
Exploring antiviral and antiparasitic activity of gold N-heterocyclic carbenes with thiolate ligands†
Igor S.
Oliveira
 a,
Marcus S. A.
Garcia
b,
Natasha M.
Cassani
c,
Ana L. C.
Oliveira
c,
Lara C. F.
Freitas
c,
Vitor K. S.
Bertolini
b,
Jennyfer
Castro
a,
Gustavo
Clauss
a,
João
Honorato
d,
Fernanda R.
Gadelha
b,
Danilo C.
Miguel
b,
Ana C. G.
Jardim
c and
Camilla
Abbehausen
*a
a,
Marcus S. A.
Garcia
b,
Natasha M.
Cassani
c,
Ana L. C.
Oliveira
c,
Lara C. F.
Freitas
c,
Vitor K. S.
Bertolini
b,
Jennyfer
Castro
a,
Gustavo
Clauss
a,
João
Honorato
d,
Fernanda R.
Gadelha
b,
Danilo C.
Miguel
b,
Ana C. G.
Jardim
c and
Camilla
Abbehausen
*a
aInstitute of Chemistry, University of Campinas, Campinas, São Paulo, Brazil. E-mail: camilla@unicamp.br
bInstitute of Biology, University of Campinas, Campinas, São Paulo, Brazil
cLaboratory of Antiviral Research (LAPAV), Institute of Biomedical Sciences, Federal University of Uberlândia, Brazil
dInstitute of Chemistry, University of São Paulo, Brazil
First published on 15th August 2024
Abstract
Gold(I) N-heterocyclic carbenes have been explored for their therapeutic potential against several diseases. Neglected tropical diseases, including leishmaniasis, Chagas disease, and viral infections, such as zika, mayaro, and chikungunya, urgently require new treatment options. The emergent SARS-CoV-2 also demands significant attention. Gold complexes have shown promise as alternative treatments for these conditions. Previously, gold(I)(1,3-bis(mesityl)imidazole-2-ylidene)Cl (AuIMesCl) demonstrated significant leishmanicidal and anti-Chikungunya virus activities. In this study, we synthesized and fully characterized a series of gold(I)(1,3-bis(mesityl)imidazole-2-ylidene)(SR) complexes, where SR includes thiolate donor species such as 1,3-thiazolidine-2-thione, 1,3-benzothiazole-2-thione, 2-mercaptopyrimidine, and 2-thiouracil. These compounds were stable in solution, and ligand exchange reactions with N-acetyl-L-cysteine indicated that complexes with SR ligands are more labile than those with chloride. Although the reactions are rapid, they reach equilibrium at varying molar ratios depending on the SR ligand. The increased lability of these compounds results in higher cytotoxicity to host cells, such as Vero E6 and bone marrow-differentiated macrophages, compared to AuIMesCl. Despite this, the compounds effectively inhibited viral replication, achieving 95.5% inhibition of Zika virus replication at 2 μM with 96% host cell viability. Although active at low concentrations (∼2 μM) against Leishmania (L.) amazonensis and Trypanosoma cruzi, their high cytotoxicity for macrophages confirmed AuIMesCl as a better candidate with a higher selectivity index. This work correlates the coordination chemistry of pyrimidines and thiazolidines with their in vitro biological activities against significant diseases.
Introduction
Gold has been used to treat diseases since ancient times.1 Robert Koch's study on gold dicyanide advanced its medical use, and Jacques Forestier's research led to the approval of gold thiolates for rheumatoid arthritis in 1978.2,3 Since then, gold's medical applications have been extensively studied.4–6 Therefore, gold(I) and gold(III) complexes show promising antitumor, antiviral, and antiparasitic activities.7–10 Approved antirheumatic drugs strongly inhibit the human immunodeficiency virus (HIV) in vitro and in patients.11–13 Auranofin reduced the virus reservoir in SIV-infected monkeys and decreased viral load in HIV-infected patients.14,15 Gold compounds also inhibited Leishmania and Trypanosoma cruzi growth in vitro and showed significant antiparasitic activity in vivo by binding to cysteine residues in trypanothione reductase.16–21 Auranofin is currently in clinical trials for amoebiasis and more than 11 clinical trials were completed, including HIV, ovarian cancer, chronic lymphocytic leukemia, non-small-cell lung cancer, and giardiasis.22Auranofin is an Au(I) non-polymeric, mononuclear, neutral, linear geometry complex, stabilized by triethylphosphine containing a trans tetraacetylthioglucose coordinated.23 It is orally absorbed, exchanging thioglucose with albumin in the bloodstream.24,25 In general, Au(I) complexes assume the linear dicoordinated structure, and they can be neutral, such as auranofin, or cationic, such as bisphosphines and bis-carbenes. Auranofin and other gold(I) compounds inhibit thioredoxin reductase (TrxR), causing redox imbalance, increasing the levels of reactive oxygen species (ROS), and leading to apoptosis.26–29 This mechanism explains its antitumor and anti-inflammatory effects and likely its antiparasitic activity, as parasites rely on a finely tuned redox balance to survive among different environments imposed by their life cycle.30–32
N-heterocyclic carbenes (NHC) have emerged as better stabilizing ligands than phosphines for gold complexes due to their similar π-accepting abilities and ease of tuning properties.8,33,34 Several Au(I)(NHC) complexes have been explored for their biological activities.35
Regarding antiviral activity, early studies demonstrated that gold(I) phosphine compounds inhibit HIV reverse transcriptase, and gold thiolates and dinuclear gold carbenes interact with viral envelope proteins, protecting cells from infection.12,36 More recently, it was shown that Auranofin inhibits severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication in human cells with an EC50 1.4 μM.37 Some Au(I)(NHC) compounds have shown promising activity against SARS-CoV-2 in the profiling of a large panel of metallodrugs, by inhibiting Spike/ACE2 receptor interaction and SARS-CoV-2 papain-like protease (PLpro).38
Despite all the good results of Au(I) based drugs, most clinical trials fail in dose-limiting toxicity. Over the years, our research has focused on the biological mechanisms of gold(I) compounds, especially ligand exchange reactions.39 Gold, with its thiophilic nature, exchanges ligands with cysteine and selenocysteine-rich molecules, including TrxR, zinc finger domains, and cysteine proteases, causing the biological effects, but also, speciating in biological milieu, puzzling the interpretation of activity and toxicity.5,40–42
Among possible ligands, here we propose thiolate-substituted thiazolidines and pyrimidines. Thiazoles and thiazolines occur naturally and have inspired their use in the synthesis of peptidomimetics, biological probes, and pharmaceuticals.43,44 It is one of the most common units in FDA-approved drugs.43 Thiazoles are a dehydrated cyclized derivative of cysteine naturally incorporated in peptide sequences by ribosomal biosynthesis. They are planar heterocycles with a strong accepting proton nitrogen, a sulfur atom with extended lone pair electron orbital.
Similarly, the pyrimidine derivatives also play a crucial role in nature and inspire drug design.45,46 They naturally occur as substituted and ring-fused compounds including nucleotides, thiamine, and alloxan. Pyrimidine derivatives turn out to be significant pharmacophore groups such as the 5-fluoroacil, anti-HIV drug zidovudine, and barbiturates. Pyrimidines are 1,3-N-substituted six-member aromatic heterocycles. They are π-deficient rings with lower basicity than pyridine. Protonation and other electrophilic additions occur in one nitrogen due to further deactivation by the second nitrogen. Thione derivatives of thiazoles and pyrimidines, depending on the substituents, coordinate well with soft and intermediate Pearson acids such as Au(I), Pd(II), and Cu(II). Due to these characteristics, we have been studying their coordination chemistry and biological applications.
Recently, we studied heteroleptic N-aryl [Au(I)(NHC)Cl] complexes as leishmanicidal agents and compared [Au(Ph3P)Cl] and [Au(IMes)Cl] (IMes = 1,3-bis(mesityl)imidazole-2-ylidene) for Chikungunya virus inhibition.5,47 Gold(I)(NHC) reduced Leishmania amastigotes by 50% at 5 μM in infected macrophages and inhibited Leishmania cysteine protease.5 The phosphine derivative was more effective against the Chikungunya virus (98% inhibition), while [Au(I)(IMes)Cl] inhibited 50% of viral replication at 10 μM, a non-cytotoxic concentration.47
We studied the ligand exchange reaction of [Au(IMes)Cl] with N-acetyl-L-cysteine (NAC), finding that 20% of [Au(IMes)(NAC)] is in equilibrium, favoring cysteine over water or DMSO.39,47 The thiophilic nature of Au(I) affects ligand exchange rates and biological activity. Here we prepared Au(I)IMes complexes with thiolate derivatives (SR) of thiopyrimidines and thiazolidines (Scheme 1) such as 1,3-thiazolidine-2-thione (HStzn), 1,3-benzothiazole-2-thione (HSbtz), pyrimidine-2-thione (HSpym) and 2-thiouracil (2-tuH) and called the series [Au(IMes)(SR)] generally. Besides its full characterization, we studied their ligand exchange reactions and biological effects on Leishmania, Trypanosoma cruzi, viruses such as Zika (ZIKV), Mayaro (MAYV), and VSV-eGFP-SARS-CoV-2-S, a psdeudotyped vesicular stomatitis virus expressing SARS-CoV-2 spike protein. The study correlates chemical properties to biological activity.
