
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of N-heterocyclic carbene (NHC) gold-fluoroalkoxide complexes†
Pierre
Arnaut
a,
Nestor
Bracho Pozsoni
a,
Fady
Nahra
 ab,
Nikolaos V.
Tzouras
*a and
Steven P.
Nolan
*a
ab,
Nikolaos V.
Tzouras
*a and
Steven P.
Nolan
*a
aDepartment of Chemistry and Centre for Sustainable Chemistry, Ghent University, Krijgslaan 281, S-3, 9000 Ghent, Belgium. E-mail: nikolaos.tzouras@ugent.be; steven.nolan@ugent.be
bMaterials & Chemistry (MATCH) unit, VITO (Flemish Institute for Technological Research), Boeretang 200, 2400 Mol, Belgium
First published on 28th June 2024
Abstract
We disclose a novel series of N-heterocyclic carbene (NHC) gold complexes with varied steric and electronic properties, bearing fluorinated alkoxide anions. Early reactivity studies involving these synthons, lead to the synthesis of various complexes of relevance to gold chemistry and catalysis.
Introduction
It is now well established that fluorinated alcohols are powerful tools in organic chemistry that combine high polarity, low nucleophilicity, and strong hydrogen-bond donor character.1,2 The presence of electron-withdrawing fluoroalkyl groups grant the alcohols unique solvating properties through hydrogen bonding networks.3,4 These can be especially useful as solvents in heterocycle synthesis,5,6 C–H functionalization,7 cross-coupling reactions,8,9 and in the controlled deprotection of functional groups in the presence of other labile protected groups.10 It is worth mentioning that among these, a specific member of this family of alcohols, hexafluoroisopropanol, (HFIP) has been deployed in numerous applications in the chemistry of peptides, as this last deprotection strategy is a key step in the total synthesis of human insulin (on N-trityl and N-Bpoc groups).11Compared to other fluorinated alcohols such as trifluoroethanol (TFE), HFIP possesses a lower boiling point and a higher melting point, making it a particularly attractive solvent since it can be easily recycled without preventing its use in synthesis at moderate temperatures (below 60 °C). HFIP can promote reactivity in three major ways: it can increase acidity through hydrogen bonding clusters, provide electrostatic stabilization of ionic species, and assist in ionization through coordination.12 The effects of hydrogen bonding are especially valuable in catalysis, whether it is for electrophile or catalyst activation.13,14 In gold catalysis, gold–chloride complexes require the use of additives such as silver or potassium salts to be rendered active.15,16 Indeed, it is only through halogen abstraction that the highly electrophilic gold cationic species is generated.17 HFIP has recently been used for this purpose and is able, through hydrogen bonding, to weaken the gold–chloride bond.18 The two most noticeable advantages of [AuCl(NHC)] complexes are their