
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Catch and release” of the CpN3 ligand using cobalt: dissociation, protonation, and C–H bond thermochemistry†
Sanju
Luhach
 ,
Roger A.
Lalancette
and
Demyan E.
Prokopchuk
*
,
Roger A.
Lalancette
and
Demyan E.
Prokopchuk
*
Department of Chemistry, Rutgers University-Newark, 73 Warren Street, Newark, NJ 07102, USA. E-mail: demyan.prokopchuk@rutgers.edu
First published on 18th June 2024
Abstract
The coordination chemistry of an amine-rich CpN3 ligand has been explored with cobalt. We demonstrate that in the presence of NaCo(CO)4, the cationic precursor [CpN3]+ yields the complex CpN3CoI(CO)2. While 2e− oxidation generates new CoIII complexes such as [CpN3Co(NCMe)3]2+ and CpN3CoI2(CO), subsequent ligand loss is facile, generating free [CpN3]+ or the protonated dication [CpN3H]2+. We have structurally characterized both these ligand release products via single crystal X-ray diffraction and obtained thermochemical C–H bond strengths via experiment and density functional theory (DFT). Upon reversible 1e− reduction, the radical cation [CpN3H]˙+ has a weak C–H BDFE of 52 kcal mol−1 in acetonitrile. Mechanistic analysis shows that [CpN3H]˙+ undergoes radical–radical disproportionation in the absence of exogenous H-atom acceptors, which is supported by deuterium isotope labelling experiments. Structural comparison of these organic molecules shows a high degree of iminium-like electron delocalization over the C–N bonds connected to the central five-membered ring.
Introduction
Cyclopentadienyl (Cp) ligands and derivatives thereof can be complexed to virtually all d-block metals and have been used extensively in homogeneous catalysis.1–4 While Cp ligands are usually considered chemically “innocent”, special cases arise where the ligand becomes chemically non-innocent, enabling Cp to act as a reservoir for protons (H+), hydrogen atoms (H˙), and hydrides (H−).5 To this end, our group recently reported that an amine rich CpN3 ligand coordinated to iron behaves as a proton acceptor, resulting in regiospecific endo-CpN3 ring protonation as an essential step during electrocatalytic H2 production.6,7 To access these Fe(CpN3) compounds, an Fe0 source such as Fe2CO9 reacts with vividly colored “cyclopentadienylium” cations ([CpN3]+),8 furnishing piano-stool [FeII(CpN3)L3]+ salts.7,9,10Cobalt complexes containing Cp ligands have also been explored for applications in chemical fuel synthesis and catalysis.11–15 Notably, cyclopentadienylcobalt complexes A and B contain diphosphine ligands with bio-inspired pendant amines in their secondary sphere,16–18 exhibiting electrocatalytic activity for H2 production and CO2-to-formate reduction (Fig. 1).19,20 In the proposed electrocatalytic cycles, the Cp ring remains chemically innocent while the nitrogen atoms in the secondary coordination sphere assist in proton transfer and/or proton-coupled electron transfer steps en route to product formation.
 | ||
| Fig. 1 Cyclopentadienylcobalt electrocatalyst system for H2 production (A, top left)19 and CO2 reduction (B, top right)20 containing pendant amines in secondary coordination sphere. This work: synthesis and reactivity of CoCpN3 complexes 1 (bottom left) and 2 (bottom right). | ||
In our efforts to expand the library of M(CpN3) complexes that exhibit chemically non-innocent behavior, we now report the synthesis and reactivity of Co(CpN3) complexes 1 and 2 (Fig. 1). While exploring their redox behavior and ligand substitution chemistry, we discovered that irreversible dissociation of [CpN3]+ is facile and results in regiospecific ligand protonation at a carbon atom within the CpN3 ring to yield the air stable organic dication [CpN3H]2+. After independently synthesizing [CpN3H]2+via protonation of [CpN3]+, we explore its C(sp3)–H bond thermochemistry in MeCN, enabling us to experimentally measure its redox potential (E°), C–H acidity (pKa) and homolytic C–H bond dissociation free energy (BDFE). Our experimentally calculated data are supported by computational (DFT) analysis, and a comparison of single crystal X-ray diffraction data and DFT-derived molecular orbitals demonstrate the highly delocalized π electron distribution for the metal-free CpN3 cations.
Results & discussion
The isopropyl-substituted cyclopentadienylium salt [CpN3][BF4], where [CpN3]+ = 6-isopropyl-1,4-dimethyl-5,7-diphenyl-2,3,4,6-tetrahydrocyclopenta[b]pyrazin-6-ylium, was prepared using a previously reported procedure8 and a more detailed description of ligand charge distribution will be described later using X-ray crystallographic data. Complex CpN3Co(CO)2 (1) was prepared by refluxing [CpN3]+ and NaCoCO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 in toluene under N2 and isolated as analytically pure orange crystals in 70% yield (Scheme 1). As anticipated, 1H NMR spectroscopic analysis reveals that metal coordination enhances the electron density at the [CpN3]+ ligand, causing the aliphatic isopropyl peaks to shift to 0.50 ppm versus 0.85 ppm in free ligand22 dissolved in CD3CN (Fig. S1†). Infrared spectral analysis shows strong carbonyl stretches at νCO = 1970 and 1901 cm−1 (KBr) which are significantly lower than the analogous CpCo(CO)2 (2028, 1967 cm−1)23 and (pentamethylcyclopentadienyl)cobalt(I) dicarbonyl, Cp*Co(CO)2 (2010, 1950 cm−1; Cp* = pentamethylcyclopentadienyl),24 further demonstrating the increased electron density at cobalt in complex 1. Single crystals of 1 suitable for X-ray diffraction show that the C–C bond distances within the CpN3 ring of 1 are equidistant, lying between 1.42 and 1.46 Å (Fig. 2). The [CpN3]+ ring binds to the metal center in an η5 coordination mode, formally oxidizing the CoI− precursor by 2e− and reducing the cationic [CpN3]+ by 2e− upon coordination.
