
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination compounds of cobalt(II) with carboxylate non-steroidal anti-inflammatory drugs: structure and biological profile†
Spyros
Perontsis
,
Antonios G.
Hatzidimitriou
and
George
Psomas
 *
*
Department of General and Inorganic Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece. E-mail: gepsomas@chem.auth.gr
First published on 28th August 2024
Abstract
Fourteen cobalt(II) complexes with the non-steroidal anti-inflammatory drugs sodium meclofenamate, tolfenamic acid, mefenamic acid, naproxen, sodium diclofenac, and diflunisal were prepared in the presence or absence of a series of nitrogen-donors (namely imidazole, pyridine, 3-aminopyridine, neocuproine, 2,2′-bipyridine, 1,10-phenanthroline and 2,2′-bipyridylamine) as co-ligands and were characterised by spectroscopic and physicochemical techniques. Single-crystal X-ray crystallography was employed to determine the crystal structure of eight complexes. The biological profile of the complexes was investigated regarding their interaction with serum albumins and DNA, and their antioxidant potency. The interaction of the compounds with calf-thymus DNA takes place via intercalation. The ability of the complexes to cleave pBR322 plasmid DNA at the concentration of 500 μM is rather low. The complexes demonstrated tight and reversible binding to human and bovine serum albumins and the binding site of bovine serum albumin was also examined. In order to assess the antioxidant activity of the compounds, the in vitro scavenging activity towards free radicals, namely 1,1-diphenyl-picrylhydrazyl and 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid), and their ability to reduce H2O2 were studied.
1. Introduction
The importance of transition metal ions in biological systems is well established for many years and many bioinorganic chemists have focused their research studies on the biological activity of metal ions and their compounds.1 Among these metal ions, cobalt is included as a trace-element essential for life.2 The main biological role of cobalt is related with vitamin B12; within this context, cobalt is involved in the synthesis of DNA and the metabolism of fatty acids and amino acids.3 For this reason, cobalt is also involved in nutrition as a supplement of vitamin B12.4 Additionally, cobalt(II) ions may interact with Z-DNA5 or cleave DNA6 and many cobalt complexes of biological interest have been reported7,8 mainly because of their potential antitumor, antiproliferative,9,10 antimicrobial,11,12 antifungal,13 antiviral14,15 and antioxidant16,17 properties.Non-steroidal anti-inflammatory drugs (NSAIDs) are a large group of medicaments used to treat pain and inflammation resulting from diseases and/or injuries.18,19 The main mechanism of action of NSAIDs is the inhibition of the cyclooxygenase-mediated production of prostaglandins.20 Moreover, NSAIDs can act synergetically with antitumor drugs,21 and exhibit cytotoxic activity against diverse cancer cell lines via mechanisms involving free radical scavenging22 or programmed cell death.23 Based on characteristic groups, NSAIDs are categorised as anthranilic acids, phenylalkanoic acids, derivatives of salicylic acid, oxicams, furanones and sulfonamides.19
The NSAIDs used in the current research are the anthranilic acids mefenamic acid (Hmef), tolfenamic acid (Htolf), and sodium meclofenamate (Na meclf), the phenylalkanoic acids naproxen (Hnap), and sodium diclofenac (Na dicl), and the salicylate derivative diflunisal (H2difl) (Fig. 1). All of them are common NSAIDs mainly used as analgesic, anti-inflammatory and antipyretic agents in various treatments. Beside the afore-mentioned general uses as NSAIDs: Hmef has mild side-effects and is also used to treat migraines and pain from dysmenorrhea;24 Htolf is also used in veterinary;25 Na meclofenamate is also administered in osteoarthritis and painful musculoskeletal disorders;26 Hnap is also used to treat rheumatoid arthritis, chronic migraine, osteoarthritis, and kidney stones,27 and its use results in milder side-effects concerning blood pressure or stomach ulcers than other NSAIDs;28,29 sodium diclofenac is administered for treating symptoms of rheumatoid arthritis and osteoarthritis;30 diflunisal, because of its relatively long half-life period of activity (∼12 h), is used to alleviate acute pain resulting from oral surgery,31 and has been recently reported to be safely administered to patients suffering from transthyretin amyloidosis cardiomyopathy.32 Furthermore, most of these NSAIDs were suggested for treatment of the COVID-19 pandemic, regarding their analgetic and antipyretic effects.33,34 Several reports are found in the literature concerning the synthesis, the characterisation, and the biological evaluation of metal complexes with NSAIDs; a plethora of manganese(II/III),35–38 iron(III),39,40 cobalt(II),16,17,41–46 nickel(II),47–49 copper(II),50–53 zinc(II),54–56 silver(I),57,58 cadmium(II),59 tin(IV),60 gold(I)61 and lanthanides(III)62 complexes with the NSAIDs under study have shown enhanced biological properties when compared to the corresponding free NSAIDs.19,63
 | ||
| Fig. 1 The syntax formulas of the NSAIDs: tolfenamic acid (Htolf), mefenamic acid (Hmef), sodium meclofenamate (Na meclf), naproxen (Hnap), sodium diclofenac (Na dicl) and diflunisal (H2difl). | ||
Taken into consideration the enhanced biological profile of metal–NSAID complexes, the significance of NSAID in medication, the biological relevance of cobalt, and as continuation of our research projects concerning Co(II)–NSAID complexes,16,17,41–43 we have prepared and characterised fourteen cobalt(II) complexes with the NSAIDs Htolf, Hmef, Na meclf, Hnap, Na dicl and H2difl in the presence or absence of N-donors 1H-imidazole (Himi), pyridine (py) and 3-aminopyridine (3-ampy) (Fig. 2(A)) or N,N′-donors 2,9-dimethyl-1,10-phenanthroline (neoc), 2,2′-bipyridine (bipy), 1,10-phenanthroline (phen) and 2,2′-bipyridylamine (bipyam) as co-ligands (Fig. 2(B)). All resultant complexes 1–14 were characterised by spectroscopic (FT-IR and UV-vis) and physicochemical techniques and the crystal structures of eight complexes were determined by single-crystal X-ray crystallography.
 | ||
| Fig. 2 The syntax formulas of the nitrogen-donor ligands: (A) the N-donor co-ligands: 1H-imidazole (Himi), pyridine (py) and 3-aminopyridine (3-ampy), and (B) the N,N′-donor co-ligands: 2,9-dimethyl-1,10-phenanthroline (neoc), 1,10-phenanthroline (phen), 2,2′-bipyridine (bipy) and 2,2′-bipyridylamine (bipyam). | ||
The in vitro biological profile of the novel compounds is related with: (i) the antioxidant activity by determining the ability to scavenge free radicals such as 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and 1,1-diphenyl-picrylhydrazyl (DPPH) and to reduce H2O2, (ii) the interaction with calf-thymus (CT) DNA monitored by cyclic voltammetry, viscosity measurements, UV-vis spectroscopy, and via competition with the intercalation marker ethidium bromide (EB) by fluorescence emission spectroscopy, (iii) the ability to cleave supercoiled circular pBR322 plasmid DNA (pDNA) examined by agarose gel electrophoretic experiments, and (iv) the affinity (calculation of binding strength and determination of binding site location) for bovine serum albumin (BSA) and human serum albumin (HSA), investigated by fluorescence emission spectroscopy.
2. Experimental
2.1. Materials–instruments–physical measurements
All chemicals and solvents were of reagent grade and were used as purchased from commercial sources without any further purification. More specifically, CoCl2·6H2O, bipy, bipyam, phen, neoc, Himi, py, 3-ampy, KOH, CT DNA, EB, BSA, HSA, ABTS, K2S2O8, nordihydroguaiaretic acid (NDGA), butylated hydroxytoluene (BHT) were purchased from Sigma-Aldrich Co; Na dicl, Htolf, Hmef, Na meclf, Hnap, H2difl, sodium warfarin, ibuprofen, DPPH from Tokyo Chemical Industry (TCI); 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (trolox) from J&K; trisodium citrate dihydrate, NaCl, NaH2PO4 from Merck; supercoiled circular pBR322 plasmid DNA from New England Bioline; Tris base, boric acid, EDTA disodium salt dehydrate, loading buffer and H2O2 (30% w/v) from PanReac Applichem; L-ascorbic acid (AA) Na2HPO4 and all solvents from Chemlab.CT DNA stock solution was prepared by the dilution of CT DNA to buffer (containing 15 mM trisodium citrate and 150 mM NaCl at pH 7.0) followed by exhaustive stirring for three days, and was kept at 4 °C for no longer than ten days. The stock solution of CT DNA gave a ratio of UV absorbance at 260 and 280 nm (A260/A280) of 1.87, indicating that the DNA used was sufficiently free of protein contamination.64 The CT DNA concentration was determined by UV absorbance at 258 nm after 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dilution using ε = 6600 M−1 cm−1.65
:20 dilution using ε = 6600 M−1 cm−1.65
Infrared (IR) spectra (400–4000 cm−1) were recorded on a Nicolet FT-IR 6700 spectrometer with samples prepared as KBr disk (abbreviations used: vs = very strong; s = strong; m = medium; Δν(COO) = vasym(COO) − vsym(COO)). UV-visible (UV-vis) spectra were recorded as nujol mulls and in solution at concentrations in the range 10−5–10−3 M on a Hitachi U-2001 dual-beam spectrophotometer. C, H and N elemental analysis was performed on a PerkinElmer 240B elemental analyzer. Molar conductivity measurements were carried out with a Crison Basic 30 conductometer. Room-temperature (RT) magnetic measurements were carried out by the Faraday method using mercury tetrathiocyanatocobaltate(II) as calibrant. Fluorescence spectra were recorded in solution on a Hitachi F-7000 fluorescence spectrophotometer. Viscosity experiments were carried out using an ALPHA L Fungilab rotational viscometer equipped with an 18 mL LCP spindle and the measurements were performed at 100 rpm.
Cyclic voltammetry studies were performed on an Eco chemie Autolab Electrochemical analyzer. Cyclic voltammetry experiments were carried out in a 30 mL three-electrode electrolytic cell. The working electrode was platinum disk, a separate Pt single-sheet electrode was used as the counter electrode and a Ag/AgCl electrode saturated with KCl was used as the reference electrode. The cyclic voltammograms of the complexes were recorded in 0.4 mM 1/2 DMSO/buffer solutions at v = 100 mV s−1 where buffer solution was the supporting electrolyte. Oxygen was removed by purging the solutions with pure nitrogen which had been previously saturated with solvent vapors. All electrochemical measurements were performed at 25.0 ± 0.2 °C.
2.2. Synthesis of the complexes
[Co(tolf)2(Himi)2], 1. Htolf (0.4 mmol, 104 mg) was used as the NSAID and Himi (0.4 mmol, 26 mg) was the N-donor used. Purple single-crystals of complex 1 suitable for X-ray crystallography were isolated after one week. Yield: 85 mg, 60%. Anal. calcd for C34H30Cl2CoN6O4 (MW = 716.49): C 57.00, H 4.22, N 11.73; found: C 57.10, H 4.30, N 11.65%. IR (KBr disk), vmax/cm−1: vasym(COO): 1581 (vs); vsym(COO): 1385 (s); Δv(COO) = 196 cm−1; ρ(C–H)Himi = 749 (m). UV-vis: as nujol mull, λ/nm: 545; in DMSO solution, λ/nm (ε/M−1 cm−1): 559 (150), 301 (15
000). μeff = 4.08 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 12 mho cm2 mol−1 in 1 mM DMSO).
[Co(mef)2(Himi)2], 2. Hmef (0.4 mmol, 96 mg) was used as the NSAID and Himi (0.4 mmol, 26 mg) was the corresponding N-donor co-ligand. Purple-coloured single-crystals of complex 2 suitable for X-ray crystallography were isolated after ten days. Yield: 95 mg, 70%. Anal. calcd for C36H36CoN6O4 (MW = 675.65): C 64.00, H 5.37, N 12.44; found: C 64.10, H 5.45, N 12.30%. IR (KBr disk), vmax/cm−1: vasym(COO): 1577 (vs); vsym(COO): 1386 (s); Δv(COO) = 191 cm−1; ρ(C–H)Himi = 747 (m). UV-vis: as nujol mull, λ/nm: 540; in DMSO solution, λ/nm (ε/M−1 cm−1): 557 (120), 334 (7000), 300 (13
000). μeff = 4.02 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 11 mho cm2 mol−1 in 1 mM DMSO).
[Co(nap)2(Himi)2], 3. The synthesis of the complex was performed in acetonitrile. Hnap (0.4 mmol, 92 mg) was used as the NSAID and Himi (0.4 mmol, 26 mg) was the N-donor used as co-ligand. Purple-coloured microcrystalline product of complex 3 was collected after a few days. Yield: 80 mg, 60%. Anal. calcd for C34H34CoN4O6 (MW = 653.60): C 62.48, H 5.24, N 8.57; found: C 62.35, H 5.20, N 8.45%. IR (KBr disk), vmax/cm−1: vasym(COO): 1606 (vs); vsym(COO): 1390 (s); Δv(COO) = 216 cm−1; ρ(C–H)Himi = 755 (m). UV-vis: as nujol mull, λ/nm: 572; in DMSO solution, λ/nm (ε/M−1 cm−1): 581 (250), 332 (6100), 320 (12
200). μeff = 4.29 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 15 mho cm2 mol−1 in 1 mM DMSO).
[Co(meclf)2(Himi)2], 4. Sodium meclofenamate (0.4 mmol, 126 mg) was used as the sodium salt of the NSAID and Himi (0.4 mmol, 26 mg) was the N-donor used as co-ligand. Purple-coloured microcrystalline product of complex 4 was collected after a few days. Yield: 95 mg, 60%. Anal. calcd for C34H28Cl4CoN6O4 (MW = 785.38): C 52.00, H 3.59, N 10.70; found: C 51.85, H 3.45, N 10.60%. IR (KBr disk), vmax/cm−1: vasym(COO): 1583 (s); vsym(COO): 1393 (s); Δv(COO) = 190 cm−1; ρ(C–H)Himi = 749 (m). UV-vis: as nujol mull, λ/nm: 559; in DMSO solution, λ/nm (ε/M−1 cm−1): 555 (100), 317 (7500), 297 (13
900). μeff = 4.30 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 11 mho cm2 mol−1 in 1 mM DMSO).
[Co(meclf)2(py)2(H2O)2]·2py, 5. Na meclf (0.4 mmol, 126 mg) was used as the sodium salt of the NSAID and pyridine (1.5 mL) was the N-donor used as co-ligand. Pink single-crystals of complex 5, suitable for X-ray crystallography were isolated after three weeks. Yield: 110 mg, 55%. Anal. calcd for C48H44Cl4CoN6O6 (MW = 1001.66): C 57.56, H 4.43, N 8.39; found: C 57.45, H 4.30, N 8.25%. IR (KBr disk), vmax/cm−1: vasym(COO): 1578 (vs); vsym(COO): 1387 (s); Δv(COO) = 191 cm−1; ρ(C–H)py = 698 (m). UV-vis: as nujol mull, λ/nm: 530, 470; in DMSO solution, λ/nm (ε/M−1 cm−1): 548 (50), 461 (95), 317 (6500), 300 (15
500). μeff = 4.35 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 12 mho cm2 mol−1 in 1 mM DMSO).
