
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese 2-phosphinophosphinine precatalysts for methanol/ethanol upgrading to isobutanol†
Daniel J.
Ward
 a,
Margot
Marseglia
a,
Daniel J.
Saccomando
b,
Gary
Walker
b and
Stephen M.
Mansell
*a
a,
Margot
Marseglia
a,
Daniel J.
Saccomando
b,
Gary
Walker
b and
Stephen M.
Mansell
*a
aInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.mansell@hw.ac.uk; Web: https://www.mansellresearch.org.uk
bLubrizol Limited, The Knowle, Nether Lane Hazelwood, Derby, Derbyshire DE56 4AN, UK
First published on 25th September 2024
Abstract
Two Mn-phosphinophosphinine complexes were synthesised from reaction of the proligand with [MnBr(CO)5] at 80 °C for 2 h; 2-diphenylphosphino-3-methyl-6-trimethylsilylphosphinine manganese tricarbonyl bromide (2TMS) and 2-diphenylphosphino-3-methyl-phosphinine manganese tricarbonyl bromide (2H). 31P{1H} NMR spectroscopy revealed characteristic chemical shifts for the phosphinine and phosphine donors bound to Mn (255.4 and 23.7 ppm for 2TMS; 234.2 and 24.8 ppm for 2H), and single crystal X-ray diffraction established the structure of the chelating complex 2TMS. Rapid reaction of both complexes with water was observed with 2TMS reacting to eventually yield a single product, syn-3TMS, from the syn-1,2-addition of water across the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C multiple bond on the bromide face, confirmed by X-ray diffraction for both an unsolvated and solvated structure, where MeOH was found to be H-bonding to the P-OH functionality. The reaction of 2R with dry methanol gave multiple products that were not in equilibrium with each other, and the molecular structure of one isomer was definitively established by X-ray diffraction as an unusual 1,4-addition product (1,4-4TMS). However, reaction of 2R with methanol in the presence of trace water showed that hydrolysis products 3R were formed preferentially. Both phosphinine complexes acted as pre-catalysts for the Guerbet upgrading of methanol/ethanol to isobutanol at 180 °C over 90 h, giving yields of isobutanol (based on moles of ethanol) of 22% for 2TMS and 27% for 2H. This is superior to known Mn dppm complexes [dppm = bis(diphenylphosphino)methane], including the 21% yield recorded for the best derivative [MnBr(κ2-PPh2C(H)PhPPh2)(CO)3] shown to date.
C multiple bond on the bromide face, confirmed by X-ray diffraction for both an unsolvated and solvated structure, where MeOH was found to be H-bonding to the P-OH functionality. The reaction of 2R with dry methanol gave multiple products that were not in equilibrium with each other, and the molecular structure of one isomer was definitively established by X-ray diffraction as an unusual 1,4-addition product (1,4-4TMS). However, reaction of 2R with methanol in the presence of trace water showed that hydrolysis products 3R were formed preferentially. Both phosphinine complexes acted as pre-catalysts for the Guerbet upgrading of methanol/ethanol to isobutanol at 180 °C over 90 h, giving yields of isobutanol (based on moles of ethanol) of 22% for 2TMS and 27% for 2H. This is superior to known Mn dppm complexes [dppm = bis(diphenylphosphino)methane], including the 21% yield recorded for the best derivative [MnBr(κ2-PPh2C(H)PhPPh2)(CO)3] shown to date.
Introduction
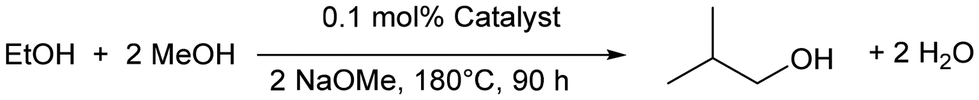
Mitigating against climate change whilst delivering the high quality of life desired by the world's population is a key challenge for the coming decades. The high energy density of liquid hydrocarbon fuels, currently derived from crude oil, is important for the transportation of people and freight and difficult to replicate by other means,1 such as hydrogen gas or electric battery power, so consequently research into sustainable biofuels has been extensive.2–4 Bio-sourced methanol and ethanol are being investigated as fuels,5,6 but they have substantial drawbacks including low energy densities, and they can also be corrosive to current engine and storage technologies.7,8 Longer chain alcohols such as n-butanol (90% the energy density of gasoline)8 and isobutanol (98% the energy density of gasoline)9 have some distinct advantages over shorter chain biofuels,10 and Guerbet chemistry can be used to couple two ethanol molecules to n-butanol11 or two molecules of methanol and one molecule of ethanol to isobutanol (Scheme 1).7,8 | ||
| Scheme 1 Production of n-butanol and isobutanol using Guerbet chemistry. MH2 can be a conventional metal dihydride or involve metal–ligand cooperativity. | ||
Ruthenium (pre)catalysts have proven to be the best for these catalytic upgrading reactions, and they work through ‘hydrogen borrowing’ pathways (Scheme 1).12 Successful catalysts often feature small bite-angle9,13–16 or tridentate pincer ligands,17–19 with ligand non-innocence potentially playing an important role.19,20 Replicating this performance with first row transition metals has proven challenging, with Mn complexes showing the most promise,19–25 but with much work still to do if the high activity and selectivity of Ru catalysts are to be matched (Scheme 2). Whilst Ru complexes of the small bite-angle bis(diphenylphosphino)methane ligand (dppm),14 and backbone-substituted derivatives,9 are excellent catalysts for ethanol upgrading,9 these ligands perform noticeably worse for Mn catalysts,20 where tridentate pincer complexes predominate.19,21,24,25
 | ||
| Scheme 2 Selected Ru and Mn pre-catalysts for ethanol upgrading reactions. | ||
Phosphinine is the P analogue of pyridine and displays markedly different coordination chemistry26 and properties as a ligand in homogeneous catalysis.27–29 Phosphinines, and aryl substituted phosphinines in particular,27,28 are typically stable to water,30,31 except when bound to electron poor transition metal centres.26,32–37 It has been established that a wider C–P–C angle of the phosphinine ring in transition metal complexes reflects a disruption to the aromaticity leading to increased susceptibility to nucleophilic attack, particularly with regards to water.35,38,39 Angles above 106° are indicative of complexes that react with water, such as the Re(I) complex [ReBr(L)(CO)3] (L = 2-(2′-pyridyl)-4,6-diphenyl-phosphinine; C–P–C = 106.3°)38 and group 9 complexes [MCp*(Cl)L][Cl] (M = Rh, Ir; C–P–C = 106.64(12)° and 106.7(3)° respectively).35 Compounds with C–P–C angles below 106° are stable to water, such as [ML(CO)4] (M = Mo, W; C–P–C = 104.34(10)° and 104.84(12)°, respectively).40