21 in toluene under N2 and isolated as analytically pure orange crystals in 70% yield (Scheme 1). As anticipated, 1H NMR spectroscopic analysis reveals that metal coordination enhances the electron density at the [CpN3]+ ligand, causing the aliphatic isopropyl peaks to shift to 0.50 ppm versus 0.85 ppm in free ligand22 dissolved in CD3CN (Fig. S1†). Infrared spectral analysis shows strong carbonyl stretches at νCO = 1970 and 1901 cm−1 (KBr) which are significantly lower than the analogous CpCo(CO)2 (2028, 1967 cm−1)23 and (pentamethylcyclopentadienyl)cobalt(I) dicarbonyl, Cp*Co(CO)2 (2010, 1950 cm−1; Cp* = pentamethylcyclopentadienyl),24 further demonstrating the increased electron density at cobalt in complex 1. Single crystals of 1 suitable for X-ray diffraction show that the C–C bond distances within the CpN3 ring of 1 are equidistant, lying between 1.42 and 1.46 Å (Fig. 2). The [CpN3]+ ring binds to the metal center in an η5 coordination mode, formally oxidizing the CoI− precursor by 2e− and reducing the cationic [CpN3]+ by 2e− upon coordination.
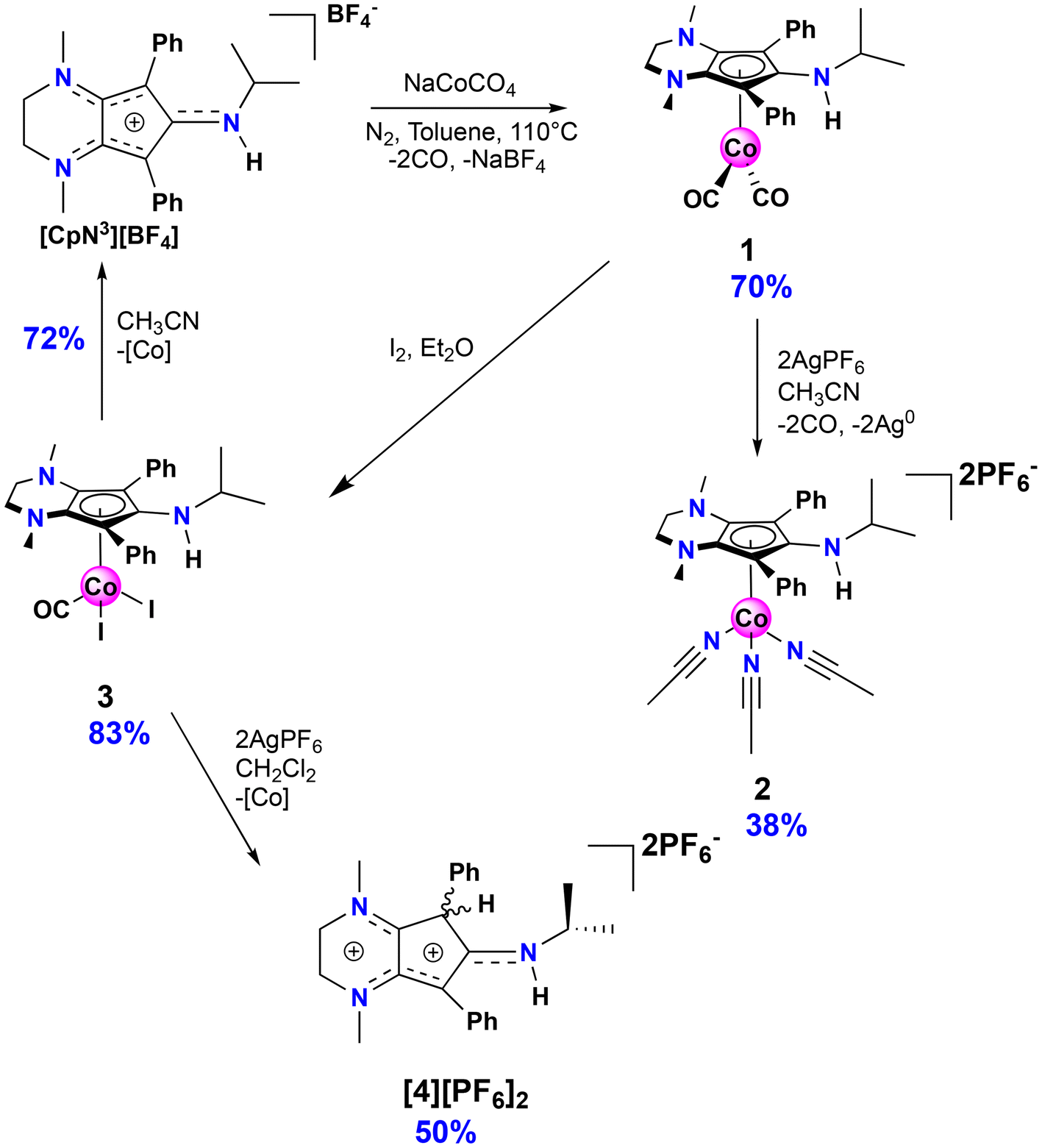 | ||
| Scheme 1 Syntheses of compounds 1–4. | ||
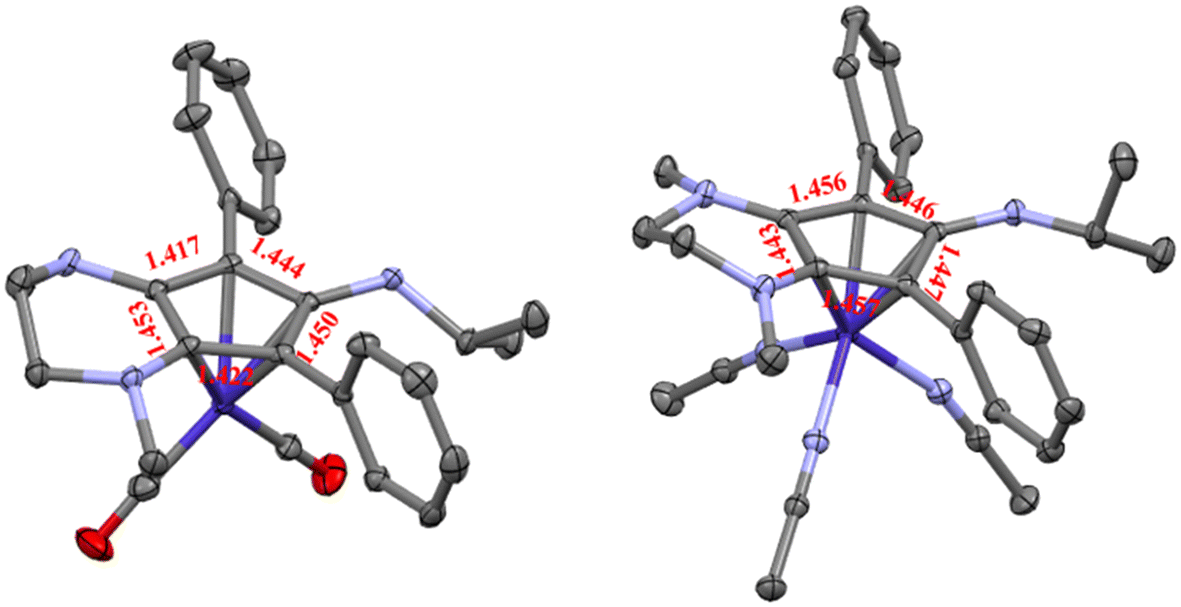 | ||
| Fig. 2 X-Ray crystallographic structures for complex 1 (left) and 2 (right) with 50% probability ellipsoids. Distances are shown in Angstroms (Å) and hydrogens/counteranions have been omitted for clarity. | ||
Electrochemical analysis of 1via cyclic voltammetry (CV) in MeCN shows a redox wave at E1/2 = 0.46 V vs. Fc+/0 when starting at 0 V and sweeping in the anodic direction (Fig. 3). Further sweeping in the cathodic direction reveals an irreversible reduction at Epc = −1.20 V followed by an oxidation peak at Epa = −0.50 V vs. Fc+/0. The observed oxidation and reduction peaks at −0.50 V and −1.20 V, respectively, are interdependent because the reduction peak does not appear if the voltage sweep is terminated before the oxidation event (Fig. S17†). Based on these observations, we hypothesize that 1e− oxidation of 1 triggers a series of chemical events that lead to the formation of a new redox-active species centered at E1/2 = 0.46 V with a corresponding reduction feature at −1.20 V vs. Fc+/0. Using slower scan rates (100 mV s−1) makes the minor features around −0.4 V and 0 V more prominent, suggesting that multi-step (electro)chemical processes are occurring in solution (Fig. S18†).
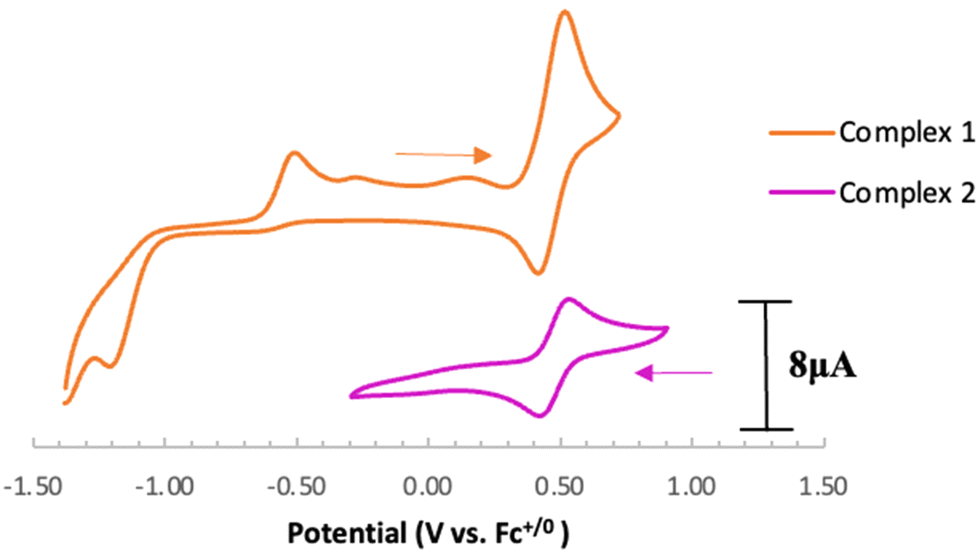 | ||
| Fig. 3 Cyclic voltammograms of complex 1 and 2 at 800 mV s−1. Conditions: Ar, MeCN solvent, 0.1 M [Bu4N][PF6], 1.0 mM analyte, PEEK-encased glassy carbon working electrode, Type 2 glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode in a frit-separated (CoralPor) glass compartment containing solvent and electrolyte. Initial scan direction and starting position indicated with arrows. | ||