{[Co(dicl)2(3-ampy)(H2O)3][Co(dicl)2(H2O)4]}, 6 (6a6b). Sodium diclofenac (0.4 mmol, 127 mg) was used as the sodium salt of the NSAID and 3-ampy (0.2 mmol, 19 mg) was the corresponding N-donor co-ligand. Pink single-crystals of complex 6, suitable for X-ray crystallography, were isolated after two weeks. Yield: 95 mg, 60%. Anal. calcd for C30.5H30Cl4CoN3O7.5 (MW = 759.33): C 48.24, H 3.98, N 5.53; found: C 48.50, H 3.95, N 5.69%. IR (KBr disk), vmax/cm−1: vasym(COO): 1578 (vs); vsym(COO): 1381 (s); Δv(COO) = 197 cm−1; ρ(C–H)3-ampy = 809 (m). UV-vis: as nujol mull, λ/nm: 545, 475; in DMSO solution, λ/nm (ε/M−1 cm−1): 525 (40), 485 (35), 291 (8100). μeff = 3.93 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 12 mho cm2 mol−1 in 1 mM DMSO).
500). μeff = 3.97 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 15 mho cm2 mol−1 in 1 mM DMSO).
[Co(tolf)2(neoc)], 8. Htolf (0.4 mmol, 104 mg) was used as the NSAID and neoc (0.2 mmol, 40 mg) was the N,N′-donor co-ligand used. In order to isolate single-crystals of the complex, the reaction solution was subjected to vapor diffusion with diethylether. Red-coloured single-crystals of complex 8, suitable for X-ray crystallography, were isolated after one week. Yield: 100 mg, 65%. Anal. calcd for C42H34Cl2CoN4O4 (MW = 788.59): C 63.97, H 4.35, N 7.10; found: C 63.80, H 4.25, N 7.00%. IR (KBr disk), vmax/cm−1: vasym(COO): 1580 (vs); vsym(COO): 1406 (s); Δv(COO) = 174 cm−1; ρ(C–H)neoc = 729 (m). UV-vis: as nujol mull, λ/nm: 537, 475; in DMSO solution, λ/nm (ε/M−1 cm−1): 550 (80), 490 (65), 296 (17
000). μeff = 4.27 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 16 mho cm2 mol−1 in 1 mM DMSO).
[Co(mef)2(neoc)], 9. Hmef (0.4 mmol, 96 mg) was used as the NSAID and neoc (0.2 mmol, 40 mg) was the N,N′-donor co-ligand used. Vapor diffusion with diethylether of the reaction solution resulted in the formation of brownish single-crystals of complex 9, suitable for X-ray crystallography after ten days. Yield: 90 mg, 60%. Anal. calcd for C44H40CoN4O4 (MW = 747.76): C 70.68, H 5.39, N 7.49; found: C 70.55, H 5.28, N 7.35%. IR (KBr disk), vmax/cm−1: vasym(COO): 1581 (vs); vsym(COO): 1401 (s); Δv(COO) = 180 cm−1; ρ(C–H)neoc = 732 (m). UV-vis: as nujol mull, λ/nm: 555, 480; in DMSO solution, λ/nm (ε/M−1 cm−1): 560 (55), 485 (35), 348 (6400), 311 (14
200). μeff = 4.31 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 16 mho cm2 mol−1 in 1 mM DMSO).
[Co(nap)2(neoc)], 10. The synthesis of the complex was performed in acetonitrile. Hnap (0.4 mmol, 92 mg) was used as the NSAID and neoc (0.2 mmol, 40 mg) was the N,N′-donor co-ligand used. Purple single-crystals of complex 10, suitable for X-ray crystallography, were isolated after one week. Yield: 95 mg, 65%. Anal. calcd for C42H38CoN2O6 (MW = 725.71): C 69.51, H 5.28, N 3.86; found: C 69.40, H 5.20, N 3.70%. IR (KBr disk), vmax/cm−1: vasym(COO): 1575 (vs); vsym(COO): 1393 (s); Δv(COO) = 182 cm−1; ρ(C–H)neoc = 731 (m). UV-vis: as nujol mull, λ/nm: 582, 480; in DMSO solution, λ/nm (ε/M−1 cm−1): 590 (75), 475 (50), 333 (5900), 318 (13
500). μeff = 4.24 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 15 mho cm2 mol−1 in 1 mM DMSO).
[Co(meclf)2(phen)], 11. Na meclf (0.4 mmol, 126 mg) was used as the sodium salt of the NSAID and phen (0.2 mmol, 36 mg) was the corresponding N,N′-donor co-ligand. Pink microcrystalline product of complex 11 was collected after a few days. Yield: 100 mg, 60%. Anal. calcd for C40H28Cl4CoN4O4 (MW = 829.43): C 57.92, H 3.40, N 6.75; found: C 57.80, H 3.25, N 6.65%. IR (KBr disk), vmax/cm−1: vasym(COO): 1580 (vs); vsym(COO): 1416 (s); Δv(COO) = 164 cm−1; ρ(C–H)phen = 728 (m). UV-vis: as nujol mull, λ/nm: 562, 485; in DMSO solution, λ/nm (ε/M−1 cm−1): 550 (40), 476 (55), 316 (6900), 292 (16
700). μeff = 4.30 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 12 mho cm2 mol−1 in 1 mM DMSO).
[Co(meclf)2(bipy)], 12. Na meclf (0.4 mmol, 126 mg) was used as the sodium salt of the NSAID and bipy (0.2 mmol, 31 mg) was the corresponding N,N′-donor co-ligand. Brownish microcrystalline product of complex 12 was collected after a few days. Yield: 95 mg, 60%. Anal. calcd for C38H28Cl4CoN4O4 (MW = 805.41): C 56.67, H 3.50, N 6.96; found: C 56.55, H 3.40, N 6.85%. IR (KBr disk), vmax/cm−1: vasym(COO): 1579 (vs); vsym(COO): 1415 (s); Δv(COO) = 164 cm−1; ρ(C–H)bipy = 763 (m). UV-vis: as nujol mull, λ/nm: 565, 475; in DMSO solution, λ/nm (ε/M−1 cm−1): 560 (20), 485 (50), 328 (7100), 289 (17
500). μeff = 4.19 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 5 mho cm2 mol−1 in 1 mM DMSO).
[Cao(meclf)2(bipyam)], 13. Na meclf (0.4 mmol, 126 mg) was used as the sodium salt of the NSAID and bipyam (0.2 mmol, 34 mg) was the corresponding N,N′-donor co-ligand. Orange microcrystalline product of complex 13 was collected after two weeks. Yield: 105 mg, 65%. Anal. calcd for C38H29Cl4CoN5O4 (MW = 820.43): C 55.63, H 3.56, N 8.54; found: C 55.50, H 3.45, N 8.40%. IR (KBr disk), vmax/cm−1: vasym(COO): 1586 (vs); vsym(COO): 1420(s); Δv(COO) = 166 cm−1; ρ(C–H)bipyam = 770 (m). UV-vis: as nujol mull, λ/nm: 543, 469; in DMSO solution, λ/nm (ε/M−1 cm−1): 548 (15), 480 (65), 318 (7800), 296 (13
700). μeff = 4.25 BM at RT. The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 11 mho cm2 mol−1 in 1 mM DMSO).
[Co2(difl)2(neoc)2]·MeOH, 14. For the synthesis of complex 14, a methanolic solution of H2difl (0.2 mmol, 50 mg) containing KOH (0.4 mmol, 0.4 mL of 1 M) was stirred for 1 h at RT. The reaction solution was added into the methanolic solution of solution (∼5 mL) of CoCl2·6H2O (0.2 mmol, 48 mg) simultaneously with neoc (0.2 mmol, 40 mg). Brown well-shaped single-crystals of complex 14, suitable for X-ray crystallography, were isolated after three days. Yield: 60 mg, 55%. Anal. calcd for C55H40Co2F4N4O7 (MW = 1062.80): C 62.16, H 3.79, N 5.27; found: C 62.00, H 3.65, N 5.15%. IR (KBr disk), vmax/cm−1: vasym(COO): 1588 (vs); vsym(COO): 1398 (s); Δv(COO) = 190 cm−1; ρ(C–H)neoc = 730 (m). UV-vis: as nujol mull, λ/nm: 548, 467; in DMSO solution, λ/nm (ε/M−1 cm−1): 555 (35), 475 (75), 319 (6500), 296 (11
000). The complex is soluble in DMSO and DMF and is non-electrolyte (ΛM = 13 mho cm2 mol−1 in 1 mM DMSO).
2.3. X-ray structural determination
Single-crystals of the complexes suitable for crystal structure analysis were mounted at room temperature on a Bruker Kappa APEX2 diffractometer equipped with a Triumph monochromator using Mo Kα (λ = 0.71073 Å, source operating at 50 kV and 30 mA) radiation. Unit cell dimensions were determined and refined by using the angular settings of at least 188 high intensity reflections (>10σ(I)) in the range 10° < 2θ < 20°. Intensity data were recorded using φ and ω-scans. All crystals presented no decay during the data collection. The frames collected were integrated with the Bruker SAINT Software package66 using a narrow-frame algorithm. Data were corrected for absorption using the numerical method (SADABS) based on crystal dimensions.67 The structures were solved using SUPERFLIP68 incorporated in Crystals. Data refinement (full-matrix least-squares methods on F2) and all subsequent calculations were carried out using the Crystals version 14.61 build 6236 program package.69 All non-hydrogen non-disordered atoms were refined anisotropically. For the disordered atoms, their occupation factors were first refined under fixed isotropic parameters. Their isotropic displacement factors were finally refined under the fixed occupation factors previously observed. Hydrogen atoms bonded to non-disordered atoms were located from difference Fourier maps and refined using soft constraints at idealised positions riding on the parent atoms with isotropic displacement parameters Uiso(H) = 1.2Ueq(C) and 1.5Ueq (–NH and –OH hydrogens) at distances C–H 0.95 Å, N–H 0.84 Å and O–H 0.82 Å. Both NH and OH hydrogen atoms were allowed to rotate. For the disordered hydrogen atoms with disordered parent atoms, their positions were selected to fulfil the previous demands as well as the hydrogen bonding demands when necessary.Crystallographic data for the complexes are presented in Tables S1–S4.† Further details on the crystallographic studies as well as atomic displacement parameters are given as ESI† in the form of cif files.
2.4. In vitro biological activity studies
In order to study in vitro the biological activity (i.e., antioxidant activity and interaction with DNA or albumins) of complexes 1–14, the complexes were initially dissolved in DMSO (1 mM). Mixing of such solutions with the aqueous buffer solutions of the biomacromolecules (DNA or albumins) never exceeded 5% DMSO (v/v) in the final solution, which was needed due to low aqueous solubility of most compounds. Control experiments with DMSO were performed and no significant effect on the measurements was observed.The antioxidant activity of the complexes was evaluated by determining their ability to scavenge DPPH and ABTS free radicals (expressed as percentage of radical scavenging, DPPH% and ABTS%, respectively) and to reduce H2O2. The interaction of the complexes with CT DNA was investigated by UV-vis spectroscopy, viscosity measurements, and cyclic voltammetry and via the evaluation of their EB-displacing ability which was monitored by fluorescence emission spectroscopy. The serum albumin (BSA or HSA) binding studies were performed by tryptophan fluorescence quenching experiments in the absence or presence of the albumin site-markers warfarin and ibuprofen. Detailed procedures regarding the in vitro study of the biological activity of the complexes are given in the ESI (sections S1–S4†).
3. Results and discussion
3.1. Synthesis and characterisation
The complexes were synthesised in high yield via the aerobic reaction of a methanolic solution of the corresponding deprotonated NSAID with CoCl2·6H2O and the corresponding N-donor co-ligand (Himi, py or 3-ampy) in a 1:2:2 Co2+:NSAID−1:(N-donor) ratio for complexes 1–6. In a similar manner, the use of corresponding N,N′-donor co-ligand (neoc, bipy, phen or bipyam) in a 1:2:1 Co2+:NSAID−1:(N,N′-donor) ratio afforded complexes 8–13. For complex 14, a 1:1:1 Co2+:difl−2:neoc ratio was used, while a 1:2 Co2+:meclf−1 ratio yielded complex 7.
The complexes were characterised by IR and UV-vis spectroscopy, RT-magnetic measurements and X-ray crystallography (for eight of the fourteen complexes X-ray crystal structures were determined). The proposed formulas of the complexes were based on the results of elemental analysis. The complexes are stable in the air, soluble in DMSO and DMF and insoluble in most organic solvents and H2O.
According to the molar conductivity values (ΛM = 5–16 mho cm2 mol−1 for 1 mM DMSO solution), the complexes are non-electrolytes in DMSO solution (in the case of a 1:1 electrolyte, the ΛM value of a 1 mM DMSO solution should be higher than 70 mho cm2 mol−1)70 and do not dissociate in solution keeping their integrity. From the magnetic measurements of the mononuclear complexes 1–13 at RT, the obtained μeff values were in the range 3.93–4.35 BM and are higher than the expected spin-only value (μeff = 3.87 BM) as expected for mononuclear high-spin Co(II) complexes.71
IR spectroscopy was used to confirm the existence of the NSAID ligands and nitrogen-donor co-ligands in the complexes. In the IR spectra of the complexes, two intense bands attributable to the characteristic stretching vibrations of their carboxylate group, i.e. the antisymmetric (vasym(COO)) and the symmetric (vsym(COO)) were observed in the regions 1578–1606 cm−1 and 1381–1420 cm−1, respectively. The parameter Δv(COO) is usually used to suggest the coordination mode of the carboxylato group of a ligand when compared to its salt. For complexes 1–7, and 14, the values of Δv(COO) were in the range 190–216 cm−1 and are higher than that of the corresponding NSAID salt revealing a monodentate coordination mode.72 For complexes 8–13, bidentate chelating mode was concluded since the Δv(COO) values were found in the range 164–182 cm−1 and were lower were than Δv(COO) of the corresponding NSAID salt.72 Furthermore, the co-existence of the corresponding nitrogen-donor co-ligands in the complexes was verified by the presence of the characteristic bands attributed to the out-of-plane ρ(C–H) vibrations found at 747–755 cm−1 for ρ(C–H)Himi in 1–4, 698 cm−1 for ρ(C–H)py in 5, 809 cm−1 for ρ(C–H)3-ampy in 6, 729–732 cm−1 for ρ(C–H)neoc in 8–10 and 14, 728 cm−1 for ρ(C–H)phen in 11, 763 cm−1 for ρ(C–H)bipy in 12, and 770 cm−1 for ρ(C–H)bipyam in 13.72 Such features regarding the coordination of the carboxylato group of the NSAID ligands and the presence of the nitrogen-donor co-ligands are in good agreement with the crystal structures of the complexes determined and discussed in section 3.2.
The electronic (UV-vis) spectra of the complexes were recorded as nujol mull and in DMSO solution and were found similar suggesting that the complexes retain their structure in solution. In the visible region of the spectrum: for complexes 1–4, one band assigned to d–d transition (4A2 → 4T1(P)) was observed in the range 555–581 nm (ε = 100–250 M−1 cm−1) which is typical for tetrahedral Co(II) complexes,71 and for complexes 5–14, two low-intensity d–d bands were observed in region 525–590 nm (ε = 10–80 M−1 cm−1) and 461–490 nm (ε = 35–95 M−1 cm−1) attributable to 4T1g(F) → 4A2g and 4T1g(F) → 4T1g(P) transitions,71 respectively. It may be noted that the ε values of the d–d transition bands for the four-coordinate Co(II) complexes 1–4 are higher than those of the six-coordinate Co(II) complexes 5–14, as expected.71 In addition, one or two bands in the range 289–334 nm assigned to intra-ligand transitions were observed in the UV region of the spectra.