The location and nature of substituents on the phosphinine ring remains important. For example, a 2,6-bis(diphenylphosphino)phosphinine decomposed under air in solution within hours but a 2,5-bis(diphenylphosphino)phosphinine was Soxhlet-extracted into hexane under air without decomposition.41 2-Diphenylphosphino-3-methyl-6-trimethylsilylphosphinine (1TMS; see Scheme 4 for structure) was shown to react slowly with atmospheric oxygen over many weeks to generate the phosphine-oxide-substituted phosphinine (cf. the slow oxidation of PPh3 in solution) and another unidentified species where the phosphinine ring had reacted,42 but 1TMS did not react with MeOH, EtOH or iPrOH.43 An intriguing reversible reaction with water was recently discovered with 2,3,5,6-tetrapyridylphosphinines (Scheme 3), but no reaction was observed with MeOH and EtOH.30 [ReBr(L)(CO)3] was also found to react reversibly with water. Although the reaction with water was relatively slow at room temperature (after 1 h some starting material was still present), [ReBr(L)(CO)3] did convert completely to water-addition products.38 However, heating the sample could regenerate [ReBr(L)(CO)3], and after 7 h at 100 °C a ratio of 75% [ReBr(L)(CO)3]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25% [ReBr(L·H2O)(CO)3] was observed; upon cooling back to room temperature, the equilibrium shifted back to the water addition products.38 The reaction of phosphinines with water can generate P(V) as a 1,2-dihydrophosphinine oxide or P(III) as a phosphinous acid,44 PR2OH (Scheme 3).37,41,45 Additional ligand reactivity and potential metal–ligand cooperativity makes their use in catalysis intriguing.41,46 Compounds of type C (Scheme 3) have been synthesised as zwitterions (e.g., E)34 and evidence for a derivative of rearomatized D was observed in a reaction of a bis(phosphinimino)phosphinine with FeCl2/H2O/HCl (F; Scheme 3).41
:25% [ReBr(L·H2O)(CO)3] was observed; upon cooling back to room temperature, the equilibrium shifted back to the water addition products.38 The reaction of phosphinines with water can generate P(V) as a 1,2-dihydrophosphinine oxide or P(III) as a phosphinous acid,44 PR2OH (Scheme 3).37,41,45 Additional ligand reactivity and potential metal–ligand cooperativity makes their use in catalysis intriguing.41,46 Compounds of type C (Scheme 3) have been synthesised as zwitterions (e.g., E)34 and evidence for a derivative of rearomatized D was observed in a reaction of a bis(phosphinimino)phosphinine with FeCl2/H2O/HCl (F; Scheme 3).41
 | ||
| Scheme 3 Reaction of water with free phosphinines and metal complexes, and potential routes for metal–ligand cooperation; py = pyridyl or 4-Me-pyridyl; Mes = 2,4,6-Me3C6H2. | ||
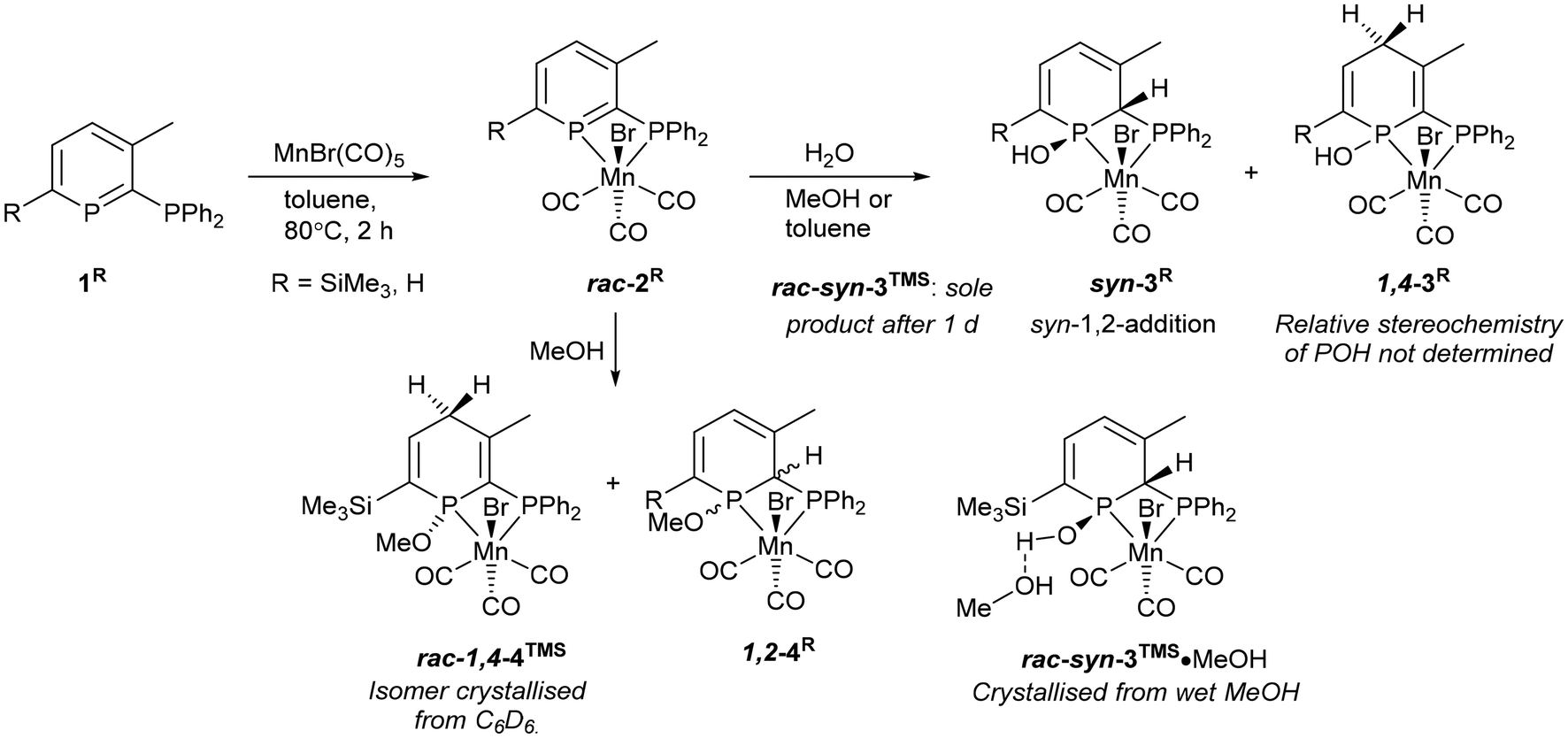 | ||
| Scheme 4 Synthesis and reactions of Mn 2-phosphinophosphinine complexes. | ||
Our group has established the use of unconventional phosphinophosphinine complexes in catalysis,41,43,47–49 which primarily utilised the favourable properties of small bite-angle ligands.13 Complex A (cis-[RuCl2(1TMS)2]; Scheme 2) was shown to be a good pre-catalyst for transfer hydrogenation and the upgrading of ethanol/methanol to isobutanol (Scheme 2),43 and was broadly comparable to cis-[RuCl2(dppm)2] for the production of isobutanol,16 but A catalysed transfer hydrogenation at room temperature whereas cis-[RuCl2(dppm)2] was inactive even at 82 °C.16,41 Subsequent research showed that the chloride ligands in A could be exchanged for hydrides, and that this ruthenium dihydride complex was a competent catalyst for the acceptorless dehydrogenative coupling of benzyl alcohol to benzyl benzoate.49 The potential ligand non-innocence of phosphinines offers additional pathways for improved catalytic performance over conventional phosphines for first row transition metal complexes, such as metal–ligand cooperation featuring reactions of the electrophilic P centre, (de)aromatisation of the phosphinine ring or H-bonding/deprotonation of POH functionalities.43 Hydroxyl functionalities have been shown to be important in H-borrowing catalysis,50 as was observed for a Ru 6,6′-dihydroxy-2,2′-bipyridine catalyst.51 Mn phosphinine complexes, in particular, are still relatively underexplored,52–59 with no new examples reported since 2010,26 so in this paper we describe the synthesis and characterisation of Mn 2-phosphinophosphinine complexes, their reaction with water and methanol, and their application in Guerbet alcohol upgrading.
Results and discussion
Reactions of donor-substituted phosphinines with unsymmetrical metal fragments (such as [MnBr(CO)3]), with water or alcohols, and especially the combination of both of these possibilities gives rise to a large number of possible isomers (Fig. 1). For the reactions of 2-phosphinophosphinine complexes with water or alcohols, the addition of the more electronegative oxygen atom to the more electropositive phosphorus (χ = 2.19) rather than carbon atom (χ = 2.55) is favoured. However, although 1,2-addition appears to be preferred from previous observations,41 a product arising from formal 1,6-addition has been observed before.45 1,4-Addition was thought to be less likely, although, as is determined in this research, cannot be discounted (see below). Multiple isomers are generated upon coordination of these phosphacyclohexadienes to unsymmetrical metal fragments, including enantiomers, making absolute assignment difficult except in the case of characterisation by single crystal X-ray diffraction.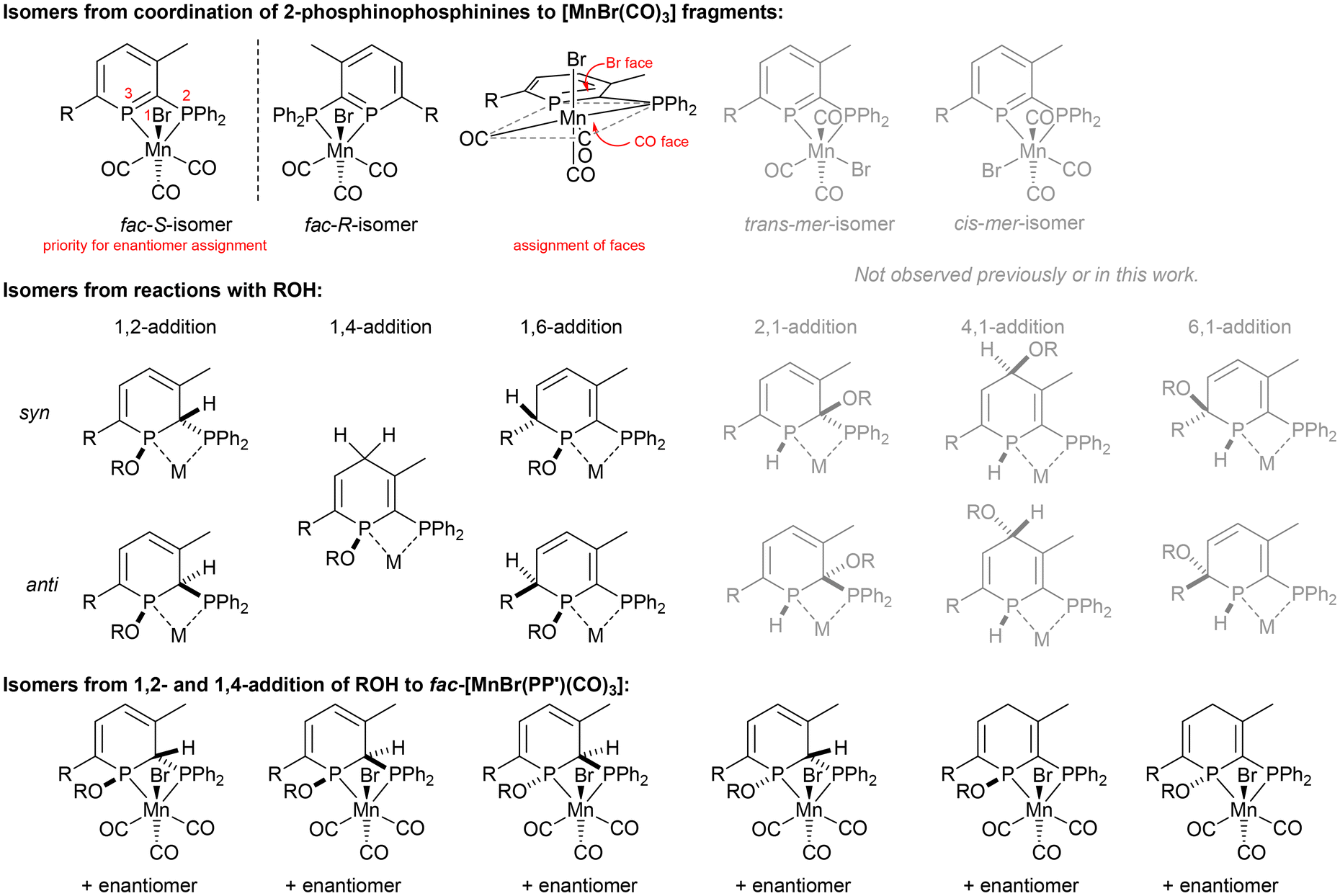 | ||
| Fig. 1 Selected isomers generated from reactions of 2-phosphinophosphinine (PP′) complexes. | ||
Mn 2-phosphinophosphinine complexes 2R were synthesised by heating the proligands 1R (R = TMS or H) with [MnBr(CO)5] at 80 °C for two hours (Scheme 4). While the formation of four isomers from this reaction is possible (an enantiomeric pair of fac isomers and two mer isomers with Br either trans or cis to the phosphinine; Fig. 1), only a racemic mixture of the fac isomer was observed. Crystallisation from a mixture of toluene and hexane gave 2TMS in 48% yield, whereas the poorer solubility of 2H made purification and isolation more difficult resulting in a 22% yield after precipitation from toluene solution using pentane. 31P{1H} NMR spectroscopy revealed two broad resonances at 255.4 and 23.7 ppm for 2TMS and 234.2 and 24.8 ppm for 2H without any P–P coupling being observed, most likely due to the quadrupolar nature of the attached Mn centre (100% I = 5/2). These chemical shifts are characteristic of a metal-coordinated phosphine (24/25 ppm) and a phosphinine (255/234 ppm) and are similar to the resonances observed in cis-[RuCl2(1TMS)2] (A, Scheme 2: 235 and 230 ppm for phosphinine P atoms, 2.3 and −2.4 ppm for phosphine P atoms).43 For 2TMS, the SiMe3 group was still present, as observed by a 29Si{1H} NMR doublet-of-doublets resonance at −0.92 ppm (cf. −0.95 and −0.13 ppm in A) and a 1H NMR singlet at 0.33 ppm; all other 1H NMR signals were observed as expected. 13C{1H} NMR spectra showed a complicated mixture of multiplet resonances that were assigned for 2TMS with the aid of HSQC and HMBC experiments, although the carbonyl resonances could not be observed. IR spectroscopy for 2TMS showed CO stretches at 2019, 1968, 1938 and 1912 cm−1, which is lower than those in [MnBr(tetramethyl-2,2′-biphosphinine)(CO)3] (G; 2038, 1982, 1944 cm−1)55 as expected due to increased π-backdonation to the carbonyl ligands in 2TMS. 2TMS was found to be light stable, unlike G where solutions were found to be very light sensitive.55 Single crystals grown from hexane/toluene confirmed the molecular structure of 2TMS (two different molecules in the asymmetric unit; only one enantiomer shown in Fig. 2).
