
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Host–guest chemistry of tridentate Lewis acids based on tribenzotriquinacene†
Maurice
Franke
 ,
Tobin
Ens
,
Andreas
Mix
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Norbert W.
Mitzel
*
,
Tobin
Ens
,
Andreas
Mix
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Norbert W.
Mitzel
*
Universität Bielefeld, Fakultät für Chemie, Lehrstuhl für Anorganische Chemie und Strukturchemie (ACS), Centre for Molecular Materials (CM2), Universitätsstraße 25, D-33615 Bielefeld, Germany. E-mail: mitzel@uni-bielefeld.de; Web: https://www.uni-bielefeld.de/fakultaeten/chemie/ac3-mitzel/
First published on 1st July 2024
Abstract
Flexible poly-Lewis acids (PLA) based on the tribenzotriquinacene (TBTQ) scaffold have been synthesised. Hydrosilylation of 4b,8b,12b-triallyltribenzotriquinacene and subsequent exchange of the chlorine substituents with weaker coordinating triflate groups afforded a novel triple silyl-functionalised PLA. By regioselective hydroboration of triallyl–TBTQ with various organoboranes, PLAs with different Lewis acidities were obtained. The synthesised PLAs were combined with neutral bases in host–guest experiments. DOSY NMR spectroscopy was performed to elucidate the complexation process in solution. These experiments revealed a highly dynamic interaction between the boron-functionalised PLA and triazine. However, the addition of one equivalent of tris(dimethylphosphino(methyl))phenylsilane led to the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct, which was confirmed by diffusion experiments.
:1 adduct, which was confirmed by diffusion experiments.
Introduction
Host–guest chemistry has become an extensively studied area of supramolecular chemistry since Pedersen's publication of crown ether–potassium complexes.1 Crown ethers consist of several linked Lewis base functions and are therefore called poly-Lewis bases. In contrast, the less well studied poly-Lewis acids consist of several Lewis acid functions linked to an organic backbone. A useful distinction is that between (semi-)flexible,2 rigid2e,3 and cyclic2e,4 PLAs, as the flexibility influences the complexation potential towards Lewis base guest molecules. While rigid PLAs are able to recognise molecules of a specific size3c,d,5 due to the defined spatial arrangement of the Lewis acid functions, PLAs with more flexible spacing are accessible to a greater variety of Lewis bases and subsequent transformations.Besides mercury,4c,6 silicon2d,7 and tin,2e,3e,4d the elements of the third main group (B,2b,c,3b,d,5b,c,8 Al,9 Ga,10 In11) are often used as acceptor sites. Today, there are various examples of organic backbones with different properties of the resulting PLAs. Previous work in the field of poly-Lewis acids by our group included the Lewis acid functionalisation of the trisilacyclohexane scaffold7d,8a,10e,g and extensive studies of the anthracene molecule5c,7c,9c,d,11b,12 as backbone for bi- and tetradentate poly-Lewis acids as well as for poly-pnictogen-bonding host-systems,13 which are the subject of current research. Recently, we reported on the preparation of novel derivatives based on C3v-symmetric tribenzotriquinacene (TBTQ)14 which was first synthesised by Kuck et al. in 1984.15 We introduced various silyl substituents via Grignard reactions and hydrosilylation. However, the attached Lewis acid functions showed no reactivity towards Lewis basic donor molecules, due to the low Lewis acidity of the silyl functions, as we already observed for other bi- and tridentate silicon-based poly-Lewis acids.2c,7a,c,e Although fluoride ions were readily complexed with trifluorosilyl-substituted PLAs,2d no further reactions with Lewis basic molecules such as pyridine were observed. Synthetic protocols to increase the Lewis acidity of silicon atoms by introducing electron-withdrawing groups have been reported by Greb et al. with the synthesis of bis(catecholato)silanes.16 After the introduction of weakly coordinating anions such as carboranes17 and triflate18 silicon atoms also exhibited an increased Lewis acidity, which has found application in organic catalysis. Recently, we reported the reactivity of bidentate silyl–triflates in host–guest chemistry.7b Diffusion NMR experiments provided insight into host–guest aggregation in solution. Comparison of the diffusion coefficients of the corresponding PLA, the Lewis base and the formed adducts provides detailed information about the aggregate.12a,16b,19 However, due to the complexity of aggregation in solution it is still difficult to make a statement about the composition of the adducts. Further elucidation by X-ray diffraction experiments provides information about the constitution in the solid phase. In this work we present the synthesis of a silyl–TBTQ derivative with increased Lewis acidity, similar to that we previously reported. In addition, we present novel boron-functionalised poly-Lewis acids and their application in host–guest chemistry.
Results and discussion
Syntheses of tridentate poly-Lewis acids
The triallyl–TBTQ compound 1, that is required for subsequent reactions was prepared according to literature data by a SnCl4 catalysed reaction of 4b,8b,12b-tribromotribenzotriquinacene with allyltrimethylsilane.20 The following hydrosilylation reaction was carried out in analogy to the reaction conditions we used in previous work to obtain the silyl–TBTQ derivative 2.14 Conversion with silver triflate in the absence of light afforded the triflate substituted TBTQ derivative 3 (Scheme 1).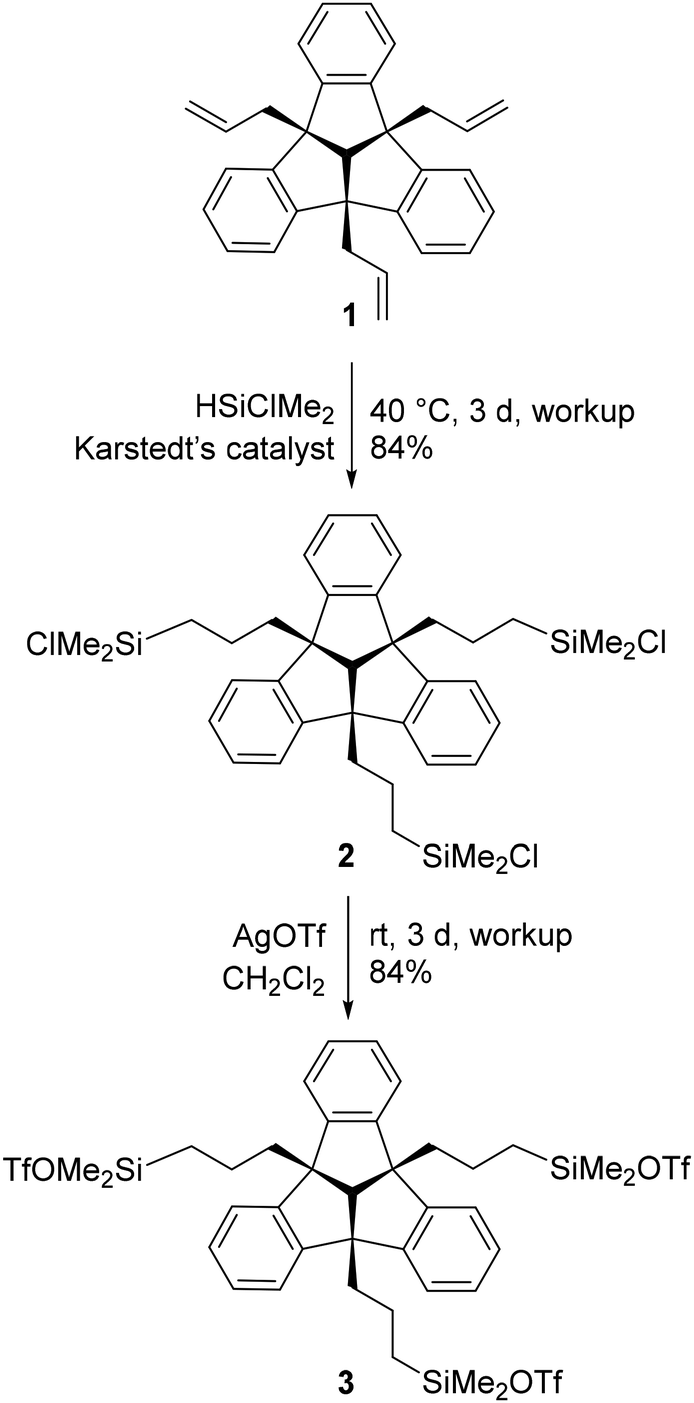 | ||
| Scheme 1 Syntheses of the trisilyl–TBTQ compounds 2 and 3, starting from triallyl–TBTQ 1. | ||
Both products 2 and 3 were obtained as colourless solids in good yields (84%) and were characterised by multinuclear NMR spectroscopy, sc-XRD and elemental analysis. The 1H NMR spectrum proved the regioselectivity of the hydrosilylation reaction and the formation of the anti-Markovnikov product 2. The chemical shift of 31.2 ppm in the 29Si{1H} NMR spectrum is consistent with previously synthesised TBTQ–silyl compounds. A single crystal of 2 suitable for X-ray diffraction experiments was obtained, by cooling a saturated solution of 2 in n-hexane to −30 °C (Fig. 1). The molecular structure of 2 confirms the triple hydrosilylation of triallyl–TBTQ 1. The spatial arrangement of the silyl substituents results in a chalice-shaped molecule.
