
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterisation of copper(I) complexes with relevance to intramolecular Ullmann O,S-arylation†
Xiaodong
Jin
 ab,
Bao-Nguyen T.
Nguyen
a and
Robert P.
Davies
*a
ab,
Bao-Nguyen T.
Nguyen
a and
Robert P.
Davies
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: r.davies@imperial.ac.uk
bDepartment of Chemistry, Xi'an Jiaotong-Liverpool University, Suzhou 215123, China
First published on 12th July 2024
Abstract
Copper-catalysed intramolecular Ullman arylation has been frequently used to synthesise benzoxazoles and benzothiazoles. Despite widespread use, investigations into the mechanism and speciation of copper-containing complexes relevant to the catalytic pathway have remained relatively limited. Accordingly, this study aims to elucidate the structural details of potential copper(I) intermediates through the analysis of their solid-state structures using X-ray crystallography, while also investigating the reactivities of these complexes. Five novel copper complexes are reported which are formed prior to the aryl halide activation step and feature distinct aggregation modes based on either Cu4N4O4C4 or Cu4N4S4C4 clusters.
Introduction
Benzoxazoles and benzothiazoles represent a valuable class of benzene-fused heterocycle that are often biologically active and prevalent in natural products and pharmaceuticals.1–6 Their widespread use and importance has spurred a variety of synthetic routes to these compounds. Among these methods, copper-catalysed intramolecular arylation has garnered particular attention due to its high reactivity, cost-effectiveness in catalyst and ligand use, and its operation under mild conditions.7,8 Various catalytic systems have been reported for this process including DMEDA/CuI,9 1,10-phenanthroline/CuI,10–13 TMEDA/Cu salts,14 oxazolidin-2-one/CuI,15 methyl 2-methoxybenzoate/CuI,16 8-hydroxyquinoline/CuI,17 an N-heterocyclic carbene/CuCl,18 and trizoles/CuI19–21 as well as a number of Cu(II) complexes22–25 and CuO nanoparticles26 (Scheme 1). Although significant progress has been made in developing more robust and efficient copper catalytic systems, the mechanism of the reaction and the identity of copper complexes in the catalytic cycle remains little explored.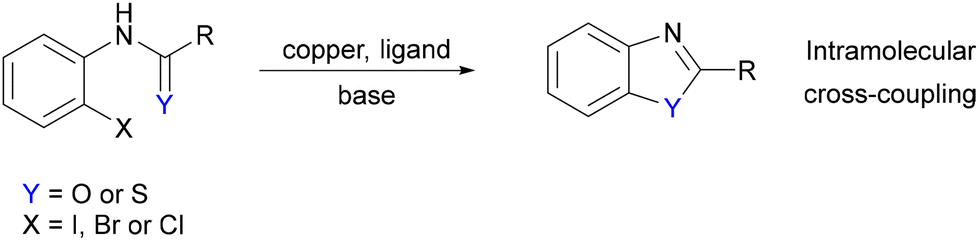 | ||
| Scheme 1 Copper-catalysed intramolecular arylation. | ||
The research reported herein aims to gain additional insight into the mechanism of copper(I)-catalysed intramolecular O- and S-arylation by analysing the solid-state structures of potential intermediary copper(I) species. These structures, prepared under air-free conditions and characterised by X-ray crystallography, offer insight into the range of interactions present between the metal centre and substrate, and builds upon previous studies in our group on the related intramolecular N-arylation reaction for the synthesis of benzimidazoles.27
Experimental
Materials
Anhydrous solvents such as dichloromethane, tetrahydrofuran, hexane, and toluene were obtained from a solvent tower using the PureSolv solvent purification system, degassed under N2, and stored over molecular sieves. Anhydrous DMSO and dioxane were purchased in Sure/Seal™ bottles and used directly from the bottle. Deuterated solvents such as DMSO-d6 and chloroform-d were purchased from Sigma-Aldrich or VWR and dried over 4 Å molecular sieves under N2 before use. Copper(I) iodide (99.999% trace metals basis, powder) purchased from Sigma-Aldrich was stored in a glove box under N2. All ligands and aryl halides were purchased from Sigma-Aldrich or VWR and used as received without further purification. For reactions that required air-free techniques, all glassware was pre-dried at 120 °C overnight and loaded in a glove-box under nitrogen. The experiments were carried out under a protective atmosphere of N2.General procedure for the synthesis of copper(I) complexes
