
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent molecular systems based on carborane-perylenediimide conjugates†
Ruben
Rodriguez-Madrid‡
 ab,
Sohini
Sinha‡
a,
Laura
Parejo
b,
Jordi
Hernando
*b and
Rosario
Núñez
*a
ab,
Sohini
Sinha‡
a,
Laura
Parejo
b,
Jordi
Hernando
*b and
Rosario
Núñez
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193, Bellaterra, Barcelona, Spain. E-mail: rosario@icmab.es
bDepartament de Química. Universitat Autònoma de Barcelona, E-08193 Bellaterra, Barcelona, Spain. E-mail: jordi.hernando@uab.cat
First published on 11th October 2024
Abstract
This study presents the successful synthesis of two perylenediimide (PDI)-based ortho-carborane (o-carborane) derivatives, PDI–CB1 and PDI–CB2, through the insertion of decaborane into alkyne-terminated PDIs (PDI1 and PDI2). The introduction of o-carborane groups did not alter the optical properties of the PDI units in solution compared to their carborane-free counterparts, maintaining excellent fluorescence quantum yields of around 100% in various solvents. This was achieved by using a methylene linker to minimize electronic interaction between PDI and o-carborane, and by incorporating bulky o-carborane groups at imide- position to enhance solubility and prevent π–π stacking-induced aggregation. Aggregation studies demonstrated that PDI–CB1 and PDI–CB2 have greater solubility than PDI1 and PDI2 in both nonpolar and aqueous solvents. Despite the steric hindrance imparted by the o-carborane units, the solid state emission of PDI–CB1 and PDI–CB2 was affected by aggregation-caused fluorescence quenching. However, solid PDI–CB1 preserved bright red excimer-type emission, which persisted in water-dispersible nanoparticles, indicating potential for application as a theranostic agent combining fluorescence bioimaging with anticancer boron neutron capture therapy (BNCT) due to its high boron content.
Introduction
During the last years the use of carboranes1 and metallocarboranes2 for the preparation of luminescent molecules and materials has raised great attention.3 As the intrinsic emissive properties of these boron clusters are often poor, the most common strategy explored in the field consists in the conjugation of well-known fluorophores to carborane and metallocarborane units.3 Different advantages arise from this approach. On the one hand, the resulting systems benefit from the superior chemical and thermal resistance of boron clusters,1 their high capacity to self-assemble4 and their improved solubility and cellular uptake.5 In addition, their rigid 3D geometry can be exploited to control interchromophoric interactions between nearby fluorophores6 and, as a result, it might lead to aggregation-induced emission (AIE),3c,7 while preventing aggregation-caused quenching (ACQ).8 Finally, modulation of the luminescence from the attached emitters can be accomplished both in solution and in the solid state thanks to the particular electronic properties of carboranes and metallacarboranes.9,10However, all these advantages come at the expense of an important limitation: most fluorophore-carborane and fluorophore-metallacarborane conjugates, with a few exceptions,11 have shown low emission efficiencies in solution due to the quenching effects caused by the boron cluster via photoinduced charge transfer (CT).3,9,10,12 This is clearly illustrated by the case of 1,2-dicarba-closo-carborane (o-carborane, o-CB), which is by far the most used building block for the preparation of these systems. Because of its electron-withdrawing nature, o-CB deactivates the emission from nearby electron-rich fluorophores (e.g., anthracene,9e–g fluorene9i,k,l), which results in low luminescence signals in solution.3,9 Though this effect can lead to intramolecular CT states displaying intense AIE upon aggregation for sensing and smart material applications,9 it is a severe restraint if highly emissive o-carborane-emitter pairs are to be developed in solution – e.g., for theranostic agents that combine fluorescence diagnosis with anticancer boron neutron capture therapy (BNCT).12a,b,13
To overcome this drawback and prevent luminescence quenching in fluorophore-carborane conjugates, two important design principles must be considered: the electronic properties of the emitter and boron cluster, and the nature and length of the linker through which they are attached. Owing to their bright fluorescence and n-type semiconductor character, perylenediimides (PDI)14 are promising candidates to obtain highly emissive tethers with o-CB in solution, as photoinduced CT between these two units should be disfavored. In addition, the bulkiness of the carborane unit must allow enhancing the solubility of the appended PDIs, which is typically low in most solvents.14 In spite of these potential advantages, only several examples of PDI–o-CB conjugates have so far been described, where the steric effects imparted by the boron cluster were exploited to prevent ACQ and produce luminescent PDI solid materials with relevant sensing, two-photon absorption and electro(fluoro)chromic properties.6f,8a,b,d,15 However, most of these compounds showed moderate fluorescence efficiencies in solution (Φf ∼0.1–0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6f,8b,d,15b,c) that lie clearly below the high emission quantum yields of their constituting PDI dyes (Φf ∼114), a detrimental behavior caused by a combination of factors: (a) enhanced intra- and intermolecular PDI–PDI interactions, which are very sensitive to concentration and solvent conditions; and (b) partial intramolecular charge transfer between the PDI and o-CB units, which is favored by the use of π-conjugated linkers (e.g., phenyl and phenylacetylene groups) and polar solvents.
