
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel dinuclear open paddle-wheel-like copper complexes involving π-stacking on the basis of chiral binaphthyl phosphoric acid {(R)-PhosH}: structural, magnetic and optical properties†
Humberto A.
Rodríguez
 a,
Daniel A.
Cruz
a,
Víctor
Lavín
b,
Juan I.
Padrón
a and
Pablo
Lorenzo-Luis
*c
a,
Daniel A.
Cruz
a,
Víctor
Lavín
b,
Juan I.
Padrón
a and
Pablo
Lorenzo-Luis
*c
aInstituto de Productos Naturales y Agrobiología, Consejo Superior de Investigaciones Científicas (IPNA-CSIC), 38206, La Laguna, Tenerife, Islas Canarias, Spain. E-mail: jipadron@ipna.csic.es
bDepartamento de Física, MALTA-Consolider Team, and IUdEA, Universidad de La Laguna, Apartado de Correos 456, San Cristóbal de La Laguna, Santa Cruz de Tenerife E-38200, Spain. E-mail: vlavin@ull.es
cÁrea de Química Inorgánica, Departamento de Química, Universidad de La Laguna, C/Astrofísico Francisco Sánchez 3, 38071 La Laguna, Spain. E-mail: plorenzo@ull.es
First published on 17th October 2024
Abstract
This research embarked on the study of a new binaphthyl phosphate scaffold of copper. There are two independent neutral complexes in the asymmetric unit: Cu1/Cu2 (I) and Cu3/Cu4 (II) from a similar structure to paddle-wheel-like, with one arm formed by an intra-hydrogen bond between the water molecule bonded to the copper and the phosphine oxide (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) moieties. Moreover, in the first complex two water and one ketone molecule complete the coordination sphere of the two-copper metals, instead, in the second one, one water and two ketone molecules. The experimental value obtained for the effective paramagnetic moment indicates that there is no appreciable interaction between the copper(II) cations and that they behave as paramagnetic ions over the entire temperature range explored (5–350 K).
O) moieties. Moreover, in the first complex two water and one ketone molecule complete the coordination sphere of the two-copper metals, instead, in the second one, one water and two ketone molecules. The experimental value obtained for the effective paramagnetic moment indicates that there is no appreciable interaction between the copper(II) cations and that they behave as paramagnetic ions over the entire temperature range explored (5–350 K).
The development of organic–inorganic hybrid materials has been associated with the evolution of new coordination topologies. The control of network parameters through a careful selection of metal ions and organic bridging ligands offers new possibilities for developing new functional materials with useful properties, such as magnetic devices, biomolecules, sensors and catalysts.1 Additionally, the consideration of molecular chirality as a property inherent to the presence of stereogenic elements leads to the development of applications in which this chirality can be exploited, such as medicine, asymmetric catalysis or C–C bond formation reactions, allowing new advances in organic synthesis.2,3 Perhaps the best-understood example of axially chiral molecule has been the 1,1′-binaphthyl-2,2′-diol unit (BINOL) and its phosphoric acid {(±)-PhosH} derivative in enantiomerically pure forms.4–7 Owing to the characteristics of the phosphate acid group, BINOL strongly binds to P(V) through two P–O–Csp2 bonds, generating a tetradentate structure, which creates a more compact and restricted chiral environment (Scheme 1a).8 Unlike other catalysts (e.g., carboxylic, sulfonic or sulfinic acids), here the acid function is highly influenced by the chiral pocket, which ensures high enantioselectivities.9 It is worth noting that the coordination ability of chiral phosphoric acids (CPAs) towards transition metal has been receiving considerable attention owing to the variety of bridging modes of the phosphoric group, allowing the formation of new coordination compounds (Scheme 1b) and thus playing a critical role in modern coordination chemistry.6,10,11
 | ||
| Scheme 1 (a) PO43− moiety interlocked with the {(R)-PhosH} ligand. (b) Coordination modes via M–π-Lewis acids. The asterisk indicates the P-stereogenic center. | ||
The most dominant sustainable metals, considering their ability to bind organic entities by means of strong coordinated bonds and their capacity, in many cases, to vary in oxidation states are iron, copper or zinc.10 On the basis of ligand binding sites, Cu(II) has one unpaired electron in the d-orbital and has been labelled as a chameleon in coordination chemistry.12
However, using chiral phosphate {(R)-PhosH} as a ligand in coordination reactions is challenging because of its low flexibility in coordination patterns; the dual acid–base activation of the phosphate group in interactions with metals (Scheme 1b); and the type of solvent used, whether polar protic or aprotic. These solvents are crucial for both coordination and the formation of a network of hydrogen bonds with neighbouring hydrogen bond donors.13,14 Therefore, designing and predicting crystallization processes often require extensive experimentation.
