
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear tricarbonylrhenium(I) complexes: impact of regioisomerism on the photoluminescence properties†
Stéphen
Le Garrec
 a,
David
Martins-Bessa
a,
Mariusz
Wolff
bc,
Béatrice
Delavaux-Nicot
d,
Sonia
Mallet-Ladeira
e,
Charles-Louis
Serpentini
f,
Eric
Benoist
a,
Florence
Bedos-Belval
*a and
Suzanne
Fery-Forgues
*a
a,
David
Martins-Bessa
a,
Mariusz
Wolff
bc,
Béatrice
Delavaux-Nicot
d,
Sonia
Mallet-Ladeira
e,
Charles-Louis
Serpentini
f,
Eric
Benoist
a,
Florence
Bedos-Belval
*a and
Suzanne
Fery-Forgues
*a
aSPCMIB, CNRS UMR 5068, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France. E-mail: florence.bedos@univ-tlse3.fr; suzanne.fery-forgues@univ-tlse3.fr
bInstitut für Funktionelle Materialien und Katalyse, Universität Wien, Währinger Straße 38-42, 1090 Wien, Austria
cInstitute of Chemistry, University of Silesia in Katowice, Szkolna 9th Street, 40-006 Katowice, Poland
dLaboratoire de Chimie de Coordination, CNRS (UPR 8241), Université de Toulouse (UPS, INPT), 205 route de Narbonne, 31077 Toulouse Cedex 4, France
eService Diffraction des Rayons X, Institut de Chimie de Toulouse, ICT-UAR 2599, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France
fLaboratoire SOFTMAT, CNRS UMR 5623, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France
First published on 26th August 2024
Abstract
Dinuclear Re(I) complexes have proportionally been much less studied than mononuclear analogues. In particular, very little information is available about their solid-state emission properties. In this work, two structural isomers of dinuclear complexes (Bi-Re-metaPhe and Bi-Re-paraPhe), which differ by the relative position of the coordination spheres on a central phenyl ring, were synthesized and compared with each other and with the parent mononuclear compound (Mono-Re-Phe), from a theoretical and experimental point of view. In solution, the electronic, electrochemical and spectroscopic properties of the dinuclear complexes were almost identical, and rather close to those of the monomer. In the solid state, the photoluminescence (PL) efficiency of dimers was not higher than that of the monomer, but a clear mechanoresponsive luminescence (MRL) effect appeared only for the former ones. The positional isomerism influenced the amplitude of this effect, as well as the aggregation-induced emission (AIE) properties in a water-acetonitrile mixture. This study reveals the importance of positional isomerism to modulate the emission properties in the solid state. It also shows the advantage of dinuclear structures to access new MRL-active materials.
Introduction
Over the last two decades, tricarbonylrhenium(I) complexes have attracted significant attention due to their stability, very low toxicity and attractive spectroscopic properties, which made them popular bio-imaging agents and sensors.1 Their coordination chemistry is clearly dominated by mononuclear species. However, many examples of complexes incorporating two rhenium centers have been reported. They can be divided into two categories. Some have little or no electronic conjugation between the coordination spheres. They can be seen as covalent assemblies of mononuclear complexes, whose physicochemical properties are the sum of the properties of each fragment and the linker. This makes them valuable probes for precise targeting in imaging microscopy,2,3 as well as potential candidates in the fields of anticancer drugs4–7 and photodynamic therapy,8 anion detection,9 liquid crystals,10 and photocatalysis.11,12 The second type of complex is characterized by the electronic conjugation between the two metal centers through an organic ligand. This allows the emergence of new chemotherapeutic properties.13,14 New electrochemical and spectroscopic properties also appear,15–23 which may be of interest for applications such as organic catalysis,24–27 electrochemical devices,28,29 sensing,30 and bio-imaging.28,31–33 For photochemically-active complexes, the photoproduction of species like 1O2 and CO is also increased with respect to mononuclear species.34–36 From a general point of view, the spectroscopic properties of dinuclear tricarbonylrhenium(I) complexes in solution attract more and more attention, although they are probably much less studied than those of luminescent iridium complexes,37–43 for example. Regarding specifically the solid-state emission properties, which are highly sought after for applications in the field of photoluminescent materials, bio-sensing and security devices,44–48 only some rare dinuclear tricarbonylrhenium(I) complexes have been reported to date.16,17,49,50 However, significant variations compared to mononuclear complexes can be expected, due to the modified electron system and to a change in the solid-state molecular arrangement and intermolecular bonds, which play a major role in photoluminescence (PL).51Recently, our team has developed original mononuclear tricarbonylrhenium(I) complexes that are potentially useful for applications in the field of photoluminescent materials.52–59 Besides strong solid-state luminescence enhancement (SLE)51 and aggregation-induced emission (AIE)60 effect, some of them showed unprecedented mechanoresponsive luminescence (MRL) properties.56–59 In the present work, the structure of one of the best performing complexes (Re-Phe, Fig. 1) based on the (3-(2-pyridyl)-1,2,4-triazole) (pyta) ligand, has been selected. The analogous mononuclear complex Mono-Re-Phe, in which an alkyl chain was introduced for solubility reasons, and the new dinuclear tricarbonylrhenium(I) complexes (Bi-Re-paraPhe and Bi-Re-metaPhe) merging two of these units were designed and synthesized (Fig. 1). The two dimers differ by the position of their pyta moieties on the central phenyl ring. The comparison with parent mononuclear complex Mono-Re-Phe allowed to better understand the influence of the dinuclear structure on their electronic, electrochemical and spectroscopic properties. Moreover, subtle changes in molecular geometry and intermolecular interactions in the solid state can be expected between the dimers, possibly resulting in new physicochemical properties. The impact of regioisomerism upon the PL properties was therefore specifically investigated. To the best of our knowledge, such a study is unprecedented in the literature. No example of cis/trans positional isomerism with respect to a photoswitchable double bond was found either, while such changes have been reported to govern the spectroscopic behavior of mononuclear species.61,62 In a somewhat distant area, it has been shown that cis/trans conformational isomerism has no effect on the photocatalytic properties of an anthracene-based dinuclear Re(I) complex in solution.36 Studies dealing with the diastereoisomerism of anticancer dinuclear Re(I) complexes incorporating chiral centers are still rare and clearly belong to another field of research.14 The present study revealed that our three complexes in solution show rather conventional behavior. In contrast, in the solid-state, unpredictable MRL properties emerged only for dimers, while regioisomerism allowed the emission efficiency and AIE properties to be modulated. This is the first study in this field, and it could open the door to a variety of new multimetallic complexes.
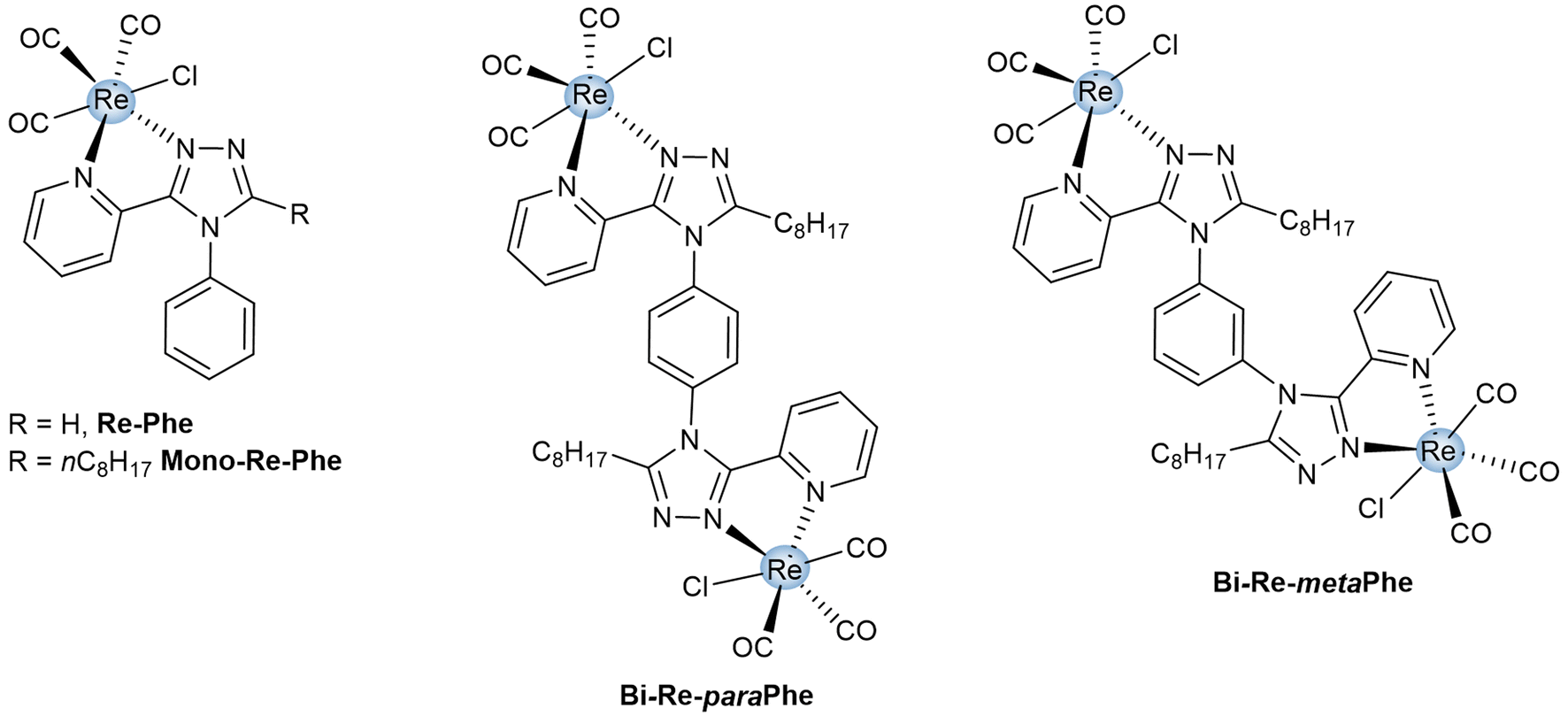 | ||
| Fig. 1 Chemical structures of dimeric complexes Bi-Re-paraPhe and Bi-Re-metaPhe, and reference monomeric complexes Re-Phe and Mono-Re-Phe. | ||
Results and discussion
Synthesis and characterization
The procedure used to synthesize the three new complexes is described in Scheme 1. The meaning of abbreviations is reported in the ESI, and the chemical characterization of the compounds is given in the Experimental section. In brief, picolinohydrazide was reacted with nonanoic acid in the presence of HATU to afford the oxadiazole derivative. The latter was then condensed with aniline or the relevant phenylenediamine in the presence of a catalytic amount of p-TsOH in refluxing 1,2-dichlorobenzene to give the corresponding ligands L-Phe, L-paraPhe and L-metaPhe respectively, with a fair to good yield after purification. The ligands were then reacted with [Re(CO)5Cl] in refluxing methanol to afford the corresponding tricarbonylrhenium(I) complexes in good yields (83%, 76% and 73%, for Mono-Re-Phe, Bi-Re-paraPhe and Bi-Re-metaPhe, respectively).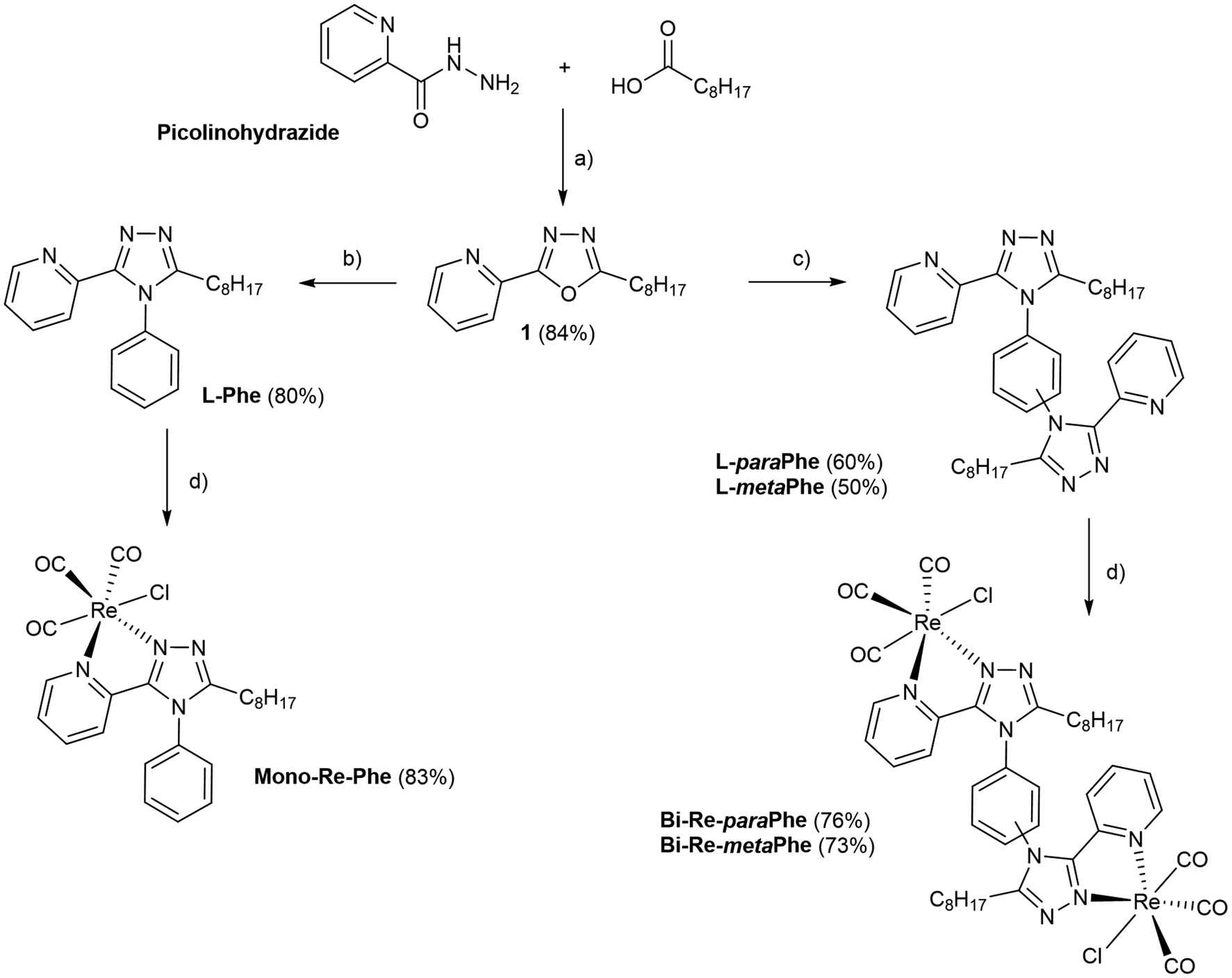 | ||
| Scheme 1 Synthesis of complexes Mono-Re-Phe, Bi-Re-paraPhe and Bi-Re-metaPhe. Conditions and reagents: (a) DIPEA, HATU, acetonitrile, 6 h, r.t.; and then DIPEA, p-TsCl, 16 h, r.t. (b) Aniline, p-TsOH, 1,2-dichlorobenzene, 20 h, 180 °C. (c) Phenylenediamine, p-TsOH, 1,2-dichlorobenzene, 20 h, 180 °C. (d) [Re(CO)5Cl], methanol, 16 h, 65 °C. | ||
All synthesized molecules were characterized by 1H and 13C NMR spectroscopy, high resolution mass spectrometry and infrared spectroscopy (Fig. S1–23†). The purity of the complexes was checked by elemental microanalysis. It is noteworthy that for the dimeric complexes Bi-Re-paraPhe and Bi-Re-metaPhe, the NMR signals corresponding to the H3 and H4 pyridine protons were split (Fig. S16 and 19†). This indicates the presence of at least two conformers, the identification of which is the topic of a forthcoming publication.