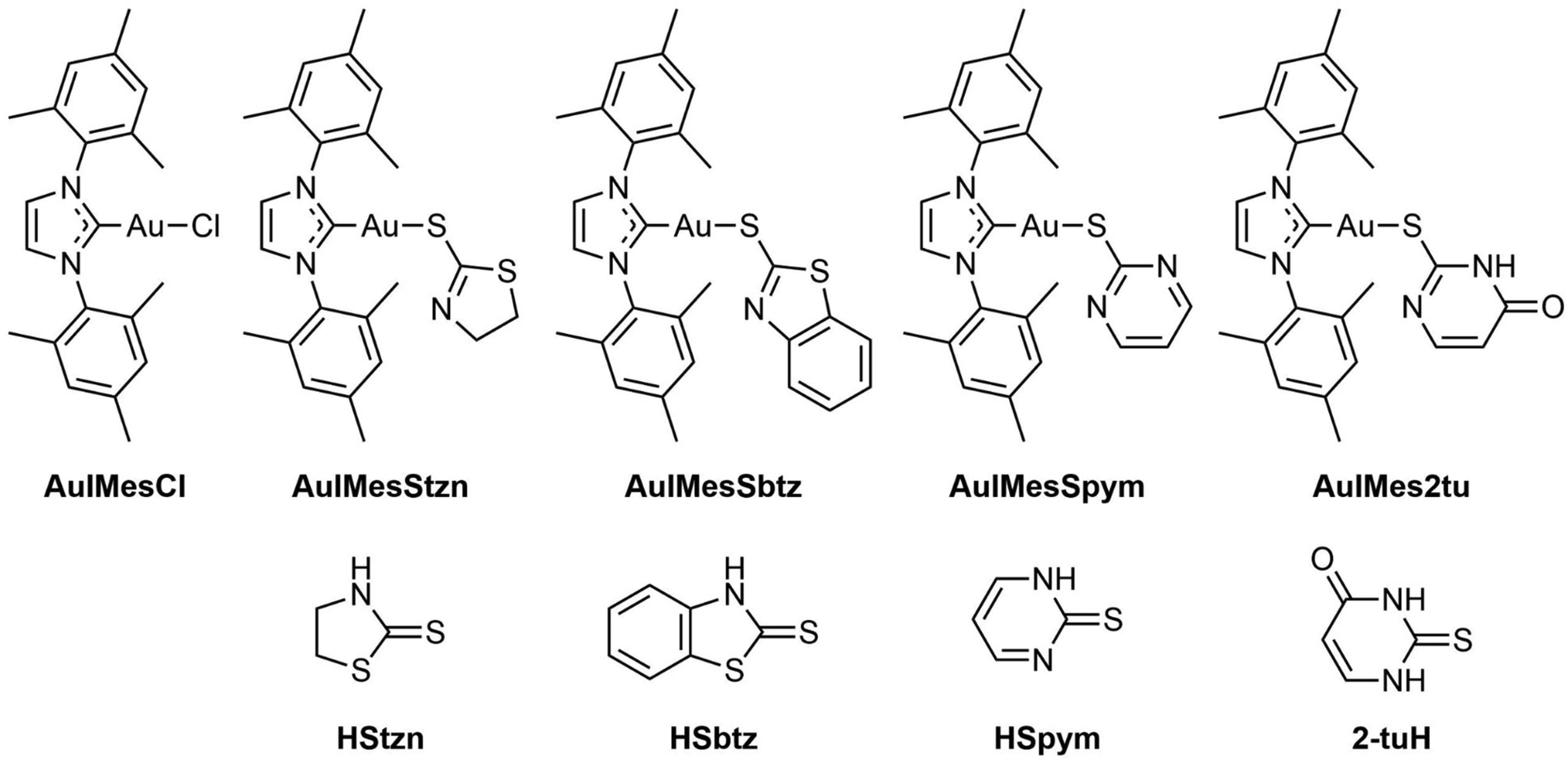 | ||
| Scheme 1 Structural formulas of the AuIMesSR complexes and ligands in the thione form (protonated). | ||
Results and discussion
Ligands SR undergo tautomeric equilibrium
The ligand precursors HSR used in this work exhibit a dynamic tautomeric equilibrium in solution.48–52 Single crystal structure determination of the ligands consistently reveals a thiazolidine-2-thione or pyrimidine-2-thione as the crystallized isomer.53–56 The C–S bond exhibits an intermediate bond distance laying between 1.66–1.68 Å. Experimental and theoretical studies affirm that the thione configuration predominates in the solid state, owing to enhanced stabilization facilitated by intramolecular NH⋯S bonding, which decreases the C–S bond order and distance. This stabilization significantly elevates the energy barrier for interconversion. Nonetheless, Fourier-transform infrared spectroscopy (FTIR) analysis indicates the coexistence of the thiol tautomer alongside the thione in the solid phase.49,57 In solution, however, the equilibrium can be influenced by the solvent environment. Proton nuclear magnetic resonance (1H NMR) spectroscopy conducted on HStzn, HSbtz, HSpym, and 2-tuH in DMSO-d6 at 25 °C (see Fig. S1†) reveals that the thiol/thione equilibrium is shifted to the thione isomer. Notably, previous research by our group has demonstrated the solvent-dependent nature of this equilibrium.50 Polar solvents promote the prevalence of the thione tautomer, with DMSO-d6 expected to predominantly stabilize the thione form.50 Consistently, the same predominance of the thione tautomer is observed under these experimental conditions, underscoring its similar interconversion dynamics compared to other ligands in the study. Here, we calculated the difference in energies (ΔG) between isomers by DFT, and we found HStzn, 2-tuH, HSbtz, and HSpym have favored thione tautomer by 56.4, 48.8, 39.6, and 11.6 kJ mol−1, respectively.An important parameter of these ligands is their pKSH. We measured pKSH by potentiometric titration of the ligands in aqueous solution. The ligands were dissolved in a basic solution, and in this condition, they assumed a thiolate form. The titration with HCl gives the pKSH that further isomerizes to thione after protonation. The thiazolidines are more acidic than pyrimidines with pKSH values of 2.0 and 3.5 for HSbtz and HStzn, respectively. The pyrimidines are 5.9 and 7.2 for 2-tuH and HSpym, respectively. It will be further discussed that the behavior of thiazolidines and pyrimidines are significantly different, and they need to be compared as isolated families.