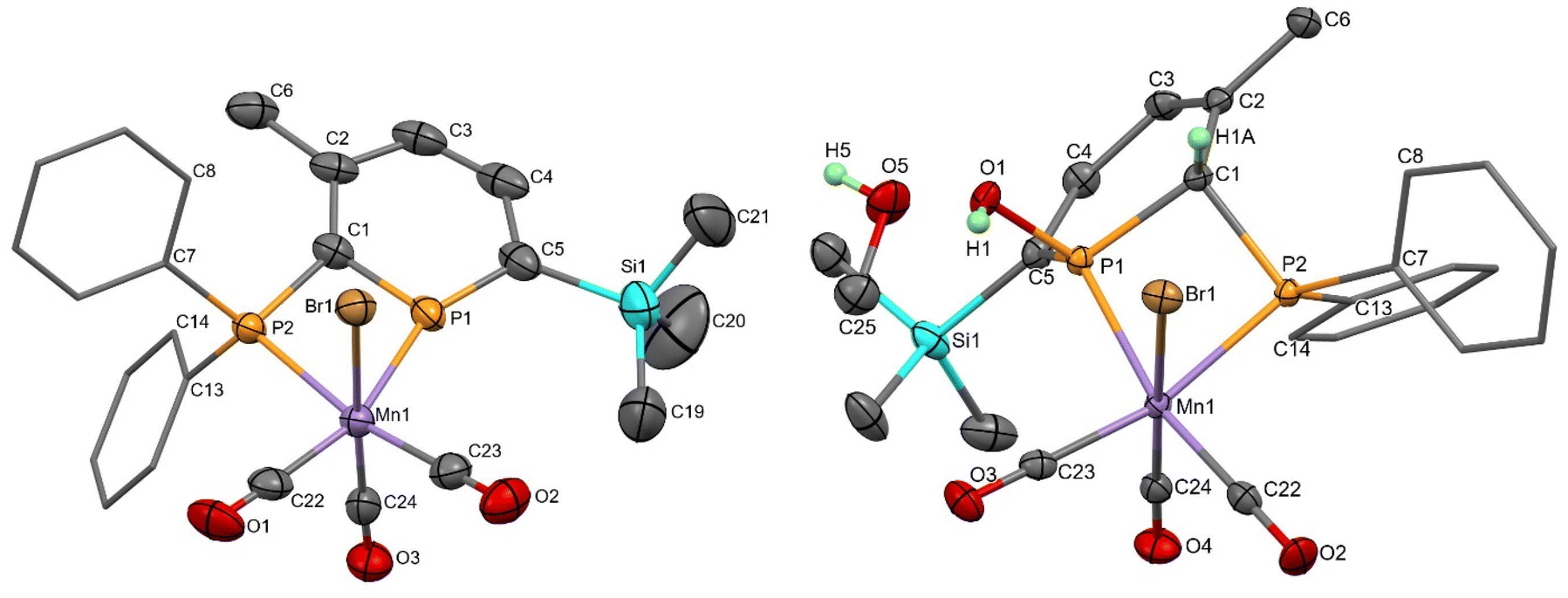 | ||
| Fig. 2 Molecular structure of 2TMS (left; only one of two molecules in the asymmetric unit shown) and syn-3TMS·MeOH (right; only one position of the MeOH molecule shown), with thermal ellipsoids at the 50% probability level. Ph rings are displayed as caped sticks and only selected H atoms are shown for clarity. | ||
Reactions with water
Both 2R complexes are very sensitive to water. 2TMS reacted immediately to initially give two products (Scheme 4), with complete conversion to phosphinous acid syn-3TMS over the course of a day (Fig. S8†), definitively characterised by X-ray crystallography (see below). 31P{1H} NMR spectroscopy showed a pair of doublets for syn-3TMS (92.3 and 21.0 ppm, 2JP–P = 35 Hz) indicative of loss of aromatisation of the phosphinine ring. The intermediate complex showed similar chemical shifts (85.9 and 39.8 ppm) but as singlets indicating disruption of the aromaticity and also the coupling between the P atoms. 29Si(1H) and 1H NMR spectroscopy demonstrated that the SiMe3 group was intact for syn-3TMS, and loss of aromaticity was also seen in the chemical shifts for the phosphacycle-2-C (13C{1H} NMR: dd at 64.9 ppm), phosphacycle-2-H/4-H (overlapping 1H NMR multiplets at 5.43 ppm) and phosphacycle-5-H atoms (dd at 6.51 ppm). IR spectroscopy of syn-3TMS showed CO stretching frequencies at 2016, 1947 and 1910 cm−1. Single crystals of syn-3TMS grown from CH2Cl2/pentane (see ESI†) and MeOH revealed the molecular structure of the product arising from syn-1,2-addition of water, with methanol H-bonding to the OH in syn-3TMS·MeOH (Fig. 2).
2H reacted with water to form two products that did not react further (Scheme 4). The major product was observed as 31P{1H} NMR signals at 80.1 and 19.5 ppm (br. d, 2JP–P = 40 Hz), and the minor product as broad singlets at 88.7 and 34.9 ppm in a ratio of approximately 3:1 (Fig. S35†). With the structure of syn-3TMS established by crystallography, the major product of 2H reacting with water was assigned as the analogous syn-1,2-addition complex syn-3H due to the similar 31P{1H} NMR signals and presence of 2JP–P coupling. Both the minor product and intermediate formed from water reacting with 2H and 2TMS respectively featured no 2JP–P coupling and similar 31P NMR chemical shifts (Fig. S15†) so it is probable that they represent the same isomer. Although products arising from the anti-1,2-addition of water have been observed for pyridyl phosphinine pro-ligands30 and complexes35 previously, there was no definitive evidence for anti-3TMS as being the correct assignment here. 1H NMR spectroscopy provided key evidence that the intermediate formed from 2TMS featured a diastereotopic methylene group as two mutually coupled, roofed doublets that integrate together as 2 H were observed at 3 ppm, away from the typical aromatic or alkene region. This resonance disappears with time and only resonances for syn-3TMS were observed after 16 h at room temperature. 1,4-Addition of ROH is the only pathway that leads to a methylene group, and hence the intermediate for 2TMS reacting with water is assigned as 1,4-3TMS, and the minor product for 2H reacting with water is similarly assigned as 1,4-3H (Scheme 4). The 1H NMR spectrum of a 3:1 mixture of syn-3H:1,4-3H showed a number of multiplets in the alkene region as well as a multiplet at 2.4 ppm assigned to the diastereotopic methylene group. The fortuitous crystallisation and characterisation of a 1,4-addition product from MeOH reacting with 2TMS supports this conclusion (see below).
