as a key intermediate in (photo)electrocatalytic CO2 reduction processes.59–61 We, therefore, were curious if the interaction between the electron-rich pyridine 1 and CO2 would be detectable. Subjecting an anhydrous toluene-d8 solution of 1 to an atmosphere of 0.8 bar of 13CO2 did not lead to the precipitation of the corresponding CO2 adduct, although CO2 complexes were isolated for NHIs with alkyl groups at the exocyclic nitrogen atom.53 Furthermore, the NMR analysis of the toluene solution gave no evidence of an interaction of 1 with CO2 at room temperature. Since polar solvents were shown to stabilize zwitterionic CO2 adducts,53,62 the experiment was repeated using anhydrous DMF-d7 and a higher CO2 pressure. Indeed, the 1H and 13C NMR resonances of 1 shift upon pressurizing the DMF-d7 solution with 4 bar CO2. Further evidence for the presence of the short-lived 1–CO2 complex in solution was provided by a variable temperature NMR study showing characteristic deshielding of the pyridine resonances in the 1H NMR spectra (see the ESI and Fig. S20† for details). It should be noted that this is the first spectroscopic evidence of a pyridine–CO2 complex. To study how water influences the reaction of 1 with CO2, a toluene solution containing 1 and stoichiometric amounts of water was pressurized with 4 bar CO2, which resulted in the immediate formation of the bicarbonate salt [1H]+HCO3− as a voluminous white precipitate. The same behavior is observed for tertiary amines which are important sorbents in the context of CO2 capture,63 while pyridine is not basic enough for such CO2 capture reactions. Without a CO2 atmosphere, the bicarbonate salt [1H]+HCO3− decomposes slowly at ambient temperature into 1, water and CO2 (Scheme 1). Pressurizing 1 and 1.5 eq. of water in the more polar solvent DMF-d7 with 4 bar CO2 led to the broadening and shifting of the H2O resonance in the 1H NMR spectrum. In addition, the CO2 resonance is broadened in the 13C NMR spectrum. Both indicate the reversible formation of [1H]+HCO3− in DMF solution (see the ESI and Fig. S16 and S17† for details). Collectively, these experiments indicate that 1 forms reversible complexes with CO2via low energy barrier processes, which makes it a promising nucleophile for CO2 activation protocols.64–66
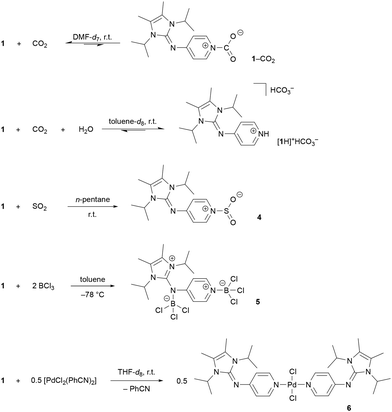 |
| | Scheme 1 Synthesis of compounds 4–6 and formation of the 1–CO2 adduct and the bicarbonate salt [1H]+HCO3−. | |
SO2 is more acidic than CO2 and the formation of the pyridine adduct therefore is energetically favored by ΔG = −0.82 kcal mol−1 in CCl4.67 While the SO2 adduct of the unsubstituted pyridine is not isolable, the more basic DMAP forms a room temperature stable SO2 adduct.68 Pressurizing a solution of 1 in n-pentane with 2 bar SO2 led to the immediate precipitation of the SO2 adduct 4 as a light-yellow solid in quantitative yield. The pyridine 1H resonances get deshielded upon SO2 complexation (4: 7.95 ppm, 6.39 ppm; 1: 7.84 ppm, 6.17 ppm). An SCXRD study confirmed that SO2 is bound to the pyridine nitrogen atom (Fig. 4). The S atom is in a trigonal-pyramidal coordination environment with an N–S bond length of 1.983 Å, an average S–O bond length of 1.421 Å and an O–S–O bond angle of 116.5°. As expected, the structural parameters of the SO2 moiety of 4 and of the DMAP-SO2 adduct (S–N: 1.991 