
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analogous carbene-stabilised [MI-(η6-tol)]+ cations (M = Fe, Co, Ni): synthetic access and [carbene·MI]+ transfer†
Annika
Schulz
and
Terrance J.
Hadlington
 *
*
Lehrstuhl für anorganische Chemie mit Schwerpunkt neue Materialien, School of Natural Sciences, Technische Universität München, Lichtenberg Strasse 4, 85747 Garching. E-mail: terrance.hadlington@tum.de
First published on 18th September 2024
Abstract
A series of low-coordinate cationic 3d metal(I) complexes of the general formula [IPr·M(η6-tol)]+ is reported (M = Fe, Co, Ni; IPr = [(H)CN(Dip)C:]; Dip = 2,6-iPr2-C6H3), employing the weakly coordinating [BArF4]− counter-anion. The central metal in these complexes is stabilised solely by neutral carbene (i.e. IPr) and arene (i.e. toluene) ligands, making them rare examples of such cationic 3d metal(I) complexes, the electronic nature of which is explored by SQUID magnetometry. The utility of these species in [IPr·MI]+ transfer chemistry is demonstrated through the addition of a further equivalent of IPr, leading to formally two-coordinate cationic complexes, [(IPr2)·MI]+.
Low-valent, low-coordinate 3d transition metal (TM) complexes have garnered significant interest owing to their typically high reactivity,1–4 and more recently their potential as single-molecule magnets.5,6 Whilst historically cyclopentadienyl and carbonyl ligands have dominated the space of organometallic TM chemistry,7–10 their high electron donor number and small size, respectively, do not favour low coordination numbers. Here, N-heterocyclic carbenes (NHCs) have played a key role,1,11–13 allowing for the synthesis of stable electron deficient TM complexes through strong σ-donation and bulky, tunable steric properties. This has shone particularly true for Ni,14 as pioneered by Hartwig and co-workers, whereby a large NHC ligand drives catalytic turn-over in, for example, hydrogenative arylether scission,15,16 and alkene hydroarylation using well-defined Ni0 pre-catalysts.17 Significant efforts have thus gone towards for the synthesis of reactive low-valent NHC-stabilised 3d metal complexes. Key examples are the dvtms-bound M0 complexes [NHC·M(dvtms)] (Fig. 1(a); NHC = IPr, M = Fe, Ni; IPr = [(H)CN(Dip)C:]; Dip = 2,6-iPr2-C6H3; NHC = IMes, M = Co; IMes = [(H)CN(Mes)C:]; Mes = 2,4,6-Me3-C6H2; dvtms = 1,3-divinyltetramethyldisiloxane),18–20 which are both effective catalysts as well as [NHC·M0] transfer reagents.3,21 This latter point has been thoroughly explored by Deng and co-workers, for example in the exploration of low-coordinate Fe and Co imido species.22–24 Indeed, the same group later reported the related Mn0 complex,25 in addition to vtms-coordinated examples for Fe and Co (vtms = vinyltrimethylsilane),22,24 which show improved reactivity due to the ease of loss of vtms. Beyond alkene-stabilised systems, arene complexes have also seen some attention (e.g.Fig. 1(b) and (c)), which have potential benefits due to the chemical innocence of an arene leaving group. In this regard, Hillhouse, Cundari and co-workers demonstrated the straightforward access to a nickel-imide featuring a two-coordinate Ni centre.26 More recently, Deng and co-workers reported the cationic CoI complex [IPr·Co{(η6-C6H5)BPh3}], in which the coordinated [BPh4]− ion can be displaced by various different neutral ligands.27
 | ||
| Fig. 1 Key reported examples of (a) (dvtms)-complexes of NHC·M0 moieties, (b) simple arene complexes of NHC·Ni0 moieties, and (c) a cationic arene complex of CoI; (d) this work, demonstrating the facile access to arene adducts of cationic [NHC·MI] moieties, and their utility in [NHC·MI]+ group transfer. | ||
In our work, we have utilized the M0 species [IPr·M(vtms)2] as [IPr·M0] transfer reagents in forming a range of heavier triylene- and tetrylene-ligated 3d metal complexes (M = Fe, Ni).28–31 We sought to develop an analogous family of cationic MI systems, viz. [IPr·M]+, which may be useful to the organometallic chemistry community as ubiquitous [NHC·MI]+ transfer reagents. Herein we describe our efforts in this direction. Specifically, we report the synthesis of FeI, CoI, and NiI cations, stabilised by the bulky carbene IPr, and toluene. The electronic nature of these species is elucidated using SQUID magnetometry, and their utility as synthons of the [IPr·MI]+ fragment is demonstrated through their reaction with a further equivalent of carbene.
