
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability of metal–metal interactions in transmetallation intermediates based on electronics of bridging arene ligands determined through pyridine titrations†
Rana
Marghalani
 and
Eric S.
Cueny
*
and
Eric S.
Cueny
*
Department of Chemistry, Boston University, 590 Commonwealth Ave., Boston, MA 02215, USA. E-mail: ecueny@bu.edu
First published on 27th July 2024
Abstract
In this contribution, we prepare the dinuclear complex [(CNCF)(PPh3)Pt–Au(PPh3)]+ (2-F) supported by an electron deficient derivative of 2,6-diphenylpyridine (CNC), 2,6-di(4-fluorophenyl)pyridine (CNCF). Solution state spectroscopic data and solid-state structural data reveals formation of the desired dinuclear complex occurs and that it remains intact in solution. The solid state structure of 2-F, compared to [(CNC)(PPh3)Pt–Au(PPh3)]+ (2), reveals a substantial change in the C–Au–P bond angle. We postulated that this change in bond angle arises due to a weaker interaction between [(PPh3)Au]+ and (CNCF)Pt(PPh3) (1-F) vs. (CNC)Pt(PPh3) (1). Through pyridine titration experiments, we demonstrate that the interaction is indeed weaker between [(PPh3)Au]+ and 1-Fvs. 1. Cyclic voltammetry (CV) experiments confirm that 1-F is less electron rich than 1. DFT calculations demonstrate that the HOMO of 1 and 1-F is not dz2, helping explain the differences in electrochemical behavior of 1 and 1-F and bonding between 1 and 1-F with [(PPh3)Au]+.
Transmetallation, the exchange of ligands between two metal centers, is a critical step in both catalysis and organometallic synthesis.1 The role of transmetallation in catalytic cross coupling has made understanding the mechanism of transmetallation particularly important.2 Mechanistic studies of transmetallation have focused on reactions between Sn- and B-based reagents with Pd- and Pt-complexes because of their relevance to the Stille and Suzuki–Miyaura coupling reactions respectively.3,4 Hartwig and coworkers found an open coordination site is necessary for efficient transmetallation using certain Pd-complexes and Sn-reagents.5 Transmetallation between Pd-complexes and boronic acids are accelerated with a Pd–OH vs. Pd–halide.3,6
By contrast, comparatively little is understood about the mechanism and thermodynamic driving force for transmetallation between two transition metal complexes,7 though it is known in the Sonogashira reaction8 and has been proposed in other cooperative catalytic reactions.9–13 Stahl and coworkers convincingly demonstrate transmetallation of an aryl group between two palladium species upon C–H activation via kinetic isotope effect (KIE) studies.14 Other research groups have explored stoichiometric transmetallation between two organometallic complexes.15–20 However, one of the challenges in developing a deeper understanding of transmetallation is the stability of the proposed intermediate along the pathway of transmetallation. Often, the proposed intermediate in transmetallation has a low kinetic barrier to formation of the final exchange product making it challenging to detect and directly probe its structure.20 Despite this challenge, several research groups have examined dinuclear complexes that may play a role in understanding transmetallation reactions.21–36
Martin and coworkers used the CNC ligand (CNC = 2,6-diphenylpyridine) bound to Pt (1) to study transmetallation intermediates.37 One such intermediate is generated by reaction of 1 with [(PPh3)Au]+ to yield 2 where the Au binds to the Pt and the ipso carbon of the phenyl ring. While metallophilic interactions between Pt and Au likely play a role in the stability of this complex, the simultaneous binding to the Pt–phenyl ring suggests that the electronics of the arene ring also play a role in stabilizing such a dinuclear complex.
We chose to study a fluorinated derivative of the CNC ligand (CNCF) to establish the role of electron withdrawing groups in the stability of dinuclear Pt/Au complexes. For efficient cooperative catalysis with transmetallation as the key step, a delicate interplay of kinetic barriers between and thermodynamic stabilities of reaction intermediates is at work (Fig. 1). If the intermediate of transmetallation (e.g.2 or 2-F) is highly stable, this will decrease the concentration of active catalyst in a cooperative catalytic reaction. An analogy can be drawn to the use of chain transfer reagents in alkene polymerization where highly stable bimetallic complexes between the polymerization catalyst and chain transfer reagent inhibit propagation as intermediate formation decreases the concentration of the propagating catalyst species.38–44 Thus, understanding the strength of such dinuclear complexes as a function of metal identity (as Martin and coworkers have previously described)31,37,45,46 as well as electronics of the bridging arene ligand (reported here) are vital to the development of cooperative catalytic reactions.