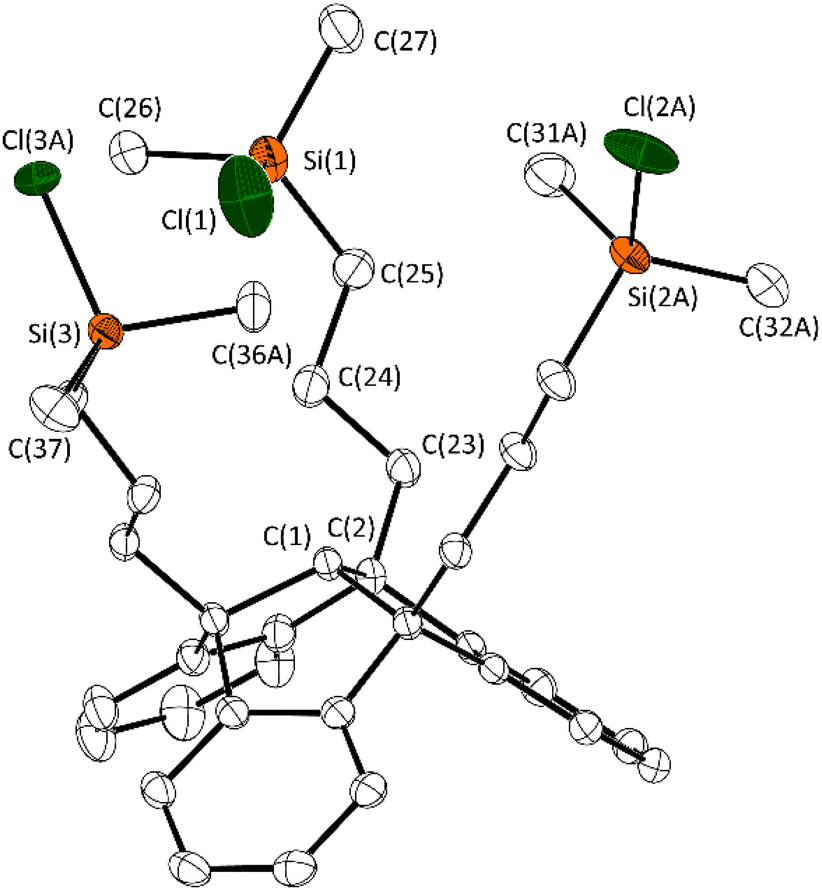 | ||
| Fig. 1 Molecular structure of 2 in the crystalline state. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms and minor occupied disordered parts are omitted for clarity. For further details, see the ESI.† Selected bond lengths [Å] and angles [°]: C(1)–C(2) 1.563(2), C(2)–C(23) 1.543(2), C(23)–C(24) 1.529(3), Si(1)–C(25) 1.864(2), Si(1)–C(26) 1.870(2), Si(1)–C(27) 1.843(2), Si(1)–Cl(1) 2.084(8); C(25)–Si(1)–Cl(1) 107.2(1), C(26)–Si(1)–Cl(1) 105.4(1), C(27)–Si(1)–Cl(1) 106.4(1). | ||
After reaction with silver triflate, all 1H NMR signals are low-field shifted compared to the corresponding signals of compound 2. This is in agreement with the 29Si{1H} chemical shift of 43.9 ppm observed for 3. In contrast to 2, which is surprisingly stable to air and moisture, the rapid formation of trifluoromethanesulfonic acid, evident from 1H NMR spectra, shows the hydrolysis sensitivity of 3. Single crystals were obtained by cooling a saturated solution of 3 in n-hexane to −30 °C and were analysed by X-ray diffraction (Fig. 2).
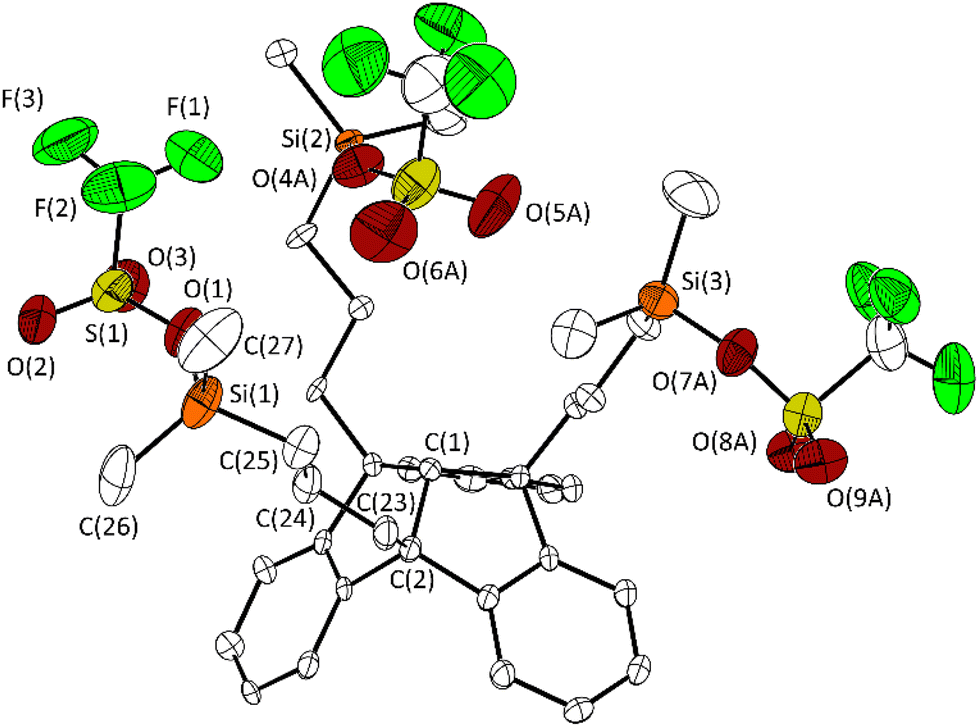 | ||
| Fig. 2 Molecular structure of 3 in the crystalline state. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms and minor occupied disordered parts are omitted for clarity. Only one of the two crystallographically independent molecules is shown. For further details, see the ESI.† Selected bond lengths [Å] and angles [°]: C(1)–C(2) 1.562(6), C(2)–C(23) 1.541(7), C(23)–C(24) 1.532(7), C(24)–C(25) 1.520(8), Si(1)–C(25) 1.856(5), Si(1)–C(26) 1.844(9), Si(1)–C(27) 1.842(9) Si(1)–O(1) 1.754(5), O(1)–S(1) 1.522(5); C(25)–Si(1)–O(1) 102.0(3), C(26)–Si(1)–O(1) 109.2(3), C(27)–Si(1)–O(1) 105.4(4). | ||
The molecular structure of 3 confirms the triple exchange of the chloro substituents with triflate groups. The bond Si(3)–O(7A) (1.688(7) Å) is significantly shorter than the bonds Si(1)–O(1) and Si(2)– O(4A) (1.754(5), 1.769(6) Å). The distance Si(3)⋯O(5A) is 3.612(9) Å, just within the sum of the van der Waals radii (rvdW(Si) + rvdW(O) = 3.62 Å).