6f,8b,d,15b,c) that lie clearly below the high emission quantum yields of their constituting PDI dyes (Φf ∼114), a detrimental behavior caused by a combination of factors: (a) enhanced intra- and intermolecular PDI–PDI interactions, which are very sensitive to concentration and solvent conditions; and (b) partial intramolecular charge transfer between the PDI and o-CB units, which is favored by the use of π-conjugated linkers (e.g., phenyl and phenylacetylene groups) and polar solvents.
Herein we hypothesize that the introduction of carborane clusters as bulky groups in the imide positions of PDIs through a rationally selected linker will prevent intermolecular π–π interactions, thus improving (a) the solubility in organic solvents and (b) the photophysical properties both in solution and solid state to be used as fluorescent dyes. With this aim, compounds PDI–CB1 and PDI–CB2 were designed (Scheme 1), which contain: (a) two different, very bright PDI dyes (Φf ∼0.9–114), and (b) a non-conjugated methylene linker between the PDI and o-CB units, which were tethered through the N-imide PDI positions to minimize the effects on the spectral properties of the fluorophore.14 In the case of PDI–CB1, a pristine PDI emitter was used bearing two different lateral N-imide substituents: an o-CB group and a branched alkyl chain. To further magnify the effect of the boron cluster on solubility, two terminal o-CB groups were introduced in PDI–CB2, whose 1,6,7,12-tetrachlorinated PDI core is also known to enhance dissolution by minimizing intermolecular π–π interactions.14 The photophysical properties of these systems have been analyzed in different solvents and in the solid state. Furthermore, nanostructures of the carboranyl-containing PDIs and their properties have also been studied.
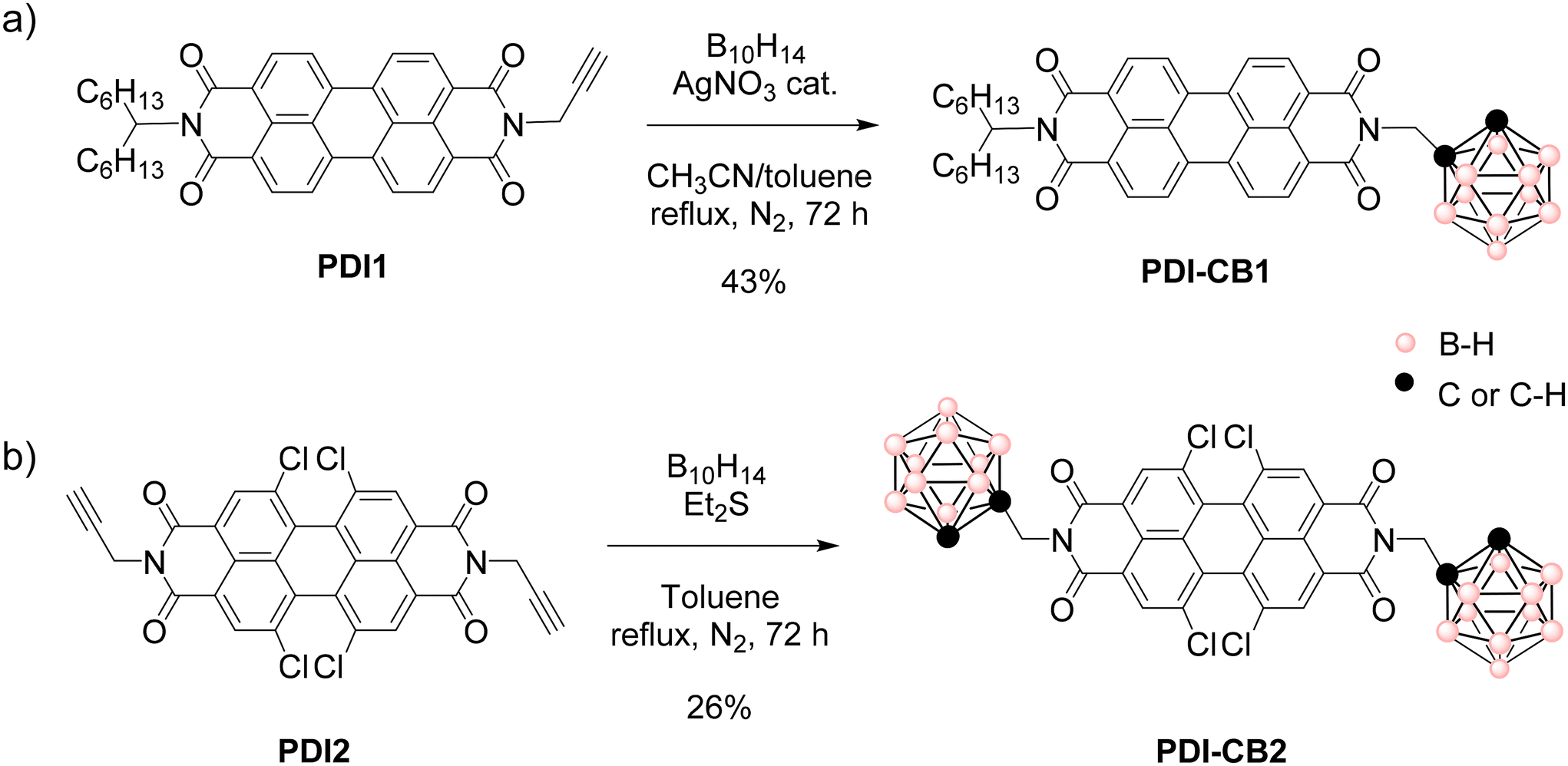 | ||
| Scheme 1 Insertion reactions to obtain (a) PDI–CB1 and (b) PDI–CB2 from B10H14 and N-propargylated PDIs PDI1 and PDI2. | ||
Results and discussion
Synthesis of perylenediimide-carborane conjugates
For the synthesis of PDI–CB1 and PDI–CB2, we followed a classical approach for the preparation of o-carborane derivatives: the reaction of commercially available decaborane (B10H14) with acetylenic compounds to construct the closed structure of the o-CB cluster,16 as previously described for other PDI–o-CB conjugates6f,8a,b,d,15 (Scheme 1). With this aim, we used well-known procedures for the preparation of the N-propargylated PDI derivatives PDI117 and PDI218 from the commercial perylenetetracarboxylic dianhydride precursor (Scheme S1 in the ESI†).
As shown in Scheme 1a, the insertion reaction of B10H14 to the acetylene group of PDI1 to afford PDI–CB1 was performed with AgNO3 as a catalyst and acetonitrile as a Lewis base.19 After optimization of the reaction procedure using different reaction conditions (see Table S1 in ESI†), this led to the target PDI–CB1 conjugate in 43% yield. By contrast, when the same optimized conditions were employed to produce PDI–CB2 from PDI2 and B10H14, a very low yield was obtained (15%). For this reason, we applied a different approach for the construction of the o-carborane scaffold of PDI–CB2,20 without silver salts, and using SEt2 as a Lewis base instead of acetonitrile (Scheme 1b). Finally, target compound PDI–CB2 was obtained with higher yield (26%).