A 4 mL vial was charged with mesitylcopper(I) (0.1 mmol) and aryl halides (0.1 mmol) fully dissolved in dry toluene (1.0 mL). The vial was placed inside a larger vial (20 mL) containing dry hexane (0.5 mL) and left at room temperature. Colourless crystals started to form in the inner vial after 24 h, and were isolated after 72 h.General procedure for the copper(I)-catalysed O-,S-arylation
A sample vial was charged with Cs2CO3 (0.15 mmol) and aryl halide (0.1 mmol) followed by addition of CuI solution (5 mol%), phenanthroline ligand when used (10 mol%) and internal standard naphthalene (0.05 mmol). The reaction was then stirred at 80 °C in DMSO-d6 (1 mL) for 6 h. The mixture was cooled to room temperature and the clear organic layer was transferred into Young's type NMR tube under an N2 atmosphere. The conversion and yield were determined by 1H NMR and are tabulated in the results and discussion section.Results and discussion
Isolation and characterisation of copper(I) complexes
In order to investigate possible mechanistic pathways for copper(I)-catalysed O- and S-arylations, a study into the solid-state structures of potential copper-containing intermediates was conducted. The precursors 2-halogenbenzanilide (1), 2-halogenthiobenzanilide (2) and (2-halogenphenyl)-benzyl-thiourea (3) were synthesised following published protocols.28,29 With the starting materials in hand, single crystals were obtained from the reaction between the aryl halides and mesitylcopper(I), followed by recrystallisation of the resulting copper(I) complexes from toluene/hexane (Scheme 2). | ||
| Scheme 2 Synthesis of copper(I) complexes. | ||
The reaction employing 1a as the reagent led to the isolation of a tetrameric complex Cu41a′4 (where 1a′ is deprotonated 1a). Through X-ray crystallography, the structure of Cu41a′4 was shown to adopt an unusual central Cu4N4O4C4 16-membered folded ring, characterised by a slightly distorted rhombic Cu4 core (Cu⋯Cu distances in the range of 2.6124(19) to 2.7445(6) Å; sum of interior angles 359.38°, Fig. 1a). The amide motif was deprotonated, giving a delocalized negative charge within the N–C–O unit. This was evident through the shorter C–N bonds (range from 1.3140(40) to 1.3257(40) Å) and longer C–O bonds (ranging from 1.2710(38) to 1.2837(34) Å) in Cu41a′4, when compared to 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 (C–N: 1.3462(24) Å, C
30 (C–N: 1.3462(24) Å, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: 1.2305(18) Å).
O: 1.2305(18) Å).
 | ||
| Fig. 1 (a) Solid-state structure of Cu41a′4; thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (b) Different views of the Cu4N4O4C4 16-membered ring. (c) Cu(I) complexes containing a Cu4N4O4C4 16-membered ring.31–33 | ||
The Cu41a′4 structure contains two distinct copper(I) sites: one in which the copper is coordinated to two nitrogen atoms (Cu1 and Cu2 in Fig. 1a) and the other coordinated to two oxygen atoms (Cu3 and Cu4). Each Cu atom resides in a near-linear coordination environment, evident from N–Cu–N bond angles of 171.77° and 178.00°, and O–Cu–O bond angles of 162.82° and 165.72°. While several copper(I) complexes featuring planar Cu4 arrangements and Cu4N4O4C4 16-membered rings have been reported previously,31–33 these tetrameric structures exhibited heteroligated N–Cu–O copper centres (Fig. 1c), in contrast to the distinct homoligated N–Cu–N and O–Cu–O units observed in this case.
