DOI:
10.1039/D4DT01458H
(Paper)
Dalton Trans., 2024,
53, 11903-11913
Structure–activity relationship of anticancer and antiplasmodial gold bis(dithiolene) complexes†
Received
17th May 2024
, Accepted 25th June 2024
First published on 26th June 2024
Abstract
Monoanionic gold bis(dithiolene) complexes were recently shown to display activity against ovarian cancer cells, Gram-positive bacteria, Candida strains and the rodent malaria parasite, P. berghei. To date, only monoanionic gold(III) bis(dithiolene) complexes with a thiazoline backbone substituted with small alkyl chains have been evaluated for biomedical applications. We now analyzed the influence of the length and the hydrophobicity vs. hydrophilicity of these complexes’ alkyl chain on their anticancer and antiplasmodial properties. Isomer analogues of these monoanionic gold(III) bis(dithiolene) complexes, this time with a thiazole backbone, were also investigated in order to assess the influence of the nature of the heterocyclic ligand on their overall chemical and biological properties. In this report we present the total synthesis of four novel monoanionic gold(III) bis(dithiolene) complexes with a long alkyl chain and a polyoxygenated (PEG) chain aiming to improve their solubility and biological properties. Our results showed that the complexes with a PEG chain showed promising anticancer and antiplasmodial activities beside improved solubility, a key parameter in drug discovery and development.
Introduction
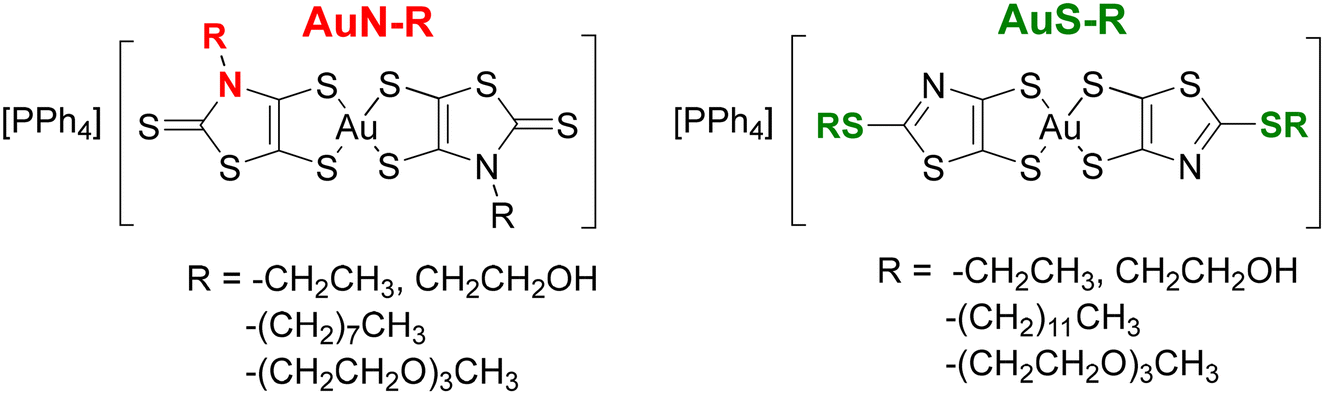
Monoanionic gold bis(dithiolene) complexes have been the focus of much attention as precursors of highly conducting neutral radical gold complexes.1–3 Once oxidized, these molecules lead to neutral radical species, which form, in most cases, uniform stacks with numerous strong intermolecular interactions that allow for electron delocalization in the solid state,4–8 to give single-component conductors,9–11 some with metallic behavior.12 Notwithstanding their interest for the elaboration of single component molecular materials, the biological properties of gold complexes have recently received particular notice.13 The investigation on the anticancer properties of gold complexes was primarily focused on auranofin, a gold(I) phosphine derivative whose broad spectrum of activity motivated the synthesis of a diversity of gold(I) complexes.14,15 On the other hand, gold(III) complexes have also been investigated as potential anticancer agents, some due to their square-planar geometry, similar to that of cisplatin.16,17 The latter is the first metal-based anticancer drug used in clinical treatment, and still frequently used for the treatment of various types of cancer.18,19 The monoanionic gold bis(dithiolene) complexes belong to the family of gold(III) complexes and some of these complexes exhibit anticancer activity similar or superior to that of cisplatin.20 For instance, we recently demonstrated the potential activity of monoanionic gold(III) bis(dithiolene) complexes against ovarian cancer cells, Gram-positive bacteria, Candida strains and hepatic infection by the rodent malaria parasite P. berghei.21 The most active monoanionic complexes, [Au(R-thiazdt)2]−, were built from the N-alkyl-1,3-thiazoline-2-thione-4,5-dithiolate ligand.21 Various modifications have been performed on these monoanionic complexes, differing in the substituent added to the nitrogen atom of the thiazoline ring, the nature of the chalcogen atoms within the metallacycles (sulfur vs. selenium), the exocyclic sulfur replacement, and the counter-ion employed (Ph4P+ or Et4N+).22 These studies showed that most of these monoanionic gold complexes exhibit strong anticancer activities, similar to the gold-based reference drug, auranofin. The most promising complexes for anticancer and antiplasmodial activities are those with a Ph4P+ counter ion, [Ph4P][Au(R-thiazdt)2] called AuN–R in this paper for an easier reading. Moreover, these complexes were also shown to exhibit low toxicity in the zebrafish embryo model.22 A natural next step of this research consists in investigating the influence of the nature of the chain connected to the complex on their properties and activity. To this end, we prepared gold bis(dithiolene) complexes substituted with long alkyl and polyethylene glycol (PEG) chains in order to improve their solubility and cellular uptake (AuN–R, Chart 1). In addition, we also investigated the bis(2-alkylthio-1,3-thiazole-4,5-dithiolate) gold complexes, herein called AuS–R, which can be considered as S-alkyl isomers of the N-alkyl gold complexes, AuN–R. The thiazole ring is an aromatic ring, based on the delocalization of a lone pair of electrons from the sulfur atom,23 which is present in a large number of widely used drugs.24 Until now, we have not explored the anticancer and antiplasmodial activities of this AuS–R family of complexes. This study was performed to assess the effect of the nature of the heterocyclic ring, AuS–R vs. AuN–R, on the biological activity of these complexes. We now report the synthesis, the electrochemical and spectro-electrochemical investigations carried out on eight monoanionic gold bis(dithiolene) complexes. We also investigated their activity against A2780, a tumorigenic cisplatin-sensitive ovarian cancer cells, and some selected complexes were screened for their ability to inhibit the infection of human hepatoma cells (Huh7) by Plasmodium berghei rodent malaria parasites. The influence of the nature and length of the alkyl chain and polyethylene glycol chain on the cellular uptake, anticancer and antiplasmodial activities of the newly synthesized molecules is discussed.
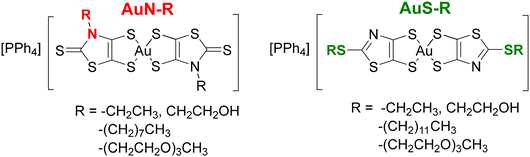 |
| | Chart 1 Target monoanionic gold bis(1,2-dithiolene) complexes. | |
Results and discussion
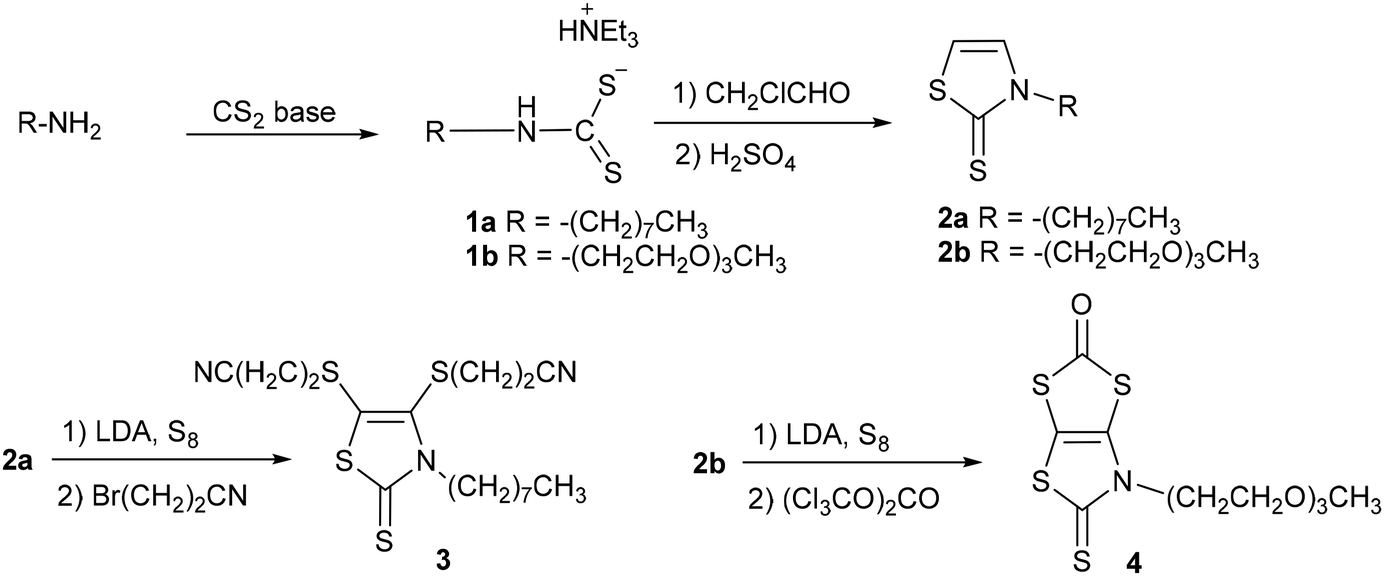
Two synthetic procedures were developed for the synthesis of the proligands required to obtain the target monoanionic gold complexes with a thiazoline and a thiazole backbone, AuN–R and AuS–R respectively. The synthesis of the N-alkylated-1,3-thiazoline-2-thione dithiolene proligands 3 and 4 was performed starting from the primary amine, either the octyl amine or the 2-(2-(2-methoxyethoxy)ethoxy)ethan-1-amine, as outlined in Scheme 1. First, the primary amine was converted into thiazoline-2-thione 2a–b, employing a chemical strategy we previously used for the synthesis of various N-substituted-1,3-thiazoline-2-thiones.25 The primary amine reacts in basic medium (NEt3 for 1a and KOH for 1b) with carbon disulfide to afford the corresponding dithiocarbamate salt 1a–b. Alkylation of the dithiocarbamate salt 1 with chloroacetaldehyde followed by cyclisation and dehydration with sulfuric acid led to the thiazoline-2-thione 2a–b. The protected dithiolene ligand 3, with a long alkyl chain, was formed after the bismetallation of 2a with LDA (lithium di-isopropyl amide) followed by the addition of sulfur and bromopropionitrile, while protected dithiolene ligand 4, with a PEG chain, was synthesized through the bismetalation of 2b with LDA followed by the addition of sulfur and triphosgene.