Crystal structures
X-Ray quality crystals of complexes Mono-Re-Phe and Bi-Re-metaPhe were grown at the interface of a chloroform solution topped with pentane, and from slow evaporation of a DCM solution, respectively. No suitable crystal could be obtained from the Bi-Re-paraPhe complex. However, crystals of ligands L-paraPhe and L-metaPhe, grown from slow evaporation of acetone and acetone/CH2Cl2 solutions, respectively, were examined to get an idea of the molecular conformation. The crystallographic data of Re-Phe57 were further processed (Fig. S24–27 and Comment S1†) and used for comparison. Selected crystallographic data are collected in the Experimental section and in Tables S1 and 2.†The coordination spheres of complexes Mono-Re-Phe and Bi-Re-metaPhe exhibit a quasi-octahedral geometry (Fig. 2). Distortion parameters are listed in Tables S3 and 4.† The rhenium atom is coordinated to three carbonyl groups in a fac configuration, one chlorine atom, the pyridyl N1 atom and the triazole N2 atom. For Bi-Re-metaPhe, the geometry of both metallic centers is exactly the same. Distances and angles of the coordination spheres are close to those found for this type of complexes. The pyridyl-triazole moieties are almost planar. The phenyl ring is close to orthogonal with respect to the pyta moiety, with a dihedral α angle of 82.6° for Mono-Re-Phe (more pronounced than for the analogue deprived of the alkyl chain (α ∼ 68°)57), and two identical α angles of 81.9° for Bi-Re-metaPhe. It must be noted that for the latter complex, the crystal obtained represents only one of the possible conformations, where the two pyta moieties face each other in an antisymmetric way, each being arranged orthogonally with respect to the central phenyl ring. This doubly-twisted conformation was visible on the structure of ligand L-metaPhe, where the triazole rings of the two molecules form distinct α angles of 85.5° and 66.0° (81.3° and 68.2° for the second molecule of the cell). The examination of the crystal structure of ligand L-paraPhe also shows that the triazole rings form α angles of 70.5° and 74.6° with the central phenyl ring. According to DFT calculations (vide infra), the torsion angles in Bi-Re-paraPhe in solution should be close to orthogonal (Table S12†). So, in the absence of X-ray data, it can be assumed that a doubly twisted conformation is retained for the Bi-Re-paraPhe complex.
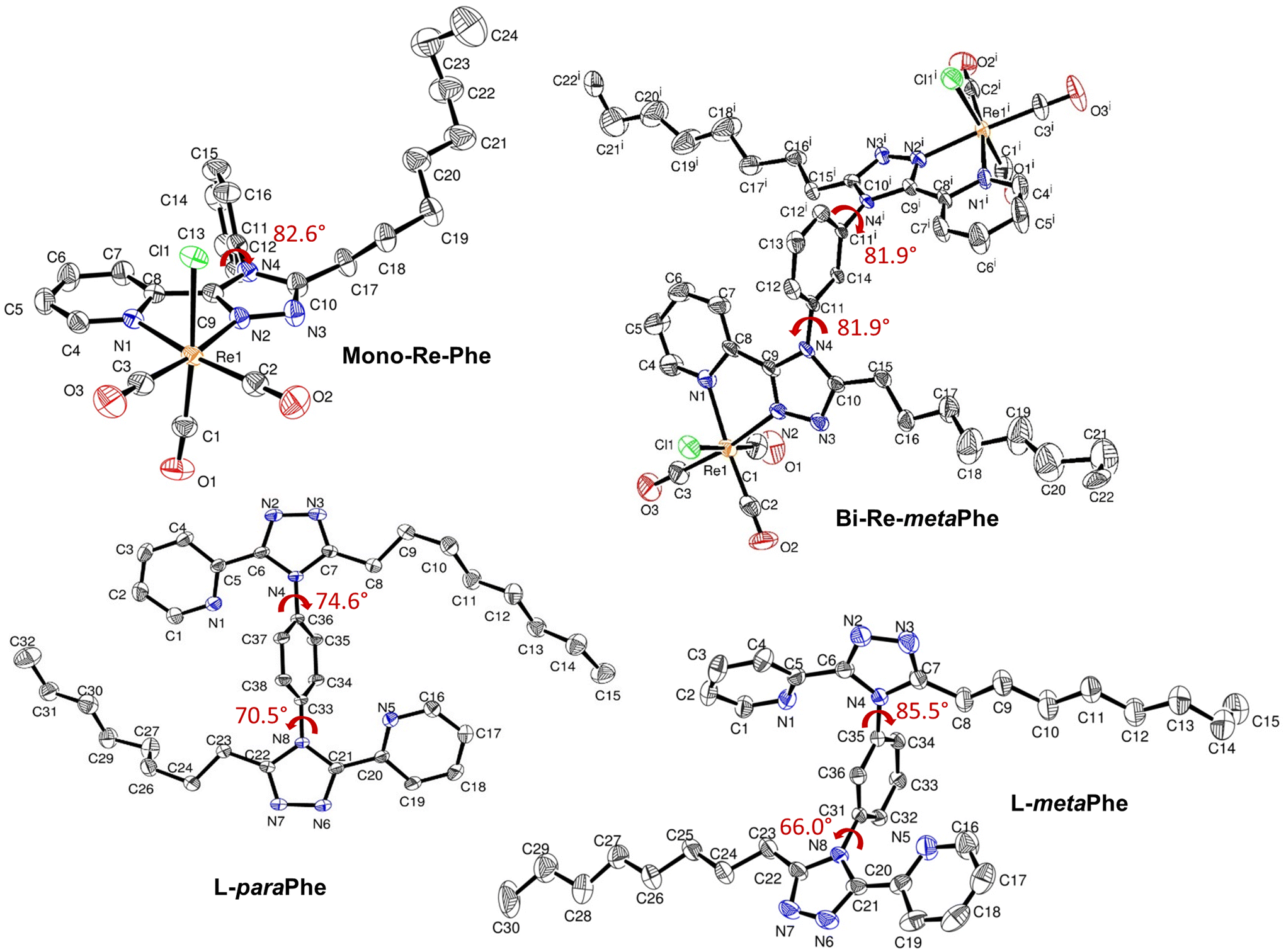 | ||
| Fig. 2 Molecular structures of complexes Mono-Re-Phe and Bi-Re-metaPhe, and ligands L-paraPhe and L-metaPhe, with the dihedral angle α indicated in red ink. Hydrogen atoms are not represented for the sake of clarity. Displacement ellipsoids are drawn at 50% probability. Symmetry code for Bi-Re-metaPhe: (i) 1 − x, y, ½ − z. | ||
For complex Mono-Re-Phe, two enantiomers, which differ by the position of the organic ligand with respect to the chlorine atom, coexist in the crystal cell in identical proportions. The same is true for Bi-Re-metaPhe, each molecule of which is constituted by two enantiomers of the same type. Both crystal structures are stabilized by C–H⋯O and C–H⋯Cl short contacts, which can be considered as weak hydrogen bonds (Fig. 3, Table S5†). Additionally, both structures show C–H⋯π interactions (Table S6†), but no obvious π–π stacking interactions were detected, contrary to what was observed for Re-Phe (Table S7†). This comparison shows the value of the alkyl chain to separate the aromatic moieties. More precisely, as regards the whole arrangement, molecules of Mono-Re-Phe are displayed on four distinct planes, with opposite orientations (Fig. S28†). Two neighboring molecules in the antiparallel arrangement form cyclic dimers R22(12) through C5–H5(pyr)⋯Cl1 interactions, which are further connected through C16–H16(Phe)⋯O1(CO) interactions to afford a 2D network along the ab plane (Fig. 3a and S29†). Regarding Bi-Re-metaPhe, all molecules are aligned along the same direction (Fig. S30†). They form antiparallel dimers (Fig. S30b†). Strong C7–H7(pyr)⋯π(Phe) (Fig. S31†) and N3(trz)⋯H12(Phe) interactions are detected between neighboring molecules. Remarkably, the presence of the alkyl chain and DCM molecules play an important role in structuring the network. Molecules of Bi-Re-metaPhe connect via intermolecular C22–H22A(CH3)octyl⋯O3(CO) interactions along the ab plane. One DCM molecule is connected to two molecules of complex via intermolecular C15–H15B(CH2)octyl⋯Cl2 and C22–H22B(CH3)octyl⋯Cl3 interactions forming an infinite 1D ladder chain which propagates along the ac plane. The same DCM molecule is also connected to a third molecule of complex through intermolecular C23–H23B(DCM)⋯O2(CO) interactions, leading to a 2D network (Fig. 3b). It can be anticipated that molecules of Bi-Re-metaPhe, which arrange as layers, have more facility to glide on each other upon mechanical forces than those of Mono-Re-Phe, the packing of which is much denser.
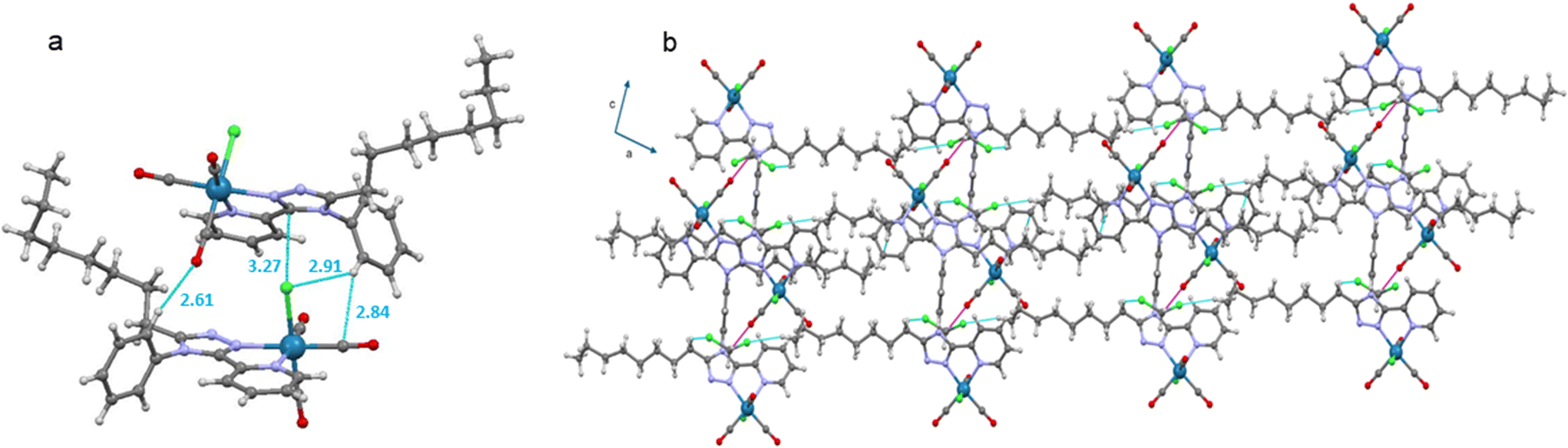 | ||
| Fig. 3 (a) Molecular view of Mono-Re-Phe with distances of short contacts in Å. (b) Representation of one-dimensional chains and two-dimensional network in Bi-Re-metaPhe formed by connection of molecules through DCM (in an up/down–up/down fashion) via intermolecular C15–H15B(CH2)octyl⋯Cl2(DCM) and C22–H22B(CH3)octyl⋯Cl3(DCM) interactions (cyan color), and C23–H23B(DCM)⋯O2(CO) interactions (magenta color). | ||
The analysis of Hirshfeld surfaces (HS)63 was performed to visualize and quantify the intermolecular interactions in the crystal lattice of complexes Re-Phe, Mono-Re-Phe and Bi-Re-metaPhe. In Fig. 4a–c, where HS are plotted over dnorm, the red spots represent the regions of the surface where intermolecular contacts are strong, and the blue areas illustrate the domains where neighboring atoms are too far away to interact with each other. It can be seen that the red regions concentrate on the part of the fat chain close to triazole ring for Mono-Re-Phe (Fig. 4b) and Bi-Re-metaPhe (Fig. 4c), while they are distributed more homogeneously on the surface of Re-Phe (Fig. 4a). On the shape-index representation (Fig. 4d), blue triangles represent the convex regions formed by carbon atoms present in the molecule inside the surface, while red triangles represent concave regions due to the carbon atoms of the π-stacked molecule above it. Adjacent red and blue triangles that highlight π–π stacking interactions were only observed for Re-Phe. The percentage contributions of different interactions to the HS were quantified (Fig. S32–34 and Comment S2†) and compared (Fig. 4e). This approach confirms that the number of intermolecular interactions involving the aromatic moieties was decreased in the presence of the alkyl chain.