Based on the observed CV features for 1, we pursued the chemical oxidation of 1 with a commercially available oxidant, AgPF6 (E′° = 0.04 V, MeCN).25 Upon mixing 1 and two equiv. AgPF6 in MeCN, a fuchsia-colored solution formed, which after workup and X-ray crystallization revealed the tris-MeCN substituted CoIII complex [CpN3Co(NCMe)3][PF6]2 (2) in 38% yield with concomitant CO release (Scheme 1 and Fig. 2). Notably, free ligand [CpN3]+ is formed as the major side-product as observed by 1H NMR, and varying standard reaction optimization parameters (time, temperature, reagent concentrations, order of addition) does not change the product distribution. The crystalline product is obtained by slow diffusion of diethyl ether into a concentrated acetonitrile solution, however minor impurities also precipitate, precluding the isolation of analytically pure product (Fig. S3 and S4†). Paramagnetic impurities also appear to coprecipitate with the crystalline product, resulting in peak broadening in 1H NMR spectra if single crystals are not adequately segregated from the powdered mixture. Using other oxidants such as “magic blue” (tris(4-bromophenyl) ammoniumyl hexafluoroantimonate) or FcPF6 result in lower product yields and purities of 2. However, to our delight, CVs of 2 in the cathodic direction reveal a redox wave at E1/2 = 0.47 V, indicating a CoIII/II redox couple which is consistent with our interpretation of the CV data for 1 (Fig. 3). Voltammograms collected beyond −0.72 V also reveal that unknown redox-active impurities are present in solution (Fig. S19†). Therefore, our synthetic and voltametric data show that the oxidation of 1 in acetonitrile leads to irreversible CO loss and formation of the isolable redox-active solvento complex 2.
We pursued ligand substitution reactions using complex 2 in the presence of common bidentate ligands such as 1,2-bis(diphenylphosphino)ethane (dppe), 1,2-bis(diethylphosphino)ethane (depe), and bipyridne (bipy), hoping that solvent dissociation would lead to the formation of new complexes with the general formula [CpN3Co(NCMe)L2]2+. In every case, clean and rapid ligand dissociation immediately generates [CpN3]+ in solution, and using dppe enabled us to crystallize the known CoII bis(diphosphine) complex [Co(dppe)2(NCMe)]2+.26 These results suggest that reduction of the metal center is thermodynamically and kinetically preferred, along with 2e− oxidation and dissociation of [CpN3]+, instead of coordinating stronger σ-donor chelates to CpN3CoIII. Using a relatively electron-poor L-type donor, 2-isocyano-1,3-dimethylbenzene, also resulted in the formation of [CpN3]+.