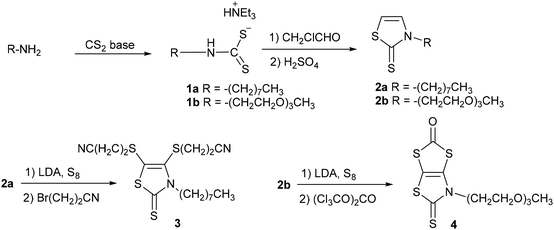 |
| | Scheme 1 Synthesis of proligands 3 and 4. | |
The chemical route depicted in Scheme 2, starting from the N-tBu protected dithiolene ligand 5, was used for the synthesis of the other class of proligands.26 Indeed, it has been shown that it is possible to convert the N-tBu thiazoline-2-thione core into the 2-alkylthio-1,3-thiazole in the presence of various electrophiles, such as an iodoalkyl chain.27,28 The plausible mechanism would be that after alkylation of the exocyclic sulfur atom of 5, leading to an intermediate N-tBu-2-thiomethyl-1,3-thiazolium iodide salt, the elimination of t-BuI leads to the formation of the aromatic thiazole ring. Therefore, in order to reach proligands 6a–b we used the 1-iodododecane and the 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane (PEG–Br) as alkylating agent. Compound 6a was obtained by heating a solution of 5 in the presence of an excess of 1-iododecane in DMSO at 55 °C overnight, while compound 6b was formed by simply reacting 5 with PEG–Br in refluxing acetonitrile overnight. Of note, purification of 6b requires the previous removal of the excess of the alkylating reagent, which can easily be achieved by the addition of petroleum ether, as the reactant, but not proligand 6b, is soluble in this solvent. Both proligands are obtained as viscous oil in 84 and 82% yields for 6a and 6b, respectively.
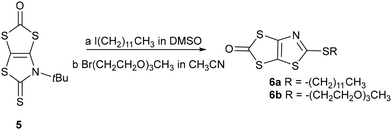 |
| | Scheme 2 Synthesis of proligands 6a–b. | |
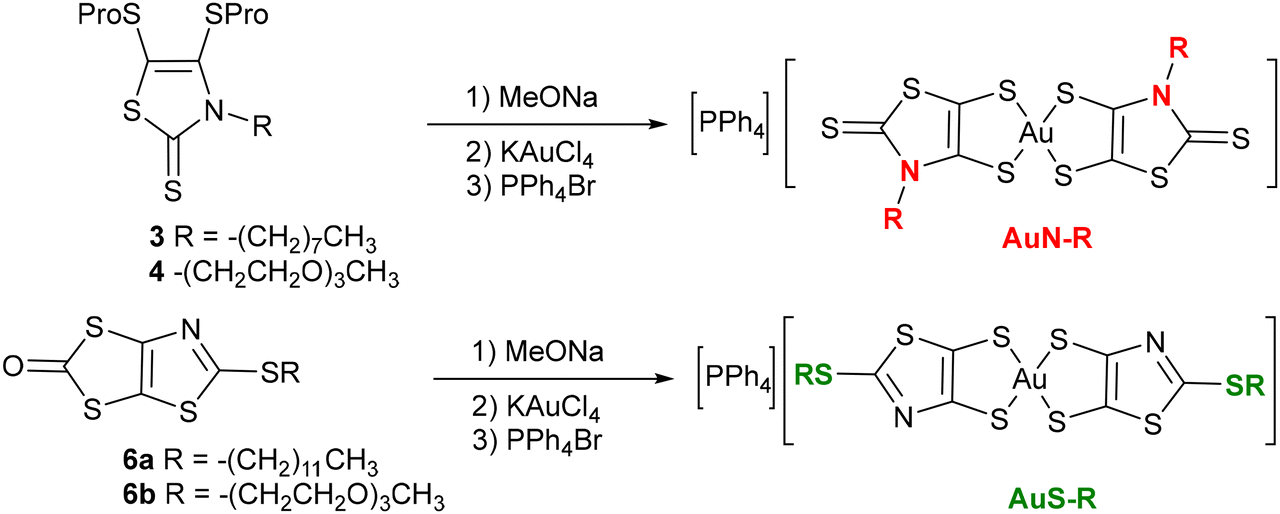
Having obtained these four novel proligands, the two proligands substituted with an alkyl chain, 3 and 6a, and the two proligands substituted with a PEG chain, 4 and 6b, we investigated the formation of the corresponding gold bis(dithiolene) complexes. We used the same strategy to achieve the synthesis of the four monoanionic gold complexes i.e., AuN–C8, AuN–PEG, AuS–C12, and AuS–PEG. It consists in the initial deprotection of the dithiolene ligand in the presence of a nucleophilic base, followed by the addition of the gold salt and the counterion. Regardless of the protecting group, sodium methanolate was used to deprotect the dithiolene ligand, followed by the subsequent addition of KAuCl4 and PPh4Br to the medium (Scheme 3). Besides AuN–C8, which was obtained as powder sample, the three other complexes, AuN–PEG, AuS–C12, and AuS–PEG, were obtained as greenish dark oil. As expected, the PEG substituted complexes are more soluble than their short-alkyl chain analogues such as AuN–Et and AuS–Et. For instance, AuN–Et and Aus–Et dissolve in DMSO in the range of 4 g L−1 while the PEG-substituted complexes, AuS–PEG and AuN–PEG, dissolve in this solvent at 25 g L−1. Single crystals of the monoanionic complex AuN–C8 were obtained by slow evaporation of a concentrated solution of this complex in acetonitrile.
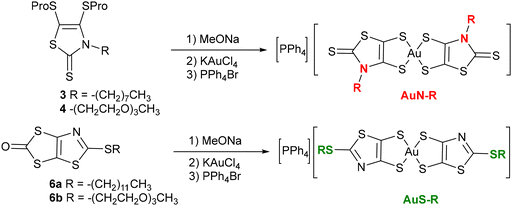 |
| | Scheme 3 Synthesis of the monoanionic gold complexes AuN–R and AuS–R. | |
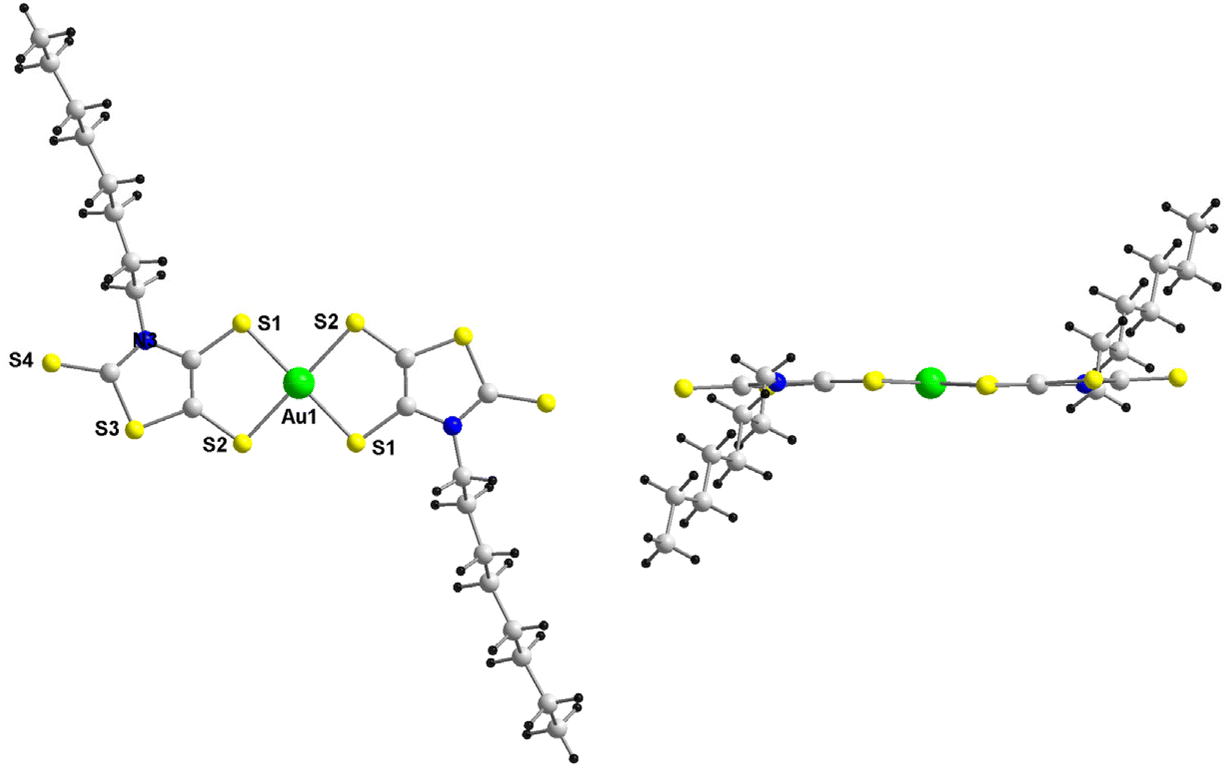
The AuN–C8 complex crystallizes in the monoclinic system, space group C2/c. The molecular structure of the monoanion is depicted in Fig. 1. This complex, located on an inversion center, crystallizes in the trans configuration with a square geometry around the gold atom as often encountered with this family of complexes with a dissymmetrical ligand. The octyl groups are located above and below the plane of the complex.