3.2. Structural characterisation of the complexes
Continuous attempts in order to obtain single-crystals of the complexes suitable for X-ray crystallography were successful for eight of the complexes, namely complexes 1, 2, 5–9 and 13. For the rest six complexes, their structural characterisation was based on the other techniques used.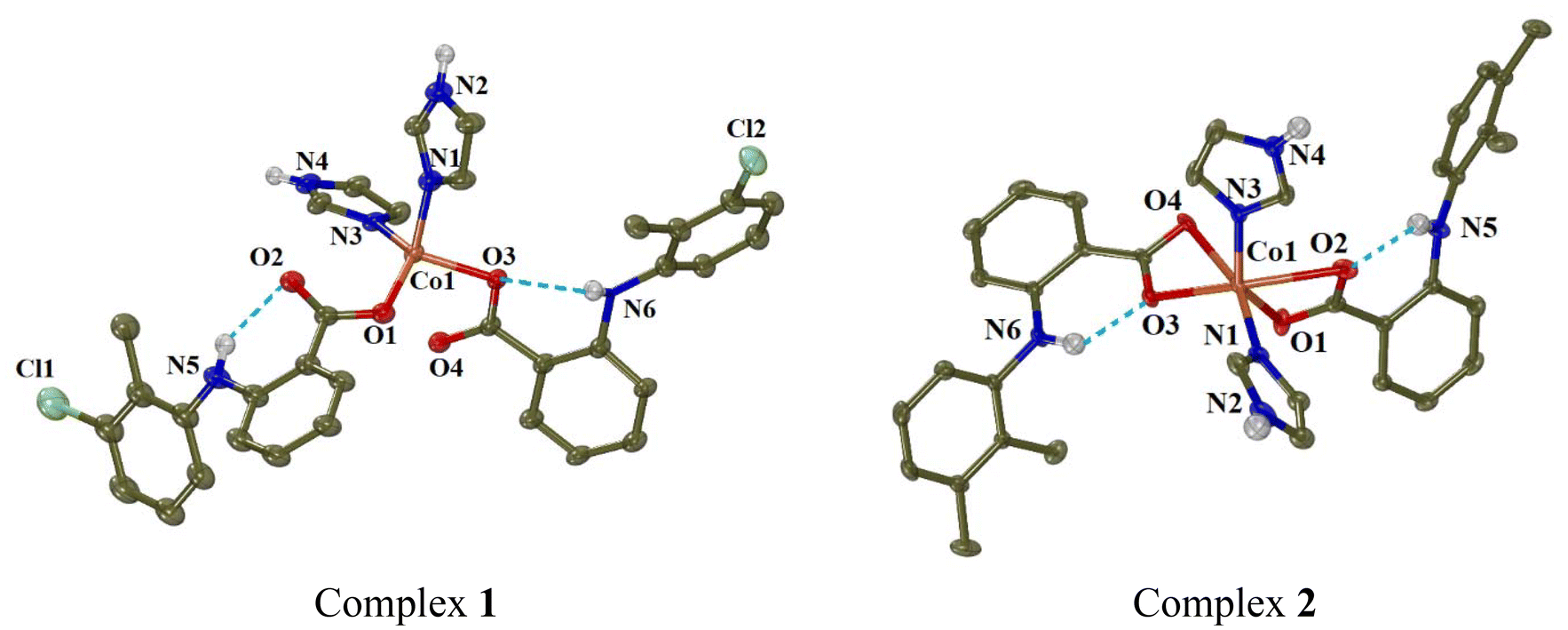 | ||
| Fig. 3 Molecular structures of complexes 1 and 2. Methyl and aromatic hydrogen atoms are omitted for clarity. The intra-ligand H-bonds are shown in light-blue dot lines. | ||
In complex 1, the tolfenamato ligands are coordinated to cobalt(II) ion in a monodentate manner through O1 and O3 atoms with Co–O distances 1.980(2) Å and 2.0078(18) Å, respectively. The distances of the non-coordinated carboxylato oxygen atoms O2 and O4 from Co1 are 2.985 Å and 2.559 Å, respectively. The two imidazole ligands are neutral and coordinated to Co1 through N1 and N3 atoms (Co–N = 2.048(2) Å and 2.025(2) Å, respectively). Quite similar is the arrangement of the NSAID and imidazole ligands around Co(II) ion in complexes [Co(dicl)2(Himi)2]42 and [Co(indo)2(Himi)2]73 where Hindo is the NSAID indomethacin.
In order to check the geometry around four-coordinated Co1, the following structural parameters were calculated: (a) the tetrahedrality (it can be determined from the angle formed by the two planes enclosing the cobalt ion and two adjacent coordinated atoms; in case of strictly square planar complexes (D4h symmetry), the tetrahedrality equals 0°; for tetrahedral complexes (D2d symmetry), the tetrahedrality is 90°),74 and (b) the tetrahedral index as introduced by Yang (τ4 = (360° − (α + β))/(360° − 2 × 109.5°), where α and β are the largest angles around the metal)75 or as introduced by Okuniewski (τ′4 = ((β − α)/(360° − 109.5°)) + ((180° − β)/(180° − 109.5°)) where β > α are the largest angles of the coordination sphere).76 For complex 1, the tetrahedrality (as determined by the dihedral angle of planes formed by atoms O1, Co1, N1 and O3, Co1, N3) has a value of 87.80° (which is close to 90°) and the tetrahedral indexes are close to 1 (τ4 = 0.85 and τ′4 = 0.81); all these values support tetrahedral geometry for the chromophore CoO2N2.
In complex 2, the mefenamato ligands are bound to Co1 ion in an asymmetric bidentate mode with two short Co–O bond distances (2.042(2) Å and 2.025(2) Å) and two much longer distances (2.420(3) Å and 2.435(3) Å). In such a case, Co1 could be considered six-coordinate, its coordination sphere (CoN2O4) is completed by N1 and N3 of the imidazole ligands (2.035(2)–2.051(3) Å) and its geometry would be described as a distorted octahedron (with the largest angles O4–Co1–N1 = 162.02(9)°, O2–Co1–O3 = 156.90(9)° and O1–Co1–N3 = 143.01(11)° revealing the distortion). However, the Co1–O2 and Co1–O4 distances can be considered too long for a bonding distance; subsequently, Co1 is four-coordinate, and the structural parameters tetrahedrality, τ4 and τ′4 have values 79.23, 0.80 and 0.68, respectively, suggesting a distorted tetrahedral geometry around Co1, which is in good agreement with electronic spectra discussed in section 3.1. It should be noted that a structure of complex [Co(mef)2(Himi)2] has been recently reported.46 That structure is similar to that of complex 2, with the difference that the reported structure was centrosymmetric.46
The structures of both complexes are further stabilised by the formation of intraligand and intermolecular hydrogen-bonds between the amino hydrogen of the NSAID (tolfenamato or mefenamato) ligands and their carboxylato oxygens in the same ligand or in neighbouring complexes (Table S6†).
Complexes 3 and 4 are isostructural with complexes 1, 2, [Co(dicl)2(Himi)2]42 and [Co(indo)2(Himi)2],73 and are expected to have similar structural features: they are mononuclear (μeff = 4.29–4.40 BM), with the naproxen and meclofenamato ligands, respectively, monodentately bound to Co(II) ion via a carboxylato oxygen atom (Δv(COO) = 190–216 cm−1) and contain two coordinated imidazole co-ligands (ρ(C–H)Himi = 749–755 cm−1). The Co(II) ions are four-coordinate with a CoN2O2 chromophore and a distorted tetrahedral geometry (Fig. 4).
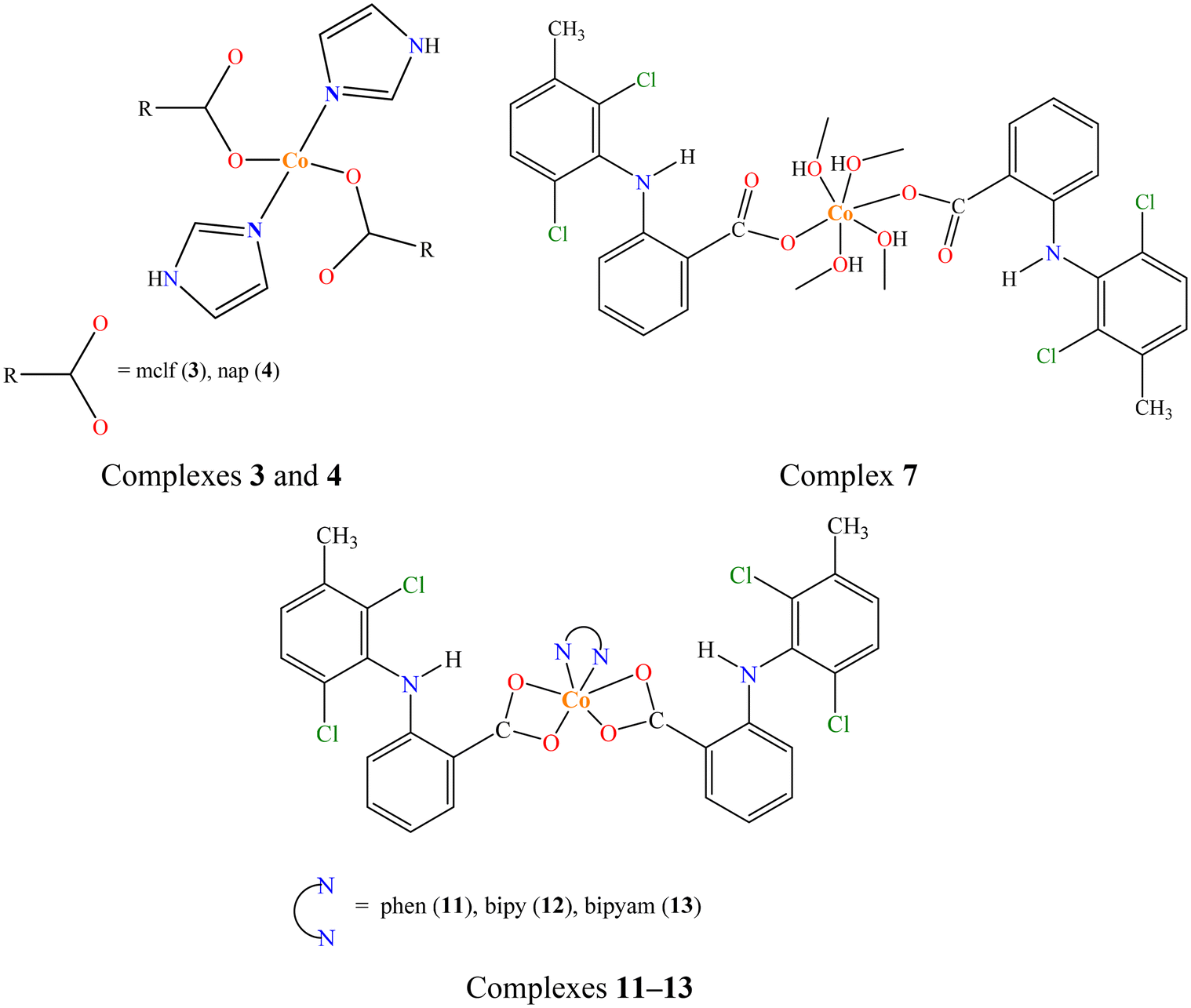 | ||
| Fig. 4 Proposed structures for complexes 3, 4, 7 and 11–13. | ||
Complex 5 (CCDC 2336777†) crystallised in triclinic crystal system and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S2†). In complex 5, the meclofenamato ligands are coordinated monodentately to Co(II) ion. Two pyridine solvate molecules were also found in the unit cell. The molecular structure of complex 5 is depicted in Fig. 5 and selected bond distances and angles are cited in Table S7.†
space group (Table S2†). In complex 5, the meclofenamato ligands are coordinated monodentately to Co(II) ion. Two pyridine solvate molecules were also found in the unit cell. The molecular structure of complex 5 is depicted in Fig. 5 and selected bond distances and angles are cited in Table S7.†
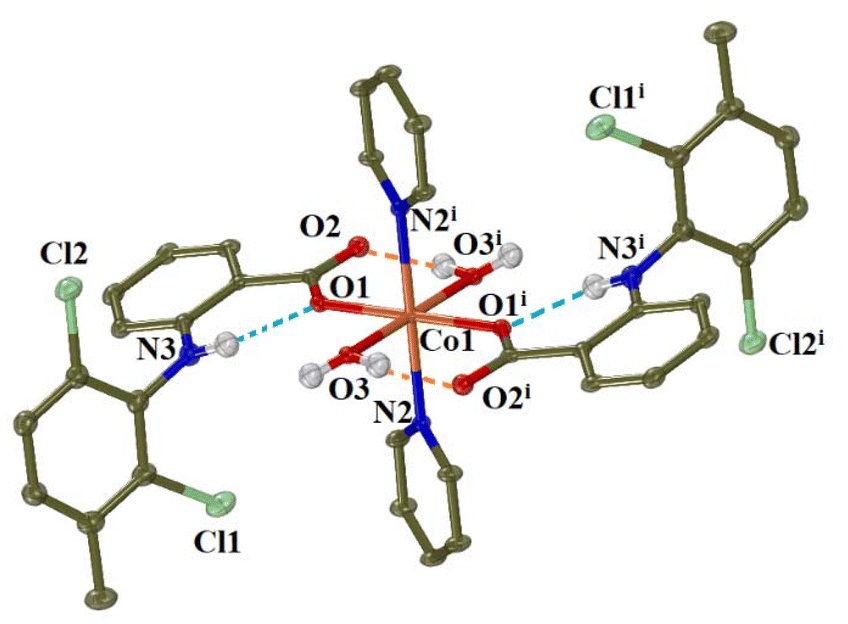 | ||
| Fig. 5 Molecular structure of complex 5 (symmetry code: (i) −x + 1, −y + 1, −z + 1). Aromatic hydrogen atoms, and solvate molecules are omitted for clarity. The intra-ligand and intramolecular H-bonds are shown in light-blue and orange dot lines, respectively. | ||
The structure of complex 5 is centrosymmetric, with the cobalt(II) ion (Co1) being on the centre of symmetry and bound to two meclofenamato, two pyridine and two aqua ligands having a distorted octahedral geometry. The basal plane of the octahedron consists of the carboxylate oxygen atoms O1 and O1i and the aqua oxygen atoms O3 and O3i while the pyridine nitrogen atoms N1 and N1i are located at the axial positions at 2.201(3) Å. In the equatorial CoO4 plane of the octahedron, the bond distances Co–Ocarb (Co1–O1 = 2.094(2) Å) and Co–Oaqua (Co1–O3 = 2.097(2) Å) are similar. The uncoordinated carboxylate oxygen atoms O2 and O2i are lying at Co1⋯O2 distance of 3.38 Å. Similar arrangement of the NSAID, pyridine and aqua ligands around the cobalt(II) ion was observed in the structures of complexes [Co(dicl)2(py)2(H2O)2]42 and [Co(nap)2(py)2(H2O)2],16 although the latter is not centrosymmetric.