Å, S–O: 1.446 Å, O–S–O: 114.75°)68 are very similar. It is noteworthy that despite the excess SO2 used in the reaction, only one molecule of SO2 is coordinated by the pyridine moiety, suggesting a significant basicity difference between the two nitrogen donor atoms of 1. Consistent with this regioselectivity, calculations on a benzimidazolin-2-ylidenamino pyridine show that the MCA of the pyridine-N is more exothermic by 15.71 kcal mol−1 than that of the imino-N atom.69 Pyridine 1 was therefore reacted with the strong Lewis acid BCl3 to investigate whether both nitrogen donor sites are available for coordination. Addition of BCl3 to a toluene solution of 1 at –78 °C led to the precipitation of 5 as a white solid (Scheme 1), which was isolated in low yield by filtration. The adduct 5 is sparingly soluble in polar solvents like DCM and fluorobenzene. Coordination of the imino-N atom to the bulky BCl3 moiety hampers rotation around the exocyclic C–N bond in 5, as indicated by the two doublets in the 1H NMR spectrum at 1.53 ppm and 1.40 ppm of the diastereotopic CH3 groups. Moreover, the pyridine 1H resonances are significantly deshielded (5: 8.92 ppm, 7.45 ppm; 1: 7.84 ppm, 6.17 ppm). The 11B NMR spectrum shows two singlets at 7.7 ppm and 5.8 ppm in the range of the resonance of the pyridine-BCl3 adduct (δ = 8.1 ppm).70 The solid-state structure of 5 was established from an SCXRD study confirming the ambident character of the NHI-substituted pyridine (Fig. 4). Both boron atoms are in a tetrahedral coordination environment and exhibit shorter N–B bond lengths (N1–B1: 1.541 Å, N4–B2: 1.583 Å) than Me3N–BCl3 (1.610 Å).71 We next explored the formation of PdII complexes with electron-rich pyridines. Treatment of [PdCl2(PhCN)2] with two equivalents of 1 in THF led to the selective formation of complex trans-[PdCl2(1)2] (6), which was obtained as an air-stable yellow solid in quantitative yield (Scheme 1). The solid-state structure of 6 displays a square planar geometry at PdII with the pyridine ligands in the trans position (Fig. 4). The selective formation of the pyridine-bound PdII complex 6 prompted us to assess the donor strength of 1 and 2 using the Huynh Electronic Parameter (HEP).72–74 This method utilizes the 13C NMR chemical shift of the carbene C atom in complexes trans-[PdBr2(BiPr)(L)] (BiPr = 1,3-dimethylbenzimidazolin-2-ylidene) as a probe to determine the donor strength of the ligand L. Treatment of 1 and 2 with the PdII dimer [PdBr2(BiPr)]2 cleanly gave the corresponding complexes trans-[PdBr2(BiPr)(1)] (7) and trans-[PdBr2(BiPr)(2)] (8) (vide supra). The 1H NMR spectra of 7 and 8 show one set of signals for the pyridine ligands. The methyl resonances of the iPr groups of 1 are magnetically equivalent, consistent with coordination of the pyridine-N atom to the PdII center. SCXRD studies confirm the trans arrangement of the pyridine ligands with Pd1–N4 bond lengths of 2.088 Å (7) and 2.101 Å (8), that are slightly elongated compared to that of 6 (2.023 Å). The 13C resonance of the carbene C atom was detected at 163.2 ppm (7) and 163.1 ppm (8), which suggests that 1 and 2 are stronger donors than pyridine (160.0 ppm)72 and DMAP (161.9 ppm)75 (Fig. 5). The TEP values calculated using the method reported by Gusev76 show the same trend (1: 2061.28 cm−1, 2: 2062.52 cm−1, DMAP: 2065.42 cm−1).