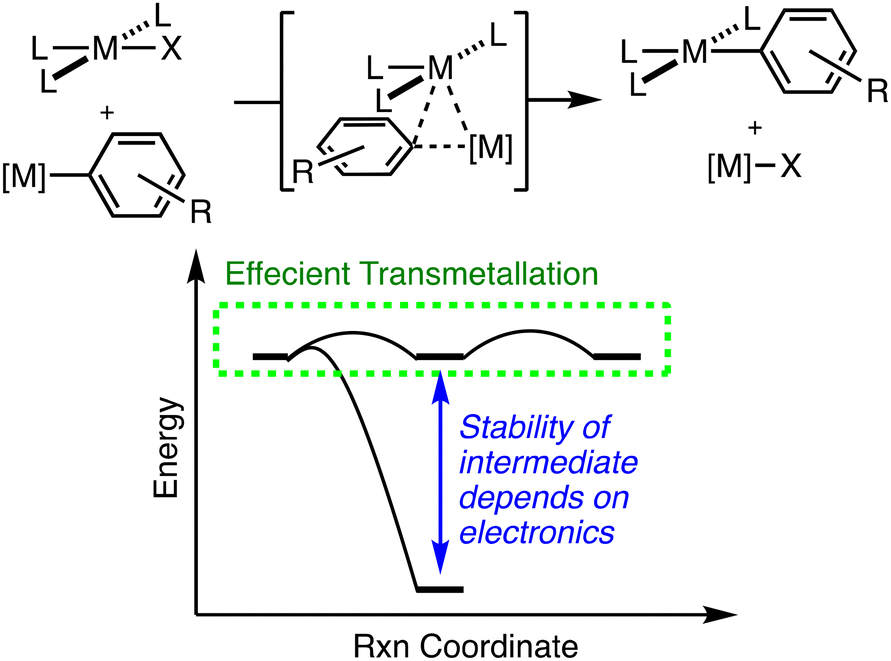 | ||
| Fig. 1 (Top) Transmetallation of an arene group between two organometallic complexes. (Bottom) Hypothetic free energy surface for transmetallation. The stability of the transmetallation intermediate is dependent on the electronics of the arene ring. | ||
Herein, we describe the synthesis of 2-F and compare its structure and stability to the known complex 2. First, we describe the differences between the solid-state structure of 2 and 2-F. Then, we use pyridine titration experiments to probe the thermodynamic stability of these two complexes. We also use cyclic voltammetry (CV) to determine the anodic peak potential of both (CNCR)Pt(PPh3), when R = H and F. We correlate the oxidation potential determined by CV experiments to the thermodynamic stability of the dinuclear [(CNCR)(PPh3)Pt–Au(PPh3)]+ complexes.
