
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of NiAl-hydride heterometallics: perturbing electron density within Al–H–Ni subunits†
Aleida G.
Gonzalez
a,
Fernando
Gonzalez
a,
Edgardo
De Leon
a,
Kaitlyn M.
Birkhoff
 b,
Sam
Yruegas
b,
Haoyuan
Chen
*cd and
Manar M.
Shoshani
*ae
b,
Sam
Yruegas
b,
Haoyuan
Chen
*cd and
Manar M.
Shoshani
*ae
aSchool of Integrated Biological and Chemical Sciences, University of Texas Rio Grande Valley, Brownsville, Texas 78520, USA
bDepartment of Chemistry, Rice University, Houston, Texas 77005, USA
cDepartment of Physics and Astronomy, University of Texas Rio Grande Valley, Edinburg, Texas 78539, USA
dDepartment of Chemistry, Southern Methodist University, Dallas, Texas 75275, USA. E-mail: haoyuan@smu.edu
eDepartment of Chemistry, University of Kansas, Lawrence, Kansas 66045, USA. E-mail: manar.shoshani@ku.edu
First published on 22nd August 2024
Abstract
Heterometallic hydride complexes are of growing interest due to their potential to contribute to highly active insertion-based catalysis; however, methods to modulate electron density within this class of molecules are underexplored. Addition of ancillary ligands to heterotrimetallic NiAl2H2 species (1) results in the formation of heterobimetallic NiAl-hydride complexes with varying phosphine donors (2-(L)2). Incorporation of sigma donating ancillary ligands of increasing strength led to contractions of the Ni–Al distances correlated to a strengthening of a back donation interaction to the Al–H sigma antibonding orbital, most prominently present in 2-(PMe3)2. Demethylation of the aryl ether from 2-(PMe3)2 provides access to a novel anionic nickel–aluminum complex (3) with a maintained bridged hydride moiety between Ni and Al. Increased negative charge in complex 3 results in an elongation of the Ni–Al interaction. Combined crystallographic, spectroscopic, and computational studies support a 3-center interaction within the Al–H–Ni subunits and were used to map the degree of Ni–H character of the series within the Al–H–Ni bonding continuum.
Introduction
Heterometallic complexes bearing redox-active and redox-inactive metal centers have been of growing interest over the past few decades.1,2 This interest is, in part, correlated to the desire to impart cooperative reactivity towards organic substrates which is predominantly described through either (1) a redox-inactive metal coordinating and guiding substrates to further activation and functionalization at the transition metal center, or (2) the redox inactive metal center binding directly to the transition metal as a sigma acceptor to generate a more electrophilic transition metal center.3,4 Both modes of cooperativity have been well-documented and have led to progress in a multitude of noteworthy reactions including, but not limited to, N-heterocycle C–H activation,5 CO2 reduction,6–8 olefin hydrogenation,9 and dihydrogen activation.10,11 Reactivity by incorporating transition metals and Lewis-acidic metalloid boron in a single system has shown a wide array of success in bond activation and catalysis.12–16The intermediacy of metal-hydrides in a plethora of catalytic cycles is widely acknowledged17 and multimetallic hydride moieties, specifically, are responsible for a multitude of impressive bond transformations.18 Indeed within homogenous catalysis, examples with late transition metals and main group hydride additives have exhibited success in inert-bond activation; yet the ill-defined nature of the reaction conditions limits the mechanistic insight one can glean in relation to cooperative reactivity.19 Examples of reactivity from well-defined heterometallic hydride complexes are still emerging20 which is, in part, due to synthetic challenges in accessing well-defined heterometallic hydride complexes with discrete nuclearity. Moreover, reliance on the self-assembly of metal ions on ligands to generate related species continues to impede progress.21–23 Dependence on monodentate ligands in this domain offers poor control over nuclearity of multimetallic hydride complexes.24 Complexes including precious metals such as Rh and Ir with bridging aluminum hydrides are known,25–27 as well as recent examples with base metals.28,29
Given the dearth of examples where the hydride moieties are bridged between heterometallic centers, studies on electronic structure to better understand the impact of generating heterometallic complexes of this sort are limited. Fortunately, examples of well-defined heterometallic hydrides of this nature have been reported recently, select examples are shown in Fig. 1. Complex I features a trimetallic Ni–Mg complex with 4 bridged hydrides by Xu et al., capable of catalytic insertion chemistry with unsaturated substrates.30,31 Complexes II and III by the Crimmin group employ main group beta-diketiminate moieties to bridge Al and Mg hydrides to Fe and Pd centers, respectively.32–34 The electronic structure of these complexes has been thoroughly studied to gain an understanding of the nature of the heterometallic subunits. Complex II is reported to facilitate ortho-directed C–H bond activation of N-heterocycles.
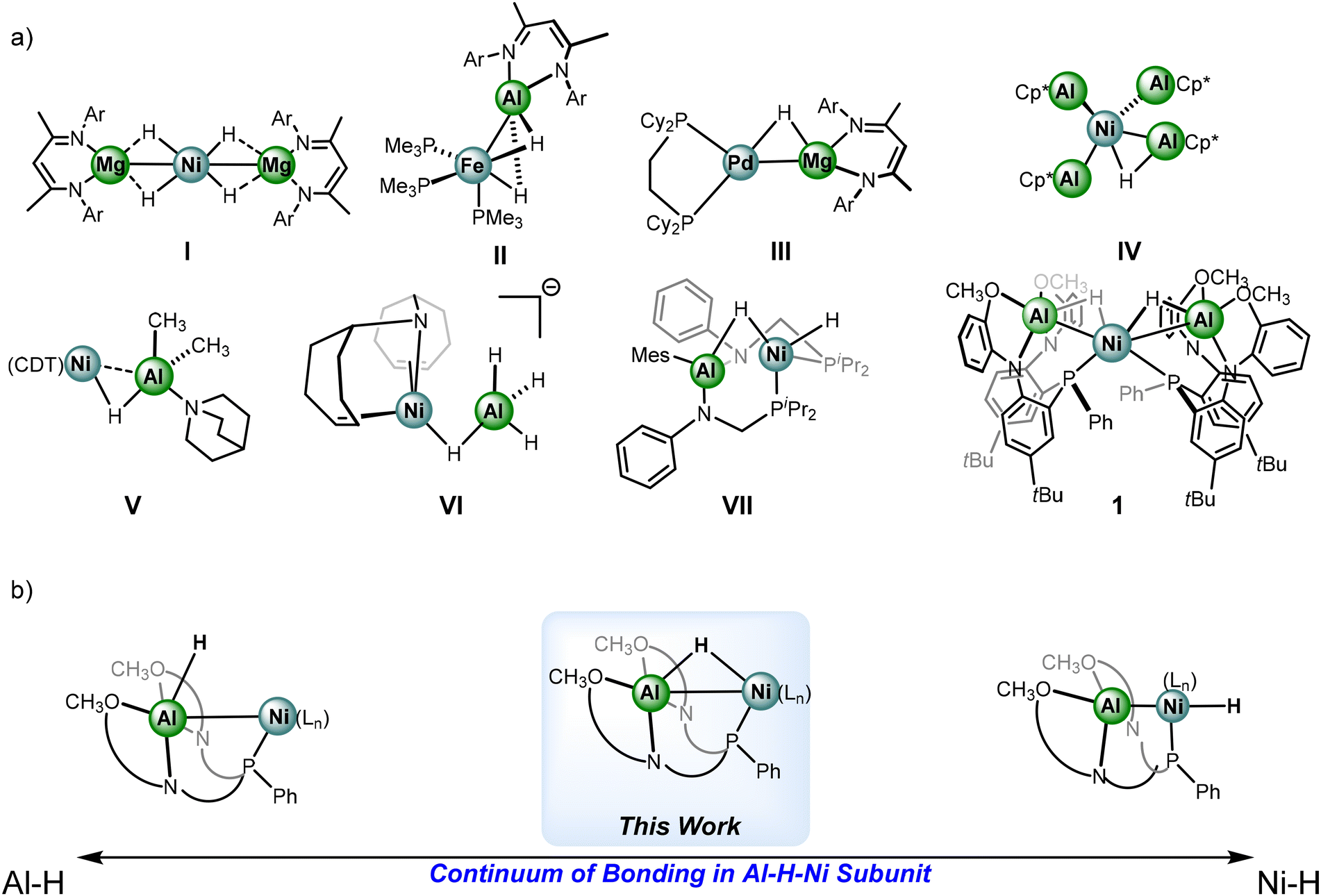 | ||
| Fig. 1 (a) Select examples of heterometallic hydride complexes with first row transition metals. (b) Extremes of sigma bonding within an Al–H and Ni–H. | ||
Examples with bridging hydrides between Ni and Al centers are highly limited; complexes IV, V, VI are the currently reported crystallographically characterized examples by other groups.35–37 Developing a thorough understanding of the capability of heterometallic hydrides, transcending the classical substrate preactivation and directing nature is crucial. Recently, facile anionic group transfer was reported for transition metal-aluminum containing species, including halides, alkyls, and hydride transfers.38 Complex VII is a rare report of an Al–H–Ni generated in situ via addition of dihydrogen to a nickel-alane complex.39 The system demonstrates facile reversibility between alane and aluminyl states through auxiliary substrate coordination. The system used in complex VII outlines potential novel pathways for future reactivity leveraging Lewis-acidic metals in redox activity and electron storage.
Our group recently reported complex 1, a NiAl2H2 catalyst capable of hydrofunctionalization of N-heterocycles.40 Complex 1 exhibits a markable increase in activity of hydrofunctionalization catalysis in comparison to the alane precursor which underscores the crucial nature of generating multimetallic hydrides to amplify insertion-based reactivity. In the interest of delineating the bonding picture in complexes related to the examples in Fig. 1a, Fig. 1b demonstrates a scale that denotes the extremes of the bonding continuum within the Al–H–Ni subunit of complex 1, ranging from an Al–H alane to a Ni–H with a highly sigma-donating and trans-influencing aluminyl moiety.41 While complex 1 lies somewhere in between the extremes, and there are accompanying parameters that offer guidance such as the Ni–Al distances and the geometry at Ni, further investigations towards understanding how to quantify or designate complexes within the context of the scale are needed. Indeed, the groups of Aldridge and Crimmin have thoroughly studied transition metal-main group hydride complexes, including examples of transition metal-alanes, and have provided a blueprint in assessing degrees of activation within main group hydrides.42,43 The interactions are described through the Dewar–Chatt–Duncanson model where the main group hydride fragment donates electron density to an empty d-orbital and back donation from a filled dπ-orbital to an Al–H sigma-antibonding orbital;27,44 notably the latter interaction has been described a minimal contributor in a series of reported examples.45 Given the lack of known examples featuring an Al–H–Ni subunit, and their growing interest given potential in catalysis, further work is needed to elucidate the electronic structure.46 Herein, we discuss the synthesis of new heterometallic complexes with an Al–H–Ni subunit and efforts to perturb and monitor the electron density distribution within the framework.
Results and discussion
Synthesis and characterization of neutral NiAl heterobimetallic hydride complexes
In the solid-state, complex 1 features a Ni center supported by two phosphine donors of the metalloligands as well as two Al–H fragments, the latter in a 3-center bonding motif. Our group desired to explore the continuum by probing complex 1 through examining the impact of addition of ancillary ligands to the primary coordination sphere and monitoring the impact in electronic structure.We envisioned the fate of the coordination site of the added ancillary ligands may be instructive to the nature of the hydride moiety, specifically towards where on the continuum of sigma-binding the hydride of derivatives of complex 1 resides. For instance, addition of an ancillary ligand (L) to complex 1 could result in coordination of L at Ni to generate an Ni-alane with greater Al–H character (A – Scheme 1). Alternatively, L could coordinate to Al and subsequently result in the formation of a species with greater Ni hydride character and a coordinated aluminyl center (B – Scheme 1), analogous to anionic group transfers reported by Lu et al.39 As discussed above, access to Ni-aluminyl complexes has been recently shown to be relatively facile and the rules that govern interconversion are of growing interest. In the event of pathway B, the retention of the trimetallic core would hinder a trans arrangement of the hydride moiety to the aluminyl. Additionally, ancillary ligands could also result in dissociation of the metalloligand itself to form heterobimetallic complexes (C – Scheme 1). Though option C provides the least decisive description towards the nature of the hydride moiety, one can still glean relevant information based on the comparative electronic properties of the ancillary ligand, and the impact towards the Al–H–Ni subunit.