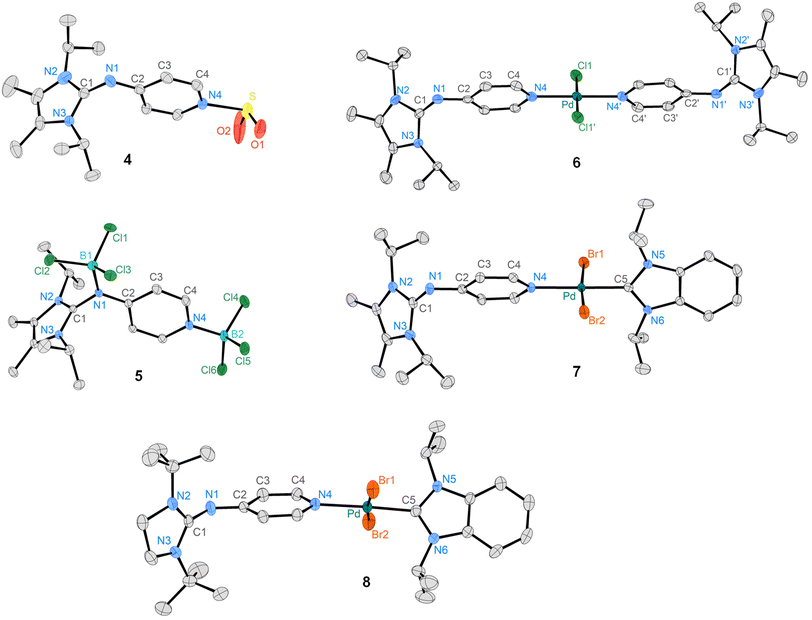 |
| | Fig. 4 Molecular structures of compounds 4–8 with ellipsoids set at 50% probability. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: 4: N4–S 1.9826(13), S–O1 1.423(2), S–O2 1.418(2), O1–S–O2 116.47(11); 5: N1–B1 1.541(3), N4–B2 1.583(3); ∅ B1–Cl 1.859, ∅ B2–Cl 1.843; 6: Pd–N4 2.023(2), Pd–Cl1 2.3061(5), Cl1–Pd–N4 89.56(2); 7: Pd–N4 2.088(2), Pd–C5 1.954(2), Pd–Br1 2.423(3), Br1–Pd–N4 92.55(5); 8: Pd–N4 2.101(2), Pd–C5 1.958(3), Pd–Br1 2.436(4), N4–Pd–Br1 93.17(7). | |
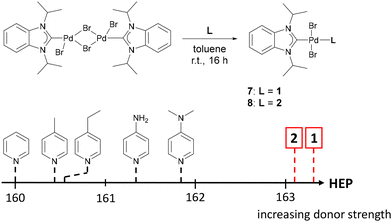 |
| | Fig. 5 The Huynh electronic parameter (HEP) scale for different pyridines. The chemical shifts of the 13C{1H} carbene resonance of the BiPr ligand are listed in Table 2. | |
Collectively, these results show that the introduction of NHI substituents at the para position is more effective than dialkylamino groups in increasing the donor properties of pyridine. The TEP and HEP both take the σ and π contributions of the ligand into account for ranking the donor strength. To gauge the σ donor ability of 1 and 2, we therefore calculated the methyl cation affinities (MCA) using the method of Zipse and coworkers.16,35,77 Comparison of the MCA of 1 and 2 with other substituted pyridines gives the same qualitative trend as observed for the HEP and TEP (Table 2). Notably, 1 and 2 both have considerably higher MCA than the acyclic guanidine derivative II and the organic superbase DBN (1,5-diazabicyclo(4.3.0)non-5-en).