A series of hydrogen bonds contribute to further stabilisation of the structure of complex 5; intra-ligand H-bonds are developed between the amino H atoms and the coordinated carboxylato oxygens O1, intramolecular H-bonds are formed between aqua H atoms and the non-coordinated carboxylato oxygen atoms O2, and intermolecular H-bonds between aqua H atoms and the nitrogen atoms of solvate pyridines (Table S6†).
Complex 6 (CCDC 2336778†) crystallised in monoclinic crystal system and C2/c space group (Table S2†). The asymmetric unit comprises the half of a cobalt(II) complex with the metal atom on a special position, one fully occupied diclofenac ligand, three aqua ligands with occupation factors of ½ and finally one more aqua ligand as well as an aminopyridine ligand with occupations of ¼. From the four complexes present in the unit cell, two of them were found to have the general formula [Co(dicl)2(3-ampy)(H2O)3] (namely 6a) and the rest two were found formulated as [Co(dicl)2(H2O)4] (namely 6b). The molecular structures of the mononuclear complexes 6a and 6b are given in Fig. 6 and selected bond distances and angles are summarised in Table S8.†
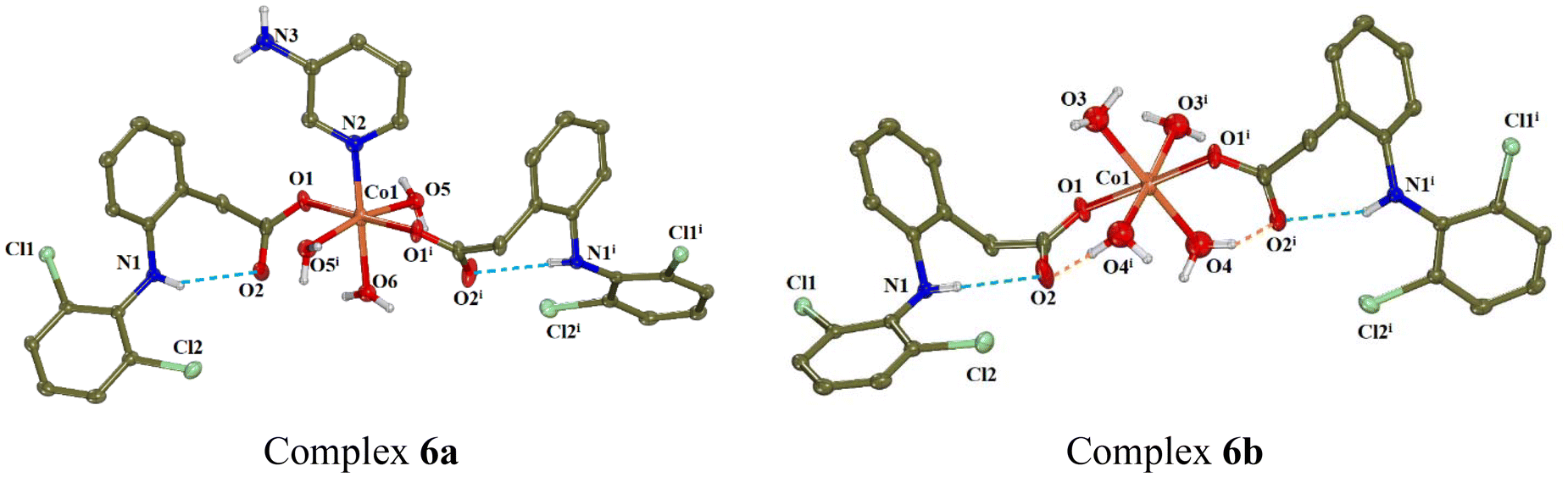 | ||
| Fig. 6 Molecular structures of complexes 6a and 6b (symmetry code: (i) −x + 1, y, −z + 1/2). Methyl and aromatic hydrogen atoms are omitted for clarity. The intra-ligand and intramolecular H-bonds are shown in light-blue and orange dot lines, respectively. | ||
In both complexes 6a and 6b, the cobalt(II) ions are six-coordinated and the diclofenac ligands are coordinated in a monodentate fashion. In the case of 6a, the Co(II) ion is bound to two carboxylato oxygen atoms O1 and O1i, three aqua oxygen atoms O5, O5i and O6 and the aromatic nitrogen atom N2 (aqua O6 and the 3-aminopyridine ligand being disordered over two positions with equal occupation factors). In the case of 6b, the coordination sphere comprises the oxygen atoms of the diclofenac ligands O1 and O1i and four aqua oxygen atoms O3, O3i, O4 and O4i. So the chromophores are CoNO5 in 6a and CoO6 in 6b, resulting in a distorted octahedral geometry around Co1 in both cases. The bonding distances around Co1 ion in both cases are in the range 2.065(12)–2.170(14) Å, while the non-coordinated diclofenac O2 atoms are lying at 3.280 Å away from Co1.
The non-coordinated carboxylato oxygen O2 of diclofenac ligand participates in the formation of intraligand H-bonds with the diclofenac amino H atoms and intramolecular H-bonds with the aqua H atoms contributing to further stabilisation of the structure. Further intermolecular H-bonds are developed between aqua H atoms and the coordinated carboxylato oxygens O1 from adjacent molecules (Table S6†).
space group (Tables S3 and S4†). The molecular structures of complexes 8–10 are depicted in Fig. 7 and selected bond distances and angles are summarised in Table S9.†
 | ||
| Fig. 7 Molecular structures of complexes 8–10. Methyl and aromatic hydrogen atoms are omitted for clarity. The intra-ligand H-bonds are shown in light-blue dot lines. | ||
These three structures bear similarities and are discussed together. In the mononuclear complexes, the NSAID ligands (tolfenamato in 8, mefenamato in 9, and naproxen in 10) are coordinated to cobalt(II) ion in an asymmetric bidentate chelating mode through their two carboxylato oxygen atoms. The six-coordinate Co(II) is surrounded by two NSAID ligands and one bidentate neoc ligand completing a CoN2O4 chromophore and shows a distorted octahedral geometry (largest angle 166.11(8)° in 8, 164.11(6)° in 9, and 160.99(9)° in 10) where two N and four O atoms lying at the vertices of the octahedron. In complexes 8 and 9, the asymmetric bidentate mode is obvious from the differences in the Co1–O bond distances; for each NSAID ligand, there is a short Co1–O distance (2.0630(15)–2.1105(19) Å) and a longer one (2.1869(17)–2.2472(15) Å). In complex 10, the case is similar for one naproxen ligand (Co1–O1 = 2.031(2) Å and Co1–O2 = 2.260(2) Å), while for the second naproxen ligand the Co1–O bond distances are much closer (2.151(2) Å and 2.172(2) Å). In all three complexes, the Co1–N bond distances are in the range 2.0966(18)–2.126(2) Å. The structures of complexes 8 and 9 are further stabilised by the development of intraligand hydrogen-bonds between the amino group of tolfenamato and mefenamato ligands, respectively, and their coordinated carboxylato group (Table S6†).
The structures of complexes 8 and 9 are similar but not identical with those previously reported by Smolkova et al.44,45 However, there are some crystallographic differences. Complex 8 crystallised in different crystal system and space group from the reported complex [Co(tolf)2(neoc)] (orthorhombic crystal system and Pna21 space group) which contained three crystallographically independent units.45 For the reported complex [Co(mef)2(neoc)], three differently coloured and shaped crystal polymorphs were isolated and characterised,44 which was not observed for complex 9. In addition, the structures were resolved in slightly different temperatures. The arrangement of the ligand atoms around cobalt(II) ion observed in complexes 8–10 is similar with that reported for isostructural complexes of the formula [Co(NSAID)2(N,N′-donor)], such as [Co(dicl)2(bipy)], [Co(dicl)2(bipyam)] and [Co(dicl)2(phen)],42 [Co(flufenamato)2(bipyam)] and [Co(mef)2(bipyam)],43 [Co(tolf)2(bipyam)],41 and [Co(fenamato)2(neoc)].45
Complexes 11–13 are expected to have similar structural features with complexes 8–10 and those reported in the literature.41–45 They are mononuclear complexes (μeff = 4.19–4.30 BM) with the meclofenamato ligands bound to Co(II) ions in an asymmetrical bidentate chelating mode (Δv(COO) = 164–166 cm−1) and contain a N,N′-donor (phen in 11 (ρ(C–H)phen = 728 cm−1), bipy in 12 (ρ(C–H)bipy = 763 cm−1), and bipyam in 13 (ρ(C–H)bipyam = 770 cm−1)) as co-ligand. The Co(II) ions are six-coordinate with CoN2O4 chromophore and distorted octahedral geometry (Fig. 4).
Complex 14 (CCDC 2336782†) crystallised in monoclinic crystal system and C2/c space group (Table S4†). The molecular structure of complex 14 is shown in Fig. 8 and selected bond distances and angles are summarised in Table S10.† A methanol solvate molecule was found in the unit cell.
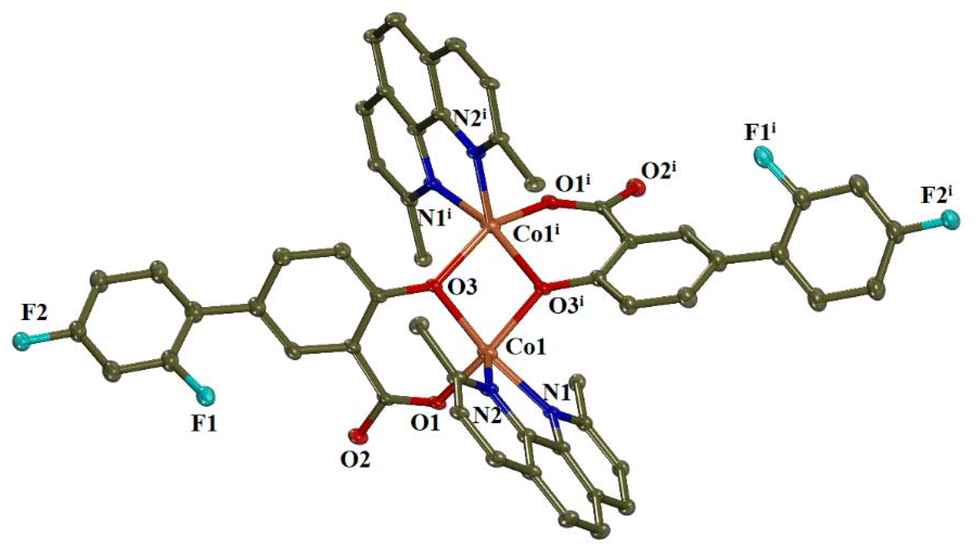 | ||
| Fig. 8 Molecular structure of complex 14. Methyl and aromatic hydrogen atoms, as well as solvate methanol molecules are omitted for clarity. | ||
In this centrosymmetric dinuclear Co(II) complex, the centre of symmetry is lying on the middle of the interatomic distance between the two cobalt(II) ions. The diflunisal ligands are doubly deprotonated and are coordinated to the cobalt ions in a tridentate bridging mode, i.e. via the phenolato oxygen atom O3 which bridges the two cobalt ions Co1 and Co1i and a carboxylato oxygen atom O1 which participate in the formation of a six-membered chelate ring. Although such coordination mode is rather usual for salicylato ligands,77–83 this is the first case reported for diflunisal ligands being in a tridentate bridging mode. There are also two complexes bearing doubly deprotonated diflunisal ligands which are coordinated in a bidentate chelating mode; an iron(III) dinuclear complex where the diflunisal ligands are terminal40 and a gallium(III) mononuclear compound.84 It should be also noted that a series of dinuclear copper(II) complexes with the general formula [Cu2(salicylato)2(N,N′-donor)2] have similar structural features with complex 14.81,85,86
Each Co(II) is five-coordinate with a CoN2O3 chromophore formed by two bridging phenolato oxygen atoms, one carboxylato oxygen atom and two nitrogen atoms from the neoc ligands. The geometry around cobalt(II) ions can be described as distorted square pyramid, according to the value of the trigonality index τ5 = (159.05(9)° − 144.43(10)°)/60° = 0.244 (τ5 = (φ1 − φ2)/60°, with φ1 and φ2 being the largest angles in the coordination sphere; τ5 = 0 is found for a perfect square pyramid and τ5 = 1 is calculated for a perfect trigonal bipyramid87). Based on the largest angles around Co1, atoms O1, O3, O3i, and N1 form the basal plane of the square pyramid, and N2 lies on the apical position. The Co1–Ocarboxylato bond distance is the shortest one in the coordination sphere (1.967(2) Å) while the Co1–Ophenolato and the Co1–Nneoc bond distances are of similar lengths (2.051(2)–2.083(3) Å).
The cobalt(II) ions are bridged by two phenolato oxygen atoms forming a parallelogram (Co1–O3–Co1i–O3i) with sides of 2.051(2) and 1.997(2) Å and angles of 78.62(11)° and 100.56(10)°. The interatomic Co1⋯Co1i separation distance is 3.114 Å and is in the range (2.923–3.4753 Å) found for two Co(II) bridged by two oxo-atoms.88–92 The solvate methanol molecules are stabilised via the formation of hydrogen bonds (Table S6†).