 | ||
| Fig. 4 (a–c) Hirshfeld surfaces (HS) plotted over dnorm for Re-Phe (a), Mono-Re-Phe (b) and Bi-Re-metaPhe (c). (d) HS plotted over shape index for Re-Phe with adjacent triangles revealing the π–π stacking surrounded by the black circle. (e) Percentage contributions of different interactions to the HS for the three complexes. | ||
Electronic properties
Computational studies were made using the density functional theory (DFT) and time-dependent DFT (TD-DFT) methods considering the complexes in dichloromethane continuum (Tables S8–25,†Fig. 5 and Fig. S35–41†). Calculations were repeated for Re-Phe so that the data were fully comparable with those of the new complexes. As expected, the alkyl chain has little influence on the electronic properties and complexes Re-Phe and Mono-Re-Phe are very similar, so that only the latter one was discussed below. For Mono-Re-Phe and Bi-Re-metaPhe, the calculated bond lengths and angles were in excellent agreement with the X-ray experimental data (Tables S8 and 9†). It is also the case for the FT-IR spectra of all complexes (Fig. S41†). The composition of the frontier molecular orbitals (Tables S14–16, and Fig. S35–38†) revealed that the electronic density of the three highest occupied molecular orbitals (HOMO, H−1 and H−2) of Mono-Re-Phe is localized on the rhenium atom and carbonyl ligands, as well as on the chlorine atom for the two first ones, as is commonly the case for tricarbonylrhenium(I) complexes. This electronic distribution was also found for orbitals HOMO to H−5 of the dinuclear Re complexes. Regarding the lowest unoccupied molecular orbitals, the LUMO and L+1 of the three complexes are centered on the pyta moiety. Significant differences between the three complexes appear for upper and lower orbitals. For instance, the electronic density of L+2 is almost totally concentrated on the phenyl ring of Mono-Re-Phe (96%), while it is distributed between the phenyl ring and the pyta moiety of Bi-Re-paraPhe (87/12) and Bi-Re-metaPhe (66/34). Regarding the energy levels, the HOMO–LUMO gap was slightly decreased when passing from Mono-Re-Phe (4.03 eV), to Bi-Re-paraPhe (3.98 eV), and then to Bi-Re-metaPhe (3.96 eV). The HOMO, H−1 and H−2 of the dinuclear complexes have very close energy levels, as well as H−4 and H−5, and this is also the case for their LUMO and L+1. Actually, these orbitals are almost degenerate with a very small energy difference due to the symmetric arrangement of the complexes. In contrast, for the mononuclear compound, these energy levels vary more importantly.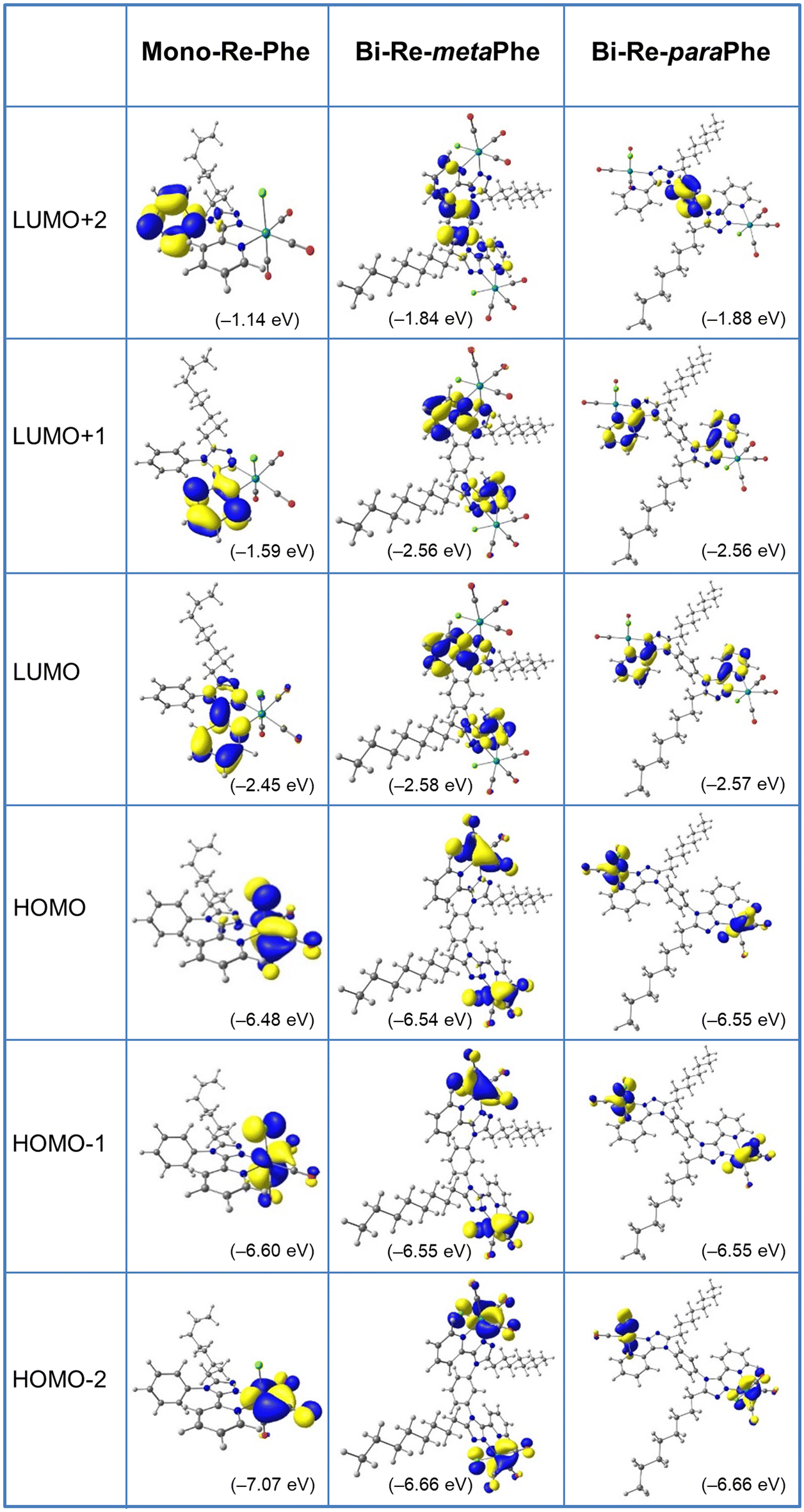 | ||
| Fig. 5 Isodensity plots (isovalue = 0.03 e bohr−3) and energy levels of the first frontier molecular orbitals, for complexes Mono-Re-Phe, Bi-Re-paraPhe, and Bi-Re-metaPhe in dichloromethane, according to DFT calculations at the PBE1PBE/LANL2DZ level of theory. | ||
For the three complexes, the lowest energy transitions correspond to a shift of the electronic density from the coordination sphere to the organic ligand (Tables S18–20†). They are therefore of metal-to-ligand charge transfer (MLCT), halogen-to-ligand charge transfer (XLCT) and ligand-to-ligand charge transfer (LLCT) type. The most significantly-active transitions are H−1 → LUMO at 382.8 nm for Mono-Re-Phe, H−3 → L+1/H−2 → L (389.6 nm) for Bi-Re-metaPhe, and H−3 → L/H−2 → L+1 (388.7 nm) for Bi-Re-paraPhe. Many high-energy transitions are of intra-ligand and ligand-to-ligand charge transfer (ILCT/LLCT) type. It is noteworthy that the most active transitions involve orbitals that are very similar for the three complexes.
The natural population analysis (NPA) (Table S24†) showed that the calculated charge on the rhenium atoms is −0.99(e) whatever the complex. The positively charged carbon atoms of the carbonyl ligands accept as much as ∼0.74/0.77/0.76(e) from the Re atoms, while the negatively charged nitrogen atoms N(1), N(2) and chlorine atom Cl(1) donate as much as ∼0.39(e), ∼0.23(e) and ∼0.46(e) to Re atoms, respectively. In summary, the mono and bimetallic complexes are very close from an electronic point of view, and great similarities can be expected in the experimental properties in solution.
Electrochemistry
The electrochemical behavior of the new complexes was studied by cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) measurements in dichloromethane at room temperature. The results are displayed in Table 1, Fig. 6 and Fig. S42–53.† The Mono-Re-Phe complex had the electrochemical characteristics of a Re(I) chlorotricarbonyl compound with a (3-(2-pyridyl)-1,2,4-triazole) substituted in the N position by a phenyl group in an orientation almost perpendicular to the pyta ligand (Table 1). Indeed, comparison with its bidentate ligand and its non-alkyl-substituted counterpart allowed us to clearly attribute its first oxidation process to a monoelectronic oxidation process centered on the Re(I) moiety, and its first reduction process to that of its substituted pyta moiety. Moreover, the value of the latter process situated at −1.39 V was around 100 mV more cathodic than that of complex Re-Phe,57 indicating the influence of the electron-donating alkyl group. In contrast, the Re center was not very sensitive to this change, while a Eox1/Ered1 intensity ratio of 1 was well observed in CV at 0.2 V s−1 as for Re-Phe (Fig. 6a).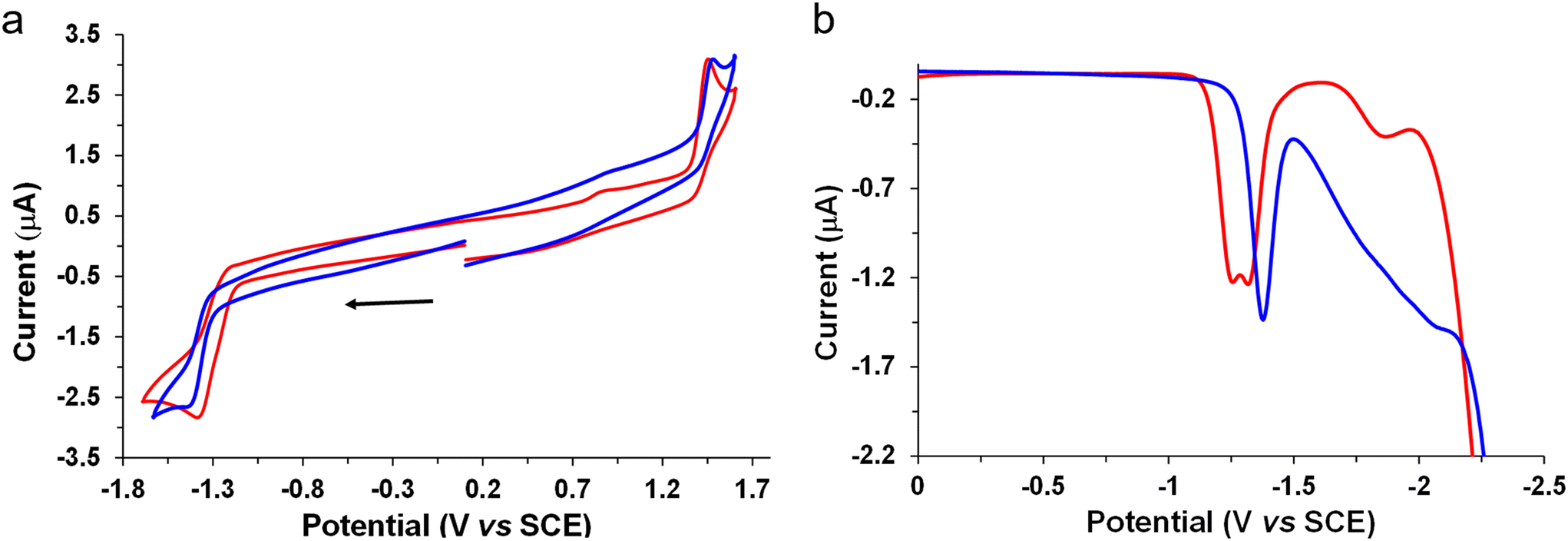 | ||
| Fig. 6 (a) Cyclic voltammograms of the first oxidation and reduction processes of complexes Mono-Re-Phe (2 × 10−3 M, blue line) and Bi-Re-paraPhe (1 × 10−3 M, orange line) at 0.2 V s−1 (left); (b) their OSWVs cathodic scans (right) on a Pt working electrode in CH2Cl2 + 0.1 M n[Bu4N][BF4] at room temperature. | ||
| Oxidation | Reduction | ||||
|---|---|---|---|---|---|
| E 2 | E 1 | E 1 | E 2 | E 3 | |
| a OSWVs were obtained using a sweep width of 20 mV, a frequency of 20 Hz, and a step potential of 5 mV. b Potential values in Volts vs. SCE (Fc+/Fc is observed at 0.55 V ± 0.01 V vs. SCE). c From ref. 57. d One-electron quasi-reversible process at 1.0 V s−1. e More reversible process at 10 V s−1 (Fig. S44†), with a 1/1 intensity ratio for Eox1/Ered1 at 0.2 V s−1 (Fig. S43 right†). f Two close reduction processes in 1/1 intensity ratio whatever the SW rate conditions (Fig. S48†). These processes give one more reversible process at 50 V s−1 in CV (Fig. S47†). g Very small intensity process. h Ill defined (Fig. S52†). | |||||
| Re-Phe | 1.78 | 1.46 | −1.29d | −1.78 | |
| L-Phe | 1.99 | 1.70 | — | — | — |
| Mono-Re-Phe | 1.75 | 1.44 | −1.39e | — | |
| L- paraPhe | — | 1.70 | — | — | — |
| Bi-Re-paraPhe | 1.75 | 1.43 | −1.24f | −1.33f | −1.88g |
| L- metaPhe | 1.70h | — | — | — | |
| Bi-Re-metaPhe | 1.75 | 1.43 | −1.24f | −1.34f | −1.88g |
Remarkably, the two bimetallic rhenium complexes presented the same electrochemical signatures. In OSSW, in the anodic part, two irreversible oxidation processes were observed. The first one was clearly assigned to the oxidation of both Re centers whose potential value (1.43 V) is very close to that of the mononuclear compounds. No electronic communication was detected between these metallic centers which behave as independent redox centers. In CV, a broad peak was observed for the reduction of the pyta moieties, and further examination of this process by OSWV allowed the detection of two very close reduction processes of equal intensity centered at −1.29 V, whatever the experimental conditions (Fig. 6b and Fig. S48†). Globally, in reduction, the bimetallic complexes are less electron rich than their mononuclear counterpart. Their reduction potential is characteristic of a preferred perpendicular arrangement of their pyta moieties with the central phenyl ring, as already observed in this family of complexes,52,53,56–58 and confirmed by X-ray study. The splitting of the reduction process by around 70 mV is likely due to the presence of the two possible enantiomers in syn and anti-conformations as already mentioned in the literature for other tricarbonylrhenium(I) derivatives.64–66
For the bimetallic complexes, exhaustive electrolyses in reduction mainly led to decomposition reactions. In contrast, those performed at the potential of the first oxidation process allowed the quantification of a possible two-electron oxidation process. Interestingly, in CVs, comparison of the intensity of their redox processes with those of Mono-Re-Phe clearly highlighted their difference and confirmed the number of electrons involved in their respective redox processes (Fig. 6a).