Table 2 Methyl cation affinity (MCA) and Huynh electronic parameter (HEP) of different pyridines. MCA values were calculated at the MP2(FC)/6-31+G(2d,p)//B98/6-31G(d) level of theory
| Lewis base (LB) |
MCA (kJ mol−1) |
δ C
carbene
(ppm) |
|
Measured in CDCl3 and internally referenced to the solvent residual signal at 77.7 ppm relative to TMS.
Ref. 16
Ref. 74
Ref. 75
|
| Pyridine |
518.7b |
160.1c |
| 4-Picoline |
532.8b |
160.4d |
| 4-Ethylpyridine |
— |
160.5d |
| 4-Aminopyridine |
— |
161.3d |
| DMAP |
581.2b |
161.9d |
| 4-Tmg-pyridine(II) |
597.5b |
— |
| 9-Azajulolidene(I) |
602.4b |
— |
| DBN |
611.3b |
— |
|
1
|
659.2 |
163.2 |
|
2
|
624.8 |
163.1 |
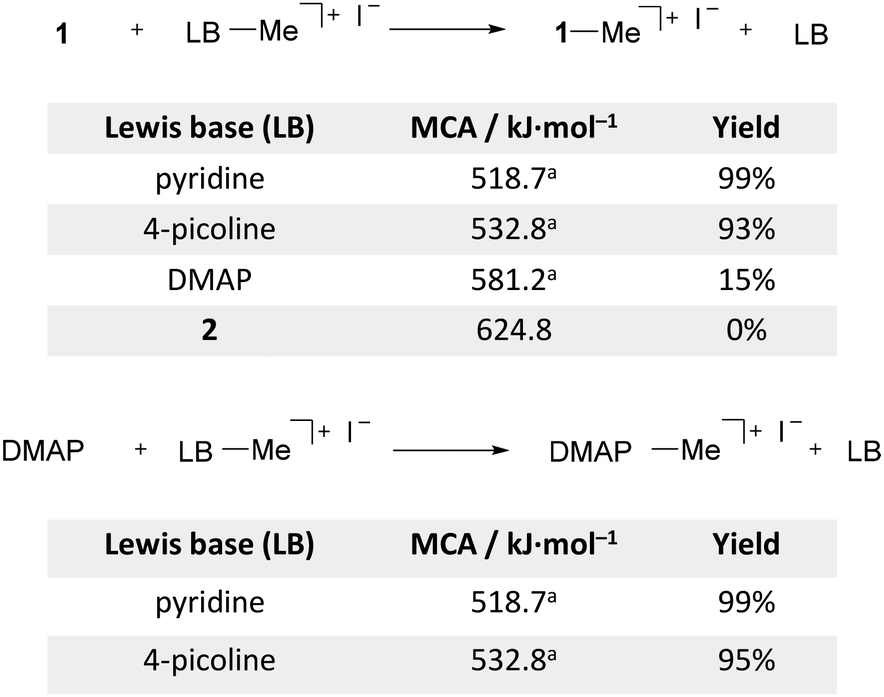
The large differences in the calculated MCA inspired us to experimentally investigate the transfer of methyl groups between pyridine derivatives (Scheme 2). Several methodologies have been reported for the N-dealkylation of pyridinium salts,78 which typically involve heating of the salt in the presence of a base including pyridine,79N-methylimidazole and triphenylphosphine.80 In the case of pyridinium iodide salts, the demethylation was shown to be a two-step process.81,82 The first and rate-determining step involves an SN2 substitution with the iodide counter anion acting as the nucleophile to generate pyridine and iodomethane. The second and faster step is the subsequent methylation of the sequestration reagent with the liberated iodomethane. We studied the methyl cation transfer by heating MeCN-d3 solutions containing stoichiometric mixtures of 1 and the methylated pyridinium iodide salts in sealed NMR tubes. The reaction process was monitored by 1H NMR spectroscopy. This showed that prolonged heating of the reaction mixture to 160 °C was required to achieve appreciable transfer rates. Demethylation of the pyridinium and 4-picolinium salts takes place in high yields using 1 or DMAP as a methyl scavenger. 1 and DMAP show similar reaction rates (see the ESI† for details), which agrees with the abovementioned two-step mechanism in which the iodine counterion is the active demethylation agent. Accordingly, methyl transfer from the [DMAP·Me]+I− salt to 1 is very slow with only 15% conversion after one week, and no sign of demethylation was detected in the case of [2·Me]+I−. The rate of methyl transfer thus depends primarily on the MCA of the methylated base and is independent of the ΔMCA between the bases in the reaction mixture.