The attempted reaction between aryl bromide 1b and mesityl copper(I) did not yield single crystals suitable for X-ray diffraction analysis. However, the reaction of the thio analog, 2a, with mesitylcopper(I) led to the formation of a copper(I) complex, the structure of which was elucidated through X-ray crystallography, revealing a tetrameric solid-state structure, Cu42a′4. This structure comprises a central Cu4N4S4C4 cluster (Fig. 2a and c). In contrast to Cu41a′4, Cu42a′4 exhibits a tetrahedral Cu4 core (Cu⋯Cu distances spanning 2.5170(26) to 2.9198(40) Å). Comparison of the bonding parameters between Cu42a′4 and 3-chlorothiobenzanilide34 (the solid-state structure of 2a is currently unreported), highlighted the deprotonation of the thioamide and subsequent delocalization of the negative charge over the N–C–S unit (C–N: range 1.288(4)–1.300(4) Å; C–S: 1.755(3)–1.767(3) Å) compared to C–N 1.342 Å and CS 1.671 Å in 3-chlorothiobenzanilide.
 | ||
| Fig. 2 (a) Solid-state structure of Cu42a′4 (a) and Cu42b′4 (b); thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (c) Different views of the Cu4N4S4C4 16-membered ring of Cu42a′4. (d) Cu(I) complexes containing a Cu4N4S4C4 16-membered ring.35–39 | ||
Each copper atom within the Cu4 core is bonded to one nitrogen and two sulfur atoms, originating from three independent 2a′ molecules, thus giving a trigonal planar N–Cu-(S)2 coordination motif. For example, Cu1 coordinates with N1, S2 and S4, exhibiting N1–Cu1–S2 angle of 113.92°, N1–Cu1–S4 angle of 116.52°, and S2–Cu1–S4 129.46°. The summation of these angles (359.91°) is consistent with the planarity of the N–Cu-(S)2 unit. Correspondingly, the summed angles around Cu2, Cu3 and Cu4 are 359.84°, 360.00° and 359.96° respectively. The four copper centres are arranged in a tetrahedral configuration, exhibiting the shortest Cu⋯Cu distance of 2.5170(26) Å, which is shorter than the corresponding distances found in Cu41a′4 (shortest Cu⋯Cu distance: 2.6124(19) Å). Furthermore, when aryl bromide 2b was employed instead of aryl chloride 2a, a tetramer structure Cu42b′4 was obtained (Fig. 2b). The structure is analogous to Cu42a′4 being also centred on a Cu4N4S4C4 polyhedron cluster and a tetrahedron Cu4 core. The distances and angles closely resemble those observed in Cu42a′4 (see ESI†). Several copper(I) complexes containing tetrahedron Cu4 arrangements and Cu4N4S4C4 16-membered rings have previously been reported and are summarised in Fig. 2d.35–39 It is notable how a change from an amide (1a/b) to thioamide (2a/b) results in a significant change in the coordination geometries around the copper centres, likely facilitated by the increased tendency of the larger, softer sulphur atom to act as in a bridging capacity to copper(I) when compared to the amide oxygen atom in 1a/b.