It is noteworthy that the dimerization of the mononuclear compound using its phenyl moiety as connected unit in meta or para position had more impact on its reduction potential (or on its LUMO energy level) than on its oxidation potential (or on its HOMO energy level). These results are in total agreement with theoretical data. Indeed, for the three compounds, the HOMO on one hand, and the LUMO on the other hand, are of similar nature. Moreover, the value of the energy level of the respective HOMOs is nearly the same (±0.07 eV), and that of the LUMOs is also close (±0.12 eV). In comparison with the mononuclear compound, a slight stabilization of these orbitals is observed when forming the dinuclear compounds leading to a smaller energy gap, as clearly highlighted experimentally by electrochemistry.
Finally, the values of the electrochemical HOMO–LUMO gap (Eelg) found experimentally for Mono-Re-Phe and the two bimetallic complexes, around 2.55 eV, 2.46 and 2.46 eV, respectively, also fit very well with the calculated Ecalc gap values 2.67, 2.62 and 2.62 eV, highlighting good correlations with the theoretical study (Table S26†).
UV-visible absorption and emission properties
The three complexes in dilute dichloromethane solutions were very pale yellow in daylight. Their absorption spectra (Fig. 7 and Table 2) showed intense bands between 200 and 300 nm, attributed to a combination of ILCT and MLCT transitions. At low-energy, a band of moderate intensity with MLCT character peaked around 379 nm for Mono-Re-Phe and above 391 nm for Bi-Re-paraPhe and Bi-Re-metaPhe. With respect to the monomer, the molar extinction coefficient of the dimers was multiplied by a little more than two, because of the two chromophoric units and the extension of the electron conjugated system due to the linker.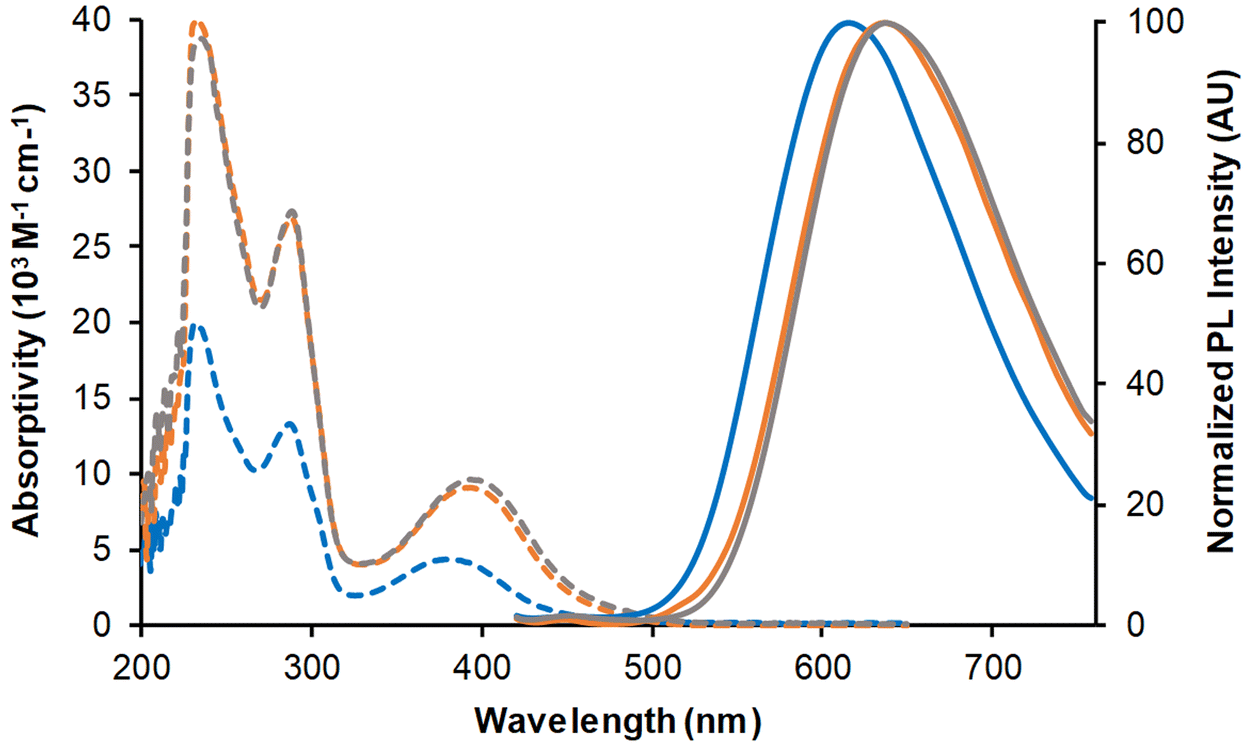 | ||
| Fig. 7 UV-vis absorption spectra (dotted lines) and normalized emission spectra (solid lines) of complexes Mono-Re-Phe (blue lines), Bi-Re-paraPhe (orange lines), and Bi-Re-metaPhe (grey lines) in aerated dichloromethane. Concentrations: ∼5 × 10−5 M (monomer) and 2.5 × 10−5 M (dimers) for absorption, ∼1.1 × 10−5 M (monomer) and 5.1 × 10−6 M (dimers) for emission. Excitation in the low-energy absorption band. | ||
| Compound | CH2Cl2 solutions | Pristine powders | Ground powders | THF-fumed powders | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ abs (nm) | ε (M−1 cm−1) | λ P (nm) | Φ P | τ (ns), [χ2] | λ PL (nm) | Φ PL | τ PL (ns), (f) [χ2]a | λ PL (nm) | Φ PL | τ PL (ns), (f) [χ2]a | λ PL (nm) | Φ PL | |
| a A short lifetime with weak contribution was also detected. The full data are given in the ESI (Fig. S55 and 56†). | |||||||||||||
| Mono-Re-Phe | 232 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
616 | 2.1 × 10−2 | 114 [1.21] | 550 | 0.37 | 105 (2.9) | 552 | 0.34 | — | — | — |
| 287 | 13400 |
1119 (96.8) | |||||||||||
| 379 | 4400 | [1.14] | |||||||||||
| Bi-Re-paraPhe | 231 | 40400 |
636 | 1.4 × 10−2 | 72 [1.40] | 560 | 0.27 | 164 (11.6) | 582 | 0.13 | 229 (31.1) | 564 | 0.20 |
| 289 | 28000 |
934 (87.8) | 873 (66.6) | ||||||||||
| 391 | 9900 | [1.16] | [1.13] | ||||||||||
| Bi-Re-metaPhe | 236 | 39000 |
638 | 1.8 × 10−2 | 71 [1.39] | 552 | 0.34 | 195 (11.5) | 586 | 0.08 | 233 (30.2) | 564 | 0.15 |
| 289 | 28200 |
778 (87.9) | 875 (67.8) | ||||||||||
| 394 | 9700 | [1.15] | [1.16] | ||||||||||
When illuminated by UV light (365 nm), the three complexes in dichloromethane solutions emitted weak orange-red light. The emission spectra showed a single unresolved band, peaking around 616 nm for the Mono-Re-Phe complex and ∼636 nm for the dimers. The experimental emission maxima were only slightly below those calculated by TD-DFT and DFT (Table S22†), considering the involvement of the lowest 3MLCT state. Emission decays were monoexponential (Fig. S54†). The lifetimes, around 114 ns for the monomer and 72 ns for the dimers, confirmed that emission is due to phosphorescence. The quantum yields were moderate, in the 2 × 10−2 range, and were increased by 64%, 24% and 16% for Mono-Re-Phe, Bi-Re-paraPhe and Bi-Re-metaPhe, respectively, when passing from aerated to argon-bubbled solutions. The bimetallic complexes are therefore less sensitive to the presence of dissolved oxygen than their parent complex, which can be explained by their shorter lifetime.
The microcrystalline powders of the Mono-Re-Phe and Bi-Re-metaPhe complexes emitted strong yellow light, with quantum yields up to 0.37 (Table 2 and Fig. 8). The Bi-Re-paraPhe complex emitted ocher light, with slightly lower quantum yield (0.27). In every case, the emission decays were much longer than in solution, indicating strong stabilization of the molecules in the solid state. They also were multiexponential (Fig. S55 and 56†), which reveals the heterogeneity of the microenvironment. The lifetimes associated with preponderant amplitude were just below or above the microsecond range. They could be attributed to bulk molecules. Shorter lifetimes between 100 and 200 ns, with a contribution reaching 11% for the bimetallic complexes, could be assigned to molecules close to the surface or involved in crystal defects. By comparison with solutions, the three complexes therefore show strong SLE effect, mainly due to molecular stiffening in the solid state, which reduces the possibilities of non-radiative deactivation.