stability and ease of synthesis via the weak base route,14 which is now routinely deployed for a plethora of gold-NHC complexes.19–21 In comparison, gold-alkoxide complexes have been shown to be very unstable in solution and can be easily converted to their hydroxide equivalent when placed in contact with water.22 As for the NHC gold hydroxides themselves, the use of strong bases remains unavoidable at this time for their synthesis.19,22,23
The chemistry of gold complexes has been the subject of intense research over the past decades.24,25 The earlier examples of gold-fluoroalkoxide species from Komiya and co-workers permitted the condensation of benzaldehyde as well as the ring opening of thiiranes with gold–phosphine complexes.26,27 The ability of these complexes to abstract protons from a wide range of substrates is the driving force behind their reactivity, to generate the corresponding carbonyl organogold compounds.28,29 The incorporation of NHC ligands later led to the isolation of the first gold-hydroxide complex,30 which was then engaged in the carboxylation of oxazole through C–H activation, among many other synthetic and catalytic applications.31 More recently, the role of gold-phenoxides in the hydrophenoxylation of alkynes has been described for both mono32 and dual activation.33 NHC gold-aryloxides can be also synthesized from NHC gold–chloride and enable the hydration of phenylacetylene.34 Yet despite these remarkable advances, the synthesis and reactivity of NHC gold-polyfluoroalkoxide complexes has remained underexplored. Therefore, we investigated such systems to seek potential benefits of gold-fluoroalkoxide species from a practical perspective.
Results and discussion
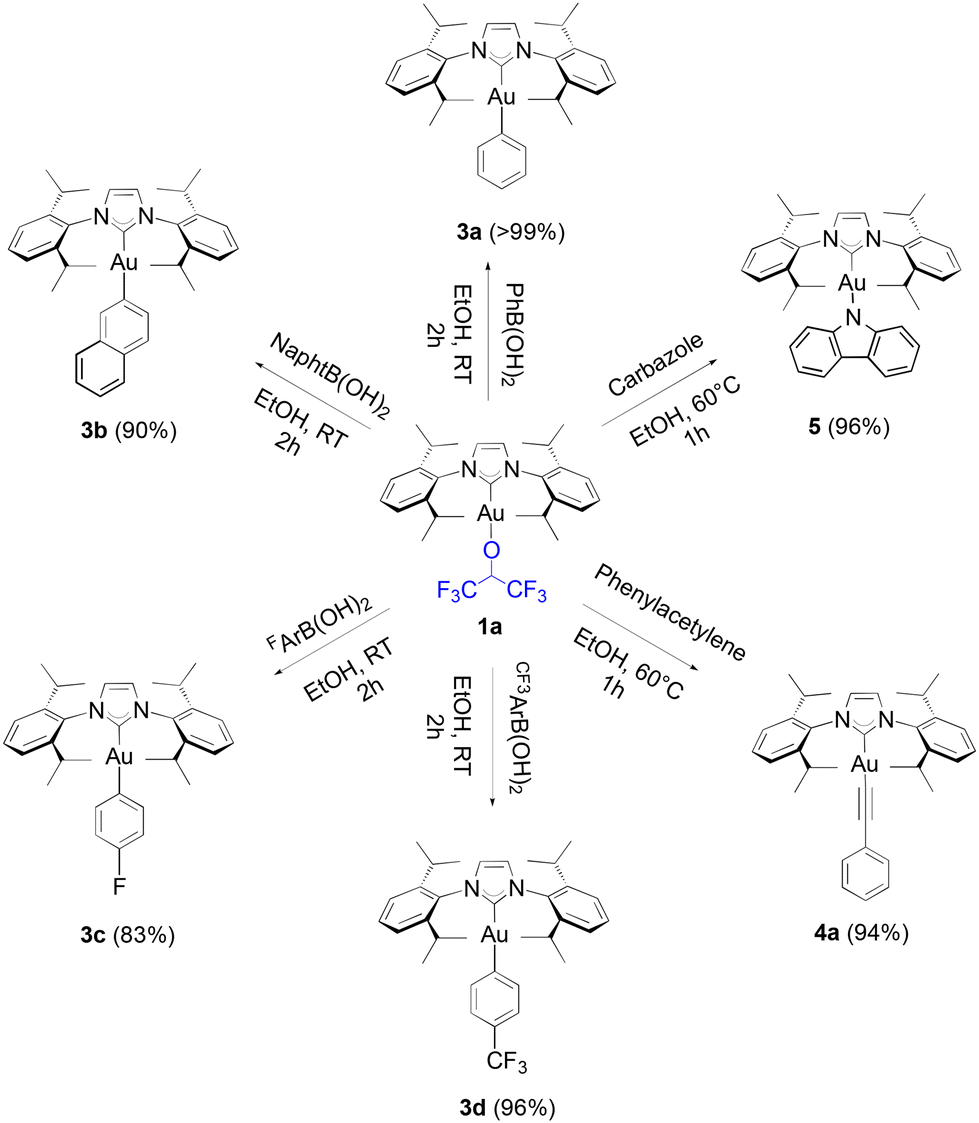
Our study begins with the use of a standard NHC ligand, N,N′-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) bound to gold. As we have previously disclosed,14 [Au(IPr)(OCH(CF3)2)] (1a) can be generated at room temperature from [Au(IPr)(OH)] in benzene within one hour. An even more straightforward route makes use of [Au(IPr)Cl] and 3 equivalents (eq.) of K2CO3, stirred at room temperature in dry ethanol for thirty minutes with 1.1 eq. of HFIP. This weak base route can also be performed on a gram-scale in high yields (see, Experimental section). It should be noted that no reaction is observed in the absence of base. Additionally, alternate bases such as NEt3 and NaOAc proved to be inactive in leading to the desired product. To explore the efficacy of the weak base route on other fluorinated alcohols, the use of trifluoroethanol (TFE) was examined overnight under the same conditions and led to the formation of 2a (eqn (1)) in high yield.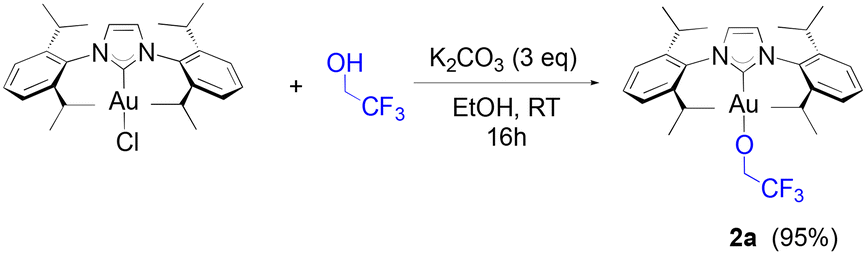 | (1) |