The hydroboration reaction is a well-established way of functionalising the organic backbone with Lewis acidic boryl-groups. To investigate the effect of Lewis acidity and steric demand in host–guest chemistry, we synthesised different tridentate boron Lewis acids. PLA 4 was obtained by conversion of 1 with dicyclohexylborane, as a colourless solid in quantitative yield (Scheme 2).
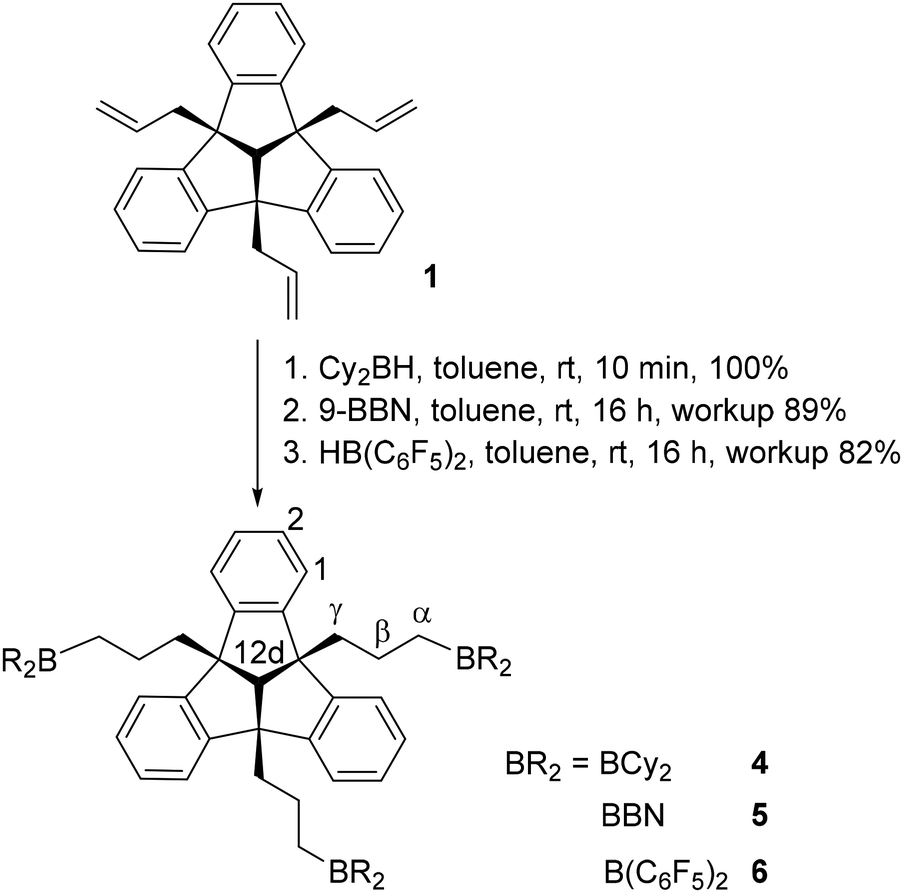 | ||
| Scheme 2 Syntheses of triboryl–TBTQ compounds 4, 5 and 6. | ||
The 1H NMR spectrum of 4 shows the characteristic multiplets of the TBTQ backbone, the signals of the propyl groups and of the hydrogen atoms of the cyclohexyl substituents. The 11B NMR chemical shift of 84.3 ppm is in the range of similar compounds.21
Regioselective hydroboration of triallyl–TBTQ 1 in anti-Markovnikov position with 9-borabicyclo[3.3.1]nonane (9-BBN) afforded the flexible boron–PLA 5 (Scheme 2). The 1H NMR signal pattern is consistent with that of compound 4 (Table 1). The 11B NMR chemical shift of 89.5 ppm is in the expected region. Since PLAs 4 and 5 are expected to have relatively low Lewis acidity,22 due to the electron donating alkyl-groups, we introduced stronger electron withdrawing pentafluorophenyl substituents at the boron functions. The hydroboration was carried out in analogy to the syntheses of 4 and 5 with Piers’ borane and afforded the bis(pentafluorophenyl)boryl substituted PLA 6 (Scheme 2). The increased Lewis acidity of 6 compared to compounds 2–5 has an effect on the chemical shifts of the signals of 6 in the 1H NMR spectrum (Table 1).
| Compound | H1 | H2 | H12d | γ-H | β-H | α-H |
|---|---|---|---|---|---|---|
| 2 | 7.07 | 7.29 | 3.78 | 2.30 | 1.56 | 0.81 |
| 3 | 7.10 | 7.30 | 3.67 | 2.24 | 1.44 | 0.78 |
| 4 | 7.07 | 7.33 | 3.81 | 2.30 | 1.59 | 1.37 |
| 5 | 7.08 | 7.36 | 3.97 | 2.36 | 1.25 | 1.54 |
| 6 | 6.96 | 7.09 | 3.45 | 2.18 | 1.55 | 2.08 |
The signals of the TBTQ backbone of 6 are high-field shifted. In contrast, the signal of the hydrogen atom adjacent to the boron atom (α-H) is low-field shifted. The observed 11B NMR signal at 74.3 ppm and the 19F chemical shifts are consistent with those of similar compounds.7c,10e,g
Host–guest-experiments
We carried out host–guest experiments by combining the new PLAs 3–6 with (multidentate) Lewis bases to give adducts as shown in Fig. 3. In preliminary tests we used pyridine to demonstrate the reactivity of the PLAs 3–6 towards neutral bases by forming the 1:3 adduct (Fig. 3A). The formation of this type of adduct (3·3Py, 4·3Py, 5·3Py, 6·3Py) was observed for each PLA. As expected, all 1H NMR signals of the complexed pyridine are shifted, compared to those of the free pyridine. The adduct formation was further confirmed by 19F NMR (3·3Py and 6·3Py) and 11B NMR spectroscopy (4·3Py, 5·3Py and 6·3Py). The 19F resonance of 3·3Py at −79.1 ppm is high-field shifted relative to that of the free PLA 3 and is in the region of the chemical shift of uncomplexed triflate.7b Relative to those of the free PLA, the 11B resonances of 4·3Py, 5·3Py and 6·3Py are high-field shifted (δ = 8.0 ppm for 4·3Py, 0.3 ppm for 5·3Py and 2.9 ppm for 6·3Py) into the region typically found for tetra-coordinated boron atoms. A single crystal of the adduct 5·3Py was obtained upon concentration of the reaction mixture and was analysed by X-ray diffraction experiments (Fig. 4).