Synthesis and structural characterization
The current study undertook the synthesis of AuIMesSR through a methodology involving the substitution of chloride ligands in AuIMesCl within an alkaline methanolic solution, drawing upon established protocols detailed in the literature (Scheme 2).58,59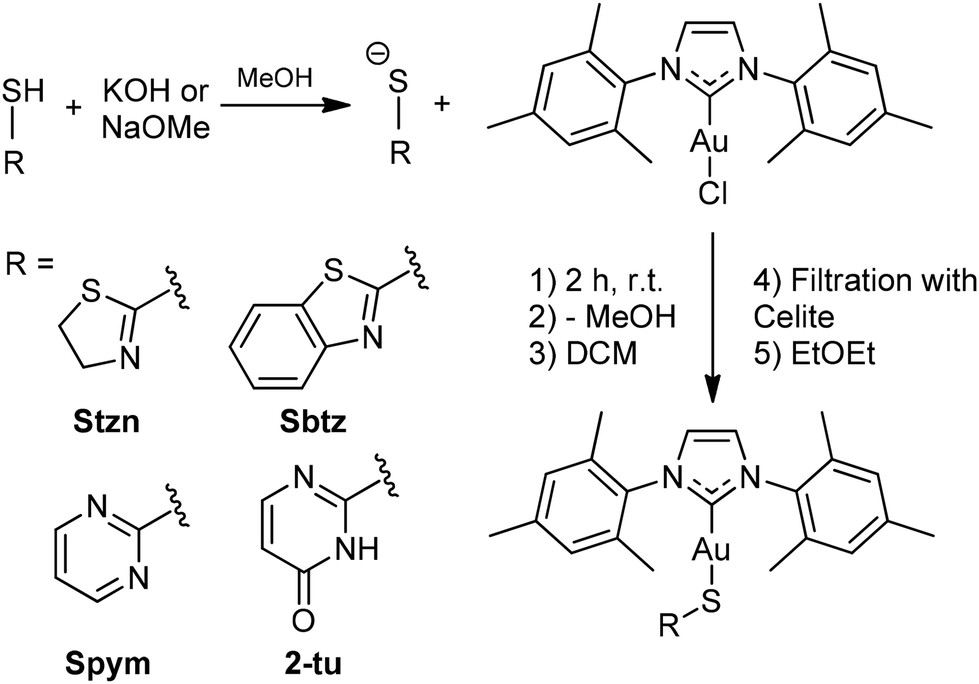 | ||
| Scheme 2 General synthesis route of the complexes AuIMesSR. Ligands were basified and further reacted with AuIMesCl in methanol. After the removal of methanol, compounds were extracted in dichloromethane, filtered in Celite for residue removal, and precipitated by the addition of diethyl ether. | ||
The HSR ligand-precursors were deprotonated in methanol to generate the requisite thiolate species, which serves as a nucleophile displacing the chloride ligand from AuIMesCl. The single crystals of AuIMesSpym and AuIMes2tu complexes were achieved through the controlled evaporation of methanolic solutions at 5 °C. While single crystals of the AuIMesStzn and AuIMesSbtz complexes were obtained from the precipitation procedure with diethyl ether.
Subsequent characterization employing techniques such as 1H and 13C{1H} NMR, 2D NMR correlation experiments as {1H, 13C} HSQC and {1H, 13C} HMBC (Fig. S3–S22†), high-resolution mass spectrometry (Fig. S23–S26†) and elemental analysis (ESI† Experimental) confirmed the formation and high purity of the target products, identified as [AuIMesSR]. The alkaline methanolic environment was found to facilitate the coordination of the thiolate group with Au(I), with the ligands adopting the thiolate form, as supported by the absence of a broad NH signal in the 1H NMR spectra and corroborated by single crystal X-ray diffraction data.
Notably, mass spectrometric analysis conducted on the complexes dissolved in methanol/water solutions containing 0.1% (v/v) formic acid consistently yielded prominent signals corresponding to the molecular ion [AuIMesSR + H+]+. Additional fragments assigned to [HSR + H+]+ and [Au2(IMes)2SR]+ were detected, albeit in lower abundance across all complexes, attributed to artifacts associated with electrospray ionization processes.
Single crystal X-ray diffraction was used to elucidate the molecular structures. The obtained ORTEPs and crystallographic data can be found in Fig. S27–S30 and Table S1.†Fig. 1 depicts the ORTEP diagram of AuIMesSR complexes. AuIMesSbtz and AuIMesSpym crystalize in a triclinic unit cell and space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , while AuIMesStzn crystalize in orthorhombic P212121 and AuIMes2tu in monoclinic P21/c. The main bond distances and angles are reported in Table 1. Another characteristic of these complexes is the presence of disorder in the ligands or asymmetric units containing two similar molecules with different ligand conformations. This results from molecular fluxionality, where the ligands exhibit dynamic behavior, leading to varying conformational positions within the crystal structure.
, while AuIMesStzn crystalize in orthorhombic P212121 and AuIMes2tu in monoclinic P21/c. The main bond distances and angles are reported in Table 1. Another characteristic of these complexes is the presence of disorder in the ligands or asymmetric units containing two similar molecules with different ligand conformations. This results from molecular fluxionality, where the ligands exhibit dynamic behavior, leading to varying conformational positions within the crystal structure.
 | ||
| Fig. 1 ORTEP diagram of the molecular structure of (A) AuIMesStzn, (B) AuIMesSbtz, (C) AuIMesSpym, and (D) AuIMes2tu obtained by single crystal X-ray diffraction, ellipsoids 50% probability. | ||
| Compounds | Au–C (Å) | Au–S (Å) | S–C (Å) | C–Au–S (°) |
|---|---|---|---|---|
AuIMesCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60 60 |
1.998(5) | 2.2756(12) (Au–Cl) | — | 180.0 (C–Au–Cl) |
| AuIMesStzn | 2.001(5) | 2.2964(10) | 1.736(11) | 179.32(18) |
| AuIMesSbtz | 1.998(3) | 2.2936(8) | 1.745(2) | 176.61(8) |
| AuIMesSpym | 1.992(3) | 2.2847(8) | 1.755(3) | 171.00(8) |
| AuIMes2tu | 1.989(3) | 2.2914(7) | 1.743(3) | 179.98(8) |
The ligands consistently coordinate to gold via the thiolate group with characteristic Au–S bond distances across all complexes. The longer S–C bond distance in the complexes, compared to the free ligands (1.66–1.68 Å) demonstrates a typical C–S single bond in the coordinated ligand. Moreover, the nitrogen in the thiazolidines assumes a typical double bond N–C distance (1.25–1.28 Å). The coordination geometry is slightly distorted from the linear geometry in all complexes as indicated by C–Au–S angles, remarkably for AuIMesSpym.
Except for AuIMesStzn, the crystal structures reveal the presence of two complexes within each asymmetric unit. Additionally, in the case of AuIMes2tu, intermolecular π-stacking interactions are observed between the pyrimidine ring and the mesityl group at 3.476(±0.008) Å within the asymmetric unit. Furthermore, intermolecular hydrogen bonding is identified between the oxygen atom of 2-tu ligand and the NH group of molecules belonging to distinct asymmetric units. The Au–S distance presented by the complexes agrees with the values reported for triphenylphosphine derivatives of the Stzn and Sbtz.58
Ligands SR decrease carbene shielding in comparison to chloride and increase lipophilicity
The 13C{1H} NMR chemical shift of the carbene serves as a valuable parameter for assessing the strength of M–C bonds. De Frémont et al. conducted a comparative analysis of the carbene 13C{1H} chemical shifts in [Au(NHC)Cl] complexes employing various NHC ligands, revealing that unsaturated arylated NHC exhibit superior donor capabilities compared to alkylated unsaturated ones, which in turn are more effective donors than saturated NHC ligands.61 By keeping the NHC constant, it becomes feasible to assess the electronic parameters of the ligand situated trans to the NHC moiety, inferring its donor strength. Building upon this framework, Huynh and collaborators showcased in their work on trans-[PdBr2(iPr2-bimy)L]n complexes how the 13C{1H} NMR chemical shift of the carbene experiences a downfield shift contingent upon the donating ability of the ancillary ligand, denoted as L.62Table 2 presents the 13C{1H} chemical shifts of the IMes-coordinated carbene alongside the corresponding Au–C distances derived from single-crystal X-ray diffraction analysis and pKSH of the ligands.P), and percentage of buried volume (%VBur) for IMes e SR ligands. 13C NMR was acquired in Bruker 500 MHz (125 MHz for 13C), using DMSO-d6 as solvent, and adjusted by residual solvent signal at 39.52 ppm
| Compounds | δ 13C | Au–C (Å) | pKSH | logP ± SD |
%VBur |
|---|---|---|---|---|---|
| AuIMesCl | 170.75 | 2.01 | — | 0.82 ± 0.09 (ref. 7) | — |
| AuIMesSbtz | 179.47 | 1.999 | 5.02 ± 0.30 | 0.81 ± 0.02 | 59.5 |
| AuIMes2tu | 180.00 | 1.993 | 5.52 ± 0.15 | 1.00 ± 0.07 | 59.5 |
| AuIMesStzn | 180.26 | 2.001 | 3.98 ± 0.16 | 1.27 ± 0.05 | 59.8 |
| AuIMesSpym | 181.63 | 1.989 | 6.91 ± 0.23 | 1.37 ± 0.21 | 58.6 |
Chloride and thiolate ligands are known to be π-donor ligands, however, the sulfur present in thiolate has a soft base character and higher affinity for gold, a soft acid, and generates a stronger bond with this metal ion than the chloride ligand. This electronic effect leads a downfield shift of 13C{1H} NMR carbene signals in the structures of AuIMesSR series in comparison to the carbene of the precursor AuIMesCl (Table 2), demonstrating that the thiolate ligands have a lower capacity to inject electronic density into the gold(I) center than chloride.61 Interestingly, within the subset of thiolates, the variance is small, indicating a relatively uniform donor capacity across these ligands. Consequently, this similarity in behavior does not exert a notable influence on the Au–C bond distances. However, differences can be noted by looking at thiazolidines and pyrimidines separately. The most basic ligand of the pair is the strongest donor.