Several [Au(NHC)] core structures were subjected to this simple protocol and results are presented in Scheme 1. Changing the backbone from electron withdrawing to electron donating groups with IPrCl (1c) and IPrMe (1d) had no negative impact on isolated yields, but SIPr (1b) could only be isolated with 75% isolated yield. Increasing the bulk of the ligand from IPr to IPr* (1f) did not noticeably affect the yield. The molecular structure of 1d is presented in Fig. 1 with a selection of bond distances and angles. In the extreme case of the exceedingly bulky IPr# however, steric effects drastically hindered reactivity, and no improvement was made even when other solvents, such as THF, were used. As for N-alkyl-bearing ligands, the ItBu complex 1h was obtained with 98% yield. The fluoroalkoxide complex can also be accessed in high yield through this method when a tertiary phosphine ligand is used (1j).
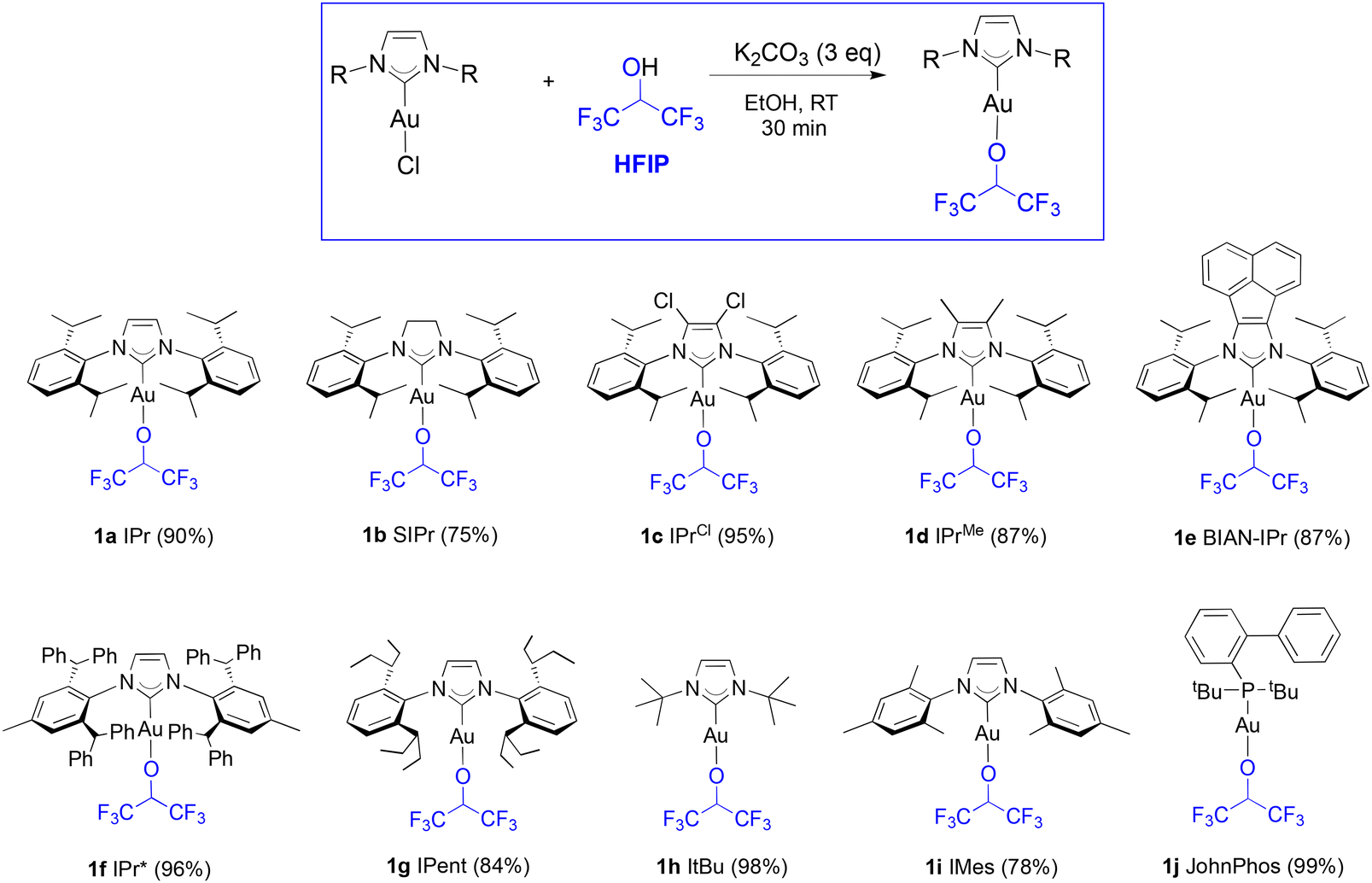 | ||
| Scheme 1 Synthetic route to various NHC and phosphine bearing gold-fluoroalkoxide complexes. | ||
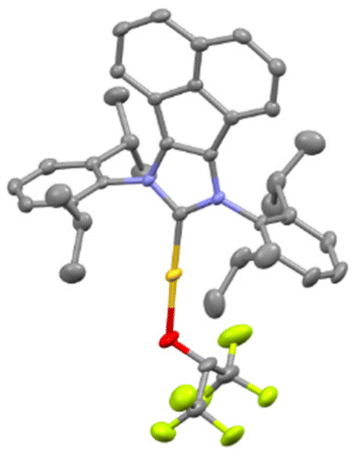 | ||
| Fig. 1 X-ray molecular structure of 1e showing thermal displacement ellipsoids at the 50% probability level. Hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (deg): Au–O 2.017(1), Au–C 1.948(1); O–CHFIP 1.372(1); N–C–N 106.29, CNHC–Au–O 175.89 (13). CCDC 2311517.† | ||
Boronic acids were initially tested in our reactivity study for the arylation of the gold-fluoroalkoxide complexes. When 1a was reacted with phenyl, 2-naphthyl, 4-fluorophenyl and 4-trifluoromethylphenyl boronic acids, the desired gold-aryl derivatives (3a, 3b, 3c and 3d, respectively) were obtained in excellent yields in reaction times of two hours at room temperature (Scheme 2).
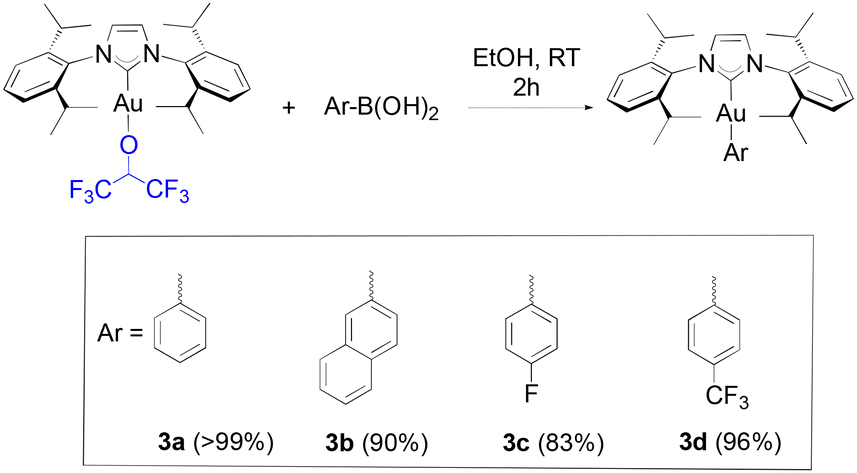 | ||
| Scheme 2 Scope of boronic acids synthesized in this study with isolated yields given in parentheses. | ||
To our knowledge, short reaction times for these transformations had only been achieved thus far by relying on the use of strong bases such as KOH on the gold hydroxide.35 Following the weak base route from [Au(IPr)Cl], the synthesis of these same products requires between 4 to 24 hours.36,37 However, no desired product was observed from the reaction of 1a with 4-methoxyphenyl boronic acid, and the same result was observed on 4-tolyl boronic acid. Finally, under identical conditions to HFIP, TFE gave a noticeably lower, but still acceptable yield with phenylboronic acid (eqn (2)).
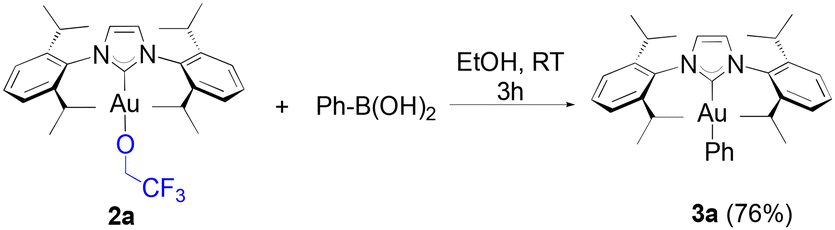 | (2) |
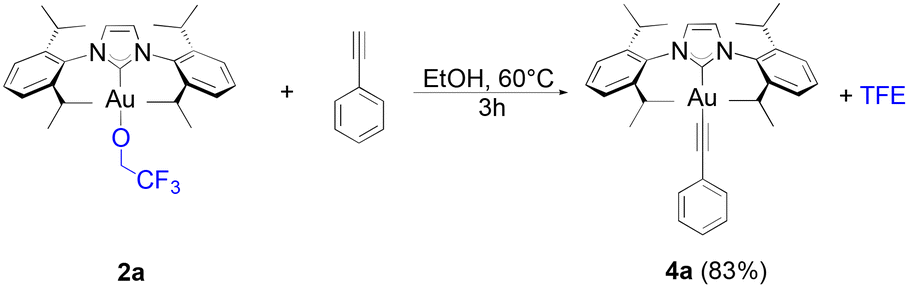
Alkynyl complexes can be easily accessed from [Au(IPr)Cl], 2 eq. of phenylacetylene and 3 eq. of sodium acetate in ethanol at room temperature yielding the product in a 93% yield.38 When 1a was used as the starting gold synthon, a 75% yield was reached under the same conditions. Improved results were achieved at higher temperatures (60 °C) with a 94% yield obtained of 4a after one hour (Scheme 3).