The molecular structures of 2TMS and syn-3TMS feature six coordinate, octahedral Mn centres bonded to a diphosphorus ligand, three carbonyl ligands and one bromide. A comparison of 2TMS and syn-3TMS·MeOH (Table 1; syn-3TMS is similar) shows the aromaticity in 2TMS where P–C bond lengths are between 1.706(5) and 1.727(5) Å and C–C bond lengths are between 1.391(9) and 1.412(8) Å. This is absent in syn-3TMS with longer P–C bonds (1.797(2)–1.852(2) Å in syn-3TMS·MeOH) and alternating C–C and CC bonds now observed (e.g., 1.494(3), 1.343(3), 1.458(3), and 1.352(3) Å in syn-3TMS·MeOH). The Mn–phosphinine bond lengths (2.3050(14) and 2.2805(14) Å) are shorter than the Mn–phosphine bond lengths, as observed previously for group 6 complexes,48 but the Mn–P bond lengths in G [MnBr(tetramethyl-2,2′-biphosphinine)(CO)3] were even shorter (2.259(1) and 2.254(1) Å).55 The selective syn-addition of water was confirmed by the presence of a hydrogen atom on the carbon connecting the two P atoms. syn-3TMS·MeOH shows a molecule of MeOH hydrogen bonding to the OH functionality, which is absent in the unsolvated structure of syn-3TMS. Despite the large excess of methanol in the crystallisation of syn-3TMS, the water addition product was clearly favoured. The C1–P1–C5 angles in the two molecules of 2TMS present in the asymmetric unit were 107.1(3) and 107.6(2)°, which is in agreement with the finding that angles larger than 106° result in sensitivity to water.38
| 2TMS, molecule 1 | 2TMS, molecule 2 | syn-3TMS·MeOH | syn-3TMS | 1,4-4TMS | |
|---|---|---|---|---|---|
| Mn1–P1 | 2.3050(14) | 2.2805(14) | 2.3031(5) | 2.286(4) | 2.3033(5) |
| Mn1–P2 | 2.3597(14) | 2.3934(14) | 2.3200(5) | 2.331(4) | 2.3617(5) |
| Mn1–Br1 | 2.5183(9) | 2.5009(10) | 2.5216(4) | 2.536(2) | 2.5378(3) |
| P1–C1 | 1.727(5) | 1.706(5) | 1.852(2) | 1.857(14) | 1.8171(18) |
| C1–C2 | 1.404(7) | 1.403(6) | 1.494(3) | 1.499(18) | 1.343(2) |
| C2–C3 | 1.412(8) | 1.395(7) | 1.343(3) | 1.336(19) | 1.499(3) |
| C3–C4 | 1.391(9) | 1.391(7) | 1.458(3) | 1.511(19) | 1.481(3) |
| C4–C5 | 1.409(8) | 1.404(7) | 1.352(3) | 1.384(19) | 1.352(3) |
| C5–P1 | 1.708(5) | 1.708(5) | 1.797(2) | 1.769(14) | 1.8021(19) |
| P1–O1 | — | — | 1.6075(14) | 1.612(9) | 1.6184(13) |
| P1–Mn1–P2 | 69.34(5) | 69.21(5) | 71.45(2) | 72.28(13) | 71.134(17) |
| P1–C1–P2 | 97.1(2) | 97.6(2) | 92.74(9) | 93.8(6) | 97.18(8) |
| C1–P1–C5 | 107.1(3) | 107.6(2) | 103.7(1) | 106.2(6) | 103.77(8) |
Reactions with methanol
Reactions of 2R with MeOH32,34,37 dried over activated 3 Å molecular sieves showed the formation of new products that no longer feature the aromatic phosphinine moiety, but not selectively as multiple species were observed by 31P{1H} NMR spectroscopy. Although selective syn-addition of MeOH to the external PC double bond in PdCl2 and PtCl2 pyridyl-phosphinine complexes has been observed previously,37 experiments involving water addition to [ReBr(L)(CO)3] gave four products from syn- and anti-1,2-addition to both the Br and CO face (cf., Fig. 1).3831P{1H} NMR spectroscopy revealed that at least four diphosphorus complexes were formed from reactions of 2TMS with dry MeOH (Fig. S23†). If methanol addition was selective to the internal PC–P multiple bond (1,2-addition seen for water in the formation of syn-3TMS and for two Ru 2-phosphinophosphinine complexes previously),41 then syn- and anti-addition of water to both enantiomers of the starting material from the Br and CO faces would produce four pairs of enantiomers,38 however, other isomers are possible, including from 1,4-addition (see below). The ratio of isomers formed was found to vary in two separate experiments of different concentrations, and a small intensity fifth product was also observed (Fig. S25†). Integration of 31P{1H} NMR spectra obtained with a long relaxation delay and inverse gated decoupling to minimise integration errors gave ratios of approximately 1:1:2:4.2:0 (first experiment) and 1:0.5:2:2.4:0.2 (second experiment), which is evidence that the isomers are not in equilibrium with each other.
With only comparisons to the reactions of 2TMS with water to help aid assignment, we were greatly aided by the fortuitous crystallisation of one isomer from benzene solution. Single crystal X-ray diffraction revealed the unexpected 1,4-addition of MeOH to the carbonyl face of the molecule (Fig. 3, 1,4-4TMS). The localised C1–C2 and C4–C5 double bonds were easily identified by their short lengths (1.343(2) and 1.352(3) Å) whilst the H atoms on the functionalised phosphacyclohexadiene ring (H3A, H3B and H4) were located and their positions freely refined. Both enantiomers were present (space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). Previously, 1,4-addition of phosphinines has been observed from [4 + 2] cycloaddition reactions with unsaturated reagents forming phosphabarrelenes.60,61
). Previously, 1,4-addition of phosphinines has been observed from [4 + 2] cycloaddition reactions with unsaturated reagents forming phosphabarrelenes.60,61
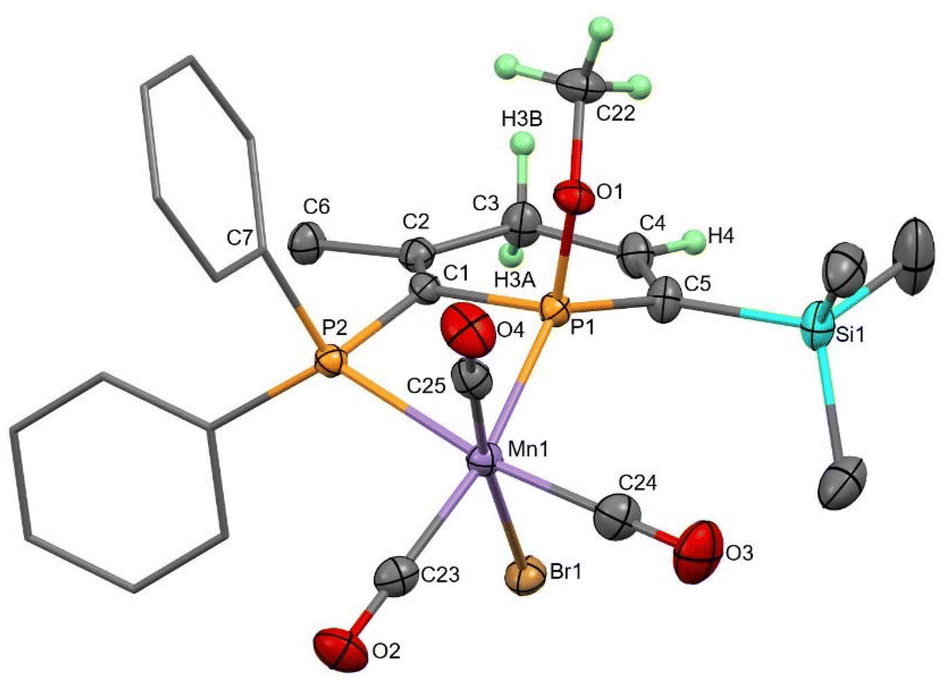 | ||
| Fig. 3 Molecular structure of 1,4-4TMS with thermal ellipsoids at the 50% probability level. Ph rings are displayed as caped sticks and only selected H atoms are shown for clarity. | ||
Redissolving the crystals of 1,4-4TMS in C6D6 allowed characterisation of this complex by 31P{1H} and 1H NMR spectroscopy. 31P resonances at 106.3 and 36.8 ppm were observed, along with low intensity resonances from two other isomers as impurities, and so clearly this isomer does not rearrange in solution to the other isomers generated in the reaction. In fact, heating to 80 °C for 4 h caused no change to the spectra, and variable temperature and exchange NMR spectroscopic experiments showed no evidence of interconversion between isomers. 1,4-4TMS was the second most abundant isomer formed in the reaction of 2TMS with MeOH.
The 31P{1H} NMR spectrum of the supernatant solution left after crystallisation of 1,4-4TMS revealed a decrease in intensity for the resonances associated with 1,4-4TMS (Fig. S26†), again indicating that the isomers are not in equilibrium, and no change was observed in integral values after 16 h at room temperature or heating to 80 °C. The most abundant isomer displayed broad resonances at 95.5 and 25.3 ppm, very similar to syn-3TMS resulting from the syn-1,2-addition of water (92.3 and 21.0 ppm). We therefore assign the major species as arising from syn-1,2-addition of MeOH. The remaining species could arise from anti-1,2-addition on the same face, syn- and anti-1,2-addition from the opposite face or one of the species could arise from the 1,4-addition of MeOH to the bromide face. We note that for the reaction of water with [ReBr(L)(CO)3], two pairs of isomers were found to be disfavoured,38 but absolute assignment proved challenging.