Results and discussion
In seeking reactive [IPr·MI]+ transfer reagents, we sought the one-electron oxidation of known [IPr·M0] reagents. To this end, the oxidation of [IPr·M(η2-vtms)2] systems (M = Fe, Co Ni) with Fc[BArF4] (Fc = ferrocene; ArF = 3,5-CF3C6H3)32 in toluene rapidly led to the deposition of deep purple (Fe), green (Co), or yellow (Ni) oily solids (see ESI† for details), beneath an orange solution (Scheme 1). 1H NMR spectroscopic analysis of the supernatant reaction solutions in all cases indicated the formation of free Fc and vtms, and disappearance of signals relating to the IPr ligand. Single crystals of compounds 1 (Fe), 2 (Co), and 3 (Ni) could be grown from the described coloured oils by layering of their fluorobenzene solutions with pentane, revealing that in all cases toluene complexes, [IPr·M(η6-tol)][BArF4], are formed. All species crystallise in the P21/n space group, and are essentially isostructural (Fig. 2). The CNHC–TM distance contracts on moving from Fe to Ni (dC1Fe1 = 2.034(3) Å; dC1Co1 = 1.994(3) Å; dC1Ni1 = 1.929(3) Å), whilst the opposite trend is observed for the average CTol distance (Fe: 2.149 Å; Co: 2.151 Å; Ni: 2.196 Å). Notably, and again in all cases, a ring-tilt of the toluene ligand is observed, relative to the [IPr·M] plane. This is primarily indicated by the non-linear NHC–M–Tolcentroid angle, which is most extreme for Co and Ni (Fig. 2 inset). This is further borne out by generally shorter M–C33 (Fe: 2.142 Å; Co: 2.128 Å; Ni: 2.161 Å) and −C34 (Fe: 2.139 Å; Co: 2.118 Å; Ni: 2.211 Å) contacts when compared with related C30 (Fe: 2.148 Å; Co: 2.182 Å; Ni: 2.212 Å) and C31 (Fe: 2.153 Å; Co: 2.175 Å; Ni: 2.311 Å) contacts. Now, when taking the centroids [C34–C29] and [C33–C32] as binding points for the M⋯Tol interaction, one finds what can be considered a pseudo-Y-shaped geometry at Fe, Co, and Ni, again most prominently for the latter two metals. That is, angles at M pertain to planarity (∑∠@Fe = 355.97°; ∑∠@Co = 359.69°; ∑∠@Ni = 359.95°), C1–M–centroid angles lie between 142° and 157°, and centroid–M–centroid angle are between 60° and 62°. A similar effect was noted in [IPr·Co(η6-PhBPh3)] reported by Deng et al.,27 as well as in IPr·CoCp.33 In the latter case, this phenomenon was presumed to be a result of steric hindrance; given that this tilting effect increases across the series Fe–Ni for analogous complexes, however, this is clearly an electronic effect. As such, and particularly for Co and Ni complexes 2 and 3, we propose a formal η4-tol binding mode in these species. All systems show clear π-stacking between their toluene ligand and one [ArF] group of the [BArF4]− counter-ion, with Tolcentroid–ArFCpara distances of 3.560 Å (Fe), 3.589 Å (Co), and 3.582 Å (Ni). In all cases, no significant increase in C–C bond lengths are observed in the toluene ligands relative to a ‘free’ arene (average C–C bond length in 1: 1.40 Å; in 2: 1.39 Å; in 3 1.39 Å), which would suggest weak binding of these ligands. | ||
| Scheme 1 Synthesis of cationic toluene complexes of Fe (1), Co (2), and Ni (3). Dip = 2,6-iPr2-C6H3; ArF = 3,5-(CF3)2-C6H3; Fc = ferrocene. | ||