The study of lipophilicity is essential to correlate how these species are distributed in biological environments (cell membranes, blood plasma and others). The values are expressed as partition coefficient (logP) and it were determined by Shake Flask method according to OECD guidelines.63 Species with logP < 0 values are considered hydrophilic species, while species with logP > 0 values have affinity with lipophilic species as organic solutions. Table 2 shows the logP values of the complexes quantified by Au content by ICP-OES. These values express the contrast of the increased lipophilicity of the AuIMes2tu, AuIMesStzn and AuIMesSpym complexes in relation to the precursor AuIMesCl, showing that thiolates ligands SR increase the lipophilicity slightly. On the other hand, the AuIMesSbtz complex showed a partition coefficient equal to that of the precursor AuIMesCl.
The SR ligand affects the exchange equilibrium with N-acetyl-L-cysteine (NAC)
The study of the coordination of NAC by 1H NMR is a simple and elegant model that can reveal the speciation of complexes in the presence of cysteine-rich biomolecules. Free NAC CH3 signal is a singlet seen at 1.875 ppm in DMSO-d6. After coordination with gold, as represented in Fig. 2A the CH3 chemical shift is 1.767 ppm, allowing us to calculate the percentage of coordinated NAC.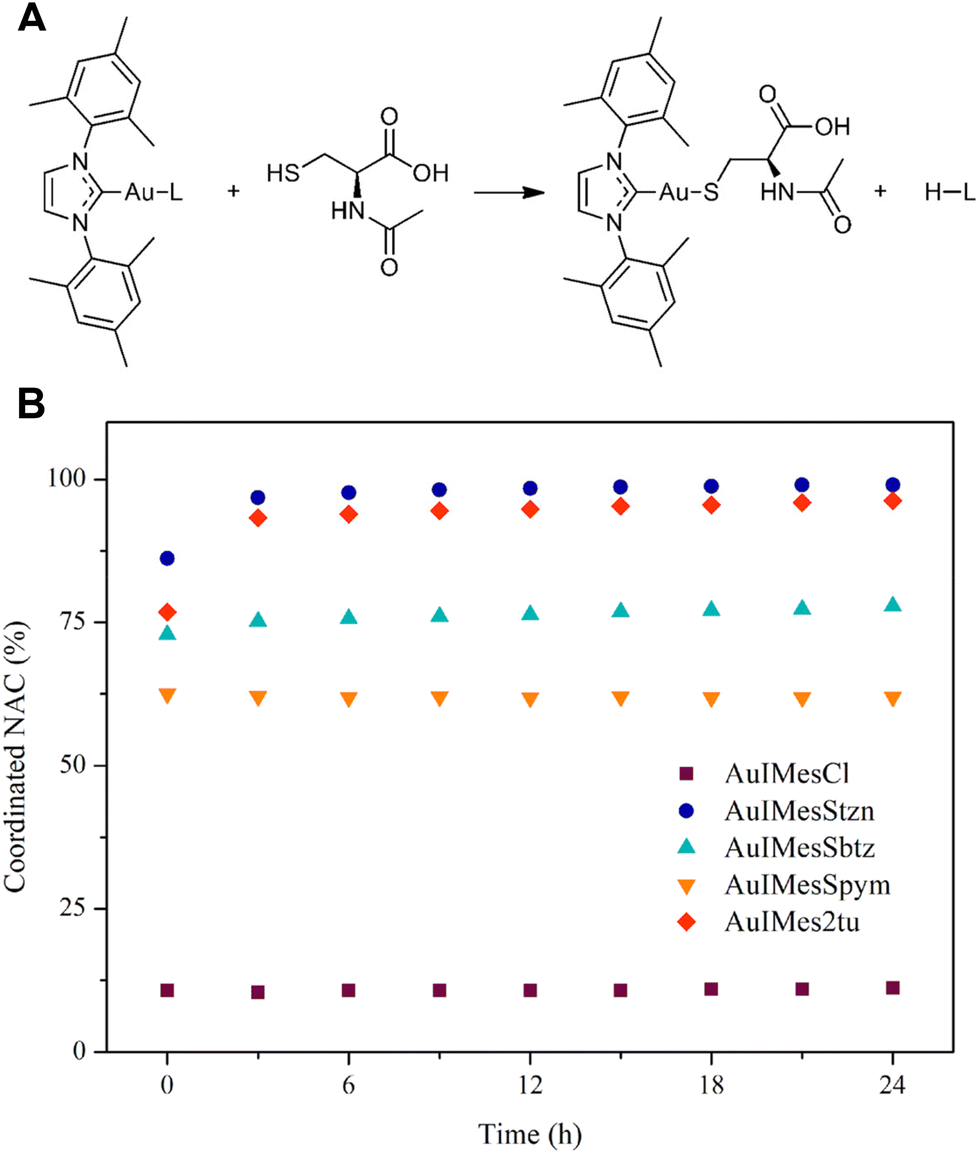 | ||
Fig. 2 Evaluation of ligand exchange of AuIMesSR by N-acetyl-L-cysteine. (A) Ligand exchange equation. (B) %Coordinated NAC with time in a stoichiometric reaction of NAC and AuIMesSR in DMSO-d6 and monitored by 1H NMR with time. The amount of NAC was obtained by integration of 1H NMR spectra at 1.875 ppm for free NAC (NACF) and 1.767 ppm for coordinated NAC (NACC). 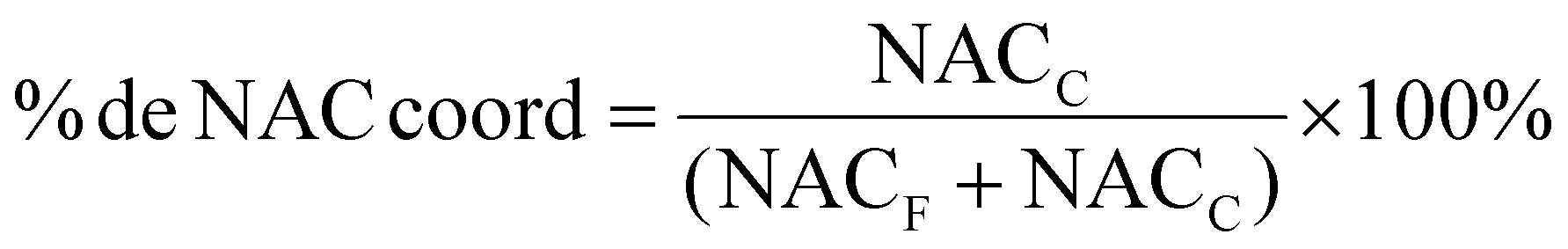 . . | ||
The compounds react fast with NAC establishing an equilibrium in solution. Our group had previously compared the exchange reaction of AuIMesCl and Au(Ph3P)Cl with NAC, showing that the Ph3P favors the reaction toward the coordinated NAC better than the IMes. All the gold compounds we have studied in our group react fast with NAC, precluding achieving kinetic data using this technique. However, depending on the ligands, the equilibrium is shifted for one side or the other, and it can be thermodynamically determined. Previously, we conducted Density Functional Theory (DFT) calculations on the exchange reaction between AuIMesCl and cysteine, in thiolate form (Cys−).64 The results indicated a thermodynamically favored reaction with a kinetic barrier of 66.7 kJ mol−1, using DMSO as an implicit solvent. However, the equilibrium does not favor NAC coordination, with only approximately 11% of AuIMesCl exchanging chloride for NAC in DMSO-d6 (Fig. 2). This suggests that a new model is required, incorporating NAC in its protonated form, as the energy of deprotonation of the SH group cannot be neglected. This approach will allow us to compare the experimental NMR results with theoretical predictions.
Fig. 2B shows that SR ligands favor substitution in comparison to chloride and significative differences were found among the complexes. The compounds AuIMesStzn and AuIMes2tu are almost completely converted to AuIMesNAC, while AuIMesSbtz and AuIMesSpym reached the equilibrium with 78% and 62% of NAC coordinated respectively.