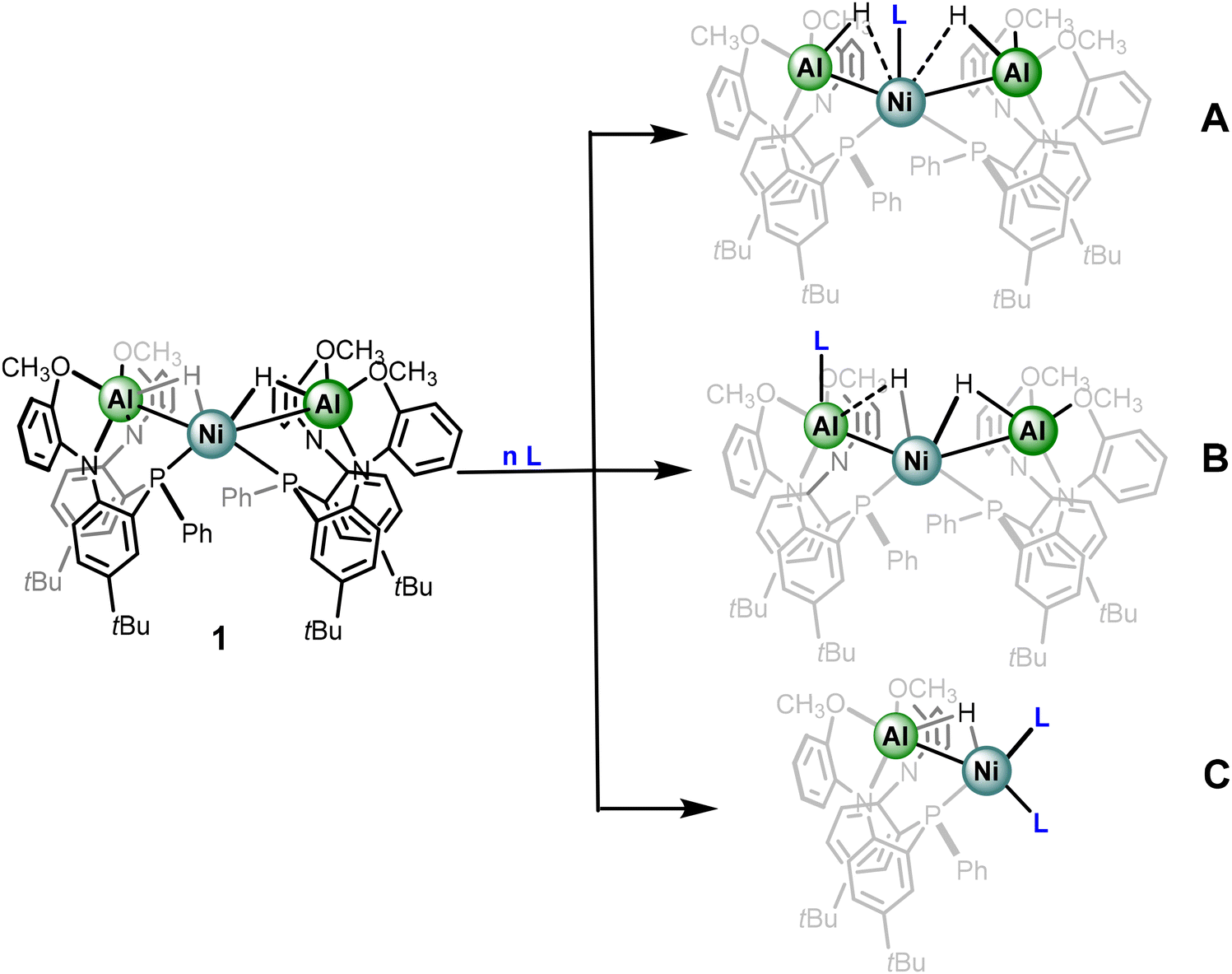 | ||
| Scheme 1 Potential coordination sites of ancillary ligands to complex 1. | ||
Given that addition of Ni(COD)2 to metalloligand LAlH results in the formation of complex 1, rather than a COD-coordinated species, ancillary ligands serving as strong sigma donors were pursued in our study. Addition of excess triphenylphosphine to complex 1 produces a new complex bearing a 31P{1H} NMR spectrum consistent with a heterometallic complex with two triphenyl phosphine ligands and one metalloligand, indicating the formation of the heterobimetallic species 2-(PPh3)2 (Scheme 2). Specifically, a triplet at δ −12.48 corresponding to the metalloligand phosphine and a broad resonance at δ 29.26 are observed. The latter resonance sharpens to a doublet at 55 °C with corresponding coupling constants of 9.6 Hz. Both 1H and 13C{1H} NMR spectra are consistent with the formation of a heterobimetallic hydride complex including the observation of a hydride resonance at δ −1.78, a slight upfield shift from the hydride resonance of complex 1. The resonance appears as a broad doublet JHP of 53.3 Hz, which is consistent with the multiplicity of complex 1. While the expectation is to observe a doublet of triplets from additional coupling to the phosphorus centers of the triphenylphosphine moieties, it is likely that the broadening from coupling to quadrupolar 27Al obfuscates observation of further coupling. Exposing complex 1 to conditions to balance the reaction, including four equivalents of PPh3 and one equivalent of Ni(COD)2 results in quantitative conversion to 2-(PPh3)2. Separately, conditions were identified to synthesize 2-(PPh3)2 directly from the LAlH precursor (Scheme 2).
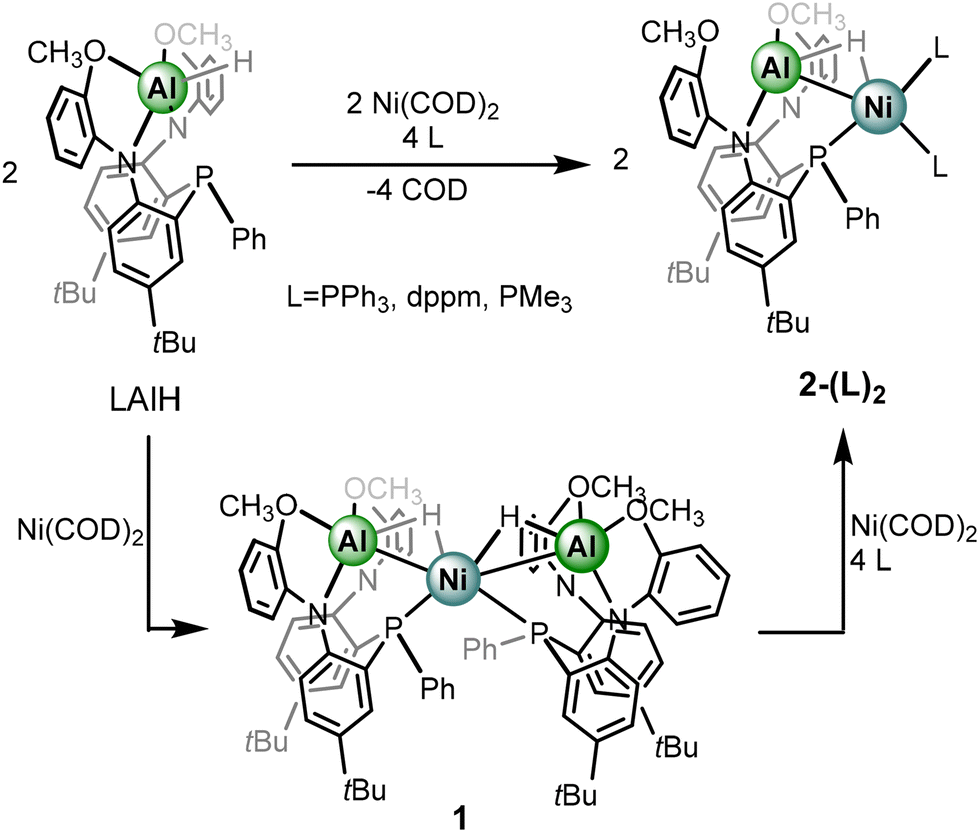 | ||
| Scheme 2 Synthesis of complexes 2-(L)2. | ||
The solid-state structure of 2-(PPh3)2 was obtained by single-crystal X-ray diffraction and features several noteworthy features (Fig. 2a). The heterobimetallic features a Ni–Al distance of 2.322(1) Å, representing a contraction compared to the Ni–Al distance of 2.363(3) Å in complex 1. The shortening is reasoned through a strengthening of the Ni–Al interaction as the two PPh3 ligands may serve as more electron donating in comparison to the metalloligand in 1. As a result, a more electron rich Ni center would be better equipped to donate to the Al–H sigma antibonding orbital. Furthermore, in 2-(PPh3)2 only one of the methoxy groups is coordinated to the Al center which may be the result of the Ni–Al interaction strengthening and the Lewis-acidic Al center requiring less coordinative stabilization. However, the 1H NMR data features only one methoxy resonance which could indicate a fast exchange of methoxy coordination to Al, or that in solution, both donors are coordinated. The hydride ligand was located on the electron difference map and indicates that the bridged nature between the Ni and Al centers is maintained. The Ni–H distances are comparable to that of 1 but the Al–H distance shows a slight elongation; however, the crystallographically determined M–H distances provide limited insight. With respect to the hydride ligand and the three phosphine donors, a τ′4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 47 value of 0.92 about Ni is calculated which is consistent with tetrahedral geometry, expected for a Ni-alane, rather than square planar geometry for a Ni-hydride-aluminyl. However, given the sigma-complex bonding assignment, determining the τ value from bond angles at some point along the Al–H bond rather than the hydride center itself is more appropriate. Nevertheless, a distorted tetrahedral geometry is the most appropriate descriptor for Ni in 2-(PPh3)2. The impact on electron count and oxidation state by sigma-acceptors has been discussed at length elsewhere.48,49 Though option C from Scheme 1 seems to dominate when L = PPh3, examining the consistency of this result with further ancillary ligands to potentially probe the Al center for coordination, as well as further interrogating the impact of the Al–H–Ni subunit via coordination at the Ni site, became of increased interest.
47 value of 0.92 about Ni is calculated which is consistent with tetrahedral geometry, expected for a Ni-alane, rather than square planar geometry for a Ni-hydride-aluminyl. However, given the sigma-complex bonding assignment, determining the τ value from bond angles at some point along the Al–H bond rather than the hydride center itself is more appropriate. Nevertheless, a distorted tetrahedral geometry is the most appropriate descriptor for Ni in 2-(PPh3)2. The impact on electron count and oxidation state by sigma-acceptors has been discussed at length elsewhere.48,49 Though option C from Scheme 1 seems to dominate when L = PPh3, examining the consistency of this result with further ancillary ligands to potentially probe the Al center for coordination, as well as further interrogating the impact of the Al–H–Ni subunit via coordination at the Ni site, became of increased interest.