Reactions of water with 1,4-4TMS and the other isomers resulting from MeOH addition were attempted. The results were complex because the various isomers were observed to react with water at different rates. 31P NMR resonances for 1,4-4TMS disappeared immediately when a drop of degassed water was added whereas the major isomer (resonances at 95.5 and 25.3 ppm assigned to syn-1,2-addition of water) was the slowest to react requiring 80 °C for 4 h for complete consumption (Fig. S30†). Whilst there are resonances that appear in similar regions to syn-3TMS, the match is not exact and multiple products are observed demonstrating that the reaction does not proceed simply to give syn-3TMS.
Reactions of 2H with dry MeOH gave spectra with poorer signal-to-noise, likely due to the reduced solubility of species without the SiMe3 group, but comparisons with reactions of 2TMS showed four similar 31P{1H} resonances between 120 and 90 ppm and four resonances between 40 and 10 ppm in agreement with the formation of four isomers, along with free ligand, 1H, and additional resonances that could not be identified (Fig. S38†). Dimerisation in addition to reaction with water was noted previously for [Cr(1H)(CO)4], and indicates that further reactivity is possible for this smaller ligand without steric protection from the SiMe3 group.48
Reactions of 2R with non-dry MeOH gave 3R. Thus, it has been established that 2R in contact with alcohols will react, and, if any water is present, the water addition product 3R will be formed. Comparisons with the analogous cis-[RuCl2(1TMS)2] precatalyst A are beneficial to enable modes of phosphinine ligand reactivity with alcohols to be established. Whilst A did not react with isopropanol at 70 °C for 48 h,43A was found to react slowly with a 100-fold excess of dry MeOH at room temperature in C6D6 over the course 2 d, but only with one phosphinine moiety, the other remaining as an aromatic phosphinine donor (multiplet resonance at 240 ppm; Fig. S40†). Heating to 80 °C generated further reactivity giving rise to a complicated mixture of products.
Guerbet catalysis
The Mn phosphinophosphinine complexes 2R were tested as precatalysts for the catalytic upgrading of ethanol/methanol to isobutanol (3R is immediately formed upon contact with non-dry methanol) and compared with the dppm analogue 5 (Fig. 4). A catalytic screen at 180 °C for 2 h (2TMS and 5), 20 h and 90 h (2TMS, 2H and 5) established that longer time periods are required compared to typical Ru pre-catalysts (e.g., 20 h for cis-[RuCl2(dppm)2] and A; Scheme 2)16,43 as previously established for Mn complexes.20 Runs 1, 2 and 3 for 2TMS, runs 4 and 5 for 2H and runs 6, 7 and 8 for 5 (Table 2) show increasing conversion of ethanol and increasing yield of isobutanol with time, with higher conversions and yields for 2H and 2TMS. The intermediate 1-propanol was the only other product in the liquid fraction, as identified by GC-FID calibrated with pure standards. The results at 90 h for 2TMS and 2H were most promising with high conversions of ethanol (run 3: 59% for 2TMS and run 5: 57% for 2H) and relatively good yields of isobutanol (22% and 27% for 2TMS and 2H respectively) compared to the dppm complex 5 (run 8: 39% conversion of EtOH, 3% yield of isobutanol; lit.: 7% yield).20 The yields for 2R are even higher than for the phenyl-substituted dppm analogue 6 after 90 hours20 and, for 2H, the same as pincer complex 7, albeit only 24 h was required for this catalyst (Scheme 2).20 The reaction uses 200 mol% base62 and generates solid by-products as well as the liquid fraction containing the desired products. Sodium formate has been observed previously as the main solid by-product,20,25 and this was observed by 1H and 13C NMR analysis in D2O (see ESI†). A much smaller amount of sodium acetate was also observed, but the full mass-balance for this reaction has not been determined. The selectivities reported in Table 1 are calculated from the amount of ethanol converted to isobutanol; another way to report selectivity is the percentage of isobutanol in the liquid fraction,20 which is usually much higher. For 2TMS and 2H, the selectivity in the liquid fraction was excellent with 1-propanol, the intermediate in the reaction (Scheme 1), as the only by-product (Table 2). Also, reactions with 2TMS and 2H both gave high residual gas pressures in the autoclave, most likely from acceptorless dehydrogenation (AD) of the alcohols.49 This was tested by venting the gas through CDCl3 at 0 °C. 1H NMR spectroscopy revealed a signal at 4.62 ppm that fits with the reported chemical shift for dihydrogen,63 which was lost upon bubbling the solution with nitrogen. IR spectroscopy showed no stretch at 2143 cm−1 for free CO. This points towards potential future applications of these species in AD catalysis. | ||
| Fig. 4 Precatalysts tested for methanol, ethanol condensation to isobutanol. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Runa | Catalyst | Time (h) | EtOH conversionb/% | Turnover numberc | Isobutanol yield/% | 1-Propanol yield/% | Isobutanol selectivityd/% |
| a Conditions: ethanol (1.0 mL, 17 mmol), methanol (10.0 mL, 247 mmol), [Mn] precatalyst (0.017 mmol, 0.1 mol%) and NaOMe64 (34.3 mmol, 200 mol%); mol% is based on ethanol substrate. Temperature = 180 °C. b Total conversion of ethanol and yield of isobutanol determined by GC analysis of the liquid phase. c Turnover number (TON) is based on mmol of ethanol converted to isobutanol/mmol of [Mn] catalyst; N.B. this does not take into account that the catalyst needs to participate in a second cycle coupling the intermediate n-propanol with methanol, so, in a sense, the TON could be considered to be double the value stated. d Selectivity to isobutanol = mmol of isobutanol/mmol of ethanol converted × 100. | |||||||
| 1 | 2TMS | 2 | 8 | — | <1 | <1 | — |
| 2 | 2TMS | 20 | 29 | 4 | 4 | 3 | 15 |
| 3 | 2TMS | 90 | 59 | 220 | 22 | 4 | 38 |
| 4 | 2H | 20 | 28 | 143 | 14 | 4 | 51 |
| 5 | 2H | 90 | 57 | 267 | 27 | 2 | 47 |
| 6 | 5 | 2 | <1 | — | <1 | <1 | — |
| 7 | 5 | 20 | 20 | 11 | 1 | <1 | 6 |
| 8 | 5 | 90 | 39 | 26 | 3 | <1 | 7 |
Mechanistic discussion
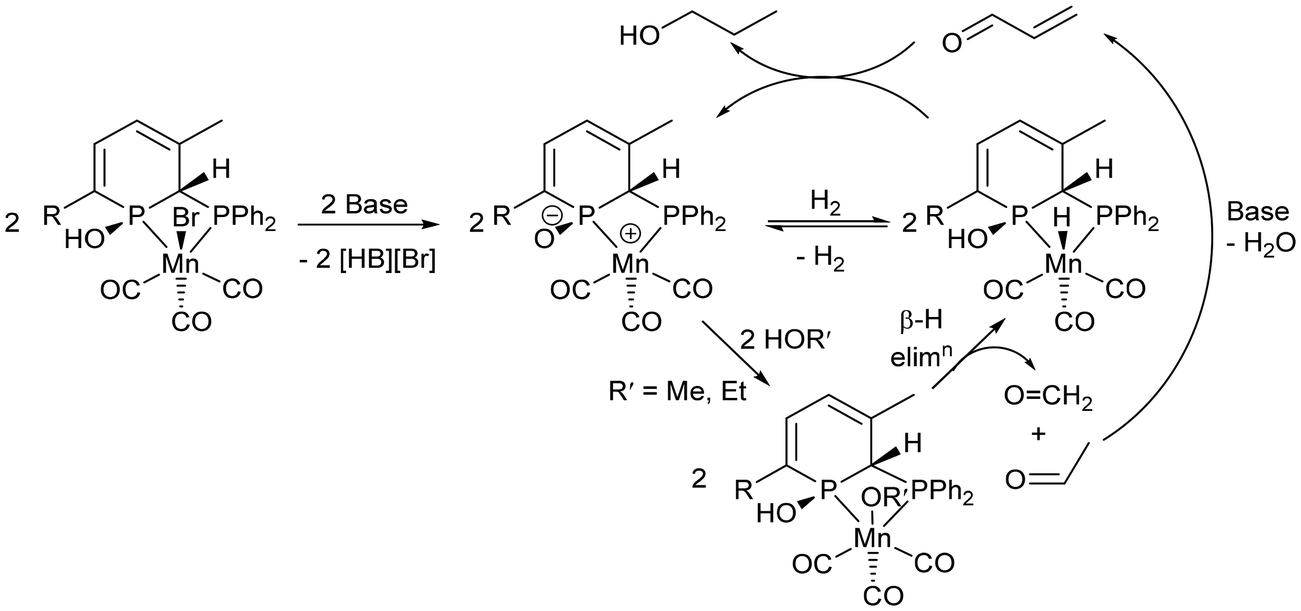
Since pioneering work by Beller65 and Milstein in 2016,66 pincer complexes of manganese have proven to be competent catalysts for (de)hydrogenation catalysis. For bis(phosphino)amine bromide tricarbonyl manganese pre-catalysts (Scheme 2), activation with base results in deprotonation of the central amine donor and loss of bromide to give a Mn-amide complex, which undergoes heterolytic cleavage of H2.65 Liu and co-workers proposed similar Mn amine/amide intermediates in the Guerbet coupling of ethanol to n-butanol,21 and Jones and co-workers tested a Mn-amide complex in catalysis and found it comparable, although with a slightly lower catalytic activity, to the Mn-amine pre-catalyst.24 A detailed proposal for the catalytic cycle of the more complicated MeOH/EtOH upgrading to isobutanol using Mn pincer catalysts wasn't given.25 Milstein and co-workers identified the deprotonation of a methylene group between a phosphine donor arm and the central pyridine ring of a pincer ligand, which led to disruption of the pyridine aromaticity, as part of their proposed catalytic cycle.66Previous work with Mn dppm catalysts has shown that it has proven difficult to investigate catalyst initiation and the nature of active species in this system,20 and therefore a detailed mechanism has not been derived. [MnBr(κ2-dppm)(κ1-dppm)(CO)2] was active at loadings of 0.1 mol% to give an 11% yield of isobutanol, whereas [MnBr(κ2-dppm)(CO)3] gave a 7% yield of isobutanol (180 °C, 90 h), so additional ligand may be beneficial.20 However, [MnBr(κ2-dppm)2(CO)] required a higher loading of 0.3 mol% to achieve a 14% isobutanol yield. Monitoring the reaction of [MnBr(κ2-dppm)(κ1-dppm)(CO)2] with NaOMe in MeOH by 31P{1H} NMR spectroscopy revealed the formation of free dppm and [MnBr(κ2-dppm)(CO)3] indicating that ligand redistribution is likely to play a role under catalytic conditions.20 Thus identifying speciation and nature/quantity of the ligands in a catalytic cycle has proven to be non-trivial. Ligand non-innocence and the presence of acidic H atoms on the dppm backbone may also play a key role as adding NaN(SiMe3)2 to [MnBr{κ2P,P-Ph2PC(H)(R)PPh2}(CO)3] (R = H, Me, Ph) led to deprotonation and coordination of the carbon backbone under elimination of NaBr to give [Mn{κ3P,C,P-Ph2PC(R)PPh2}(CO)3].67 Reaction with H2 proceeded across the Mn–C bond to give [MnH{κ2P,P-Ph2PC(H)(R)PPh2}(CO)3] most cleanly for R = Me, Ph.67 Further studies revealed additional mechanistic complexity of apparently simple reactions with these species.68,69
With this in mind, it is too early to give a detailed proposal of how 3TMS and 3H – and the many other potential isomers of the different species involved – participate in the catalytic cycle. From previous studies of catalysts, ligand non-innocence has proved to be an essential component, and this could take the form of zwitterionic complexes of type C (Scheme 3) in our system. Heterolytic cleavage of H2 would be possible, as well as reactions with alcohols to give carbonyls that would undergo aldol condensation (Scheme 5). Future work will involve the synthesis of hydride49,70 analogues of 2R and 3R and the zwitterionic complexes related to them through loss of hydrogen. Their role as cooperative complexes in catalysis will then be assessed.