3.3. Antioxidant ability of the complexes
Free radicals are species with unpaired electron(s) and play a crucial role in inflammatory process. Radicals may transfer their unpaired electron(s) to adjacent molecule(s), and this process may ignite a series of chain reactions which are further responsible for diverse undesired side-effects in the organisms such as inflammations, swelling, or even cancer.63 The anti-inflammatory activity and potential subsequent anticancer effects of NSAIDs and their compounds are often connected with their activity against free radicals; they can either inhibit or capture these radicals. As a result, compounds with antioxidant properties might play an important role towards inflammation and can be pioneers for developing effective pharmaceuticals.93Within this context and taking into consideration the antioxidant efficacy of the free NSAIDs used as ligands in the complexes under study, the potential antioxidant activity of complexes 1–14 was evaluated through their ability to scavenge DPPH and ABTS radicals and to reduce H2O2. The results are summarised in Table S11† and depicted in Fig. 9, and were compared with the activity of well-known reference compounds NDGA, BHT, trolox and L-ascorbic acid (AA).93,94
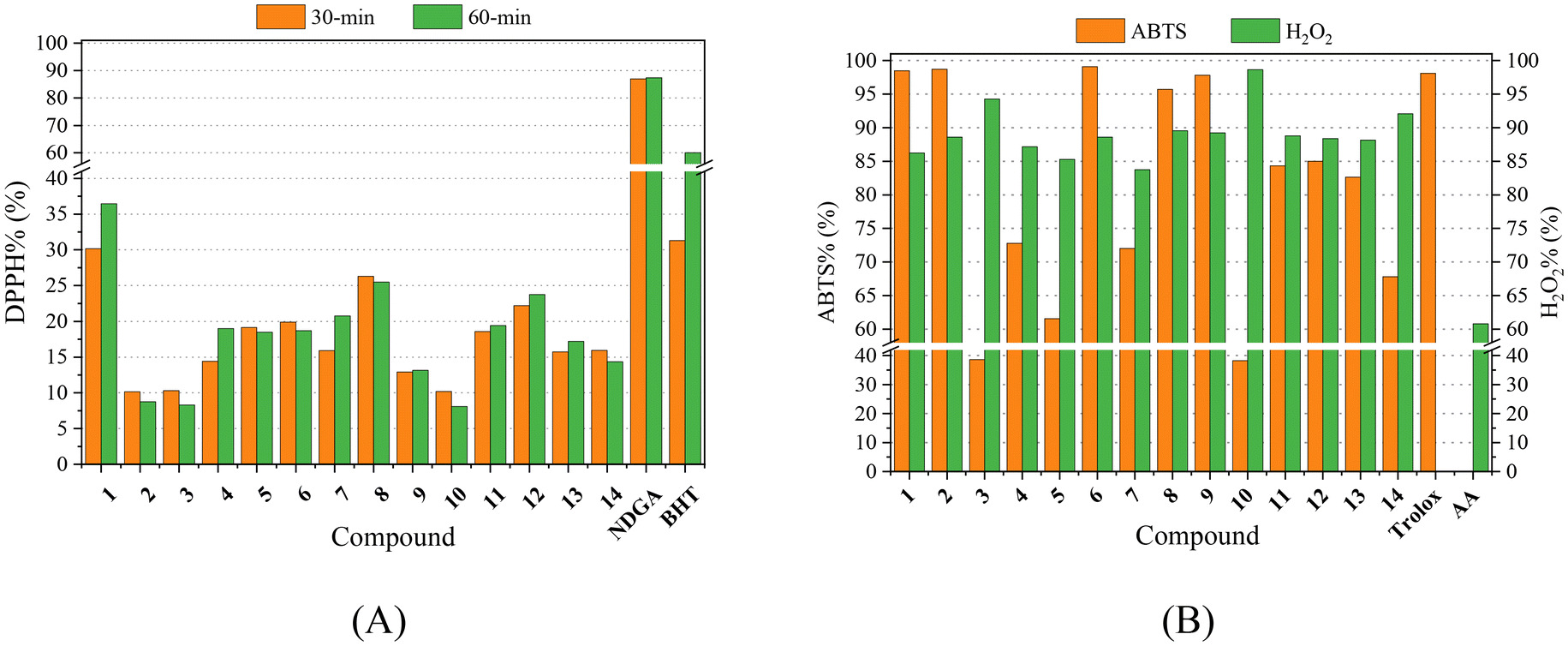 | ||
| Fig. 9 (A) % DPPH-scavenging ability (DPPH%) for the compounds after 30-min and 60-min incubation. (B) % ABTS radical scavenging activity (ABTS%) and H2O2 reducing activity (H2O2%) for the compounds. | ||
The neutralisation of DPPH radicals by antioxidants gains continuous recognition due to its potential activity against cancer, aging process and/or inflammation. The DPPH radical is a stable free radical which exhibits an intense absorption band at λmax = 517 nm.50 The ability of the compounds to scavenge DPPH radicals was found to be mainly time-independent, as shown after measuring the absorbance after 30 min and 60 min reaction, except for complexes 1 and 7 whose activity increased by ∼5–6% upon time. Almost all complexes were more active towards DPPH than the corresponding free NSAIDs. However, when compared with the reference compounds NDGA and BHT, complexes 1–14 showed much lower DPPH-scavenging activity (Table S11,†Fig. 9). Among the complexes under study, [Co(tolf)2(Himi)2] (complex 1) exhibited the highest DPPH-scavenging activity (DPPH% = 30.18–36.44%) which was close to the short-term (30 min) activity of the reference compound BHT (31.30 ± 0.10%).
The overall antioxidant activity of compounds is related to their ability to scavenge the cationic ABTS radical (ABTS˙+). This process is based on the discoloration of a dark green solution containing the cationic radical ABTS˙+ resulting from the oxidation caused by a hydrogen-donating antioxidant agent.51 In general, the activity of complexes 1–14 towards ABTS radicals is higher than the activity of the corresponding free NSAIDs (Fig. 9). When compared with the reference compound trolox, the activity of most complexes is moderate-to-high with complexes 1, 2, 6 and 9 being the most active compounds (ABTS% = 97.81–99.07%) approaching the ABTS-scavenging activity of trolox (= 98.10 ± 0.48%).
Hydrogen peroxide is an oxidizing agent capable to generate dangerous hydroxyl radicals (˙OH) when interacting with transition metal ions causing high oxidative stress. Therefore, scavengers of ˙OH or reductants of H2O2 may relieve oxidative stress and inhibit the production of reactive oxygen species.95 The interaction of complexes 1–14 with H2O2 was studied revealing that all complexes are more active (H2O2% = 83.73–98.64%, Fig. 9) than the corresponding free NSAIDs and L-ascorbic acid (= 60.80 ± 0.20%) which is the reference compound.96 Complex 10 ([Co(nap)2(neoc)]) was the most active complex towards H2O2 (H2O2% = 98.64 ± 0.90%).
In total, all complexes 1–14 exhibited significant activity especially towards the ABTS radicals and hydrogen peroxide which is comparable with other metal–NSAIDs complexes.16,17,37–39,41–43,47–49,51,54,55,63,97,98 When comparing the antioxidant activity of complexes 1–14 with that of the reported Co(II)–NSAID complexes,16,17,41–43,73 we may conclude that complex 1 is among the top-three DPPH-scavengers (DPPH%: 42.42 ± 0.13% for [Co(nap)2(phen)(H2O)2];16 36.8% for [Co(mef)2(phen)(MeOH)2];17 36.44 ± 0.11% for 1), and complexes 1, 2, 6 and 9 are among the most active ABTS-scavengers (ABTS%: 99.07 ± 0.07% for 6; 98.71 ± 0.14% for 2; 98.48 ± 0.13% for 1; 97.81 ± 0.06% for 9; 96.7% for [Co(mef)2(py)2(MeOH)2]17).
3.4. Interaction of the complexes with DNA
Coordination compounds can usually interact with DNA covalently (covalent binding to N atoms of DNA-bases) or noncovalently (via intercalation, electrostatically interactions, and/or major/minor groove binding) and/or may induce cleavage of the DNA-helix.99,100 The interaction of herein reported complexes 1–14 with CT DNA (which is linear DNA) was studied by viscosity measurements, cyclic voltammetry, UV-vis spectroscopy, and via their ability to displace EB which was monitored by fluorescence emission spectroscopy. In addition, the interaction of the complexes with plasmid DNA (which is circular DNA) was studied via gel electrophoresis.| Compound | λ (nm) (ΔA/A0 (%)a, Δλ (nm)b) | K b (M−1) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a “+” denotes hyperchromism, “−” denotes hypochromism. b “+” denotes red-shift, “−” denotes blue-shift. c “elim” = eliminated. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 1 | I: 301 (−4, 0) | 9.79(±0.28) × 104 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 2 | I: 300 (+3, +2), II: 334 (−10, 0) | 3.58(±0.24) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 3 | I: 320 (+2, +2), II: 332 (−1, 0) | 3.51(±0.24) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 4 | I: 297 (+12, +4), II: 317 (+8, +2) | 3.15(±0.08) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 5 | I: 300 (+10, +2), II: 317 (+5, 0) | 1.14(±0.14) × 106 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 6 | I: 291 (+4, +2) | 1.62(±0.08) × 106 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 7 | I: 299 (+10, +3), II: 321 (+1, 0) | 3.16(±0.32) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 8 | I: 296 (+2.5, +1) | 4.80(±0.13) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 9 | I: 311 (+20, +4), II: 348 (−40, elimc) | 4.14(±0.12) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 10 | I: 318 (−15, +2), II: 333 (−4, +1) | 6.55(±0.12) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 11 | I: 292 (−6, −2), II: 316 (−4, 0) | 4.60(±0.22) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 12 | I: 289 (+1, −3), II: 328 (−11, 0) | 3.18(±0.09) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 13 | I: 296 (−4, +2), II: 318 (−8, 0) | 1.01(±0.17) × 106 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 14 | I: 296 (−4.5, 0), II: 319 (−12, 0) | 4.59(±0.18) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Htolf41 | 305 (+40, +5) | 5.00(±0.10) × 104 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hmef17 | 324 (+10, 0) | 1.05(±0.02) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hnap16 | 325 (+22, 0) | 2.67(±0.22) × 104 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Na meclf51 | 302 (−12, −1) | 1.51(±0.12) × 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Na dicl52 | 295 (−7.5, 0) | 3.16(±0.14) × 104 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H2difl43 | 295 (+15, +2) | 3.08(±0.15) × 103 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The Wolfe–Shimer equation (eqn (S1)†)102 and the plots [DNA]/(εA − εf) versus [DNA] (Fig. S2†) were used for the calculation of the Kb values of the complexes. Almost all complexes 1–14 presented higher DNA-binding constants than the corresponding free NSAIDs (Table 1) with complex 6 being the tightest DNA-binder among the complexes according to its highest Kb value (= 1.62(±0.08) × 106 M−1). The Kb values of the complexes were also higher than that of the classic intercalator EB (Kb = 1.23 × 105 M−1).103 The DNA-binding constants of all complexes were found in the range reported for similar Co(II)–NSAID complexes in the literature.16,17,41–43,73 Comparing the DNA-binding constants of the Co(II)–NSAID complexes, complexes 5, 6 and 13 are among the tightest DNA–binders bearing Kb values higher than 106 M−1 (Kb = 1.01(±0.17) × 106 M−1 for 13; 1.14(±0.14) × 106 M−1 for 5; 1.14 × 106 M−1 for [Co(tolf)2(MeOH)4];41 1.62(±0.08) × 106 M−1 for 6; 2.28 × 106 M−1 for [Co(tolf)2(phen)(MeOH)2];41 3.03 × 106 M−1 for [Co2(indo)4(bipy)2(H2O)];73 3.57 × 106 M−1 for [Co3(fluf)6(bipy)2] (Hfluf the NSAID flufenamic acid);43 9.41 × 106 M−1 for [Co(dicl)2(bipy)];42 1.67 × 107 M−1 for [Co(dicl)2(MeOH)4]42).
Cyclic voltammetry is used to acquire additional information for the DNA–interaction profile of coordination compounds. In general, certain changes in the cyclic voltammograms of the complexes in the presence of CT DNA may shed light to the interaction mode between the coordination compounds and CT DNA. More specifically, the positive shift of a potential is evidence of intercalation, while a negative shift occurs in case of electrostatic interactions with DNA.104
The cyclic voltammograms of compounds 1–14 (0.33 mM) were recorded in the absence and presence of CT DNA solution (representatively shown for compounds 1 and 12 in Fig. 10). The cathodic potential (Epc) and the anodic potential (Epa) concerning the redox couple Co(II)/Co(I) for the complexes 1–14 and their shifts in presence of CT DNA are summarised in Table 2. The positive shifts of the potentials of the redox couple Co(II)/Co(I) for the complexes (up to +77 mV) may suggest an intercalating mode of interaction with CT DNA (Table 2). However, the presence of a negative shift is also observed in some cases. In conclusion, these features cannot lead to safe conclusions about the interaction mode between the complexes and CT DNA, although the intercalation is the most possible interaction mode which may be accompanied by external interactions. The ratio of the DNA–binding constants for the reduced (Kred) and oxidised forms (Kox) of the cobalt ions (Kred/Kox) was calculated with eqn (S2)†105 and is used to evaluate the redox equilibrium. For most complexes, the ratio Kred/Kox is higher than 1 (Table 2) revealing that CT DNA may interact selectively with the reduced form of the complexes.105
 | ||
| Fig. 10 Cyclic voltammograms of complexes 1 and 12 (0.33 mM, 1/2 DMSO/buffer), in the absence or presence of CT DNA. Scan rate = 100 mV s−1. Supporting electrolyte = buffer solution. The arrows show the changes upon addition of CT DNA. | ||
:2 DMSO:buffer solution in the absence (Epc(f), Epa(f)) and presence (Epc(b), Epa(b)) of CT DNA as well as their shifts (ΔEpc, ΔEpa). Ratio of equilibrium binding constants (Kr/Kox)
| Complex | E pc(f) | E pc(b) | ΔEpcc | E pa(f) | E pa(b) | ΔEpaf | K red/Kox | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a E pc(f) = Epc in DMSO/buffer in the absence of CT DNA. b E pc(b) = Epc in DMSO/buffer in the presence of CT DNA. c ΔEpc = Epc(b) − Epc(f). d E pa(f) = Epa in DMSO/buffer in the absence of CT DNA. e E pa(b) = Epc in DMSO/buffer in the presence of CT DNA. f ΔEpa = Epa(b) − Epa(f). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 1 | −780 | −765 | +15 | −500 | −480 | +20 | 1.35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 2 | −755 | −700 | +55 | −440 | −480 | −40 | 1.14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 3 | −714 | −708 | +6 | −530 | −520 | +10 | 1.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 4 | −765 | −700 | +65 | −475 | −530 | −55 | 1.09 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 5 | −770 | −700 | +70 | −445 | −495 | −50 | 1.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 6 | −730 | −715 | +15 | −503 | −500 | +3 | 1.17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 7 | −785 | −710 | +75 | −450 | −495 | −45 | 1.29 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 8 | −719 | −699 | +20 | −415 | −445 | −30 | 0.92 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 9 | −740 | −745 | −5 | −460 | −450 | +10 | 1.04 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 10 | −706 | −711 | −5 | −537 | −525 | +12 | 1.06 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 11 | −721 | −711 | +10 | −528 | −514 | +14 | 1.23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 12 | −762 | −700 | +62 | −440 | −525 | −85 | 0.83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 13 | −777 | −700 | +77 | −445 | −520 | −75 | 1.02 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex 14 | −670 | −675 | −5 | −492 | −482 | +10 | 1.04 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The measurement of DNA-viscosity is usually employed to monitor the interaction mode between compounds and CT DNA. The viscosity of the DNA-solution is measured because it is sensitive to changes in the relative length of the DNA chain constituting viscometry a reliable method to clarify the DNA-interaction mode. The presence of a compound that binds externally to CT DNA (i.e. electrostatically or groove-binding) will probably induce a slight bending of the double DNA-helix, without significantly affecting its length (in some cases, a slight shortening may occur) and subsequently resulting in negligible changes (usually slight decrease) of the DNA-viscosity. A classic intercalation of the compound in-between DNA-bases will result in an increase in the length of the DNA helix because of the increase of the separation distance between DNA-base pairs in order to host the inserting compound, and subsequently the viscosity of CT DNA will exhibit an increase.106 Furthermore, in the case of DNA-cleavage induced by the compound, the formation of much shorter fragments with significantly shorter relevant length will be revealed through a significant decrease of DNA-viscosity.
In the present study, the viscosity of a CT DNA solution (0.1 mM) was measured in the presence of incrementally increasing amounts of compounds 1–14 (up to r = [compound]/[DNA] = 0.35). For all compounds 1–14, the viscosity increased (Fig. 11) suggesting intercalation as the most possible mode of their interaction with CT DNA.107 In some cases, the lowering of DNA-viscosity at the early steps of incremental addition of the compounds (r values up to 0.15) indicated an initial external interaction which aided to closer approach followed by intercalation.