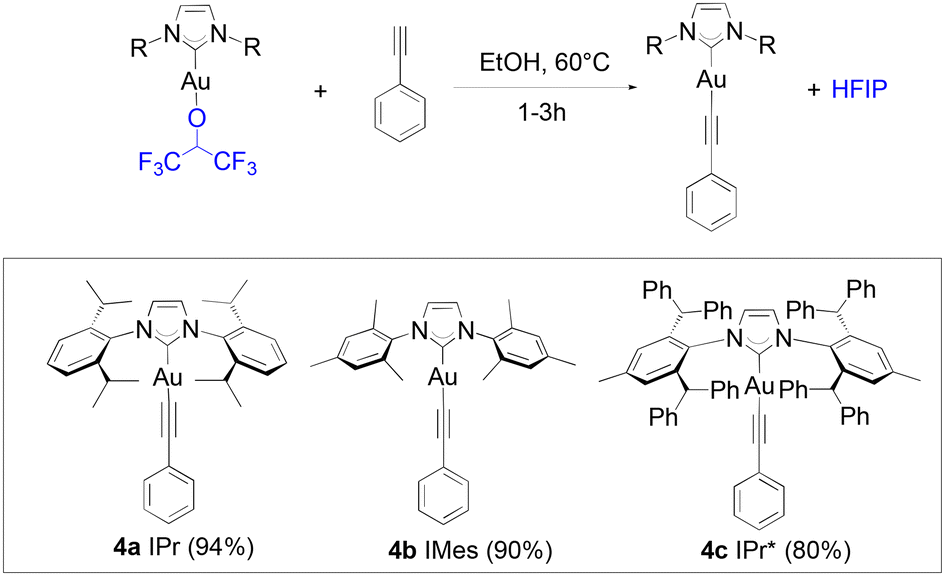 | ||
| Scheme 3 Scope and reaction conditions for the synthesis of gold-acetylide complexes. | ||
The lower reactivity of 1a compared to that displayed by [Au(IPr)(OH)] is overcome by using a slightly higher reaction temperature. The same is observed when 1i reacts with phenylacetylene to yield 4b, in one hour. When using 1f, only a 50% conversion is observed within the first hour (or up to 80% with 3 eq. of alkyne) in the alkynylation reaction. The slower kinetics must be caused by the significant higher steric hindrance of the bulky IPr* compared to that of IPr. Nevertheless, high yields can still be obtained for 4c after three hours, and further improved to 85% with the use of an excess of the alkyne (3 eq.).
 | (3) |
Using 2a as the starting gold complex requires slightly longer reaction times than when 1a is used and full conversion is not observed after 60 minutes (eqn (3)). This is surprising as TFE, being more electron rich than HFIP, would be expected to be more reactive. Considering that HFIP has a lower boiling point than TFE (58 °C compared to 74 °C), the evaporation of HFIP during the reaction might be the reason behind the shift in equilibrium in favour of the gold-alkynyl bond formation.
Another example of the reactivity of 1a is exemplified with a carbazole substrate resulting in the formation of complex 5 in a 96% yield in ethanol within one hour, but did require heating at 60 °C, whereas the weak base route can be performed at room temperature but requires a 24-hour reaction time. It should be noted that this gold complex 5 appears extremely versatile as a photocatalyst in cycloaddition reactions (Scheme 4).38–41
 | ||
| Scheme 4 Examples of the use of 1a as synthon. | ||
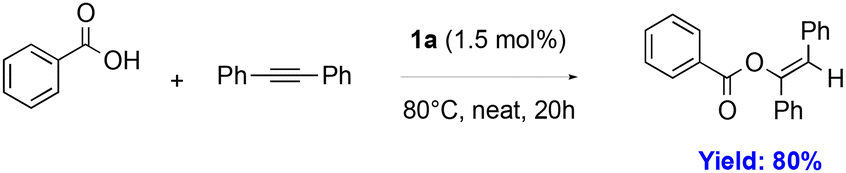
To confirm the catalytic efficacy of our well-defined [Au(NHC)(OCH(CF3)2)] complexes, 1a was deployed in the addition of carboxylic acids to alkynes (eqn (4)) resulting in an 80% yield. This compares to NMR yields of 87% using the [Au(IPr)(cbz)] (cbz = carbazolyl) catalyst under identical conditions.42
 | (4) |
The weak base route is routinely used in the synthesis of [Au(NHC)Cl] complexes and now enables access to a variety of bench-stable gold fluoroalkoxide complexes. These fluoroalkoxide complexes display Csp–H/N–H bond activation properties and permit the synthesis of aryl-, alkynyl- and amido gold complexes. A remarkable feature of the gold-fluoroalkoxides is their generally high solubility which also extends to non-polar organic solvents, such as benzene and toluene. In the context of organometallic synthesis, this feature can be advantageous, especially along with their stability and ease of access. Therefore, these complexes possess valuable properties that are complementary to those of the gold hydoxides, even though the latter display higher and wider reactivity. The presence of stable gold-alkoxide moieties will make these well-defined and easily synthesized complexes invaluable synthetic and mechanistic probes that will permit further advances in gold chemistry and catalysis.
Experimental synthetic procedures
Synthesis of 1a
[Au(IPr)Cl] (200 mg, 0.32 mmol) is charged in a 4 mL vial and suspended in 1 mL of ethanol with 37 μL of HFIP (0.35 mmol, 1.1 eq.) in the presence of K2CO3 (134 mg, 0.96 mmol, 3 eq.). The reaction is left at room temperature under magnetic stirring overnight for 30 minutes. After this time, the solvent is evaporated, the residue is microfiltered with 8 mL of toluene and filtered through basic alumina. The solution is then concentrated on the rotary evaporator until it becomes a viscous oil. The product is triturated with pentane (5 mL) and isolated as a microcrystalline white solid upon decantation and drying under vacuum in 90% yield (220 mg). Spectroscopic data match those found in the literature.14Gram-scale synthesis of 1a
Identical protocol as above starting from [Au(IPr)Cl] (1.00 g, 1.61 mmol) and K2CO3 (667 mg, 4.83 mmol, 3 eq.) in a 10 mL vial with 5 mL of ethanol and 339 μL of HFIP (3.22 mmol, 2 eq.). 1a is thus obtained as a microcrystalline solid in a 93% yield (1.13 g).1H NMR (300 MHz, CDCl3) δ 7.53–7.47 (t, J = 7.7 Hz, 2H), 7.29 (d, J = 7.8 Hz, 4H), 7.17 (s, 2H), 4.23 (hept, J = 6.4 Hz, 1H), 2.53 (hept, J = 6.9 Hz, 4H), 1.32 (d, J = 6.9 Hz, 12H), 1.21 (d, J = 6.9 Hz, 12H).