Typically, the {(R)-PhosH} ligand binds differently depending on the nature of the metal. It binds poorly to M–π-Lewis metals (MII = Fe, Co and Cu)15 but binds well through phosphoric acid oxygens, alternating two metal atoms with two phosphinate groups (O–P–O) in the case of M–π-Lewis metals (MII = Rh and Pd; MIII = Yb).14,16,17
Having in mind the above considerations, we present the synthesis and X-ray structure of a novel dinuclear [{Cu2(μ-η2-(R)-Phos-κ2O:κO′)3(η1-(R)-Phos-κO)}(H2O)x(C3H6O)y] (x,y = 2,1 (I); x,y = 1,2 (II)) complex 1·(C3H6O)2, together with their thermal, magnetic and optical properties. Focusing on copper as a metal, our first attempts at the coordination reaction between {(R)-PhosH}-binaphthyl phosphoric acid and copper(II) triflate resulted in a silver-blue precipitate, which was dissolved in dichloromethane and the same amount of ketone and stored in a refrigerator (+4 °C) to give cyan-yellow crystals suitable for X-ray structure determination. It is noteworthy that the difficulties in obtaining other complexes in the metal series (Sc–Zn) have limited the study of their properties. In fact, all our attempts to achieve an acceptable degree of sample purity were unsuccessful (Table S1†). Complex 1·(C3H6O)2 crystallizes in the centrosymmetric triclinic space group P1 (Tables S3 and S4†). The asymmetric unit shows two independent neutral complexes in 1·(C3H6O)2 (Fig. 1a and Fig. S4†): Cu1/Cu2 (I) and Cu3/Cu4 (II). The Flack parameter was refined to a value of −0.004(7) confirming the absolute structure (Table S3†). Both complexes show a μ-η2-κ2O:κO′ coordination mode [through phosphinate groups O–P–O towards Cu1/Cu2 and Cu3/Cu4, respectively] forming (Cu2P2O4) 8- and (Cu2PO2) 5-membered fused chelate rings. The value for the dihedral angle between the equatorial plane (Cu–O–P–O–Cu–O–P–O) and that lies out of this plane (Cu–O–P–O–Cu) is 87.58(3)° [Cu1/Cu2 (I)] and 85.62(4)° [Cu3/Cu4 (II)]. The equatorial phosphorus atoms do not deviate significantly from those that form the five-member-plane envelope [P2: −2.615(2), P4: −2.608(2), P6: 2.603(2) Å], whereas the maximum deviation between Cu3/Cu4 (II) complex is −2.628(2) Å for P8 (Fig. S5†). The complexes are stacked in an ABABA sequence interlinked by π⋯π interactions among the aromatic rings (Fig. 1b), which are extended in two directions forming an interlayer separation (3.73 and 3.79 Å) in agreement with those reported in the literature (3.8 Å).18 It should be noted that each Cu1 and Cu3 cations within these layers are linked to the η1-κO coordination mode from a similar structure to paddle-wheel-like with one arm formed by an intra-hydrogen bond between the water molecule bonded to the copper and the phosphine oxide (PO) moiety (Fig. 1b): [(D–H⋯A/Å: D⋯A, H⋯A), 1 + x, 1 + y, 1 + z]: O2W–H2WA⋯O14: 2.583(8), 1.836(5); O4W–H4WB⋯O54: 2.532(6), 1.941(5)]. The latter values move away from the upper limit of the distance range between two oxygen atoms (vdW radii 3.04 Å), indicating a strong interaction due to the participation of the polarized phosphine oxide (vide infra).4 On the other hand, the apical P1 and P5 atoms (Fig. 1a) are shifted by −2.984(2) and −2.887(2) Å, respectively, from the mean equatorial plane toward the shorter axially position [Cu1–O13 1.945(5), Cu3–O53 1.917(4)].