o-Carboranyl derivatives PDI–CB1 and PDI–CB2 were characterized by 1H, 13C{1H} and 11B{1H} NMR, FT-IR spectroscopies and mass spectrometry (MS) (Fig. S7–S14 in the ESI†). In the 1H NMR of PDI–CB1 and PDI–CB2, we observed: (a) the disappearance of the narrow resonance at around 2.25 ppm corresponding to the alkyne proton of precursors PDI1 and PDI2; (b) the appearance of a new resonance at 4.21 ppm and 4.18 ppm for PDI–CB1 and PDI–CB2, respectively, which arise from the Ccluster–H (CCB–H) of the carborane units introduced in these compounds; and (c) a very broad 1H NMR signal between 3.0–1.0 ppm, which corresponds to the B–H protons of the o-CB clusters that cannot be well resolved because of boron-proton coupling. Furthermore, the IR-ATR spectrum of both PDI–CB1 and PDI–CB2 showed the broad band characteristic of B–H stretching at around 2580 cm−1, further indicating that the insertion reaction between the PDI precursors and B10H14 had occurred. Finally, it must be mentioned that, in the 13C{1H} NMR spectrum of PDI–CB2, the signals of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and some of the aromatic carbons were split into two, a feature that has been previously observed for symmetric PDIs bearing very bulky groups at the N-imide positions that hinder free rotation of the C–N bond.21
O group and some of the aromatic carbons were split into two, a feature that has been previously observed for symmetric PDIs bearing very bulky groups at the N-imide positions that hinder free rotation of the C–N bond.21
Photophysical properties of perylenediimide-carborane conjugates in solution
The UV-Vis absorption and fluorescence properties of the newly designed PDI–CB1 and PDI–CB2 were first evaluated in solution. The absorption spectra of these compounds and carboranyl-free perylenediimide dyes PDI1 and PDI2 were recorded in acetonitrile at low concentrations (c < 1.0 × 10−5 M, Fig. 1a, Table 1 and Table S2 in the ESI†). All of them showed similar absorption bands, exhibiting several peaks in the visible range of 400–525 nm, which correspond to different vibronic transitions between the ground electronic state (S0) and the first excited state (S1) of their PDI core.14a,b By contrast, no absorption signals were observed for the o-carborane units of PDI–CB1 and PDI–CB2, as they do not absorb at λabs > 200 nm.3 The fact that the absorption spectra of the PDI units in PDI–CB1 and PDI–CB2 very much resemble those of the starting, carboranyl-free compounds PDI1 and PDI2 can be rationalized on the basis of their electronic structure: the HOMO and LUMO orbitals of PDI chromophores have nodes at the imide nitrogen atoms; as a consequence, derivatization at the N-imide positions does not significantly modify their energies.14a,b However, changes can occur by functionalization of their bay positions due to two main factors:14a,b (a) the electronic effects imparted by the substituents