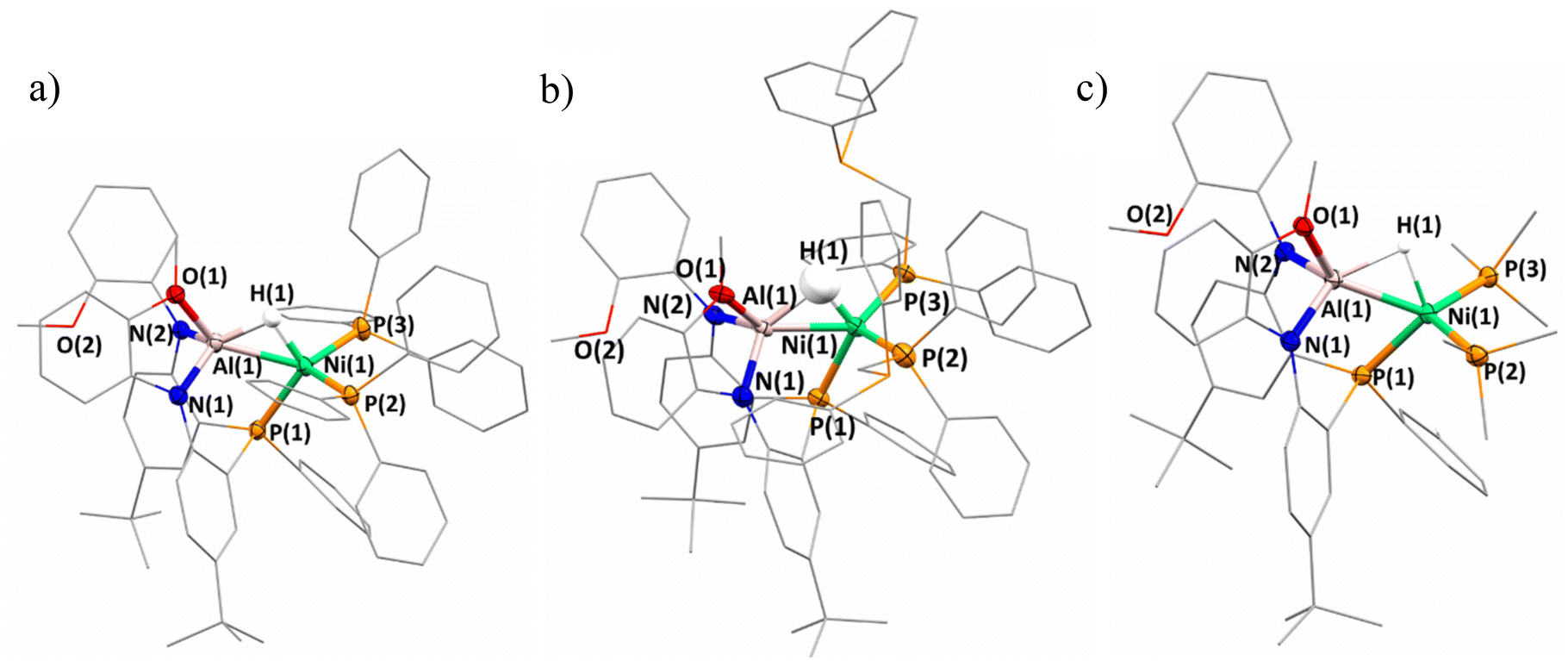 | ||
| Fig. 2 Solid-state structures of (a) 2-(PPh3)2, (b) 2-(dppm)2, and (c) 2-(PMe3)2. Hydrogen atoms on ligands are excluded for clarity. Mixed wireframe/ellipsoid view used for clarity. | ||
The chelating phosphine, bis-diphenylphosphinomethane (dppm) was employed as an auxiliary donor. We sought to explore whether the phosphine centers would serve as a chelate between Ni and Al, bind one equivalent to the Ni or Al centers in a chelating fashion, or coordinate two equivalents at Ni in a terminal fashion, akin to 2-(PPh3)2.50 Indeed, addition of dppm to complex 1 along with Ni(COD)2 quantitatively formed the heterometallic analogue, 2-(dppm)2 with the phosphine ligands coordinating in a non-chelate fashion. Similar to 2-(PPh3)2, complex 2-(dppm)2 is preferentially and reasonably synthesized directly from metalloligand LAlH. Complex 2-(dppm)2 features three resonances in the 31P{1H} NMR spectrum: a triplet at δ −12.35, a corresponding doublet at δ −27.05, and a broad resonance at δ 20.72 integrating in a 1:2:2 ratio, respectively. The triplet and doublet resonances correspond to the phosphorus centers in the coordination sphere of Ni, the triplet belonging to the metalloligand phosphorus center and doublet to the bound dppm phosphorus center. Lastly, the broad resonance corresponds to the unbound dppm phosphorus centers. Variable-temperature 31P{1H} NMR was employed and displays decoalescence of both dppm resonances at −20 °C amounting to a total of 5 resonances for complex 2-(dppm)2 (Fig. S2 and S3†). The 31P{1H} NMR spectrum at 60 °C exhibits sharpening of all resonances as well as resolving of coupling features (Fig. S2 and S4†). The 1H NMR spectrum shows the presence of the bridged hydride at δ −2.10, which once again appears as a broad doublet and is slightly upfield in comparison to the hydride resonance in 1 and 2-(PPh3)2.
The solid-state structure confirms the solution-state assignment demonstrating that 2-(dppm)2 (Fig. 2b) features a heterobimetallic core with two dppm ligands bound terminally to the Ni center. The terminal coordination of the dppm ligand observed rather than binding between the Ni and Al center is relatively unsurprising given the resultant strain of a potential chelate due to the single atom bridge. The Ni center takes on a distorted tetrahedral geometry with respect to the metalloligand phosphine, bound phosphine centers from dppm ligands, and across the Al–H bond. The Ni–H and Al–H distances range from 1.43–1.47(12) Å and 1.55–1.72(12) Å respectively. Notably, the hydride moiety could only be located in two of the four molecules within the asymmetric unit. The distorted tetrahedral geometry about Ni underscores a preference for the Ni-alane designation rather than a Ni-aluminyl. The Ni–Al distances range from 2.269–2.275(4) Å which is a contraction from both 1 and 2-(PPh3)2. Similar to 2-(PPh3)2, only one methoxy ligand is bound to the Al center.
Given the consistency in the observation of route C from Scheme 1, perturbing the electron density within the Al–H–Ni subunit by adding ancillary ligands of increasing sigma donation became of greater interest. Thus far, increasingly sigma donating ligands resulted in (1) a Ni–Al contraction in the solid-state and (2) a progressively upfield chemical shift for the hydride moiety. Substitution of more electron rich phosphine ligands to known heterometallic complexes have also demonstrated an impact in accessing challenging reactivity.51 Addition of two equivalents of trimethylphosphine to LAlH and Ni(COD)2 results in the quantitative formation of 2-(PMe3)2. The assignment is supported by multinuclear NMR spectroscopy with a doublet and triplet resonance in the 31P{1H} NMR spectrum. The triplet at δ −9.18 and doublet at δ −21.11, correspond to the metalloligand phosphorus center and trimethyl phosphine phosphorus centers, respectively. A JHP coupling constant of 9.1 Hz is observed between the two phosphorus environments. The 1H NMR spectra also support the assignment of the formation of the heterobimetallic 2-(PMe3)2 species. The hydride resonance once again is observed as an apparent broad doublet, the JHP coupling constant is 54.5 Hz. Gratifyingly, the hydride resonance shifts substantially upfield to δ −3.28 in comparison to complexes 1, 2-(PPh3)2, and 2-(dppm)2.
The solid-state structure of 2-(PMe3)2 was also obtained and a significantly shortened Ni–Al interaction is observed (2.261(1) Å) which is among the shortest Ni–Al distances reported (Fig. 2c).52 The hydride was located on the electron difference map Ni–H and Al–H distances of 1.61–1.67(4) Å and 1.67–1.71(4) Å, respectively. Similar to the other complexes discussed in the 2-(L)2 series, a slight distortion of tetrahedral geometry is observed when considering the three phosphine ligands and hydride by the Ni center (τ′4 = 0.87). Once more, the solid-state structure shows only one of the methoxy ligands remains bound to the Al, again supporting that coordination of the methoxy ligands to the Al center is dependent on the Lewis-acidity of Al.
Synthesis and characterization of an anionic NiAl heterobimetallic hydride complex
The comparative spectral and structural parameters of 1 and 2-(L)2 demonstrate a trend that more sigma donating ligands result in a stronger Ni–Al interaction, as indicated by the bond length contractions. Furthermore, a correlation is also observed relating increasingly upfield hydride chemical shifts with more electron-rich ancillary ligands. Motivated by leaning further on this scale, strongly sigma-donating N-heterocyclic carbene ligands were then employed. Addition of 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (iPrMe) to complex 2-(PMe3)2 was investigated to access a carbene coordination complex while minimizing need for metalloligand dissociation. Monitoring the reaction through NMR spectroscopy revealed clean conversion to a single heterometallic species with resonances consistent with incorporation of the N-heterocycle moiety as well as PMe3 ligands. Remarkably, rather than a carbene coordinated species, an anionic NiAl-hydride species, complex 3, is formed. The carbene ligand facilitates demethylation from one of the methoxy groups to form an Al-phenolate and a methylated imidazolium counter cation (Scheme 3). Though carbene ligands have been reported to generate phenolates through demethylation53 the observed reactivity is somewhat surprising given the presence of the hydride ligands, which one may have expected to be particularly prone to abstraction. The selective formation of complex 3 highlights that the bridged hydride bears a stabilizing role within these frameworks. Furthermore, formation of 3 demonstrates that access to anionic states within catalytic insertion chemistry should be considered and warrants exploration in mechanistic studies. The choice of carbene and heterometallic combination seems to be rather consequential as comparative reactivity of iPrMe with complex 1 produced an indiscernible mixture of complexes via NMR spectroscopic studies. Potential rationale is the formation of a mixture of anionic species as well as carbene coordination complexes. The variance in reactivity between 1 and 2-(PMe3)3 towards the carbene ligand is intriguing and could be correlated to an increased dissociative nature of LAlH as opposed to PMe3. Furthermore, addition of bulkier carbene ligand, IPr, to complex 1 shows surprisingly no evidence of reactivity.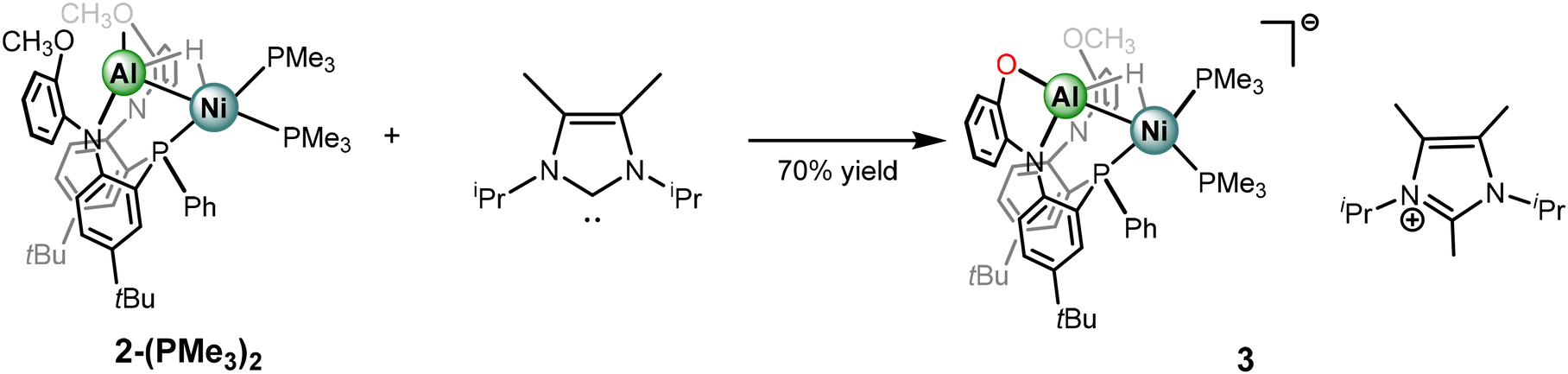 | ||
| Scheme 3 Synthesis of anionic NiAl hydride complex 3. | ||
Despite the deviation from the ligand substitution reactions, isolation of complex 3 presented a unique opportunity to interrogate the impact of negative charge on the Al–H–Ni subunit. Complex 3 bears only slight solubility in aromatic solvents and thus more polar solvents were employed for characterization. The 31P{1H} NMR spectrum in THF-D8 shows that complex 3 is desymmetrized and three resonances are observed at δ −0.65, −16.60, and −25.55 each a doublet of doublets with corresponding coupling constants for J-coupling to the neighboring phosphorus centers. The 1H NMR spectrum is also consistent with the desymmetrized nature of complex 3, and features resonances for both the anionic NiAl-hydride complex as well as the methylated imidazolium fragment. Intriguingly, the hydride resonance appears at approximately δ −4.07 as a broad apparent doublet and bears a JHP coupling constant of 44.8 Hz, the coupling is better resolved in a crude NMR spectrum in C6D6 (Fig. S1–S14†). The chemical shift of the hydride resonance in complex 3 presents the most upfield of the Al–H–Ni series within the study.