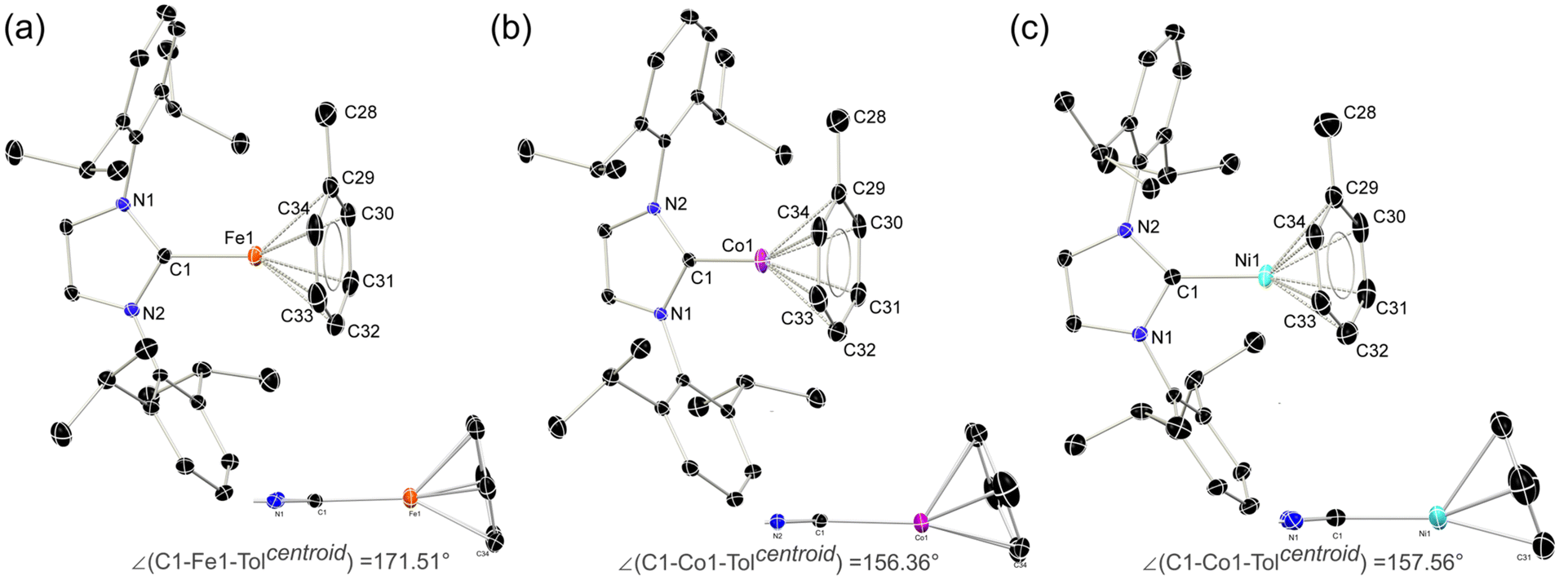 | ||
| Fig. 2 Molecular structure of the cationic part in (a) 1, (b) 2, and (c) 3, with thermal ellipsoids at 30% probability, and hydrogen atoms removed for clarity. Inset: side-on view of the NHC–M–tol binding, demonstrating an increased tilt-angle in the C1–M–Tolcentroid centroid on moving from M = Fe to M = Ni. | ||
All complexes exhibit highly broadened signals in their 1H NMR spectra, over a wide shift range (see ESI†), so demonstrating paramagnetic character. This is expected for 1 and 3, which are d7 and d9 respectively. In addition, closely related [IPr·Co(η6-PhBPh3)], recently reported by Deng et al.,27 is open-shell (i.e. high-spin) and paramagnetic, despite being d8 as per 2. The described solid-state structures give potential information regarding the electronic nature of 1–3, whereby Y-shaped geometries lead to five non-degenerate d-orbitals.34 SQUID magnetometry yields room temperature μeff values of 4.19, 3.98, and 2.16μB for 1, 2, and 3, respectively. For 1, this a clear indication of a high-spin FeI system, with S = 3/2, the observed value being only slightly higher than the spin-only value of 3.88μB. Equally, 3 demonstrates a near ideal μeff for an S = 1/2 system, fitting the proposed d9 electronic configuration. Complex 2, on the other hand, has a significantly higher μeff than expected for an S = 1, high-spin CoI system, with a theoretical spin-only value of 2.83μB. This effect most likely arises from a significant spin–orbit coupling in this species and an incomplete coupling of orbital angular momentum.35 Interestingly, the Evans method-derived μeff for the same system as a solution in D8-THF falls to 2.85μB, very close to the theoretical spin-only value, which may suggest some quenching of spin–orbit coupling in solution. Of course, we cannot discount that THF coordination may play a role in a geometry change for this species in solution. Looking deeper into the magnetisation of these species, the magnetic susceptibility (χM) of 1 and 3 follows clear Curie–Weiss paramagnetic behavior (Fig. 3(d) and (f)), whilst that for 2 is indicative of antiferromagnetic coupling (θCW = −24.2 K; Fig. 3(e)). For all systems, a linear increase in magnetization with increasing field strength is observed at 300 K (Fig. S7, S15 and S23 in ESI†). Density Functional Theory (DFT) optimised structures for simplified modifications of 1–3, i.e. [IXyl·M(η6-benz)] (1′, M = Fe; 2′; M = Co; 3′, M = Ni; IXyl = [(H)CN(Xyl)C:]; Xyl = 1,6-Me2-C6H3; Fig. S32–S34 in ESI†), yields structures in keeping with the general form observed in the X-ray crystal structures, for S = 3/2, S = 1, and S = 1/2 spin states for Fe, Co, and Ni, respectively. That is, a tilted and ‘slipped’ arene-M binding mode is observed, yielding what may be described as Y-shaped coordination at M. Mulliken spin densities are localised at the M-centres in all cases (Fe: 3.00; Co: 2.00; Ni: 0.96), suggesting minimal delocalisation to the NHC or arene ligands.