 | ||
| Fig. 1 (a) View of the two independent neutral complexes (Cu1/Cu2 (I) and Cu3/Cu4 (II)): outlook of the (Cu2P2O4) 8- and (Cu2PO2) 5-membered rings (turquoise green and aqua blue, respectively). (b) Packing diagram of 1·(C3H6O)2 showing weak interactions between coordinated water and solvent ketone molecules (D–H⋯A/Å: D⋯A, H⋯A): O1W–H1WA⋯O41C: 2.701(2), 1.846(2); O1W–H1WB⋯O51C: 2.725(3), 2.146(2); O4W–H4WA⋯O61C: 2.659(2), 3.046(3). The layers are stacked in the –ABABA– sequence with an interlayer separation (3.73 and 3.79 Å). | ||
The values of the PO distances are found to be 1.481 Å, slightly shorter than P–O (1.492 Å) and ca. 0.13 Å smaller than that found for P–O–Csp2 bonds. Concerning the phosphinate groups, O–P–O is slightly larger than the tetrahedral (ca. 5°), probably because of the involvement of oxygen in the bridge formation. The mean value is in line with that found for Cu3(PO4)2 (1.546 Å, 109.45°),19 comparable with that for those complexes earliest report containing {(R)-PhosH} ligand.14,15 Instead, the metal ion does not deviate significantly from the coordinated plane (complex I Cu1: 0.002 and Cu2: 0.005 Å; complex II Cu3: 0.001 and Cu4: 0.006 Å). Besides, the average deviation from the mean equatorial plane is 0.061 Å, the smaller deviation being for Cu1, 0.038 Å (Fig. S5†). The Cu⋯Cu contacts are significantly larger than the sum of the corresponding vdW radii of the two copper atoms (mean value 3.61 vs. 2.80 Å). Moreover, in the first complex two water and one ketone molecule complete the coordination sphere of the two-copper metals, instead, in the second one, one water and two ketone molecules (Fig. 1a).
The coordination around Cu2+ moiety can be described from trigonal–bipyramidal (TBP, for pure environments τ = 1) to square-pyramid (SP, for pure environments τ = 0) geometry environment according to the Addison parameter tau (τ5),20 of 0.49 and 0.48 [Cu1/Cu2 (I)] and 0.66 and 0.28 [Cu3/Cu4 (II)] (Table S4 and Fig. S5†). From a comparison between the coordination spheres of Cu4 and Cu2 atoms, which exhibit the same coordination pathway, a larger tendency toward square-pyramid (SP) geometry (τ5 = 0.28 vs. 0.48) is evidenced. As in Cu2, the apical bond distance is significantly shorter than the basal ones (Table S4†). Without influencing the distortion of the coordination center, it can be concluded that the steric and electronic properties cannot be considered as separate factors. In fact, the larger distortion of Cu4 could not be evidenced by the torsion angle of the equatorial plane (P8–O84–Cu4–O64) of −23.4° vs. −24.4° (P2–O24–Cu2–O64) in Cu2 (Fig. S5†). Therefore, the degree of trigonality (τ5) index together with the spin–orbit interaction, lifts the degenerations of the energy levels, adding complexity to the spectral profiles (vide infra).21
From a practical standpoint note that by careful soft vacuum process of the crystalline sample 1·(C3H6O)2 we obtained a completely anhydrous phase (hereafter 1), which remained stable under normal conditions of temperature and humidity. A straightforward preliminary test to confirm the fully anhydrous phase was the TG-analysis curve between 45 to 260 °C (Fig. 2, path a), which displays a complicated first weight loss with two overlapped stages (see DTG curve), attributable to the loss of water and ketones coordinated molecules followed of a likely loss of the π⋯π interactions. The involvement of these water molecules