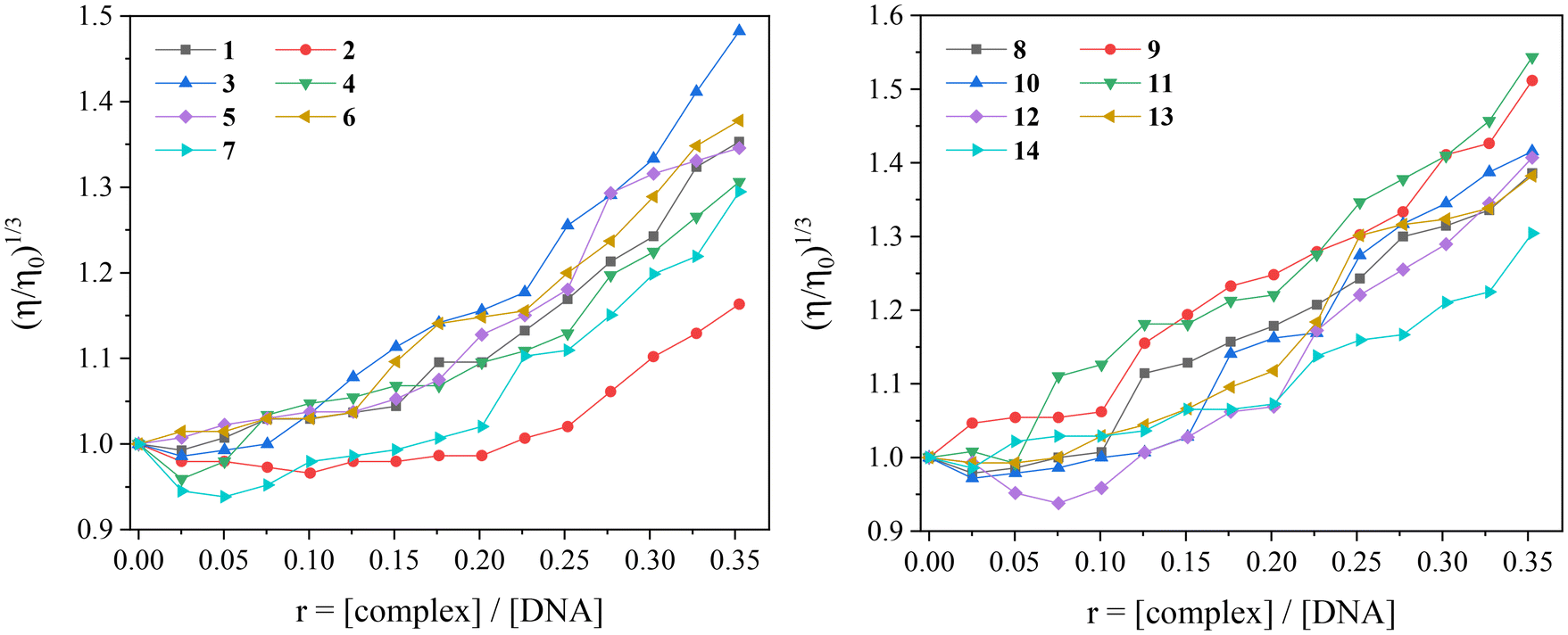 | ||
| Fig. 11 Relative viscosity (η/η0)1/3 of CT DNA (0.1 mM) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) in the presence of complexes 1–14 at increasing amounts (r = [complex]/[DNA]). | ||
Ethidium bromide is a typical DNA-intercalator since its planar phenanthridine ring inserts in-between two adjacent DNA-bases. As a fluorescent compound, EB exhibits an intense emission band at 592–594 nm when bound to CT DNA. For this reason, the changes of the fluorescence emission spectra of the EB–DNA adduct induced by the addition of a compound are monitored to investigate its potential interaction mode with CT DNA. The EB–DNA emission band at 592 nm will decrease upon addition of a compound which binds to CT DNA as strongly as or more strongly than EB does. A slight or negligible decrease in fluorescence emission indicates an inability to displace EB from the EB–DNA adduct and is usually observed for compounds that do not act as intercalators.108
In the current study, the fluorescence emission spectra of the EB–DNA adduct (which was formed after 1 h pretreatment of EB (40 μM) and CT DNA (40 μM) in buffer solution) were recorded in the presence of increasing amounts of solutions of complexes 1–14 and the occurring changes were monitored. The addition of the complexes in the EB–DNA solution led to a significant decrease in fluorescence emission band at 594 nm (Fig. 12). In total, the decrease of the intensity of the emission band attributed to the EB–DNA adduct was significant (up to 74.5% of the initial fluorescence, Table 3, Fig. S3†), and may be assigned to the displacement of EB from the EB–DNA adduct.
 | ||
| Fig. 12 Fluorescence emission spectra (λexcitation = 540 nm) for EB–DNA conjugate ([EB] = 40 μM, [DNA] = 40 μM) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH = 7.0) in the absence and presence of increasing amounts (up to r = [complex]/[DNA] = 0.21) of complex (A) 1 and (B) 8. The arrows show the changes of intensity upon increasing amounts of the complexes. | ||
| Compound | ΔI/I0 (%) | K SV (M−1) | k q (M−1 s−1) |
|---|---|---|---|
| Complex 1 | 74.5 | 3.50(±0.06) × 105 | 1.52(±0.03) × 1013 |
| Complex 2 | 66.8 | 1.89(±0.04) × 105 | 8.21(±0.02) × 1012 |
| Complex 3 | 41.2 | 2.71(±0.06) × 104 | 1.18(±0.03) × 1012 |
| Complex 4 | 50.9 | 1.01(±0.02) × 105 | 4.39(±0.09) × 1012 |
| Complex 5 | 72.6 | 2.04(±0.06) × 105 | 8.87(±0.03) × 1012 |
| Complex 6 | 43.3 | 1.09(±0.02) × 105 | 4.75(±0.07) × 1012 |
| Complex 7 | 53.2 | 1.08(±0.03) × 105 | 4.68(±0.12) × 1012 |
| Complex 8 | 69.6 | 4.55(±0.08) × 105 | 1.98(±0.04) × 1013 |
| Complex 9 | 43.4 | 5.50(±0.10) × 104 | 2.39(±0.04) × 1012 |
| Complex 10 | 42.0 | 3.52(±0.05) × 104 | 1.53(±0.02) × 1012 |
| Complex 11 | 49.4 | 1.01(±0.02) × 105 | 4.38(±0.08) × 1012 |
| Complex 12 | 49.3 | 9.87(±0.20) × 104 | 4.29(±0.09) × 1012 |
| Complex 13 | 51.6 | 5.68(±0.10) × 105 | 2.47(±0.42) × 1013 |
| Complex 14 | 68.9 | 5.42(±0.13) × 105 | 2.36(±0.06) × 1013 |
| Htolf41 | 74.0 | 1.15(±0.04) × 106 | 5.00(±0.17) × 1013 |
| Hmef17 | 80.0 | 1.58(±0.06) × 105 | 6.87(±0.26) × 1012 |
| Hnap16 | 82.0 | 1.47(±0.04) × 105 | 6.39(±0.17) × 1012 |
| Na meclf51 | 80.1 | 8.20(±0.26) × 104 | 3.57(±0.11) × 1012 |
| Na dicl52 | 65.0 | 2.47(±0.06) × 105 | 1.07(±0.03) × 1013 |
| H2difl43 | 65.0 | 8.59(±0.35) × 105 | 3.73(±0.15) × 1013 |
The ability of the compounds to induce quenching of the fluorescence emission EB–DNA band is evaluated through the value of the Stern–Volmer constants (KSV) which were calculated with the Stern–Volmer equation (eqn (S3)†) and the corresponding Stern–Volmer plots (Fig. S4†). All complexes show relatively high KSV values (Table 3), with complex 13 bearing the highest Stern–Volmer constant (KSV = 5.68(±0.10) × 105 M−1). In addition, the quenching constants (kq) of the complexes were calculated with eqn (S4)† (the value of fluorescence lifetime of EB–DNA system (τ0) is equal to 23 ns109), and have significantly higher values (Table 3) than the value of 1010 M−1 s−1, indicating a static quenching mechanism which may confirm the formation of a new adduct between CT DNA and each complex.
The potential DNA-cleaving activity of complexes 1–13 (Fig. S5,† Lane 2–14) was revealed only as single-stranded (ss) nicks in the supercoiled DNA forming relaxed circular DNA (Form II), since no signal (double-stranded (ds) nick) attributed to the formation of linear DNA (Form III) was observed. The percentages of the cleavage (shown in Fig. S5†) were calculated with eqn (S5) and (S6).† At the high concentration of 500 μM used in the experiment, the compounds present low-to-negligible ability to cleave DNA with complex 8 being the only active among the compounds showing a 35% of cleavage (Fig. S5,† Lane 9). Although the reports concerning the pDNA-cleavage ability of metal–NSAID complexes are quite few,110 it seems that herein reported Co(II)–NSAID complexes cannot induce significantly the cleavage of pDNA.
3.5. Interaction of the complexes with albumins
The interaction of complexes 1–14 with the albumins was studied by monitoring the quenching of tryptophan fluorescence emission band in the presence of incremental addition of the complexes. The fluorescence emission spectra of the albumins (3 mM) in buffer solution were recorded in the range 300–500 nm for λexcitation = 295 nm (Fig. S6†). The incremental addition of complexes 1–14 resulted in a decrease of the intensity of the corresponding albumin emission band (λmax,emission = 340 nm for HSA and 345 nm for BSA), while in the case of complex 14 an additional emission band (as previously reported for diflunisal and its complexes)112 appeared at 415 nm (Fig. S6†). All complexes induced significant quenching of the albumin emission band which was more pronounced in the case of BSA than HSA (Fig. S7 and S8†). Such quenching may be attributed to re-arrangement of the SA secondary structure or its modifications as a result from the association with the complexes.108 For the quantitative evaluation of this interaction, the fluorescence emission spectra (with λexcitation = 295 nm) of the free complexes were subtracted from the overall spectra. The influence of the inner-filter effect on the measurements was assessed with eqn (S7),†113 and was found negligible to affect the measurements.
The Stern–Volmer constant (Ksv), the SA-quenching constant (kq) and the SA-binding constant (K) of the compounds were calculated with the Stern–Volmer and Scatchard equations (eqn (S3), (S4) and (S8)†) and plots (Fig. S9–S12†) considering that the fluorescence lifetime of tryptophan in SAs (τ0) has the value of 10−8 s.108 The calculated kq values of complexes 1–14 are significantly higher (in most cases by two-to-three orders) than the value of 1010 M−1 s−1 (Table 4) and indicate that the quenching takes place via a static mechanism108 which subsequently may confirm the interaction of the complexes with the albumins.
| Compound | k q(BSA) (M−1 s−1) | K (BSA) (M−1) | k q(HSA) (M−1 s−1) | K (HSA) (M−1) |
|---|---|---|---|---|
| Complex 1 | 1.08(±0.06) × 1014 | 2.40(±0.29) × 106 | 3.87(±0.14) × 1012 | 6.45(±0.20) × 104 |
| Complex 2 | 3.92(±0.29) × 1013 | 8.37(±0.10) × 105 | 1.41(±0.07) × 1012 | 9.44(±0.30) × 104 |
| Complex 3 | 3.70(±0.11) × 1012 | 2.47(±0.07) × 105 | 3.93(±0.17) × 1011 | 1.92(±0.07) × 104 |
| Complex 4 | 8.74(±0.44) × 1013 | 1.01(±0.04) × 106 | 1.47(±0.07) × 1012 | 1.32(±0.07) × 105 |
| Complex 5 | 7.57(±0.52) × 1013 | 2.36(±0.11) × 106 | 1.08(±0.04) × 1012 | 1.80(±0.08) × 105 |
| Complex 6 | 1.98(±0.08) × 1013 | 6.44(±0.19) × 105 | 1.35(±0.04) × 1012 | 4.68(±0.16) × 104 |
| Complex 7 | 9.26(±0.58) × 1013 | 1.19(±0.04) × 106 | 1.38(±0.08) × 1012 | 8.36(±0.39) × 104 |
| Complex 8 | 9.99(±0.42) × 1013 | 1.56(±0.37) × 106 | 4.43(±0.17) × 1012 | 1.12(±0.37) × 105 |
| Complex 9 | 8.14(±0.21) × 1013 | 7.90(±0.24) × 105 | 3.93(±0.10) × 1012 | 9.59(±0.50) × 104 |
| Complex 10 | 4.16(±0.17) × 1012 | 3.01(±0.14) × 104 | 8.14(±0.31) × 1011 | 1.98(±0.06) × 104 |
| Complex 11 | 7.16(±0.40) × 1013 | 7.67(±0.38) × 105 | 1.89(±0.08) × 1012 | 7.63(±0.31) × 104 |
| Complex 12 | 6.35(±0.39) × 1013 | 1.11(±0.05) × 106 | 1.80(±0.04) × 1012 | 1.73(±0.07) × 104 |
| Complex 13 | 1.03(±0.05) × 1014 | 9.24(±0.51) × 105 | 2.05(±0.10) × 1012 | 8.13(±0.41) × 104 |
| Complex 14 | 3.14(±0.20) × 1013 | 6.09(±0.15) × 105 | 1.42(±0.05) × 1012 | 5.38(±0.21) × 104 |
| Htolf41 | 2.18(±0.12) × 1013 | 1.60(±0.14) × 105 | 6.10(±0.38) × 1012 | 3.12(±0.25) × 105 |
| Hmef17 | 2.78(±0.20) × 1013 | 1.35(±0.22) × 105 | 7.13(±0.34) × 1012 | 1.32(±0.15) × 105 |
| Hnap16 | 1.18(±0.06) × 1012 | 5.35(±0.42) × 103 | 1.24(±0.09) × 1012 | 3.27(±0.30) × 104 |
| Na meclf51 | 4.84(±0.32) × 1013 | 1.78(±0.11) × 106 | 2.98(±0.31) × 1013 | 1.05(±0.03) × 106 |
| Na dicl52 | 8.11(±0.34) × 1012 | 3.55(±0.03) × 105 | 1.81(±0.17) × 1012 | 1.63 × 105 |
| H2difl43 | 1.53(±0.08) × 1013 | 1.93(±0.15) × 105 | 2.67(±0.16) × 1012 | 1.22(±0.07) × 105 |
The values of the SA-binding constants of complexes 1–14 are of the 104–106 M−1 magnitude (Table 4) and fall within the range reported for many metal(II)–NSAID complexes.16,17,37–43,47–49,51,54,55,63,97,98 Complexes 1 and 5 possess the highest K values for BSA among the compounds under study, while the highest HSA-binding constant was found for complex 5 (Table 4). Therefore, complex 5 is found to exhibit the highest affinity for both albumins among the herein studied complexes. A comparison with the albumin-binding constants of the reported Co(II)–NSAID complexes16,17,41–43,73 may reveal that complexes 1–14 under study, especially complexes 1 and 5, bear some of the highest BSA-binding constants (>2 × 106 M−1) among such complexes. The K values observed for complexes 1–14 are significantly lower than the value of the association constants of avidin with various compounds (ca. 1015 M−1) which is considered the borderline for reversible noncovalent interactions. So, the SA-binding constants of the complexes suggest their reversible binding to the albumins, and the potential to be transferred by the albumins, and get released at their specific targets.114
The incremental addition of each complex in a solution containing BSA and the site-marker (warfarin or ibuprofen) resulted in a quenching of the initial fluorescence emission band (representatively shown in Fig. 13). A decrease of the K value in presence of the site-marker indicates that the binding of the compound to albumin is influenced by the presence of this marker and can be attributed to the competition for the same binding site.100,116 The SA-binding constants of the compounds in the presence of warfarin or ibuprofen were calculated (with the Scatchard equation (eqn (S8)†) and plots (Fig. S13 and S14†)) and their values (Table 5) are compared with those determined in the absence of any site-marker in order to check for the preferable drug site.