To better understand this phenomenon, we use 1H NMR and 13C{1H} NMR of the converted solution of AuIMes2tu and NAC to check the carbene chemical shift from IMes in the AuIMesNAC complex (Fig. S31 and S32†). The carbene chemical shift in the AuIMesNAC complex is 183.48 ppm, which demonstrates a deshielding of the carbene in comparison to AuIMesSR complexes in all cases, suggesting a stronger bond Au–S in AuIMesNAC. When compared to the values reported in Table 2, the thiolate form of NAC exhibits a better donor ability than the SR ligands, implying a thermodynamic favorability for the exchange reaction.
The optimized compounds by DFT allowed the prediction of the free Gibbs energy of the reaction by simply calculating the energy differences between reagents and products. The results corroborate experimental observation when cysteine reacts with the complex producing the compound AuIMesNAC and the thione tautomer derived from the SR ligand or HCl in the case of AuIMesCl. The reaction of AuIMesCl is endergonic by 59 kJ mol−1, probably an overestimated value as the solvation effects were implicit and dissociation of HCl in DMSO was neglected. Interestingly the reaction of AuIMesSpym was also endergonic by 6 kJ mol−1 corroborating the experimental results and the lower predicted energy for tautomerization. The compounds based on ligands Stzn, 2-tu, and Sbtz were predicted to react exergonically with NAC by −30.7, −21.7, and −15.7 kJ mol−1, respectively, corroborating the experimental results.
To investigate the role of steric effects in the exchange reaction with NAC, we calculated the percentage of buried volume (%VBur) for ligands IMes and SR in the AuIMesSR complexes series (Fig. S33†). The data (Table 2) demonstrate similar %VBur values for the complexes, which reflect the similar steric demand shared by this series. The complex AuIMesStzn has the greatest value, 59.8, and AuIMesSpym the lowest value, 58.6.
The SR ligand changes considerably the binding affinities of AuIMesSR for BSA
BSA was used as a model transporter protein due to its similarity to HSA.65 It has long been reported that the interaction of Au(I) compounds with HSA is not only by non-covalent interaction but also by exchanging ligands with the Cys-34 close to one of the binding sites of HSA.66–69 Auranofin is reported to exchange its thiosugar by Cys-34 HSA in the bloodstream of patients, and the same with BSA. Moreover, non-covalent interactions can also take place between the compound and BSA or HSA.The intrinsic fluorescence of BSA is attributed to the amino acid residues tryptophan and phenylalanine in its structure.70 Among these, the tryptophan residues on its surface exert the greatest influence on this property. The suppression of BSA protein fluorescence occurs through various interaction mechanisms with the titrant, such as substrate binding, conformational changes in the protein structure, or even denaturation.71,72 The emission spectra of BSA titrated with AuIMesSR compounds are presented in the ESI (Fig. S33†). For comparative analysis, the results of fluorescence suppression data were treated according to the Stern–Volmer (Fig. S35†) and Scatchard equations73 (Fig. S36†). Table 3 describes the main parameters.
log[Q], [Q] concentration of the quencher F0 steady-state BSA fluorescence intensity and F steady-state BSA fluorescence intensity in the presence of the quencher. Concentration BSA 20 μM, [Au(IMes)L] 1.2–9.6 μM in DMSO. Temperature 25 °C (298 K)
| Compound | K SV (104 L mol−1) | k q (1012 L mol−1 s−1) | R 2 | K b (104 L mol−1) | R 2 | n |
|---|---|---|---|---|---|---|
| AuIMesStzn | 2.05 ± 0.09 | 3.62 | 0.987 | 13.2 | 0.993 | 1.16 |
| AuIMesSbtzFIGURE | 2.57 ± 0.02 | 4.45 | 0.999 | 3.32 | 0.999 | 1.03 |
| AuIMesSpym | 3.07 ± 0.08 | 5.31 | 0.995 | 350.0 | 0.978 | 1.40 |
| AuIMes2tu | 2.57 ± 0.06 | 4.45 | 0.995 | 13.6 | 0.994 | 1.14 |
|
AuIMesCl47 |
0.70 | 1.21 | 0.891 | 3.5 | — | 0.95 |
Fluorescence quenching can happen through two different mechanisms: static, caused by the formation of a ground-state complex between fluorophore and quencher, or dynamic, which results from collisional encounters between the excited-state fluorophore and the quencher. In the concentrations studied, the data fit shows high linearity demonstrating that Stern–Volmer is an adequate model. The values of kq were obtained using the fluorescence lifetime τ0 5.78 × 10−9 s,74 a general average value for biomacromolecules. They are larger than the maximum scatter collision quenching constants of various kinds of quenchers to biopolymers (2.0 × 1010 mol L−1 s−1),75 suggesting AuIMesSR quench the BSA fluorescence by a static mechanism.
The binding constant Kb is a useful parameter to describe the binding ability of the molecule to the protein. It can give information on pharmacokinetics and pharmacodynamics properties of compounds. A high degree of binding can prolong the drug action, decrease the concentration of the free drug, and affect biodistribution. The results show the ligand SR affects the BSA affinity considerably. The range 104–106 shows a high influence of the SR in BSA binding. While chloride and Sbtz have Kb values in order of 104, Spym elevated the binding constant to 106. The exchange equilibrium with NAC does not correlate directly with Kb, although the Stzn and 2-tu, which are completely exchanged by NAC, present both a similar constant. It is not possible to infer the reason for the different binding by this study only, but it is an interesting property that can be modulated by these ligands. It might include a mix of non-covalent and coordination interactions to different extents.
AuIMesSR does not affect the CT-DNA structure in circular dichroism (CD)
The CD spectrum of calf thymus DNA (CT-DNA) in the range of 190–340 nm exhibits two characteristic bands, with one negative band around 247 nm, related to the right-handed helix conformation of the B form, and another positive band at 275 nm, attributed to the stacking of nitrogenous bases in DNA. The CD spectra of CT-DNA in the presence of the gold complexes explored here in different concentrations are presented in the ESI (Fig. S37†). The compounds change the intensity of the positive band, but not in a concentration dependent manner. This result suggests interference with the stacking and a mode of interaction by non-covalent binding. The main helix structure is not affected, and the non-concentration dependence of the changes demonstrates that it is not a significant interaction.AuIMesSR inhibit a broad spectrum of viruses at low concentrations
The antiviral activity of the complexes was evaluated here against viruses from three different viral families: an arbovirus from Togaviridae, MAYV expressing the nanoluciferase gene reporter; an arbovirus from Flaviviridae, ZIKVPE243;76 and a pseudotyped vesicular stomatitis virus (VSV) expressing eGFP as a marker of infection, in which the glycoprotein gene (G) was replaced by the spike protein (S) of SARS-CoV-2 (VSV-eGFP-SARS-CoV-2-S).77 The investigation of inhibition was performed in one concentration, determined through the cell viability in the host cells Vero E6. The viability of the compounds AuIMesSR and AuIMesCl towards Vero E6 cells was first measured by treating the cells with each complex at 2, 10, and 50 μM, and performing the MTT assay (Table S2†). The results showed that the highest non-cytotoxic concentration tested was 2 μM for AuIMesSR and 10 μM for AuIMesCl. The effect of compounds on the virus replication was evaluated by the infection of Vero E6 cells with the respective viruses, followed by the treatment with compounds. Viral replication rates were assessed by measuring the Focus of Forming Unit (FFU) 24 hours post-infection (h.p.i.) for 72 h.p.i. for ZIKV and VSV-eGFP-SARS-CoV-2-S; and by quantifying the activity of the nanoluciferase reporter at 24 h.p.i. for MAYV-nanoluc. In parallel, cell viability was evaluated by MTT assay. The results are presented in Fig. 3. | ||
| Fig. 3 Antiviral activities of the series of compounds AuIMesSR. (A) ZIKV. Vero E6 cells were seeded at a density of 5 × 103 cells per well in 96-well plates for 24 h, and then infected with ZIKVPE243 at a multiplicity of infection (MOI) of 0.01 in the presence of each compound at the established non-cytotoxic concentration for 72 h. Non-infected cells were also treated with the non-cytotoxic concentration of each compound and cell viability was determined by MTT assay. The infected cells were fixed with 4% (v/v) formaldehyde, washed with PBS [1×], and added with a blocking buffer for immunofluorescence assay. FFU were counted using fluorescence microscopy of EVOs cell imaging systems. (B) MAYV. Vero E6 cells at a density of 2 × 104 cells per well were seeded in 48-well plates. After 24 h, cells were infected with MAYV-nanoluc in the presence of each compound. After 48 h cell were lysed and viral replication was quantified by measuring nanoluciferase activity using Renilla-luciferase assay. (C) VSV-eGFP-SARS-CoV-2-S. 1 × 104 Vero E6 cells were cultured in 96-well plates 24 h before infection. Cells were infected with VSV-eGFP-SARS-CoV-2-S (MOI of 0,001) in the presence of compounds at non cytotoxic concentration for 2 hours. Then, cells were washed and incubated with fresh media for 24 hours in a humidified incubator with 5% CO2 at 37 °C. FFU were counted using fluorescence microscopy of EVOs cell imaging systems, detecting eGFP expression. Mean ± SD values of three independent experiments, each measured in triplicate. DMSO was used as untreated control. P < 0.05 was considered statistically significant. (**) p < 0.01, (***) p < 0.001, and (****) p < 0.0001. | ||
The cytotoxicity of the series AuIMesSR is higher than the precursor AuIMesCl limiting the concentration for viral inhibition evaluation to 2 μM (Table S2†) while the precursor is viable to this cell line up to 10 μM. Cell viabilities of the series AuIMesSR vary from 88% to 96% at 2 μM. However, even at this lower concentration, the compounds presented an excellent inhibition of viral replication in general (Fig. 3). Regarding the inhibition of ZIKV, the compounds AuIMesSbtz and AuIMes2tu significantly inhibited 96% and 94% of the viral replication at 2 μM, respectively, while the precursor AuIMesCl inhibited 70% at 10 μM (Fig. 3A). Though the other compounds were less efficient they also performed well in the low concentration, presenting an inhibition of 88.3% and 55% for AuIMesStzn and AuIMesSpym, respectively. Concerning MAYV inhibition, all the complexes similarly inhibited the replication, varying in values between 80 to 90% (Fig. 3B). Contrasting the ZIKV inhibition, AuIMesSpym inhibited MAYV replication at 90% at 2 μM, the best value in the series. All the compounds presented better values than the precursor AuIMesCl at 10 μM (76.2%). It is important to emphasize that besides ZIKV and MAYV are arboviruses, they are classified in different viral families, which means that the replication processes for these viruses varies, and therefore, different antiviral profiles can be expected. However, based on the results presented here the series of AuIMesSR compounds significantly inhibited ZIKV and MAYV.