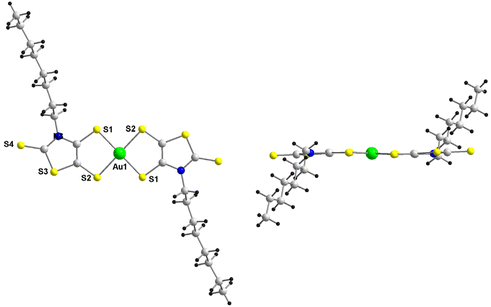 |
| | Fig. 1 Molecular structure of the monoanionic AuN–C8 without the Ph4P+ counterion. | |
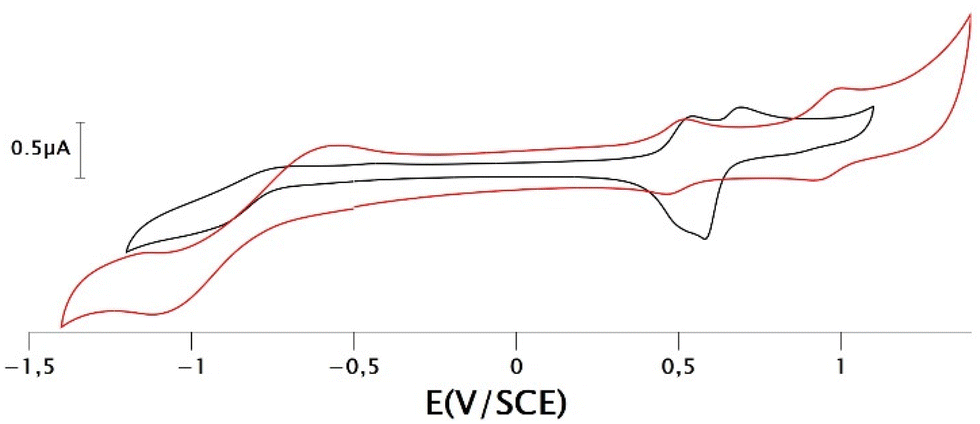
The electrochemical characteristics of the gold bis(dithiolene) complexes have been examined by cyclic voltammetry (CV) in CH2Cl2 containing 0.1 M of Bu4NPF6 as supporting electrolyte. The redox potential of the ferrocene/ferrocenium couple was used as an internal reference. The results are summarized in Table 1. Redox potentials (E) are also given in V vs. Saturated Calomel Electrode (SCE) and are collected in Table S1.† For comparison purposes, we listed in this table the redox properties of the gold complexes substituted with shorter alkyl chains, AuN–R and AuS–R with R = Et, EtOH. Two types of CVs were observed, depending on the class of ligand coordinated to the metal ion, either N-alkylated complexes AuN–R or S-alkylated complexes AuS–R (Fig. 2). For the N-alkylated complexes gold complex, AuN–C8 and AuN–PEG, two close oxidation processes assigned to the oxidation of the monoanionic complex into the neutral radical one and then to the monocationic complex were observed upon anodic scan. On the reverse scan, a sharp cathodic peak of higher intensity than the anodic peak was observed. This indicates that an adsorption phenomenon occurs with this class of ligand. In fact, the shape of this CV is very similar to the one obtained for gold complexes substituted on the nitrogen atoms with smaller alkyl chain, such as for example AuN–Et.4 For the other class of ligand with the thiazole core, such as AuS–C12 and AuS–PEG, two well separated reversible oxidation processes were observed upon oxidation. Moreover, the presence of the thiazole backbone induced a larger potential difference between these two oxidation systems, indicating a larger stability window of the neutral radical species for AuS–R vs. AuN–R. This could plausibly be assigned to the nature of the heterocycle, as changes in the bond lengths occur, during the oxidation of monoanionic gold complexes, mainly on the metallacycle rings. This observation is reminiscent of what has been observed for gold complexes with dithiolene thiophene ligands, where it has been found that the aromatic ring stabilizes the neutral complex.21 Herein, the shape of the voltammogram is very different from the one observed with the shorter alkyl chain, for which only one pseudo reversible system was observed upon oxidation, with a sharp cathodic peak resulting from electrodeposition upon oxidation.12 Actually, in this series only AuS–tBu exhibited a cyclic voltammogram similar to that of the complexes with a long alkyl chain.29 The reversible systems obtained suggests that AuS–C12 and AuS–PEG are less susceptible to settles on the electrode surface as it was observed for the gold complexes belonging to the same series with shorter alkyl chain (Fig. S1 and S2†). An irreversible reduction process was observed on the cathodic scan for these four gold complexes regardless of the ligand. This reduction corresponds to the reduction of the monoanionic complex into the dianionic species.
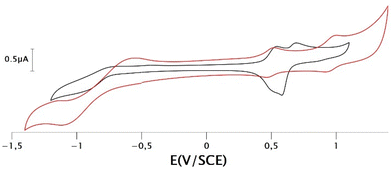 |
| | Fig. 2 Cyclic voltammograms of complexes AuN–PEG (black curve) and AuS–PEG (red curve), in 0.1 M [CH2Cl2][Bu4NPF6], scan rate 100 mV s−1. | |
Table 1 Redox potentials (E in V) are given vs. Fc/Fc+ couple and absorption maxima λmax (nm) for the NIR absorptions of the investigated monoanionic gold complexes
| |
E
1pc
|
E
2pa/pc
|
E
3pa/pc
|
λ
max (nm) |
| Neutral |
Monocation |
|
Irreversible process.
Adsorption Epa/Epc, Epa and Epc: anodic and cathodic peak potentials.
|
| AuN–C8 |
−1.50a |
0.14/0.09 |
0.37/0.19 |
2080 |
— |
| AuN–PEG |
−1.34a |
0.14/0.13b |
0.28/0.18b |
2030 |
— |
| AuN–Et |
−1.40a |
0.15/0.09b |
0.31/0.21b |
— |
— |
| AuN–EtOH |
−1.39a |
0.07/0.05b |
0.27/0.06b |
— |
— |
| AuS–C12 |
−1.41a |
0.11/−0.06 |
0.70/0.51b |
1734 |
1030 |
| AuS–PEG |
−1.50a |
0.12/0.06 |
0.60/0.53 |
1700 |
1050 |
| AuS–Et |
−1.44a |
0.08/0.04b |
— |
— |
— |
| AuS–EtOH |
−1.52a |
0.05/−0.12b |
— |
— |
— |
| AuS–tBuS29 |
−1.58a |
0.18/0.02 |
0.65/0.58 |
1614 |
1038 |
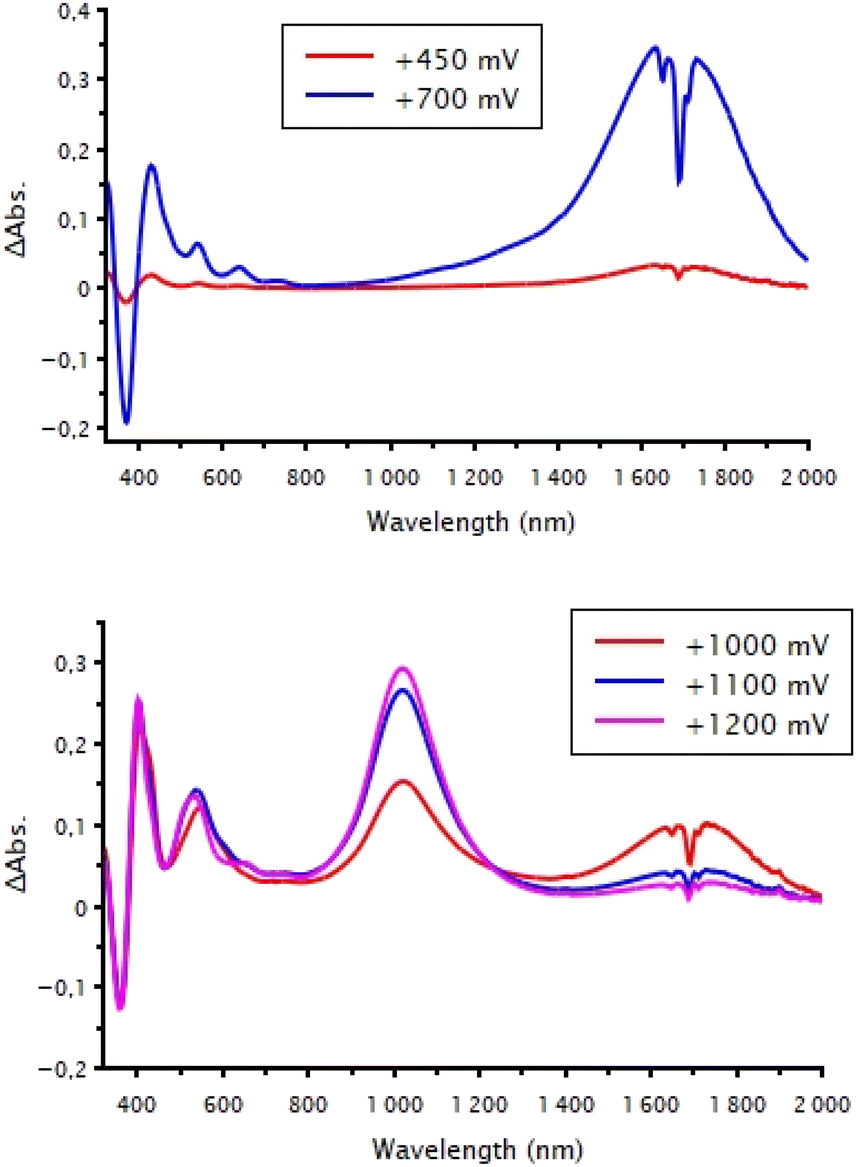
All monoanionic complexes present absorption bands in the UV-vis region only. We performed spectroelectrochemical investigations on the four novel monoanionic complexes, i.e., AuN–C8, AuN–PEG, AuS–C12, and AuS–PEG, in order to determine the absorption spectra of the neutral species. The UV-vis-NIR spectroscopic investigations of the monoanionic complexes were carried out at room temperature in dichloromethane solution (C = 5 × 10−5 M) containing 0.2 M of Bu4NPF6 as supporting electrolyte. Two different behaviors can be observed depending on the class of ligand. Indeed, upon gradual oxidation of the monoanionic complexes AuN–R, a broad absorption band appears at 2030–2080 nm, which corresponds to the spectral signature of the generated neutral species in the short wavelength infrared (SWIR) (Table 1). No further evolution of the spectra is observed following the continuation of gradual oxidation. This could be due to adsorption of the oxidized species on the platinum grid. Of note, a closer look at the working platinum grid shows that the neutral complex covers the surface of the electrode, preventing the investigation of the monocationic species. This is consistent with the adsorption noted upon the electrochemical investigation. For the AuS–R complexes, the evolution of the absorption spectra shows respectively the signature of the neutral species growing at 1700–1734 nm followed by the concomitant disappearance of this absorption band at 1700 nm and the appearance of a novel absorption band centered at 1030–1050 nm (Fig. 3). This absorption band at higher energy corresponds to the spectral signature of the monocationic complex generated in the medium. This spectral evolution upon gradual oxidation of AuS–C12, and AuS–PEG is uncommon for neutral radical gold bis(dithiolene) complexes. Actually, besides these two complexes and AuS–tBu,29 only one example reported in the literature shows a similar behavior.30 Due to a slight adsorption of the electrogenerated species on the platinum grid upon oxidation we were unable to estimate the molar absorption coefficients of neither the neutral nor the monocationic complexes. An interesting feature is the red shift observed for the neutral AuN–R gold complexes with a thiazoline ligand compared to those of the AuS–R with a thiazole ligand thus highlighting the influence of the ligand, aromatic or not, on the absorption properties.