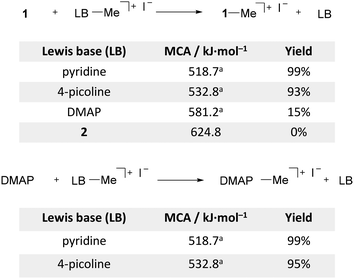 |
| | Scheme 2 Methyl cation transfer experiments using 1 (top) and DMAP (bottom) as a sequestration reagent. NMR yields after heating the MeCN-d3 solutions in sealed NMR tubes at 160 °C for 7 days. aref. 16 | |
Conclusion
In conclusion, we find that pyridines bearing N-heterocyclic imine substituents at the para position, which so far have only been examined with respect to their catalytic potential in acylation reactions of alcohols, show promise as strong donor ligands in coordination chemistry. The experimental determination of the HEP and the calculation of the TEP and MCA show that pyridines 1 and 2 are considerably stronger donor ligands than the aminopyridine DMAP or the acyclic 4-guanidinyl pyridine II. The descriptors indicate that the π donor ability of the N-heterocyclic imine substituent has a significant impact on the electronic properties of the pyridine, which provides leverage for tuning the stereoelectronic properties of pyridines. Reactivity studies with various Lewis acids show that 1 can act as an ambident ligand, with the pyridine nitrogen atom being preferentially coordinated. In addition, the transfer of methyl groups between different pyridines was investigated. This showed that 1 and DMAP can be used for the demethylation of pyridine derivatives at rates that depend primarily on the MCA of the latter. Furthermore, NHI-substituted pyridine derivatives are currently being developed in our laboratories with a focus on applications for small molecule activation and catalysis.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
J. H. F. and L. F. B. W. carried out experiments for the synthesis of 1 and 2 and their respective PdII complexes and Lewis acid adducts 4–8. J. H. F investigated the CO2 adduct formation and methyl cation transfer reactions. P. R. performed the calculations of the MCA and TEP values for 1 and 2. L. F. B. W., K. W. and M. S. performed the SCXRD studies. F. D. directed the investigations. The manuscript was written and edited by J. H. F. and F. D. with contributions from L. F. B. W. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We sincerely thank the Tiroler Wissenschaftsförderung (TWF, F.45075) for funding to realize this project and to the German Academic Scholarship Foundation for a PhD fellowship (L. F. B. W.). We thank assoc. Prof. Dr Christoph Kreutz for the measurements of the variable temperature NMR spectra.
References
- W. Steglich and G. Höfle, Angew. Chem., Int. Ed. Engl., 1969, 8, 981 (
Angew. Chem.
, 1969
, 81
, 1001
) CrossRef CAS.
- G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1978, 17, 569–583 (
Angew. Chem.
, 1978
, 90
, 602–615
) CrossRef.
- A. Hassner, L. R. Krepski and V. Alexanian, Tetrahedron, 1978, 34, 2069–2076 CrossRef CAS.
- A. C. Spivey and S. Arseniyadis, Angew. Chem., Int. Ed., 2004, 43, 5436–5441 CrossRef CAS PubMed.
- E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129 RSC.
-
Applications of Dialkylaminopyridine (DMAP) Catalysts in Organic Synthesis, ed. M. Ramiah and E. F. V. Scriven, 2003 Search PubMed.
- G. C. Fu, Acc. Chem. Res., 2000, 33, 412–420 CrossRef CAS PubMed.