Motivated by the success of using the diiodo reagent CpCoIIII2(CO) as a synthon for preparing new redox-active cobalt complexes,11–13 exposure of 1 to I2 in diethyl ether cleanly generates CpN3CoI2CO (3) as an analytically pure brownish-black solid that precipitates from solution (Scheme 1). Similar to 1, IR spectroscopic analysis shows a significantly more activated CO ligand (νCO = 2000 cm−1, KBr) in 3 when compared to CpCoIIII2(CO) (2035 cm−1).27 Surprisingly, halide abstraction in the presence of AgPF6 in DCM results in the clean and consistent release of protonated free ligand [CpN3H][PF6]2, ([4][PF6]2), a dicationic product containing a proton at the phenyl-substituted carbon of the CpN3 ring (Scheme 1). Although air-free conditions were used in combination with thoroughly dried solvents and reagents, the origin of the proton remains unclear. A maximum yield of 50% suggests that partial decomposition may lead to the formation of 42+ (Scheme 1). The introduction of H+ on the [CpN3]+ ring desymmetrizes the molecule and restricts free rotation about the C–N bond of the N-isopropyl moiety due to its enhanced iminium-like character, generating chemically inequivalent methyl resonances in NMR spectra (Fig. S5†). Gratifyingly, the BF4− salt of dication 42+ can be independently prepared in quantitative yield by simply protonating [CpN3]+ with HBF4·Et2O in DCM, generating [CpN3H][BF4]2, ([4][BF4]2) (Scheme 2). Single crystals suitable for X-ray diffraction were obtained via vapor diffusion of diethyl ether into a concentrated acetonitrile solution and with metrical parameters revealing a delocalized positive charge distributed across three iminium-like N–C bonds and one alkene-like C–C bonds, respectively (Fig. 5 and Table 1). Moreover, dissolving complex 3 in MeCN releases unprotonated free ligand, [CpN3]+ (Fig. S11†), and unidentified cobalt-containing byproducts suggesting that solvent identity plays some role in ligand protonation.
 | ||
| Scheme 2 Syntheses of compounds 4 and 5. | ||
| [CpN3]+ | [CpN3H]2+ | [CpN3H2]+ | |
|---|---|---|---|
| C(1)–C(2) | 1.465(3) | 1.516(2) | 1.511(2) |
| C(2)–C(3) | 1.368(4) | 1.503(2) | 1.556(2) |
| C(3)–C(4) | 1.513(3) | 1.467(2) | 1.514(2) |
| C(4)–C(5) | 1.373(4) | 1.392(2) | 1.414(2) |
| C(5)–C(1) | 1.452(3) | 1.420(2) | 1.396(2) |
| C(1)–N(1) | 1.314(3) | 1.301(2) | 1.326(2) |
| C(3)–N(2) | 1.340(3) | 1.284(2) | 1.451(2) |
| C(4)–N(3) | 1.341(3) | 1.326(2) | 1.308(2) |
| C(1)–C(2)–C(3) | 105.4(2) | 99.5(1) | 100.5(1) |
| C(2)–C(3)–C(4) | 108.5(2) | 110.7(1) | 102.9(1) |
| C(3)–N(2)–C(6) | 121.0(2) | 123.4(1) | 110.7(1) |
| C(4)–N(3)–C(7) | 121.8(2) | 124.5(1) | 123.1(1) |
| C(3)–C(4)–C(5) | 109.2(2) | 107.6(1) | 109.0(1) |
| C(4)–C(5)–C(1) | 105.1(2) | 109.0(1) | 107.5(1) |
| C(5)–C(1)–C(2) | 111.1(2) | 111.8(1) | 112.6(1) |
| C(5)–C(1)–N(1) | 127.9(2) | 125.0(1) | 124.7(2) |
Intrigued by these results, we sought to study the C(sp3)–H proton-coupled electron transfer (PCET) thermochemistry28,29 of the protonated compound [4][BF4]2, as we have recently shown that [CpN3]+ ring protonation is essential during H2 production electrocatalysis using CpN3Fe(CO)2 complexes.6,7 To determine the C(sp3)–H bond acidity of 42+, we found via trial and error that reactions with aniline generate an equilibrium mixture of 42+, [CpN3]+, aniline, and anilinium BF4− (pKMeCNa = 10.62)30–32 in solution (Fig. S9 and S10†). Using equilibrium 1H NMR spectroscopic data at room temperature, we determined that the pKMeCNa(C–H) = 11.7 ± 0.1 for compound 42+ (see the ESI for details†). This experimentally derived pKa is only four units more acidic than the computed C–H pKa of 15.7 for the endo-protonated electrocatalytic intermediate [(endo-CpN3H)Fe(NCMe)(CO)2]+.7 The CV of [4]2+ in MeCN shows two partially reversible redox curves at ca. −0.72 V and −1.0 V vs. Fc+/0 (Fig. 4). We surmise that the first reduction peak is 1e− reduction of [4]2+ to the radical cation [CpN3H]˙+ ([4]˙+) while the second reduction yields the neutral CpN3H (4), neither of which we have been able to experimentally isolate to date. If the switching potential in CV experiments is set to −0.90 V, the first reduction becomes more reversible, with E1/2(42+/+) = −0.72 V and ipa/ipc = 0.76. Using these data enables us to calculate the PCET thermochemistry for [CpN3]+, [4]2+, and [4]˙+ as shown in Scheme 3. A weak C(sp3)–H BDFE of 51.8 kcal mol−1 is determined for [4]˙+ (Scheme 3), which is similar in magnitude to the M–H BDFEs of CpCr(CO)2(PPh3)H28,33 (52.7 kcal mol−1) and (dppm)V(CO)4H28,34 (51.1 kcal mol−1) in MeCN. To further validate these data, the DFT calculated values in parentheses in Scheme 3 are in excellent agreement with experimental data (see the ESI for computational details†). We also computed the C–H pKa of the radical cation [4]˙+, which would generate the neutral radical species [CpN3]˙ upon release of H+. Our computations indicate that the decrease in overall charge increases the pKa of [4]˙+ to 26.2 in acetonitrile, making it about 14 orders of magnitude less acidic than [4]2+.