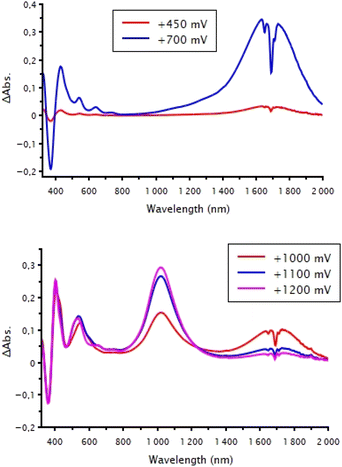 |
| | Fig. 3 Differential spectroelectrochemical spectrums of AuS–PEG (top) from the monoanionic to the neutral state and (bottom) from the neutral to the monocationic state in 0.2 M [CH2Cl2][Bu4NPF6]. | |
Assessment of complexes activity against the ovarian cancer cells
The anticancer activity of the complexes under study was evaluated in the A2780 cells representative of an epithelial ovarian cancer cell type, the most fatal gynecological malignancy.31 The pH of the cell medium is approximately 7.5. The complexes are stable in this pH range. Cells were exposed to increasing concentrations of the complexes (0.01–50 μM) for 24 h, at 37 °C. IC50 values were calculated from dose–response curves using the colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Our results show that the compounds cytotoxic profile depends on the nature of the heterocycle, thiazoline (AuN–R) vs. thiazole (AuS–R), as well as the length and presence or not of oxygen atoms within the alkyl chain (Table 2). For instance, if we take into account only the isomeric forms, that is to say AuN–R vs. AuS–R with the same alkyl chain R (R = Et, EtOH and PEG), the thiazoline ring in AuN–R increases the cytotoxic activity compared to the thiazole ring in AuS–R. If we compare the activity of short alkyl chain with the one of long alkyl chain in both series, AuN–R/AuS–R, the presence of long alkyl chain decreases significantly the cytotoxicity of the complexes by two order of magnitude. On the other hand, both complexes with the PEG chain showed higher cytotoxicity than the complexes with only alkyl chains (AuS–PEG vs. AuS–C12 and AuN–PEG vs. AuN–C8). This tends to indicate that it is not the length of the alkyl chain but rather the nature, hydrophobic vs. hydrophilic, that induces this difference. All complexes, excluding AuN–C8, are much more active than the chemotherapeutic drug cisplatin. Moreover, except for AuN–C8 and AuS–C12, the tested complexes presented activities within the same range or even superior to that of the gold standard auranofin. Also, as expected, the complexes containing a PEG chain showed improved solubility, which is in line with the results obtained by some of us with the use of block copolymer micelles (BCMs) formulations encapsulating related gold complexes.32
Table 2 IC50 values (μM) calculated for the Au(III) bis(dithiolene) complexes with the A2780 ovarian cancer cells after 24 h exposure
| Complex |
IC50 (μM) |
|
Results from ref. 22.
|
| AuN–Et |
0.70 ± 0.2 |
| AuS–Et |
0.87 ± 0.4 |
| AuN–EtOH |
0.25 ± 0.03 |
| AuS–EtOH |
2.61 ± 0.9 |
| AuN–C8 |
71.3 ± 2.0 |
| AuS–C12 |
17.8 ± 6.2 |
| AuN–PEG |
1.27 ± 0.3 |
| AuS–PEG |
1.70 ± 0.7 |
| Cisplatin |
21.1 ± 5.0a |
| Auranofin |
2.01 ± 0.7a |
Cellular uptake analysis
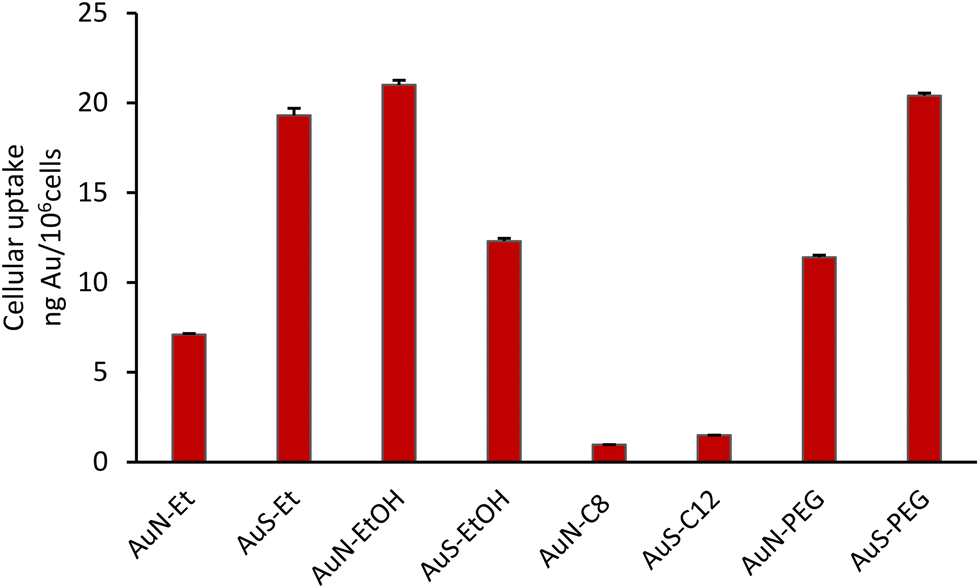
The cellular uptake of the complexes by the A2780 cells was evaluated to assess a possible relationship with their cytotoxic activity. All the complexes were incubated with the cells at 50 μM concentration for 3 h, except AuN–EtOH, that due to its higher cytotoxicity was evaluated at 20 μM. The gold content was determined by PIXE (proton induced X-ray emission) in the bulk cell pellets after exposure with the complexes. As can be observed from Fig. 4, the highest uptake of complexes AuS–Et, AuN–EtOH and AuS–PEG by the A2780 cells, followed by those of complexes AuS–EtOH, AuN–PEG and AuN–Et, do not follow the same trend with their cytotoxic activity (Table 2). AuS–C12 and AuN–C8 showed very low uptake that is in line with their poor cytotoxicity in particular for AuN–C8. Thus, it can be seen that with a longer alkyl chain as in AuS–C12 and AuN–C8 compared to AuS–Et and AuN–Et, the increase in hydrophobicity limits the cellular uptake and, as a consequence, the cytotoxicity of these complexes. Contrariwise, the hydrophilic character brought by the PEG chain induces an increase of the cellular uptake of AuN–PEG and AuS–PEG. Among all these complexes, it is noteworthy that the AuN–EtOH complex, with the hydroxyethyl group exhibit the highest uptake together with the strongest cytotoxicity. These results suggest that different parameters should be taking into account when the cytotoxicity and cellular uptake values are concerned such as the incubation time, rate of uptake of the compound and/or the molecular cellular targets involved.
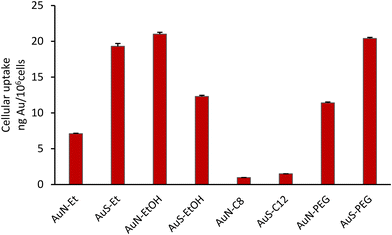 |
| | Fig. 4 Total cellular uptake of gold in the A2780 cells upon exposure at 50 μM of each complex and at 20 μM for AuN–EtOH after 3 h incubation. Results (ng Au per 106 cells) are the mean ± SD of two independent experiments. | |
In vitro assessment of complexes activity against the hepatic stage of P. berghei infection
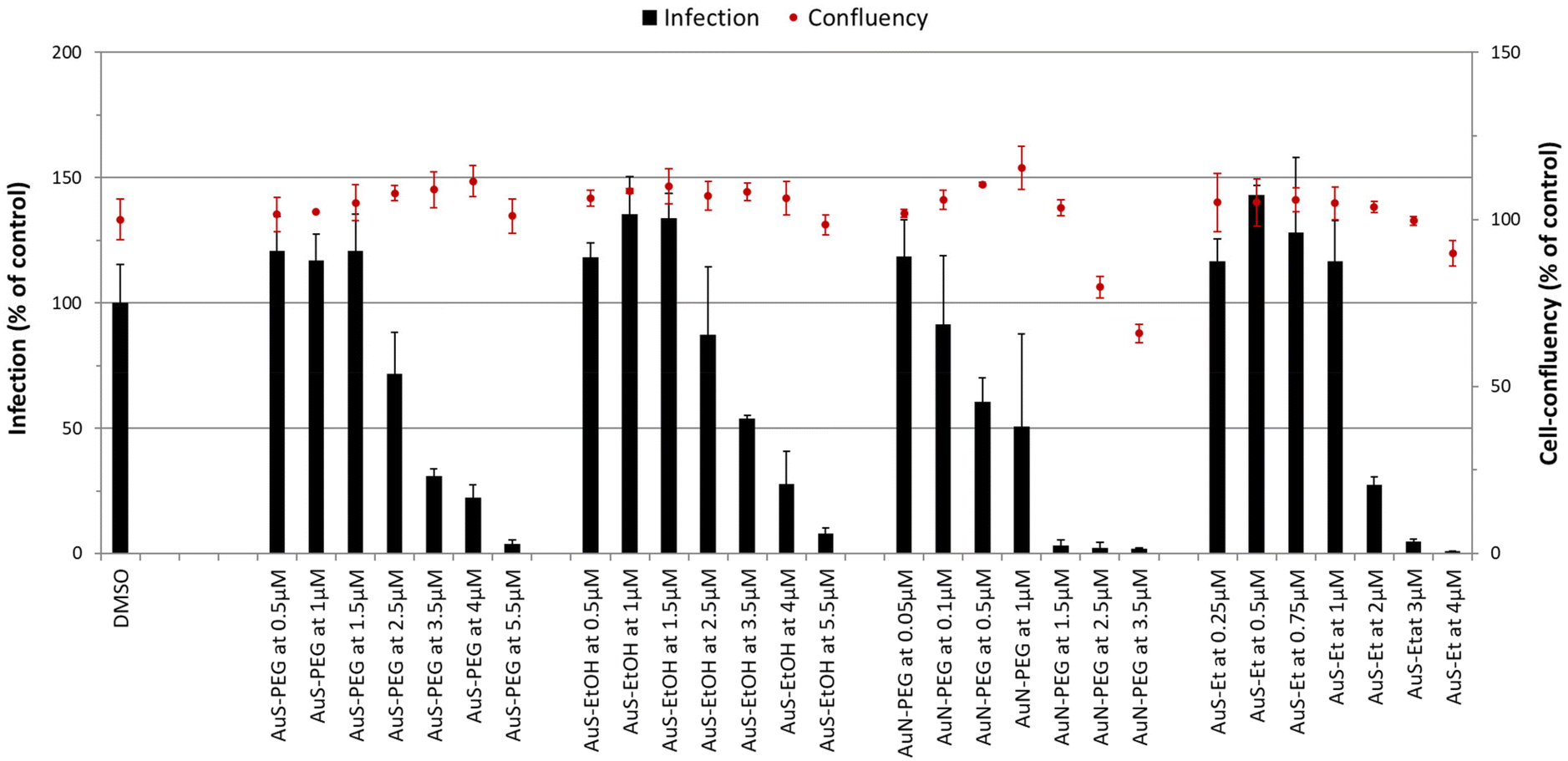
The antiplasmodial activity of selected complexes AuS–PEG, AuS–EtOH, AuN–PEG and AuS–Et was assessed against the obligatory, yet clinically silent hepatic stage of Plasmodium infection, employing Huh-7 cells infected with rodent P. berghei sporozoites. The standard antiplasmodial drug primaquine was used as a positive control,33 while DMSO employed at a percentage equivalent to that of the highest compound concentrations tested was used as a negative control. All four complexes reduced infection at concentrations below 5 μM, with complexes AuN–PEG and AuS–Et displaying the strongest potency against infection (Table 3, Fig. 5). IC50 determinations revealed that complex AuN–PEG displayed the strongest activity against hepatic infection by the malaria parasite, with an estimated IC50 of 0.92 ± 0.29 μM, while AuS–EtOH was the least active of the four, with an estimated IC50 of 3.64 ± 0.16 μM (Table 3).