Synthesis of 1b
Identical protocol as 1a starting from [Au(SIPr)Cl] (100 mg, 0.16 mmol) and K2CO3 (66 mg, 0.48 mmol, 3 eq.) with 1 mL of ethanol and 18 μL of HFIP (0.17 mmol, 1.1 eq.). 1b is thus obtained as a microcrystalline solid in a 75% yield (92 mg).1H NMR (300 MHz, CDCl3) δ 7.41 (t, J = 7.7 Hz, 2H), 7.25 (d, J = 5.4 Hz, 4H), 7.22 (s, 2H), 4.17–4.08 (m, 1H), 4.05 (s, 4H), 3.02 (hept, J = 6.9 Hz, 4H), 1.40 (d, J = 6.8 Hz, 12H), 1.33 (d, J = 6.9 Hz, 12H).
13C NMR (100 MHz, CDCl3) δ 189.41 (Au–C), 146.3 (CAr), 133.9 (CHAr), 129.7 (CAr), 124.3 (CHAr), 76.2 (p, JCF = 30.3 Hz, CH(CF3)), 53.1 (NCimid), 28.7 (CH(CH3)2), 24.4 (CH(CH3)2), 24.1 (CH(CH3)2).
19F NMR (500 MHz, CDCl3) δ −76.59 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C29H41AuN3: 628.2966. Found: 628.2952.
Synthesis of 1c
Same protocol as 1a from [Au(IPrCl)Cl] (50 mg, 0.07 mmol) and K2CO3 (30 mg, 0.21 mmol, 3 eq.) with 0.5 mL of ethanol and 8 μL of HFIP (0.07 mmol, 1.1 eq.). 1c is thus obtained as a microcrystalline solid in a 95% yield (56 mg).1H NMR (300 MHz, CDCl3) δ 7.55 (td, J = 7.8, 1.9 Hz, 2H), 7.32 (d, J = 7.8 Hz, 4H), 4.20 (p, J = 6.3 Hz, 1H), 2.45 (h, J = 6.9 Hz, 4H), 1.33 (dd, J = 6.8, 1.6 Hz, 12H), 1.25 (d, J = 6.9 Hz, 12H).
13C NMR (100 MHz, CDCl3) δ 167.7 (Au–C), 146.1 (CAr), 131.6 (CHAr), 131.5 (CAr), 124.6 (CHAr), 118.8 (NCimid), 77.1 (p, JCF = 30.5 Hz, CH(CF3)), 29.2 (CH(CH3)2), 24.2 (CH(CH3)2), 23.7 (CH(CH3)2).
19F NMR (500 MHz, CDCl3) δ −76.48 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C29H37AuCl2N3: 694.2030. Found: 694.2013.
Synthesis of 1d
Same protocol as 1a starting from [Au(IPrMe)Cl] (100 mg, 0.15 mmol) and K2CO3 (63 mg, 0.46 mmol, 3 eq.) with 1 mL of ethanol and 17 μL of HFIP (0.16 mmol, 1.1 eq.). 1d is thus obtained as a microcrystalline solid in a 87% yield (104 mg).1H NMR (300 MHz, CDCl3) 7.48 (td, J = 7.8, 2.1 Hz, 2H), 7.28 (d, J = 7.3 Hz, 4H), 4.19 (hept, J = 6.4 Hz, 1H), 2.43 (tt, J = 13.9, 6.9 Hz, 4H), 1.94 (d, J = 6.1 Hz, 6H), 1.33 (d, 12H), 1.22 (d, 12H).
13C NMR (100 MHz, CDCl3) δ 163.8 (Au–C), 145.9 (CAr), 132.6 (CAr), 130.5 (CHAr), 126.1 (CHAr), 124.4 (NCimid), 76.9 (p, JCF = 29.9 Hz, CH(CF3)), 28.8 (CH(CH3)2), 25.2 (CH(CH3)2), 24.7 (CH(CH3)2), 23.6 (CH3), 23.5 (CH3).
19F NMR (500 MHz, CDCl3) δ −76.41 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C31H43AuN3: 654.3123. Found: 654.3104.
Synthesis of 1e
Identical protocol as 1a starting from [Au(BIAN-IPr)Cl] (100 mg, 0.13 mmol) and K2CO3 (55 mg, 0.40 mmol, 3 eq.) with 1 mL of ethanol and 16 μL of HFIP (0.14 mmol, 1.1 eq.). 1e is thus obtained as a microcrystalline solid in a 87% yield (102 mg). Crystals suitable for single crystal X-ray diffraction were grown via vapor diffusion of hexane into a saturated solution of the product in benzene.1H NMR (300 MHz, C6D6) δ 7.32 (dd, J = 8.4, 7.2 Hz, 2H), 7.23 (dd, J = 8.1, 1.0 Hz, 4H), 6.87 (d, J = 7.0 Hz, 2H), 6.82 (t, J = 7.2 Hz, 2H), 6.79 (d, J = 6.4 Hz, 2H), 4.78 (p, J = 6.4 Hz, 1H), 2.94 (hept, J = 6.7 Hz, 4H), 1.45 (d, J = 6.9 Hz, 12H), 0.96 (d, J = 6.9 Hz, 12H).
1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 8.3 Hz, 2H), 7.49–7.42 (t, J = 7.8 Hz, 2H), 7.32–7.26 (m, 2H), 7.26 (d, J = 7.8 Hz, 4H), 7.11 (s, 1H), 6.88 (d, J = 7.0 Hz, 2H), 4.14 (p, J = 6.4 Hz, 1H), 2.66 (hept, J = 6.7 Hz, 4H), 1.22 (d, J = 6.9 Hz, 12H), 0.95 (d, J = 6.9 Hz, 12H).
13C NMR (100 MHz, CDCl3) δ 172.4 (Au–C), 145.7 (CAr), 138.1 (CAr), 133.0 (CBIAN), 130.9 (CHAr), 129.9 (CBIAN), 128.6 (CHBIAN), 127.9 (CHBIAN), 125.5 (CBIAN), 124.6 (CHAr), 121.3 (CBIAN), 76.9 (p, JCF = 30.0 Hz, CH(CF3)), 29.1 (CH(CH3)2), 24.2 (CH(CH3)2), 24.1 (CH(CH3)2).
19F NMR (500 MHz, CDCl3) δ −76.35 (s, CF3).
Synthesis of 1f
Same protocol as 1a starting from [Au(IPr*)Cl] (100 mg, 0.08 mmol) and K2CO3 (36 mg, 0.26 mmol, 3 eq.) with 1 mL of ethanol and 10 μL of HFIP (0.09 mmol, 1.1 eq.). 1f is thus obtained as a microcrystalline solid in a 96% yield (107 mg).1H NMR (300 MHz, CDCl3) δ 7.22–7.11 (m, 32H), 6.86–6.82 (m, 12H), 5.65 (s, 2H), 5.30 (s, 4H), 4.32 (p, J = 6.3 Hz, 1H), 2.24 (s, 6H).