The solid-state structure of complex 3 was obtained and confirmed the assignment of the anionic nickel–aluminum species and a methylated imidazolium cation (Fig. 3). The anion in complex 3 can be described within the extremes as either a nickel aluminate or a nickelate-alane given the literature precedent for accessing both aluminate54 and nickelate complexes in heterometallic species.55,56 Both the solid-state structure of complex 3 and the chemical shift support the retention of the hydride moiety in a bridged nature. The Ni–Al distance of 2.333(1) Å demonstrates that complex 3 undergoes an elongation in comparison to 2-(PMe3)2. Despite the Ni–Al distance elongation in complex 3, the complexes within this study feature a Ni–Al bond distance that are less than the sum of the covalent radii of Ni and Al.57 Complex 3 establishes the only observation where the chemical shift of the hydride moiety continues to trend upfield while a elongation of the Ni–Al interaction is observed (Fig. 4). A table comparing the solid-state structural patterns of complexes 1, 2-(L)2, and 3 is shown in Table 1. Complex 3 demonstrates a mild distortion from tetrahedral geometry at Ni with respect to the hydride moiety and three phosphine centers with the largest distortion amongst the class of molecules (τ′4 = 0.80).
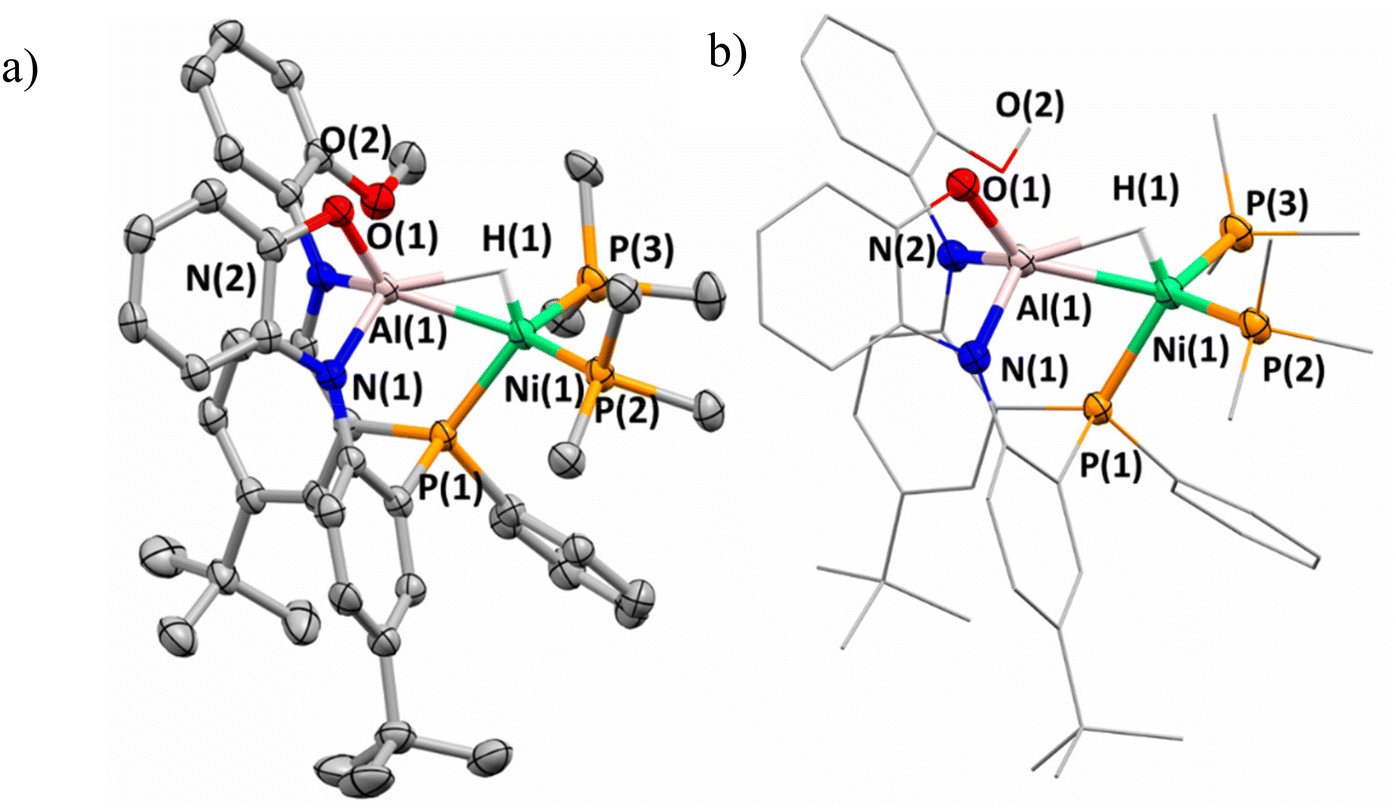 | ||
| Fig. 3 (a) Solid-state structure of anionic portion of complex 3. Cation and hydrogen atoms on ligands are excluded for clarity. (b) Mixed wireframe/ellipsoid structure of complex 3 shown for clarity. | ||
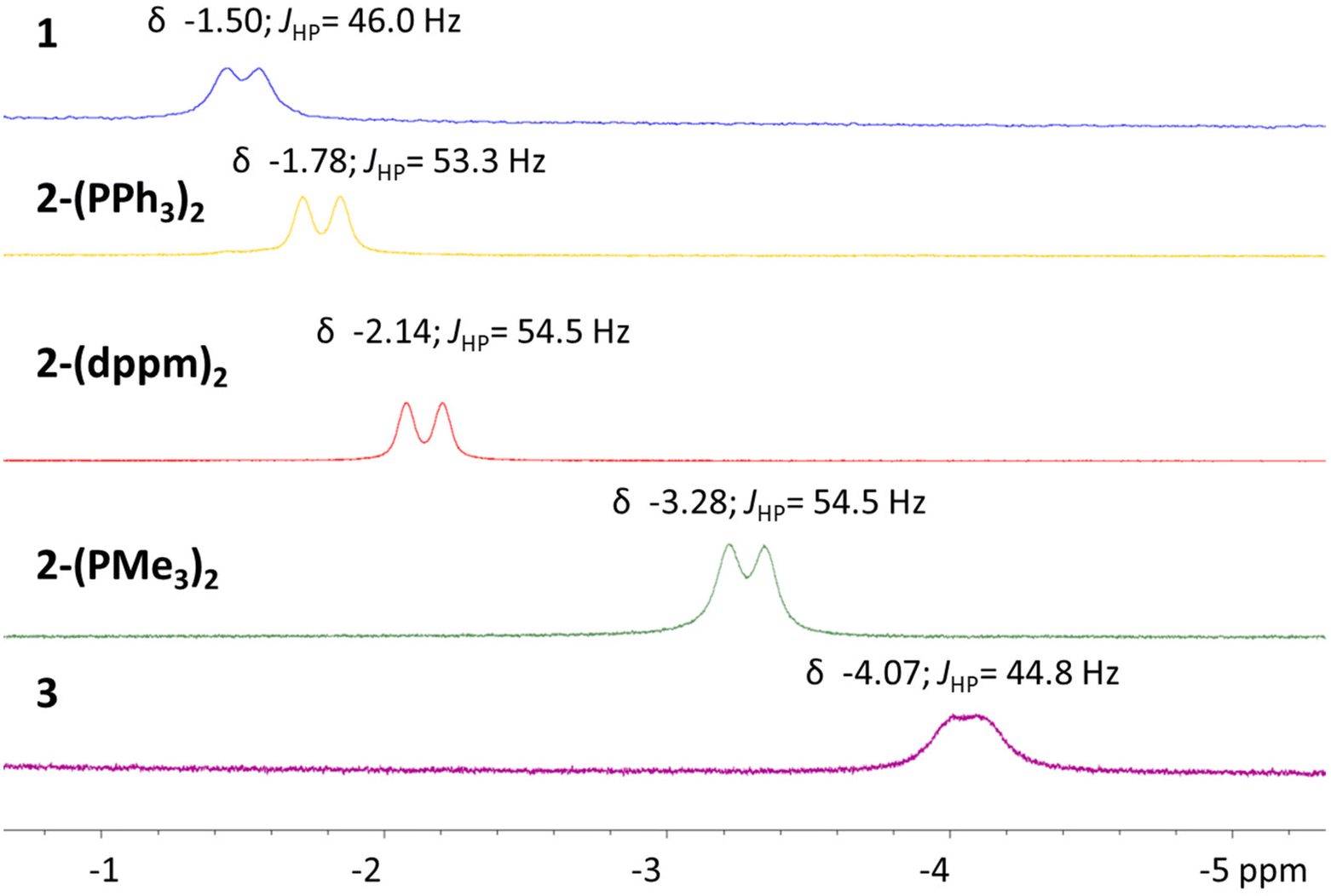 | ||
| Fig. 4 Stacked 1H NMR spectra in hydride region for complexes 1, 2-(L)2, and 3. | ||
| 1 | 2-(PPh3)2 | 2-(dppm)2 | 2-(PMe3)2 | 3 | |
|---|---|---|---|---|---|
| a Values in brackets determined from DFT-optimized structures. b τ 5 value. | |||||
| Ni–Al (Å) | 2.3629(7) | 2.3222(16) | 2.269–2.276(4) | 2.2503(7) | 2.3336(11) |
| 2.3683(7) | 2.2613(7) | 2.3358(11) | |||
| Ni–H (Å) | 1.57(3) | 1.54(4) | 1.43–1.47(13) | 1.61(4) | 1.19(3) |
| 1.60(3) | 1.67(4) | 1.31(3) | |||
| Al–H (Å) | 1.59(3) | 1.74(4) | 1.55–1.72(12) | 1.68(4) | 1.88(3) |
| 1.61(3) | 1.71(4) | 1.75(3) | |||
| Ni–P(1) (Å) | 2.1580(6) | 2.1832(14) | 2.169–2.175(2) | 2.1410(6) | 2.1311(10) |
| 2.1635(6) | 2.1449(6) | 2.1329(10) | |||
| Ni–(P2/P3) (Å) | — | 2.2095(14) | 2.171–2.186(2) | 2.1570(7) | 2.1661(11) |
| 2.1915(16) | 2.1612(7) | 2.1614(11) | |||
| 2.1674(7) | 2.1559(11) | ||||
| 2.1706(7) | 2.1677(10) | ||||
| Ni τ′4 | 0.89(0.86)a | 0.92(0.87) | 0.90(0.86) | 0.87(0.84) | 0.80(0.82) |
| Al τ′4 | 0.23(0.37)b | 0.76(0.80) | 0.73(0.72) | 0.70(0.69) | 0.78(0.80) |
| δ Al–H–Ni | −1.50 | −1.78 | −2.14 | −3.28 | −4.07 |
Electronic structure studies
The study of the series of NiAl-hydride complexes reveals an intriguing trend, the inclusion of more electron rich ancillary ligands, as well as negative charge, results in increasingly upfield chemical shifts of the hydride moieties. The principles that govern chemical shift are non-trivial and convoluted;58 however, diamagnetic Ni-hydrides often have chemical shifts in the range of −5 to −38 ppm, (with the majority not lying within the extremes)59 while diamagnetic aluminum hydride species have chemical shifts typically ranging from 3–6 ppm.60–62 The chemical shifts of the complexes reported here show an intermediary of the two, consistent with a designation of sigma-complexes.59 Moreover, comparison of the Tolman parameters of similar ancillary ligands63,64 employed in the 2-(L)2 series are in agreement with the upfield chemical shifts belonging to more donating ligands; though, this comparison excludes complex 1 and complex 3.The described trend could be explained by consideration of the 3-center bonding motif within the Al–H–Ni subunit. One could argue that predominant interactions within the subunit are donation of electron density from an Al–H sigma-bonding orbital to the Ni center and backdonation from the Ni center to the Al–H sigma antibonding orbital, as described in the introduction. Notably, more electron rich Ni centers would be best suited for a strengthened back donation interaction. Within the series of complexes discussed here, the strength of the backbonding interaction would delineate the degree of Ni–H character observed, akin to arguments for dihydrogen coordinated intermediates prior to formation of metal-dihydrides.65 Similar arguments for Mg-hydrides and Zn-hydrides to Pd centers have been depicted previously.34 However, unlike the prototypical dihydrogen coordination, the interaction is asymmetric; where the accepting orbital from the main group-hydride is of greater main group-element character. Consequently, donation and backdonation interactions are likely not evenly distributed. Physical methods to quantify the electron distribution within an Al–H–Ni subunit are not well developed, mostly due to the lack of known examples. Although Al is spin active, the quadrupolar nature limits the insight one can glean from Al–H coupling constants, compared to those of silane-coordinated sigma species.66 In this series of complexes, the only observable coupling constant for the hydrides is to the metalloligand phosphine center as confirmed through 1H{31P} NMR experiments.40 Nevertheless, the chemical shift of the hydride moiety trends progressively upfield across the series (Fig. 4). A finding consistent with a gradual shift towards Ni–H character across the continuum of bonding within the subunit.