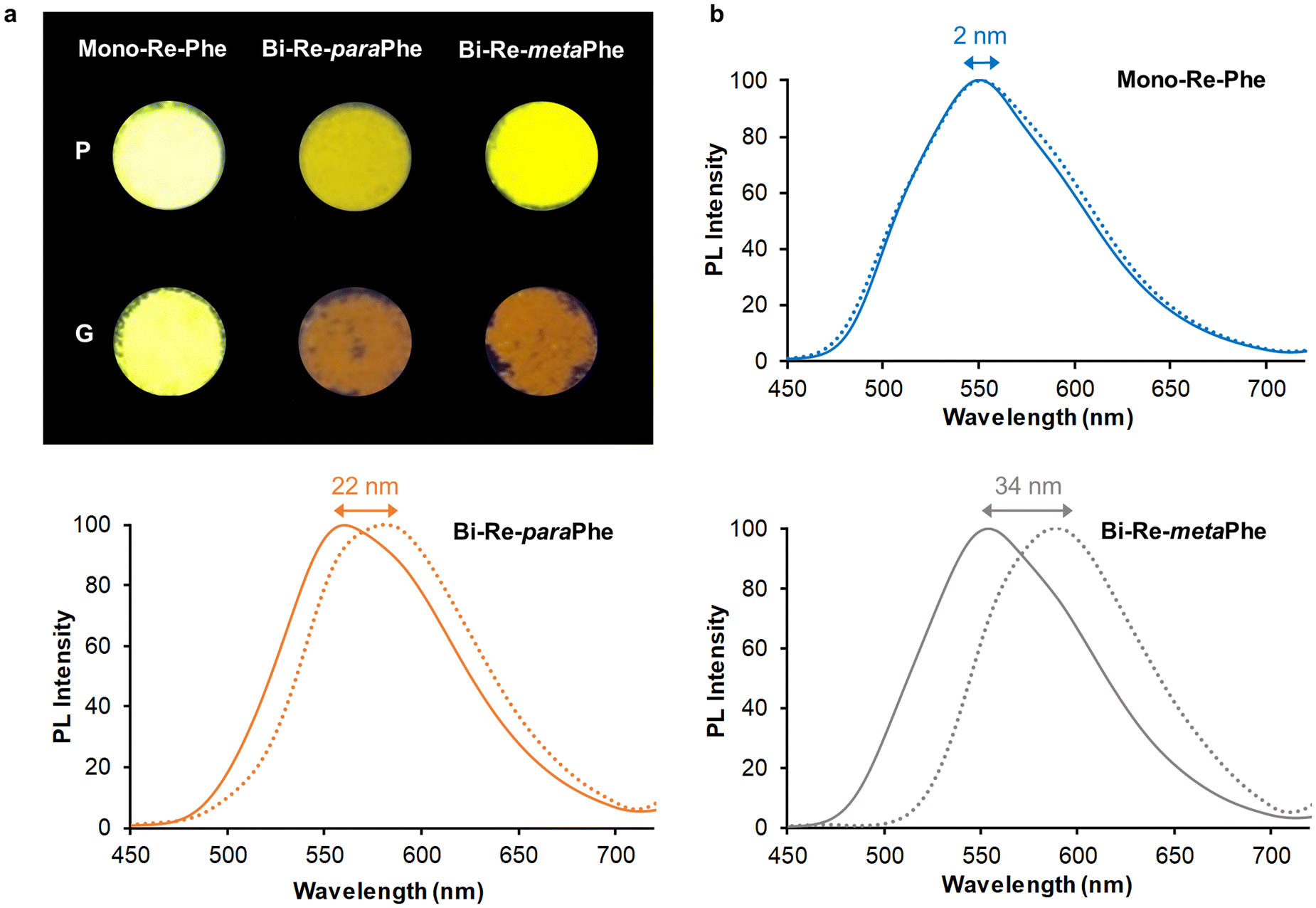 | ||
| Fig. 8 (a) Image of the pristine and ground powders of the three complexes illuminated by UV light at 365 nm. (b) Normalized photoluminescence spectra of Mono-Re-Phe (blue lines), Bi-Re-paraPhe (orange lines), and Bi-Re-metaPhe (grey lines), as microcrystalline pristine powders (solid lines) and ground powders (dotted lines). The wavelength shift between maxima is indicated. Excitation at 380 nm. | ||
Mechanoresponsive luminescence was investigated. To do so, the pristine powders were ground using a mortar and a pestle. Very few emission changes were detected for Mono-Re-Phe, as was also the case for Re-Phe.57 In contrast, the dimer ground powders emitted orange light. The red shift of emission was very significant, ranging from 22 nm for Bi-Re-paraPhe to 34 nm for Bi-Re-metaPhe. Meanwhile, the relative amplitude of the short lifetime was increased with respect to that of the long lifetime, which may indicate the decrease of the population of bulk, well-packed molecules. Fuming the ground powders with THF vapors allowed the partial recovery of the initial photoluminescence. The examination of the samples of Bi-Re-paraPhe and Bi-Re-metaPhe by powder X-Ray diffraction (pXRD) analysis showed that grinding has induced partial amorphization of the pristine microcrystalline sample, and that crystallinity was mostly recovered after THF fuming, which promotes the mobility of molecules (Fig. S57†). A mechanoresponsive luminescence (MRL) behavior thus appears only for dimers, and its amplitude depends on the substitution pattern. For Bi-Re-metaPhe, it is most likely that the MRL effect is promoted by the loose crystal packing mode and easy sliding of crystalline layers on each other upon grinding. Knowing the crystalline arrangement of Bi-Re-paraPhe would make it possible to say whether this is also the case for this compound.
Finally, it was interesting to see if our three complexes that display strong SLE effect as powders also lead to a valuable aggregation-induced emission (AIE) effect in aqueous medium,60 knowing that the formation of aggregates in this medium is not always compatible with good light emission. In a very conventional way, the AIE behavior was investigated by increasing the water proportion in an acetonitrile solution of the complexes, which were used at similar concentrations for optimal comparison. For Mono-Re-Phe, the weak red emission centered at 624 nm in acetonitrile abruptly became a strong green-yellow emission with maximum at 546 nm when the water fraction reached 90%. Under the same conditions, the red emission of Bi-Re-paraPhe and Bi-Re-metaPhe at around 628 nm was progressively shifted to the yellow (568 nm) and orange (592 nm) regions, respectively, with a neat increase of intensity (Fig. 9). The intensity of the PL signal at the maximum wavelength of the most intense band was multiplied by 41, 27 and 10, respectively, for Mono-Re-Phe, Bi-Re-paraPhe and Bi-Re-metaPhe. All these spectroscopic changes were accompanied by the appearance of small particles, visible to the naked eye in some samples. The examination under the fluorescence microscope showed for Mono-Re-Phe the presence of strongly-emissive rod-like microcrystals, which measured about 10–20 μm × 2 μm and agglomerated. The suspensions of Bi-Re-paraPhe contained numerous yellow-emitting agglomerates of very thin particles. Those of Bi-Re-metaPhe mainly contained agglomerates of particles that emitted weakly in the orange, together with very rare microcrystals. In the three cases, no particles were clearly visible in the samples containing the highest proportion of water. The red-shifted emission and intensity decrease observed for these samples suggested the formation of possibly amorphous, ultra-small particles. The AIE effect was therefore clear in every case. However, this effect was much shaper for Mono-Re-Phe, which appears to be the most suitable complex for AIE-related applications. An explanation could be that the monomer readily gives microcrystals in the presence of water, while the more hydrophobic dimers lead to much smaller particles, where molecules are much less protected from water. Additionally, it is interesting to see that Bi-Re-paraPhe gives a better AIE effect than Bi-Re-metaPhe, whereas the opposite could have been expected given the examination of the solid-state emission properties of the microcrystalline powders. This indicates that particles formed in contact with water do not necessarily have the same nature as the microcrystals grown in a solvent, and that subsequent interactions with water are difficult to predict.
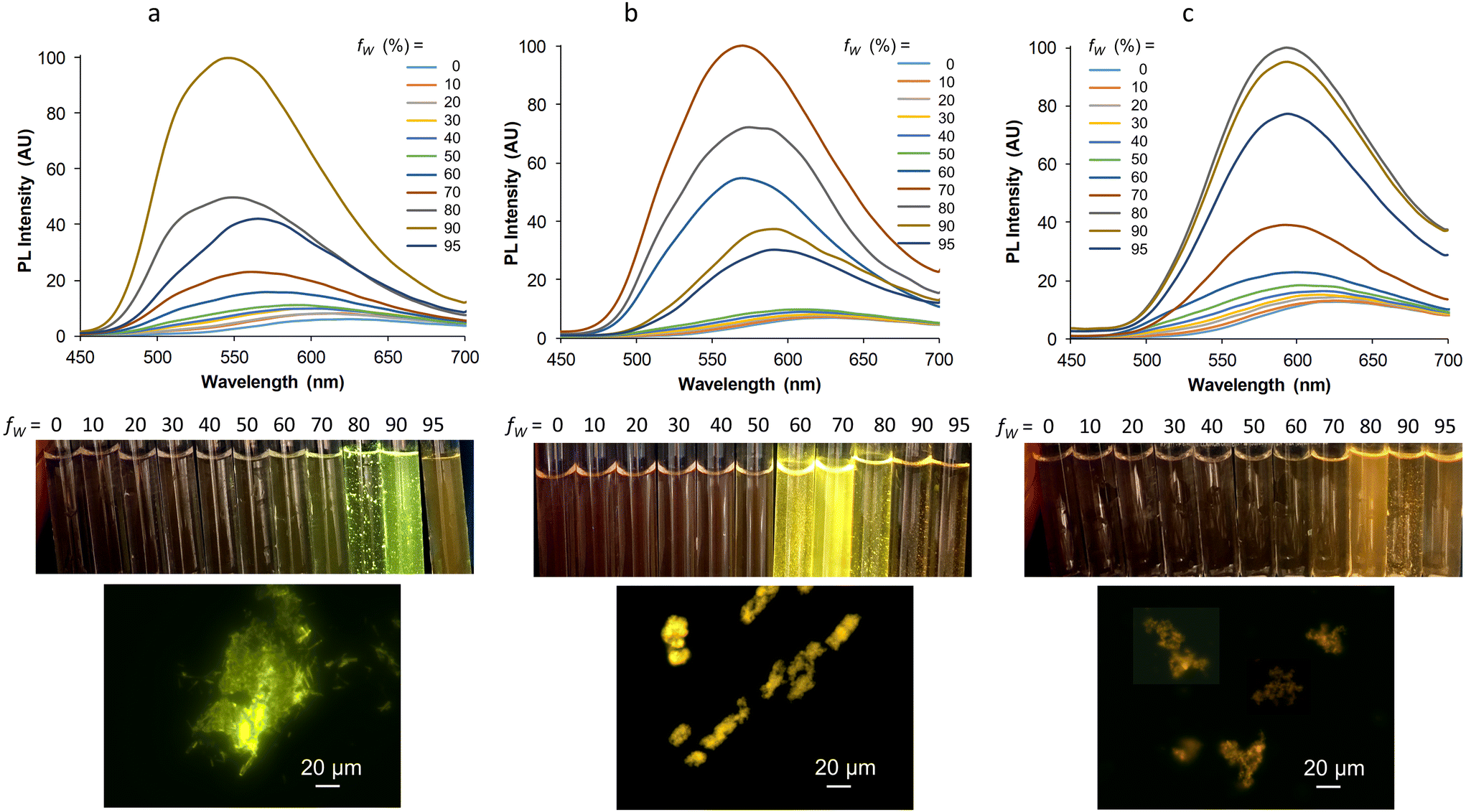 | ||
| Fig. 9 From top to bottom: emission spectra of Mono-Re-Phe (a), Bi-Re-paraPhe (b), and Bi-Re-metaPhe (c) at ∼1.5 × 10−5 M in acetonitrile solutions containing from 0 to 95% water, λex = 364 nm; corresponding samples illuminated at 365 nm; fluorescence microscopy images of the suspensions at fw = 80% or 90%. All samples were observed 3 h after preparation. | ||
Conclusion
The three compounds were efficiently synthesized via a three-step procedure. In solution, the dinuclear structure was shown to induce minor electronic, electrochemical and spectroscopic variations compared with the mononuclear one. In the solid state, all complexes exhibited clear solid state luminescence enhancement, and significant differences appeared between them. The presence of second metal center did not improve the PL efficiency. The mononuclear derivative even seemed to be more advantageous for applications linked to AIE properties. In contrast, MRL was only observed for the dinuclear complexes. Our previous studies showed that this property appears when replacing chloride by bromide or iodide as ancillary ligands, or introducing a bulky substituent such as an adamantyl or a benzoxazole moiety on the phenyl ring.56–59 All these complexes form layers in the crystal network, with the inclusion of several solvent molecules. Similarly, the large flat molecules of Bi-Re-metaPhe organize as a two-dimensional network, thanks to the presence of their alkyl chain and to the crucial role of intermolecular interactions with the solvent. The present work confirms that this type of arrangement is associated with MRL effect. Regarding the influence of regioisomerism, it was observed that the PL efficiency, AIE behavior and the amplitude of the MRL phenomena varied greatly between the two dinuclear complexes. As almost no differences were observed in solution, it can be deduced that those observed in the solid state come from a difference of packing, resulting from the substitution pattern. To the best of our knowledge, the effect of positional isomerism on solid-state emission properties was shown here for the first time.Our study highlights the potential of dinuclear Re(I) complexes in the field of AIE and MRL-active photoluminescent materials. As the research was limited to just three complexes, expanding the investigation to include a broader range of complexes is necessary to validate these findings. Additionally, exploring other multinuclear structures could provide further insights. For instance, we have recently shown the interest of mononuclear complexes based on triazolylidene.55 This type of ligand could easily lead to di-, tri- and tetranuclear complexes.67,68 We can also learn from relatively close polynuclear tricarbonylmanganese complexes.69,70 Complexes incorporating four71 and five72 tricarbonylrhenium(I) centers, as well as multinuclear Re(I) boxes, some of which have already shown their value for solid-state emission, can also be an endless source of inspiration.23,73–75
Experimental section
General methods and synthesis
All purchased chemicals were of the highest purity commercially available and used without further purification. Analytical grade solvents were used as received. Unless otherwise noted, all experiments were carried out under an argon atmosphere. Reactions were monitored by TLC on silica gel Alugram® Xtra SIL G/UV254. Column chromatography was performed on Machery-Nagel silica gel.NMR, mass and infrared spectra were obtained in the relevant ‘Services communs de l'Institut de Chimie de Toulouse, Université de Toulouse III-Paul-Sabatier’. 1H- and 13C-NMR spectra were recorded on Bruker Avance 300 MHz spectrometers. Attributions of the signals were made using 2D NMR data (HSQC and HMBC). Signals are described as follow: bs, broad singlet; s, singlet; d, doublet; t, triplet; q: quadruplet; quint: quintuplet; m, multiplet. App = Apparent. HRMS data were recorded on a Xevo G2 QTOF (Waters) instrument. Fourier transform infrared (FT-IR) spectra were obtained on a Nexus Thermonicolet apparatus with DTGS as the detector. Melting points (Mp) were obtained on a Mettler Toledo apparatus.