Alternatively, the potential activity of these compounds on S protein of SARS-CoV-2 was evaluated using the pseudotyped VSV-eGFP-SARS-CoV-2-S, which represents a useful tool for studying emerging and highly pathogenic enveloped viruses in level 2 biosafety facilities.
In general, the compounds were less active against VSV-eGFP-SARS-CoV-2-S in comparison to the other viruses evaluated here (Fig. 3C). We can highlight the AuIMesSpym and AuIMesStzn, which inhibited 76% and 70%, respectively, both at 2 μM. The other compounds inhibited in the same range as the precursor 52–60%. Taking into consideration that by using VSV-eGFP-SARS-CoV-2-S the effects of the compounds were evaluated only on early stages of the virus infection and focusing on the presence of S protein into the VSV particle, the interference of these compounds on post-entry stages of SARS-CoV-2 needs to be further investigated in a future study.
Compounds are active against Leishmania amazonensis and Trypanosoma cruzi
The series of compounds were evaluated against promastigotes of Leishmania amazonensis and epimastigotes of Trypanosoma cruzi in vitro (Table 4). In general, the compounds have a toxic effect on the parasites with an EC50 around 2 μM. The compounds AuIMesSbtz and AuIMesSpym were evaluated against T. cruzi showing contrasting results. AuIMesSbtz presented the lowest EC50 of the series, while AuIMesSpym presented the highest. The limiting point of application is the cytotoxicity in primary macrophages, which is around 10 μM, decreasing considerably the selectivity index, however, we considered that in vitro infections should be performed using lower concentrations (up to 5 μM) of the best candidates to assess their activity against intracellular amastigotes (Fig. 4). Infection rates and intracellular parasite burden were determined and AuIMes2tu led to a reduction in the infection rate when incubated at the higher concentration tested (22.3%; Fig. 4A), and pronounced results in the second parameter, being able to decrease the parasite burden in ∼30% when incubated with 5.0 μM of the complex (Fig. 4B). | ||
| Fig. 4 Reduction of in vitro infection by AuIMesStzn and AuIMes2tu. BMDM infected with promastigotes of L. amazonensis at a ratio of 5:1 incubated with AuIMesStzn and AuIMes2tu for 24 h. Each bar refers to the percentage of reduction of the infection rate (A) and parasite burden per macrophage (B) in relation to untreated control infections (100%). The results presented are representative of two independent assays performed in triplicate. Ordinary One Way ANOVA was applied as a statistical test comparing treated groups with the untreated control: *p < 0.05; **p < 0.01; ***p < 0.001. | ||
| Compound | L. amazonensis EC50 (μM) | T. cruzi EC50 (μM) | BMDM CC50 (μM) | SI L. amazonensis |
|---|---|---|---|---|
| AuIMesStzn | 1.93 ± 0.09 | 2.33 ± 0.55 | 10.3 | 5.3 |
| AuIMesSbtz | — | 1.50 ± 0.38 | — | — |
| AuIMesSpym | — | 5.80 ± 0.41 | — | — |
| AuIMes2tu | 2.03 ± 0.21 | 2.13 ± 0.44 | 11.3 | 5.6 |
|
AuIMesCl5 |
1.57 ± 0.41 | — | 21.81 ± 1.07 | 13.9 |
Conclusions
This work described the preparation and full characterization of a set of AuIMesSR compounds where SR are derived from 1,3-thiazolidine-2-thione (HStzn), 1,3-benzothiazolidine-2-thione (HSbtz), 2-thiopyrimidine (HSpym) and 2-thiouracil (2-tuH), and IMes is the NHC 1,3-bis(mesityl)imidazole-2-ylidene. This series was idealized expecting to have a different reactivity toward sulfur-rich biomolecules in comparison with AuIMesCl, expressed in this work by the exchange with the model amino acid, NAC. We have been investigating Au(NHC) compounds, especially AuIMesCl, as a chemotype for the design of gold-based antiparasitic and antivirals. As the SR ligands mimic biomolecules functional groups, we looked for the correlation of the reactivity and chemical behavior, mainly the ligand exchange reactions, with biological activity.The results showed that SR ligands bind gold through the sulfur atom in typical Au–S single bond distance and assume the thiolate tautomer conformation after coordination, while in solid and solution state the ligand-precursors are predominantly the thione tautomer. The SR ligands act as better donors than chloride, which promotes stronger bonds. It would be expected that the SR ligands would be less labile than chloride in reaction with sulfur-rich biomolecules, but the reverse was observed. The nature of SR ligands considerably affects the thermodynamics of the ligand exchange reaction with NAC, and AuIMesSR complexes are more reactive towards NAC than AuIMesCl. The AuIMesSpym is the less reactive in the series, reaching 62% exchange in the equilibrium, while gold complexes AuIMesStzn and AuIMes2tu exchange the ligand by NAC almost completely at a fast rate. In contrast, AuIMesCl exchanges only 11% of chloride by NAC at the equilibrium in DMSO-d6. DFT calculations could predict the experimental results, elucidating the favored reaction. This trend is almost followed by the BSA binding constant with one outlier, the AuIMesSpym, which needs further investigation to understand the significantly higher BSA binding constant (106).
The high reactivity towards NAC of AuIMesSR might explain the higher cytotoxicity found for the host cells (Vero E6 and BMDM) for the series when compared to AuIMesCl, impairing the selectivity index in the antiparasitic evaluation, but not in the antiviral activity. The higher antiviral activity, even in lower concentrations (non-cytotoxic to host), makes the AuIMesSR better antiviral candidates than AuIMesCl, with promising inhibition (90–95%) at 2 μM. This result moves one step forward the field in the search for ligands to design gold-based pharmaceuticals. Despite the apparent low selectivity observed in leishmanicidal assays, intracellular amastigotes of L. amazonensis were inhibited by ∼30% after 24 h incubation, preserving host cell viability. In this sense, our results point to a great perspective for future evaluation of the new gold complex AuIMes2tu against Leishmania parasites.