 | ||
| Fig. 3 Structure motifs of a tridentate PLA with monodentate guests (A), tridentate guests (B) and intermolecular linkage via bidentate guests (C). | ||
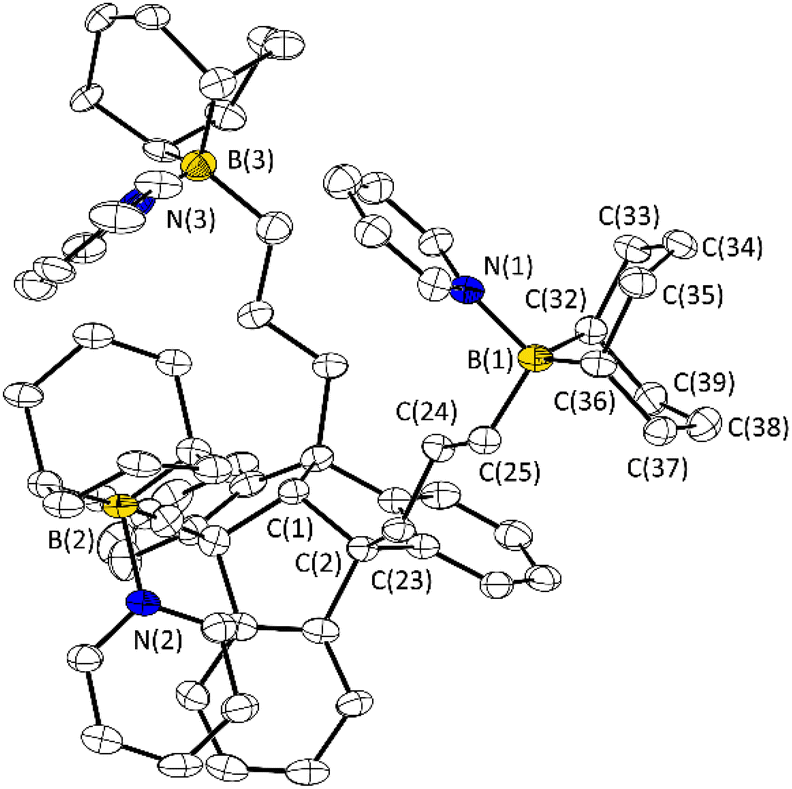 | ||
| Fig. 4 Molecular structures of 5·3Py in the crystalline state. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms, solvent benzene molecules and minor occupied disordered parts are omitted for clarity. For further details, see the ESI.† Selected bond lengths [Å] and angles [°]: C(1)–C(2) 1.566(7), C(2)–C(23) 1.531(7), C(23)–C(24) 1.524(6), C(24)–C(25) 1.536(6), B(1)–C(25) 1.626(7), B(1)–C(32) 1.626(7), B(1)–C(36) 1.631(7) B(1)–N(1) 1.646(7); C(25)–B(1)–N(1) 104.2(4), C(25)–B1(1)–C(32) 114.9(4), C(25)–B(1)–C(36) 113.4(4), C(32)–B(1)–C(36) 103.9(4). N(1)–B(1)–C(36) 110.0(4). | ||
The molecular structure of 5·3Py confirms the complexation of three pyridine molecules, resulting in the 1:3 adduct with tetra-coordinated boron atoms. The B–N bond lengths are comparable to similar structures. Due to the flexibility of the propyl spacers, the spatial arrangement of the boron functions is variable, making PLA 5 a suitable host for guest molecules of different sizes.
Compared to the 1H NMR spectra of 4·3Py and 5·3Py, the chemical shifts of the resonances of excess and complexed pyridine in 6·3Py differ significantly, indicating a less dynamic complexation due to the increased Lewis acidity of the boron atoms caused by the electron withdrawing pentafluorophenyl substituents.
Considering the molecular structure of 5·3Py in Fig. 4, a complexation of tridentate guests is conceivable in the cavity spanned between the boron atoms or in the outer sphere to form adducts as shown in Fig. 3B. We therefore investigated the host–guest interactions of the PLAs 4–6 with the tridentate nitrogen base 1,3,5-triazine.
The addition of triazine to a solution of 4 in C6D6, did not result in the formation of an adduct, due to the low Lewis acidity of the cyclohexyl-substituted boron atom. In contrast, the 1H NMR spectra indicated complexation of triazine by PLA 5. We also observed a broadened, high-field shifted 11B resonance of the adduct at 27.8 ppm. We suggest that this is an averaged signal of complexed and partially free boron functions of PLA 5, due to a rapid exchange of triazine. Apparently, the spatial orientation of the Lewis base functions does not fit well with that of the Lewis acidic moieties. To gain further insight into the complexation process of 5 and triazine we performed an NMR titration experiment by treating 5 with an increasing concentrations of the guest compound triazine (Fig. 5).
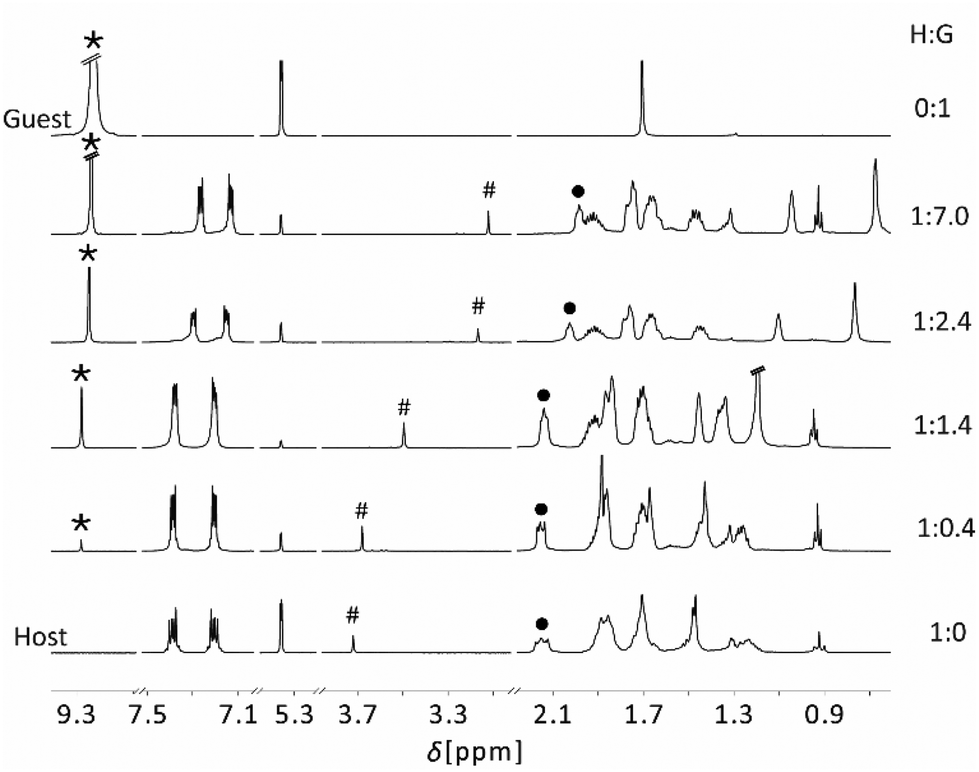 | ||
| Fig. 5 Series of the 1H NMR spectra of mixtures of host compound 5 with increasing concentration of triazine and free triazine (300 MHz, 298 K). | ||
Fig. 5 shows the 1H NMR spectra of the non-complexed PLA 5 and of triazine together with spectra of the PLA 5 (host, H) with an increasing concentration of triazine (guest, G). For a simplified view we only consider the signals of triazine (*), the hydrogen atom in the centre of the TBTQ backbone (H12d, #) and of the γ-H of the propyl group (•). Addition of small amounts of triazine (0.4 eq.) only results in a slight shift of H12d (#) and γ-H (•) to higher field. The triazine signal (*) is low-field shifted compared to free triazine, due to the complete complexation of guest molecules. Further addition of guest (1.4 eq.) leads to significantly shifted signals of H12d (#) and γ-H (•), while the triazine signal (*) experienced no further shift. This is consistent with for the formation of a 1:1 adduct, possibly with dynamic exchange of the guest between the Lewis acidic functions of the host molecule, as suggested by the 11B NMR data. Increasing the guest concentration to 2.4 eq., the resonances of H12d (#) and γ-H (•) receive a strong high-field shift, while the guest signal (*) approaches the shift of free triazine. This is in agreement with the formation of a 1:3 adduct in which only one of the nitrogen atoms of triazine is complexed. For higher guest concentrations, only minor changes are observed, as all Lewis acidic functions of the host component are complexed by guest molecules.