introduced on the HOMO and LUMO energies, which can lead to absorption (and emission) spectral shifts; and (b) the steric congestion caused when two non-hydrogen substituents are attached to neighboring bay positions, which makes the PDI core take twisted conformations that result in absorption (and emission) broadening with less defined vibronic peaks and lower molar absorptivities. As already reported,14a,b the latter is the main factor affecting the absorption of chlorinated PDI derivatives such as PDI2, a situation that is also herein reproduced for PDI–CB2.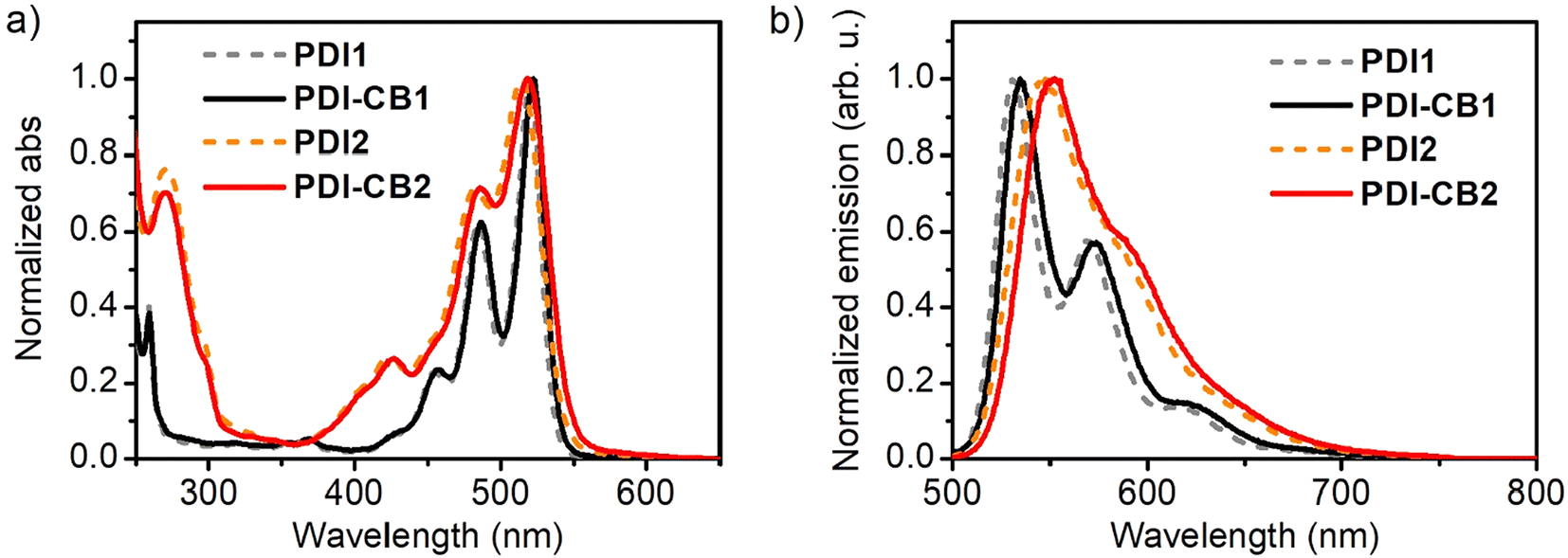 | ||
| Fig. 1 (a) Absorption (c = 5.0 × 10−6 M) and (b) emission spectra (c = 2.0 × 10−6 M, λexc = 445 nm) of PDI–CB1, PDI–CB2 and PDI dyes PDI1 and PDI2 in acetonitrile. They were all normalized respect to their spectral maximum. | ||
| Compound | λ maxabs (nm) | ε (L mol−1 cm−1) | λ maxf (nm) | Φ f |
|---|---|---|---|---|
| a Molar absorption coefficients at the spectral maximum. | ||||
| PDI1 | 520 | 67351 |
530 | 1.0 |
| PDI–CB1 | 522 | 64295 |
535 | 1.0 |
| PDI2 | 514 | 35384 |
546 | 0.89 |
| PDI–CB2 | 519 | 33011 |
552 | 0.90 |
The emission spectra for all the compounds were also measured in acetonitrile (Fig. 1b, Table 1 and Table S2 in the ESI†). In all the cases, the characteristic PDI emission signals were observed in the range λf = 500–700 nm, which correspond to different vibronic bands from the S1 → S0 transition of the PDI unit.14a,b Notably, a ∼16 nm bathochromic shift in emission as well as a slight spectral broadening and decrement in Φf were measured for PDI2 and PDI–CB2 relative to the non-chlorinated dyes PDI1 and PDI–CB1, a series of changes that are normally attributed to the electronic and steric effects imparted by the chlorine substituents introduced.14a,b In contrast, very minor differences were observed between the spectra of PDI–CB1 and PDI–CB2 and their precursors PDI1 and PDI2. Therefore, the introduction of o-carborane clusters at the N-imide positions did not alter the emission bands from the nearby PDI fluorophores, except for a small spectral red-shift observed. Very minor changes were also registered for the fluorescent quantum yields of these compounds, as both PDI–CB1 and PDI–CB2 preserved the high Φf values of the starting PDI dyes PDI1 and PDI2 in acetonitrile. Therefore, no electronic interaction takes place between the excited states of the PDI units and the nearby o-carborane clusters introduced in the conjugates that could affect their emission efficiency. This is a striking difference with respect to other compounds where o-carborane groups are directly attached to other fluorophores different from PDI, where intramolecular charge transfer occurs from the dye (donor unit) to boron clusters (acceptor unit) and leads to significant fluorescence emission quenching (FEQ).3,9–12 As anticipated above, two main factors should account for this result: (a) the electron-deficient nature of PDIs, which make them poor donors in charge transfer processes; and (b) the introduction of a CH2 linker between the dye and o-carborane units in PDI–CB1 and PDI–CB2.