The structure across the series of complexes was evaluated for changes at both Al and the Ni centers. As discussed above, the expectation for a Ni-alane would result in a tetrahedral geometry while the Ni-aluminyl would likely adapt a square planar geometry. Thus, the evaluation of the τ′4 value at Ni across the series serves as an informative metric to guide the level of deviation from tetrahedral Ni-alane. Indeed, analysis of τ′4 across the series of complexes reported here show an increasing deviation from 1 (perfectly tetrahedral) across the series of heterobimetallic complexes examined, ranging from 0.92 to 0.80, consistent with the trend in chemical shift of the hydride ligand, a finding again supporting a gradual shift to more Ni–H character (Table 1). Given the major structural differences and bidentate nature in metalloligand LAlH in comparison to the phosphine donors examined, it is not surprising heterotrimetallic complex 1 slightly deviates from the trend. Comparison of τ values is more complicated at Al given the increased coordination number at complex 1. Across the heterobimetallic complexes there seems to be a consistent deviation from tetrahedral geometry with exception to complex 3. This deviation is potentially associated to the anionic nature and the overall structural difference that a phenolate moiety would impose rather than an aryl ether. Given that both τ′4 values at Ni and Al utilize bond angles determined from the hydride location, the τ′4 value was also calculated from the DFT-optimized structures (discussed below); the results show strong consistency with the trends discussed above.
To deepen our understanding within this argument, density functional theory (DFT) and Quantum Theory Atoms-In-Molecules (QTAIM) calculations were performed to interrogate the electronic structure, while also gaining more reliable metrics regarding the metal hydride bond lengths. The bond lengths from DFT-optimized structures correlate well with experimental results (Table S3-1†) with exception to metal–hydride bond lengths in 2-(dppm)2, 2-(PPh3)2 and 3; in these cases the DFT-calculated structure is deemed a better representation of the hydride position given challenges in predicting hydride locations accurately through X-ray diffraction. Representative examples highlighting interactions within the core are observed in the HOMO and HOMO−6 of 2-(PMe3)2 (Fig. 5). The HOMO of 2-(PMe3)2 specifically demonstrates contributions from all three components of the subunit; Ni (64%), Al (3.6%), and H (6.4%). The primary contributors from Al are the 3s (2.03%) and 2py (0.99%) orbitals, while the Ni orbital contributors are a hybrid mixture mostly consistent with 3d (∼55.3%) with some 4p (∼9%) character. Orbital mixing into the ligand framework renders further evaluation difficult to deconvolute; however, the frontier orbitals demonstrate significant contributions from the Ni centers across the series of complexes, their details of which are displayed upon in Tables S3-3 to S3-7.†
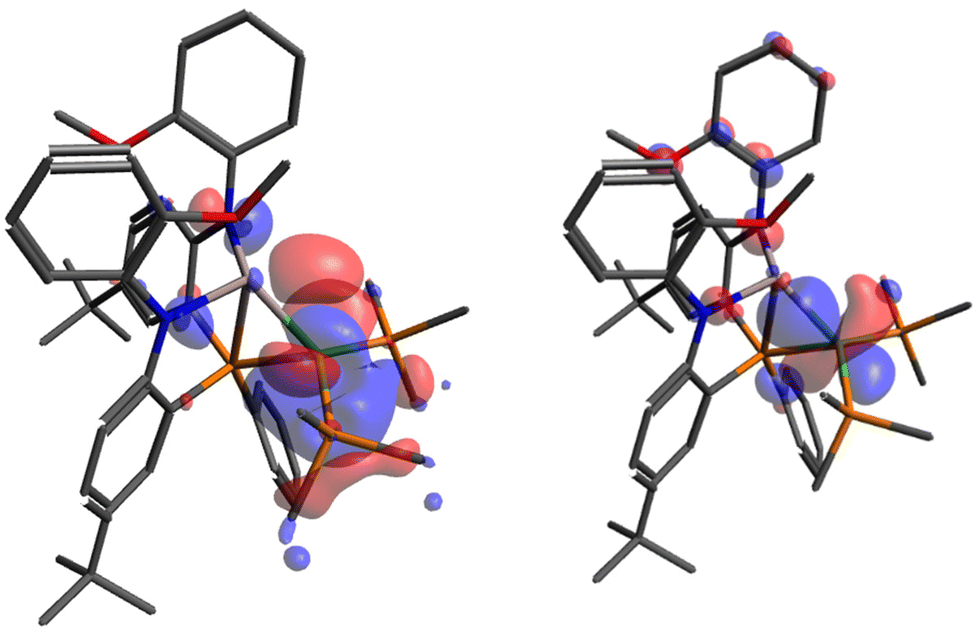 | ||
| Fig. 5 Pictorial representation of HOMO (left) and HOMO (−6) (right) of 2-(PMe3)2. Isosurface value set to 0.03 e A−3. Hydrogen atoms are omitted for clarity. | ||
Table 2 summarizes key metrical bonding parameters within the Al–H–Ni subunits of complexes 1, 2-(L)2, and 3 including the computationally determined bond lengths, Wiberg Bond Indices (WBI), and QTAIM (3, −1) critical points. The bond orders of Ni–Al, Ni–H and Al–H are all relatively close and range between 0.30 and 0.52, the values themselves are consistent with known heterometallic hydride complexes and provide more support for the classification of a 3-center bonding motif within the series of complexes.34 Examining the WBI values across the series of complexes with varying ancillary ligands demonstrate moderate changes and are generally in agreement that the stronger sigma-donating ancillary ligands leads to an increase in the Ni–Al bonding interaction. Furthermore, the decrease in the Ni–Al WBI for complex 3 is consistent with the longer bond length in both the crystallographically determined and the DFT-optimized structures. Studies probing metal–metal interactions with Rh and varying metals have shown similar consistency with an increased WBI value correlated to a shorter crystallographically determined Rh–M bond.67 Despite the diminished Ni–Al bonding interaction in 3, an increase for the Ni–H WBI is observed, and in agreement with a trend correlating slight increases in the Ni–H WBI to increasingly upfield hydride chemical shifts. The computational studies were further buttressed with Quantum Theory of Atoms-In-Molecules (QTAIM) analysis to identify the (3, −1) bond critical points (BCPs) within the Al–H–Ni subunit (Table S3-2†).68,69 Gratifyingly, BCPs with ρ(r) values ranging from 0.06 to 0.09 were observed between Al–H and Ni–H bonds substantiating the 3-center bonding motif.20 Critical points were not identified between Ni and Al centers; consistent with known TaCu-hydride complexes with strong metal–metal interactions.70
| Metric | 1 | 2-(PPh3)2 | 2-(dppm)2 | 2-(PMe3)2 | 3 | |
|---|---|---|---|---|---|---|
| Key bond lengths of DFT optimized structures (Å) | Ni–Al | 2.321, 2.341 | 2.264 | 2.278 | 2.254 | 2.343 |
| Ni–H | 1.627, 1.659 | 1.636 | 1.637 | 1.658 | 1.621 | |
| Al–H | 1.700, 1.707 | 1.713 | 1.730 | 1.727 | 1.734 | |
| Wiberg bond indices | Ni–Al | 0.30, 0.31 | 0.35 | 0.36 | 0.39 | 0.34 |
| Ni–H | 0.47, 0.48 | 0.48 | 0.49 | 0.49 | 0.52 | |
| Al–H | 0.36, 0.39 | 0.37 | 0.37 | 0.40 | 0.37 | |
| ρ(r) (electron density at bond critical points) | Ni–Al | — | — | — | — | — |
| Ni–H | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | |
| Al–H | 0.07 | 0.06 | 0.06 | 0.07 | 0.06 | |
Intriguingly, the difference in NPA charges with Ni and Al suggest strong polarization between the two metal centers (Table S3-2†). The high negative charge at Ni, specifically, is consistent with reported examples describing partial anionic character at the transition metal center,26,71–73 and contrasts the slightly positive values, at Ni, of heterometallic Ni–H–Mg complexes. Crucially, the NPA charge distribution data should be interpreted with caution as NPA charges have been shown to overestimate negative charge in heterometallic systems.74 QTAIM derived charges decrease substantially in comparison to the NPA-derived charges; a finding analogous to reported FeAl and FeCu complexes.74
Early reports studying late metal hydrides and phosphine ancillary ligands outline that altering the electron rich nature of the phosphine centers influenced the hydricity of the moiety and is presented through an upfield chemical shift.75,76 The hydricity amongst the heterometallic series of complexes were evaluated through DFT calculations with solvation in acetonitrile.77 Generally, more electron rich phosphine donors were found to lead to a more hydridic hydride ligand (Table S3-8†); consistent with literature examples of nickel bisphosphine systems.78,79 For example, complex 2-(PPh3)2 features a 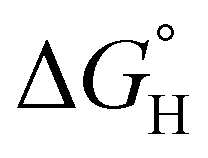 of 59.3 kcal mol−1 which is considered moderate amongst neutral Ni species; however, 2-(PMe3)2 demonstrates a heightened hydricity (
of 59.3 kcal mol−1 which is considered moderate amongst neutral Ni species; however, 2-(PMe3)2 demonstrates a heightened hydricity (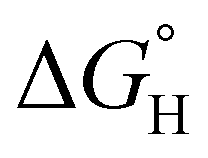 of 54.1 kcal mol−1). Amongst the series of complexes, 3 proved to be the strongest hydride donor by far
of 54.1 kcal mol−1). Amongst the series of complexes, 3 proved to be the strongest hydride donor by far 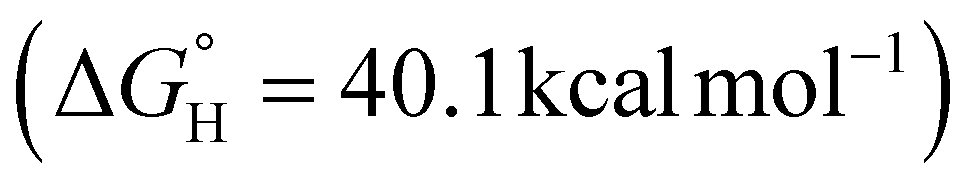 , which the anionic charge of the system is likely accountable for.80 Further studies to correlate hydricity in separate reactivity studies are underway. Lastly, IR spectroscopy was examined to interrogate the impact that the sigma donating ability of the ligands bear on the metal-hydride stretches (Fig. S5-1 to S5-5†). The bridging nature of the hydrides likely result in broadening of the metal-hydride stretches. However, DFT calculations indicate a gradual decrease in the M–H wavenumber for ligands with stronger donating ability among the neutral heterometallics (Table S5-1†).