The thiourea substrates 3a/b are of particular interest due to the presence of the additional NH group and the reported selectivity for the benzothiazole product over the mercaptobenzimidazole by-product on treatment with the Cu-catalyst. Thus, reaction of 3a with mesitylcopper(I) led to the formation of a tetrameric aggregate, Cu43a′4, whose solid-state structure was determined through X-ray crystallography (Fig. 3a). Cu43a′4 shares similarities with Cu42a′4, also containing a central Cu4 tetrahedron (Cu⋯Cu distances in range 2.5742(24) to 2.8690(13) Å). Comparison to the solid-state structure of 3a obtained via recrystallisation of 3a from toluene/hexane (see ESI†), showed deprotonation of the Nα–H of thiourea motif and charge delocalisation over the Nα–C–S unit. This was evidenced by the reduction in C–N bond lengths from 1.3565(39) Å in 3a to 1.3062(67)–1.3074(71) Å in Cu43a′4, along with an increase in C–S bond lengths from 1.6907(31) Å in 3a to 1.7636(54)–1.7771(47) Å in Cu43a′4. Furthermore, the amine group in the γ-position was not deprotonated and does not form any interactions with copper metal centres (3a: C–Nγ = 1.3338(50) Å; Cu43a′4: C–Nγ = 1.3386(67)–1.3442(61) Å). This structural insight suggests a potential explanation for the selectivity observed between S-arylation and N-arylation in the Cu-catalysed cyclisation of (2-bromophenyl)-benzyl-thiourea (3a), as the copper is selectively coordinated to the Nα and S atoms. The synthesis of 2-mercaptobenzimidazoles through N-arylation usually requires an S-alkylation step to prevent the formation of C–S bonds.40 Additionally, the utilisation of aryl bromide 3b yielded a structurally similar tetrameric complex, Cu43b′4 (Fig. 3b) with similar bond distances and angles to the chloride analogue (see ESI†). Once more, the copper selectively coordinated to the Nα and S atoms, while no interaction with Nγ was observed.
 | ||
| Fig. 3 Solid-state structure of Cu43a′4 (a) and Cu43b′4 (b); thermal ellipsoids are set at 50% probability and hydrogen atoms (except Nγ–H) are omitted for clarity. | ||
Reactivity of copper(I) complexes
To validate the plausibility of these copper(I) complexes as reaction intermediates, their reactivity was investigated by heating the complexes directly in DMSO-d6 in order to obtain the final cyclised product. The resulting yields at varying temperatures are presented in Table 1, entries 1–5, along with the yields from the analogous catalytic reactions (entries 6–10). These complexes gave similar yield compared to that observed in catalytic reactions. Notably, reactions involving aryl chlorides required higher temperatures compared to those with aryl bromides as expected due to the higher aryl–halide bond strength. Furthermore, addition of an ancillary 1,10-phenanthroline ligand (L1) improved the final yields for both the stoichiometric and catalytic reactions. Addition of this ligand is thought to assist with lowering the aggregation state of the copper(I) clusters thus leading to an increase in reactivity,27 although our attempts to isolate related copper(I) complexes incorporating this ligand have so far been unsuccessful.|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Temp (°C) | Yield (%) | Yield with L1 (%) |
| The reaction was carried out by heating the Cu(I) complexes Cu4Ar4 (0.1 mmol) in DMSO-d6 (1 mL) with or without addition of L1 (0.1 mmol). The comparison experiment was carried out by heating the aryl halides (0.1 mmol) using Cs2CO3 (0.15 mmol) as base in the presence of CuI (5 mol%) and L1 (10 mol%). Yields were determined by 1H NMR using naphthalene as internal standard. | ||||
| 1 | Cu41a′4 | 130 | 26 | 96 |
| 2 | Cu42a′4 | 110 | 90 | 99 |
| 3 | Cu42b′4 | 80 | 97 | 99 |
| 4 | Cu43a′4 | 110 | 86 | 99 |
| 5 | Cu43b′4 | 80 | 96 | 99 |

|
||||
| 6 | 1a | 130 | 4 | 93 |
| 7 | 2a | 110 | 58 | 99 |
| 8 | 2b | 80 | 94 | 99 |
| 9 | 3a | 110 | 72 | 99 |
| 10 | 3b | 80 | 91 | 99 |
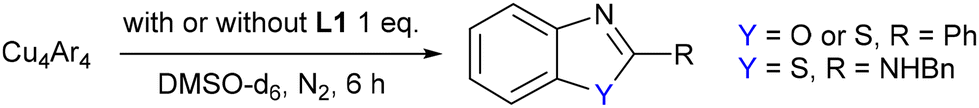
A putative mechanism based on the structural insights obtained in this work is proposed in Scheme 3. Based upon the observed distance between the copper(I) centres and aryl–halide bonds in the crystal structures, and also transposing the findings of our previously reported kinetic analysis on related copper-catalysed intramolecular N-arylation27 two different copper centres are proposed to participate in the catalytic cycle. The aggregation state of the copper complexes in solution (DMSO) at high temperatures is likely to differ from those observed in the solid-state, and hence the complexes are shown as monomeric in the figure for simplicity.