The high emission efficiency measured for PDI–CB1 and PDI–CB2 in acetonitrile also significantly improves the behavior of previously reported carborane-perylenediimide conjugates, which showed moderate Φf values (Φf ∼0.1–0.86f,8b,d,15), especially when exposed to polar solvents such as acetonitrile. In addition, their superior fluorescent properties were preserved in a wide range of organic media, and a clear decrease in emission quantum yield was only observed for PDI–CB1 and PDI–CB2 in a very high polar solvent such as DMSO (Table 2). We ascribe this situation to the particular structure of these compounds. As further discussed below, PDI–CB1 and PDI–CB2 exhibit high solubility in a variety of solvents thanks to the presence of the bulky o-carborane groups, which disfavors intermolecular PDI–PDI interactions that often detrimentally affect their emission efficiency. Moreover, partial intramolecular charge transfer between the PDI and o-carborane units that could lead to fluorescence quenching is disfavored by the use of a methylene linker instead of π-conjugated tethers (e.g., phenyl and phenylacetylene groups).
| Solvent | PDI–CB1 | PDI–CB2 |
|---|---|---|
| Cyclohexane | 0.98 | 0.91 |
| Dioxane | 1.0 | 0.92 |
| Toluene | 1.0 | 0.92 |
| Chloroform | 1.0 | 0.92 |
| Tetrahydrofuran | 1.0 | 0.91 |
| Dichloromethane | 1.0 | 0.92 |
| Acetonitrile | 1.0 | 0.90 |
| Methanol | 0.78 | 0.89 |
| Dimethylsulfoxide | 0.25 | 0.01 |
Aggregation studies of PDI–CB1 and PDI–CB2 in solution
As mentioned above, the introduction of bulky carboranyl groups on the N-imide positions of PDI fluorophores should enhance their solubility and, therefore, modify their capacity to aggregate in solution. For this reason, we performed aggregation studies for PDI–CB1, PDI–CB2, PDI1 and PDI2 by adding increasing amounts of these compounds in media that are known to be poor solvents for PDI dyes. This should favor molecular aggregation, which in the case of PDI dyes often leads to spectral changes in absorption and emission that can be monitored by UV-vis absorption and fluorescence spectroscopies.14aFor the aggregation studies, hexane was first chosen as a solvent because of the poor solubility described for PDI derivatives in this medium.14a Actually, o-carboranyl-free compounds PDI1 and PDI2 showed clear changes in absorption that are consistent with intermolecular aggregation when dissolved in hexane even at relatively low concentrations: new red-shifted absorption bands were registered (λmaxabs = 542 nm and 569 nm for PDI1, and λmaxabs = 555 nm for PDI2), while broadening and relative intensity variation of the monomeric PDI absorption bands were also observed (Fig. S1 in the ESI†). This behavior is characteristic of the molecular stacking of PDI cores via π–π interactions, which leads to new exciton absorption bands for the aggregates.14a,22
Interestingly, when increasing amounts of PDI–CB1 and PDI–CB2 were added to hexane, no changes in the absorption spectra were appreciated other than a linear increment of the monomeric absorption intensities (Fig. S2 in the ESI†). This is a clear evidence of the enhancement of their solubility relative to PDI1 and PDI2 by the introduction of bulky groups such as carboranes on the N-imide positions, which are required to hinder molecular π–π stacking and prevent aggregation.14a,22 For this reason, we chose dioxane:water mixtures to trigger the molecular aggregation of PDI–CB1 and PDI–CB2, as PDI dyes are normally highly insoluble in aqueous media. Under these conditions, we could register a clear transition from monomer-type to aggregate absorption spectra for increasing concentrations of PDI–CB1 in 3:2 dioxane:water, as proven by the appearance of a red-shifted band and the broadening and relative intensity variation of the monomer signals (Fig. 2a and Fig. S3 in the ESI†). In the case of PDI–CB2, less clear spectral changes were observed when monitoring its aggregation in 2:3 dioxane:water by UV-vis absorption measurements. In particular, a slight broadening of its absorption spectrum was registered when rising concentration (Fig. 2a and Fig. S3 in the ESI†). The lack of more defined spectral variations upon aggregation for PDI–CB2 might be due to high molecular disorder and/or large interchromophoric distance in the aggregates, which are known to hinder the occurrence of strong exciton effects.23 Actually, the latter is expected to occur for PDI–CB2 because of the presence of two bulky carboranes side groups as well as the four chlorine bay substituents.