 | ||
| Scheme 5 Reactions starting from 2R that could play a role in the catalytic cycle forming 1-propanol, the first step in the coupling of EtOH/MeOH to isobutanol. | ||
Conclusions
The first use of Mn phosphinine pre-catalysts in homogeneous catalysis has been described. These Mn 2-phosphinophosphinine complexes were easily synthesised by reaction of the proligand with MnBr(CO)5 at 80 °C for 2 h, and were shown to react immediately with water, even in trace quantities, to produce phosphinous acid metal complexes. Reactions with methanol were not selective, but single crystal X-ray diffraction experiments revealed that one of the products arises from unusual 1,4-addition of methanol to the phosphinine ring. Mn 2-phosphinophosphinine pre-catalysts gave good conversions for the Guerbet upgrading of ethanol/methanol to isobutanol at 180 °C for 90 h, with isobutanol yields superior to Mn dppm and related complexes, and similar to a Mn pincer catalyst, albeit requiring a longer reaction time.General experimental details
All reactions requiring inert condition were performed under an oxygen free nitrogen atmosphere by using standard Schlenk line techniques or by using an MBRUAN UNILab Plus glovebox, unless otherwise noted. Dry toluene, CH2Cl2 and THF were obtained from a solvent purification system (MBraun SP-300) and stored over 4 Å molecular sieves prior to use. Benzene was dried over molten potassium and distilled, or dried over activated 4 Å molecular sieves, prior to use. Methanol was dried over activated 3 Å molecular sieves. Non-dry solvents were used as received from Fisher Scientific. 2-Phosphinophosphinines 1TMS and 1H were synthesised as previously described.43,48 NMR spectra were obtained using either a AVIII400 (400 MHz) or AVIIIHD (400 MHz) spectrometer. 1H NMR spectra were recorded at 400 MHz and referenced to the residual solvent peak (7.24 for CDCl3, and 7.16 for C6D6). 13C{1H} NMR spectra were recorded at 101 MHz and referenced to the residual solvent peak (77.16 for CDCl3 and 128.06 for C6D6). FTIR was performed on a Thermo Scientific Nicolet iS5/iD5 ATR spectrometer. Mass spectrometry was conducted at the National Mass Spectrometry Facility at Swansea University using the techniques stated or using an in-house Shimadzu LCMS-2020 mass spectrometer in ASAP mode. Elemental analyses were performed at London Metropolitan University and are an average of two runs.GC analysis used a Shimazdu GC-2014 with Flame Ionisation Detector (GC-FID) using a DB-WAXETR column (1 μm film thickness, 60 m length, 0.32 mm inner diameter). Method: starting oven temperature 50 °C, hold at 50 °C for 5 minutes, heat to 250 °C at 50 °C min−1, hold at 250 °C for 8 minutes. A linear calibration curve was constructed for ethanol, isobutanol and 1-propanol made up of four calibration standards ranging from 50–100 mg mL−1 ran in duplicate.
2TMS
MnBr(CO)5 (102 mg, 0.37 mmol) and 2-phosphinophosphinine 1TMS (137 mg, 0.37 mmol) were dissolved in toluene (3 mL) and sealed in a flask under nitrogen. The reaction mixture was heated to 80 °C, bubbling was observed from loss of CO, for 2 h forming a bright orange solution. The solution was filtered and n-hexane was added (3 cm3). The solution was warmed to 80 °C briefly then placed at −25 °C for 16 h forming the product as yellow crystals (104 mg, 0.18 mmol, 48%).1 H-NMR (400 MHz, C 6 D 6 , 298 K) δ = 7.91 (m, 2 H, o-Ph), 7.68 (dd, 1 H, 3JH–P = 24.4 Hz, 3JH–H = 8.6 Hz, phosphinine 5-H), 7.54 (m, 2 H, o-Ph), 7.00 (m, 6 H, m/p-Ph), 6.55 (m, 1 H, phosphinine 4-H), 1.65 (s, 3 H, Me), 0.33 (s, 9 H, SiMe3); 31P{1H}-NMR (162 MHz, C6D6, 298 K)δ = 255.4 (br. s) and 23.7 (br. s); 13C{1H}-NMR (100.6 MHz, C6D6, 298 K)δ = 163.60 (d, 1JP–C = 18.8 Hz, 2-C-phosphinine from HMBC), 159.07 (dd, 1JP–C = 56.5 and 1JP–C = 16.2 Hz, 6-C-phosphinine from HMBC), 149.91 (dd, 2JP–C = 13 and 2JP–C = 7 Hz, 3-C-phosphinine from HMBC), 145.68 (dd, 2JP–C = 19.7 and 4JP–C = 3.4 Hz, assigned with HSQC, 5-C-phosphinine), 134.54 (d, 2JP–C = 9.6 Hz, ortho-Ph), 132.74 (d, 1JP–C = 38.0 Hz, i-Ph), 132.66 (d, 1JP–C = 37.9 Hz, i-Ph), 132.28 (d, 2JP–C = 10.8 Hz, ortho-Ph), 131.47 (d, 4JP–C = 2.7 Hz, p-Ph), 131.11 (d, 4JP–C = 2.2 Hz, p-Ph), 129.66 (d, 3JP–C = 9.8 Hz, Ar, m-Ph), 128.93 (d, 3JP–C = 10.3 Hz, Ar, m-Ph), 128.6 (peak underneath C6D5H resonance; assigned with HSQC, 4-C phosphinine), 22.42 (pseudo t, JP–C = 6.3 Hz, phosphinine-Me), −0.11 (d, JP–C = 3.6 Hz, SiMe3). CO resonances were not observed; 29Si{1H}-NMR (79.5 MHz, C6D6, 298 K)δ = −0.92 (dd, 2JP–Si = 20.6 Hz, 4JP–Si = 3 Hz); IR (ATR): 2018.5 (s, CO), 1968.1 (m, CO), 1937.5 (s, CO), 1912.2 (s, CO); Anal. calcd for C24H24BrMnO3P2Si (2TMS): C 49.25; H 4.13; N 0. Found C 50.05; H 3.49; N 0.