 | ||
| Scheme 3 Proposed mechanism for copper(I)-catalysed intramolecular O,S-arylation. | ||
Conclusions
Investigations into the solid-state structures of copper(I) complexes with selected organic amide and thioamide substrates have led to the observation of some novel copper(I) coordination complexes with unusual aggregation modes, and in addition offered insight into potential species present during intramolecular Ullmann O,S-arylation reactions. All copper(I) complexes exhibit amide deprotonation at the substrate Nα site and form tetrameric structures featuring Cu4Y4N4C4 (Y = O or S) central units. In the structures of thiourea complexes Cu43a′4 and Cu43b′4 the copper centres selectively coordinate with the Nα and S atoms, and no interactions with the Nγ atom are observed. This is congruent with the observed selectivity for S-arylation over N-arylation in the intramolecular cyclisation reaction. All copper complexes undergo intramolecular bond formation on heating in DMSO, yielding the desired cyclised products and making them viable intermediates or resting states in the intramolecular O,S-arylation reaction. Additionally, the introduction of 1,10-phenanthroline enhances the final yield, emphasising the positive impact of this ancillary ligand on the reaction outcome.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for all compounds have been deposited at Cambridge Crystallographic data centre (CCDC) under numbers 2349373–2349378 and can be obtained viahttps://www.ccdc.cam.ac.uk/structures. Additional information on the data refinements are included in the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge Dr Andrew White for help with collecting and solving the crystal structure data.References
- S. Soni, N. Sahiba, S. Teli, P. Teli, L. K. Agarwal and S. Agarwal, RSC Adv., 2023, 13, 24093–24111 RSC.
- R. S. Keri, M. R. Patil, S. A. Patil and S. Budagumpi, Eur. J. Med. Chem., 2015, 89, 207–251 CrossRef CAS PubMed.
- S. Rajasekhar, B. Maiti and K. Chanda, Synlett, 2017, 521–541 CAS.
- S. Noël, S. Cadet, E. Gras and C. Hureau, Chem. Soc. Rev., 2013, 42, 7747 RSC.
- V. Facchinetti, R. da R. Reis, C. R. B. Gomes and T. R. A. Vasconcelos, Mini-Rev. Org. Chem., 2012, 9, 44–53 CrossRef CAS.
- A. Kamal, M. A. H. Syed and S. M. Mohammed, Expert Opin. Ther. Pat., 2015, 25, 335–349 CrossRef CAS PubMed.
- Q. Cai and W. Zhou, Chin. J. Chem., 2020, 38, 879–893 CrossRef CAS.
- Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- G. Altenhoff and F. Glorius, Adv. Synth. Catal., 2004, 346, 1661–1664 CrossRef CAS.
- G. Evindar and R. A. Batey, J. Org. Chem., 2006, 71, 1802–1808 CrossRef CAS PubMed.
- R. D. Viirre, G. Evindar and R. A. Batey, J. Org. Chem., 2008, 73, 3452–3459 CrossRef CAS PubMed.
- Z. Luo, H. Wu, Y. Li, Y. Chen, J. Nie, S. Lu, Y. Zhu and Z. Zeng, Adv. Synth. Catal., 2019, 361, 4117–4125 CrossRef CAS.
- L. L. Joyce, G. Evindar and R. A. Batey, Chem. Commun., 2004, 446–447 RSC.
- N. Barbero, M. Carril, R. SanMartin and E. Domínguez, Tetrahedron, 2007, 63, 10425–10432 CrossRef CAS.
- H. Ma and X. Jiang, Synlett, 2008, 1335–1340 CAS.
- F. Wu, J. Zhang, Q. Wei, P. Liu, J. Xie, H. Jiang, B. Dai, S. Chang, C. Bolm, C. Tumanut, J. Li, G. Spraggon, J. Chang, T. Tuntland, J. L. Harris and D. S. Karanewsky, Org. Biomol. Chem., 2014, 12, 9696–9701 RSC.