First, we set out to synthesize 2-F by reaction of the known 1-F complex with the in situ generated [(PPh3)Au]+ (Scheme 1).47 We observed two peaks in the 31P NMR spectrum of this new complex at 33.8 ppm with Pt satellites (2JP–Pt = 267 Hz) and 21.5 ppm with Pt satellites (1JP–Pt = 3568 Hz) corresponding to the PPh3 ligands bound to Au and Pt respectively. Similar to complex 2, the 2JP–Pt of the PPh3 ligated to Au supports that a dinuclear complex remains intact in solution as a two bond coupling between Pt and the P of PPh3 bound to Au is observed. The 1H NMR spectrum of 2-F reveals an expected upfield shift of the ortho C–H (relative to Pt) of 0.18 ppm upon coordination of the cationic Au species. A similar shift upfield was observed for 2 of 0.21 ppm upon coordination of [(PPh3)Au]+.37
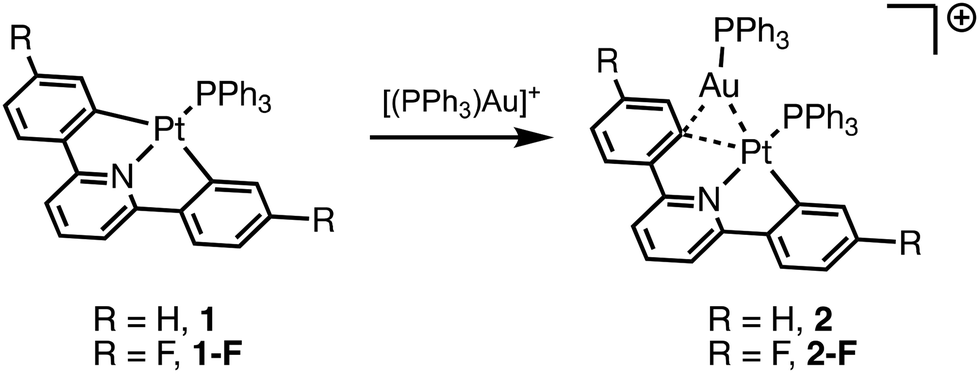 | ||
| Scheme 1 Synthesis of dinuclear complexes 2 and 2-F. | ||
The solid-state structure of 2-F (Fig. 2), obtained by single crystal X-ray diffraction, revealed differences between 1-F and 1 upon binding the [(PPh3)Au]+.37 Most prominently, the bond angles between the P–Au–C for the two complexes are substantially different; for 2-F, P–Au–C = 173.5°, whereas for 2, P–Au–C = 153.2°. Interestingly, despite this change in the P–Au–C bond angle, the degree of arene group transfer (as indicated by the Cpara–Cipso–Au angle) does not change substantially between 2 and 2-F. For 2-F, Cpara–Cipso–Au = 120.9° whereas for 2 Cpara–Cipso–Au = 120.1°. If the arene fully transferred to Au, the Cpara–Cipso–Au would reach ∼180°, with no arene Au interaction Cpara–Cipso–Au would be ∼90°.46 Other bond metrics change minimally between the proteo and fluoro substituted arenes including the Pt–Au distances, 2.7430(5) Å for 2-F and 2.7222(2) Å for 2. The Pd derivative of 2 (2-Pd) has been previously characterized by single crystal X-ray diffraction.46
 | ||
| Fig. 2 (Top) Molecular structure of the cation portion of complex 2-F. Hydrogens, DCM solvent, and triflate anion omitted for clarity. (Bottom) Molecular structure of the cation portion of complex 2.37 Hydrogens and perchlorate anion omitted for clarity. For both complexes, thermal ellipsoids are shown at the 50% probability level. | ||
Interestingly, 2-Pd shows some structural features related to both 2 and 2-F; for example, Pd–Au = 2.7422(3) Å, similar to the Pt–Au distances in 2 and 2-F. In 2-Pd, the P–Au–C = 173.8°, which is quite similar to that of 2-F (P–Au–C = 173.5°). However, the degree of arene transfer in 2-Pd is much higher than 2 and 2-F as indicated by Cpara–Cipso–Au = 143.1° vs. 120.1° and 120.9° for 2 and 2-F respectively. The higher degree of arene transfer in 2-Pd is reflective of generally stronger Pt–C bonds relative to Pd–C bonds. Clearly, there are multiple structural factors to consider when trying to understand the bonding interactions between such dinuclear, frustrated transmetallation complexes.
Previously, Martin and coworkers have used Energy Decomposition Analysis (EDA) to understand the strength of the interactions in dinuclear complexes such as 2, 2-Pd, and other related complexes.37,45 Herein, we chose to use experimental data to qualitatively determine the relative stabilities of 2vs. 2-F. We postulated that the C–Au–P bond angle difference between 2 and 2-F (with similar degrees of arene group transfer) may arise from an overall decrease in stability of the interaction between the [(PPh3)Au]+ and 1-F relative to 1. We chose to evaluate the thermodynamic strength of these interactions using pyridine titrations. We dissolved [(CNCR)(PPh3)Pt–Au(PPh3)]+ in dcm–d2 and added various equivalents of pyridine or 2-fluoropyridine. In these titration experiments, an equilibrium may be established between the dinuclear complex plus pyridine and the monometallic Pt complex and a pyridine ligated [(PPh3)Au]+ (Fig. 3).