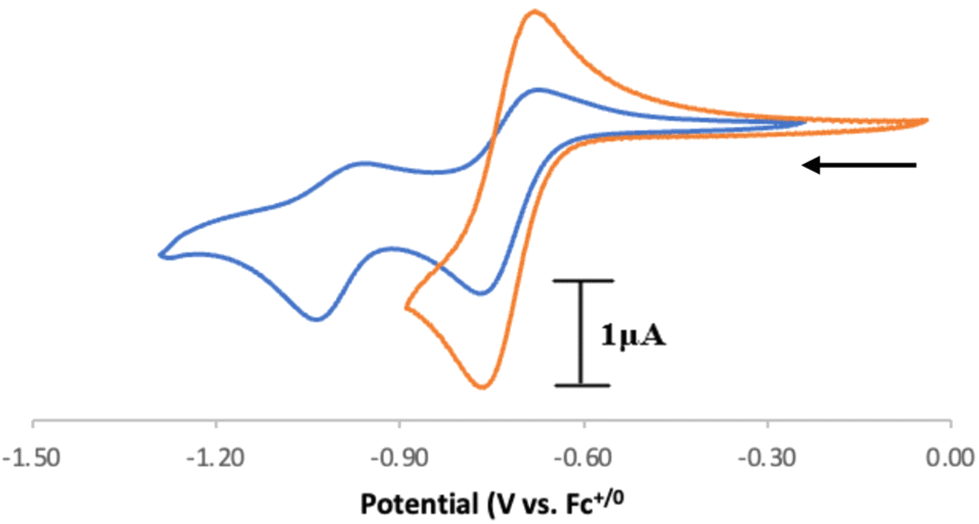 | ||
| Fig. 4 Cyclic voltammograms of [4]2+ at 100 mV s−1. The cathodic to anodic peak current ratio (ipa/ipc) is 0.47 when swept beyond −1.20 V and 0.76 when the scan direction is switched at −0.90 V. Conditions: Ar, MeCN solvent, 0.1 M [Bu4N][PF6], 1.0 mM analyte, PEEK-encased glassy carbon working electrode, Type 2 glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode in a frit-separated (CoralPor) glass compartment containing solvent and electrolyte. Initial scan direction and starting position indicated with an arrow. | ||
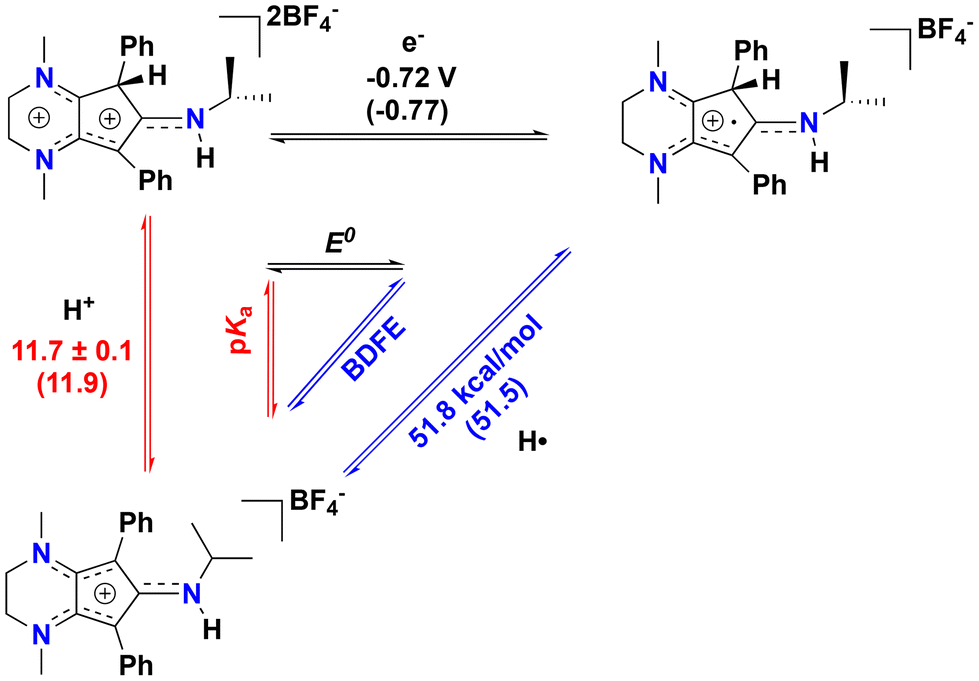 | ||
| Scheme 3 PCET thermochemistry for [CpN3]+, [4]2+, and [4]˙+a (a = DFT-computed values are in parentheses). | ||
We then turned our attention to studying the propensity of radical cation [4]˙+ to undergo H-atom transfer (HAT) reactions, as organic radical cations have been widely used in synthetic organic transformations.35 Reduction of [4][BF4]2 in the presence of one equiv. chromocene (Cp2Cr; E1/2 = −1.12 V vs. Fc+/Fc0) reveals a 1:1 ratio of [CpN3]+ and a new organic compound via1H NMR spectroscopy (Fig. S12†). X-ray diffraction of colorless single crystals reveals this compound to be [CpN3H2][BF4], ([5][BF4]), where one of the diene moieties of Cp ring has been reduced (Scheme 2, Fig. 5 and 6). While the reduction of [4]2+ using classic borohydride or aluminum hydride reagents resulted in intractable product mixtures, the hydrogenation product [5][BF4] can also be accessed via H+/e− addition to [4]2+, suggesting that H-atom transfer (HAT) is required to cleanly form [5]+ (Scheme 2).