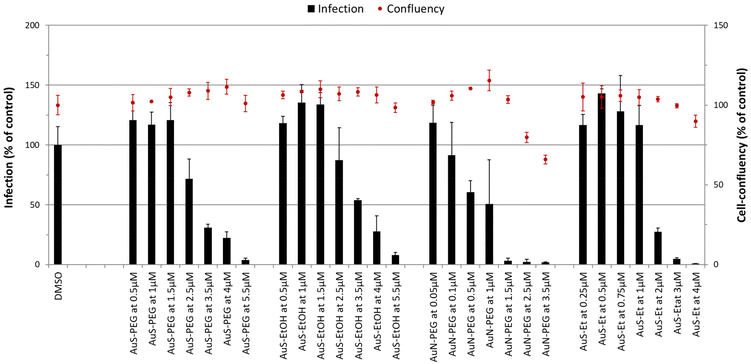 |
| | Fig. 5 Dose-dependent assessment of selected complexes against the hepatic stage of P. berghei infection. Huh-7 cells were incubated with each complex at seven different concentrations and infected with luciferase-expressing P. berghei parasites. Total parasite load (infection scale, bars) and cellular viability (cell confluency scale, red dots) are shown. Results were normalized to the negative control, n ≥ 2. IC50 values are displayed in Table 3. | |
Table 3
In vitro activity assessment (IC50, μM) of selected complexes against the hepatic stage of P. berghei infection. Primaquine was used as a positive control. Results are shown as mean ± SD of three technical replicates
| Complexes |
IC50 (μM) |
| AuS–PEG |
3.16 ± 0.13 |
| AuS–EtOH |
3.64 ± 0.16 |
| AuN–PEG |
0.92 ± 0.29 |
| AuS–Et |
1.67 ± 0.27 |
| Primaquine |
8.43 ± 3.38 |
Our results show that all complexes display higher inhibitory activity against P. berghei hepatic infection than the reference control, primaquine. The PEG chain-containing complexes presented herein offer potential advantages over previously reported BCMs-encapsulating Au(III) bis(dithiolene) complexes,32 besides their higher solubility and high cellular uptake. Indeed, whereas the antiplasmodial activity of AuS–PEG and AuN–PEG appears to be preserved in the presence of the PEG chain, the previously reported complexes encapsulated in micelles displayed lower activity against P. berghei hepatic infection than their non-encapsulated counterparts.
Conclusions
In summary, we have designed and synthesized two types of novel monoanionic gold(III) bis(dithiolene) complexes, with either thiazoline ligands, corresponding to the N-substituted complexes, AuN–R, or with thiazole ligands, corresponding to the S-substituted complexes, AuS–R. In both series, we substituted theses complexes with long alkyl chains or with PEG ones in order to analyze the influence of these substituents on the overall properties of these complexes. Compared to the same complexes substituted by shorter alkyl chains, the presence of these long chains improved the solubility of higher oxidation states as evidenced by the observation of fully reversible redox systems and the absorption spectra of the neutral and monocationic species. In this present study, we also investigated these four complexes with four of their analogues, substituted with shorter alkyl chains (R = Et, EtOH), as anticancer and antiplasmodial agents. Besides the two complexes substituted with long alkyl chains, AuN–C8 and AuS–C12, they all exhibited strong anticancer activities, similar or even higher than the reference drugs cisplatin and auranofin. This low anticancer activity for the AuN–C8 and AuS–C12 is associated to a very low cellular uptake presumably due to the hydrophobicity provided by the long chains. The replacement of these long alkyl chains with PEG substituents reduces the hydrophobicity and consequently increased the cellular uptake together with the anticancer activity close to the one noticed for gold complexes with short alkyl chains. A noticeable difference is observed between the two gold complexes series, as the AuN–R complexes are more cytotoxic than the AuS–R complexes. A closer analysis of the biological activities of the complexes with the polyoxygenated chain revealed that besides its influence on the solubility the complexes maintained their biological properties in particular their antiplasmodial activities. Therefore, the presence of PEG chains on the complexes avoids any subsequent encapsulation step into block copolymer micelles to improve their solubility. To sum up, the herein presented results indicated for this type of gold complexes their worth value as prospective anticancer and antiplasmodial drugs and the importance for further research on the path to finding alternative drugs showing a better clinical profile to the ones in clinical use.
Experimental
Chemicals and materials from commercial sources were used without further purification. All the reactions were performed under an argon atmosphere. 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm) and 13C NMR spectra were referenced to CDCl3 (77.2 ppm) and chemical shifts are reported in ppm. Melting points were measured on a Kofler hot-stage apparatus and are uncorrected. Mass spectra were recorded by the Centre Régional de Mesures Physiques de l'Ouest, Rennes. Methanol, acetonitrile and dichloromethane were dried using Inert pure solvent column device. CVs were carried out on a 10−3 M solution of complex in CH2Cl2 with NBu4PF6 0.1 M. Potentials were measured versus Saturated Calomel Electrode (SCE). Ferrocene was added directly to the solution after analysis of the analyte of interest to allow the potentials normalization in situ, relative to the ferrocene/ferrocenium couple redox potential. The spectroelectrochemical setup was performed in CH2Cl2 with Bu4NPF6 0.2 M using a Pt grid as the working electrode, a Pt wire as the counter electrode and SCE reference electrode. A Shimatzu 3600 plus spectrophotometer was employed to record the UV-vis-NIR spectra.
N-Octyl-1,3-thiazoline-2-thione 2a
To a solution of octylamine (12.92 g, 0.1 mol) in 200 mL of diethylether at 0 °C under nitrogen was added trimethylamine (15.17 g, 0.15 mol) and carbon disulfide (11.42 g, 0.15 mol). The mixture was stirred for 2 hours. The precipitate was filtered off and washed with diethylether to afford 14.85 g of dithiocarbamate salt which was used without further purification. Yield 53%, m.p. 50 °C. 1H NMR (300 MHz, CDCl3) δ 0.80 (t, 3H, CH3, 3J = 7.1 Hz), 1.20 (m, 6H, 3CH2), 1.32 (t, 9H N(CH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3)3, 3J = 7.1 Hz), 1.52 (m, 2H, CH2), 1.68 (m, 2H, CH2), 2.99 (m, 2H, CH2), 3.22 (q, 6H N(C2CH3)3, 3J = 7.1 Hz), 3.48 (m, 2H, CH2), 7.61 (m, 1H, NH), 8.44 (broad, 1H, NH). To a suspension of dithiocarbamate salt (14.85 g, 52.0 mmol) in acetonitrile (200 mL) was added chloroacetaldehyde (50% solution in water, 6.7 mL, 52.0 mmol). The mixture was stirred 12 hours at room temperature under nitrogen. The volume was reduced to approximatively 1/5 in vacuo and the solution was slowly added to a flask containing 15 mL of H2SO4 at 0 °C. The mixture was stirred 20 min at 0 °C, hydrolyzed with 100 mL of water, extracted with CH2Cl2 (3 × 80 mL), washed with water until aqueous phase reach neutral/basic pH and dried over MgSO4. The concentrated solution was purified by chromatography on silica gel using CH2Cl2 as eluant to afford 2a as pale brown oil (9.8 g). Yield 83%; 1H NMR (300 MHz, CDCl3) δ 0.85 (t, 3H, CH3, 3J = 7.1 Hz), 1.28 (m, 10H, 5CH2), 1.78 (m, 2H, CH2), 4.13 (m, 2H, NCH2), 6.59 (d, 1H, CH, 3J = 4.6 Hz), 7.05 (d, 1H, CH, 3J = 4.6 Hz); 13C NMR (75 MHz, CDCl3) δ 14.1, 22.6, 26.5, 28.4, 29.1, 31.7, 50.0, 111.0, 131.3, 187.2; HRMS (ESI) calcd for C11H20NS2 [M + H]+: 230.10317. Found: 230.1030; anal calcd for C11H19NS2: C, 57.59; H, 8.35; N, 6.11; S, 27.95. Found: C, 57.91; H 8.42; N, 6.27; S, 28.07.
3)3, 3J = 7.1 Hz), 1.52 (m, 2H, CH2), 1.68 (m, 2H, CH2), 2.99 (m, 2H, CH2), 3.22 (q, 6H N(C2CH3)3, 3J = 7.1 Hz), 3.48 (m, 2H, CH2), 7.61 (m, 1H, NH), 8.44 (broad, 1H, NH). To a suspension of dithiocarbamate salt (14.85 g, 52.0 mmol) in acetonitrile (200 mL) was added chloroacetaldehyde (50% solution in water, 6.7 mL, 52.0 mmol). The mixture was stirred 12 hours at room temperature under nitrogen. The volume was reduced to approximatively 1/5 in vacuo and the solution was slowly added to a flask containing 15 mL of H2SO4 at 0 °C. The mixture was stirred 20 min at 0 °C, hydrolyzed with 100 mL of water, extracted with CH2Cl2 (3 × 80 mL), washed with water until aqueous phase reach neutral/basic pH and dried over MgSO4. The concentrated solution was purified by chromatography on silica gel using CH2Cl2 as eluant to afford 2a as pale brown oil (9.8 g). Yield 83%; 1H NMR (300 MHz, CDCl3) δ 0.85 (t, 3H, CH3, 3J = 7.1 Hz), 1.28 (m, 10H, 5CH2), 1.78 (m, 2H, CH2), 4.13 (m, 2H, NCH2), 6.59 (d, 1H, CH, 3J = 4.6 Hz), 7.05 (d, 1H, CH, 3J = 4.6 Hz); 13C NMR (75 MHz, CDCl3) δ 14.1, 22.6, 26.5, 28.4, 29.1, 31.7, 50.0, 111.0, 131.3, 187.2; HRMS (ESI) calcd for C11H20NS2 [M + H]+: 230.10317. Found: 230.1030; anal calcd for C11H19NS2: C, 57.59; H, 8.35; N, 6.11; S, 27.95. Found: C, 57.91; H 8.42; N, 6.27; S, 28.07.