Author contributions
ISO investigation, formal analysis (chemical and biophysical data) and writing; MSAG investigation, formal analysis (anti-Leishmania investigation) and writing; NMC, ALCO and LCFF investigation, formal analysis (antiviral investigation) and writing. VKSB investigation, formal analysis (anti-T. cruzi investigation) and writing. JC investigation, formal analysis (pK, logP) and writing. GC formal analysis (DFT calculations), and writing; JH formal analysis (crystal structure) and writing; FRG supervision, data curation and writing (review and editing); DCM supervision, data curation and writing (review and editing); ACGJ supervision, data curation and writing (review and editing); CA conceptualization, data curation, writing – review and editing, funding acquisition and supervision.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for AuIMes2tu, AuIMesSbtz, AuIMesStzn and AuIMesSpym has been deposited at the CCDC under 236392, 2363393, 2363394 and 2363395.†Conflicts of interest
No conflicts of interest to declare.Acknowledgements
This study was supported by São Paulo Research Foundation (FAPESP) Grants # 2022/02618-0, 2023/06493-0, and 2023/12657-6 and Brazilian Council for Scientific and Technological Development (CNPq) Grant # 404668/2021-6. We thank CAPES (Brazilian Ministry of Education) (Doctorate scholarship # 88887.713008/2022-00) and the CENAPAD Project #638 for computational resources and support. We thank the Unicamp Found for the support of Research and Extension (FAEPEX) Grant #2391/23. ACGJ is grateful to FAPEMIG (Minas Gerais Research Foundation APQ-01487-22 and APQ-04686-22), CNPq (409187/2023-2) and to CAPES (Prevention and Combat of Outbreaks, Endemics, Epidemics and Pandemics—Finance Code #88881.506794/2020-01 and—Finance Code 001). NMC is grateful to CAPES for scholarship #88887.703845/2022-00.References
- Z. Huaizhi and N. Yuantao, Gold Bull., 2001, 34, 24–29 CrossRef.
- T. G. Benedek, J. Hist. Med. Allied Sci., 2004, 59, 50–89 CrossRef PubMed.
- W. F. Kean, F. Forestier, Y. Kassam, W. W. Buchanan and P. J. Rooney, Semin. Arthritis Rheum., 1985, 14, 180–186 CrossRef CAS PubMed.
- Y. Lu, X. Ma, X. Chang, Z. Liang, L. Lv, M. Shan, Q. Lu, Z. Wen, R. Gust and W. Liu, Chem. Soc. Rev., 2022, 51, 5518–5556 RSC.
- L. B. Rosa, C. Galuppo, R. L. A. Lima, J. V. Fontes, F. S. Siqueira, W. A. S. Júdice, C. Abbehausen and D. C. Miguel, J. Inorg. Biochem., 2022, 229, 111726 CrossRef CAS PubMed.
- R. E. F. de Paiva, A. Marçal Neto, I. A. Santos, A. C. G. Jardim, P. P. Corbi and F. R. G. Bergamini, Dalton Trans., 2020, 49, 16004–16033 RSC.
- A. Balfourier, J. Kolosnjaj-Tabi, N. Luciani, F. Carn and F. Gazeau, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22639–22648 CrossRef CAS PubMed.
- B. Dominelli, J. D. G. Correia and F. E. Kühn, J. Organomet. Chem., 2018, 866, 153–164 CrossRef CAS.
- J. H. Kim, E. Reeder, S. Parkin and S. G. Awuah, Sci. Rep., 2019, 9, 12335 CrossRef PubMed.
- M. Gil-Moles, S. Türck, U. Basu, A. Pettenuzzo, S. Bhattacharya, A. Rajan, X. Ma, R. Büssing, J. Wölker, H. Burmeister, H. Hoffmeister, P. Schneeberg, A. Prause, P. Lippmann, J. Kusi-Nimarko, S. Hassell-Hart, A. McGown, D. Guest, Y. Lin, A. Notaro, R. Vinck, J. Karges, K. Cariou, K. Peng, X. Qin, X. Wang, J. Skiba, Ł. Szczupak, K. Kowalski, U. Schatzschneider, C. Hemmert, H. Gornitzka, E. R. Milaeva, A. A. Nazarov, G. Gasser, J. Spencer, L. Ronconi, U. Kortz, J. Cinatl, D. Bojkova and I. Ott, Chem. – Eur. J., 2021, 27, 17928–17940 CrossRef CAS PubMed.
- P. N. Fonteh, F. K. Keter and D. Meyer, BioMetals, 2010, 23, 185–196 CrossRef CAS PubMed.
- T. Okada, B. K. Patterson, S.-Q. Ye and M. E. Gurney, Virology, 1993, 192, 631–642 CrossRef CAS PubMed.
- P. Fonteh and D. Meyer, Metallomics, 2009, 1, 427–433 CrossRef CAS.
- D. L. Shapiro and J. R. Masci, J. Rheumatol., 1996, 23, 1818–1820 CAS.
- M. G. Lewis, S. DaFonseca, N. Chomont, A. T. Palamara, M. Tardugno, A. Mai, M. Collins, W. L. Wagner, J. Yalley-Ogunro, J. Greenhouse, B. Chirullo, S. Norelli, E. Garaci and A. Savarino, AIDS, 2011, 25, 1347–1356 CrossRef CAS PubMed.
- E. R. Sharlow, S. Leimgruber, S. Murray, A. Lira, R. J. Sciotti, M. Hickman, T. Hudson, S. Leed, D. Caridha, A. M. Barrios, D. Close, M. Grögl and J. S. Lazo, ACS Chem. Biol., 2014, 9, 663–672 CrossRef CAS.
- L. G. Tunes, R. E. Morato, A. Garcia, V. Schmitz, M. Steindel, J. D. Corrêa-Junior, H. F. dos Santos, F. Frézard, M. V. de Almeida, H. Silva, N. S. Moretti, A. L. B. de Barros and R. L. do Monte-Neto, ACS Infect. Dis., 2020, 6, 1121–1139 CrossRef CAS PubMed.
- A. Ilari, P. Baiocco, L. Messori, A. Fiorillo, A. Boffi, M. Gramiccia, T. Di Muccio and G. Colotti, Amino Acids, 2012, 42, 803–811 CrossRef CAS PubMed.
- P. I. da S. Maia, Z. A. Carneiro, C. D. Lopes, C. G. Oliveira, J. S. Silva, S. de Albuquerque, A. Hagenbach, R. Gust, V. M. Deflon and U. Abram, Dalton Trans., 2017, 46, 2559–2571 RSC.
- I. Winter, J. Lockhauserbäumer, G. Lallinger-Kube, R. Schobert, K. Ersfeld and B. Biersack, Mol. Biochem. Parasitol., 2017, 214, 112–120 CrossRef CAS PubMed.
- L. B. Rosa, R. L. Aires, L. S. Oliveira, J. V. Fontes, D. C. Miguel and C. Abbehausen, ChemMedChem, 2021, 16, 1682–1696 CrossRef PubMed.
- Search for: Auranofin | Card Results | ClinicalTrials.gov, https://clinicaltrials.gov/search?intr=Auranofin, (accessed May 22, 2024).
- A. E. Finkelstein, D. T. Walz, V. Batista, M. Mizraji, F. Roisman and A. Misher, Ann. Rheum. Dis., 1976, 35, 251–257 CrossRef CAS PubMed.
- J. R. Roberts, J. Xiao, B. Schleisman, D. J. Parsons and C. F. Shaw, Inorg. Chem., 1996, 35, 424–433 CrossRef CAS PubMed.
- C. F. Shaw, A. A. Isab, M. T. Coffer and C. K. Mirabelli, Biochem. Pharmacol., 1990, 40, 1227–1234 CrossRef CAS PubMed.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692–1707 CrossRef CAS.
- Y. Omata, M. Folan, M. Shaw, R. L. Messer, P. E. Lockwood, D. Hobbs, S. Bouillaguet, H. Sano, J. B. Lewis and J. C. Wataha, Toxicol. In Vitro, 2006, 20, 882–890 CrossRef CAS PubMed.
- M. G. Karaaslan, A. Aktaş, C. Gürses, Y. Gök and B. Ateş, Bioorg. Chem., 2020, 95, 103552 CrossRef CAS PubMed.
- G. Bjørklund, L. Zou, J. Wang, C. T. Chasapis and M. Peana, Pharmacol. Res., 2021, 174, 105854 CrossRef PubMed.
- T. Battista, G. Colotti, A. Ilari and A. Fiorillo, Molecules, 2020, 25, 1924 CrossRef CAS PubMed.
- M. Beig, F. Oellien, L. Garoff, S. Noack, R. L. Krauth-Siegel and P. M. Selzer, PLoS Neglected Trop. Dis., 2015, 9, e0003773 CrossRef.