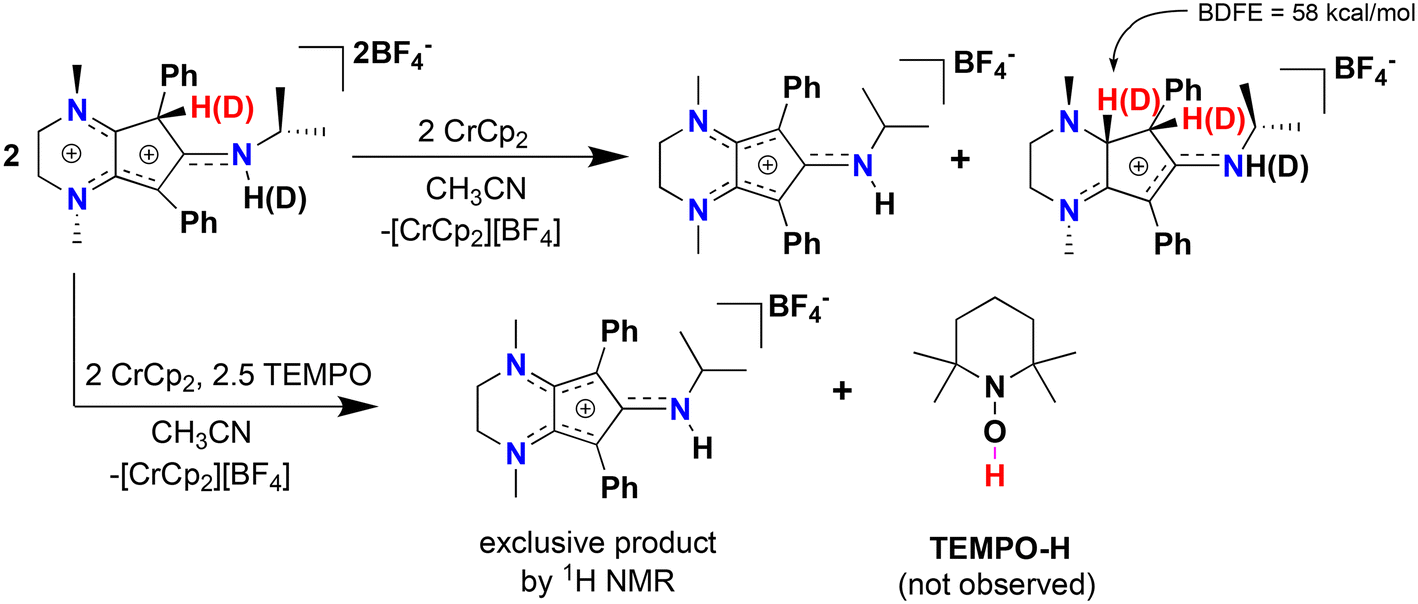 | ||
| Fig. 5 Top: reductive disproportionation of [CpN3H][BF4]2 ([4][BF4]2) to give [CpN3]+ and [CpN3H2][BF4] ([5][BF4]) with a computed BDFE of 58 kcal mol−1 for the newly formed C–H bond. Bottom: reduction in the presence of excess TEMPO. | ||
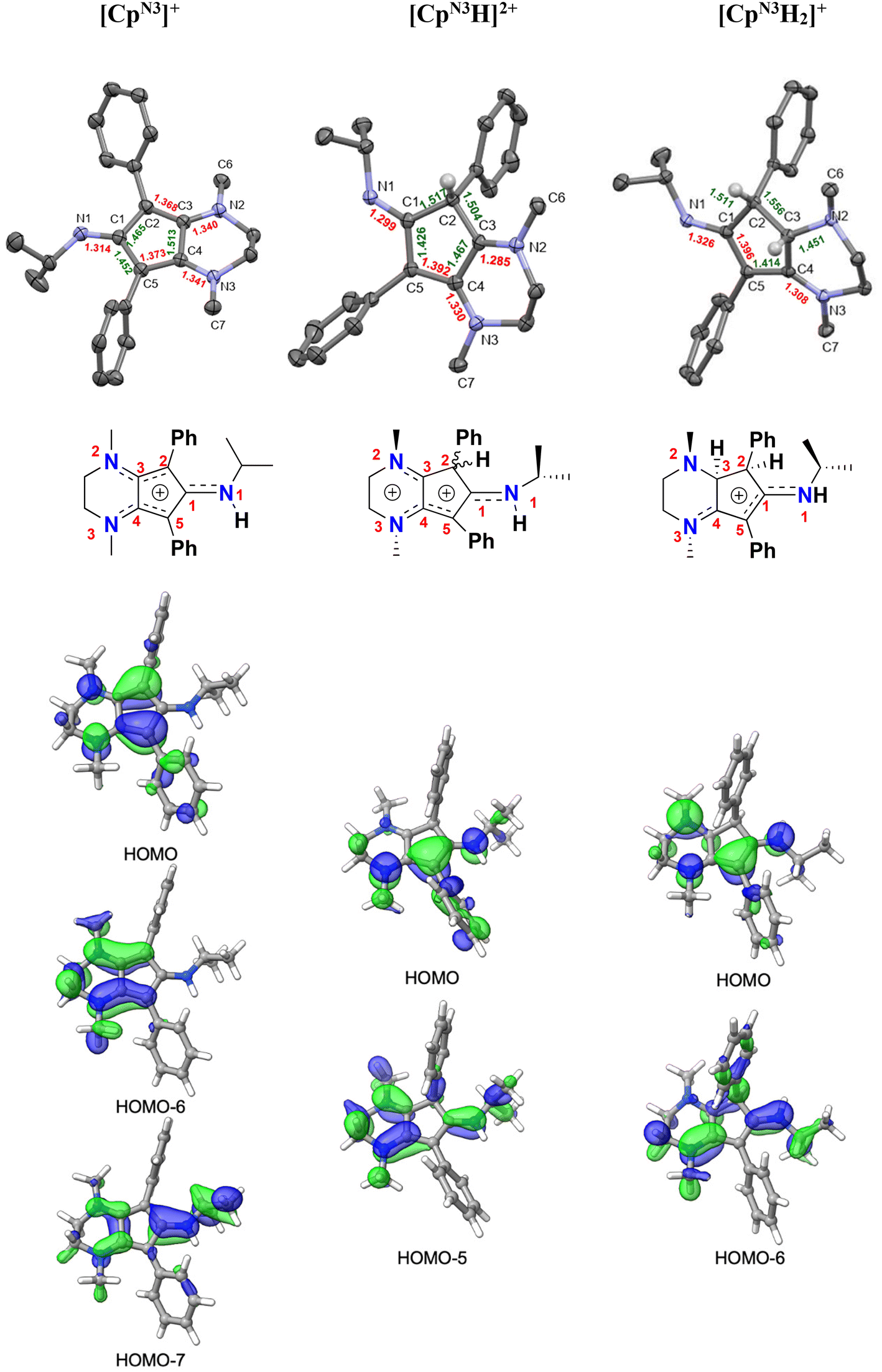 | ||
| Fig. 6 Top: X-ray crystallographic structures of [CpN3][B(C6F5)4] (left), [CpN3H][BF4]2 ([4][BF4]2, middle), and [CpN3H2][PF6] ([5][PF6]; right) with 50% probability ellipsoids. Co-crystallized solvent molecules, counterions, and most hydrogens have been omitted for clarity. Middle: structural cartoons showing charge delocalization based on metrical parameters and typical interatomic distances for organic compounds (see Table 1 and discussion). Bottom: selected molecular orbitals for all three molecules, showing the main π-type MOs responsible for electron delocalization involving the CpN3 ring (isovalue = 0.04). | ||
We propose that [5][BF4] forms through bimolecular HAT involving radical–radical disproportionation of [4]˙+ to produce equimolar amounts of [5][BF4] and [CpN3]+. To support this hypothesis, we prepared the deuterated analogue [CpN3D][BF4]2 by adding two drops of MeOD to a stirring solution of [4][BF4]2 in CD3CN, resulting in 95% H–D exchange at the ring carbon and full deuteration of the secondary amine (Fig. 5 and Fig. S13†). Addition of 1 equiv. chromocene to this product results in the deuteration at one additional site within the Cp ring of [5][BF4], which is wholly consistent with an intermolecular HAT event (Fig. S14 and S15†). Moreover, DFT calculations support the feasibility of HAT to [5]+via[4]˙+ on thermochemical grounds, as the BDFE(C–H) for [5]+ is calculated to be 58.0 kcal mol−1. We then tried this same reaction in the presence of TEMPO, expecting that the weak C(sp3)–H BDFE of 51.8 kcal mol−1 for 4˙+ would quantitatively generate TEMPO-H (BDFE(O–H) = 66 kcal mol−1, MeCN)36 and [CpN3]+. However, with only one equiv. TEMPO and Cp2Cr in solution, a 3:1 ratio of [CpN3]+:[5]+ is observed suggesting that the kinetics of bimolecular HAT are comparable to the HAT kinetics involving TEMPO. Gratifyingly, reacting [4][BF4]2 with 5 equiv. TEMPO and one equiv. Cp2Cr does exclusively generate [CpN3]+via1H NMR spectroscopy (Fig. 5 and S16†), although we could not clearly identify signals belonging to TEMPO-H in these spectra. Furthermore, compound [5]+ can be independently synthesized via 2H+/2e− reduction of [CpN3]+ and isolated as the PF6− salt ([CpN3H2][PF6]) in 32% yield (Scheme 2, Fig. S7 and S8†).