, which the anionic charge of the system is likely accountable for.80 Further studies to correlate hydricity in separate reactivity studies are underway. Lastly, IR spectroscopy was examined to interrogate the impact that the sigma donating ability of the ligands bear on the metal-hydride stretches (Fig. S5-1 to S5-5†). The bridging nature of the hydrides likely result in broadening of the metal-hydride stretches. However, DFT calculations indicate a gradual decrease in the M–H wavenumber for ligands with stronger donating ability among the neutral heterometallics (Table S5-1†).
Conclusions
With the aim of gaining a thorough understanding of the nature of the hydride moiety in 1, a series of complexes were synthesized and characterized that feature an Al–H–Ni subunit. The series of complexes discussed above demonstrate the relative ease of synthetic access to sigma complexes of alanes to Ni centers, as well as the surprising stability of the resultant Al–H–Ni subunits. The observation of metalloligand dissociation in cases where phosphine ancillary ligands were introduced underscores that full hydride migration to either metal center has, thus far, not been accessible while maintaining the core trimetallic structure in 1. Nevertheless, key insights were garnered through examining the impact of the coordinated ancillary ligands towards the remaining Al–H–Ni subunits in the 2-(L)2 complexes. Namely, the introduction of increasingly strong sigma donating phosphine ligands perturb the electron distribution within the Al–H–Ni subunit of the neutral heterometallic species, which is evidenced by progressively contracted Ni–Al bond distances as well as more upfield chemical shifts for the hydride moieties. Furthermore, the Al–H–Ni subunit proved to persist despite changes in the charge of the molecule as imposed in anionic complex 3. Complexes 1 and 2-(L)2 abide by the principles described in the sigma-bonding continuum (Fig. 1; bottom) relating the shortening of a Ni–Al distance with more Ni–H character. Conversely, complex 3, which according to spectroscopic and computational studies bears the most Ni–H character in the series, forces a deviation from the accepted trend.Computational studies support the 3-center bonding nature of the Al–H–Ni subunit and outline mild changes across the series but a general agreement in the correlation of strength of the sigma-donor leading to an increased Ni–Al interaction. Cumulatively, crystallographic, spectroscopic, and computational studies support that, with respect to the discussed bonding continuum, the hydride moiety shifts slightly towards more Ni–H character and becomes increasingly hydridic with more electron donating ancillary ligands as well as the introduction of negative charge to the framework. Although option C, from Scheme 1, is the dominant pathway observed in this study, ongoing efforts in accessing options A and B are underway. Additional studies are focused on examining the impact of varying ancillary ligands in insertion chemistry and identifying methodology to spectroscopically determine the nature of the hydride moieties in Al–H–Ni systems.
Experimental
General procedures
All air- and water-sensitive compounds were manipulated under N2 using standard Schlenk or glovebox techniques. The solvents for air- and moisture-sensitive reactions were dried over activated 3 Å molecular sieves and the anhydrous nature was confirmed by addition of persistence of ketyl radical upon addition. Deuterated solvents were purchased from Millipore Sigma; C6D6, and C7D8 was dried as mentioned above. Potassium was purchased from Millipore Sigma and used without further purification. 1,3-Diisopropylthiourea, 3-hydroxy-2-butanone, and 1-hexanol, triphenylphosphine, trimethylphosphine, and bis-diphenylphosphinomethane were purchased from Oakwood Chemical and Millipore Sigma and used without further purification once purity was confirmed by NMR spectroscopy. 1,3-Bis(isopropyl)-4,5(dimethyl)imidazol-2-ylidene81 and 140 were synthesized according to the literature procedure. All 1H, 13C, and 31P spectra of organic and organometallic compounds were recorded on a Bruker 400 MHz spectrometer. 1H and 13C chemical shifts are reported relative to residual solvent resonances. 31P{1H} NMR spectra were referenced to external 85% H3PO4 at δ 0.00. IR spectroscopy was carried out on a Thermo Scientific Nicolet iS5 FTIR spectrometer.Synthesis of complex 2-(PPh3)2
Method A: in a N2 filled glovebox, a solution of 1 (20.0 mg, 0.015 mmol) in 0.6 mL of C6D6 in a 20 mL scintillation vial was added four equivalents of triphenyl phosphine (16.3 mg, 0.060 mmol) and an equivalent of Ni(COD)2 (4.1 mg, 0.015 mmol) simultaneously. Over the course of an hour, the solution turned dark orange and multinuclear NMR indicated the quantitative formation of complex 2-(PPh3)2. The volatiles of the reaction mixture were removed and triturated with n-pentane (3 × 1 mL). The crude orange solid was washed with n-pentane (1 mL) and diethyl ether (1 mL) and then extracted with benzene and filtered through a pad of Celite. The bright orange solution was evaporated yielding analytically pure 2-(PPh3)2 (31.8 mg, 0.026 mmol) in 86.7% yield. Method B: to a solution of LAlH (500 mg, 0.78 mmol) in 10 mL of benzene in a 50 mL round bottom flask was added one equivalent of Ni(COD)2 (215 mg, 0.78 mmol) and two equivalents of triphenylphosphine (409 mg, 1.56 mmol) simultaneously resulting in the formation of a dark yellow solution. The solution was stirred for 0.25 hours at which point all volatiles were removed in vacuo. The dark yellow crude mixture was triturated with n-pentane (3 × 5 mL) and then washed with pentane (10 mL) and diethyl ether (10 mL) and then extracted with benzene (12 mL) and filtered through a Celite pad in the filter frit. The benzene solution of 2-(PPh3)2 was evaporated resulting in analytically pure 2-(PPh3)2 (410.0 mg, 0.36 mmol) in 46% yield. Single-crystal X-ray quality crystals were grown by vapor diffusion of n-pentane to a concentrated solution of 2-(PPh3)2 in benzene. 1H NMR (400 MHz, C6D6, 298 K): δ 7.57 (d, 3JHH = 9.4 Hz, 2H, PhH), 7.36 (t, 3JHH = 8.0 Hz, 12H, PhH), 7.24 (broad m, W1/2 = 14.4 Hz, 2H, PhH), 6.94–6.88 (overlapping m, 21H, PhH), 6.69 (broad m, W1/2 = 12.4 Hz, 4H, PhH), 6.49 (apparent t, 3JHH = 8.1 Hz, 2H, PhH), 6.32 (broad multiplet, W1/2 = 12.3 Hz, 2H, PhH), 2.96 (s, 6H, OCH3), 1.15 (s, 18H, C(CH3)3), −1.78 (apparent d, JHP = 53.3 Hz, 1H, Al–H–Ni). 31P{1H} NMR (242.87 MHz, C6D6, 298 K): δ 29.26 (broad s, W1/2 = 40.3 Hz, 2P, PPh3), −12.48 (t, 2JPP = 11.2 Hz, 1P, PPhAr). 31P{1H} NMR (242.87 MHz, C7D8, 328 K): δ 29.01 (d, 2JPP = 9.6 Hz, 2P, PPh3), −12.40 (t, 2JPP = 9.6 Hz, 1P, PPhAr). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ 151.9 (overlapping m, aryl-C), 140.34 (2nd order multiplet, AXX′Y, aryl-C), 137.58 (d, JCP = 5.4 Hz, aryl-C), 137.47 (s, aryl-C), 136.51 (dt, 1JCP = 37.6 Hz, JCP = 3.5 Hz, aryl-C), 133.71 (2nd order multiplet, AXX′Y, aryl-C), 133.09 (d, JCP = 11.1 Hz, aryl-C), 129.54 (broad s, W1/2 = 15.4 Hz, aryl-C), 128.23 (s, aryl-C), 127.28 (overlapping m, aryl-C), 126.95 (d, JCP = 8.4 Hz, aryl-C), 126.13 (s, aryl-C), 125.39 (s, aryl C), 123.57 (broad s, W1/2 = 24.8 Hz, aryl-C), 122.40 (s, aryl-C), 120.69 (broad s, W1/2 = 181.5 Hz, aryl-C), 120.25 (s, aryl-C), 115.20 (s, JCP = 5.8 Hz, aryl-C), 109.46 (s, aryl-C), 55.07 (s, OCH3), 33.85 (s, C(CH3)3), 31.31 (s, C(CH3)3). C76H74AlN2NiO2P3 theoretical: C% 74.45, H% 6.08, N% 2.28. Determined C% 74.48, H% 6.28, N% 2.04.Synthesis of complex 2-(dppm)2
To a solution of LAlH (450.0 mg, 0.70 mmol) in 4 mL in benzene was added bis-diphenylphosphinemethane (478.3 mg, 1.24 mmol) followed by addition of a solution of Ni(COD)2 (171.1 mg, 0.62 mmol) in 4 mL of benzene. Over the course of four hours, the color of the solution changed from yellow to mahogany. All volatiles were removed in vacuo and the crude mixture was triturated with n-pentane (3 × 4 mL) and then washed with pentane (10 mL) and diethyl ether (7 mL) and then extracted with benzene (5 mL). The benzene fraction was evaporated resulting in analytically pure 2-(dppm)2 in 41.4% yield (379.0 mg, 0.26 mmol). Crystals of sufficient quality for single-crystal X-diffraction were grown from benzene/HMDSO slow evaporation. 1H NMR (400 MHz, C6D6, 298 K): δ 7.62 (d, 3JHH = 8.4 Hz, 2H, PhH), 7.53 (d, 3JHH = 10.0 Hz, 2H, PhH), 7.29–6.65 (overlapping m, PhH), 6.45 (t, 3JHH = 8.5 Hz, 2H, PhH), 6.37 (d, 3JHH = 7.8 Hz, 2H, PhH), 3.17 (dd, 2JHP = 41.5 Hz, 2JHP = 14.5 Hz, 4H, PCH2P), 3.09 (s, 6H, OCH3), 1.15 (s, 18H, C(CH3)3), −2.14 (apparent d, JHP = 54.5 Hz, W1/2 = 80.6 Hz, 2H, Al–H–Ni). 31P{1H} NMR (242.87 MHz, C6D6, 298 K): δ 20.75 (broad s, W1/2 = 115.3 Hz, 2P, M-PPh2CH2PPh2), −12.36 (t, 2JPP = 12.0 Hz, 1P, PPhAr), −27.06 (apparent d, 2JPP = 41.0 Hz, 2P, M-PPh2CH2PPh2). 31P{1H} NMR (242.87 MHz, C7D8, 253 K): δ 26.78 (broad m, W1/2 = 395.3 Hz, 1P, M-PPh2CH2PPh2), 15.46 (broad m, W1/2 = 395.3 Hz, 1P, M-PPh2CH2PPh2), −12.50 (t, 2JPP = 11.0 Hz, 1P, PPhAr), −25.92 (broad m, W1/2 = 395.3 Hz, 1P, M-PPh2CH2PPh2), −29.40 (broad m, W1/2 = 395.3 Hz, 1P, M-PPh2CH2PPh2). 31P{1H} NMR (242.87 MHz, C7D8, 333 K): δ 20.98 (2nd order multiplet, AA′XX′Y, 2P, M-PPh2CH2PPh2), −11.46 (t, 2JPP = 11.0 Hz, 1P, PPhAr), −27.06 (2nd order multiplet, AA′XX′Y, 2P, M-PPh2CH2PPh2). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ 152.17 (overlapping broad m, aryl-C), 141.24 (2nd order multiplet, aryl-C), 139.36 (2nd order multiplet, aryl-C), 138.54 (broad m, aryl-C), 137.81 (s, aryl-C), 137.53 (broad m, W1/2 = 13.4 Hz, aryl-C), 137.02 (dt, JCP = 38.2 Hz, JCP = 4.7 Hz, aryl-C), 133.24 (broad d, JCP = 11.7 Hz, aryl-C), 132.82 (dd, JCP = 20.2 Hz, JCP = 7.7 Hz, aryl-C), 132.65 (doublet of m, JCP = 98.2 Hz, aryl-C), 132.47 (d, JCP = 12.7 Hz, aryl-C), 129.58 (broad m, W1/2 = 19.6 Hz, aryl-C), 128.23 (s, aryl-C), 127.35–127.09 (overlapping m, aryl-C), 126.95 (d, JCP = 8.7 Hz, aryl-C), 125.93 (s, aryl-C), 125.41 (s, aryl-C), 123.53 (broad s, W1/2 = 64.4 Hz, aryl-C), 122.50 (s, aryl-C), 120.19 (broad s, W1/2 = 63.5 Hz, aryl-C), 115.30 (d, JCP = 5.6 Hz, aryl-C), 109.39 (s, aryl-C), 55.21 (s, OCH3), 33.31 (s, C(CH3)3), 32.86 (2nd order multiplet AXX′YZ, Ni–Ph2P–CH2–PPh2), 31.30 (s, C(CH3)3). C91H91AlN2NiO2P5 theoretical: C% 73.59, H% 6.18, N% 1.82. Determined C% 73.25, H% 6.27, N% 1.97.Synthesis of complex 2-(PMe3)2