:5 to 90:10 v/v) to give the expected compound 1 (1.56 g, 84% yield) as a brown oil.
1H NMR (300 MHz, CDCl3): δ (ppm) = 8.73 (ddd, 1H, J6–5 = 4.8 Hz, J6–4 = 1.7 Hz, J6–3 = 0.9 Hz, H6), 8.20 (dtapp, 1H, J3–4 = 7.9 Hz, J3–5 and 3–6 = 1.0 Hz, H3), 7.84 (td, 1H, J4–3 and 4–5 = 7.8, J4–6 = 1.7 Hz, H4), 7.41 (ddd, 1H, J5–4 = 7.6 Hz, J5–6 = 4.9, J5–3 = 1.2 Hz, H5), 2.92 (t, 2H, J8–9 = 7.8 Hz, H8), 1.83 (quint, 2H, J9–8 and 9–10 = 7.5 Hz, H9), 1.17–1.43 (m, 10H, H10, H11, H12, H13, H14), 0.78–0.86 (m, 3H, H15). 13C NMR (75 MHz, CDCl3): δ (ppm) = 168.2 (C7), 164.0 (C1), 150.2 (C6), 143.8 (C2), 137.3 (C4), 125.7 (C5), 123.0 (C3), 31.8/29.1/22.7 (C10, C11, C12, C13, C14), 26.6 (C9), 25.6 (C8), 14.1 (C15). ESI+ HRMS m/z 260.1763 ([M + H]+ calcd for C15H22N3O1: 260.1763).
:3 to 95:5 v/v), to afford the expected L-Phe ligand as a grey solid (310 mg, 80% yield).
1H NMR (300 MHz, acetone-d6): δ (ppm) = 8.25 (ddd, 1H, J6–5 = 4.8 Hz, J6–4 = 1.8 Hz, J6–3 = 1.0 Hz, H6), 8.10 (dtapp, 1H, J3–4 = 8.0 Hz, J = 1.1 Hz, H3), 7.87 (tdapp, 1H, J4–5 and 4–3 = 7.8 Hz, J4–6 = 1.8 Hz, H4), 7.52–7.54 (m, 3H, Hb, Hd, Hf), 7.36–7.39 (m, 2H, Hc, He), 7.30 (ddd, 1H, J5–4 = 7.6 Hz, J5–6 = 4.8 Hz, J5–3 = 1.2 Hz, H5), 2.60 (t, 2H, J8–9 = 7.8 Hz, H8), 1.64 (quint, 2H, J9–8 and 9–10 = 7.6 Hz, H9), 1.16–1.35 (m, 10H, H10, H11, H12, H13, H14), 0.84–0.88 (m, 3H, H15). 13C NMR (75 MHz, acetone-d6): δ (ppm) = 156.3 (C7), 152.5 (C2), 148.7 (C6), 147.8 (C1), 136.6 (C4), 136.2 (Ca), 129.1 (Cb, Cf), 128.8 (Cd), 127.7 (Cc, Ce), 123.6 (C5), 123.3 (C3), 31.6/28.9/22.4 (C10, C11, C12, C13, C14), 26.9 (C9), 24.8 (C8), 13.5 (C15). ESI+ HRMS m/z 335.2238 ([M + H]+ calcd for C21H27N4: 335.2236).
:3 to 90:10 v/v).
1H NMR (300 MHz, acetone-d6): δ (ppm) = 8.32 (ddd, 2H, J6–5 and 6′–5′ = 4.8 Hz, J6–4 and 6′–4′ = 1.8 Hz, J6–3 and 6–3′ = 1.0 Hz, H6, H6′), 8.15 (dtapp, 2H, J3–4 and 3′–4′ = 8.0 Hz, J = 1.1 Hz, H3, H3′), 7.92 (tdapp, 2H, J4–5, 4–3, 4′-5′ and 4′–3′ = 7.8 Hz, J4–6 and 4′–6′ = 1.8 Hz, H4, H4′), 7.56 (s, 4H, Hb, Hc, He, Hf), 7.36 (ddd, 2H, J5–4 and 5′–4′ = 7.6 Hz, J5–6 and 5′–6′ = 4.8 Hz, J5–3 and 5′–3′ = 1.2 Hz, H5, H5′), 2.72 (t, 4H, J8–9 and 8′–9′ = 7.7 Hz, H8, H8′), 1.68 (quint, 4H, J9–8, 9–10, 9′–8′ and 9′–10′ = 7.5 Hz, H9, H9′), 1.21–1.40 (m, 20H, H10, H11, H12, H13, H14, H10′, H11′, H12′, H13′, H14′), 0.81–0.90 (m, 6H, H15, H15′). 13C NMR (75 MHz, acetone-d6): δ (ppm) = 157.2 (C7, C7′), 153.3 (C2, C2′), 149.5 (C6, C6′), 148.5 (C1, C1′), 137.8 (C4, C4′), 137.7 (Ca, Cd), 129.5 (Cb, Cc, Ce, Cf), 124.7 (C5, C5′), 124.2 (C3, C3′), 32.6/30.0/23.4 (C10, C10′, C11, C11′, C12, C12′, C13, C13′, C14, C14′), 27.9 (C9, C9′), 25.8 (C8, C8′), 14.4 (C15, C15′). ESI+ HRMS m/z 591.3930 ([M + H]+ calcd for C36H47N8: 591.3924).
:3 to 90:10 v/v).
1H NMR (300 MHz, acetone-d6): δ (ppm) = 8.29 (bsapp, 2H, H6, H6′), 8.15 (dapp, 2H, J3–4 and 3′–4′ = 7.8 Hz, H3, H3′), 7.90 (tapp, 2H, J4–5, 4–2, 4′-5′ and 4′–2′ = 7.8 Hz, H4, H4′), 7.69 (m, 1H, He), 7.57 (m, 2H, Hd, Hf), 7.55 (m, 1H, Hb), 7.36 (ddapp, 2H, J = 7.0, J = 3.8 Hz, H5, H5′), 2.64 (t, 4H, J8–9 and 8′–9′ = 7.5 Hz, H8, H8′), 1.67 (quint, 4H, J9–8, 9–10, 9′–8′ and 9′–10′ = 7.4 Hz, H9, H9′), 1.16–1.35 (m, 20H, H10, H11, H12, H13, H14, H10′, H11′, H12′, H13′, H14′), 0.83–0.87 (m, 6H, H15, H15′). 13C NMR (75 MHz, acetone-d6): δ (ppm) = 157.3 (C7, C7′), 153.3 (C2, C2′), 149.6 (C6, C6′), 148.5 (C1, C1′), 138.0 (Ca, Cc), 137.8 (C4, C4′), 130.7 (Ce), 129.2 (Cd, Cf), 128.5 (Cb), 124.7 (C5, C5′), 124.2 (C3, C3′), 32.6/30.0/23.4 (C10, C11, C12, C13, C14, C10′, C11′, C12′, C13′, C14′), 27.7 (C9, C9′), 25.7 (C8, C8′), 14.4 (C15, C15′). ESI+ HRMS m/z 591.3924 ([M + H]+ calcd for C36H47N8: 591.3939).
:3 to 95:5 v/v), to give Mono-Re-Phe as a yellow solid (95 mg, 83% yield).
1H NMR (300 MHz, acetone-d6): δ (ppm) = 9.11 (ddd, 1H, J6–5 = 5.5 Hz, J6–4 = 1.6 Hz, J6–3 = 0.8 Hz, H6), 8.04 (tdapp, 1H, J4–5 and 4–3 = 8.0 Hz, J4–6 = 1.6 Hz, H4), 7.84–7.87 (m, 4H, Hb/c/e/f), 7.74–7.77 (m, 1H, Hd), 7.71 (ddd, 1H, J5–4 = 7.8 Hz, J5–6 = 5.5 Hz, J5–3 = 1.3 Hz, H5), 6.97 (dtapp, 1H, J3–4 = 8.1 Hz, J = 1.2 Hz, H3), 2.66–2.78 (m, 2H, H8), 1.73 (quint, 2H, J9–8 and 9–10 = 7.6 Hz, H9), 1.20–1.40 (m, 10H, H10–14), 0.84–0.88 (m, 3H, H15). 13C NMR (75 MHz, acetone-d6): δ (ppm) = 198.2 (CO), 197.3 (CO), 189.0 (CO), 158.9 (C7), 155.1 (C2), 154.4 (C6), 145.3 (C1), 139.9 (C4), 132.5 (Ca), 131.8/131.1/127.8 (Cb, Cd, Ce, Cf), 127.5 (C5), 122.4 (C3), 31.6/28.8/22.4 (C10, C11, C12, C13, C14), 26.5 (C9), 24.4 (C8), 13.5 (C15). ESI+ HRMS m/z 605.1562 [M − Cl]+ calcd for C24H26N4O3Re: 605.1560. IR (ATR) νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: 2018, 1900, 1886 cm−1. Anal. calcd (%) for C24H26ClN4O3Re: C 45.03, H 4.09, N 8.75; found: C 46.24, H 4.32, N 8.05.
O: 2018, 1900, 1886 cm−1. Anal. calcd (%) for C24H26ClN4O3Re: C 45.03, H 4.09, N 8.75; found: C 46.24, H 4.32, N 8.05.
1H NMR (300 MHz, acetone-d6): δ (ppm) (mixture of isomers): 9.16 (ddd, 2H, J6–5 and 6′–5′ = 5.5 Hz, J6–4 and 6′–4′ = 1.6 Hz, J6–3 and 6′–3′ = 0.8 Hz, H6, H6′), 8.31–8.47 (m, 4H, Hb, Hc, He, Hf), 8.25 (tdapp, 1H, J4/4′–5 and 4/4′–3 = 8.0 Hz, J4/4′–6 = 1.5 Hz, H4, H4′), 8.06 (tdapp, 1H, J4/4′–5 and 4/4′–3 = 7.9 Hz, J4/4′–6 = 1.6 Hz, H4, H4′), 7.75–7.80 (m, 2H, H5, H5′), 7.52–7.55 (m, 1H, H3, H3′), 7.20–7.23 (m, 1H, H3, H3′), 2.78–3.02 (m, 4H, H8, H8′), 1.72–1.91 (m, 4H, H9, H9′), 1.19–1.50 (m, 20H, H10, H11, H12, H13, H14, H10′, H11′, H12′, H13′, H14′), 0.82–0.89 (m, 6H, H15, H15′). 13C NMR (75 MHz, acetone-d6): δ (ppm) (mixture of isomers): 199.0 (CO), 198.1 (CO), 189.9 (CO), 159.8, 159.7 (C7, C7′), 156.1 (C1, C1′), 155.6, 155.5 (C6, C6′), 146.0, 145.9 (C2, C2′), 141.1, 141.0, 140.6 (C4, C4′), 136.3 (Cd), 132.3, 132.1, 132.0 (Cb, Cc, Ce, Cf), 128.7, 128.6 (C5, C5′), 124.2, 124.1, 123.6 (C3, C3′), 32.6, 30.0, 23.4, 23.3 (C10, C11, C12, C13, C14, C10′, C11′, C12′, C13′, C14′), 27.5, 27.4 (C9, C9′), 25.6, 25.5 (C8, C8′), 14.4 (C15, C15′). ESI− HRMS m/z 1201.1919 [M − H]− calcd for C42H45Cl2N8O6Re2: 1201.1932. IR (ATR) νCO: 2018, 1907, 1894, 1866 cm−1. Anal. calcd (%) for C42H46Cl2N8O6Re2: C 41.96, H 3.86, N 9.32; found: C 41.81, H 3.40, N 9.21.
1H NMR (300 MHz, acetone-d6): δ (ppm) (mixture of isomers): 9.07–9.15 (m, 2H, H6, H6′), 8.32–8.54 (m, 4H, Hb, Hd, He, Hf), 8.21 (tdapp, 1H, J4/4′–5 and 4/4′–3 = 8.0 Hz, J4/4′–6 = 1.4 Hz, H4, H4′), 8.13 (dqd, 1H, J = 8.0 Hz, J = 4.2 Hz, J = 1.5 Hz, H4, H4′), 7.69–7.80 (m, 2H, H5, H5′), 7.52–7.58 (m, 1H, H3, H3′), 7.21–7.26 (m, 1H, H3, H3′), 2.74–3.03 (m, 4H, H8, H8′), 1.75–1.85 (m, 4H, H9, H9′), 1.18–1.47 (m, 20H, H10, H11, H12, H13, H14, H10′, H11′, H12′, H13′, H14′), 0.83–0.90 (m, 6H, H15, H15′). 13C NMR (75 MHz, acetone-d6): δ (ppm) (mixture of isomers): 199.0 (CO), 198.1 (CO), 189.8 (CO), 159.8 (C7, C7′), 156.2, 156.1 (C1, C1′), 155.6, 155.5, 155.4 (C6, C6′), 146.1, 146.0, 145.9 (C2, C2′), 140.9, 140.8, 140.6 (C4, C4′), 135.6, 135.4, 135.3 (Ca, Cc), 135.0, 134.8, 132.7, 132.6, 132.5 (Cb, Cf), 129.6, 129.4, 129.1, 128.8, 128.7 (C5, C5′), 124.0, 123.9, 123.8, 123.7 (C3, C3′), 32.6, 32.5, 30.0, 23.3 (C10, C11, C12, C13, C14, C10′, C11′, C12′, C13′, C14′), 27.3, 27.2 (C9, C9′), 25.7, 25.6 (C8, C8′), 14.4 (C15, C15′). ESI− HRMS m/z 1197, 1913 ([M − H]− calcd for C42H45Cl2N8O6Re2: 1197.1898). IR (ATR) νCO: 2019, 1937, 1903, 1877 cm−1. Anal. calcd (%) for C42H46Cl2N8O6Re2: C 41.96, H 3.86, N 9.32; found: C 41.00, H 3.62, N 8.94.