 | ||
| Fig. 3 (Top) Titrations of 2 (R = H) and 2-F (R = F) with pyridine (R1 = H) and 2-fluoropyridine (R1 = F). Titrations were monitored by 1H NMR spectroscopy (see ESI† for details). We used the peak associated with the proton alpha to Pt in 2 & 2-F and monitored the change in ppm based on the equivalents of pyridine or 2-fluoropyridine added. We normalized the ppm change upon pyridine (or 2-fluoropyridine addition) plotting the normalized Δppm on the y-axis. Here, the value of zero on the y-axis corresponds to no change in the ppm shift (i.e. the chemical shift of the starting 2 or 2-F). The value of one on the y-axis corresponds to complete dissociation of [(PPh3)Au]+ from the Pt (i.e. the chemical shift of 1 or 1-F). (Bottom left) Titration of 2 or 2-F with pyridine. (Bottom right) Titration of 2 or 2-F with 2-fluoropyridine. | ||
Based upon a qualitative comparison of these two complexes, it is clear that [(PPh3)Au]+ binds less strongly to the more electron deficient 1-F than 1. While one equivalent of pyridine completely displaces [(PPh3)Au]+ from 2-F, nearly 10 equivalents of pyridine are required to completely displace [(PPh3)Au]+ from 2 based upon 1H NMR spectroscopic analysis. Using 2-fluoropyridine as the titrant, 2 is only minimally affected; however, an equilibrium is established where [(PPh3)Au]+ is partially displaced from 2-F by 2-fluoropyridine. It is worth noting that 2-fluoropyridine is a weaker base than pyridine by several orders of magnitude revealing how weak of an interaction exists between [(PPh3)Au]+ and 1-F. Importantly, control reactions (see ESI† for details) demonstrate that pyridine and 2-fluoropyridine do not react with 1 or 1-F and appear to bind readily to [(PPh3)Au]+. Based on the ppm shift change upon addition of pyridine and 2-fluoropyridine, we can calculate the Keq for these titration experiments (see ESI† for details).48,49 The affinity of pyridine and 2-fluoropyridine for the [(PPh3)Au]+ fragment of 2-F is higher than for 2 by two orders of magnitude (Fig. 3).
Both titration results were initially surprising. We anticipated that pyridine would more easily displace [(PPh3)Au]+ from both 2 and 2-F. The stability of 2, however, suggests that the Pt–C bond of 1 serves as a reasonably good ligand for the cationic gold species as it competes with pyridine for ligation with [(PPh3)Au]+. While 2 has been studied computationally and through variable temperature NMR spectroscopy,37 a more direct experimental measurement of the stability of 2 has not been reported previously. Based on the stability of 2, it was surprising that 2-F was readily displaced by pyridine and equilibrium established with 2-fluoropyridine (a substantially weaker base than pyridine) as the only difference between these complexes is the meta (relative to Pt) fluoro groups of 2-F.
Based on the electron withdrawing nature of fluorine, we expect that the Pt–C bond of 1-F is likely less electron rich than 1, leading to a dinculear complex 2-F with lower overall stability than 2. Using CV, we established that 1-F is less electron rich than 1 as judged by the relative oxidation potentials of these two complexes. The onset of oxidation of 1-F occurs ∼200 mV more anodic relative to 1. However, neither 1 nor 1-F exhibit fully reversible electrochemical behavior as determined by scan rate dependent CV studies precluding a more detailed analysis. These electrochemical results support our hypothesis that the electron deficient nature of the CNCF ligand plays a substantial role in the decreased stability of the dinuclear complex 2-Fvs. 2.
Given that both 1 and 1-F are d8 complexes and nearly square planar in geometry, one might expect that the HOMO is the dz2 orbital. The dz2 should be minimally impacted by the electronics of the arene rings. The results from CV studies suggest, however, that the HOMO is impacted by the electronics of the arene ring. The expectation that the dz2 orbital is the HOMO arises from a sigma only view of bonding in transition metal complexes, whereas the extended π structures of CNC and CNCF may alter the HOMO of complexes 1 and 1-F. DFT calculations (see ESI† for details) performed at various levels of theory (B3LYP, PBE, PBE0, and M06, see ESI† for details) reveal that the HOMO is a dπ* orbital for both 1 and 1-F with significant contributions from the ligand π orbitals, which likely explains why the oxidation potential of 1 and 1-F are different by ∼200 mV.