To a solution of LAlH (700.0 mg, 1.088 mmol) in 7 mL in toluene was added a solution of Ni(COD)2 (214 mg, 0.77 mmol) and 1.55 mL of 1.0 M trimethylphosphine solution (1.55 mmol) in 3 mL of toluene, resulting in the formation of a dark brown solution. The solution was stirred for 48 hours at which point all volatiles were removed in vacuo. The dark brown crude mixture was triturated with n-pentane (3 × 5 mL) and washed thoroughly with pentane (15 mL), diethyl ether (10 mL) and then extracted with benzene (10 mL). The benzene fraction was evaporated resulting in the isolation of an analytically pure bright orange powder (291.0 mg, 0.341 mmol) in 44% yield. The powder is then dissolved in a 50:50 mixture of n-pentane and diethyl ether and recrystallized at −40 °C to afford X-ray quality bright yellow single crystals of complex 2-(PMe3)2. 1H NMR (400 MHz, C6D6, 298 K): δ 7.91 (apparent t, 3JHH = 8.9 Hz, 4H, PhH), 7.32(d, 3JHH = 8.0 Hz, 2H, PhH), 7.20 (apparent t, 3JHH = 8.0 Hz, 2H, PhH), 7.02–7.14 (overlapping multiplets, 5H, PhH), 6.77 (apparent t, 3JHH = 7.4 Hz, 2H, PhH), 6.69 (apparent t, 3JHH = 7.4 Hz, 2H, PhH), 6.36 (d, 3JHH = 7.9 Hz, 2H, PhH), 3.07 (s, 6H, OCH3), 1.23 (s, 18H, C(CH3)3), 0.96 (s, 18H, P(CH3)3), −3.28 (apparent d, JHP = 54.5 Hz, W1/2 = 93.6 Hz, 1H, Al–H–Ni). 31P{1H} NMR (242.87 MHz, C6D6, 298 K): δ −9.18 (t, 2JPP = 9.1 Hz, 1P, PPhAr), 21.11 (d, 2JPP = 9.1 Hz, 2P, PMe3). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ 152.41 (d, 2JCP = 14.5 Hz, aryl-C), 152.00 (s, aryl-C), 139.60 (dt, 1JCP = 36.3 Hz, 2JCP = 5.6 Hz, aryl-C), 138.37 (broad s, aryl-C), 137.44 (d, 2JCP = 5.3 Hz, aryl-C), 132.46 (d, 2JCP = 11.6 Hz, aryl-C), 129.08 (s, aryl-C), 126.91 (d, 2JCP = 8.5 Hz, aryl-C), 126.42 (s, aryl-C), 125.43 (s, aryl-C), 123.03 (broad s, aryl-C), 122.55 (broad s, aryl-C), 121.70 (dt, 1JCP = 44.1 Hz, 2JCP = 9.1 Hz, aryl-C), 119.60 (s, aryl-C), 115.69 (d, 2JCP = 6.0 Hz, aryl-C), 110.16 (s, aryl-C), 55.21 (s, OCH3), 33.86 (s, C(CH3)3), 31.38 (s, C(CH3)3), 21.91 (td, 1JCP = 11.1 Hz, 2JCP = 4.5 Hz, P(CH3)3). C46H62AlN2NiO2P3. Theoretical: C% 64.73, H% 7.32, N% 3.28. Determined C% 64.82, H% 7.42, N% 2.97.
Synthesis of complex 3
To a solution of 2-(PMe3)2 (200.0 mg, 0.24 mmol) in 4 mL in benzene was added one equivalent of 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (42.7 mg, 0.24 mmol) as a solution in benzene (4 mL) at room temperature. Over the course of one hour, the solution darkened and a yellow precipitate formed. The suspension was transferred to a pipette filter with Celite and the solid precipitate was washed with diethyl ether (5 mL) and the precipitate was then extracted with 10 mL of tetrahydrofuran. The THF solution was then evaporated to yield 69.8% of analytically pure complex 3 (169.1 mg, 0.16 mmol). X-ray quality crystals were grown via vapor diffusion of hexamethyldisiloxane to a solution of 3 in THF. 1H NMR (400 MHz, C4D8O, 298 K): δ 7.72–7.64 (overlapping m, 3H, PhH), 7.54 (dd, 3JHH = 6.1 Hz, 3JHH = 8.2 Hz, 1H, PhH), 7.36 (apparent t, 3JHH = 6.2 Hz, 1H, PhH), 7.22 (apparent t, 3JHH = 7.2 Hz, 2H, PhH), 7.14 (apparent t, 3JHH = 7.2 Hz, 1H, PhH), 7.05 (d, 3JHH = 7.1 Hz, 1H, PhH), 6.95 (apparent t, 3JHH = 10.7 Hz, 2H, PhH), 6.82 (apparent t, 3JHH = 7.5 Hz, 1H, PhH), 6.77–6.63 (overlapping multiplets, 3H, PhH), 6.43 (d, 3JHH = 7.2 Hz, 2H, PhH), 6.30 (t, 3JHH = 7.3 Hz, 1H, PhH), 6.06 (apparent t, 3JHH = 7.2 Hz, 1H, PhH), 5.99 (apparent t, 3JHH = 7.2 Hz, 1H, PhH), 4.37 (sep, 3JHH = 7.2 Hz, 2H, CH(CH3)2), 3.48 (s, 3H, OCH3), 2.06 (overlapping s, 9H, imidazolium CH3), 1.25 (d, 3JHH = 7.2 Hz, 12H, CH(CH3)2), 1.23 (s, 9H, C(CH3)3), 1.10 (s, 9H, C(CH3)3), 0.85 (d, 3JHH = 4.2 Hz, 9H), 0.72 (d, 3JHH = 4.2 Hz, 9H), −4.07 (apparent d, JHP = 44.8 Hz, W1/2 = 115.2 Hz, 1H, Al–H–Ni). 31P{1H} NMR (242.87 MHz, C4D8O, 298 K): δ −0.65 (dd, 2JPP = 32.5 Hz, 2JPP = 6.4 Hz, 1P, PPhAr), −16.60, (dd, 2JPP = 30.5 Hz, 2JPP = 6.4 Hz, 1P, P(CH3)3), −25.45 (dd, 2JPP = 30.5 Hz, 2JPP = 32.5 Hz, 1P, P(CH3)3). 13C{1H} NMR (101 MHz, C4D8O, 298 K): δ 158.06 (s, aryl-C), 157.90 (d, JCP = 17.5 Hz, aryl-C), 156.07 (s, aryl-C), 150.53 (d, JCP = 12.0 Hz, aryl-C), 143.21 (ddd, JCP = 26.4 Hz, JCP = 10.9 Hz, JCP = 1.8 Hz, aryl-C), 142.07 (s, aryl-C), 141.76 (s, aryl-C), 139.13 (s, aryl-C), 134.74 (d, JCP = 5.5 Hz, aryl-C), 132.80 (d, JCP = 12.4 Hz, aryl-C), 132.36 (d, JCP = 4.6 Hz, aryl-C), 132.18 (s, aryl-C), 130.33 (s, ary-C), 127.48 (ddd, JCP = 42.9 Hz, JCP = 13.3 Hz, JCP = 8.5 Hz, aryl-C), 125.95 (d, JCP = 8.8 Hz, aryl-C), 125.66 (s, aryl-C), 125.05 (s, aryl-C), 124.66 (s, aryl-C), 124.24 (s, aryl-C), 121.99 (s, aryl-C), 120.95 (s, aryl-C), 119.92 (s, aryl-C), 116.45 (dd, JCP = 19.0 Hz, JCP = 17.5 Hz, aryl-C), 116.16 (d, JCP = 6.3 Hz, aryl-C), 116.08 (s, aryl-C), 113.66 (s, aryl-C), 113.41 (d, JCP = 5.6 Hz, aryl-C), 113.24 (s, aryl-C), 112.41 (s, aryl-C), 111.16 (s, aryl-C), 54.44 (s, CH(CH3)2), 50.38 (s, OCH3), 33.69 (s, C(CH3)3), 33.35 (s, C(CH3)3), 31.37 (s, C(CH3)3), 31.11 (s, C(CH3)3), 22.50 (ddd, JCP = 12.6 Hz, JCP = 7.4 Hz, JCP = 5.2 Hz, P(CH3)3), 21.69 (ddd, JCP = 13.2 Hz, JCP = 7.8 Hz, JCP = 5.5 Hz, P(CH3)3), 20.27 (s, CH(CH3)2), 10.24 (s, iPrNCCH3), 8.84 (s, NCCH3CCH3N). C57H82AlN4NiO2P3 theoretical: C% 66.22, H% 7.99, N% 5.42. Determined C% 66.20, H% 7.99, N% 5.23.Computational details
The initial starting point geometries were adapted from the corresponding crystallographically obtained structures and optimized to a stationary point, followed by analytical frequency calculations to confirm that no imaginary frequencies were present. Geometry optimization was performed for all the complexes using the BP86 density functional.82,83 A mixed basis set scheme was employed, in which H, C, N, O and P were described by the def2-SVP basis set84 while Al and Ni were described by the larger def2-TZVP basis set.85 Vibrational analysis was then performed on the same level to verify that all the optimized structures are true stationary points with no imaginary frequencies. Gibbs free energies obtained from vibrational analysis were used in the calculation of hydricity, which is defined as the reaction free energy of a compound losing a H-anion. Natural Bond Orbital (NBO) analysis86 was carried out to calculate the Natural Population Analysis (NPA) atomic charges87 and Wiberg bond indices (WBIs).88 All calculations were performed using Gaussian 16, Rev. C01.89 The wavefunction data of the optimized complexes was then used to perform the Quantum Theory of Atoms-In-Molecules (QTAIM) analysis to obtain the bond critical points (BCPs, or (3, −1) CPs) and AIM charges using the Multiwfn68 and AIMALL90 packages. The Cartesian coordinates of all DFT-optimized structures are provided separately as part of the ESI.†Author contributions
M. M. S. and H. C. designed the project, supervised the research, and authored the manuscript. A. G. G., F. G., E. D., and M. M. S. conducted all synthetic experimentation. A. G. G. and H. C. conducted computational experimentation. K. B. and S. Y. carried out crystallographic experimentation and structure solutions.Data availability
The data supporting this article have been included as part of the ESI.† Complete details of the X-ray structures can be obtained from the Cambridge Crystallographic Data Centre at https://www.ccdc.cam.ac.uk. The CCDC accession numbers for the reported complexes are 2362005, 2362006, 2362008, and 2362009.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
A. G. G. acknowledges the Dean's Graduate Fellowship for funding. F. G. acknowledges the Engaged Scholars and Artist Award for funding. M. M. S. acknowledges NSF-CHE-2316582 for financial support of this work. The authors acknowledge the Texas Advanced Computing Center (TACC) under award no. CHE23024 and CHE22010. K. M. B. and S. Y. acknowledge the Cancer Prevention and Research Institute of Texas (Grant No. RR220055) for financial support of this work. Hadi D. Arman is acknowledged for crystallographic assistance. We thank Sergio Gonzalez-Eymard for assistance with IR spectroscopy.References
- Q. Wang, S. H. Brooks, T. Liu and N. C. Tomson, Chem. Commun., 2021, 57, 2839–2853 RSC.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS.