in an extended hydrogen-bonding network (vide supra) can justify this overlapping step. It is noteworthy that, after this first double overlap step, great stability of the remaining network is observed up to 343 °C (see DTG curve). One strong peak as an exothermic process {[DSC]peak at 341 °C} corresponds to the loss of the {(R)-Phos−} moiety.15 Comparing this thermal behaviour, a similar trend was observed for the binaphthyl phosphoric acid {(R)-PhosH} (Fig. S6†). This well-defined thermal process between 45 to 343 °C, is correlated to the loss of three water and three ketone coordinated molecules and a binaphthyl phosphoric acid moiety per formula unit (calcd 17.66% vs. found 17.02%). Finally, a strong peak with the mass loss was observed {Fig. 2, path b, [Tpeak]DTG = 405 °C}, which is due to the start of a thermolysis process of remaining complex and resulting in the formation of Cu2P2O7.22
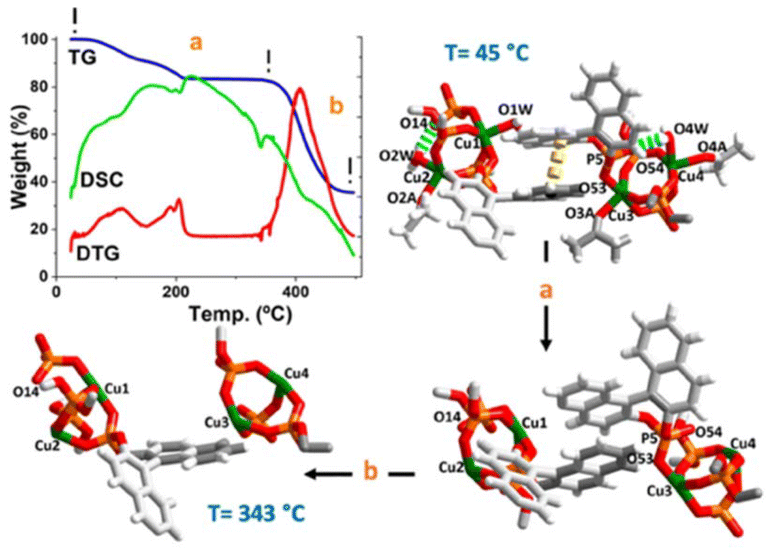 | ||
| Fig. 2 TG/DTG-DSC curves of 1: network from overlapped and stability stages (path a); network from the beginning of the pyrolytic phase (path b). TG = mass loss (percent); DTG = percent per minute and DSC = ΔT (microvolts) (↓exo). | ||
According to the magnetization and its relationship to the  -field, complex 1 revealing clear paramagnetic characteristics. Notably, there is a discernible linear dependency of
-field, complex 1 revealing clear paramagnetic characteristics. Notably, there is a discernible linear dependency of  on
on  , particularly evident at 300 K (Fig. S7†), with a positive slope. It is noteworthy that despite the presence of diamagnetic components alongside coppers, they did not affect the overall positive experimental slope, thus confirming its paramagnetic nature.
, particularly evident at 300 K (Fig. S7†), with a positive slope. It is noteworthy that despite the presence of diamagnetic components alongside coppers, they did not affect the overall positive experimental slope, thus confirming its paramagnetic nature.
Regarding the M–H measurements at low temperature (5 K), there was a tendency towards magnetization saturation at high fields approaching a paramagnetic moment close to 4 Bohr magnetons at 7 Tesla. This phenomenon at high fields could be influenced by the presence of a small ferromagnetic impurity at very low levels, suggesting long-term ordering at low temperatures and high fields. This could also indicate a slight superparamagnetic contribution. The theoretical value of the paramagnetic moment for 4 Cu2+ (d9) cations in the “spin-only” approximation, S = 1/2, is 3.46μB. The experimental effective paramagnetic moment was calculated from the experimental data of M versus T using the Curie–Weiss Law: 3.1 ± 0.2μB/f.u. The fact that the experimental value is slightly lower than the theoretical one is consistent, being compatible with the “spin-only” approximation, with a negligible orbital contribution to the paramagnetic moment. The paramagnetic Curie–Weiss temperature obtained from the fits, Θ between +8 and +40 K, shows a value close to 0 K, consistent with fundamentally paramagnetic behaviour. The linear fitting of the inverse of the magnetic susceptibility was performed for both the ZFC and FC curves across various temperature ranges, with fittings at higher temperatures being more representative. From the range of values obtained, the error in the experimental value of the paramagnetic moment was estimated (Fig. S8†).