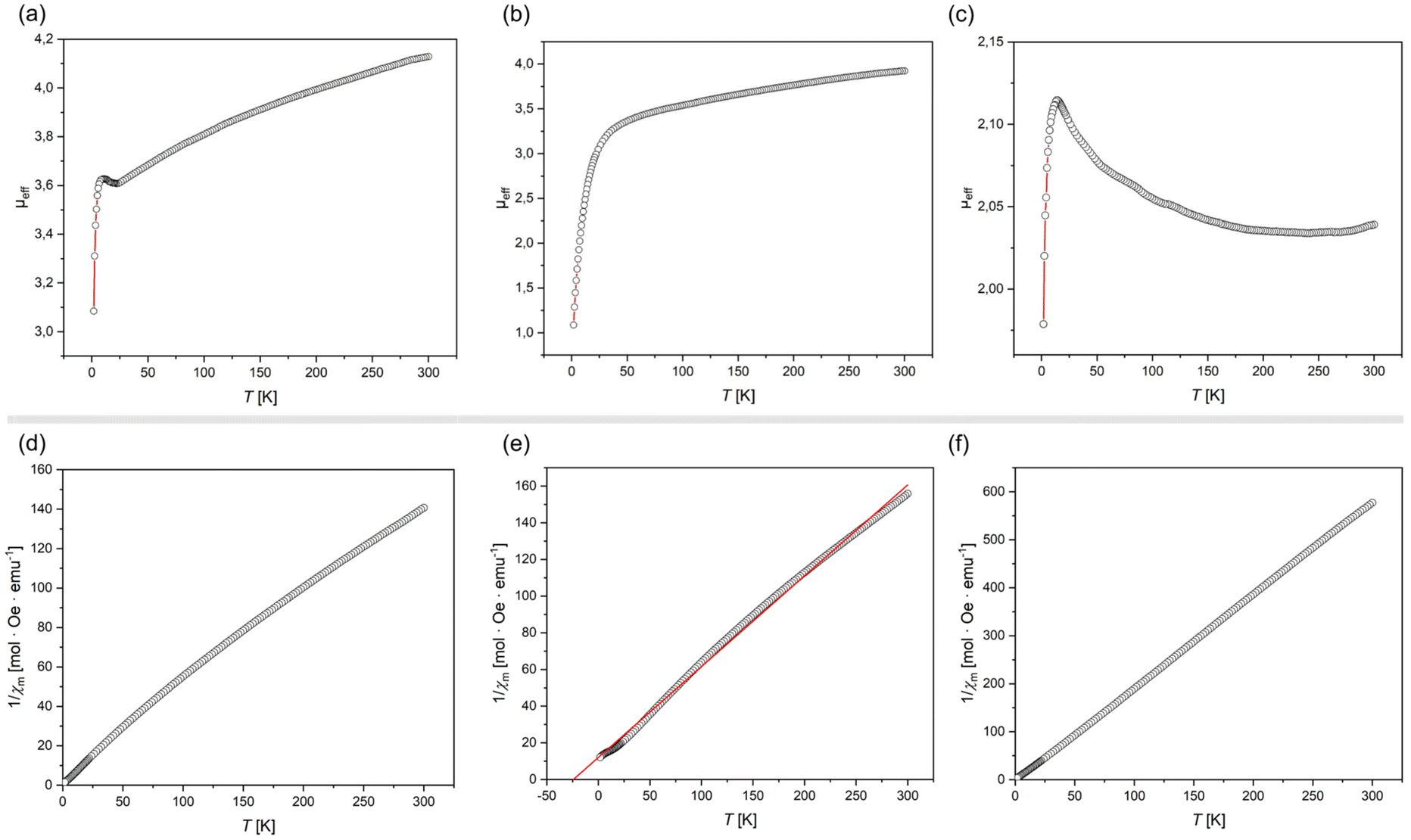 | ||
| Fig. 3 Plots of (a)–(c) μeffvs. T, and (d)-(e) 1/χMvs. T, for complexes 1–3. | ||
Following the isolation of compounds 1–3, we aimed to investigate their utility as [IPr·M]+ transfer reagents. To this end, a further equivalent of IPr was added to those species, targeting two-coordinate MI cations, examples of which have seen interest in the literature as single-molecule magnets.36,37
Addition of IPr to Et2O solutions of 1–3 led to dramatic colour changes in the formation of [(IPr)2M][BArF4] complexes (M = Fe (4), Co (5), and Ni (6)), which can be isolated as deep red, orange, and colourless crystalline solids, respectively, in good yields of between 69 and 74% (Scheme 2). All could be crystallographically characterised, with their molecular structures shown in Fig. 4. As for toluene complexes 1–3, complexes 4–6 are isostructural, crystallising in the P21/n space group and equal cell parameters. Still, slight differences are observed in their molecular structures: 5 and 6 feature essentially linear CNHC–M–CNHC angles of 178.4(2) and 178.8(2)°, respectively, and are similar to their reported counterparts.36,38–40 In contrast, this central CNHC–M–CNHC angle