Crystallography
Crystallographic data were collected at low temperature (193K) with an Oxford Instruments Cryostream 700+ Series device using MoKα radiation (wavelength = 0.71073 Å) on a Bruker AXS D8-Venture diffractometer equipped with a multilayer TRIUMPH X-ray mirror and a Photon III-C14 detector. Phi- and omega-scans were used. The space group was determined on the basis of systematic absences and intensity statistics. An empirical absorption correction was employed.76 The structures were solved using an intrinsic phasing method (ShelXT).77 All non-hydrogen atoms were refined anisotropically using the least-square method on F2.78 Hydrogen atoms were refined isotropically at calculated positions using a riding model. Selected crystallographic data are collected in Table 3.| Mono-Re-Phe | Bi-Re-metaPhe | L- paraPhe | L- metaPhe | |
| Empirical formula | C24H26ClN4O3Re | C42H46Cl2N8O6Re2·2 CH2Cl2 | C36H46N8 | C36H46N8·½ C3H6O |
| Formula weight | 640.15 | 1372.04 | 590.81 | 619.84 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Triclinic |
| Space group | Pbca | C2/c | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Unit cell dimensions | ||||
| a (Å) | 11.4066(7) | 30.087(2) | 20.3002(15) | 9.1394(9) |
| b (Å) | 17.3844(10) | 12.0833(12) | 8.9187(6) | 20.290(2) |
| c (Å) | 24.7532(15) | 15.2938(15) | 20.5373(14) | 20.328(2) |
| α (°) | 90 | 90 | 90 | 75.123(4) |
| β (°) | 90 | 95.995(3) | 116.516(3) | 79.003(4) |
| γ (°) | 90 | 90 | 90 | 82.681(4) |
| Volume (Å3) | 4908.5(5) | 5529.7(9) | 3327.2(4) | 3564.1(6) |
| Z | 8 | 4 | 4 | 2 |
| Density (calculated) (Mg m−3) | 1.732 | 1648 | 1.179 | 1.155 |
| Crystal size (mm3) | 0.200 × 0.040 × 0.040 | 0.100 × 0.040 × 0.040 | 0.500 × 0.080 × 0.080 | 0.200 × 0.200 × 0.020 |
| Reflections collected | 200053 |
137719 |
103159 |
102101 |
| Independent reflections | 9826 | 6392 | 11042 |
17775 |
| R int | 0.0520 | 0.0646 | 0.0771 | 0.0668 |
| Restraints/parameters | 233/374 | 227/375 | 0/399 | 889/1089 |
| Final R1 index I > 2σ(I) | 0.0233 | 0.0614 | 0.0620 | 0.0709 |
| wR2 (all data) | 0.0470 | 0.1566 | 0.1798 | 0.2390 |
| Largest diff. peak and hole (e Å−3) | 1.020 and −1.072 | 1.964 and −1.916 | 0.493 and −0.251 | 0.490 and −0.393 |
| CCDC | 2365135 | 2365136 | 2365138 | 2365137 |
Hirshfeld surfaces (HS) were mapped over dnorm and shape-index. 3D Hirshfeld surfaces and 2D fingerprint plots (FP)79 were generated with high resolution based on the crystallographic information file (CIF) using Crystal Explorer 17.5 software.80 The normalized contact distance (dnorm) was defined according to eqn (1), where di and de are the distances from the surface to the nearest atom interior and exterior to the surface, respectively, and rvdw is the van der Waals radii of the corresponding atoms:
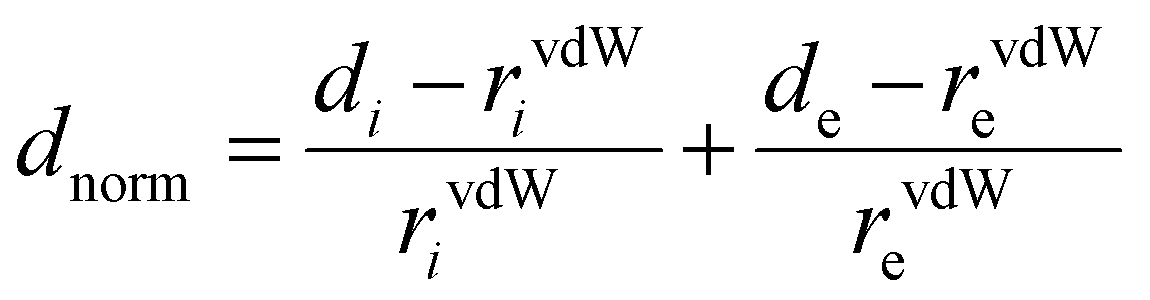 | (1) |
On the dnorm point representation, intermolecular contacts shorter than the sum of the van der Waals radii (dnorm < 0) of the interacting atoms are denoted as red spots on the surfaces, whereas longer than the sum of the van der Waals radii (dnorm > 0) of the interacting atoms are represented by blue regions. The van der Waals contacts (dnorm = 0) are colored in white. 2D fingerprint plots provide correlation between di and de.
Powder X-ray diffraction measurements were performed in the LPCNO-INSA laboratory of Toulouse on a Malvern Panalytical Empyrean diffractometer, equipped with a Co anticathode source (wavelength 1.789 Å), a Bragg–Brentano HD primary optics, and a Pixcel1D linear detector. The measurements were made using a monocrystalline silicon sample holder with no background noise. Each diffraction pattern was obtained using a theta-theta symmetric scan with a pitch of 0.02° and an exposure time of 1s per step.
Computational details
The GAUSSIAN16 program package81 was employed for all calculations (the geometry optimization, the ground-state and excited-state electronic structures, and optical spectra) with the aid of the ChemCraft visualization program.82 The ground state (S0), the first excited state (S1) and the lowest triplet state (T1) geometries of the complexes were fully optimized with the restricted and unrestricted density functional theory (R-DFT and U-DFT) method using the Perdew–Burke–Ernzerhof PBE1PBE functional with no symmetry constraints.83 In all calculations, the “double-ζ” quality basis set LANL2DZ with Hay and Wadt's relative effective core potential ECP (outer-core [(5s25p6)] electrons and the (5d6) valence electrons)84,85 was employed for the Re atom. The 6-311+G** basis set for H, C, N and O atoms was used.86 The solvent effect (dichloromethane, ε = 8.93) was simulated using the Self-Consistent Reaction Field (SCRF) under the Conductor Polarizable Continuum Model (CPCM).87–89 The vibrational frequencies calculations were performed using the optimized structural parameters of the complexes, to confirm that each optimized structure represents a local minimum on the potential energy surface and all eigenvalues are non-negative. The optimized Cartesian coordinates are included in the ESI part (see Tables S27–38†). On the optimized ground and excited state geometries, the absorption and emission properties were calculated by the time dependent density functional theory (TD-DFT) method at the PBE1PBE/LANL2DZ/6-311+G** level. These methods have already shown good agreement with experimental studies for different rhenium(I) complexes.90Electrochemistry
The electrochemical properties of the complexes were determined by cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) in dichloromethane. The solutions used during the electrochemical studies were typically 1 × 10−3 M in complex, and 0.1 M in supporting electrolyte. The supporting electrolyte n[Bu4N][BF4] (Fluka, 99% electrochemical grade) was used as received and simply degassed under Argon. Dichloromethane was dried using an MB SPS-800 solvent purification system just prior to use. The measurements were carried out with an Autolab PGSTAT100 potentiostat controlled by GPES 4.09 software. Experiments were performed at room temperature (r.t.) in a homemade airtight three-electrode cell connected to a vacuum/Ar line. The reference electrode consisted of a saturated calomel electrode (SCE) separated from the solution by a bridge compartment. The counter electrode was a Pt wire of ca. 1 cm2 apparent surface. The working electrode was a Pt microdisk (0.5 mm diameter). Before each measurement, the solutions were degassed by bubbling Ar and the working electrode was polished with a polishing machine (Presi P230). Under these experimental conditions, Fc+/Fc is observed at +0.55 ± 0.01 V vs. SCE. OSWVs were obtained using an amplitude of 20 mV, a frequency of 20 Hz, and a step potential of 5 mV.Spectroscopy
Spectroscopic measurements in solutions were conducted at 20 °C in a temperature-controlled cell. UV-visible absorption spectra and emission spectra in solutions were measured with a Xenius SAFAS spectrofluorometer using cells of 1 cm optical pathway. All emission spectra were corrected. The emission quantum yields in solution (Φ) were determined using the classical formula:| Φx = (As × Ix × nx2 × Φs)/(Ax × Is × ns2) | (2) |
For AIE measurements, a small volume (30 μL) of a concentrated solution of complex in acetonitrile was injected in 2.97 mL of various acetonitrile/water mixtures. The samples were left to stand under stirring in the dark, and then they were sonicated for 5 min before optical measurement, so that they were as homogeneous as possible. Absorbance variations due to scattering by microparticles, and in particular the baseline deviation, were taken into account for measuring the extinction coefficient value, the fluorescence quantum yields of the suspension in the water/acetonitrile 90:10 v/v mixture, and the magnitude of the AIE effect.
Solid state spectra were recorded on a HORIBA Fluorolog 3-2iHR320 spectrofluorometer and were corrected. The absolute photoluminescence quantum yield values (ΦP) were determined using the Xenius SAFAS spectrofluorometer provided with an integrating sphere, by a method based on the one developed by De Mello et al.,92 as described elsewhere.51 The error was estimated to be about 20%.
The emission decay curves were recorded using the time-correlated single-photon counting (TCSPC) method on the HORIBA Fluorolog 3-2iHR320 spectrofluorometer used at right-angle geometry and equipped with a NanoLED-370 (λex = 371 nm). The absorbance of solutions and suspensions at λex was lower than 0.1. The solid sample was deposited on a quartz holder. Photons were detected through a monochromator by means of a Hamamatsu R928 photomultiplier. Emission was recorded near the maximum with a bandpass of 10–15 nm. The instrumental response was recorded at 371 nm. All analyses were recorded using the Datastation v2.7 software. The decay curves were analyzed with reconvolution and global non-linear least-squares minimization method using DAS6 v6.8 software. The rate constants for radiative (kr) and nonradiative (knr) decay were calculated using the following equation:
| kr = Φ/τ and knr = (1 − Φ)/τ | (3) |
Fluorescence microscopy was performed with a Leitz Laborlux D fluorescence microscope equipped with an Andor Luca camera (λex ∼ 450–490 nm, λem > 500 nm).
Author contributions
Stéphen Le Garrec: investigation, writing original draft. David Martins-Bessa: investigation. Mariusz Wolff: investigation, formal analysis, writing original draft. Béatrice Delavaux-Nicot: investigation, writing original draft. Sonia Mallet-Ladeira: investigation. Charles-Louis Serpentini: investigation. Eric Benoist: project administration, funding acquisition, supervision, writing – review and editing. Florence Bedos-Belval: conceptualization, methodology, supervision, writing – original draft. Suzanne Fery-Forgues: conceptualization, methodology, supervision, writing – original draft.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds Mono-Re-Phe, Bi-Re-metaPhe, L-paraPhe and L-metaPhe have been deposited at CCDC under numbers 2365135, 2365136, 2365138, 2365137, respectively, and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DFT calculations were carried out using resources provided by Wrocław Centre for Networking and Supercomputing (https://www.wcss.wroc.pl), Poland. M. W. thanks Université de Toulouse III for offering him a visiting professor position in September 2023. We are grateful to Mr Nicolas Ratel-Ramond (LPCNO-INSA of Toulouse) for the measurement of powder XRD patterns, and to Dr Alix Sournia-Saquet and Mr Alain Moreau (LCC) for their help in electrochemical measurements.References
- L. C.-C. Lee, K.-K. Leung and K. K.-W. Lo, Dalton Trans., 2017, 46, 16357–16380 RSC.
- R. G. Balasingham, F. L. Thorp-Greenwood, C. F. Williams, M. P. Coogan and S. J. A. Pope, Inorg. Chem., 2012, 51, 1419–1426 CrossRef CAS PubMed.
- R.-R. Ye, C.-P. Tan, M.-H. Chen, L. Hao, L.-N. Ji and Z.-W. Mao, Chem. – Eur. J., 2016, 22, 7800–7809 CrossRef CAS PubMed.
- S. A. Sharma, N. Vaibhavi, B. Kar, U. Das and P. Paira, RSC Adv., 2022, 12, 20264–20295 RSC.
- C. A. Kumar, S. Karthikeyan, B. Varghese, V. Veena, N. Sakthivel and B. Manimaran, J. Organomet. Chem., 2014, 766, 86e94 Search PubMed.
- B. Ramakrishna, R. Nagarajaprakash, V. Veena, N. Sakthivel and B. Manimaran, Dalton Trans., 2015, 44, 17629–17638 RSC.
- N. Montesdeoca, R. L. Borkar, M. Sathiyendiran and J. Karges, Chemistry, 2024, 30, 202400217 CrossRef PubMed.
- Z.-Y. Pan, D.-H. Cai and L. He, Dalton Trans., 2020, 49, 11583–11590 RSC.
- D. Pelleteret, N. C. Fletcher and A. P. Doherty, Inorg. Chem., 2007, 46, 4386–4388 CrossRef CAS PubMed.