13C NMR (100 MHz, CDCl3) δ 167.6 (Au–C), 143.0 (CAr), 142.6 (CAr), 141.0 (CAr), 140.2 (CAr), 133.8 (CHAr), 130.2 (CHAr), 129.8 (CHAr), 129.4 (CHAr), 128.6 (CHAr), 128.4 (CHAr), 126.8 (CHAr), 126.7 (CHAr), 123.2 (NCHimid), 77.2 (p, JCF = 30.2 Hz, CH(CF3)2), 21.9 (CH3).
19F NMR (500 MHz, CDCl3) δ −76.23 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C71H59AuN3: 1150.4375. Found: 1150.4610.
Synthesis of 1g
Same protocol as 1a starting from [Au(IPent)Cl] (100 mg, 0.16 mmol) and K2CO3 (70 mg, 0.50 mmol, 3 eq.) with 1 mL of ethanol and 19 μL of HFIP (0.18 mmol, 1.1 eq.). 1g is thus obtained as a microcrystalline solid in a 84% yield (59 mg).1H NMR (300 MHz, CDCl3) δ 7.48 (t, J = 7.8 Hz, 2H), 7.20 (d, J = 7.7 Hz, 4H), 7.03 (s, 2H), 4.15 (hept, J = 6.4 Hz, 1H), 2.15 (p, J = 7.2 Hz, 4H), 1.92–1.58 (m, 12H), 1.55–1.39 (m, 4H), 0.93 (t, J = 7.4 Hz, 12H), 0.78 (t, J = 7.4 Hz, 12H).
13C NMR (100 MHz, CDCl3) δ 167.3 (Au–C), 143.4 (CAr), 136.8 (CHAr), 130.2 (CAr), 124.8 (CHAr), 123.6 (NCimid), 76.5 (p, JCF = 29.9 Hz, CH(CF3)), 42.9, (CH(CH2)2), 29.1, (CH2(CH3)2) 28.2 (CH2(CH3)2), 12.6 (CH2CH3).
19F NMR (500 MHz, CDCl3) δ −76.52 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C37H55AuN3: 738.4062. Found: 738.4043.
Synthesis of 1h
Same protocol as 1a starting from [Au(ItBu)Cl] (100 mg, 0.24 mmol) and K2CO3 (100 mg, 0.72 mmol, 3 eq.) with 1 mL of ethanol and 28 μL of HFIP (0.26 mmol, 1.1 eq.). 1h is thus obtained as a microcrystalline solid in a 98% yield (131 mg).1H NMR (300 MHz, CDCl3) δ 7.06 (s, 2H), 4.77 (hept, J = 6.4 Hz, 1H), 1.86 (s, 18H).
13C NMR (100 MHz, CDCl3) δ 159.7 (Au–C), 116.5 (CAr), 77.7 (p, JCF = 30.1 Hz, CH(CF3)), 59.0 (C(CH3)), 31.9 (CH3), 31.6 (CH3).
19F NMR (500 MHz, CDCl3) δ −75.89 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C13H23AuN3: 418.1557. Found: 418.1547.
Synthesis of 1i
Same protocol as 1a starting from [Au(IMes)Cl] (100 mg, 0.18 mmol) and K2CO3 (76 mg, 0.55 mmol, 3 eq.) with 1 mL of ethanol and 21 μL of HFIP (0.20 mmol, 1.1 eq.). 1i is thus obtained as a microcrystalline solid in a 78% yield (97 mg).1H NMR (300 MHz, CDCl3) δ 7.08 (s, 2H), 6.99 (s, 4H), 4.30 (hept, J = 6.4 Hz, 1H), 2.35 (s, 6H), 2.08 (s, 12H).
13C NMR (300 MHz, CDCl3) δ 165.9 (Au–C), 139.8 (CAr), 134.9 (CAr), 134.5 (CAr), 129.4 (CHAr), 122.2 (NCHimid), 21.2 (CH3), 17.7 (CH3).
19F NMR (500 MHz, CDCl3) δ −76.23 (s, CF3).
HRMS (ESI): m/z [M] calcd for C42H48AuN4: 805.3544. Found: 805.3520.
Synthesis of 1j
Same protocol as 1a starting from [Au(JohnPhos)Cl] (100 mg, 0.18 mmol) and K2CO3 (78 mg, 0.56 mmol, 3 eq.) with 1 mL of ethanol and 21 μL of HFIP (0.20 mmol, 1.1 eq.). 1j is thus obtained as a microcrystalline solid in a 99% yield (123 mg).1H NMR (300 MHz, CDCl3) δ 7.86 (td, J = 7.6, 1.7 Hz, 1H), 7.47 (m, 5H), 7.29 (dd, J = 4.3, 2.1 Hz, 1H), 7.19–7.14 (m, 2H), 4.35 (pd, J = 6.4, 2.8 Hz, 1H), 1.40 (d, J = 15.5 Hz, 9H), 1.35 (d, J = 15.4 Hz, 9H).
13C NMR (100 MHz, CDCl3) 133.6 (d, J = 2.7 Hz), 133.4 (d, J = 3.1 Hz), 133.3 (d, J = 7.8 Hz), 130.6 (d, J = 2.4 Hz), 130.6 (d, J = 2.3 Hz), 129.5, 129.3, 128.8, 128.4, 128.3, 126.8 (d, J = 6.8 Hz), 126.7 (d, J = 7.1 Hz), 76.4 (p, J = 28.6 Hz), 31.0 (d, J = 6.7 Hz), 30.8 (d, J = 6.3 Hz).
31P NMR (300 MHz, CDCl3) δ 54.71 (s, P).
19F NMR (500 MHz, CDCl3) δ −75.58 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C22H30AuNP: 536.1781. Found: 536.1772.
Synthesis of 2a
Same protocol as 1a starting from [Au(IPr)Cl] (200 mg, 0.32 mmol) and K2CO3 (133 mg, 0.96 mmol, 3 eq.) with 1 mL of ethanol and 25 μL of TFE (0.35 mmol, 1.1 eq.). 2 is thus obtained as a microcrystalline solid in a 95% yield (210 mg).1H NMR (300 MHz, CDCl3) δ 7.52–7.46 (t, J = 7.8 Hz, 2H), 7.28 (d, J = 7.9 Hz, 4H), 7.14 (s, 2H), 3.90 (q, J = 9.3 Hz, 2H), 2.55 (hept, J = 6.9 Hz, 4H), 1.35 (d, J = 6.9, 4.8 Hz, 12H), 1.21 (d, J = 6.9, 1.1 Hz, 12H).