We observed a similar complexation behaviour of triazine with PLA 6. As expected, the triazine signal of 6·triazine is further low-field shifted, compared to the signal of 5·triazine, due to the stronger Lewis acidity of 6. Also, the averaged signal of complexed and free Lewis acidic functions of 6 with a chemical shift of 40.4 ppm in the 11B NMR spectrum is consistent with the host–guest experiment of 5 and triazine.
To elucidate the effect of the Lewis basic function on the complexation with boron PLAs, we also carried out an exemplary host–guest experiment with PLA 5 and PMe3 and further experiments with the tridentate phosphorus Lewis base tris(dimethylphosphinomethyl)phenylsilane (TrisPhos, (Me2PCH2)3SiPh). As expected, the conversion of PLA 5 with PMe3 resulted in the formation of the corresponding 1:3 adduct 5·PMe3. In addition to the TBTQ signals, the doublet of the PMe3 is also shifted in the 1H NMR spectrum. The chemical shifts of −4.1 ppm in the 11B NMR and of 15.6 ppm in the 31P{1H} NMR spectrum are indicative of the formation of the adduct 5·PMe3. The addition of one equivalent of TrisPhos to a solution of 5 in CDCl3 resulted in the selective formation of the 1:1 adduct 5·TrisPhos, according to the NMR spectroscopy (Fig. 6).
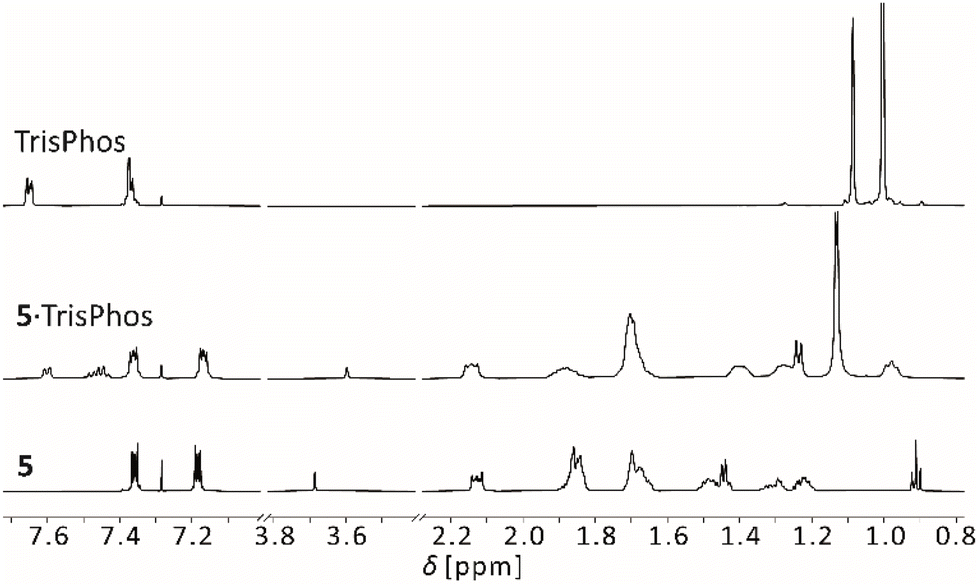 | ||
| Fig. 6 Sections of the 1H NMR spectra of TrisPhos, PLA 5 (600 MHz, 293 K) and the adduct 5·TrisPhos (500 MHz, 298 K) in CDCl3. | ||
We observed a shift for all signals of PLA 5 and TrisPhos. Compared to 5·triazine, the observed signals of 5·TrisPhos are narrow, especially those of the guest, indicating a much slower or no exchange dynamics and therefore a comparatively stable 1:1 adduct. Apparently, the higher flexibility of TrisPhos compared to triazine allows the adjustment of the Lewis base functions to the spatial orientation of the Lewis acidic moieties. The chemical shifts of 9.1 ppm in the 11B and −27.2 ppm in the 31P{1H} NMR spectrum are consistent with an adduct formation.
In contrast, there is a more dynamic complexation process between TrisPhos and 4 in CDCl3. The 1H, 31P{1H} and 11B NMR spectra show strongly broadened signals, due to the lower Lewis acidity of the cyclohexyl-substituted boron function and the associated small equilibrium constant of the adduct formation.
Although, the addition of TrisPhos to a solution of 6 in CDCl3 resulted in a shift of the 1H NMR signals, the many signals of host and guest indicate the formation of different adduct species or oligomers. Due to the high Lewis acidity of 6, the reversibility of this adduct formation is limited.
In order to elucidate the data obtained from the NMR titration of 5 with triazine and the host–guest experiment of 5 with TrisPhos, we have further studied the adduct formation in more detail by means of 1H DOSY NMR experiments. According to the Stokes–Einstein (SE) equation, the experimentally accessible diffusion coefficient D is inversely proportional to the hydrodynamic radius (rH) of the diffusing particle.24 A comparison of D of PLA 5 and the corresponding Lewis base with those of the host–guest adducts should therefore provide information about the aggregate formed in solution. The van der Waals volumes (VvdW) of host, guest and adduct give a first indication about the dimensions of the hydrodynamic volumes (VH). Table 2 shows the van der Waals volumes (VvdW) calculated using the method reported by Abraham et al.,25 the measured and corrected diffusion coefficients, the hydrodynamic radii (rH) and volumes (VH) calculated using the Stokes–Einstein equation.
| Compound | V vdW [Å3] | V XRD [Å3] | D [10−10 m2 s−2] | r H [Å] | V H [Å3] |
|---|---|---|---|---|---|
| Host/guest | Host/guest | Host/guest | |||
| a Calculated by a modified formula, that is suitable for small molecules. For further details see the ESI.† | |||||
| 5 | 945 | — | 6.17/— | 6.14/— | 967/— |
| Triazine | 115 | 9826 |
18.7/— | 2.03a/— | 138a/— |
| TrisPhos | 390 | — | 9.14/— | 4.95a/— | 508a/— |
| 5·triazine | 1060 | — | 5.54/7.16 | 6.83/5.29 | 1335/620 |
| 5·TrisPhos | 1335 | — | 5.25/7.16 | 7.22/7.04 | 1574/1463 |
The diffusion NMR experiment of PLA 5 and one equivalent triazine reveals a different composition of the aggregate than we initially assumed from the 1H NMR spectrum. The hydrodynamic volume VH determined from the host component 5 in the adduct 5·triazine, results in a larger value (1335 Å3) than the sum of those of the single components: VH of host 5 is 967 Å3 and that of free triazine is 138 Å3. The VH of the guest part (620 Å3) in 5·triazine indicates a fast exchange of triazine molecules and a small equilibrium constant, due to the presence of free guest molecules. The DOSY experiment sees only an average in time of the bound and free triazine molecules and their VH is therefore between that of the free triazine and that measured for the component 5 in the adduct. This dynamic process is in agreement with the slightly broadened signals in the 1H and 11B NMR spectra.
In contrast, the 1H DOSY NMR experiment of 5 with TrisPhos is in accordance with the NMR data we discussed above and confirms the formation of a 1:1 adduct. The determined hydrodynamic volumes of the adduct 5·TrisPhos (1574 Å3 (host part), 1463 Å3 (guest part)) are approximately the sum of the hydrodynamic volume of the host 5 (967 Å3) and of TrisPhos (508 Å3).
Further host–guest experiments were carried out with PLA 3 (H) and the bidentate base 3,3′-bipyridine (G), to obtain a 2:3 adduct ([H2G3]6+) with a cage-like structure as shown in Fig. 3C. The aggregation process was followed by 1H DOSY NMR titration experiments: we gradually added 3,3′-bipyridine to a solution of 3 in acetonitrile; in addition, the diffusion coefficient of the triflate component was followed by 19F DOSY NMR. From these D values, we calculated the hydrodynamic radii (rH) for host and guest (1H) and triflate (19F) and calculated their hydrodynamic volumes (VH). Since acetonitrile has already been shown to be a suitable solvent for ionic compounds formed during the adduct formation,7b it was also used in this experiment.