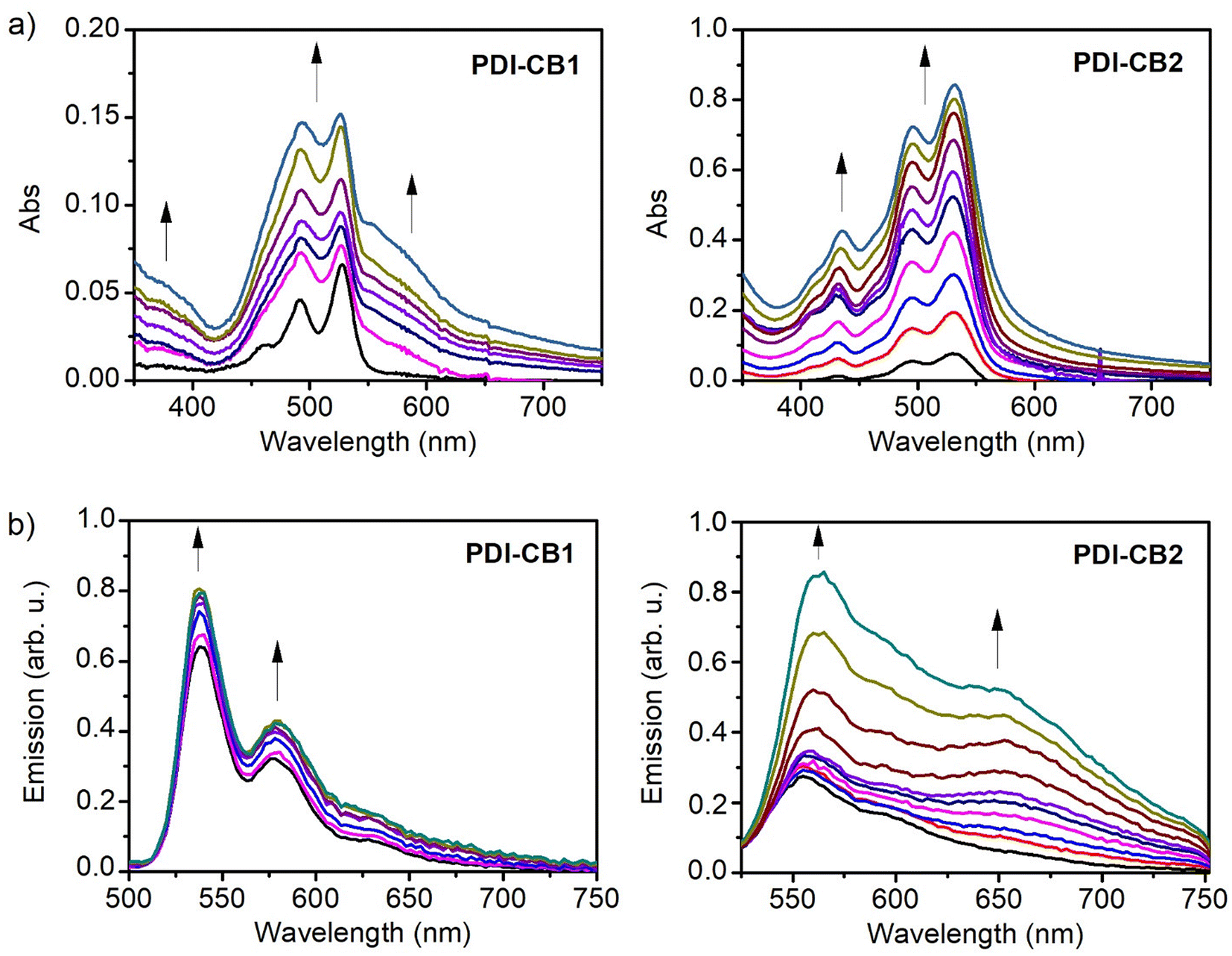 | ||
| Fig. 2 (a) Variation of the absorption spectrum of PDI–CB1 and PDI–CB2 upon consecutive additions of: 5 μL of a stock solution of PDI–CB1 in dioxane (c = 0.4 × 10−3 M) to 2 mL of a 3:2 dioxane:water mixture (v = 5, 10, 15, 20, 25, 30 and 35 μL); and 15 μL of a stock solution of PDI–CB2 in dioxane (c = 0.41 × 0−3 M) to 2 mL of a 2:3 dioxane:water mixture (v = 15, 30, 45, 60, 75, 90, 105, 120, 135 and 150 μL). (b) Variation of emission spectra (λexc = 445 nm) of PDI–CB1 and PDI–CB2 upon consecutive additions of: 5 μL of a stock solution of PDI–CB1 in dioxane (c = 0.4 × 10−3 M) to 2 mL of a 3:2 dioxane:water mixture (v = 5, 10, 15, 20, 25, 30 and 35 μL); and 15 μL of a stock solution of PDI–CB2 in dioxane (c = 0.41 × 0−3 M) to 2 mL of a 2:3 dioxane:water mixture (v = 15, 30, 45, 60, 75, 90, 105, 120, 135 and 150 μL). In all the graphs arrows indicate the direction of the spectral changes observed upon consecutive additions of PDI–CB1 or PDI–CB2. | ||
Further proof of the aggregation of PDI–CB1 and PDI–CB2 in solution was obtained using emission measurements (Fig. 2b). When adding increasing amounts of these compounds to dioxane:water mixtures (cPDI–CB1 = 1.0 × 10−6 M–6.9 × 10−6 M, cPDI–CB2 = 3.0 × 10−6 M–2.8 × 10−5 M), low-to-moderate increments of PDI monomer emission at λabs ∼535 (PDI–CB1) or 550 nm (PDI–CB2) were observed, which indicates that PDI–o-CB conjugate molecules efficiently aggregate as their concentration raises. This effect is particularly evident for PDI–CB1, as also illustrated by the complementary absorption measurements shown in Fig. 2a. As for the emission of the aggregates formed, it can be correlated with the growth of the new broad and red-shifted band appearing at λf > 600 nm, which is of very low intensity for PDI–CB1 and more clearly visible for PDI–CB2. This type of emission is typical for the so-called PDI excimers, which are excited dimers formed in π-stacks of PDI fluorophores that are typically low-emissive.12a This feature, together with other interchromophoric interactions expected to take place in the aggregates (e.g., emission quenching by photoinduced electron transfer), explains why the monomer emission bands are still