N-PEG-1,3-thiazoline-2-thione 2b
Under inert atmosphere, to a suspension of potassium hydroxide (0.48 g, 8.52 mmol) finely ground and the 2-(2-(2-methoxyethoxy)ethoxy)ethan-1-amine (1.39 g, 8.52 mmol) in a mixture of 30 mL of diethyl ether and 30 mL of acetonitrile at 0 °C was added dropwise CS2 (0.52 mL, 8.52 mmol). The suspension was stirred for 3 hours. The precipitate was filtered off, washed with diethyl ether and dried under a vacuum to afford the product 1b as a yellowish powder (1.32 g). This salt was used without further purification for the synthesis of the thiazoline-2-thione ring. Yield 56%; Mp 48 °C; 1H NMR (300 MHz, D2O): δ 3.79 (m, 10H, 5CH2), 3.71 (m, 2H, H2), 3.46 (s, 3H, CH3); 13C NMR (75 MHz, D2O): δ 47.2, 58.1, 68.3, 69.5, 69.6, 69.6, 71.1, 211.9. Under inert atmosphere, to a suspension of dithiocarbamate salt 1b (1.32 g, 4.76 mmol) in 50 mL of MeCN, was added as solution of chloroacetaldehyde, 50% in water (0.64 mL, 5.00 mmol). The suspension was stirred overnight. 4/5 of MeCN was evaporated and the solution was added dropwise to 4 mL of H2SO4, at 0 °C. The solution was stirred for 20 minutes before hydrolyzed with 60 mL of water. The product was extracted three times with CH2Cl2 and washed with water until aqueous phase reach neutral/basic pH. The organic phase was dried over MgSO4, filtered off and concentrated. Purification by flash chromatography (CH2Cl2/diethyl ether gradient from 100/0 to 50/0) give the product as a brown oil (1.09 g); yield 87%; 1H NMR (300 MHz, CDCl3): δ 3.37 (s, 3H, CH3), 3.53 (m, 2H, CH2), 3.60 (m, 6H, 3CH2), 3.82 (m, 2H, NCH2), 4.38 (m, 2H, NCH2C2), 6.52 (d, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, 3J = 4.6 Hz), 7.28 (d, 1H, CH, 3J = 4.6 Hz); 13C NMR (75 MHz, CDCl3): δ 49.8, 59.1, 68.4, 70.6, 70.6, 72.1, 109.9, 133.4, 187.7; HRMS (ASAP) calcd for C10H17NO3NaS2+ [M + Na]+: 286.05421, found 286.0547.
CH, 3J = 4.6 Hz), 7.28 (d, 1H, CH, 3J = 4.6 Hz); 13C NMR (75 MHz, CDCl3): δ 49.8, 59.1, 68.4, 70.6, 70.6, 72.1, 109.9, 133.4, 187.7; HRMS (ASAP) calcd for C10H17NO3NaS2+ [M + Na]+: 286.05421, found 286.0547.
4,5-Biscyanoethylthio-N-octyl-1,3 thiazole-2-thione 3
To a −10 °C cooled solution of thiazoline 2a (2 g, 8.7 mmol) in 50 mL of dry THF under nitrogen was added a solution of LDA prepared from diisopropylamine (1.84 mL, 13.1 mmol) and n-BuLi 1.6 M in hexane (8.2 mL, 13.1 mmol) in 15 mL of dry THF. After stirring for 30 min at −10 °C, sulphur S8 (419 mg, 13.1 mmol) was added and the solution was stirred for an additional 30 min. A solution of LDA (diisopropylamine 2.45 mL, 17.5 mmol and n-BuLi 1.6 M in hexane 10.9 mL, 17.5 mmol) in 20 mL of dry THF was added. The mixture was stirred for 3 hours and S8 (558 mg, 17.5 mmol) was added. After 30 min bromopropionitrile (7.7 mL, 87.0 mmol) was added. The reaction mixture was stirred overnight. The solvent was evaporated in vacuo. The concentrated solution was purified by chromatography on silica gel using CH2Cl2 as eluant to afford 3 as a brown powder (2.01 g). Yield 15%; Mp 58 °C; 1H NMR (300 MHz, CDCl3) δ 0.85 (t, 3H, CH3, 3J = 7.1 Hz), 1.28 (m, 10H, 5CH2), 1.70 (m, 2H, CH2), 2.71 (m, 4H, C2CH2CN), 3.10 (m, 4H, CH2C2CN), 4.31 (m, 2H, NCH2); 13C NMR (75 MHz, CDCl3) δ 14.1, 18.7, 22.6, 26.7, 27.8, 29.0, 29.1, 31.7, 31.8, 32.4, 49.7, 117.3, 125.5, 135.9, 187.3; HRMS (ESI) calcd for C17H25N3NaS4 [M + Na]+: 422.08235. Found: 422.0822; anal. calcd for C17H25N3S4: C, 51.09; H, 6.31; N, 10.51; S, 32.09. Found: C, 51.32; H 6.35; N, 10.21; S, 31.88.
1,3 dithiol-2-one/N-PEG thiazoline 4
Under inert atmosphere, to a three necked flask containing a solution of N-PEG thiazoline 2b (312 mg, 1.18 mmol) and sulfur (134 mg, 4.16 mmol) in 7 mL of dry THF at −15 °C was added dropwise LDA, freshly prepared under inert atmosphere at 0 °C from butyllithium 1.6 M in hexane (2.6 mL, 4.16 mmol) and diisopropylamine (0.62 mL, 4.16 mmol) in 3 mL of dry THF. The solution was stirred for 1 hour and a solution of triphosgen (521 mg, 1.72 mmol) in 2 mL of dry THF was added. The temperature was maintained at −15 °C and the solution stirred for 1 hour, then warmed to room temperature for 30 minutes. The reaction was quenched with 25 mL of water, the THF was evaporated and the product was extracted with CH2Cl2 3 times, dried over MgSO4, filtered off and concentrated. Purification by flash chromatography on silica gel (CH2Cl2/ethyl acetate from gradient 100/0 to 90/10) give the 4 as a brown oil (244 mg). Yield 58%. 1H NMR (300 MHz, CDCl3): δ 3.37 (s, 3H, CH3), 3.52 (m, 2H, CH2), 3.60 (m, 6H, 3CH2), 3.86 (m, 2H, NCH2), 4.35 (m, 2H, NCH2C2); 13C NMR (75 MHz, CDCl3): δ 52.3, 59.2, 67.9, 70.7, 70.8, 71.0, 72.1, 103.1, 128.8, 187.6, 187.8; HRMS (ASAP) calcd for C11H16NO4S4 [M + H]+: 353.99567, found 353.9954; anal. calcd for C11H15NO4S4 + 1/3 CH3CO2Et: C, 38.69; H, 4.65. Found: C, 38.50; H 4.69.
1,3 dithiol-2-one/SC12 thiazole 6a
Under inert atmosphere, to a flask containing a solution of dithiolone 5 (250 mg, 0.95 mmol) in 3 mL of degassed DMSO was added 1-iodododecane (2.34 mL, 9.5 mmol). The solution was stirred at 55 °C overnight. The concentrated solution was purified by flash chromatography on silica gel (CH2Cl2/petroleum ether, 0/100 to 40/60) to afford 6a as a brown oil (302 mg). Yield 84%; 1H NMR (300 MHz, CDCl3) δ 0.87 (t, 3H, CH3, 3J = 7.1 Hz), 1.27 (m, 16H, 8CH2), 1.44 (m, 2H, CH2), 1.77 (m, 2H, CH2), 3.23 (t, 2H, SCH2); 13C NMR (75 MHz, CDCl3) δ 14.1, 22.7, 28.7, 29.0, 29.1, 29.3, 29.4, 29.5, 29.6, 31.9, 35.2, 76.6, 77.1, 77.5, 115.5, 142.1, 167.5, 190.5; HRMS (ESI) calcd for C16H25NONaS4 [M + Na]+: 398.07112. Found: 398.0710; anal. calcd for 2[C16H25NOS4] + DMSO: C, 52.27; H, 7.22; N, 3.59; S, 32.83. Found: C, 52.56; H, 6.93; N, 3.14; S, 33.58.
1,3 dithiol-2-one/S-PEG thiazole 6b
Under inert atmosphere, to a flask containing a solution of dithiolone 5 (300 mg, 1.14 mmol) in 3 mL of MeCN is added PEG–Br (2 mL, 11.5 mmol). The solution was stirred at 80 °C overnight. The mixture was then concentrated and purified by flash chromatography on silica gel (CH2Cl2/ethyl acetate gradient from 100% CH2Cl2 to 80% CH2Cl2) to afford a mixture of proligand 6b and starting PEG–Br. The mixture was washed with petroleum ether and the supernatant was removed. The oil obtained was dried under vacuum to afford 6b as a brown oil (330 mg). Yield 82%; 1H NMR (300 MHz, CDCl3): δ 3.38 (s, 3H, CH3). 3.45 (t, CH2, 3J = 6.3 Hz), 3.55 (m, 2H, CH2), 3.65 (m, 6H, 3CH2), 3.82 (t, 2H, SCH2C2, 3J = 6.3 Hz); 13C NMR (75 MHz, CDCl3): δ 34.7, 59.2, 69.5, 70.7, 70.7, 70.7, 72.1, 116.0, 142.1, 167.0, 190.7; HRMS (ASAP) calcd for C11H16NO4S4 [M + H]+: 353.99567, found 353.9953; anal. calcd for C11H15NO4S4: C, 37.38; H, 4.28; N, 3.96; S, 36.28. Found: C, 37.85; H, 4.50; N, 3.99; S, 35.72.