For a more precise interpretation of the diffusion data, additional measurements of the free PLA 3 were performed, as well as of the adduct of 3 with p-phenylpyridine (3·3pPhPy), as highly similar reference compound for a monodentate acting 3,3′-bipyridine; in this way we intended to simulate the saturation of the Lewis acidic functions by formation of a [HG3]3+ adduct as shown in Fig. 3A and to obtain its corresponding hydrodynamic volume (1721 Å3, Table 3). It is noteworthy, that we observed a deviation of the hydrodynamic volume of 3 (VH = 1331 Å3) determined by 1H DOSY NMR from that determined using 19F DOSY NMR (for the triflate substituents of 3, VH = 567 Å3). Consistently, VT NMR experiments of 3 also show deviations for the triflate component: with increasing temperature, we determined a decrease in the hydrodynamic volume of 3, indicating a partial dissociation of the triflate substituents from the core of 3.
| Compound | Obs. nucl. | r H [Å] | V H [Å3] | V XRD |
|---|---|---|---|---|
| 3 | 1H | 5.13 | 1331 | 1244 |
| 19F | 6.31 | 567 | ||
| 3·3pPhPy | 1H | 7.43 | 1721 | — |
| 19F | 3.30 | 150 |
Fig. 7 shows the calculated hydrodynamic volumes (VH) of host, guest and triflate for the corresponding equivalents of 3,3′-bipyridine, as well as the hydrodynamic volumes of the individual components, derived from the DOSY measurements of the host 3 and the reference compound 3·3pPhPy. In addition it shows the potentially formed adducts and their corresponding expected hydrodynamic volumes (VH,exp) calculated from the determined values of the reference compound. Without guest component, we observe a deviation of the hydrodynamic volumes (VH) of host and triflate in the diffusion NMR measurement of the free PLA 3, as mentioned above.
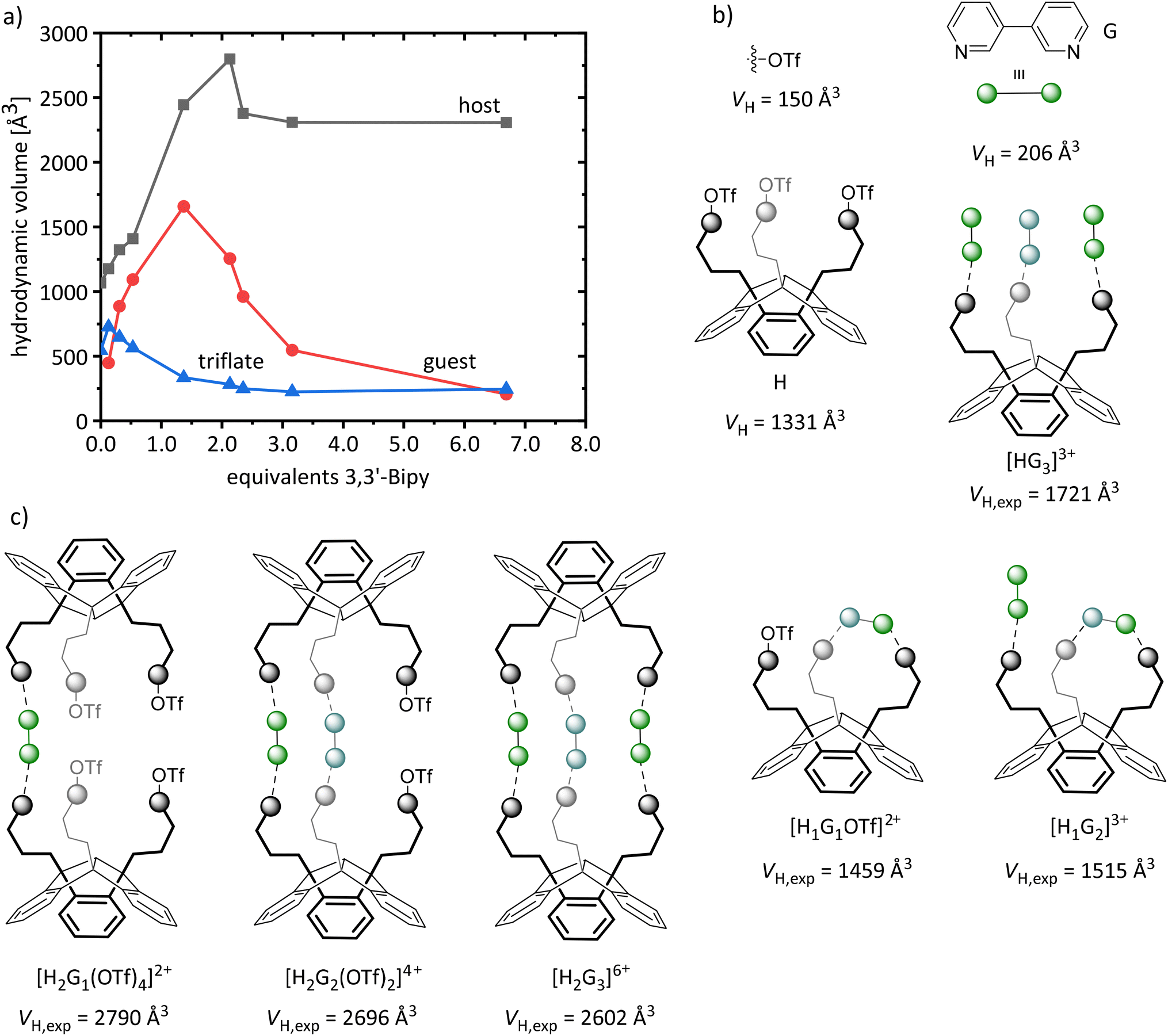 | ||
| Fig. 7 (a) Calculated hydrodynamic volumes (VH) of host (■, 3), guest (●, 3,3′-bipyridine) and triflate (▲) component, derived from the 1H and 19F DOSY NMR experiments as a function of the equivalents of the guest (3,3′-Bipy); (b) determined hydrodynamic volumes (VH) of host, guest triflate and the HG33+ adduct derived from the DOSY measurements of the host 3 and the reference compound 3·3pPhPy; (c) potentially formed adducts and their expected hydrodynamic volume (VH,exp) in the DOSY titration experiment of 3 and 3,3′-bipyridine. | ||
As expected, the hydrodynamic volume (VH) of triflate decreases with increasing guest concentration. Remarkably, the VH of host and guest already differ at low guest concentrations, indicating a small equilibrium constant and the presence of free guest molecules. Addition of only 1.3 eq. of the guest leads to a rapid increase of the VH of the host (2512 Å3), which corresponds to a [H2Gx](x+1)+ adduct. Further addition of guest component (2.13 eq.) leads to a VH,max. of 2798 Å3. Considering a VH of 1721 Å3 for [HG3]3+, 1331 Å3 for H and 150 Å3 for triflate (Table 3), we expect a VH,max. of approximately 2602 Å3 for [H2G3]6+ (Fig. 7). The deviation is most possibly a result of a mixture of different [H2Gx](x+1)+ aggregates and partially non-complexed host and guest molecules resulting from the fast exchange. Even the intramolecular complexation of 3,3′-bipyridine by two Lewis acidic sites of one host molecule ([H1G1OTf]2+, [H1G2]3+; Fig. 7) must be taken into consideration here.
The addition of 6.9 eq. of the guest leads to a final hydrodynamic volume of 2307 Å3 (H) and a continuous decrease of the VH of the guest (206 Å3) as increasingly non-complexed guest compound becomes the predominant component in solution. Remarkably, even for high guest concentrations the aggregates of the composition [H2Gx](x+1)+ are present in solution. The formation of larger oligomers or chain structures can be ruled out as the aggregates formed showed a high solubility throughout the entire titration experiment.