In addition to having X-ray structural data for [4]2+ and [5]+, we were also fortunate to obtain X-ray quality crystals of [CpN3]+via anion exchange with B(C6F5)4−, enabling us to compare the metrical parameters for all three salts with respect to typical interatomic distances for organic compounds (Fig. 6 and Table 1).37,38 Crystallographic analysis of [CpN3]+ indicates that C(2)–C(3) and C(4)–C(5) are far closer to an average Csp2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Csp2 bond length (1.330 Å) while C(1)–C(2), C(3)–C(4) and C(5)–C(1) are more consistent with singly-bonded Csp2–Csp2 atoms (1.460 Å). Moreover, the C–N bonds to the five-membered ring are all substantially shorter than expected for Csp2–Nsp3 atoms (1.468 Å) and harbor more CN double bond character when compared to typical C1sp2N1sp2 distances (1.301 Å). These combined factors lead us to depict the positive charge delocalization of [CpN3]+ with dashed lines in all Figures and Schemes throughout this article. Similarly in case of [4]2+, C(4)–C(5) bond distances match well with average Csp2Csp2 bond lengths (1.330 Å) while C(1)–C(5) and C(3)–C(4) are closer to an average singly-bonded Csp2–Csp2 atoms (1.460 Å). The bond lengths for C(3)–N(2) C(4)–N(3) and C(1)–N(1) are closer to typical Csp2Nsp2 distances (1.301 Å). For [5]+, C(1)–N(1) and C(4)–N(3) have a Csp2Nsp2 double bond character (1.301 Å) and the C(5)–C(1) bond lengths are closer to average literature Csp2Csp2 (1.330 Å) distance. Molecular orbital DFT analyses generally agree with this assessment (Fig. 6, bottom). There are three low-lying π-type MOs mainly responsible for the delocalized electron density in [CpN3]+ (HOMO, HOMO−6, HOMO−7), two MOs in [4]2+ (HOMO, HOMO−5), and two MOs in [5]+ (HOMO, HOMO−6).
Csp2 bond length (1.330 Å) while C(1)–C(2), C(3)–C(4) and C(5)–C(1) are more consistent with singly-bonded Csp2–Csp2 atoms (1.460 Å). Moreover, the C–N bonds to the five-membered ring are all substantially shorter than expected for Csp2–Nsp3 atoms (1.468 Å) and harbor more CN double bond character when compared to typical C1sp2N1sp2 distances (1.301 Å). These combined factors lead us to depict the positive charge delocalization of [CpN3]+ with dashed lines in all Figures and Schemes throughout this article. Similarly in case of [4]2+, C(4)–C(5) bond distances match well with average Csp2Csp2 bond lengths (1.330 Å) while C(1)–C(5) and C(3)–C(4) are closer to an average singly-bonded Csp2–Csp2 atoms (1.460 Å). The bond lengths for C(3)–N(2) C(4)–N(3) and C(1)–N(1) are closer to typical Csp2Nsp2 distances (1.301 Å). For [5]+, C(1)–N(1) and C(4)–N(3) have a Csp2Nsp2 double bond character (1.301 Å) and the C(5)–C(1) bond lengths are closer to average literature Csp2Csp2 (1.330 Å) distance. Molecular orbital DFT analyses generally agree with this assessment (Fig. 6, bottom). There are three low-lying π-type MOs mainly responsible for the delocalized electron density in [CpN3]+ (HOMO, HOMO−6, HOMO−7), two MOs in [4]2+ (HOMO, HOMO−5), and two MOs in [5]+ (HOMO, HOMO−6).