AuN–C8
To a dry two necked flask containing thiazoline-2-thione 3 (250 mg, 0.62 mmol) was added a solution of NaOMe (5.0 mmol) in dry methanol (prepared from 115 mg of Na in 10 mL of dry MeOH). The solution was stirred 1 hour and a solution of KAuCl4 (118 mg, 0.31 mmol) in 5 mL of dry MeOH was added. The reaction mixture was stirred 5 hours at room temperature and PPh4Br (130 mg, 0.31 mmol) was added. The mixture was stirred at room temperature for 12 hours and the precipitate was filtered off and recrystallized in Me3CN to afford the monoanionic complex as green crystals (220 mg). Yield 63%; Mp 130 °C; 1H NMR (300 MHz, CDCl3) δ 0.87 (m, 6H, CH3), 1.20 (m, 20H, 10CH2), 1.72 (m, 4H, CH2), 4.03 (m, 4H, 2NCH2), 7.60 (m, 8Ar), 7.74 (m, 8Ar), 7.88 (m, 4Ar); 13C NMR (75 MHz, CDCl3) δ 14.1, 22.6, 26.8, 27.5, 29.2, 31.8, 47.8, 47.9, 76.6, 77.0, 77.4, 112.4, 116.8, 118.0, 130.7, 130.9, 132.2, 132.4, 134.3, 134.4, 135.9, 191.3, 191.4; UV-vis (CH2Cl2) λmax(nm) (ε[L mol−1 cm−1]) = 364 (38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 110); HRMS (ESI) calcd for C68H88N4PS16Au2 [2A−, C+]−: 1897.16148. Found: 1897.1619; anal. calcd for C46H54AuN2PS8: C, 79.36; H, 4.86; N, 2.50; S, 22.92. Found: C, 49.33; H, 4.85; N, 2.63; S, 23.42.
110); HRMS (ESI) calcd for C68H88N4PS16Au2 [2A−, C+]−: 1897.16148. Found: 1897.1619; anal. calcd for C46H54AuN2PS8: C, 79.36; H, 4.86; N, 2.50; S, 22.92. Found: C, 49.33; H, 4.85; N, 2.63; S, 23.42.
AuN–PEG
Under inert atmosphere, to a two necked flask containing proligand 4 (160 mg, 0.45 mmol) was added a solution of NaOMe (3.2 mmol) prepared from 88 mg of Na in 8.5 mL of dry MeOH under inert atmosphere. The solution was stirred 1 hour and a solution of KAuCl4 (85 mg, 0.23 mmol) in 5.5 mL of dry MeOH was added. The reaction mixture was stirred 5 hours at room temperature and PPh4Br (95 mg, 0.23 mmol) was added. The mixture was stirred overnight. The supernatant was removed and concentrated to dryness. The crude was dissolved in CH2Cl2 and washed with water twice. Organic layer was dried over MgSO4, filtered off and concentrated to afford the product as a greenish black oil (192 mg). Yield 74%; 1H NMR (300 MHz, CDCl3): δ 3.38 (s, 6H, CH3), 3.56 (m, 4H, 2CH2), 3.65 (m, 8H, 4CH2), 3.71 (m, 4H, 2CH2), 3.81 (m, 4H, NCH2), 4.30 (m, 4H, NCH2C2), 7.60 (m, 8H, Ar), 7.74 (m, 8H, Ar), 7.89 (m, 4H, Ar); 13C NMR (75 MHz, CDCl3): δ 46.4, 59.2, 67.0, 70.2, 70.3, 70.4, 71.7, 112.8, 112.9, 117.0, 118.1, 128.6, 128.7, 130.8, 131.0, 132.1, 132.2, 132.5, 133.1, 134.4, 134.5, 136.0, 192.5; UV-vis (CH2Cl2) λmax(nm) (ε[L mol−1 cm−1]) = 364 (50600); HRMS (ESI) calcd for C64H80N4O12PS16Au2 [2A−, C+]−: 2033.03786. Found: 2033.0371; anal. calcd for C44H50AuN2O6PS8 + 0.5 pentane: C, 45.65; H, 4.61; N, 2.29. Found: C, 45.95; H, 4.31; N, 2.17.
AuS–C12
To a dry two necked flask containing dithiolone 6a (184 mg, 0.49 mmol) was added a solution of NaOMe (3.91 mmol) in dry methanol (prepared from 90 mg of Na in 4 mL of dry MeOH). The solution was stirred 1 hour and a solution of KAuCl4 (93 mg, 0.25 mmol) in 5 mL of dry MeOH was added. The reaction mixture was stirred 5 hours at room temperature and PPh4Br (103 mg, 0.25 mmol) was added. The mixture was stirred at room temperature for 12 hours. The supernatant was removed and the oil was washed 2 times with methanol to afford the complex as a brown oil (100 mg); yield 17%; 1H NMR (300 MHz, CDCl3) δ 0.85 (t, 6H, CH3, 3J = 7.1 Hz), 1.24 (m, 36H, 18CH2), 1.65 (m, 4H, 2CH2), 2.98 (m, 4H, 2SCH2), 7.63 (m, 16H, Ar), 7.79 (m, 4H, Ar); 13C NMR (75 MHz, CDCl3) δ 14.1, 22.7, 28.7, 29.1, 29.3, 29.5, 29.5, 29.6, 29.6, 31.9, 36.4, 36.4, 76.6, 77.1, 77.5, 116.9, 118.1, 130.6, 130.8, 134.4, 134.5, 135.6, 135.6, 150.3, 166.6; UV-vis (CH2Cl2) λmax(nm) (ε[L mol−1 cm−1]) = 360 (12330); HRMS (ESI) calcd for C84H120N4PS16Au2 [2A−, C+]−: 2121.41188. Found: 2121.4123.
AuS–PEG
Under inert atmosphere, to a dry two necked flask containing proligand 6b (152 mg, 0.43 mmol) was added a solution of NaOMe (3.2 mmol) prepared from 83 mg of Na in 8 mL of dry MeOH under inert atmosphere. The solution was stirred 1 hour and a solution of KAuCl4 (80 mg, 0.22 mmol) in 5 mL of dry MeOH was added. The reaction mixture was stirred 5 hours at room temperature and PPh4Br (90 mg, 0.22 mmol) was added. The mixture was stirred overnight. The supernatant was removed and concentrated to dryness. The crude was dissolved in DCM and washed with water twice. Organic layer was dried over MgSO4, filtered off and concentrated to afford the product as a brown oil (199 mg); yield 78%; 1H NMR (300 MHz, CDCl3): δ 3.19 (m, 4H, SCH2), 3.35 (s, 6H, CH3), 3.53 (m, 4H, 2CH2), 3.62 (m, 12H, 6CH2), 3.71 (m, 4H, SCH2C2), 7.61 (m, 8H, Ar), 7.71 (m, 8H, Ar), 7.83 (m, 4H, Ar); 13C NMR (75 MHz, CDCl3): δ 35.5, 59.2, 69.8, 70.3, 70.5, 71.8, 116.9, 118.1, 127.0, 127.1, 130.7, 130.9, 134.4, 134.6, 135.7, 135.8, 150.1, 150.3, 165.5, 165.6; UV-vis (CH2Cl2) λmax(nm) (ε[L mol−1 cm−1]) = 360 (12060). HRMS (ESI) calcd for C64H80N4O12PS16Au2 [2A−, C+]−: 2033.03731. Found: 2033.0372. Anal. calcd for C44H50AuN2O6PS8: C, 44.51; H, 4.24; N, 2.36. Found: C, 45.17; H, 4.47; N, 2.68.
Crystallography
Suitable crystals for X-ray diffraction single crystal experiment were selected and mounted with a cryoloop on the goniometer head of a D8 Venture (Bruker-AXS) diffractometer equipped with a CMOS-PHOTON70 detector, using Mo-Kα radiation (λ = 0.71073 Å, multilayer monochromator). Crystal structure was solved by dual-space algorithm using SHELXT program,34 and then refined with full-matrix least-squares methods based on F2 (SHELXL program).35 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. Crystallographic data for AuN–C8 (C22H34AuN2S8·C24H20P), M = 1119.33, monoclinic, space group C2/c, a = 34.043(4), b = 8.3744(9), c = 17.891(2) Å, β = 110.075(4)°, V = 4790.6(9) Å3, Z = 4, T = 150(2) K, d = 1.552 g cm−3, μ = 3.487 mm−1. A final refinement on F2 with 5478 unique intensities and 265 parameters converged at ωR(F2) = 0.0997 (RF = 0.0423) for 4140 observed reflections with I > 2σ. In the checkcif procedure, no A-type meaningful alerts has remained. X-ray crystallographic file have been deposited at the Cambridge Crystallographic Data Centre (CCDC 2352881†).
Cellular uptake analysis
The concentrations of gold were determined by particle-induced X-ray emission (PIXE) technique, installed at the Van de Graaff accelerator of Instituto Superior Técnico (IST), in A2780 cells incubated with the complexes during 3 h at equimolar concentrations of 50 μM and 20 μM for AuN–EtOH. The cell pellets were obtained by centrifugation after washing the cells with PBS to remove the culture medium, freeze-dried and digested using suprapure reagents, nitric and hydrochloric acids (1:3 molar ratio) together with yttrium (Y) (100 mg l−1) as an internal standard. The procedure combined ultrasound cycles of 30 min at 40 °C and microwave-assisted acid digestion (350 W, 3 × 10 s). The detailed methodology was previously described.21 The elemental concentrations were obtained in μg Au g−1 and converted to ng Au per 106 cells.
Anticancer activity
The anticancer activity of the gold complexes was evaluated in the ovarian cancer cells A2780 (Sigma-Aldrich) sensitive to cisplatin. Cells were grown in RPMI supplemented with 10% fetal bovine serum and were maintained in a 5% CO2 incubator in a humidified atmosphere at 37 °C as previously described.21 Briefly, stock solutions of all compounds were prepared in DMSO, and then diluted in complete medium to a prepare work solutions in the range 0.01–50 μM. The complexes are stable in DMSO, as no evolution of the 1HNMR spectra is observed over a period of 66 h (Fig. S33†). The cellular viability was measured by the MTT assay, which relies on the cellular reduction of a tetrazolium salt to its formazan crystals by metabolically active cells. For the assays, cells were seeded in 96-well plates at a density of 2 × 104 cells in 200 μL medium and incubated overnight to adhere. After, the medium was discarded and 200 μL of a serial dilution of the complexes in fresh medium was added to the cells and leave to incubate. After 24 h at 37 °C, the medium was discarded and the cells were incubated with 200 μL of a MTT solution (1.2 mM) in PBS. After incubation for 3 h and upon removal of the MTT solution, the purple formazan crystals formed were dissolved in DMSO. The absorbance at 570 nm was measured using a plate spectrophotometer (Power Wave Xs, Bio-Tek) and the IC50 values were calculated with GraphPad Prism.