Conclusions
Various flexible poly-Lewis acids based on the tribenzotriquinacene (TBTQ) scaffold have been synthesised in hydrometallation reactions of 4b,8b,12b-triallyltribenzotriquinacene with hydrochlorosilanes and hydroboranes. The Lewis acidity of the introduced silyl functions was increased by substituting their chloro substituents with weaker coordinating triflate groups. Host–guest experiments with pyridine demonstrated the reactivity of the new PLAs. The complexation behaviour of the boron-functionalised PLAs was exemplarily studied for the host–guest interaction of the boryl-substituted PLA 5 with triazine and tris(dimethylphosphinomethyl)phenylsilane (TrisPhos) in 1H DOSY NMR experiments. This proved to be a suitable method for the analysis of host–guest aggregates, as it revealed a contrasting composition of the formed adduct to that suggested by the 1H NMR spectra. It also emphasises the high dynamics involved in the aggregation processes. The complexation of TrisPhos with PLA 5 proved to be less dynamic and the determined hydrodynamic volume corresponds to the formation of a 1:1 adduct, which is consistent with the 1H NMR data. The complexation of the boron-functionalised PLAs 4–6 also showed the influence of the Lewis acidity on the constitution of the adducts. Further diffusion experiments were carried out with the tris(triflatosilyl)-substituted PLA 3 and 3,3′-bipyridine. The titration experiment showed again the complexity of host–guest reactions in solution. The determined hydrodynamic volume of the host component indicates a linkage of two host molecules by up to three guests, while the formation of larger oligomers can be ruled out. The dynamics of the aggregation process do not allow more detailed conclusions to be drawn about the number of guest molecules bound. Overall, the results show that we often observe a counterintuitive behaviour of the aggregations processes and not only the simplest possible or highest symmetry adducts are formed.
Experimental section
General remarks
4b,8b,12b-Tribromotribenzotriquinacene and 4b,8b,12b-triallyltribenzotriquinacene (1) were synthesised according to literature protocols.20,23 All reagents for host–guest experiments were dried under vacuum or freshly distilled before use. All reactions involving oxidation- or hydrolysis-sensitive substances were carried out using standard Schlenk techniques or in glove boxes under inert nitrogen or argon atmospheres. Solvents were freshly dried and degassed (benzene, toluene and n-hexane, dried over Na/K alloy; n-pentane and Et2O, dried over LiAlH4; dichloromethane over calcium hydride and THF dried over potassium). NMR spectra were recorded on a Bruker Avance III 300, Bruker Avance III 500 HD and Bruker Avance NEO 600 instrument at 298 K and 293 K (DOSY) respectively. Chemical shifts (δ) were measured in ppm with respect to the solvents (C6D6: 1H NMR δ = 7.16 ppm, 13C NMR δ = 128.06 ppm; CDCl3: 1H NMR δ = 7.26 ppm, 13C NMR δ = 77.16 ppm; CD2Cl2: 1H NMR δ = 5.32 ppm, 13C NMR δ = 53.84 ppm, CD3CN: 1H NMR δ = 1.94 ppm, 13C NMR δ = 1.32 ppm, 118.26 ppm) or referenced externally (11B: BF3·Et2O, 19F: CFCl3; 29Si: SiMe4; 31P: 85% H3PO4,). Elemental analyses were performed using a HEKAtech EURO EA instrument. Assignments of the NMR-signals are based on the IUPAC guidelines and are shown in Scheme 3.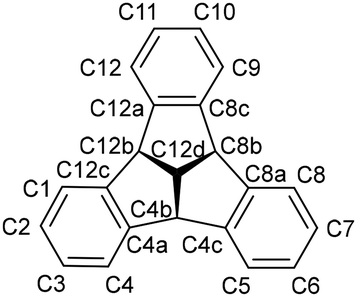 | ||
| Scheme 3 Numbering scheme of the tribenzotriquinacene skeleton for NMR assignments. | ||
1H-NMR (300 MHz, C6D6) δ = 7.30 (m, 6H, H2/H3/H6/H7/H10/H11). 7.10 (m, 6H, H1/H4/H5/H8/H9/H12), 3.67 (s, 1H, H12d), 2.24 (m, 6H, CH2CH2CH2Si), 1.44 (m, 6H, CH2CH2CH2), 0.78 (t, 3JH,H = 7.9 Hz, 6H, CH2CH2CH2Si), 0.08 (s, 18H, Si(CH3)2OTf) ppm. 13C{1H}-NMR (75.5 MHz, C6D6) δ = 148.6 (C4a/C4c/C8a/C8c/C12a/C12c), 128.1 (C2/C3/C6/C7/C10/C11), 123.2 (C1/C4/C5/C8/C9/C12), 67.8 (C12d), 64.1 (C4b/C8b/C12b), 45.2 (CH2CH2CH2Si), 19.1 (CH2CH2CH2Si), 16.7 (CH2CH2CH2Si), 0.8 (Si(CH3)2OTf) −1.8 (Si(CH3)2) ppm (resonances for CF3 were not observed). 19F-NMR (282 MHz, C6D6) δ = −77.4 ppm. 29Si{1H}-NMR (99 MHz, C6D6) δ = 43.9 ppm. Elemental analysis calcd (%) for C40H49F9O9S3Si3 (1025.24): C 46.86 3 H 4.82 S 9.38; found C 47.58 H 4.75 S 8.69.
General procedure for the host–guest experiments
10–15 mg of the host compound was dissolved in a NMR tube fitted with a PTFE tap in C6D6, CD2Cl2, CDCl3 or CD3CN and the corresponding guest compound was added. The host–guest mixture was then analysed by 1H NMR (3–7), 11B NMR (4–6) and 19F NMR spectroscopy (2 and 6).Data availability
The data published in this contribution are available as ESI,† submitted with the manuscript.Crystallographic data have been deposited with the Cambridge Crystal Structure Database (CCDC).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Deutsche Forschungsgemeinschaft (DFG, grant no. MI477/39-1, project number 424957011). The authors thank Barbara Teichner for performing elemental analyses.References
- C. J. Pedersen and H. K. Frensdorff, Angew. Chem., Int. Ed. Engl., 1972, 11, 16–25 CrossRef CAS PubMed.
- (a) W. Uhl, F. Hannemann, W. Saak and R. Wartchow, Eur. J. Inorg. Chem., 1998, 921–926 CrossRef CAS; (b) D. F. Shriver and M. J. Biallas, J. Am. Chem. Soc., 1967, 5, 1078 CrossRef; (c) F. Schäfer, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2021, 3265–3271 CrossRef; (d) J. Horstmann, M. Niemann, K. Berthold, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2017, 46, 1898–1913 RSC; (e) M. M. Naseer and K. Jurkschat, Chem. Commun., 2017, 53, 8122–8135 RSC.
- (a) P. Jutzi, J. Izundu, H. Sielemann, B. Neumann and H.-G. Stammler, Organometallics, 2009, 28, 2619–2624 CrossRef CAS; (b) V. C. Williams, W. E. Piers, W. Clegg, M. R. J. Elsegood, S. Collins and T. B. Marder, J. Am. Chem. Soc., 1999, 121, 3244–3245 CrossRef CAS; (c) C. R. Wade and F. P. Gabbaï, Z. Naturforsch., 2014, 69B, 1199–1205 CrossRef; (d) H. E. Katz, J. Org. Chem., 1985, 50, 5027 CrossRef CAS; (e) R. Altmann, K. Jurkschat, M. Schürmann, D. Dakternieks and A. Duthie, Organometallics, 1998, 17, 5858–5866 CrossRef CAS.