- G. C. Fu, Acc. Chem. Res., 2004, 37, 542–547 CrossRef CAS PubMed.
- U. Ragnarsson and L. Grehn, Acc. Chem. Res., 1998, 31, 494–501 CrossRef CAS.
- L. J. Gooßen and A. Döhring, Synlett, 2004, 263–266 CrossRef.
- I. Held, P. von den Hoff, D. S. Stephenson and H. Zipse, Adv. Synth. Catal., 2008, 350, 1891–1900 CrossRef CAS.
- S. K. Chaudhary and O. Hernandez, Tetrahedron Lett., 1979, 20, 99–102 CrossRef.
- P. Patschinski, C. Zhang and H. Zipse, J. Org. Chem., 2014, 79, 8348–8357 CrossRef CAS PubMed.
- M. Marin-Luna, P. Patschinski and H. Zipse, Chem. – Eur. J., 2018, 24, 15052–15058 CrossRef CAS PubMed.
- E. Larionov, F. Achrainer, J. Humin and H. Zipse, ChemCatChem, 2012, 4, 559–566 CrossRef CAS.
- Y. Wei, G. N. Sastry and H. Zipse, J. Am. Chem. Soc., 2008, 130, 3473–3477 CrossRef CAS PubMed.
- C. Lindner, R. Tandon, B. Maryasin, E. Larionov and H. Zipse, Beilstein J. Org. Chem., 2012, 8, 1406–1442 CrossRef CAS PubMed.
- I. Despotović and R. Vianello, Chem. Commun., 2014, 50, 10941–10944 RSC.
- N. de Rycke, F. Couty and O. R. P. David, Chem. – Eur. J., 2011, 17, 12852–12871 CrossRef CAS PubMed.
- M. Baidya and H. Mayr, Chem. Commun., 2008, 1792–1794 RSC.
- M. R. Heinrich, H. S. Klisa, H. Mayr, W. Steglich and H. Zipse, Angew. Chem., Int. Ed., 2003, 42, 4826–4828 CrossRef CAS PubMed.
- I. Held, S. Xu and H. Zipse, Synthesis, 2007, 1185–1196 CAS.
- S. Singh, G. Das, O. V. Singh and H. Han, Org. Lett., 2007, 9, 401–404 CrossRef CAS PubMed.
- R. Tandon, T. Unzner, T. A. Nigst, N. de Rycke, P. Mayer, B. Wendt, O. R. P. David and H. Zipse, Chem. – Eur. J., 2013, 19, 6435–6442 CrossRef CAS PubMed.
- I. Held, E. Larionov, C. Bozler, F. Wagner and H. Zipse, Synthesis, 2009, 2267–2277 CAS.
- N. A. Richard, C. K. Khor, S. M. Hetherington, S. L. Mills, A. Decken and C. A. Dyker, Chem. – Eur. J., 2020, 26, 17371–17375 CrossRef CAS PubMed.
- N. A. Richard, G. D. Charlton and C. A. Dyker, Org. Biomol. Chem., 2021, 19, 9167–9171 RSC.
- E. Anders, U. Korn and A. Stankowiak, Chem. Ber., 1989, 122, 105–111 CrossRef CAS.
- J. Helberg, T. Ampßler and H. Zipse, Chem. Commun., 2020, 85, 5390–5402 CAS.
- S. H. Dempsey, A. Lovstedt and S. R. Kass, J. Org. Chem., 2023, 88, 10525–10538 CrossRef CAS PubMed.
- N. M. C. Schmidlin, M. Lõkov, I. Leito and T. Böttcher, Chem. – Eur. J., 2018, 24, 16851–16856 CrossRef CAS PubMed.
- N. M. C. Schmidlin, V. Radtke, A. Schmidt, M. Lõkov, I. Leito and T. Böttcher, Z. Anorg. Allg. Chem., 2022, 648, e202200136 CrossRef CAS.