Antiplasmodial activity
The in vitro activity of AuS–PEG, AuS–EtOH, AuN–PEG and AuS–Et against the hepatic stages of rodent P. berghei malaria parasites was evaluated by a bioluminescence method, as previously described.36,37 Briefly, a human hepatoma cell line (Huh-7) provided by Cenix Bioscience GmbH and cultured in Roswell Park Memorial Institute 1640 (RPMI) medium supplemented with 1% (v/v) penicillin/streptomycin (Gibco), 10% (v/v) fetal bovine serum (Gibco), 1% (v/v) glutamine (Gibco), 1% (v/v) non-essential amino acids (Gibco) and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; Gibco), pH 7 (cRPMI) was maintained at 37 °C and 5% CO2. Compound stock solutions were prepared in dimethyl sulfoxide (DMSO) at a final concentration of 10 mM and stored at −20 °C. One day prior to infection, 1 × 104 Huh-7 cells were seeded in 96-well plates and incubated at 37 °C with 5% CO2. Compounds were serially diluted from stock solutions in cRPMI infection medium supplemented with 0.8 μg mL−1 fungizone and 50 μg mL−1 gentamicin. Three technical replicates were produced per test concentration. For IC50 determination, seven concentrations of each compound were prepared. One hour prior to infection, the culture medium was replaced by cRPMI containing appropriate compound dilutions and each well was subsequently infected with 1 × 104 firefly luciferase-expressing P. berghei sporozoites, harvested and purified from the salivary glands of infected female Anopheles stephensi mosquitoes reared at iMM's insectary. Following centrifugation at 1800g for 5 min, plates were incubated for 48 h under the same conditions, after which time the effect of each compound on cellular viability was assessed by the AlamarBlue assay, following the manufacturer's recommendations (ThermoFisher), using a TECAN Spark microplate reader (TECAN, Switzerland). Parasite infection load was then assessed by a bioluminescence assay (Biotium, USA), as described.21
Statistical analyses
Nonlinear regression analysis was performed to fit normalized results of the dose–response curves for IC50 determinations of in vitro hepatic stage activity, using GraphPad Prism 8 (GraphPad software, USA).
Data availability
The data supporting this article have been included as part of the ESI.†
Crystallographic data has been deposited at the CCDC under CCDC number 2352881 for AuN–C8 and can be obtained directly from https://www.ccdc.cam.ac.uk/products/csd/request/request.php4.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
This work was supported by the Université de Rennes. Marques F. and Pinheiro T., thanks the financial support from Fundacão para a Ciência e Tecnologia (FCT) through project UID/Multi/04349/2019, UIDP/04565/2020, and LA/P/0140/2020 (Associate Laboratory Institute for Health and Bioeconomy (i4HB)). MP acknowledges funding from the “la Caixa” Foundation, grant No. HR21-848.
References
- T. Naito, Inorganics, 2020, 8, 53 CrossRef CAS.
- M. F. G. Velho, R. A. L. Silva, G. Brotas, E. B. Lopes, I. C. Santos, A. Charas, D. Belo and M. Almeida, Dalton Trans., 2020, 49, 13737–13743 RSC.
- B. Garreau de Bonneval, K. I. Moineau-Chane Ching, F. Alary, T. T. Bui and L. Valade, Coord. Chem. Rev., 2010, 254, 1457–1467 CrossRef CAS.
- N. C. Schiødt, T. Bjørnholm, K. Bechgaard, J. J. Neumeier, C. Allgeier, C. S. Jacobsen and N. Thorup, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 1773–1778 CrossRef PubMed.
- O. J. Dautel, M. Fourmigué, E. Canadell and P. Auban-Senzier, Adv. Funct. Mater., 2002, 12, 693–698 CrossRef CAS.
- D. Belo, H. Alves, E. B. Lopes, M. T. Duarte, V. Gama, R. T. Henriques, M. Almeida, A. Pérez-Benítez, C. Rovira and J. Veciana, Chem. – Eur. J., 2001, 7, 511–519 CrossRef CAS PubMed.
- W. Suzuki, E. Fujiwara, A. Kobayashi, Y. Fujishiro, E. Nishibori, M. Takata, M. Sakata, M. H. Fujiwara and H. Kobayashi, J. Am. Chem. Soc., 2003, 125, 1486–1487 CrossRef CAS PubMed.
- N. Tenn, N. Bellec, O. Jeannin, L. Piekara-Sady, P. Auban-Senzier, J. Íñiguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2009, 131, 16961–16967 CrossRef CAS PubMed.
-
(a) G. Yzambart, N. Bellec, G. Nasser, O. Jeannin, T. Roisnel, M. Fourmigué, P. Auban-Senzier, J. Íñiguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2012, 134, 17138–17148 CrossRef CAS PubMed;
(b) A. Filatre-Furcate, N. Bellec, O. Jeannin, P. Auban-Senzier, M. Fourmigué, J. Íñiguez, E. Canadell, B. Brière and D. Lorcy, Inorg. Chem., 2016, 55, 6036–6046 CrossRef CAS PubMed.
- A. Filatre-Furcate, T. Roisnel, M. Fourmigué, O. Jeannin, N. Bellec, P. Auban-Senzier and D. Lorcy, Chem. – Eur. J., 2017, 23, 16004–16013 CrossRef CAS PubMed.
- Y. Le Gal, T. Roisnel, P. Auban-Senzier, T. Guizouarn and D. Lorcy, Inorg. Chem., 2014, 53, 8755–8761 CrossRef CAS PubMed.
- Y. Le Gal, T. Roisnel, P. Auban-Senzier, N. Bellec, J. Íñiguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2018, 140, 6998–7004 CrossRef CAS PubMed.
- D. vd.Westhuisen, D. I. Bezuidenhout and O. Q. Munro, Dalton Trans., 2021, 50, 17413–17437 RSC.
- M. J. McKeage, L. Maharaj and S. J. Berners-Price, Coord. Chem. Rev., 2002, 232, 127–135 CrossRef CAS.
- C. Nardon, G. Boscutti and D. Fregona, Anticancer Res., 2014, 34, 487–492 CAS.
- B. Bertrand, M. R. M. Williams and M. Bochmann, Chem. – Eur. J., 2018, 24, 11840–11851 CrossRef CAS PubMed.
- S. Radisavljević and B. Petrović, Front. Chem., 2020, 8, 379 CrossRef PubMed.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- R. Paprochcka, M. Wiese-Szadkowska, S. Janciauskiene, T. Kosmalski, M. Kulik and A. Helmin-Basa, Coord. Chem. Rev., 2022, 452, 214307 CrossRef.
- S. A. Sousa, J. H. Leitão, R. A. L. Silva, D. Belo, I. C. Santos, J. F. Guerreiro, M. Martins, D. Fontinha, M. Prudêncio, M. Almeida, D. Lorcy and F. Marques, J. Inorg. Biochem., 2020, 202, 110904 CrossRef CAS PubMed.
- D. Fontinha, S. A. Sousa, T. S. Morais, M. Prudêncio, J. H. Leitão, Y. Le Gal, D. Lorcy, R. A. L. Silva, M. F. G. Velho, D. Belo, M. Almeida, J. F. Guerreiro, T. Pinheiro and F. Marques, Metallomics, 2020, 12, 974–987 CrossRef CAS PubMed.
- Y. Le Gal, A. Filatre-Furcate, D. Lorcy, O. Jeannin, T. Roisnel, V. Dorcet, D. Fontinha, D. Francisco, M. Prudencio, M. Martins, D. Soeiro, S. A. Sousa, J. H. Leitão, T. S. Morais, I. Bartolo, N. Taveira, J. F. Guerreiro and F. Marques, Int. J. Mol. Sci., 2022, 23, 7146 CrossRef CAS PubMed.
-
E. Abele, R. Abele and E. Lukevics, Chemistry of Heterocyclic Compounds, Springer, New York, 2007 Search PubMed.
-
M. A. Alam, Thiazole, a privileged scaffold in drug discovery, Privileged Scaffolds in Drug Discovery, Academic press, 2023, pp. 1–19 Search PubMed.
-
(a) N. Bellec, D. Lorcy, A. Robert, R. Carlier, A. Tallec, C. Rimbaud, L. Ouahab, R. Clerac and P. Delhaes, Adv. Mater., 1997, 9, 1052–1056 CrossRef CAS;
(b) N. Bellec, D. Lorcy, K. Boubekeur, R. Carlier, A. Tallec, S. Los, W. Pukacki, M. Trybula, L. Piekara-Sady and A. Robert, Chem. Mater., 1999, 11, 3147–3153 CrossRef CAS.
- A. Filatre-Furcate, P. Auban-Senzier, M. Fourmigué, T. Roisnel, V. Dorcet and D. Lorcy, Dalton Trans., 2015, 44, 15683–15689 RSC.
-
(a) H. Hachem, N. Bellec, M. Fourmigué and D. Lorcy, Dalton Trans., 2020, 49, 6056–6064 RSC;
(b) H. Hachem, O. Jeannin, M. Fourmigué, F. Barrière and D. Lorcy, CrystEngComm, 2020, 22, 3579–3587 RSC.
- A. Filatre-Furcate, T. Roisnel and D. Lorcy, J. Organomet. Chem., 2016, 819, 182–188 CrossRef CAS.
- H. Hachem, O. Jeannin, M. Fourmigué and D. Lorcy, Transition Met. Chem., 2023, 48, 91–98 CrossRef CAS.
- S. Kokatam, K. Ray, J. Pap, E. Bill, W. E. Geiger, R. J. LeSuer, P. H. Rieger, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2007, 46, 1100–1111 CrossRef CAS PubMed.
- O. Zaheed, S. S. McDade, K. Dean and A. McCabe, Front. Cell Dev. Biol., 2023, 11, 1104514 CrossRef PubMed.
- J. F. Santos, R. Azevedo, M. Prudêncio, F. Marques, Y. Le Gal, D. Lorcy and C. Fernandes, Pharmaceutics, 2023, 15, 1030 CrossRef CAS PubMed.
- L. Singh, D. Fontinha, D. Francisco, M. Prudêncio and K. Singh, Sci. Rep., 2022, 12, 564 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. M. Mendes, I. S. Albuquerque, M. Machado, J. Pissarra, P. Meireles and M. Prudêncio, Antimicrob. Agents Chemother., 2017, 61(2), e02005–e02016 CrossRef CAS PubMed.
- I. H. J. Ploemen, M. Prudêncio, B. G. Douradinha, J. Ramesar, J. Fonager, G.-J. van Gemert, A. J. F. Luty, C. C. Hermsen, R. W. Sauerwein, F. G. Baptista, M. M. Mota, A. P. Waters, I. Que, C. W. G. M. Lowik, S. M. Khan, C. J. Janse and B. M. D. Franke-Fayard, PLoS One, 2009, 4(11), e7881 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S33 and Table S1. CCDC 2352881. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01458h |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Dominique
Lorcy
a,
Dominique
Lorcy