bends to 170.4(2)° in the iron(I) complex 4. The CNHC–Fe distances in this complex (i.e. 2.035(4) and 2.027(4) Å) are also longer than those found in 5 (i.e. 1.990(5) and 1.971(6) Å) and 6 (i.e. 1.945(4) and 1.934(5) Å), though this follows a similar trend as observed for 1–3. All species are paramagnetic, borne out by the broad and wide-spanning 1H NMR spectra. A number of earlier reported two-coordinate cationic [(NHC)2Fe]+ systems,37,41 two-coordinate anionic FeI species [{(Me3Si)2HC}2Fe]− and [{(Me3Si)2N}2Fe]−,42,43 and indeed Y-shaped toluene complex 1 demonstrate large solution-state magnetic moments, typically attributed to high-spin (i.e. S = 3/2) FeI with significant spin–orbit coupling and large magnetic contributions from unquenched orbital angular momentum. In contrast, the corresponding value for 4 is 2.27μB. This is now closer to that expected for an S = 1/2 system, and would therefore suggest a low-spin FeI complex, apparently brought about by a simple increase in steric bulk of the NHC ligand. This suggests that further exploration of bis(NHC) iron(I) species, analgous to 4, may allow for tuning of electronic and magnetic properties. For this, toluene complex 1 seems an ideal candidate.