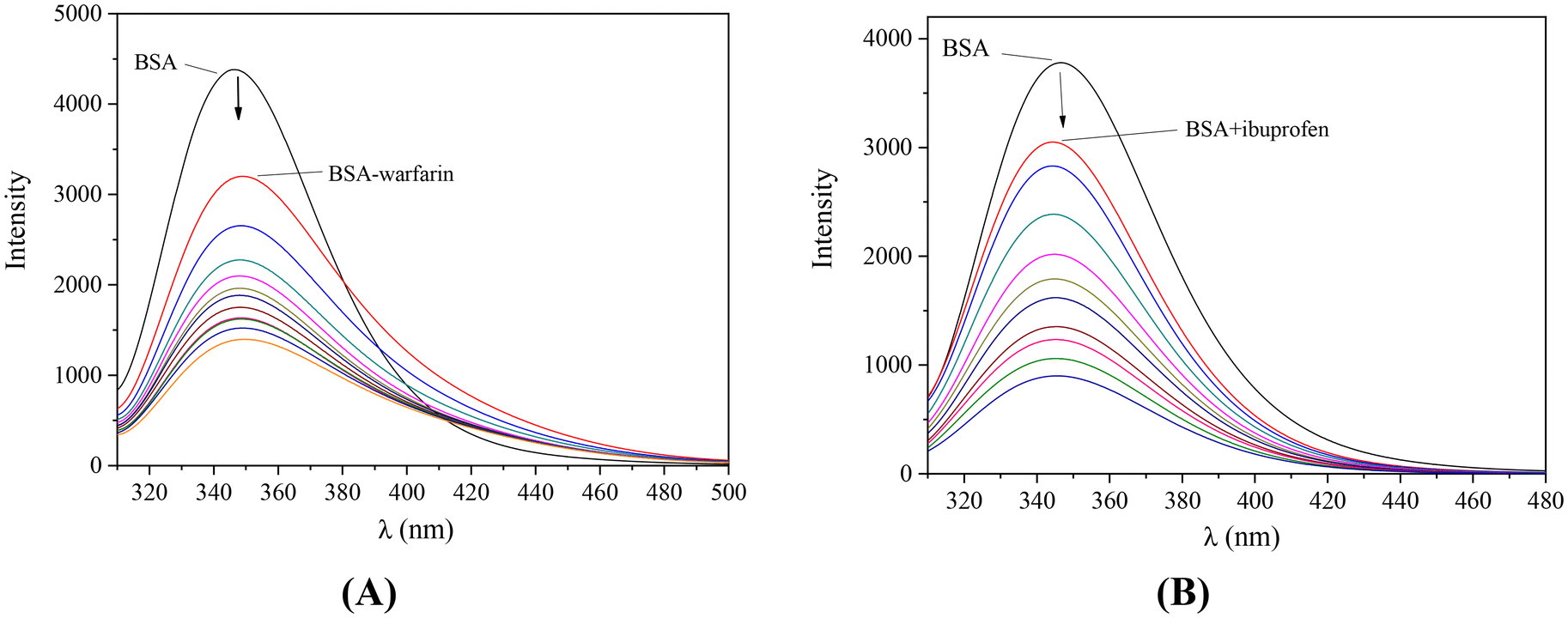 | ||
| Fig. 13 Fluorescence emission spectra (λexcitation = 295 nm) of a buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) of BSA (3 μM) (A) in the presence of warfarin (3 μM) upon addition of increasing amounts of complex 1, (B) in the presence of ibuprofen (3 μM) upon addition of increasing amounts of complex 2. The arrows show the changes of intensity upon increasing amounts of the complexes. | ||
| Complex | K (BSA) | K (BSA,ibu) | K (BSA,warf) |
|---|---|---|---|
| Complex 1 | 2.40(±0.29) × 106 | 2.29(±0.07) × 105 | 3.76(±0.15) × 105 |
| Complex 2 | 8.37(±0.10) × 105 | 9.42(±0.34) × 104 | 2.54(±0.09) × 105 |
| Complex 3 | 2.47(±0.07) × 105 | 9.31(±0.58) × 104 | 2.90(±0.28) × 104 |
| Complex 4 | 1.01(±0.04) × 106 | 6.23(±0.26) × 105 | 5.42(±0.21) × 105 |
| Complex 5 | 2.36(±0.11) × 106 | 4.60(±0.13) × 105 | 7.18(±0.23) × 105 |
| Complex 6 | 6.44(±0.19) × 105 | 2.20(±0.06) × 105 | 2.38(±0.11) × 105 |
| Complex 7 | 1.19(±0.04) × 106 | 6.51(±0.18) × 105 | 1.59(±0.06) × 105 |
| Complex 8 | 1.56(±0.37) × 106 | 3.55(±0.14) × 105 | 2.67(±0.13) × 105 |
| Complex 9 | 7.90(±0.24) × 105 | 2.21(±0.06) × 105 | 2.30(±0.09) × 105 |
| Complex 10 | 3.01(±0.14) × 104 | 8.27(±0.56) × 103 | 1.37(±0.04) × 105 |
| Complex 11 | 7.67(±0.38) × 105 | 7.13(±0.25) × 105 | 5.76(±0.20) × 105 |
| Complex 12 | 1.11(±0.05) × 106 | 9.49(±0.38) × 105 | 4.53(±0.15) × 105 |
| Complex 13 | 9.24(±0.51) × 105 | 5.25(±0.14) × 105 | 4.83(±0.15) × 105 |
| Complex 14 | 6.09(±0.15) × 105 | 4.99(±0.15) × 105 | 5.08(±0.09) × 105 |
For almost all complexes (Table 5), a significant decrease of the BSA-binding constants in the presence of both warfarin and ibuprofen was found showing that the complexes may bind to Sudlow sites I and II, respectively. However, for complexes 1, 2, 5 and 10, lower K values were calculated in the presence of ibuprofen than warfarin suggesting their preference for drug site II. On the other hand, for complexes 3, 4, 7, 8, and 11–12, a selective binding to drug site I may be suggested since the presence of warfarin led to lower K values than the presence of ibuprofen did.100,116 For complexes 6, 9, 13 and 14, the presence of both site-markers resulted in similarly decreased K values revealing the lack of selectivity for Sudlow sites I and II.
4. Conclusions
Fourteen cobalt(II) complexes with diverse carboxylate NSAIDs as ligands in the presence of a series of nitrogen-donors as co-ligands have been isolated and characterised. The crystal structures of eight out of the fourteen cobalt(II) complexes, i.e. complexes 1, 2, 5, 6, 8–10 and 14, were determined by single-crystal X-ray crystallography. In complexes 1–13, the NSAIDs are simply deprotonated acting as monodentate or bidentate chelating ligands. In complex 14, diflunisal is doubly deprotonated and is bound as a tridentate bridging ligand leading to the formation of the dinuclear complex [Co2(difl)2(neoc)2]·MeOH. This is the first structure that diflunisal is coordinated in a tridentate bridging mode.The in vitro potential biological profile of the complexes was evaluated regarding their interaction with biomacromolecules (albumins and DNA), and their antioxidant ability. The complexes may interact with CT DNA via intercalation and the highest DNA-binding constant was found for complex 6 (Kb = 1.62(±0.08) × 106 M−1). The ability of the complexes to cleave pBR322 plasmid DNA to relaxed circular DNA is rather low at the concentration of 500 μM, since only complex 8 showed noteworthy cleavage activity (up to 35%).
The binding of complexes 1–14 to bovine and human serum albumins is tight and reversible showing their potency for effective transportation towards potential biological targets of the complexes where they may get released in order to fulfil a possible biological mission. Furthermore, competitive studies with the typical site-markers warfarin and ibuprofen were employed to explore whether the complexes bind selectively to typical BSA-binding sites. A clear selectivity tendency was not revealed since almost equal numbers of the complexes prefer to bind at Sudlow's site 1 (in competition with warfarin) or Sudlow's site 2 (in competition with ibuprofen) or both sites (almost equal BSA-binding constants in the presence of warfarin and ibuprofen).
Regarding the antioxidant potency of the complexes, their ability to scavenge DPPH and ABTS free radicals and to reduce H2O2 was monitored. Almost all complexes were more active than the corresponding free NSAIDs. The DPPH-scavenging ability of the complexes is relatively low except for [Co(tolf)2(Himi)2] (complex 1) which was the most active DPPH-scavenger with relatively moderate time-dependent activity (DPPH% = 30.18–36.44%). Most complexes showed significant ability to scavenge ABTS radicals with complexes 1, 2, 6 and 9 (ABTS% = 97.81–99.07%) being as active as the reference compound trolox. Concerning the activity towards H2O2, all complexes were found more active than the reference compound L-ascorbic acid.
In conclusion, the cobalt(II)–NSAID compounds showed significant ABTS scavenging and hydrogen peroxide reduction activity and may bind tightly to CT DNA. Such biological activity is a prerequisite for potential anticancer activity. Thus, the herein reported data combined with the affinity of the compounds for albumins may reveal a group of potentially bioactive compounds deserving further biological studies.
Abbreviations
| AA | L-Ascorbic acid |
| ABTS | 2,2′-Azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) |
| 3-ampy | 3-Aminopyridine |
| BHT | Butylated hydroxytoluene |
| bipy | 2,2′-Bipyridine |
| bipyam | 2,2′-Bipyridylamine |
| BSA | Bovine serum albumin |
| CT | Calf-thymus |
| dicl− | Anion of diclofenac |
| difl−2 | Dianion of diflunisal |
| DPPH | 1,1-Diphenyl-picrylhydrazyl |
| EB | Ethidium bromide |
| fluf− | Flufenamato anion |
| Hdicl | Diclofenac |
| Hdifl− | Anion of diflunisal |
| Hfluf | Flufenamic acid |
| Himi | 1H-Imidazole |
| Hindo | Indomethacin |
| Hmeclf | Meclofenamic acid |
| Hmef | Mefenamic acid |
| Hnap | Naproxen |
| Hnif | Niflumic acid |
| HSA | Human serum albumin |
| Htolf | Tolfenamic acid |
| H2difl | Diflunisal |
| indo− | Anion of indomethacin |
| K | SA-binding constant |
| K b | DNA-binding constant |
| K ox | DNA-binding constant for the oxidised form |
| k q | Quenching constant |
| K r | DNA-binding constant for the reduced form |
| K SV | Stern–Volmer constant |
| meclf− | Meclofenamato anion |
| mef− | Mefenamato anion |
| nap− | Anion of naproxen |
| NDGA | Nordihydroguaiaretic acid |
| neoc | Neocuproine, 2,9-dimethyl-1,10-phenanthroline |
| nif− | Niflumato anion |
| NSAID | Non-steroidal anti-inflammatory drugs |
| RT | Room-temperature |
| phen | 1,10-Phenanthroline |
| py | Pyridine |
| SA | Serum albumin |
| trolox | 6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid |
| tolf− | Tolfenamato anion |
| Δν(COO) | v asym(COO) − vsym(COO) |
Data availability
The data supporting this article have been included as part of the ESI.†CCDC deposition numbers 2336775–2336782 contain the supplementary crystallographic data for the complexes. Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1039/d4dt01846j.
Conflicts of interest
There are no conflicts to declare.References
- P. Chellan and P. J. Sadler, Philos. Trans. R. Soc., A, 2015, 373, 2014018 CrossRef PubMed.
- M. A. Zoroddu, J. Aaseth, G. Crisponi, S. Medici, M. Peana and V. M. Nurchi, J. Inorg. Biochem., 2019, 195, 120–129 CrossRef CAS PubMed.
- K. Yamada, Met. Ions Life Sci., 2013, 13, 295–320 Search PubMed.
- P. J. Sadler, Adv. Inorg. Chem., 1991, 36, 1–48 CrossRef CAS.
- Y. gui Gao, M. Sriram and A. H. J. Wang, Nucleic Acids Res., 1993, 21, 4093–4101 CrossRef PubMed.
- E. L. Baldwin, J. A. W. Byl and N. Osheroff, Biochemistry, 2004, 43, 728–735 CrossRef CAS PubMed.
- M. C. Heffern, N. Yamamoto, R. J. Holbrook, A. L. Eckermann and T. J. Meade, Curr. Opin. Chem. Biol., 2013, 17, 189–196 CrossRef CAS PubMed.
- A. K. Renfrew, E. S. O'Neill, T. W. Hambley and E. J. New, Coord. Chem. Rev., 2018, 375, 221–233 CrossRef CAS.
- Z. Zhao, J. Zhang, S. Zhi, W. Song and J. Zhao, J. Inorg. Biochem., 2019, 197, 110696 CrossRef CAS PubMed.
- H. Crlikova, H. Kostrhunova, J. Pracharova, M. Kozsup, S. Nagy, P. Buglyó, V. Brabec and J. Kasparkova, JBIC, J. Biol. Inorg. Chem., 2020, 25, 339–350 CrossRef CAS PubMed.
- L. Q. Chai, L. Zhou, H. Bin Zhang, K. H. Mao and H. S. Zhang, New J. Chem., 2019, 43, 12417–12430 RSC.
- G. Balan, O. Burduniuc, I. Usataia, V. Graur, Y. Chumakov, P. Petrenko, V. Gudumac, A. Gulea and E. Pahontu, Appl. Organomet. Chem., 2020, 34, e5423 CrossRef CAS.
- A. M. Mansour and M. S. Ragab, RSC Adv., 2019, 9, 30879–30887 RSC.
- A. P. King, H. A. Gellineau, S. N. Macmillan and J. J. Wilson, Dalton Trans., 2019, 48, 5987–6002 RSC.
- E. L. Chang, C. Simmers and D. A. Knight, Pharmaceuticals, 2010, 3, 1711–1728 CrossRef CAS PubMed.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2012, 107, 54–64 CrossRef CAS PubMed.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, Dalton Trans., 2010, 39, 4517–4528 RSC.
- C. N. Banti and S. K. Hadjikakou, Eur. J. Inorg. Chem., 2016, 3048–3071 CrossRef CAS.
- G. Psomas and D. P. Kessissoglou, Dalton Trans., 2013, 42, 6252–6276 RSC.
- A. R. Amin, P. Vyas, M. Attur, J. Leszczynska-Piziak, I. R. Patel, G. Weissmann and S. B. Abramson, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7926–7930 CrossRef CAS PubMed.
- K. S. Kim, J. H. Yoon, J. K. Kim, S. J. Baek, T. E. Eling, W. J. Lee, J. H. Ryu, J. G. Lee, J. H. Lee and J. B. Yoo, Biochem. Biophys. Res. Commun., 2004, 325, 1298–1303 CrossRef CAS PubMed.