Conclusion
In summary, we have prepared new Co(CpN3) complexes that either undergo CO loss upon oxidation or [CpN3]+ ligand dissociation. Upon loss of cobalt, we observe selective protonation at the five-membered CpN3 ring to give [CpN3H]2+, which was independently prepared in high yield via protonation of [CpN3]+. Through equilibrium measurements via1H NMR spectroscopy and cyclic voltammetry, we experimentally determined the C(sp3)–H pKa and [CpN3H]2+/[CpN3H]˙+ redox potential. Using these data, we calculated the C–H BDFE of [CpN3H]˙+, which has a very weak BDFE in MeCN (52 kcal mol−1). Although H-atom transfer in the presence of excess TEMPO is feasible with in situ 1e− reduction of [CpN3H]2+, attempts to isolate this radical cation in the absence of an H-atom acceptor result in radical disproportionation to give diamagnetic products [CpN3H2]+ and [CpN3]+. With the help of single-crystal X-ray diffraction data, we were able to compare the structural parameters of all three CpN3 salts, showing that positive charge delocalization is distributed across the iminium-like C–N bonds. Work is ongoing to explore the reactivity of these CpN3 salts in the presence and absence of 3d metals.Data availability
The CCDC contains the supplementary crystallographic data for entries 2358580–2358584.† Additional raw data is available from the authors upon request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
D. E. P. thanks the National Science Foundation (NSF) for support under Grant 2055097. X-ray structural solutions were partly supported by the NSF under Grant 2018753. The authors thank David Tresp for assistance with processing X-ray structural data.References
- P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS.
- J. Mas-Roselló, A. G. Herraiz, B. Audic, A. Laverny and N. Cramer, Angew. Chem., 2021, 133, 13306–13332 CrossRef.
- P. J. Shapiro, Coord. Chem. Rev., 2002, 231, 67–81 CrossRef CAS.
- M. Enders and R. W. Baker, Curr. Org. Chem., 2006, 10, 937–953 CrossRef CAS.
- A. VanderWeide and D. E. Prokopchuk, Nat. Rev. Chem., 2023, 7, 561–572 CrossRef CAS PubMed.
- B. Goel, H. Neugebauer, A. I. VanderWeide, P. Sánchez, R. A. Lalancette, S. Grimme, A. Hansen and D. E. Prokopchuk, ACS Catal., 2023, 13, 13650–13662 CrossRef CAS.
- P. Sánchez, B. Goel, H. Neugebauer, R. A. Lalancette, S. Grimme, A. Hansen and D. E. Prokopchuk, Inorg. Chem., 2021, 60, 17407–17413 CrossRef PubMed.
- R. Gompper and H. Glöckner, Angew. Chem., Int. Ed. Engl., 1984, 23, 53–54 CrossRef.
- A. Lator, Q. G. Gaillard, D. S. Mérel, J.-F. Lohier, S. Gaillard, A. Poater and J.-L. Renaud, J. Org. Chem., 2019, 84, 6813–6829 CrossRef CAS PubMed.
- Q. G. Gaillard, L. Bettoni, D. Paris, C. Lequertier, J. F. Lohier, S. Gaillard and J. L. Renaud, Adv. Synth. Catal., 2023, 365, 3704–3712 CrossRef.
- A. W. Cook, T. J. Emge and K. M. Waldie, Inorg. Chem., 2021, 60, 7372–7380 CrossRef CAS PubMed.
- K. M. Waldie, S. Ramakrishnan, S.-K. Kim, J. K. Maclaren, C. E. Chidsey and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 4540–4550 CrossRef CAS PubMed.
- K. M. Waldie, S. K. Kim, A. J. Ingram and R. M. Waymouth, Eur. J. Inorg. Chem., 2017, 2017, 2755–2761 CrossRef CAS.
- U. Koelle and S. Paul, Inorg. Chem., 1986, 25, 2689–2694 CrossRef CAS.
- D. A. Kurtz, D. Dhar, N. Elgrishi, B. Kandemir, S. F. McWilliams, W. C. Howland, C.-H. Chen and J. L. Dempsey, J. Am. Chem. Soc., 2021, 143, 3393–3406 CrossRef CAS PubMed.
- R. M. Evans, E. J. Brooke, S. A. Wehlin, E. Nomerotskaia, F. Sargent, S. B. Carr, S. E. Phillips and F. A. Armstrong, Nat. Chem. Biol., 2016, 12, 46–50 CrossRef CAS PubMed.
- B. Mondal, J. Song, F. Neese and S. Ye, Curr. Opin. Chem. Biol., 2015, 25, 103–109 CrossRef CAS PubMed.
- M. Frey, ChemBioChem, 2002, 3, 153–160 CrossRef CAS PubMed.
- M. Fang, E. S. Wiedner, W. G. Dougherty, W. S. Kassel, T. Liu, D. L. DuBois and R. M. Bullock, Organometallics, 2014, 33, 5820–5833 CrossRef CAS.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- J.-J. Brunet, C. Sidot and P. Caubere, J. Organomet. Chem., 1981, 204, 229–241 CrossRef.
- A. Lator, Q. G. Gaillard, D. S. Merel, J.-F. Lohier, S. Gaillard, A. Poater and J.-L. Renaud, J. Org. Chem., 2019, 84, 6813–6829 CrossRef CAS PubMed.
- T. Piper, F. Cotton and G. Wilkinson, J. Inorg. Nucl. Chem., 1955, 1, 165–174 CrossRef.
- L. R. Byers and L. F. Dahl, Inorg. Chem., 1980, 19, 277–284 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- R. Ciancanelli, B. C. Noll, D. L. DuBois and M. R. DuBois, J. Am. Chem. Soc., 2002, 124, 2984–2992 CrossRef CAS PubMed.
- R. King, Inorg. Chem., 1966, 5, 82–87 CrossRef CAS.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2021, 122, 1–49 CrossRef PubMed.
- D. D. Wayner and V. D. Parker, Acc. Chem. Res., 1993, 26, 287–294 CrossRef CAS.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 6735–6748 CrossRef CAS.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- G. Kiss, K. Zhang, S. L. Mukerjee, C. D. Hoff and G. C. Roper, J. Am. Chem. Soc., 1990, 112, 5657–5658 CrossRef CAS.
- J. Choi, M. E. Pulling, D. M. Smith and J. R. Norton, J. Am. Chem. Soc., 2008, 130, 4250–4252 CrossRef CAS PubMed.
- M. Schmittel and A. Burghart, Angew. Chem., Int. Ed. Engl., 1997, 36, 2550–2589 CrossRef.
- C. F. Wise, R. G. Agarwal and J. M. Mayer, J. Am. Chem. Soc., 2020, 142, 10681–10691 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 1, 1987, S1–S19 CAS.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1–S83 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2358580–2358584. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01560f |
| This journal is © The Royal Society of Chemistry 2024 |