 | ||
| Fig. 4 Molecular structure of the cationic part in (a) 4, (b) 5, and (c) 6, with thermal ellipsoids at 30% probability, and hydrogen atoms removed for clarity. | ||
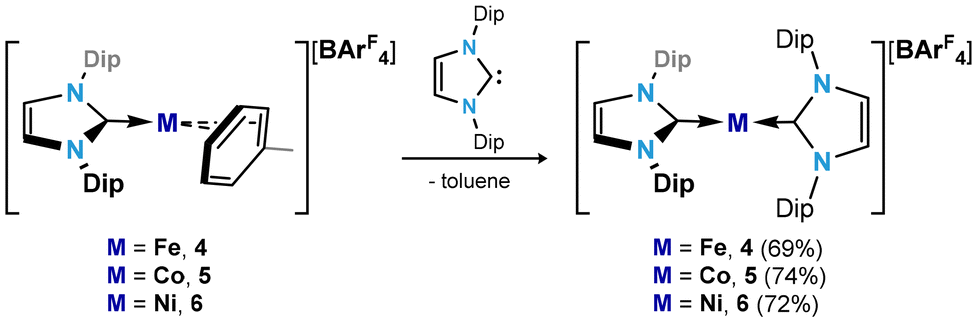 | ||
| Scheme 2 Synthesis of cationic bis(NHC) complexes of Fe (4), Co (5), and Ni (6). Dip = 2,6-iPr2-C6H3; ArF = 3,5-(CF3)2-C6H3. | ||
Conclusions
In conclusion, we have described the synthesis, structure, and electronic nature of a series of MI cations (M = Fe, Co, Ni), stabilised by an NHC and a labile toluene ligand. All systems demonstrate a high-spin, open shell ground state. All systems also feature a tilted arene binding mode, becoming more prominent on moving from Fe to Ni, and pertaining to a form η4-toluene binding mode for Co and Ni. These complexes have been utilized in the high-yielding [NHC·M]+ transfer reaction on combination with a further equivalent of carbene, leading to two-coordinate bis(NHC) FeI, CoI, and NiI cations. This study thus introduces a new readily accessible and analogous family of 3d-metal-cation transfer reagents, which we are exploring in their complexation behavior towards heavier low-valent p-block ligands.Author contributions
A. S. carried all experimental work. T. J. H. carried out computational evaluations, and conceived and supervised the project. A. S. and T. J. H. co-wrote the manuscript.Data availability
The ESI contains: Synthetic and analytical data for all new compounds; images of spectra for all new compounds; X-ray crystallographic information for structurally characterised species; computational details. In addition the Crystallographic Information Files (CIFs) for 1–6 are freely available from the CCDC (numbers 2377592–2377597†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
TJH thanks the Fonds der Chemischen Industrie (FCI) for generous funding of this research through a Liebig Stipendium, the ERC for a Starting grant (Project 101076897 – SINGAMBI), the DFG for an Independent Research grant (ProjectNr. HA9030/3-147032324, the Technical University Munich for the generous endowment of TUM Junior Fellow funds, and Prof. Fässler for his continued support. We also thank P. Mollik and J. Gilch for their help in acquiring LIFDI-MS data, and L. Kreimer for help in acquiring UV/vis data.References
- L. J. Taylor and D. L. Kays, Dalton Trans., 2019, 48, 12365–12381 RSC.
- P. P. Power, Chem. Rev., 2012, 112, 3482–3507 CrossRef PubMed.
- Y. Liu, J. Cheng and L. Deng, Acc. Chem. Res., 2020, 53, 244–254 CrossRef PubMed.
- A. Noor, Coord. Chem. Rev., 2023, 476, 214941 CrossRef.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- A. Zabala-Lekuona, J. M. Seco and E. Colacio, Coord. Chem. Rev., 2021, 441, 213984 CrossRef.
- M. Green and I. R. Butler, in Organometallic Chemistry, ed. M. Green, The Royal Society of Chemistry, 1st edn, 2004, pp. 393–444 Search PubMed.
- S. Lauk and A. Schäfer, Eur. J. Inorg. Chem., 2021, 2021, 5026–5036 CrossRef.
- P. J. Dyson and J. S. McIndoe, Transition Metal Carbonyl Cluster Chemistry, CRC Press, 2018 Search PubMed.
- H. Nakazawa, in Organometallic Chemistry, ed. H. Nakazawa and J. Koe, The Royal Society of Chemistry, 2021, pp. 43–57 Search PubMed.
- F. Glorius, N-Heterocyclic Carbenes in Transition Metal Catalysis, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, vol. 21 Search PubMed.
- H. V. Huynh, The Organometallic Chemistry of N–heterocyclic Carbenes, Wiley, 1st edn, 2017 Search PubMed.
- G. G. Zámbó, J. F. Schlagintweit, R. M. Reich and F. E. Kühn, Catal. Sci. Technol., 2022, 12, 4940–4961 RSC.
- V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef.
- N. I. Saper and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 17667–17676 CrossRef PubMed.
- N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao and J. F. Hartwig, Nat. Chem., 2020, 12, 276–283 CrossRef PubMed.
- J. Wu, J. W. Faller, N. Hazari and T. J. Schmeier, Organometallics, 2012, 31, 806–809 CrossRef.
- L. Zhang, Y. Liu and L. Deng, J. Am. Chem. Soc., 2014, 136, 15525–15528 CrossRef PubMed.
- H. Zhang, Z. Ouyang, Y. Liu, Q. Zhang, L. Wang and L. Deng, Angew. Chem., Int. Ed., 2014, 53, 8432–8436 CrossRef PubMed.