13C NMR (300 MHz, CDCl3) δ 169.4 (Au–C), 145.7 (CAr), 134.3 (CAr), 130.6 (CHAr), 124.2 (CHAr), 123.0 (NCHimid), 68.9 (q, J = 31.7 Hz, CH2(CF3)), 28.9 (CH(CH3)2), 24.3 (CH(CH3)2), 24.2 (CH(CH3)2).
19F NMR (500 MHz, CDCl3) δ −77.5 (s, CF3).
HRMS (ESI): m/z [M + CH3CN] calcd for C29H39AuN3: 626.2810. Found: 626.2800.
Reactions involving boron reagents
1H NMR (300 MHz, CDCl3) δ 7.46 (t, J = 7.8 Hz, 2H), 7.27 (d, J = 6.8 Hz, 4H), 7.14 (s, 2H), 7.09–7.05 (m, 2H), 6.99 (t, 2H), 6.85–6.79 (m, 1H), 2.67 (hept, J = 6.7 Hz, 4H), 1.41 (d, J = 6.9 Hz, 12H), 1.24 (d, J = 6.9 Hz, 12H). Spectroscopic data match those found in the literature.37
1H NMR (400 MHz, CDCl3) δ 7.56 (dd, J = 16.2, 8.1, 2H), 7.46 (m, 2H), 7.45 (t, J = 8.1 Hz, 2H), 7.30 (d, J = 7.8 Hz, 4H), 7.22 (m, 1H), 7.16 (s, 2H), 7.18–7.14 (m, 2H), 2.70 (hept, J = 6.9 Hz, 4H), 1.44 (d, J = 6.8 Hz, 12H), 1.26 (d, J = 7.0 Hz, 12H). Spectroscopic data match those found in the literature.37
1H NMR (400 MHz, CDCl3) δ 7.47 (t, J = 7.8 Hz, 2H), 7.28 (d, J = 7.8 Hz, 4H), 7.14 (s, 2H), 7.00 (m, 2H), 6.73 (dd, J = 10.3, 8.2 Hz, 2H), 2.66 (hept, J = 7.0 Hz, 4H), 1.39 (d, J = 6.9 Hz, 12H), 1.24 (d, J = 6.9 Hz, 12H). Spectroscopic data match those found in the literature.43
1H NMR (400 MHz, CDCl3) δ 7.48 (t, J = 7.8 Hz, 2H), 7.29 (d, J = 7.8 Hz, 4H), 7.20 (d, J = 7.8 Hz, 2H), 7.16 (s, 2H), 7.14 (d, J = 7.9 Hz, 2H), 2.65 (hept, J = 6.9 Hz, 4H), 1.39 (d, J = 6.8 Hz, 12H), 1.24 (d, J = 6.9 Hz, 12H).
Reactions involving non-boron reagents
1H NMR (400 MHz, CDCl3) δ 7.49 (t, J = 7.8 Hz, 2H), 7.29 (m, 2H), 7.29 (d, J = 7.8 Hz, 4H), 7.12 (s, 2H), 7.12–7.06 (m, 2H), 7.06–7.03 (m, 1H), 2.61 (hept, J = 6.8 Hz, 4H), 1.38 (d, J = 6.9 Hz, 12H), 1.21 (d, J = 6.9 Hz, 12H). Spectroscopic data match those found in the literature.38
1H NMR (300 MHz, CDCl3) δ 7.35–7.30 (m, 2H), 7.15–7.06 (m, 3H), 7.05 (s, 2H), 6.99 (s, 4H), 2.34 (s, 12H), 2.12 (s, 12H). Spectroscopic data match those found in the literature.38
1H NMR (300 MHz, CDCl3) δ 7.55–7.51 (m, 2H), 7.25–7.11 (m, 40H), 6.87 (d, J = 2.2 Hz, 3H), 6.85 (d, J = 1.0 Hz, 2H), 6.84–6.83 (m, 4H), 5.70 (s, 2H), 5.37 (s, 4H), 2.22 (s, 6H). Spectroscopic data match those found in the literature.43
1H NMR (300 MHz, CD2Cl2) δ 7.89 (d, J = 7.7 Hz, 2H), 7.65 (t, J = 7.9 Hz, 2H), 7.44 (d, J = 7.8 Hz, 4H), 7.36 (s, 2H), 7.03 (t, J = 7.6 Hz, 2H), 6.86 (t, J = 7.3 Hz, 2H), 6.73 (d, J = 8.1 Hz, 2H), 2.73 (p, J = 7.1 Hz, 4H), 1.38 (d, J = 6.9 Hz, 12H), 1.29 (d, J = 6.9 Hz, 12H). Spectroscopic data match those found in the literature.39,41
Catalytic reaction protocol
Benzoic acid (37.7 mg, 0.3 mmol), diphenylacetylene (50 mg, 0.3 mmol) and 1a (2.2 mg, 1.5 mol%) are stirred neat at 80 °C for 20 hours. After this time, the mixture is microfiltered with diethyl ether (1 mL) and concentrated under vacuum. The product, (Z)-1,2-diphenylvinyl benzoate, is triturated with pentane (5 mL) and isolated as a microcrystalline white solid with 80% yield (70 mg). Spectroscopic data match those found in the literature.42
1H NMR (300 MHz, CDCl3) δ (ppm) = 8.25 (dd, J = 8.4, 1.4 Hz, 2H, CHAr), 7.71–7.64 (m, 1H, CHAr), 7.63–7.51 (m, 6H, CHAr), 7.41–7.32 (m, 4H, CHAr), 7.23–7.20 (m, 1H, CHAr), 6.81 (s, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH).
CH).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the Research Foundation – Flanders (FWO) and the Special Research Fund (BOF) of Ghent University (starting and senior grants to S.P.N.). Umicore AG is gratefully thanked for gifts of materials.References
- X. An and J. Xiao, Chem. Rec., 2020, 20, 142–161 CrossRef CAS PubMed.
- P. Trillo, A. Baeza and C. Nájera, J. Org. Chem., 2012, 77, 7344–7354 CrossRef CAS PubMed.
- F.-X. Tian and J. Qu, J. Org. Chem., 2022, 87, 1814–1829 CrossRef CAS PubMed.
- X. Zeng, S. Liu, Z. Shi and B. Xu, Org. Lett., 2016, 18, 4770–4773 CrossRef CAS PubMed.
- S. Khaksar, J. Fluor. Chem., 2015, 172, 51–61 CrossRef CAS.