As pointed out by Qiu et al.,23 the optical properties of the Cu2+-ligand complexes cannot be explained by ligand-field theory alone, since factors related to the ligand nature, fine structure of energy levels, and complex geometry makes the interpretation of the optical spectra a difficult task. As can be seen in Fig. 3a, there are different absorption bands in the whole optical range, from NIR to UV. For clarifying purposes, they are compared to those found for the ligand, as the organic part of the complex, and the Cu3(PO4)2, as representative of the phosphate structure. Moreover, it has been also compared to that of the Na3PO4·12H2O complex (Fig. S9†). The strongly broadened absorption band centered at around 1000 nm can be ascribed to intra-configurational 2A1′ → 2E′′, E′ transitions (for pure TBP D3h environments), and have been also observed for Cu2+ ions in 5-coordinated TBP environments.21,24,25 The shift of this band to higher wavelengths (lower energies), compared to other matrices,23–25 would indicate that these Cu2+ ions are subjected to weak ligand field interactions with the five oxygen ions of their first coordination shells. Moreover, differences in the local structure of the four Cu2+ ions in the complex give rise to a superposition of different energy level diagrams that further generates an inhomogeneous broadening of these transitions, also observed for the Cu3(PO4)2, which doubles the full width at half maximum (FWHM) observed in other TBP environments.24,25 Concerning the band centered at around 325 nm, there is a clear convolution of the organic and inorganic parts of the complex, being this band is normally related to the Cu–O charge transfer band, although it may be strongly overlapped with intra-configurational 3d84s1–3d9 bands.26
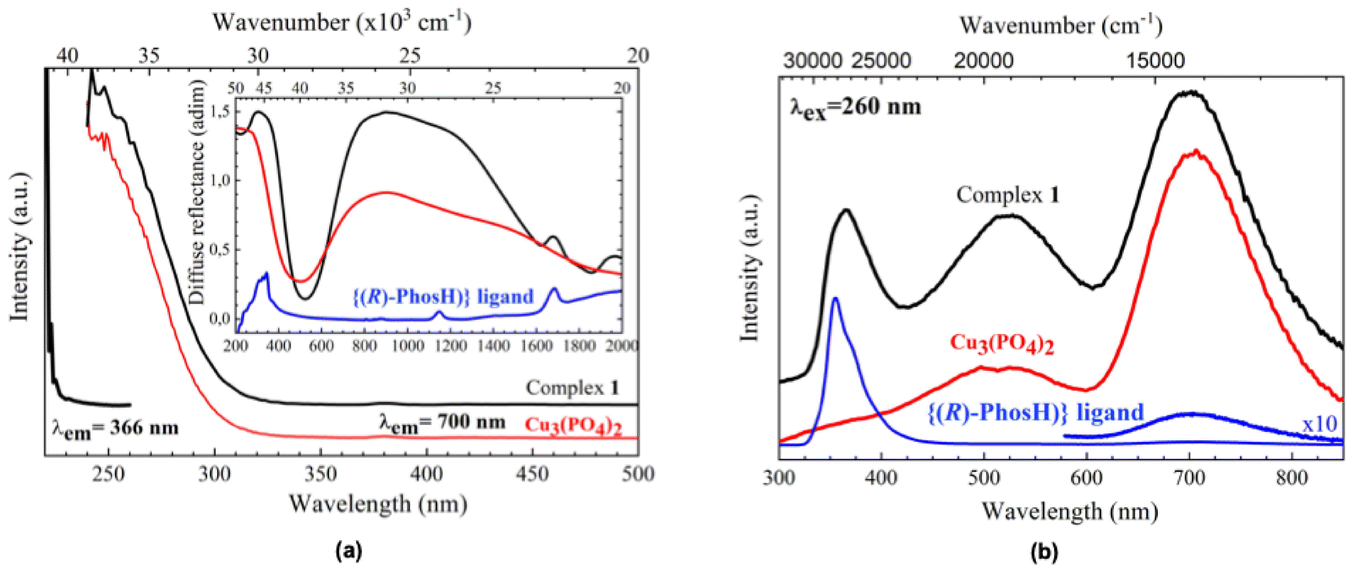 | ||
| Fig. 3 (Inset) Diffuse reflectance spectra of complex 1 (in black), the Cu3(PO4)2 matrix (as representative of the phosphate structure, in red), and the ligand (in blue). Excitation spectra of 1 (in black), monitoring emission at 366 and 700 nm, and the Cu3(PO4)2 phosphate matrix (in red), monitoring emission at 700 nm (a). Emission spectra of 1 (in black), the Cu3(PO4)2 matrix (as representative of the phosphate structure, in red), and the ligand (in blue) exciting with a xenon light source at 260 nm (b). | ||
Luminescence in the near-infrared range was observed for five-coordinate Cu2+-complexes in TBP geometry environmental,24,25 although it has not been found for 1, at least till 1800 nm.