Conclusions
In this manuscript, we have synthesized the new dinuclear complex 2-F and compared it to the previously studied 2. Solution state NMR spectroscopic data support that the Pt–Au interaction remains intact for 2-F similar to the previously studied 2. However, the interaction between Au and Pt in 2-F is much weaker than in 2 (based on pyridine titration experiments) while structural data suggests a similar degree of arene group transfer between Pt and Au in both complexes. CV experiments reveal that 1 is easier to oxidize than 1-F, which helps explain the increased stability of 2 relative to 2-F. DFT results reveal that the HOMO is the dπ* orbital of 1 and 1-F. These computations help explain both the difference in oxidation potential between 1 and 1-F and the stability of the interaction between [(PPh3)Au]+ and 1vs. 1-F.Complex 2-F and its comparison to 2, help shed light on the proposed intermediates involved in transmetallation. Martin and coworkers have established that the metal identity in these dinuclear complexes plays a major role in their structure and stability. Here, we show that the electronics of the arene ring involved in this arrested transmetallation state impacts the stability of the dinuclear complex but not the degree of arene group transfer. As the development of cooperative catalytic reactions continues where transmetallation is the key step, it is important to not only consider the metals involved in transmetallation but also the steric and electronic parameters of the ligands that undergo exchange during transmetallation.
Author contributions
R.M. synthesized and characterized 2-F, collected all titration data, collected CV data, and assisted in manuscript writing. E.S.C. performed DFT calculations and assisted in manuscript writing and editing.Data availability
The NMR spectroscopic data supporting the research in this article can be found in the ESI.† The CV chromatograms supporting the research in this article can also be found in the ESI.† Geometric coordinates used in the DFT calculations supporting this research are included with this submission. Crystallographic data for compound 2-F has been deposited at the CCDC under 2364269 and can be obtained from https://www.ccdc.cam.ac.uk/structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Boston University for funding of this project. We acknowledge the NIH S10OD028585 for the purchase of the X-ray diffractometer. We thank Dr Paul Ralifo for help with NMR spectroscopic data collection, Dr Jeffery Bacon for help with X-ray data collection, Dr James McNeely for help with DFT calculations, and Dr Maria del Carmen Piqueras for help with mass spectrometry.References
- D. V. Partyka, Transmetalation of Unsaturated Carbon Nucleophiles from Boron-Containing Species to the Mid to Late d-Block Metals of Relevance to Catalytic C–X Coupling Reactions (X = C, F, N, O, Pb, S, Se, Te), Chem. Rev., 2011, 111, 1529–1595 CrossRef CAS PubMed.
- K. Osakada and T. Yamamoto, Transmetallation of alkynyl and aryl complexes of Group 10 transition metals, Coord. Chem. Rev., 2000, 198, 379–399 CrossRef CAS.
- K. Osakada and Y. Nishihara, Transmetalation of boronic acids and their derivatives: mechanistic elucidation and relevance to catalysis, Dalton Trans., 2022, 51, 777–796 RSC.
- P. Nilsson, G. Puxty and O. F. Wendt, Reaction Mechanism of Transmetalation between Tetraorganostannanes and Platinum(II) Aryltriflate Complexes. Mechanistic Model for Stille Couplings, Organometallics, 2006, 25, 1285–1292 CrossRef CAS.
- J. Louie and J. F. Hartwig, Transmetalation, Involving Organotin Aryl, Thiolate, and Amide Compounds. An Unusual Type of Dissociative Ligand Substitution Reaction, J. Am. Chem. Soc., 1995, 117, 11598–11599 CrossRef CAS.
- B. P. Carrow and J. F. Hartwig, Distinguishing between pathways for transmetalation in Suzuki-Miyaura reactions, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed.