- J. Dong, X. Leng, D. Wang and L. Deng, ACS Catal., 2024, 14, 4001–4007 CrossRef.
- J. Du, L. Wang, M. Xie and L. Deng, Angew. Chem., Int. Ed., 2015, 54, 12640–12644 CrossRef.
- X.-N. Yao, J.-Z. Du, Y.-Q. Zhang, X.-B. Leng, M.-W. Yang, S.-D. Jiang, Z.-X. Wang, Z.-W. Ouyang, L. Deng, B.-W. Wang and S. Gao, J. Am. Chem. Soc., 2017, 139, 373–380 CrossRef PubMed.
- J. Cheng, J. Liu, X. Leng, T. Lohmiller, A. Schnegg, E. Bill, S. Ye and L. Deng, Inorg. Chem., 2019, 58, 7634–7644 CrossRef PubMed.
- J. Cheng, Q. Chen, X. Leng, Z. Ouyang, Z. Wang, S. Ye and L. Deng, Chem, 2018, 4, 2844–2860 Search PubMed.
- C. A. Laskowski, A. J. M. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2011, 133, 771–773 CrossRef PubMed.
- Q. Wang, X. Leng, D. Wang, S.-D. Bai and L. Deng, Organometallics, 2024, 43, 689–694 CrossRef.
- P. M. Keil, A. Soyemi, K. Weisser, T. Szilvási, C. Limberg and T. J. Hadlington, Angew. Chem., Int. Ed., 2023, 62, e202218141 CrossRef PubMed.
- A. Schulz, T. L. Kalkuhl, P. M. Keil and T. J. Hadlington, Angew. Chem., Int. Ed., 2023, 62, e202305996 CrossRef.
- P. M. Keil, S. Ezendu, A. Schulz, M. Kubisz, T. Szilvási and T. J. Hadlington, J. Am. Chem. Soc., 2024, 146, 23606–23615 CrossRef.
- T. Kalkuhl, I. Fernández and T. Hadlington, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-hp230.
- A. R. O'Connor, C. Nataro, J. A. Golen and A. L. Rheingold, J. Organomet. Chem., 2004, 689, 2411–2414 CrossRef.
- J. M. Andjaba, J. W. Tye, P. Yu, I. Pappas and C. A. Bradley, Chem. Commun., 2016, 52, 2469–2472 RSC.
- W. J. M. Blackaby, S. Sabater, R. C. Poulten, M. J. Page, A. Folli, V. Krewald, M. F. Mahon, D. M. Murphy, E. Richards and M. K. Whittlesey, Dalton Trans., 2018, 47, 769–782 RSC.
- S. Mugiraneza and A. M. Hallas, Commun. Phys., 2022, 5, 95 CrossRef.
- Y.-S. Meng, Z. Mo, B.-W. Wang, Y.-Q. Zhang, L. Deng and S. Gao, Chem. Sci., 2015, 6, 7156–7162 RSC.
- Y.-S. Meng, Z. Ouyang, M.-W. Yang, Y.-Q. Zhang, L. Deng, B.-W. Wang and S. Gao, Inorg. Chem. Front., 2019, 6, 1050–1057 RSC.
- S. C. Meier, A. Holz, J. Kulenkampff, A. Schmidt, D. Kratzert, D. Himmel, D. Schmitz, E. Scheidt, W. Scherer, C. Bülow, M. Timm, R. Lindblad, S. T. Akin, V. Zamudio-Bayer, B. von Issendorff, M. A. Duncan, J. T. Lau and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 9310–9314 CrossRef.
- M. W. Kuntze-Fechner, H. Verplancke, L. Tendera, M. Diefenbach, I. Krummenacher, H. Braunschweig, T. B. Marder, M. C. Holthausen and U. Radius, Chem. Sci., 2020, 11, 11009–11023 RSC.
- Z. Mo, D. Chen, X. Leng and L. Deng, Organometallics, 2012, 31, 7040–7043 CrossRef.
- Z. Ouyang, J. Du, L. Wang, J. L. Kneebone, M. L. Neidig and L. Deng, Inorg. Chem., 2015, 54, 8808–8816 CrossRef CAS.
- C. G. Werncke, P. C. Bunting, C. Duhayon, J. R. Long, S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2015, 54, 245–248 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577–581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2377592–2377597. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02372b |
| This journal is © The Royal Society of Chemistry 2024 |