However, strong UV and visible luminescence have been found for 1. Similar to the absorption spectrum, the emission is a convolution of contributions from the organic ligand, with a strong emission band centered at around 365 nm and a weak one at 700 nm, and the inorganic part, more related to the Cu2+ ions, that contributes with two strong emission bands at around 525 and 700 nm (Fig. 3b). The excitation spectra of the complex and its phosphate partner, also included in Fig. 3a and obtained by monitoring the emission at 700 nm, show similar spectral profiles, suggesting that the Cu–O charge transfer band feeds the energy levels of the excited configuration. However, the excitation spectrum monitoring the emissions at 366 and 525 nm, the latter not shown, are similar and clearly shifted to higher energies, indicating that the levels involved in the Cu2+ radiative de-excitation are more related to intra-ligand π → π* transitions of the ligand.27 In conclusion, and as a general feature, the profiles found for the excitation and emission spectra point to strong energy transfer processes between the organic and inorganic parts of the complex after UV radiation excitation. Indeed, the phosphine oxide from binaphthyl phosphate, which is composed of a low-frequency PO stretching vibration (∼1137 cm−1, Fig. S1†), can support an oxygen coordination environment for the Cu2+ ion, which could lead to enhanced emission.28
Conclusions
The BINOL-based phosphonate (CPA) was found to be an appropriate O-donor ligand for divalent first-row d-block metal centres, such as Cu(II). To the best of our knowledge, an example of metal-containing simultaneous bidentate (μ-η2-Phos-κ2O:κO′) and monodentate (η1-Phos-κO) coordination modes has not yet been reported. One of the phosphine oxides (PO) remains uncoordinated and is involved via intra-hydrogen bonds with the coordinated water molecules. The complex exhibited remarkable canted luminescence in the UV and visible ranges. The visible luminescence was attributed to the emission band of excited states located in the binaphthyl phosphate framework. Our results might be important to understand the solid-state architecture of these chiral coordinative systems and the development of new materials with chiral properties.
Author contributions
Humberto A. R., Daniel A. C.; Juan I. P., Pablo L. L.; performed, conceived and designed the experiments; Víctor L.; performed the optical measurements. All the authors contributed equally to writing the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† ESI available: A full characterization of complex 1·(C3H6O)2 is available under pdf format. CCDC 2345065.† For ESI and crystallographic data in CIF format see https://doi.org/10.1039/d4dt02044h.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Grant PID2021-126747NB-I00 was funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”. H. A. R. thanks the Spanish MCIU for an FPU scholarship. We thank the SQUID Magnetometry and Low-Temperature service of the ICMM-Madrid for the magnetic characterization. We also acknowledge the use of the SEGAI-ULL service for X-ray and thermal analysis.References
- Z. Zhang and M. J. Zaworotko, Chem. Soc. Rev., 2014, 43, 5444–5455 CAS.
- V. Sinka, I. Fernández and J. I. Padrón, J. Org. Chem., 2022, 87, 11735–11742 CAS.