2H
MnBr(CO)5 (137 mg, 0.50 mmol) and 2-phosphinophosphinine 1H (147 mg, 0.50 mmol) were dissolved in toluene (3 mL) and sealed in a flask under nitrogen. The reaction mixture was heated to 80 °C, bubbling was observed from loss of CO, for 2 h forming a bright orange solution. n-Pentane (1 mL) was added, causing a ppt. to form immediately, but would not redissolve with heating. Additional n-pentane (30 mL) was added, and the mixture was cooled to −25 °C for 16 h. The resultant yellow solid was isolated by filtration and dried under vacuum (55 mg, 0.11 mmol, 22%).1 H-NMR (400 MHz, C 6 D 6 , 298 K) δ = 7.85 (m, 2 H, o-Ph), 7.49 (m, 4 H, Ar), 7.00 (m, Ar), 6.89 (br. s, Ar), 6.39 (br s, 1 H, Ar), 1.57 (s, 3 H, Me); 31P{1H}-NMR (162 MHz, C6D6, 298 K)δ = 234.2 (br. s) and 24.8 (br. s); 13C{1H}-NMR (100.6 MHz, C6D6, 298 K)δ = 167.5 (s, Ar), 149.4 (m, Ar), 149.4 (m, Ar), 142.8 (m, Ar), 140.4 (Ar), 133.8 (d, 1JP–C = 9.5 Hz, Ar), 131.5 (d, 1JP–C = 10.5 Hz, Ar), 130.6 (dd, JP–C = 38.1 and 2.8 Hz, Ar), 129.0 (m, Ar), 21.8 (phosphinine-Me). CO resonances were not observed; Anal. calcd for C21H16BrMnO3P2 (2H): C 49.15; H 3.14; N 0. Found C 49.63; H 2.87; N 0.
syn-3TMS
Water (2 mg, 0.11 mmol, 3.2 equiv.) was added to 2TMS (20 mg, 0.034 mmol) in CDCl3 (0.7 mL). 1H and 31P{1H} NMR spectroscopy revealed an instantaneous reaction generating two species, that ultimately yielded one compound after 5 d. Removal of all volatiles under vacuum gave syn-3TMS as a pale solid (4 mg, 7 μmol, 20%).1 H-NMR (400 MHz, CDCl 3 , 298 K) δ = 7.40 (m, 2 H, Ph), 7.07 (m, 2 H, Ph), 6.93 (m, 4 H, Ph), 6.85 (m, 1 H, POH), 6.51 (dd, 1 H, 3JH–P = 36.4 Hz and 3JH–H = 6.4 Hz, phosphacyclohexadiene 5-H), 5.43 (m, 2 H, phosphacyclohexadiene 4-H and phosphacyclohexadiene 2-H) 1.18 (s, 3 H, Me), 0.08 (s, 9 H, SiMe3); 31P{1H}-NMR (162 MHz, CDCl3, 298 K)δ = 92.3 (d, 2JP–P = 35 Hz, POH) and 21.0 (d, 2JP–P = 35 Hz, PPh2); 13C{1H}-NMR (100.6 MHz, CDCl3, 298 K)δ = 145.7 (m, phosphacyclohexadiene 5-C), 135.4 (m, 4 °C phosphacyclohexadiene), 133.3 (d, JP–C = 10.7 Hz, Ph), 132.6 (d, JP–C = 10.2 Hz, Ph), 131.0 (br. S, Ph), 129.6 (d, JP–C = 35.3 Hz, Ph), 128.7 (d, JP–C = 10.2 Hz, Ph), 128.2 (d, JP–C = 9.7 Hz, Ph), 127 (m, 4 °C phosphacyclohexadiene), 124.3 (dd, JP–C = 19.3 and 7 Hz, phosphacyclohexadiene 4-C), 64.9 (dd, JP–C = 12.3 and 7.4 Hz, phosphacyclohexadiene 2-C), 24.4 (m, phosphacyclohexadiene-Me), 0.0 (s, SiMe3); 29Si{1H}-NMR (79.5 MHz, C6D6, 298 K)δ = −0.14 (d, 2JP–Si = 19.5 Hz); IR (ATR): 2015.7 (s, CO), 1946.8 (m, CO), 1909.5 (s, CO).
3H
One drop of water (excess) was added to 2H (17 mg, 0.033 mmol) in C6D6 (0.7 mL). 31P{1H} NMR spectroscopy revealed an instantaneous reaction generating two species in a ratio of approximately 3:1; this did not change with time or heating.
31 P{ 1 H}-NMR (162 MHz, C 6 D 6 , 298 K) δ = 88.7 (br. s, POH minor isomer), 80.1 (br. s, POH major isomer), 34.9 (br. s, PPh2 minor isomer) and 19.5 (br. d, 2JP–P = 40 Hz, PPh2 major isomer).
Data for the major isomer assigned as syn-3H:
1 H-NMR (400 MHz, C 6 D 6 , 298 K) δ = 7.50–7.44 (br. m, 4 H, Ph), 7.08 (br. s overlapping with C6D5H, 2 H, Ph), 6.88 (br. s, 4 H, Ph), 6.27 (br. dd, 1 H, J = 29 Hz and 12 Hz, phosphacyclohexadiene), 5.86 (m, 1 H, phosphacyclohexadiene), 5.73 (br. t, 1 H, J = 13.6 Hz, phosphacyclohexadiene), 5.07 (br. s, 1 H, POH), 1.23 (br. s, 3 H, Me).
Reaction of 2TMS with dry methanol
2TMS (19.8 mg, 0.0338 mmol, 1 equiv.) was dissolved in C6D6 (0.7 mL) and dry MeOH (11 mg, 0.343 mmol, 10 equiv.) was added. Monitoring by multinuclear NMR spectroscopy revealed the formation of multiple products. Single crystals of 1,4-4TMS formed when the reaction mixture was left at room temperature for 1 h.31 P{ 1 H}-NMR (162 MHz, C 6 D 6 , 298 K) δ = 115.6 (br. s, isomer 1), 109.8 (br. poorly resolved d, isomer 2), 106.0 (br. s, 1,4-4TMS), 96.5 (br. s, syn-1,2-addition isomer), 36.3 (br. s, 1,4-4TMS), 28.8 (br. s, isomer 1), 25.3 (br. d, J = 24 Hz, syn-1,2-addition isomer), 12.4 (br. d, J = 46 Hz, isomer 2). Peak integrals used to associate resonances from the same species.
Data for 1,4-4TMS from redissolved crystals (major species only):
1 H-NMR (400 MHz, C 6 D 6 , 298 K) δ = 8.14 (m, 2 H, Ph), 7.59 (m, 2 H, Ph), 7.15 (m overlapping with C6D5H, Ph), 6.94 (m, 4 H, Ph), 6.62 (br. d, 3JH–P = 29 Hz, 1H, phosphacyclohexadiene 5-H), 2.82 (d, 3JP–H = 11.2 Hz, 3 H, OMe), 2.34 (m, 2 H, phosphacyclohexadiene 4-H),1.15 (d, 4JP–H = 1.6 Hz, 3 H, Me), 0.42 (s, SiMe3).
31 P{ 1 H}-NMR (162 MHz, C 6 D 6 , 298 K) δ = 106.4 (br. s, POMe) and 36.8 (br. s, PPh2).
Reaction of A with MeOH
A (10 mg, 0.011 mmol) was dissolved in C6D6 (0.7 mL) and dry methanol was added (41 mg, 1.3 mmol, 100-fold excess). Monitoring by 31P{1H} NMR spectroscopy revealed a slow reaction over the course of 48 h at room temperature.31 P{ 1 H}-NMR (162 MHz, C 6 D 6 , 298 K) δ = 240.0 (m), 75.0 (m), 16.4 (m) and −10.2 (m).
Heating to 80 °C led to further reactions and the formation of a complex mixture of products.
Catalytic reactions
The precatalyst (0.017 mmol, 0.1 mol%) and NaOMe (1.85 g, 34.3 mmol, 200 mol%) were added to a clean and dry 100 cm3 Hastalloy Parr autoclave inside a glove box, which was then sealed and transferred to a N2/vacuum manifold. Methanol (10 mL) was injected into the autoclave through an inlet against a flow of nitrogen followed by ethanol (1.0 mL, 17 mmol). The autoclave was sealed and placed into a pre-heated (180 °C) aluminium heating mantle. After the reaction run time (2, 20 or 90 h), the autoclave was cooled to room temperature in an ice-water bath. The autoclave was vented to remove any gas generated during the reaction. A liquid sample was removed, filtered through a short plug of alumina (acidic) and analysed by GC (100 μL of sample, 25 μL of pentanol standard, 1.7 cm3 Et2O – sample refiltered through a glass filter paper to remove insoluble salts). Residual solids were isolated by filtration, washed with toluene and dried under vacuum. Analysis was achieved by dissolution in D2O for 1H and 13C{1H} NMR spectroscopy.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
DJW thanks Lubrizol and EaSI-cat for funding a PhD studentship. DJW and SMM thank the Royal Society of Chemistry for an RSC Research Enablement Grant (E21-8488203428) and DJW thanks the RSC for a travel grant.References
- R. A. Kerr and R. F. Service, Science, 2005, 309, 101–101 CrossRef CAS PubMed.
- M. Trincado, D. Banerjee and H. Grützmacher, Energy Environ. Sci., 2014, 7, 2464–2503 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- R. E. H. Sims, W. Mabee, J. N. Saddler and M. Taylor, Bioresour. Technol., 2010, 101, 1570–1580 CrossRef CAS PubMed.
- A. Gupta and J. P. Verma, Renewable Sustainable Energy Rev., 2015, 41, 550–567 CrossRef CAS.
- M. V. Rodionova, R. S. Poudyal, I. Tiwari, R. A. Voloshin, S. K. Zharmukhamedov, H. G. Nam, B. K. Zayadan, B. D. Bruce, H. J. M. Hou and S. I. Allakhverdiev, Int. J. Hydrogen Energy, 2017, 42, 8450–8461 CrossRef CAS.
- A. Messori, A. Gagliardi, C. Cesari, F. Calcagno, T. Tabanelli, F. Cavani and R. Mazzoni, Catal. Today, 2023, 423, 114003 CrossRef CAS.
- H. Aitchison, R. L. Wingad and D. F. Wass, ACS Catal., 2016, 6, 7125–7132 CrossRef CAS.
- F. J. Sama, R. A. Doyle, B. M. Kariuki, N. E. Pridmore, H. A. Sparkes, R. L. Wingad and D. F. Wass, Dalton Trans., 2024, 53, 8005–8010 RSC.