- G. Colotti, A. Ilari, A. Fiorillo, P. Baiocco, M. A. Cinellu, L. Maiore, F. Scaletti, C. Gabbiani and L. Messori, ChemMedChem, 2013, 8, 1634–1637 CrossRef CAS.
- C. Zhang, M.-L. Maddelein, R. W.-Y. Sun, H. Gornitzka, O. Cuvillier and C. Hemmert, Eur. J. Med. Chem., 2018, 157, 320–332 CrossRef CAS PubMed.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC.
- C. Zhang, C. Hemmert, H. Gornitzka, O. Cuvillier, M. Zhang and R. W.-Y. Sun, ChemMedChem, 2018, 13, 1218–1229 CrossRef CAS PubMed.
- R. W. Y. Sun, W. Y. Yu, H. Z. Sun and C. M. Che, ChemBioChem, 2004, 5, 1293–1298 CrossRef CAS PubMed.
- H. A. Rothan, S. Stone, J. Natekar, P. Kumari, K. Arora and M. Kumar, Virology, 2020, 547, 7–11 CrossRef CAS PubMed.
- M. Gil-Moles, U. Basu, R. Büssing, H. Hoffmeister, S. Türck, A. Varchmin and I. Ott, Chem. – Eur. J., 2020, 26, 15140–15144 CrossRef CAS PubMed.
- C. Abbehausen, C. M. Manzano, P. P. Corbi and N. P. Farrell, J. Inorg. Biochem., 2016, 165, 136–145 CrossRef CAS PubMed.
- C. Abbehausen, R. E. F. de Paiva, R. Bjornsson, S. Q. Gomes, Z. Du, P. P. Corbi, F. A. Lima and N. Farrell, Inorg. Chem., 2018, 57, 218–230 CrossRef CAS PubMed.
- L. Massai, L. Messori, N. Micale, T. Schirmeister, L. Maes, D. Fregona, M. A. Cinellu and C. Gabbiani, BioMetals, 2017, 30, 313–320 CrossRef CAS.
- C. Abbehausen, E. J. Peterson, R. E. F. de Paiva, P. P. Corbi, A. L. B. Formiga, Y. Qu and N. P. Farrell, Inorg. Chem., 2013, 52, 11280–11287 CrossRef CAS PubMed.
- J. Y. W. Mak, W. Xu and D. P. Fairlie, in Peptidomimetics I, ed. W. D. Lubell, Springer International Publishing, Cham, 2017, pp. 235–266 Search PubMed.
- A. Singh, D. Malhotra, K. Singh, R. Chadha and P. M. S. Bedi, J. Mol. Struct., 2022, 1266, 133479 CrossRef CAS.
- S. B. Patil, Heliyon, 2023, 9, e16773 CrossRef CAS PubMed.
- I. M. Lagoja, Chem. Biodivers., 2005, 2, 1–50 CrossRef CAS PubMed.
- R. L. Aires, I. A. Santos, J. V. Fontes, F. R. G. Bergamini, A. C. G. Jardim and C. Abbehausen, Metallomics, 2022, 14, mfac056 CrossRef.
- G. Bereket, C. Öğretir, M. Yaman and E. Hür, J. Mol. Struct.: THEOCHEM, 2003, 625, 31–38 CrossRef CAS.
- A. Altun, E. Kuliyev and N. M. Aghatabay, Spectrochim. Acta, Part A, 2016, 152, 181–191 CrossRef CAS PubMed.
- C. Abbehausen, R. E. F. de Paiva, A. L. B. Formiga and P. P. Corbi, Chem. Phys., 2012, 408, 62–68 CrossRef CAS.
- D. L. Ruiz, M. M. Schiavoni, S. L. Laurella, J. M. Giussi, J. J. P. Furlong and P. E. Allegretti, Spectrochim. Acta, Part A, 2011, 78, 1397–1402 CrossRef PubMed.
- Y. Liu, H. Du, G. Wang, X. Gong, L. Wang and H. Xiao, Int. J. Quantum Chem., 2010, 111, 1115–1126 CrossRef.
- A. M. Owczarzak, N. Kourkoumelis, S. K. Hadjikakou and M. Kubicki, CrystEngComm, 2013, 15, 3607 RSC.
- J. P. Chesick and J. Donohue, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 1441–1444 CrossRef CAS.
- E. R. T. Tiekink, Z. Kristallogr., 1989, 187, 79–84 CrossRef CAS.
- E. S. Raper, R. E. Oughtred and I. W. Nowell, Inorg. Chim. Acta, 1983, 77, L89–L93 CrossRef CAS.
- M. J. Nowak, H. Rostkowska, L. Lapinski, J. Leszczynski and J. S. Kwiatkowski, Spectrochim. Acta, Part A, 1991, 47, 339–353 Search PubMed.
- K. N. Kouroulis, S. K. Hadjikakou, N. Kourkoumelis, M. Kubicki, L. Male, M. Hursthouse, S. Skoulika, A. K. Metsios, V. Y. Tyurin, A. V. Dolganov, E. R. Milaeva and N. Hadjiliadis, Dalton Trans., 2009, 10446–10456 RSC.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- P. de Frémont, E. D. Stevens, M. D. Eelman, D. E. Fogg and S. P. Nolan, Organometallics, 2006, 25, 5824–5828 CrossRef.
- P. de Frémont, N. Marion and S. P. Nolan, J. Organomet. Chem., 2009, 694, 551–560 Search PubMed.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- O. OECD, Test No 107 Partit. Coeff. N-Octanolwater Shake Flask Method, 1995 Search PubMed.
- G. C. Rodrigues, M. V. F. Barrionuevo, M. A. San-Miguel and C. Abbehausen, New J. Chem., 2024, 48, 2040–2047 RSC.
- J. Steinhardt, J. Krijn and J. G. Leidy, Biochemistry, 1971, 10, 4005–4015 Search PubMed.
- J. Christodoulou, P. J. Sadler and A. Tucker, FEBS Lett., 1995, 376, 1–5 CrossRef CAS PubMed.
- J. Christodoulou, P. J. Sadler and A. Tucker, Eur. J. Biochem., 1994, 225, 363–368 CrossRef CAS PubMed.
- J. Xiao and C. F. Shaw, Inorg. Chem., 1992, 31, 3706–3710 CrossRef CAS.
- J. Xiao and C. F. Shaw, Metal-Based Drugs, 1994, 1 DOI:10.1155/MBD.1994.520.
- N. Tayeh, T. Rungassamy and J. R. Albani, J. Pharm. Biomed. Anal., 2009, 50, 107–116 CrossRef CAS PubMed.
- X. Yu, Y. Yang, L. Shiyu, Q. Yao, L. Heting, L. Xiaofang and Y. Pinggui, Spectrochim. Acta, Part A, 2011, 83, 322–328 CrossRef CAS PubMed.
- D. M. Charbonneau and H.-A. Tajmir-Riahi, J. Phys. Chem. B, 2010, 114, 1148–1155 CrossRef CAS PubMed.
- A. K. dos S. Pereira, C. M. Manzano, D. H. Nakahata, J. C. T. Clavijo, D. H. Pereira, W. R. Lustri and P. P. Corbi, New J. Chem., 2020, 44, 11546–11556 RSC.
- U. Anand, C. Jash and S. Mukherjee, J. Phys. Chem. B, 2010, 114, 15839–15845 CrossRef CAS PubMed.
- S. Afrin, Riyazuddeen, G. Rabbani and R. H. Khan, J. Lumin., 2014, 151, 219–223 CrossRef CAS.
- N. M. Cassani, I. A. Santos, V. R. Grosche, G. M. Ferreira, M. Guevara-Vega, R. B. Rosa, L. J. Pena, N. Nicolau-Junior, A. C. O. Cintra, T. P. Mineo, R. Sabino-Silva, S. V. Sampaio and A. C. G. Jardim, Int. J. Biol. Macromol., 2023, 227, 630–640 CrossRef CAS PubMed.
- J. B. Case, P. W. Rothlauf, R. E. Chen, Z. Liu, H. Zhao, A. S. Kim, L.-M. Bloyet, Q. Zeng, S. Tahan, L. Droit, M. X. G. Ilagan, M. A. Tartell, G. Amarasinghe, J. P. Henderson, S. Miersch, M. Ustav, S. Sidhu, H. W. Virgin, D. Wang, S. Ding, D. Corti, E. S. Theel, D. H. Fremont, M. S. Diamond and S. P. J. Whelan, Cell Host Microbe, 2020, 28, 475–485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and data. CCDC 2363392–2363395. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01879f |
| This journal is © The Royal Society of Chemistry 2024 |