- M.-A. Guillevic, M. E. Light, S. J. Coles, T. Gelbrich, M. B. Hursthouse and D. W. Bruce, J. Chem. Soc., Dalton Trans., 2000, 1437–1445 RSC.
- C. Bruckmeier, M. W. Lehenmeier, R. Reithmeier, B. Rieger, J. Herranz and C. Kavakli, Dalton Trans., 2012, 41, 5026–5037 RSC.
- R. Giereth, P. Lang, E. McQueen, X. Meißner, B. Braun-Cula, C. Marchfelder, M. Obermeier, M. Schwalbe and S. Tschierlei, ACS Catal., 2021, 11, 390–403 CrossRef CAS.
- F.-X. Wang, J.-H. Liang, H. Zhang, Z.-H. Wang, Q. Wan, C.-P. Tan, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2019, 11, 13123–13133 CrossRef CAS PubMed.
- A. B. Solea, G. Demirci, F. M. Harvey, A. Crochet, F. Zobi and O. M. Steiner, Dalton Trans., 2024, 53, 13743–13755 RSC.
- P. J. Ball, T. Rarog Shtoyko, J. A. Krause Bauer, W. J. Oldham and W. B. Connick, Inorg. Chem., 2004, 43, 622–632 CrossRef CAS PubMed.
- M. Yu. Petyuk, I. Yu. Bagryanskaya, O. I. Artyushin, V. K. Brel and A. V. Artem'ev, Mendeleev Commun., 2021, 31, 810–812 CrossRef CAS.
- M. Yu. Petyuk, A. S. Berezin, I. Yu. Bagryanskaya, O. I. Artyushin, V. K. Brel and A. V. Artem'ev, Inorg. Chem. Commun., 2020, 119, 108058 CrossRef CAS.
- P. Cavigli, G. Balducci, E. Zangrando, N. Demitri, A. Amati, M. T. Indelli and E. Iengo, Inorg. Chim. Acta, 2016, 439, 61–68 CrossRef CAS.
- S. Frantz, M. Sieger, I. Hartenbach, F. Lissner, T. Schleid, J. Fiedler, C. Duboc and W. Kaim, J. Organomet. Chem., 2009, 694, 1122–1133 CrossRef CAS.
- N. Saleh, D. Kundu, N. Vanthuyne, J. Olesiak-Banska, A. Pniakowska, K. Matczyszyn, V. Y. Chang, G. Muller, J. A. G. Williams, M. Srebro-Hooper, J. Autschbach and J. Crassous, ChemPlusChem, 2020, 85, 2446–2454 CrossRef CAS PubMed.
- K. S. Kisel, J. R. Shakirova, V. V. Pavlovskiy, R. A. Evarestov, V. V. Gurzhiy and S. P. Tunik, Inorg. Chem., 2023, 62, 18625–18640 CrossRef CAS PubMed.
- R. Jordan, M. Niazi, S. Schäfer, W. Kaim and A. Klein, Molecules, 2022, 27, 8159 CrossRef CAS PubMed.
- Y. Zhang, M. R. Crawley, C. E. Hauke, A. E. Friedman, T. S. Janik and T. R. Cook, Eur. J. Inorg. Chem., 2017, 34, 4055–4060 CrossRef.
- A. Wilting, T. Stolper, R. A. Mata and I. Siewert, Inorg. Chem., 2017, 56, 4176–4185 CrossRef CAS PubMed.
- L. A. Paul, S. Rajabi, C. Jooss, F. Meyer, F. Ebrahimi and I. Siewert, Dalton Trans., 2020, 49, 8367–8374 RSC.
- Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976–11987 CrossRef CAS PubMed.
- W. Yang, S. S. Roy, W. C. Pitts, R. L. Nelson, F. R. Fronczek and J. W. Jurss, Inorg. Chem., 2018, 57, 9564–9575 CrossRef CAS PubMed.
- M. Panigati, M. Mauro, D. Donghi, P. Mercandelli, P. Mussini, L. De Cola and G. D'Alfonso, Coord. Chem. Rev., 2012, 256, 1621–1643 CrossRef CAS.
- M. Mauro, C.-H. Yang, C.-Y. Shin, M. Panigati, C.-H. Chang, G. D'Alfonso and L. De Cola, Adv. Mater., 2012, 24, 2054–2058 CrossRef CAS PubMed.
- X. Xu and H. A. Xiao, J. Lumin., 2012, 132, 2251–2258 CrossRef CAS.
- A. W.-T. Choi, K. K.-S. Tso, V. M.-W. Yim, H.-W. Liu and K. K.-W. Lo, Chem. Commun., 2015, 51, 3442–3445 RSC.
- A. Palmioli, A. Aliprandi, D. Septiadi, M. Mauro and A. Bernardi, Org. Biomol. Chem., 2017, 15, 1686–1699 RSC.
- M. Proverbio, E. Quartapelle Procopio, M. Panigati, S. Mercurio, R. Pennati, M. Ascagni, R. Leone, C. La Porta and M. Sugni, Org. Biomol. Chem., 2019, 17, 509–518 RSC.
- F. Palominos, P. Mella, K. Guajardo, G. Günther, A. Vega and N. Pizarro, Photochem. Photobiol. Sci., 2024, 23, 119–132 CrossRef CAS PubMed.
- J. Muñoz, X. Rojas, F. Palominos, R. Arce, F. Cañas, N. Pizarro and A. Vega, Polyhedron, 2023, 239, 116442 CrossRef.
- N. P. Liyanage, W. Yang, S. Guertin, S. S. Roy, C. A. Carpenter, R. E. Adams, R. H. Schmehl, J. H. Delcamp and J. W. Jurss, Chem. Commun., 2019, 55, 993–996 RSC.
- G. Li, Y. Chen, J. Wang, Q. Lin, J. Zhao, L. Jia and H. Chao, Chem. Sci., 2013, 4, 4426–4433 RSC.
- E. V. Puttock, A. Sil, D. S. Yufit and J. A. G. Williams, Dalton Trans., 2020, 49, 10463–10476 RSC.
- L. Lu, M. Wang, Z. Mao, T.-S. Kang, X.-P. Chen, J.-J. Lu, C.-H. Leung and D.-L. Ma, Sci. Rep., 2016, 6, 22458 CrossRef CAS PubMed.
- B. Liu, S. Monro, L. Lystrom, C. G. Cameron, K. Colón, H. Yin, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2018, 57, 9859–9872 CrossRef CAS PubMed.
- W. Cheng, R. Sheng, Y. Liu, S. Wang, P. Chen and B. Tong, Inorg. Chem. Commun., 2021, 129, 108667 CrossRef CAS.
- M. A. Esteruelas, A. M. López, E. Oñate, A. San-Torcuato, J.-Y. Tsai and C. Xia, Organometallics, 2017, 36, 699–707 CrossRef CAS.
- E. Martínez-Vollbert, C. Philouze, I. Gautier-Luneau, Y. Moreau, P.-H. Lanoë and F. Loiseau, Phys. Chem. Chem. Phys., 2021, 23, 24789–24800 RSC.
- P. Alam, C. Climent, P. Alemany and I. R. Laskar, J. Photochem. Photobiol., C, 2019, 41, 100317 CrossRef CAS.
- L. Ravotto and P. Ceroni, Coord. Chem. Rev., 2017, 346, 62–76 CrossRef CAS.
- V. Sathish, A. Ramdass, P. Thanasekaran, K.-L. Lu and S. Rajagopal, J. Photochem. Photobiol., C, 2015, 23, 25–44 CrossRef CAS.
- L. Ma, Y. Wang, X. Wang, Q. Zhu, Y. Wang, L. Li, H.-B. Cheng, J. Zhang and X.-J. Liang, Coord. Chem. Rev., 2022, 473, 214822 CrossRef CAS.
- H. Shen, C. Xu, F. Sun, M. Zhao, Q. Wu, J. Zhang, S. Li, J. Zhang, J. W. Y. Lam and B. Z. Tang, ChemMedChem, 2022, 17, e202100578 CrossRef CAS PubMed.
- V. Sathish, A. Ramdass, Z.-Z. Lu, M. Velayudham, P. Thanasekaran, K.-L. Lu and S. Rajagopal, J. Phys. Chem. B, 2013, 117, 14358–14366 CrossRef CAS PubMed.
- E. Quartapelle Procopio, M. Mauro, M. Panigati, D. Donghi, P. Mercandelli, A. Sironi, G. D'Alfonso and L. De Cola, J. Am. Chem. Soc., 2010, 132, 14397–14399 CrossRef CAS PubMed.
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS.
- J. Wang, B. Delavaux-Nicot, M. Wolff, S. Mallet-Ladeira, R. Métivier, E. Benoist and S. Fery-Forgues, Dalton Trans., 2018, 47, 8087–8099 RSC.
- J. Wang, A. Poirot, B. Delavaux-Nicot, M. Wolff, S. Mallet-Ladeira, J. P. Calupitan, C. Allain, E. Benoist and S. Fery-Forgues, Dalton Trans., 2019, 48, 15906–15916 RSC.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, C. Meslien, N. Saffon-Merceron, C.-L. Serpentini, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Dalton Trans., 2023, 52, 5453–5465 RSC.
- C. Vanucci-Bacqué, M. Wolff, B. Delavaux-Nicot, A. M. Abdallah, S. Mallet-Ladeira, C.-L. Serpentini, F. Bedos-Belval, K. W. Fong, X. Y. Ng, M. L. Low, E. Benoist and S. Fery-Forgues, Dalton Trans., 2024, 53, 11276–11294 RSC.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, N. Saffon-Merceron, C.-L. Serpentini, N. Leygue, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Photochem. Photobiol. Sci., 2023, 22, 169–184 CrossRef CAS PubMed.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, N. Leygue, N. Saffon-Merceron, F. Alary, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Dalton Trans., 2021, 50, 13686–13698 RSC.
- A. Poirot, N. Leygue, B. Delavaux-Nicot, N. Saffon-Merceron, C. Allain, E. Benoist and S. Fery-Forgues, J. Photochem. Photobiol., A, 2023, 445, 114982 CrossRef CAS.
- J. P. Calupitan, A. Poirot, J. Wang, B. Delavaux-Nicot, M. Wolff, M. Jaworska, R. Métivier, E. Benoist, C. Allain and S. Fery-Forgues, Chem. – Eur. J., 2021, 27, 4191–4196 CrossRef CAS PubMed.
- H. Shen, C. Xu, F. Sun, M. Zhao, Q. Wu, J. Zhang, S. Li, J. Zhang, J. W. Y. Lam and B. Z. Tang, ChemMedChem, 2022, 17, e202100578 CrossRef CAS PubMed.
- V. W.-W. Yam, Y. Yang, J. Zhang, B. W.-K. Chu and N. Zhu, Organometallics, 2001, 20(23), 4 11–4918 CrossRef.
- O. S. Wenger, L. M. Henling, M. W. Day, J. R. Winkler and H. B. Gray, Inorg. Chem., 2004, 43, 2043–2048 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- W. Kaim and S. Kohlmann, Inorg. Chem., 1990, 29, 2909–2914 CrossRef CAS.
- S. Frantz, M. Sieger, I. Hartenbach, F. Lissner, T. Schleid, J. Fiedler, C. Duboc and W. Kaim, Organomet. Chem., 2009, 694, 1122–1133 CrossRef CAS.
- R. Jordan, M. Niazi, S. Schäfer, W. Kaim and A. Klein, Molecules, 2022, 27, 8159 CrossRef CAS PubMed.
- E. B. Patricio-Rangel, V. Salazar-Pereda, O. Cortezano-Arellano and D. Mendoza-Espinosa, Dalton Trans., 2022, 51, 2641–2651 RSC.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- I. Mishra, M. Priyatharsini and M. Sathiyendiran, J. Organomet. Chem., 2021, 949, 121934 CrossRef CAS.
- P. Govender, S. Pai, U. Schatzschneider and G. S. Smith, Inorg. Chem., 2013, 52, 5470–5478 CrossRef CAS PubMed.
- U. Phukon, M. Priyatharsini and M. Sathiyendiran, J. Organomet. Chem., 2020, 923, 121460 CrossRef CAS.
- R. Xu, X.-S. Wang, H. Zhao, H. Lin, Y.-B. Huang and R. Cao, Catal. Sci. Technol., 2018, 8, 2224–2230 RSC.
- B.-C. Tzeng, Y.-J. Hsiao, G.-H. Lee, H.-Y. Wang, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Dalton Trans., 2019, 48, 7946–7952 RSC.
- B. Manimaran, P. Thanasekaran, T. Rajendran, R.-T. Liao, Y.-H. Liu, G.-H. Lee, S.-M. Peng, S. Rajagopal and K.-L. Lu, Inorg. Chem., 2003, 42, 4795–4797 CrossRef CAS PubMed.
- G.-X. Jin, T. Wang, Y. Sun, Y.-L. Li and J.-P. Ma, Inorg. Chem., 2020, 59, 15019–15027 CrossRef CAS PubMed.
- SADABS, Program for data correction, Bruker-AXS Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- G. Zhurko and D. Zhurko, ChemCraft 1.6, 2011, https://www.chemcraftprog.com/index.html Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- G. Velmurugan, B. K. Ramamoorthi and P. Venuvanalingam, Phys. Chem. Chem. Phys., 2014, 16, 21157–21171 RSC.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- J. C. De Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including NMR, HRMS and IR spectra, molecular views and crystallographic data, theoretical calculations, electrochemical experiments, photoluminescence spectra and decays. CCDC 2365135–2365138. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01907e |
| This journal is © The Royal Society of Chemistry 2024 |