predominant in the spectra shown in Fig. 2b even for high PDI–CB1 and PBI–CB2 concentrations where most of these molecules are aggregated. Actually, the formation of dimly red-emitting excimers could be observed by naked eye for larger aggregates of PDI–CB1 and PDI–CB2 prepared by simply adding a few droplets of dioxane stock solutions into water (Fig. S4 in the ESI†).
Photophysical properties of perylenediimide-carborane conjugates in the solid state: from bulk powder to nanostructures
In general, PDIs produce vividly colored solutions in organic solvents with intense fluorescence. By contrast, a red-shift of the emission is typically observed in the solid state together with a dramatic decay of the fluorescence efficiency. This is due to the π-stacking of PDI cores, which gives rise to interchromophoric interactions that lead to the formation of excimers, among other types of low- (or non-) emissive species.14a,22 As a consequence, the optical properties of PDI derivatives in the solid state are very dependent of molecular packing and there is strong interest to develop compounds of this type that preserve the strong monomeric emission when aggregated, e.g., by introducing bulky groups such as boron clusters that hinder strong interchromophoric interactions at short distances upon aggregation.6f,8a,b,15bTo investigate if this behavior is accomplished for PDI–CB1 and PDI–CB2, the diffuse-reflectance absorption and emission spectra of the solid powders of these compounds were measured and compared to those in solution (Fig. 3). In the case of solid PDI–CB1, its absorption spectrum resembles those registered for its aggregates in dioxane:water mixtures, as a new red-shifted band and the broadening of the monomer bands were observed (Fig. 3a). This suggests that compact π–π stacking of the PDI units of PDI–CB1 also takes place in the solid state. As for solid PDI–CB2, a similar absorption spectrum to that of the monomer in solution was found. This is the expected behavior according to the aggregation studies performed in solution, as we found tight chromophore packing to be prevented in this case by the steric hindrance imparted by the two o-carborane side groups and the chlorine bay substituents.