- S. Stang, A. Lebkücher, P. Walter, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2012, 2012, 4833–4845 CrossRef CAS.
- S. Wiesner, A. Wagner, O. Hübner, E. Kaifer and H.-J. Himmel, Chem. – Eur. J., 2015, 21, 16494–16503 CrossRef CAS PubMed.
- P. Hommes, C. Fischer, C. Lindner, H. Zipse and H.-U. Reissig, Angew. Chem., Int. Ed., 2014, 53, 7647–7651 CrossRef CAS PubMed.
- M. Kleoff, S. Suhr, B. Sarkar, R. Zimmer, H.-U. Reissig, M. Marin-Luna and H. Zipse, Chem. – Eur. J., 2019, 25, 7526–7533 CrossRef CAS PubMed.
- S. Suhr, N. Schröter, M. Kleoff, N. Neuman, D. Hunger, R. Walter, C. Lücke, F. Stein, S. Demeshko, H. Liu, H.-U. Reissig, J. van Slageren and B. Sarkar, Inorg. Chem., 2023, 62, 6375–6386 CrossRef CAS PubMed.
- X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS.
- T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC.
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857–11860 CrossRef PubMed.
- P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem. – Eur. J., 2017, 23, 5929–5933 CrossRef CAS PubMed.
- T. Witteler, H. Darmandeh, P. Mehlmann and F. Dielmann, Organometallics, 2018, 37, 3064–3072 CrossRef CAS.
- L. F. Wilm, P. Mehlmann, F. Buß and F. Dielmann, J. Organomet. Chem., 2020, 909, 121097 CrossRef CAS.
- M. D. Böhme, T. Eder, M. B. Röthel, P. D. Dutschke, L. F. B. Wilm, F. E. Hahn and F. Dielmann, Angew. Chem., Int. Ed., 2022, 61, e202202190 (
Angew. Chem.
, 2022
, 134
, e202202190
) CrossRef PubMed.
- P. Rotering, L. F. B. Wilm, J. A. Werra and F. Dielmann, Chem. – Eur. J., 2020, 26, 406–411 CrossRef CAS PubMed.
- M. Abdinejad, L. F. B. Wilm, F. Dielmann and H. B. Kraatz, ACS Sustainable Chem. Eng., 2021, 9, 521–530 CrossRef CAS.
- P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139–1143 CrossRef CAS PubMed.
- C. H. Suresh and S. R. Gadre, J. Am. Chem. Soc., 1998, 120, 7049–7055 CrossRef CAS.
- G. S. Remya and C. H. Suresh, Phys. Chem. Chem. Phys., 2016, 18, 20615–20626 RSC.
- F. B. Sayyed and C. H. Suresh, Tetrahedron Lett., 2009, 50, 7351–7354 CrossRef CAS.
- V. U. Krishnapriya and C. H. Suresh, Organometallics, 2023, 42, 571–580 CrossRef CAS.
- R. A. Kunetskiy, S. M. Polyakova, J. Vavřík, I. Císařová, J. Saame, E. R. Nerut, I. Koppel, I. A. Koppel, A. Kütt, I. Leito and I. M. Lyapkalo, Chem. – Eur. J., 2012, 18, 3621–3630 CrossRef CAS PubMed.
- L. F. B. Wilm, T. Eder, C. Mück-Lichtenfeld, P. Mehlmann, M. Wünsche, F. Buß and F. Dielmann, Green Chem., 2019, 21, 640–648 RSC.
- J. Garnier, A. R. Kennedy, L. E. A. Berlouis, A. T. Turner and J. A. Murphy, Beilstein J. Org. Chem., 2010, 6, 73 Search PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- J. L. Doran, B. Hon and K. R. Leopold, J. Mol. Struct., 2012, 1019, 191–195 CrossRef CAS.