- S. Fustero, R. Román, J. F. Sanz-Cervera, A. Simón-Fuentes, A. C. Cuñat, S. Villanova and M. Murguía, J. Org. Chem., 2008, 73, 3523–3529 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Colobert, Org. Chem. Front., 2016, 3, 394–400 RSC.
- A. M. Wilders, J. Henle, M. C. Haibach, R. Swiatowiec, J. Bien, R. F. Henry, S. O. Asare, A. L. Wall and S. Shekhar, ACS Catal., 2020, 10, 15008–15018 CrossRef CAS.
- Z. Xu, Z. Hang, L. Chai and Z.-Q. Liu, Org. Lett., 2016, 18, 4662–4665 CrossRef CAS PubMed.
- B. Riniker, B. Kamber and P. Sieber, Helv. Chim. Acta, 1975, 58, 1086–1094 CrossRef CAS PubMed.
- P. Sieber, B. Kamber, A. Hartmann, A. Jöhl, B. Riniker and W. Rittel, Helv. Chim. Acta, 1974, 57, 2617–2621 CrossRef CAS PubMed.
- H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood and J. Aube, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- R. Gauthier, N. V. Tzouras, Z. Zhang, S. Bédard, M. Saab, L. Falivene, K. Van Hecke, L. Cavallo, S. P. Nolan and J. Paquin, Chem. – Eur. J., 28,(4), e202103886 Search PubMed.
- N. V. Tzouras, A. Gobbo, N. B. Pozsoni, S. G. Chalkidis, S. Bhandary, K. Van Hecke, G. C. Vougioukalakis and S. P. Nolan, Chem. Commun., 2022, 58, 8516–8519 RSC.
- J. Wolf, F. Huber, N. Erochok, F. Heinen, V. Guérin, C. Y. Legault, S. F. Kirsch and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 16496–16500 CrossRef CAS PubMed.
- M. Veguillas, G. M. Rosair, M. W. P. Bebbington and A.-L. Lee, ACS Catal., 2019, 9, 2552–2557 CrossRef CAS.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed.
- R. Ebule, S. Liang, G. B. Hammond and B. Xu, ACS Catal., 2017, 7, 6798–6801 CrossRef CAS PubMed.
- F. Nahra, N. V. Tzouras, A. Collado and S. P. Nolan, Nat. Protoc., 2021, 16, 1476–1493 CrossRef CAS PubMed.
- N. V. Tzouras, F. Nahra, L. Falivene, L. Cavallo, M. Saab, K. Van Hecke, A. Collado, C. J. Collett, A. D. Smith, C. S. J. Cazin and S. P. Nolan, Chem. – Eur. J., 2020, 26, 4515–4519 CrossRef CAS PubMed.
- E. A. Martynova, N. V. Tzouras, G. Pisano, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 RSC.
- F. Nahra, S. R. Patrick, A. Collado and S. P. Nolan, Polyhedron, 2014, 84, 59–62 CrossRef CAS.
- A. Gómez-Suárez, R. S. Ramón, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2012, 41, 5461 RSC.
- A. S. K. Hashmi, Chem. Rev., 2021, 121, 8309–8310 CrossRef CAS PubMed.
- B. Ranieri, I. Escofet and A. M. Echavarren, Org. Biomol. Chem., 2015, 13, 7103–7118 RSC.
- S. Komiya, T. Sone, Y. Usui, M. Hirano and A. Fukuoka, Gold Bull., 1996, 29, 131–136 CrossRef CAS.
- Y. Usui, J. Noma, M. Hirano and S. Komiya, Inorg. Chim. Acta, 2000, 309, 151–154 CrossRef CAS.
- S. Komiya, M. Iwata, T. Sone and A. Fukuoka, J. Chem. Soc., Chem. Commun., 1992, 1109 RSC.
- M. A. Cinellu and G. Minghetti, Gold Bull., 2002, 35, 11–20 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC.
- I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674–8677 CrossRef CAS PubMed.
- M. Ramos, J. Poater, N. Villegas-Escobar, M. Gimferrer, A. Toro-Labbé, L. Cavallo and A. Poater, Eur. J. Inorg. Chem., 2020, 2020, 1123–1134 CrossRef CAS.
- Y. Oonishi, A. Gómez-Suárez, A. R. Martin and S. P. Nolan, Angew. Chem., Int. Ed., 2013, 52, 9767–9771 CrossRef CAS PubMed.
- N. Ibrahim, M. H. Vilhelmsen, M. Pernpointner, F. Rominger and A. S. K. Hashmi, Organometallics, 2013, 32, 2576–2583 CrossRef CAS.
- S. Dupuy, L. E. Crawford, M. Bühl, A. M. Z. Slawin and S. P. Nolan, Adv. Synth. Catal., 2012, 354, 2380–2386 CrossRef CAS.
- R. D. Adams and G. Elpitiya, Inorg. Chem., 2015, 54, 8042–8048 CrossRef CAS PubMed.
- N. V. Tzouras, M. Saab, W. Janssens, T. Cauwenbergh, K. Van Hecke, F. Nahra and S. P. Nolan, Chem. – Eur. J., 2020, 26, 5541–5551 CrossRef CAS PubMed.
- T. Scattolin, N. V. Tzouras, L. Falivene, L. Cavallo and S. P. Nolan, Dalton Trans., 2020, 49, 9694–9700 RSC.
- N. V. Tzouras, E. A. Martynova, X. Ma, T. Scattolin, B. Hupp, H. Busen, M. Saab, Z. Zhang, L. Falivene, G. Pisanò, K. Van Hecke, L. Cavallo, C. S. J. Cazin, A. Steffen and S. P. Nolan, Chem. – Eur. J., 2021, 27, 11904–11911 CrossRef CAS PubMed.
- E. A. Martynova, T. Scattolin, E. Cavarzerani, M. Peng, K. Van Hecke, F. Rizzolio and S. P. Nolan, Dalton Trans., 2022, 51, 3462–3471 RSC.
- E. A. Martynova, V. A. Voloshkin, S. G. Guillet, F. Bru, M. Beliš, K. Van Hecke, C. S. J. Cazin and S. P. Nolan, Chem. Sci., 2022, 13, 6852–6857 RSC.
- I. Ibni Hashim, N. V. Tzouras, W. Janssens, T. Scattolin, L. Bourda, S. Bhandary, K. Van Hecke, S. P. Nolan and C. S. J. Cazin, Chem. – Eur. J., 2022, 28(47), e202201224 CrossRef CAS PubMed.
- M. M. Hansmann, F. Rominger, M. P. Boone, D. W. Stephan and A. S. K. Hashmi, Organometallics, 2014, 33, 4461–4470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2311517. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01402b |
| This journal is © The Royal Society of Chemistry 2024 |