- M. H. Perez-Temprano, J. A. Casares and P. Espinet, Bimetallic catalysis using transition and Group 11 metals: an emerging tool for C-C coupling and other reactions, Chem. – Eur. J., 2012, 18, 1864–1884 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- J. J. Hirner, Y. Shi and S. A. Blum, Organogold Reactivity with Palladium, Nickel, and Rhodium: Transmetalation, Cross-Coupling, and Dual Catalysis, Acc. Chem. Res., 2011, 44, 603–613 CrossRef CAS PubMed.
- M. M. Hansmann, M. Pernpointner, R. Dopp and A. S. Hashmi, A theoretical DFT-based and experimental study of the transmetalation step in Au/Pd-mediated cross-coupling reactions, Chem. – Eur. J., 2013, 19, 15290–15303 CrossRef CAS PubMed.
- J. Wang, L. Zhan, G. Wang, Y. Wei, M. Shi and J. Zhang, Pd-Promoted cross coupling of iodobenzene with vinylgold via an unprecedented phenyl transmetalation from Pd to Au, Chem. Commun., 2020, 56, 6213–6216 RSC.
- M. Oi, R. Takita, J. Kanazawa, A. Muranaka, C. Wang and M. Uchiyama, Organocopper cross-coupling reaction for C-C bond formation on highly sterically hindered structures, Chem. Sci., 2019, 10, 6107–6112 RSC.
- A. S. Gundogan, X. Meng, R. W. Winkel and K. S. Schanze, Platinum carbon bond formation via Cu(I) catalyzed Stille-type transmetallation: reaction scope and spectroscopic study of platinum-arylene complexes, Dalton Trans., 2015, 44, 17932–17938 RSC.
- D. Wang, Y. Izawa and S. S. Stahl, Pd-catalyzed aerobic oxidative coupling of arenes: evidence for transmetalation between two Pd(II)-aryl intermediates, J. Am. Chem. Soc., 2014, 136, 9914–9917 CrossRef CAS PubMed.
- A. L. Casado and P. Espinet, A Novel Reversible Aryl Exchange Involving Two Organometallics: Mechanism of the Gold(I)-Catalyzed Isomerization of trans-[PdR2L2] Complexes (R = Aryl, L = SC4H8), Organometallics, 1998, 17, 3677–3683 CrossRef CAS.
- N. H. Chan, J. J. Gair, M. Roy, Y. Qiu, D.-S. Wang, L. J. Durak, L. Chen, A. S. Filatov and J. C. Lewis, Insight into the Scope and Mechanism for Transmetalation of Hydrocarbyl Ligands on Complexes Relevant to C–H Activation, Organometallics, 2020, 40, 6–10 CrossRef.
- L. J. Durak and J. C. Lewis, Transmetalation of Alkyl Ligands from Cp*(PMe3)IrR1R2 to (cod)PtR3X, Organometallics, 2013, 32, 3153–3156 CrossRef CAS.
- S. E. Smith, J. M. Sasaki, R. G. Bergman, J. E. Mondloch and R. G. Finke, Platinum-Catalyzed Phenyl and Methyl Group Transfer from Tin to Iridium: Evidence for an Autocatalytic Reaction Pathway with an Unusual Preference for Methyl Transfer, J. Am. Chem. Soc., 2008, 130, 1839–1841 CrossRef CAS PubMed.
- M. H. Perez-Temprano, J. A. Casares, A. R. de Lera, R. Alvarez and P. Espinet, Strong metallophilic interactions in the palladium arylation by gold aryls, Angew. Chem., Int. Ed., 2012, 51, 4917–4920 CrossRef CAS PubMed.
- P. Villar, M. H. Pérez-Temprano, J. A. Casares, R. Álvarez and P. Espinet, Experimental and DFT Study of the [AuAr(AsPh3)]-Catalyzed cis/trans Isomerization of [PdAr2(AsPh3)2] (Ar = C6F5 or C6Cl2F3): Alternative Mechanisms and Its Switch upon Pt for Pd Substitution, Organometallics, 2020, 39, 2295–2303 CrossRef CAS.