- D. Miao, X. Shi, G. He, Y. Tong, Z. Jiang and S. Han, Tetrahedron, 2015, 71, 431–435 CrossRef CAS.
- J. I. Urzúa, R. Contreras, C. O. Salas and R. A. Tapia, RSC Adv., 2016, 6, 82401–82408 RSC.
- A. S. Singh, M. Singh, N. Mishra, S. Mishra, A. K. Agrahari and V. K. Tiwari, ChemistrySelect, 2017, 2, 154–159 CrossRef CAS.
- N. Mishra, A. S. Singh, A. K. Agrahari, S. K. Singh, M. Singh and V. K. Tiwari, ACS Comb. Sci., 2019, 21, 389–399 CrossRef CAS PubMed.
- M. Singh, P. Bose, A. S. Singh and V. K. Tiwari, ChemistrySelect, 2019, 4, 9627–9631 CrossRef CAS.
- A. Naidu and G. Sekar, Synthesis, 2010, 579–586 CAS.
- E. A. Jaseer, D. J. C. Prasad, A. Dandapat and G. Sekar, Tetrahedron Lett., 2010, 51, 5009–5012 CrossRef CAS.
- S. N. M. Boddapati, C. M. Kurmarayuni, B. R. Mutchu, R. Tamminana and H. B. Bollikolla, Org. Biomol. Chem., 2018, 16, 8267–8272 RSC.
- Y. Tang, M. Li, H. Gao, G. Rao and Z. Mao, RSC Adv., 2020, 10, 14317–14321 RSC.
- P. Saha, T. Ramana, N. Purkait, M. A. Ali, R. Paul and T. Punniyamurthy, J. Org. Chem., 2009, 74, 8719–8725 CrossRef CAS PubMed.
- X. Jin, Y. Lin and R. P. Davies, Catal. Sci. Technol., 2023, 13, 7181–7189 RSC.
- E. Feng, H. Huang, Y. Zhou, D. Ye, H. Jiang and H. Liu, J. Comb. Chem., 2010, 12, 422–429 CrossRef CAS PubMed.
- D. Bernardi, L. Ba and G. Kirsch, Synlett, 2007, 2121–2123 CrossRef CAS.
- B. T. Gowda, B. P. Sowmya, J. Kozísek, M. Tokarcík and H. Fuess, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o2906 CrossRef CAS.
- M. Berry, W. Clegg, C. D. Garner and I. H. Hillier, Inorg. Chem., 1982, 21, 1342–1345 CrossRef CAS.
- L. P. Wu, M. Yamamoto, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and M. Munakata, J. Chem. Soc., Dalton Trans., 1996, 23, 2031 RSC.
- H. Q. Do, S. Bachman, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 2162–2167 CrossRef CAS PubMed.
- G. Zhao, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o353 CrossRef CAS PubMed.
- D. M. Knotter, M. D. Janssen, D. M. Grove, W. J. J. Smeets, E. Horn, A. L. Spek and G. Van Koten, Inorg. Chem., 1991, 30, 4361–4366 CrossRef CAS.
- E. S. Raper, J. R. Creighton and W. Clegg, Inorg. Chim. Acta, 1991, 183, 179–187 CrossRef CAS.
- T. Kuroda-Sowa, M. Munakata, M. Miyazaki and M. Maekawa, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1026–1028 CrossRef.
- J.-M. Du, H.-Y. Zeng, Z.-C. Dong, G. C. Guo and J.-S. Huang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m396–m397 CrossRef PubMed.
- C. Yue, C. Yan, R. Feng, M. Wu, L. Chen, F. Jiang and M. Hong, Inorg. Chem., 2009, 48, 2873–2879 CrossRef CAS PubMed.
- S. Murru, B. K. Patel, J. Le Bras and J. Muzart, J. Org. Chem., 2009, 74, 2217–2220 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and crystallographic details. CCDC 2349373–2349378. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01418a |
| This journal is © The Royal Society of Chemistry 2024 |