- M. Navarro, J. J. Moreno, M. Pérez-Jiménez and J. Campos, Chem. Commun., 2022, 58, 11220–11235 RSC.
- J. A. Zurakowski, B. J. Austen and M. W. Drover, Trends Chem., 2022, 4, 331–346 CrossRef CAS.
- M. M. Shoshani, Cell Rep. Phys. Sci., 2023, 4(4), 101213 CrossRef CAS.
- C. Yoo and Y. Lee, Chem. Sci., 2017, 8, 600–605 RSC.
- R. C. Cammarota, M. V. Vollmer, J. Xie, J. Ye, J. C. Linehan, S. A. Burgess, A. M. Appel, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 14244–14250 CrossRef CAS PubMed.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed.
- B. L. Ramirez, P. Sharma, R. J. Eisenhart, L. Gagliardi and C. C. Lu, Chem. Sci., 2019, 10, 3375–3384 RSC.
- R. M. Charles III, T. W. Yokley, N. D. Schley, N. J. DeYonker and T. P. Brewster, Inorg. Chem., 2019, 58, 12635–12645 CrossRef.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1–40 CrossRef CAS.
- B. Wang, C. S. Seo, C. Zhang, J. Chu and N. K. Szymczak, J. Am. Chem. Soc., 2022, 144, 15793–15802 CrossRef CAS PubMed.
- W.-C. Shih and O. V. Ozerov, J. Am. Chem. Soc., 2017, 139, 17297–17300 CrossRef CAS PubMed.
- J. A. Zurakowski, B. J. Austen, M. C. Dufour, M. Bhattacharyya, D. M. Spasyuk and M. W. Drover, Dalton Trans., 2021, 50, 12440–12447 RSC.
- B. E. Cowie and D. J. Emslie, Organometallics, 2018, 37, 1007–1016 CrossRef CAS.
- L. A. Grose and D. Willcox, Chem. Commun., 2023, 59, 7427–7430 RSC.
- J. R. Norton and J. Sowa, Chem. Rev., 2016, 116, 8315–8317 CrossRef PubMed.
- T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed.
- Y.-X. Luan and M. Ye, Chem. Commun., 2022, 58, 12260–12273 RSC.
- M. Garçon, A. Phanopoulos, A. J. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2023, 62, e202213001 CrossRef.
- Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. García-Serres and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 1499–1503 CrossRef CAS.
- Q. Zhao and T. A. Betley, Angew. Chem., Int. Ed., 2011, 50, 709–712 CrossRef CAS.
- C. J. Reed and T. Agapie, J. Am. Chem. Soc., 2019, 141, 9479–9484 CrossRef CAS.
- M. M. Shoshani, J. Liu and S. A. Johnson, Organometallics, 2018, 37, 116–126 CrossRef CAS.
- S. Morisako, S. Watanabe, S. Ikemoto, S. Muratsugu, M. Tada and M. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 15031–15035 CrossRef CAS PubMed.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS.
- O. Ekkert, A. J. White, H. Toms and M. R. Crimmin, Chem. Sci., 2015, 6, 5617–5622 RSC.
- T. Steinke, C. Gemel, M. Cokoja, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2299–2302 CrossRef CAS PubMed.
- N. Gorgas, J. Brünig, B. Stöger, S. Vanicek, M. Tilset, L. F. Veiros and K. Kirchner, J. Am. Chem. Soc., 2019, 141, 17452–17458 CrossRef CAS.
- Y. Cai, S. Jiang, T. Rajeshkumar, L. Maron and X. Xu, J. Am. Chem. Soc., 2022, 144, 16647–16655 CrossRef CAS PubMed.
- Y. Cai, S. Jiang and X. Xu, Chin. J. Chem., 2024, 42(18), 2133–2139 CrossRef CAS.
- N. Gorgas, A. J. White and M. R. Crimmin, J. Am. Chem. Soc., 2022, 144, 8770–8777 CrossRef CAS.
- N. Gorgas, B. Stadler, A. J. White and M. R. Crimmin, J. Am. Chem. Soc., 2024, 146(6), 4252–4259 CrossRef CAS PubMed.
- M. Garçon, A. Phanopoulos, G. A. Sackman, C. Richardson, A. J. White, R. I. Cooper, A. J. Edwards and M. R. Crimmin, Angew. Chem., Int. Ed., 2022, 61, e202211948 CrossRef.
- T. Steinke, C. Gemel, M. Cokoja, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2299–2302 CrossRef CAS.
- K. R. Pörschke, W. Kleimann, Y. H. Tsay, C. Krüger and G. Wilke, Chem. Ber., 1990, 123, 1267–1273 CrossRef.
- B. Pribanic, M. Trincado, F. Eiler, M. Vogt, A. Comas-Vives and H. Grützmacher, Angew. Chem., 2020, 132, 15733–15739 CrossRef.
- V. T. Nguyen, Q. Lai, N. Witayapaisitsan, N. Bhuvanesh, P. Surawatanawong and O. V. Ozerov, Organometallics, 2023, 42, 3120–3129 CrossRef CAS PubMed.
- B. J. Graziano, M. V. Vollmer and C. C. Lu, Angew. Chem., 2021, 133, 15214–15221 CrossRef.
- E. De Leon, F. Gonzalez, P. Bauskar, S. Gonzalez-Eymard, D. De Los Santos and M. M. Shoshani, Organometallics, 2023, 42, 435–440 CrossRef CAS.
- N. Hara, K. Semba and Y. Nakao, ACS Catal., 2022, 12, 1626–1638 CrossRef CAS.
- M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348–1365 RSC.
- I. M. Riddlestone, J. A. Abdalla and S. Aldridge, Adv. Organomet. Chem, Elsevier, 2015, vol. 63, pp. 1–38 Search PubMed.
- I. M. Riddlestone, S. Edmonds, P. A. Kaufman, J. Urbano, J. I. Bates, M. J. Kelly, A. L. Thompson, R. Taylor and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 2551–2554 CrossRef CAS.
- O. Ekkert, A. J. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2016, 55, 16031–16034 CrossRef CAS.
- F. Gonzalez, E. De Leon and M. M. Shoshani, Synlett, 2024, 35, A–E Search PubMed.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- G. Parkin, Dalton Trans., 2022, 51, 411–427 RSC.
- A. F. Hill, Organometallics, 2006, 25, 4741–4743 CrossRef CAS.
- R. J. Puddephatt, Chem. Soc. Rev., 1983, 12, 99–127 RSC.
- N. H. Hunter, E. M. Lane, K. M. Gramigna, C. E. Moore and C. M. Thomas, Organometallics, 2021, 40, 3689–3696 CrossRef CAS.
- T. Steinke, C. Gemel, M. Cokoja, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2299–2302 CrossRef CAS PubMed.
- L. M. Toomey and J. D. Atwood, Organometallics, 1997, 16, 490–493 CrossRef CAS.
- M. G. Klimpel, R. Anwander, M. Tafipolsky and W. Scherer, Organometallics, 2001, 20, 3983–3992 CrossRef CAS.
- A. M. Borys, L. A. Malaspina, S. Grabowsky and E. Hevia, Angew. Chem., 2022, 134, e202209797 CrossRef.
- A. M. Borys and E. Hevia, Chimia, 2023, 77, 242–242 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- Y. Ruiz-Morales, G. Schreckenbach and T. Ziegler, Organometallics, 1996, 15, 3920–3923 CrossRef CAS.
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373–8426 CrossRef CAS PubMed.
- W. Liu, Y. Ding, D. Jin, Q. Shen, B. Yan, X. Ma and Z. Yang, Green Chem., 2019, 21, 3812–3815 RSC.
- C.-C. Chia, Y.-C. Teo, N. Cham, S. Y.-F. Ho, Z.-H. Ng, H.-M. Toh, N. Mézailles and C.-W. So, Inorg. Chem., 2021, 60, 4569–4577 CrossRef CAS PubMed.
- M. Bhandari, M. Kaur, S. Rawat and S. Singh, Inorg. Chem., 2023, 62, 6598–6607 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- G. Ciancaleoni, N. Scafuri, G. Bistoni, A. Macchioni, F. Tarantelli, D. Zuccaccia and L. Belpassi, Inorg. Chem., 2014, 53, 9907–9916 CrossRef CAS PubMed.
- G. J. Kubas, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6901–6907 CrossRef CAS.
- L. J. Murphy, H. Hollenhorst, R. McDonald, M. Ferguson, M. D. Lumsden and L. Turculet, Organometallics, 2017, 36, 3709–3720 CrossRef CAS.
- S. Bajo, M. G. Alférez, M. M. Alcaide, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 16833–16845 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- M. L. Maiola and J. A. Buss, Angew. Chem., Int. Ed., 2023, 62, e202311721 CrossRef CAS PubMed.
- J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed.
- O. Ekkert, A. J. P. White, H. Toms and M. R. Crimmin, Chem. Sci., 2015, 6, 5617–5622 RSC.
- S. Banerjee, M. K. Karunananda, S. Bagherzadeh, U. Jayarathne, S. R. Parmelee, G. W. Waldhart and N. P. Mankad, Inorg. Chem., 2014, 53, 11307–11315 CrossRef CAS.
- S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210–3221 CrossRef CAS PubMed.
- F. Cotton, R. Luck, D. Root and R. Walton, Inorg. Chem., 1990, 29, 43–47 CrossRef CAS.
- J. Pursiainen and T. A. Pakkanen, Acta Chem. Scand., 1989, 43, 463–470 CrossRef CAS.
- X.-J. Qi, Y. Fu, L. Liu and Q.-X. Guo, Organometallics, 2007, 26, 4197–4203 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS.
- M. R. Nimlos, C. H. Chang, C. J. Curtis, A. Miedaner, H. M. Pilath and D. L. DuBois, Organometallics, 2008, 27, 2715–2722 CrossRef CAS.
- J. R. Prat, R. C. Cammarota, B. J. Graziano, J. T. Moore and C. C. Lu, Chem. Commun., 2022, 58, 8798–8801 RSC.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, Gaussian Inc., Pittsburgh, 2003.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016.
- T. Keith, https://aim.tkgristmill.com.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2362005, 2362006, 2362008 and 2362009. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01786b |
| This journal is © The Royal Society of Chemistry 2024 |