- A. Inoue, S. Muranaka, H. Fujita, T. Kanno, H. Tamai and K. Utsumi, Free Radicals Biol. Med., 2004, 37, 1290–1299 CrossRef CAS PubMed.
- D. Ho Woo, I. S. Han and G. Jung, Life Sci., 2004, 75, 2439–2449 CrossRef PubMed.
- T. Pringsheim, W. J. Davenport and D. Dodick, Neurology, 2008, 70, 1555–1563 CrossRef CAS PubMed.
- E. Moilanen and H. Kankaanranta, Pharmacol. Toxicol., 1994, 75, 60–63 CrossRef CAS PubMed.
- A. S. Kalgutkar, B. C. Crews, S. W. Rowlinson, A. B. Marnett, K. R. Kozak, R. P. Remmel and L. J. Marnett, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 925–930 CrossRef CAS PubMed.
- L. French, Am. Fam. Physician, 2005, 71, 285–291 Search PubMed.
- F. Ruschitzka, J. S. Borer, H. Krum, A. J. Flammer, N. D. Yeomans, P. Libby, T. F. Lüscher, D. H. Solomon, M. E. Husni, D. Y. Graham, D. A. Davey, L. M. Wisniewski, V. Menon, R. Fayyad, B. Beckerman, D. Iorga, A. M. Lincoff and S. E. Nissen, Eur. Heart J., 2017, 38, 3282–3292 CrossRef CAS PubMed.
- S. E. Nissen, N. D. Yeomans, D. H. Solomon, T. F. Lüscher, P. Libby, M. E. Husni, D. Y. Graham, J. S. Borer, L. M. Wisniewski, K. E. Wolski, Q. Wang, V. Menon, F. Ruschitzka, M. Gaffney, B. Beckerman, M. F. Berger, W. Bao and A. M. Lincoff, N. Engl. J. Med., 2016, 375, 2519–2529 CrossRef CAS PubMed.
- W. J. Wechter, E. D. Murray, D. Kantoci, D. D. Quiggle, D. D. Leipold, K. M. Gibson and J. D. McCracken, Life Sci., 2000, 66, 745–753 CrossRef CAS PubMed.
- G. M. Lawton and P. J. Chapman, Aust. Dent. J., 1993, 38, 265–271 CrossRef CAS PubMed.
- G. Lohrmann, A. Pipilas, R. Mussinelli, D. M. Gopal, J. L. Berk, L. H. Connors, N. Vellanki, J. Hellawell, O. K. Siddiqi, J. Fox, M. S. Maurer and F. L. Ruberg, J. Card. Failure, 2020, 26, 753–759 CrossRef PubMed.
- D. Wojcieszyńska, H. Guzik and U. Guzik, Sci. Total Environ., 2022, 834, 155317 CrossRef PubMed.
- M. T. Kelleni, Biomed. Pharmacother., 2021, 133, 110982 CrossRef CAS PubMed.
- G. D. Geromichalos, A. Tarushi, K. Lafazanis, A. A. Pantazaki, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2018, 187, 41–55 CrossRef CAS PubMed.
- A. Tarushi, G. D. Geromichalos, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2019, 190, 1–14 CrossRef CAS PubMed.
- M. Zampakou, N. Rizeq, V. Tangoulis, A. N. Papadopoulos, F. Perdih, I. Turel and G. Psomas, Inorg. Chem., 2014, 53, 2040–2052 CrossRef CAS PubMed.
- F. Dimiza, C. P. Raptopoulou, V. Psycharis, A. N. Papadopoulos and G. Psomas, New J. Chem., 2018, 42, 16666–16681 RSC.
- F. Dimiza, A. G. Hatzidimitriou, Y. Sanakis, A. N. Papadopoulos and G. Psomas, J. Inorg. Biochem., 2021, 218, 111410 CrossRef CAS PubMed.
- F. Dimiza, A. Barmpa, A. Chronakis, A. G. Hatzidimitriou, Y. Sanakis, A. N. Papadopoulos and G. Psomas, Int. J. Mol. Sci., 2023, 24, 6391 CrossRef CAS PubMed.
- S. Tsiliou, L. A. Kefala, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, Eur. J. Med. Chem., 2012, 48, 132–142 CrossRef CAS PubMed.
- S. Perontsis, A. Dimitriou, P. Fotiadou, A. G. Hatzidimitriou, A. N. Papadopoulos and G. Psomas, J. Inorg. Biochem., 2019, 196, 110688 CrossRef CAS PubMed.
- S. Tsiliou, L. A. Kefala, A. G. Hatzidimitriou, D. P. Kessissoglou, F. Perdih, A. N. Papadopoulos, I. Turel and G. Psomas, J. Inorg. Biochem., 2016, 160, 125–139 CrossRef CAS PubMed.
- R. Smolkova, V. Zelenak, L. Smolko and M. Dusek, Z. Kristallogr. - Cryst. Mater., 2016, 231, 715–724 CrossRef CAS.
- R. Smolkova, L. Smolko, E. Samolova and M. Dusek, J. Mol. Struct., 2023, 1272, 134172 CrossRef CAS.
- G. G. Nnabuike, S. Salunke-Gawali, A. S. Patil, R. J. Butcher, J. A. Obaleye, H. Ashtekar and B. Prakash, J. Mol. Struct., 2023, 1285, 135519 CrossRef CAS.
- S. Perontsis, A. G. Hatzidimitriou, A. N. Papadopoulos and G. Psomas, J. Inorg. Biochem., 2016, 162, 9–21 CrossRef CAS PubMed.
- A. Barmpa, G. D. Geromichalos, A. G. Hatzidimitriou and G. Psomas, J. Inorg. Biochem., 2021, 222, 111507 CrossRef CAS PubMed.
- X. Totta, A. A. Papadopoulou, A. G. Hatzidimitriou, A. Papadopoulos and G. Psomas, J. Inorg. Biochem., 2015, 145, 79–93 CrossRef CAS PubMed.
- F. Dimiza, S. Fountoulaki, A. N. Papadopoulos, C. A. Kontogiorgis, V. Tangoulis, C. P. Raptopoulou, V. Psycharis, A. Terzis, D. P. Kessissoglou and G. Psomas, Dalton Trans., 2011, 40, 8555–8568 RSC.
- A. Barmpa, A. G. Hatzidimitriou and G. Psomas, J. Inorg. Biochem., 2021, 217, 111357 CrossRef CAS PubMed.
- F. Dimiza, F. Perdih, V. Tangoulis, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 476–489 CrossRef CAS PubMed.
- G. Malis, A. S. Bakali, A. G. Hatzidimitriou and G. Psomas, J. Mol. Struct., 2024, 1303, 137590 CrossRef CAS.
- A. Tarushi, X. Totta, A. Papadopoulos, J. Kljun, I. Turel, D. P. Kessissoglou and G. Psomas, Eur. J. Med. Chem., 2014, 74, 187–198 CrossRef CAS PubMed.
- A. Tarushi, Z. Karaflou, J. Kljun, I. Turel, G. Psomas, A. N. Papadopoulos and D. P. Kessissoglou, J. Inorg. Biochem., 2013, 128, 85–96 CrossRef CAS PubMed.
- A. Tarushi, C. Kakoulidou, C. P. Raptopoulou, V. Psycharis, D. P. Kessissoglou, I. Zoi, A. N. Papadopoulos and G. Psomas, J. Inorg. Biochem., 2017, 170, 85–97 CrossRef CAS PubMed.
- C. N. Banti, A. G. Hatzidimitriou, N. Kourkoumelis and S. K. Hadjikakou, J. Inorg. Biochem., 2019, 194, 7–18 CrossRef CAS PubMed.
- S. Caglar, A. Altay, B. Harurluoglu, E. K. K. Yeniceri, B. Caglar and O. Şahin, J. Coord. Chem., 2022, 75, 178–196 CrossRef CAS.
- Y. T. Wang, G. M. Tang, W. Z. Wan, Y. Wu, T. C. Tian, J. H. Wang, C. He, X. F. Long, J. J. Wang and S. W. Ng, CrystEngComm, 2012, 14, 3802–3812 RSC.
- C. N. Banti, E. I. Gkaniatsou, N. Kourkoumelis, M. J. Manos, A. J. Tasiopoulos, T. Bakas and S. K. Hadjikakou, Inorg. Chim. Acta, 2014, 423, 98–106 CrossRef CAS.
- A. Johnson, C. Olelewe, J. H. Kim, J. Northcote-Smith, R. T. Mertens, G. Passeri, K. Singh, S. G. Awuah and K. Suntharalingam, Chem. Sci., 2023, 14, 557–565 RSC.
- Y. L. Li, Q. Y. Liu, C. M. Liu, Y. L. Wang and L. Chen, Aust. J. Chem., 2014, 68, 488–492 CrossRef.
- G. Psomas, Coord. Chem. Rev., 2020, 412, 213259 CrossRef CAS.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- Bruker Analytical X–ray Systems, Inc. Apex2, Version 2 User Manual, M86-E01078, 2006 Search PubMed.
- Siemens Industrial Automation, Inc. SADABS: Area–Detector Absorption Correction, 1996 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487–1487 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- P. V. Bernhardt and G. A. Lawrance, Compr. Coord. Chem. II, 2003, 6, 1–145 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, 2008, pp. 1–408 Search PubMed.
- S. Perontsis, E. Geromichalou, F. Perdih, A. G. Hatzidimitriou, G. D. Geromichalos, I. Turel and G. Psomas, J. Inorg. Biochem., 2020, 212, 111213 CrossRef CAS PubMed.
- L. P. Battaglia, A. B. Corradi, G. Marcotrigiano, L. Menabue and G. C. Pellacani, Inorg. Chem., 1979, 18, 148–152 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- B. Chakraborty and T. K. Paine, Inorg. Chim. Acta, 2011, 378, 231–238 CrossRef CAS.
- M. L. Kirk, W. E. Hatfield, M. S. Lah, D. P. Kessissoglou, V. L. Pecoraro and C. Raptopoulou, Inorg. Chem., 1991, 30, 3900–3907 CrossRef CAS.
- S. Taktak, M. Flook, B. M. Foxman, L. Que and E. V. Rybak-Akimova, Chem. Commun., 2005, 5301–5303 RSC.
- J. A. van der Horn, B. Souvignier and M. Lutz, Crystals, 2017, 7, 377 CrossRef.
- Y. Wang and N. Okabe, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1434–m1436 CrossRef CAS.
- Y. Yang, P. Du, J. F. Ma, W. Q. Kan, B. Liu and J. Yang, Cryst. Growth Des., 2011, 11, 5540–5553 CrossRef CAS.
- S. K. Langley, N. F. Chilton, B. Moubaraki and K. S. Murray, Dalton Trans., 2011, 40, 12201–12209 RSC.
- Z. Xiao, G. Passeri, J. Northcote-Smith, K. Singh and K. Suntharalingam, Chem. – Eur. J., 2021, 27, 13846–13854 CrossRef CAS PubMed.
- L. Kuckova, K. Jomova, A. Svorcova, M. Valko, P. Segla, J. Moncol and J. Kozísek, Molecules, 2015, 20, 2115–2137 CrossRef PubMed.
- N. Palanisami, G. Prabusankar and R. Murugavel, Inorg. Chem. Commun., 2006, 9, 1002–1006 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. J. Murphy, P. M. Usov, F. J. Rizzuto, C. J. Kepert and D. M. D'Alessandro, New J. Chem., 2014, 38, 5856–5860 RSC.
- I. A. Koval, M. Huisman, A. F. Stassen, P. Gamez, M. Lutz, A. L. Spek, D. Pursche, B. Krebs and J. Reedijk, Inorg. Chim. Acta, 2004, 357, 294–300 CrossRef CAS.
- L. N. Saunders, M. E. Pratt, S. E. Hann, L. N. Dawe, A. Decken, F. M. Kerton and C. M. Kozak, Polyhedron, 2012, 46, 53–65 CrossRef CAS.
- S. Kita, H. Furutachi and H. Okawa, Inorg. Chem., 1999, 38, 4038–4045 CrossRef CAS.
- X. Meng, H. Hou, G. Li, B. Ye, T. Ge, Y. Fan, Y. Zhu and H. Sakiyama, J. Organomet. Chem., 2004, 689, 1218–1229 CrossRef CAS.
- C. Kontogiorgis, M. Ntella, L. Mpompou, F. Karallaki, P. Athanasios, D. Hadjipavlou-Litina and D. Lazari, J. Enzyme Inhib. Med. Chem., 2016, 31, 154–159 CrossRef CAS PubMed.
- S. Dairi, M. A. Carbonneau, T. Galeano-Diaz, H. Remini, F. Dahmoune, O. Aoun, A. Belbahi, C. Lauret, J. P. Cristol and K. Madani, Food Chem., 2017, 237, 297–304 CrossRef CAS PubMed.
- M. Wettasinghe and F. Shahidi, Food Chem., 2000, 70, 17–26 CrossRef CAS.
- B. M. Ali, M. Boothapandi and A. S. Sultan Nasar, Data Brief, 2020, 28, 104972 CrossRef PubMed.
- X. Totta, A. G. Hatzidimitriou, A. N. Papadopoulos and G. Psomas, New J. Chem., 2017, 41, 4478–4492 RSC.
- M. Lazou, A. G. Hatzidimitriou, A. N. Papadopoulos and G. Psomas, J. Inorg. Biochem., 2023, 243, 112196 CrossRef CAS PubMed.
- B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505–3526 RSC.
- C. Kakoulidou, P. S. Gritzapis, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, J. Inorg. Biochem., 2020, 211, 111194 CrossRef CAS PubMed.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 2002, 111, 3051–3058 CrossRef.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- A. Dimitrakopoulou, C. Dendrinou-Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618–628 CrossRef CAS PubMed.
- P. Zivec, F. Perdih, I. Turel, G. Giester and G. Psomas, J. Inorg. Biochem., 2012, 117, 35–47 CrossRef CAS PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 8901–8911 CrossRef CAS.
- J. L. Garcia-Gimenez, M. Gonzalez-Alvarez, M. Liu-Gonzalez, B. Macías, J. Borras and G. Alzuet, J. Inorg. Biochem., 2009, 103, 923–934 CrossRef PubMed.
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198–1211 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- D. P. Heller and C. L. Greenstock, Biophys. Chem., 1994, 50, 305–312 CrossRef CAS PubMed.
- S. Perontsis, C. T. Chasapis, A. G. Hatzidimitriou and G. Psomas, J. Inorg. Biochem., 2021, 223, 111534 CrossRef CAS PubMed.
- R. E. Olson and D. D. Christ, Annu. Rep. Med. Chem., 1996, 31, 327–336 CAS.
- S. Fountoulaki, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 1645–1655 CrossRef CAS PubMed.
- L. Stella, A. L. Capodilupo and M. Bietti, Chem. Commun., 2008, 4744–4746 RSC.
- O. H. Laitinen, V. P. Hytönen, H. R. Nordlund and M. S. Kulomaa, Cell. Mol. Life Sci., 2006, 63, 2992–3017 CrossRef CAS PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 CAS.
- M. Lazou, A. Tarushi, P. Gritzapis and G. Psomas, J. Inorg. Biochem., 2020, 206, 111019 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2336775–2336782. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01846j |
| This journal is © The Royal Society of Chemistry 2024 |