- (a) T. Ooi, M. Takahashi and K. Maruoka, J. Am. Chem. Soc., 1996, 118, 11307–11308 CrossRef CAS; (b) F. P. Schmidtchen, Angew. Chem., Int. Ed. Engl., 1977, 16, 720–721 ( Angew. Chem. , 1977 , 89 , 751–752 ) CrossRef; (c) T. J. Wedge and M. F. Hawthorne, Coord. Chem. Rev., 2003, 240, 111–128 CrossRef CAS; (d) A. S. Wendji, C. Dietz, S. Kühn, M. Lutter, D. Schollmeyer, W. Hiller and K. Jurkschat, Chem. – Eur. J., 2016, 22, 404–416 CrossRef CAS PubMed.
- (a) C.-H. Chen and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 1799–1804 ( Angew. Chem. , 2017 , 129 , 1825–1830 ) CrossRef CAS PubMed; (b) H. E. Katz, J. Org. Chem., 1989, 54, 2179–2183 CrossRef CAS; (c) P. Niermeier, S. Blomeyer, Y. K. J. Bejaoui, J. L. Beckmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 1965–1969 ( Angew. Chem. , 2019 , 131 , 1985–1990 ) CrossRef CAS PubMed; (d) M. Hirai and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 54, 1205–1209 ( Angew. Chem. , 2015 , 127 , 1221–1225 ) CrossRef CAS PubMed.
- (a) M. F. Hawthorne and Z. Zheng, Acc. Chem. Res., 1997, 30, 267–276 CrossRef CAS; (b) J. D. Wuest and B. Zacharie, Organometallics, 1985, 4, 410–411 CrossRef CAS; (c) J. D. Beckwith, M. Tschinkl, A. Picot, M. Tsunoda, R. Bachman and F. P. Gabbaï, Organometallics, 2001, 20, 3169–3174 CrossRef CAS; (d) O. Loveday, J. Jover and J. Echeverría, Inorg. Chem., 2022, 61(32), 12526–12533 CrossRef CAS PubMed.
- (a) A. Schwartzen, J.-H. Weddeling, J. Langosch, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2021, 27, 1821–1828 CrossRef CAS PubMed; (b) F. Schäfer, A. Mix, N. Cati, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2022, 51, 7164–7173 RSC; (c) J.-H. Lamm, J. Horstmann, J. H. Nissen, J.-H. Weddeling, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2014, 4294–4301 CrossRef CAS; (d) J. Rudlof, B. Neumann, H.-G. Stammler and N. W. Mitzel, Z. Anorg. Allg. Chem., 2021, 647, 1967–1972 CrossRef CAS; (e) M. E. Jung and H. Xia, Tetrahedron Lett., 1988, 29, 297–300 CrossRef CAS.
- (a) F. Schäfer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2021, 3083–3090 CrossRef; (b) J. Rudlof, T. Glodde, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2022, e202100842 CrossRef CAS.
- (a) W. Uhl, A. Hepp, H. Westenberg, S. Zemke, E.-U. Würthwein and J. Hellmann, Organometallics, 2010, 29, 1406–1412 CrossRef CAS; (b) W. Uhl, D. Heller, M. Rohling and J. Kösters, Inorg. Chim. Acta, 2011, 374, 359–365 CrossRef CAS; (c) N. Aders, P. C. Trapp, J.-H. Lamm, J. L. Beckmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Organometallics, 2022, 41, 3600–3611 CrossRef CAS; (d) N. Aders, J.-H. Lamm, J. L. Beckmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2022, 51, 12943–12953 RSC.
- (a) W. Uhl and M. Claesener, Inorg. Chem., 2008, 47, 4463–4470 CrossRef CAS PubMed; (b) J. Tomaschautzky, B. Neumann, H.-G. Stammler, A. Mix and N. W. Mitzel, Dalton Trans., 2017, 46, 1645–1659 RSC; (c) J. Horstmann, M. Hyseni, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2017, 56, 6107–6111 CrossRef CAS PubMed; (d) W. Uhl and M. Claesener, Inorg. Chem., 2008, 47, 729–735 CrossRef CAS PubMed; (e) E. Weisheim, L. Bücker, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2016, 45, 198–207 RSC; (f) W. Uhl, A. Hepp, H. Westenberg, S. Zemke, E.-U. Würthwein and J. Hellmann, Organometallics, 2010, 29, 1406–1412 CrossRef CAS; (g) E. Weisheim, C. G. Reuter, P. Heinrichs, Y. V. Vishnevskiy, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2015, 21, 12436–12448 CrossRef CAS PubMed.
- (a) F. P. Gabbaï, A. Schier, J. Riede and D. Schichl, Organometallics, 1996, 15, 4119–4121 CrossRef; (b) J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2010, 16, 11906–11914 CrossRef CAS PubMed.
- (a) J.-H. Lamm, P. Niermeier, A. Mix, J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2014, 53, 7938–7942 CrossRef CAS PubMed; (b) J.-H. Lamm, J. Glatthor, J.-H. Weddeling, A. Mix, J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Org. Biomol. Chem., 2014, 12, 7355–7365 RSC.
- (a) J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. Sci., 2023, 14, 13551–13559 RSC; (b) J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2023, 62, e202310439 CrossRef CAS PubMed.
- J. Tomaschautzky, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2017, 46, 1112–1123 RSC.
- D. Kuck, Angew. Chem., Int. Ed. Engl., 1984, 23, 508 CrossRef.
- (a) T. Thorwart, D. Roth and L. Greb, Chem. – Eur. J., 2021, 27, 10422–10427 CrossRef CAS PubMed; (b) D. Hartmann, T. Thorwart, R. Müller, J. Thusek, J. Schwabedissen, A. Mix, J.-H. Lamm, B. Neumann, N. W. Mitzel and L. Greb, J. Am. Chem. Soc., 2021, 143, 18784–18793 CrossRef CAS PubMed.
- (a) T. Küppers, E. Bernhardt, R. Eujen, H. Willner and C. W. Lehmann, Angew. Chem., Int. Ed., 2007, 46, 6346–6349 CrossRef PubMed; (b) J. B. Lambert and Y. Zhao, J. Am. Chem. Soc., 1996, 118, 7867–7868 CrossRef CAS; (c) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- (a) A. Hermannsdorfer and M. Driess, Angew. Chem., Int. Ed., 2020, 59, 23132–23136 CrossRef CAS PubMed; (b) S. A. Weicker and D. W. Stephan, Chem. – Eur. J., 2015, 21, 13027–13034 CrossRef CAS PubMed.
- (a) J. Tomaschautzky, B. Neumann, H.-G. Stammler, A. Mix and N. W. Mitzel, Dalton Trans., 2017, 46, 1645–1659 RSC; (b) A. Mix, J.-H. Lamm, J. Schwabedissen, E. Gebel, H.-G. Stammler and N. W. Mitzel, Chem. Commun., 2022, 58, 3465–3468 RSC.
- D. Kuck, A. Schuster, R. A. Krause, J. Tellenbröker, C. P. Exner, M. Penk, H. Bögge and A. Müller, Tetrahedron, 2001, 57, 3587–3613 CrossRef CAS.
- M. W. Drover, M. C. Dufuour, L. A. Lesperance-Nantau, R. P. Noriega, K. Levin and R. W. Schurko, Chem. – Eur. J., 2020, 26, 11180 CrossRef CAS PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- G. Markopoulos, L. Henneicke, J. Shen, Y. Okamoto, P. G. Jones and H. Hopf, Angew. Chem., Int. Ed., 2012, 51, 12884–12887 CrossRef CAS PubMed.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed.
- Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368–7373 CrossRef CAS PubMed.
- P. Coppens, Science, 1967, 158, 1577–1579 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 11B, 13C, 19F, 29Si, 31P, NMR spectra, crystallographic details. CCDC 2345286–2345288. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01558d |
| This journal is © The Royal Society of Chemistry 2024 |