- M. E. Moret, D. Serra, A. Bach and P. Chen, Transmetalation supported by a Pt(II)-Cu(I) bond, Angew. Chem., Int. Ed., 2010, 49, 2873–2877 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, A. Comas-Vives, R. R. Fayzullin, A. Lledos and J. R. Khusnutdinova, Dynamic Pd(II) /Cu(I) Multimetallic Assemblies as Molecular Models to Study Metal-Metal Cooperation in Sonogashira Coupling, Chem. – Eur. J., 2020, 26, 12168–12179 CrossRef CAS PubMed.
- R. J. Oeschger and P. Chen, Structure and Gas-Phase Thermochemistry of a Pd/Cu Complex: Studies on a Model for Transmetalation Transition States, J. Am. Chem. Soc., 2017, 139, 1069–1072 CrossRef CAS PubMed.
- D. Serra, M. E. Moret and P. Chen, Transmetalation of methyl groups supported by Pt(II)-Au(I) bonds in the gas phase, in silico, and in solution, J. Am. Chem. Soc., 2011, 133, 8914–8926 CrossRef CAS PubMed.
- G. J. Arsenault, C. M. Anderson and R. J. Puddephatt, Complexes with platinum-gold and -silver bonds: catalysis by silver(I) of alkyl exchange reactions between platinum centers, Organometallics, 1988, 7, 2094–2097 CrossRef CAS.
- S. Fuertes, C. H. Woodall, P. R. Raithby and V. Sicilia, Heteropolynuclear Pt(II)–M(I) Clusters with a C^N^C Biscyclometalated Ligand, Organometallics, 2012, 31, 4228–4240 CrossRef CAS.
- A. Martín, Ú. Belío, S. Fuertes and V. Sicilia, Luminescent Pt–Ag Clusters Based on Neutral Benzoquinolate Cyclometalated Platinum Complexes, Eur. J. Inorg. Chem., 2013, 2013, 2231–2247 CrossRef.
- J. Fornies, S. Ibanez, E. Lalinde, A. Martin, M. T. Moreno and A. C. Tsipis, Benzoquinolateplatinum(II) complexes as building blocks in the synthesis of Pt-Ag extended structures, Dalton Trans., 2012, 41, 3439–3451 RSC.
- J. Forniés, S. Ibáñez, A. Martín, M. Sanz, J. R. Berenguer, E. Lalinde and J. Torroba, Influence of the Pt→Ag Donor−Acceptor Bond and Polymorphism on the Spectroscopic and Optical Properties of Heteropolynuclear Benzoquinolateplatinum(II) Complexes, Organometallics, 2006, 25, 4331–4340 CrossRef.
- T. Yamaguchi, F. Yamazaki and T. Ito, A Helical Metal–Metal Bonded Chain via the Pt→Ag Dative Bond, J. Am. Chem. Soc., 2001, 123, 743–744 CrossRef CAS PubMed.
- M. Baya, Ú. Belío, J. Forniés, A. Martín, M. Perálvarez and V. Sicilia, Neutral benzoquinolate cyclometalated platinum(II) complexes as precursors in the preparation of luminescent Pt–Ag complexes, Inorg. Chim. Acta, 2015, 424, 136–149 CrossRef CAS.
- S. Deolka, O. Rivada-Wheelaghan, S. L. Aristizabal, R. R. Fayzullin, S. Pal, K. Nozaki, E. Khaskin and J. R. Khusnutdinova, Metal-metal cooperative bond activation by heterobimetallic alkyl, aryl, and acetylide Pt(II)/Cu(I) complexes, Chem. Sci., 2020, 11, 5494–5502 RSC.
- M. J. Karimi and S. Jamali, Synthesis and structure of the cyclometalated hetero-binuclear Pt-Au complexes with bridging 2-diphenylphosphinopyridine ligand, J. Organomet. Chem., 2015, 786, 14–20 CrossRef CAS.
- J. Campos, Dihydrogen and Acetylene Activation by a Gold(I)/Platinum(0) Transition Metal Only Frustrated Lewis Pair, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS PubMed.
- L. Kaufmann, S. A. Föhrenbacher, M. C. Krummer and B. Butschke, Investigations into the AuI and PtII Coordination Chemistry of Bidentate “cis–Spanning” Ligands, Eur. J. Inorg. Chem., 2023, 27, e202300577 CrossRef.