- D. A. Cruz, V. Sinka, P. de Armas, H. S. Steingruber, I. Fernández, V. S. Martín, P. O. Miranda and J. I. Padrón, Org. Lett., 2021, 23, 6105–6109 CrossRef CAS PubMed.
- A. Parra, S. Reboredo, A. M. M. Castro and J. Alemán, Org. Biomol. Chem., 2012, 10, 5001–5020 RSC.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- N. Brodt and J. Niemeyer, Org. Chem. Front., 2023, 10, 3080–3109 RSC.
- Q. Yue, B. Liu, G. Liao and B.-F. Shi, ACS Catal., 2022, 12, 9359–9396 CrossRef CAS.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- M. Terada, Chem. Commun., 2008, 4097–4112 RSC.
- G.-C. Fang, Y.-F. Cheng, Z.-L. Yu, Z.-L. Li and X.-Y. Liu, Top. Curr. Chem., 2019, 377(23), 1–32 CAS.
- K. Muñoz-Becerra, D. Aravena, E. Ruiz, E. Spodine, N. Soto-Donoso, V. Paredes-García and D. Venegas-Yazigi, Inorg. Chem. Front., 2017, 4, 509–520 RSC.
- D. Reinen, Comments Inorg. Chem., 1983, 2(5), 227–246 CrossRef CAS.
- S. Ciattini, F. Costantino, P. Lorenzo-Luis, S. Midollini, A. Orlandini and A. Vacca, Inorg. Chem., 2005, 44, 4008–4016 CrossRef CAS PubMed.
- R. Hrdina, L. Guénée, D. Moraleda and J. Lacour, Organometalllic, 2013, 32, 473–479 CrossRef CAS.
- M. Mccann, E. Murphy, C. Cardin and M. Convery, Polyhedron, 1991, 10, 2771–2777 CAS.
- G. Jiang, R. Halder, Y. Fang and B. List, Angew. Chem., Int. Ed., 2011, 50, 9752–9755 CAS.
- Y. Deng, C. V. Karunaratne, E. Csatary, D. L. Tierney, K. Wheeler and H. Wang, J. Org. Chem., 2015, 80, 7984–7993 CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 CAS.
- G. L. Shoemaker, J. B. Anderson and E. Kostiner, Acta Crystallogr., 1997, B33, 2969–2972 Search PubMed.
- A. G. Blackman, E. B. Schenk, R. E. Jelley, E. H. Krensked and L. R. Gahan, Dalton Trans., 2020, 49, 14798–14806 RSC.
- J. Titiš, C. Rajnák and R. Boča, Inorganics, 2022, 10(8), 116–133 Search PubMed.
- T. Prapakaran, C. I. Sathish, J. Yi, A. Vinu and R. Murugavel, Eur. J. Inorg. Chem., 2023, 26, e202300071 CAS.
- S. R. Qiu, B. C. Wood, P. R. Ehrmann, S. G. Demos, P. E. Miller, K. I. Schaffers, T. I. Suratwala and R. K. Brow, Phys. Chem. Chem. Phys., 2015, 17, 18913 CAS.
- A. N. Romanov, E. V. Haula, D. P. Shashkin and V. N. Korchak, J. Lumin., 2020, 228, 117652–117657 CAS.
- A. Möller, U. Löw, T. Taetz, M. Kriener, G. André, F. Damay, O. Heyer, M. Braden and J. A. Mydosh, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78(2), 024420 CrossRef.
- T. Aidilibike, Y. Li, J. Guo, X. Liu and W. Qin, J. Mater. Chem. C, 2016, 4, 2123–2126 RSC.
- S. Charoensuk, J. Tan, M. Sain and H. Manuspiya, Nanomaterials, 2021, 11, 2281 CrossRef CAS PubMed.
- T. Harada, Y. Hasegawa, Y. Nakano, M. Fujiki, M. Naito, T. Wada, Y. Inoue and T. Kawai, J. Alloys Compd., 2009, 488, 599–602 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A full characterization of complex 1·(C3H6O)2 is available in the pdf format. CCDC 2345065. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02044h |
| This journal is © The Royal Society of Chemistry 2024 |