- K. D. Vogiatzis, A. Mavrandonakis, W. Klopper and G. E. Froudakis, ChemPhysChem, 2009, 10, 374–383 CrossRef CAS PubMed.
- S. Y. Han, I. Chu, J. H. Kim, J. K. Song and S. K. Kim, J. Chem. Phys., 2000, 113, 596–601 CrossRef CAS.
- M. Z. Kamrath, R. A. Relph and M. A. Johnson, J. Am. Chem. Soc., 2010, 132, 15508–15511 CrossRef CAS PubMed.
- D. V. Vasilyev and P. J. Dyson, ACS Catal., 2021, 11, 1392–1405 CrossRef CAS.
- J. K. Mannisto, L. Pavlovic, T. Tiainen, M. Nieger, A. Sahari, K. H. Hopmann and T. Repo, Catal. Sci. Technol., 2021, 11, 6877–6886 RSC.
- X. Yang, R. J. Rees, W. Conway, G. Puxty, Q. Yang and D. A. Winkler, Chem. Rev., 2017, 117, 9524–9593 CrossRef CAS PubMed.
- L. J. Murphy, K. N. Robertson, R. A. Kemp, H. M. Tuononen and J. A. C. Clyburne, Chem. Commun., 2015, 51, 3942–3956 RSC.
- H. Zhou and X. Lu, Sci. China: Chem., 2017, 60, 904–911 CrossRef CAS.
- P. Sreejyothi and S. K. Mandal, Chem. Sci., 2020, 11, 10571–10593 RSC.
- J. W. Keller, J. Phys. Chem. A, 2015, 119, 10390–10398 CrossRef CAS PubMed.
- K. Huynh, E. Rivard, A. J. Lough and I. Manners, Chem. – Eur. J., 2007, 13, 3431–3440 CrossRef CAS PubMed.
- N. Patel, M. Arfeen, R. Sood, S. Khullar, A. K. Chakraborti, S. K. Mandal and P. V. Bharatam, Chem. – Eur. J., 2018, 24, 6418–6425 CrossRef CAS PubMed.
- S. Coffie, J. M. Hogg, L. Cailler, A. Ferrer-Ugalde, R. W. Murphy, J. D. Holbrey, F. Coleman and M. Swadźba-Kwaśny, Angew. Chem., Int. Ed., 2015, 54, 14970–14973 CrossRef CAS PubMed.
- P. H. Clippard, J. C. Hanson and R. C. Taylor, J. Cryst. Mol. Struct., 1971, 1, 363–371 CrossRef CAS.
- H. V. Huynh, Y. Han, R. Jothibasu and J. An Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- Q. Teng and H. V. Huynh, Dalton Trans., 2017, 46, 614–627 RSC.
- Y. Han, H. V. Huynh and G. K. Tan, Organometallics, 2007, 26, 6447–6452 CrossRef CAS.
- Q. Teng, P. S. Ng, J. N. Leung and H. V. Huynh, Chem. – Eur. J., 2019, 25, 13956–13963 CrossRef CAS PubMed.
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS.
- E. Larionov and H. Zipse, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 601–619 CAS.
- A. Ruiz, P. Rocca, F. Marsais, A. Godard and G. Quéguiner, Tetrahedron Lett., 1997, 38, 6205–6208 CrossRef CAS.
- A. R. Katritzky and S. S. Thind, J. Chem. Soc., Perkin Trans. 1, 1980, 1895 RSC.
- L. W. Deady and W. L. Finlayson, Synth. Commun., 1980, 10, 947–950 CrossRef CAS.
- L. W. Deady and O. L. Korytsky, Tetrahedron Lett., 1979, 20, 451–452 CrossRef.
- C. Yin, K. Zhong, W. Li, X. Yang, R. Sun, C. Zhang, X. Zheng, M. Yuan, R. Li, Y. Lan, H. Fu and H. Chen, Adv. Synth. Catal., 2018, 360, 3990–3998 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a