- A. L. Olson, M. Tunér and S. Verhelst, Energies, 2023, 16, 7470 CrossRef CAS.
- S. M. Cypher, M. Pauly, L. G. Castro, C. L. Donley, P. A. Maggard and K. I. Goldberg, ACS Appl. Mater. Interfaces, 2023, 15, 36384–36393 CrossRef CAS PubMed.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS PubMed.
- S. M. Mansell, Dalton Trans., 2017, 46, 15157–15174 RSC.
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005–9008 CrossRef CAS PubMed.
- R. L. Wingad, P. J. Gates, S. T. G. Street and D. F. Wass, ACS Catal., 2015, 5, 5822–5826 CrossRef CAS.
- R. L. Wingad, E. J. E. Bergström, M. Everett, K. J. Pellow and D. F. Wass, Chem. Commun., 2016, 52, 5202–5204 RSC.
- K.-N. T. Tseng, S. Lin, J. W. Kampf and N. K. Szymczak, Chem. Commun., 2016, 52, 2901–2904 RSC.
- A. M. Davies, Z.-Y. Li, C. R. J. Stephenson and N. K. Szymczak, ACS Catal., 2022, 12, 6729–6736 CrossRef CAS.
- M. Roca Jungfer, J. L. Schwarz, F. Rominger, T. Oeser, R. Paciello, A. S. K. Hashmi and T. Schaub, ChemCatChem, 2024, e202301588 CrossRef CAS.
- A. M. King, H. A. Sparkes, R. L. Wingad and D. F. Wass, Organometallics, 2020, 39, 3873–3878 CrossRef CAS PubMed.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- K. Das, S. Waiba, A. Jana and B. Maji, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC.
- E. S. Gulyaeva, E. S. Osipova, R. Buhaibeh, Y. Canac, J.-B. Sortais and D. A. Valyaev, Coord. Chem. Rev., 2022, 458, 214421 CrossRef CAS.
- N. V. Kulkarni, W. W. Brennessel and W. D. Jones, ACS Catal., 2018, 8, 997–1002 CrossRef CAS.
- Y. Liu, Z. Shao, Y. Wang, L. Xu, Z. Yu and Q. Liu, ChemSusChem, 2019, 12, 3069–3072 CrossRef CAS PubMed.
- N. T. Coles, A. S. Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 433, 213729 CrossRef CAS.
- C. Müller and D. Vogt, Dalton Trans., 2007, 5505–5523 RSC.
- C. Müller and D. Vogt, in Phosphorus Compounds. Advanced Tools in Catalysis and Material Sciences, ed. M. Peruzzini and L. Gonsalvi, Springer, Dordrecht, 2011, vol. 37 Search PubMed.
- C. Müller, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, John Wiley & Sons, Ltd, 2012, pp. 287–307 Search PubMed.
- R. O. Kopp, S. L. Kleynemeyer, L. J. Groth, M. J. Ernst, S. M. Rupf, M. Weber, L. J. Kershaw Cook, N. T. Coles, S. E. Neale and C. Müller, Chem. Sci., 2024, 15, 5496–5506 RSC.
- M. Doux, N. Mézailles, L. Ricard and P. Le Floch, Eur. J. Inorg. Chem., 2003, 2003, 3878–3894 CrossRef.
- B. Schmid, L. M. Venanzi, A. Albinati and F. Mathey, Inorg. Chem., 1991, 30, 4693–4699 CrossRef CAS.
- C. Müller, J. A. W. Sklorz, I. de Krom, A. Loibl, M. Habicht, M. Bruce, G. Pfeifer and J. Wiecko, Chem. Lett., 2014, 43, 1390–1404 CrossRef.
- I. de Krom, E. A. Pidko, M. Lutz and C. Müller, Chem. – Eur. J., 2013, 19, 7523–7531 CrossRef CAS PubMed.
- I. de Krom, L. E. E. Broeckx, M. Lutz and C. Müller, Chem. – Eur. J., 2013, 19, 3676–3684 CrossRef CAS PubMed.
- X. Chen, Z. Li and H. Grützmacher, Chem. – Eur. J., 2018, 24, 8432–8437 CrossRef CAS PubMed.
- A. Campos-Carrasco, L. E. E. Broeckx, J. J. M. Weemers, E. A. Pidko, M. Lutz, A. M. Masdeu-Bultó, D. Vogt and C. Müller, Chem. – Eur. J., 2011, 17, 2510–2517 CrossRef CAS PubMed.
- A. Loibl, M. Weber, M. Lutz and C. Müller, Eur. J. Inorg. Chem., 2019, 2019, 1575–1585 CrossRef CAS.
- P. Le Floch and F. Mathey, Coord. Chem. Rev., 1998, 178–180, 771–791 CrossRef CAS.
- I. de Krom, M. Lutz and C. Müller, Dalton Trans., 2015, 44, 10304–10314 RSC.
- R. J. Newland, M. P. Delve, R. L. Wingad and S. M. Mansell, New J. Chem., 2018, 42, 19625–19636 RSC.
- P. A. Cleaves, B. Gourlay, R. J. Newland, R. Westgate and S. M. Mansell, Inorganics, 2022, 10, 17 CrossRef CAS.
- R. J. Newland, M. F. Wyatt, R. L. Wingad and S. M. Mansell, Dalton Trans., 2017, 46, 6172–6176 RSC.
- A. Gallen, A. Riera, X. Verdaguer and A. Grabulosa, Catal. Sci. Technol., 2019, 9, 5504–5561 RSC.
- P. A. Cleaves, B. Gourlay, M. Marseglia, D. J. Ward and S. M. Mansell, Inorganics, 2022, 10, 203 CrossRef CAS.
- L. E. E. Broeckx, A. Bucci, C. Zuccaccia, M. Lutz, A. Macchioni and C. Müller, Organometallics, 2015, 34, 2943–2952 CrossRef CAS.
- R. J. Newland, J. M. Lynam and S. M. Mansell, Chem. Commun., 2018, 54, 5482–5485 RSC.
- R. J. Newland, A. Smith, D. M. Smith, N. Fey, M. J. Hanton and S. M. Mansell, Organometallics, 2018, 37, 1062–1073 CrossRef CAS.
- E. C. Trodden, M. P. Delve, C. Luz, R. J. Newland, J. M. Andresen and S. M. Mansell, Dalton Trans., 2021, 50, 13407–13411 RSC.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS.
- T. A. DiBenedetto and W. D. Jones, Organometallics, 2021, 40, 1884–1888 CrossRef CAS.
- M. Doux, N. Mézailles, L. Ricard, P. Le Floch, P. D. Vaz, M. J. Calhorda, T. Mahabiersing and F. Hartl, Inorg. Chem., 2005, 44, 9213–9224 CrossRef CAS PubMed.
- C. Elschenbroich, J. Six and K. Harms, Chem. Commun., 2006, 3429–3431 RSC.
- P. L. Floch, N. Maigrot, L. Ricard, C. Charrier and F. Mathey, Inorg. Chem., 1995, 34, 5070–5072 CrossRef.
- F. Hartl, T. Mahabiersing, P. Le Floch, F. Mathey, L. Ricard, P. Rosa and S. Záliš, Inorg. Chem., 2003, 42, 4442–4455 CrossRef CAS PubMed.
- J. Fischer, A. De Cian and F. Nief, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1067–1071 CrossRef.
- K. Waschbüch, P. Le Floch, L. Ricard and F. Mathey, Chem. Ber., 1997, 130, 843–849 CrossRef.
- F. Nief, C. Charrier, F. Mathey and M. Simalty, J. Organomet. Chem., 1980, 187, 277–285 CrossRef CAS.
- F. Mathey and P. Le Floch, Chem. Ber., 1996, 129, 263–268 CrossRef CAS.
- M. Blug, C. Guibert, X.-F. Le Goff, N. Mézailles and P. Le Floch, Chem. Commun., 2008, 203 Search PubMed.
- M. Rigo, J. A. W. Sklorz, N. Hatje, F. Noack, M. Weber, J. Wiecko and C. Müller, Dalton Trans., 2016, 45, 2218–2226 RSC.
- Experiments with cis-[RuCl2(dppm)2] have shown that 200 mol% of base is necessary as conversion and yield drops rapidly as the amount of base is reduced. D. J. Ward and S. M. Mansell, unpublished results.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- Experiments with cis-[RuCl2(dppm)2] show that NaOH works equally as well as a base. D. J. Ward and S. M. Mansell, unpublished results.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben David, N. A. Espinosa Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed.
- N. V. Kireev, O. A. Filippov, E. S. Gulyaeva, E. S. Shubina, L. Vendier, Y. Canac, J.-B. Sortais, N. Lugan and D. A. Valyaev, Chem. Commun., 2020, 56, 2139–2142 RSC.
- E. S. Osipova, E. S. Gulyaeva, N. V. Kireev, S. A. Kovalenko, C. Bijani, Y. Canac, D. A. Valyaev, O. A. Filippov, N. V. Belkova and E. S. Shubina, Chem. Commun., 2022, 58, 5017–5020 RSC.
- E. S. Osipova, S. A. Kovalenko, E. S. Gulyaeva, N. V. Kireev, A. A. Pavlov, O. A. Filippov, A. A. Danshina, D. A. Valyaev, Y. Canac, E. S. Shubina and N. V. Belkova, Molecules, 2023, 28, 3368 CrossRef CAS PubMed.
- P. A. Cleaves and S. M. Mansell, Organometallics, 2019, 38, 1595–1605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2365932–2365934 and 2380027. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02142h |
| This journal is © The Royal Society of Chemistry 2024 |