- S. Rajabi, S. Jamali, S. Naseri, A. Jamjah, R. Kia, H. Samouei, P. Mastrorilli, H. R. Shahsavari and P. R. Raithby, Pt–M (M = Au and Tl) Dative Bonds Using Bis(cyclometalated)platinum(II) Complexes, Organometallics, 2019, 38, 1709–1720 CrossRef CAS.
- M. Baya, U. Belio, I. Fernandez, S. Fuertes and A. Martin, Unusual Metal-Metal Bonding in a Dinuclear Pt-Au Complex: Snapshot of a Transmetalation Process, Angew. Chem., Int. Ed., 2016, 55, 6978–6982 CrossRef CAS PubMed.
- E. S. Cueny, H. C. Johnson and C. R. Landis, Selective Quench-Labeling of the Hafnium-Pyridyl Amido-Catalyzed Polymerization of 1-Octene in the Presence of Trialkyl-Aluminum Chain-Transfer Reagents, ACS Catal., 2018, 8, 11605–11614 CrossRef CAS.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. van Meurs, Iron Catalyzed Polyethylene Chain Growth on Zinc: A Study of the Factors Delineating Chain Transfer versus Catalyzed Chain Growth in Zinc and Related Metal Alkyl Systems, J. Am. Chem. Soc., 2004, 126, 10701–10712 CrossRef CAS PubMed.
- M. van Meurs, G. J. P. Britovsek, V. C. Gibson and S. A. Cohen, Polyethylene Chain Growth on Zinc Catalyzed by Olefin Polymerization Catalysts: A Comparative Investigation of Highly Active Catalyst Systems across the Transition Series, J. Am. Chem. Soc., 2005, 127, 9913–9923 CrossRef CAS PubMed.
- R. A. Petros and J. R. Norton, Effectiveness in Catalyzing Carboalumination Can Be Inferred from the Rate of Dissociation of M/Al Dimers, Organometallics, 2004, 23, 5105–5107 CrossRef CAS.
- K. P. Bryliakov, E. P. Talsi, A. Z. Voskoboynikov, S. J. Lancaster and M. Bochmann, Formation and Structures of Hafnocene Complexes in MAO- and AlBui3/CPh3[B(C6F5)4]-Activated Systems, Organometallics, 2008, 27, 6333–6342 CrossRef CAS.
- J. M. Camara, R. A. Petros and J. R. Norton, Zirconium-Catalyzed Carboalumination of α-Olefins and Chain Growth of Aluminum Alkyls: Kinetics and Mechanism, J. Am. Chem. Soc., 2011, 133, 5263–5273 CrossRef CAS PubMed.
- M. Bochmann and S. J. Lancaster, Monomer–Dimer Equilibria in Homo- and Heterodinuclear Cationic Alkylzirconium Complexes and Their Role in Polymerization Catalysis, Angew. Chem., Int. Ed. Engl., 1994, 33, 1634–1637 CrossRef.
- M. Baya, U. Belio, D. Campillo, I. Fernandez, S. Fuertes and A. Martin, Pt–M Complexes (M = Ag, Au) as Models for Intermediates in Transmetalation Processes, Chem. – Eur. J., 2018, 24, 13879–13889 CrossRef CAS PubMed.
- D. Campillo, D. Escudero, M. Baya and A. Martin, Heteropolymetallic Architectures as Snapshots of Transmetallation Processes at Different Degrees of Transfer, Chem. – Eur. J., 2022, 28, e202104538 CrossRef CAS PubMed.
- P. A. Shaw, J. M. Phillips, G. J. Clarkson and J. P. Rourke, Trapping five-coordinate platinum(IV) intermediates, Dalton Trans., 2016, 45, 11397–11406 RSC.
- D. J. Parks and W. E. Piers, Tris(pentafluorophenyl)boron-Catalyzed Hydrosilation of Aromatic Aldehydes, Ketones, and Esters, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- R. S. Drago, Physical Methods for Chemists, Surfside Scientific Publishers, Gainesville, FL, 2nd edn, 1992, pp. 290–291 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2364269. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01828a |
| This journal is © The Royal Society of Chemistry 2024 |