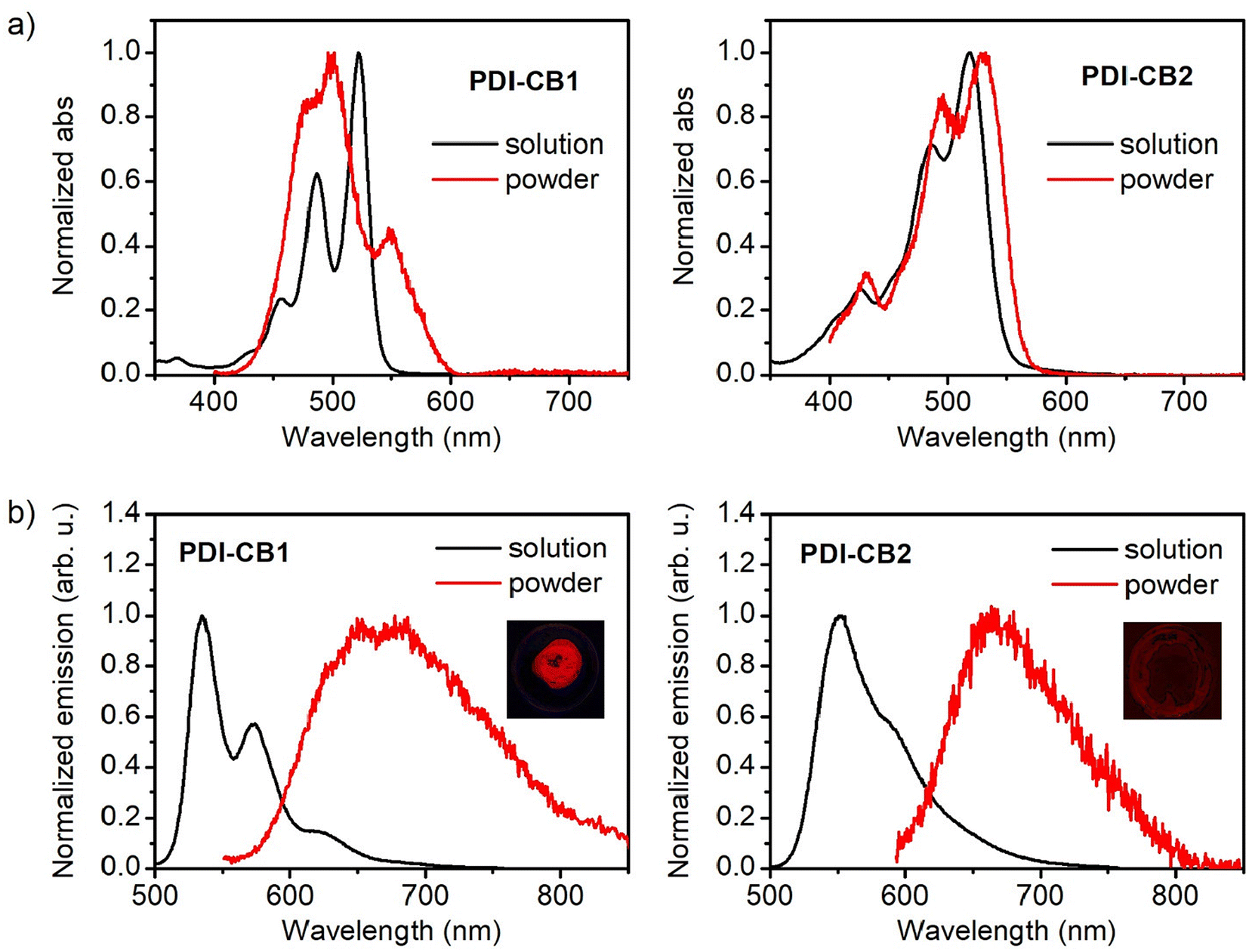 | ||
| Fig. 3 (a) Absorption spectra of the solid powders obtained from PDI–CB1 and PDI–CB2, which are compared to those measured in acetonitrile solution for both compounds. In the case of powders absorption, the Kubelka–Munk function determined from the diffuse reflectance absorption spectrum is given. For sake of comparison, all the spectra are normalized to unity at their maxima in the visible region. (b) Emission spectra (λexc = 445 nm) of the solid powders obtained from PDI–CB1 and PDI–CB2, which are compared to those measured in acetonitrile solution for both compounds. The insets in (b) show a photograph of the emission from the powder of each respective compound under irradiation at λexc = 365 nm. | ||
By contrast, the solid powders of PDI–CB1 and PDI–CB2 showed a rather similar behavior in emission: the fluorescence bands from monomeric PDI units were not measured and, instead, red excimer-type emission was registered with lower efficiency (Fig. 3b). Therefore, the introduction of o-carborane units in the N-imide positions of our PDIs could not prevent the formation of excimers due to π–π stacking upon aggregation, thus detrimentally affecting the emissive properties of these fluorophores in the solid state. However, this effect was found to differently impact the fluorescent performance of PDI–CB1 and PDI–CB2 powders. On the one hand, the emission of PDI–CB2 was mainly suppressed in the solid state, as illustrated by the image in the inset of Fig. 3b and corroborated by the Φf value measured (Φf = 0.012 ± 0.005). In fact, a similar behavior was registered for the solid powder of the reference compound PDI2 (Φf = 0.010 ± 0.006). On the other hand, a much brighter red emission was detected for PDI–CB1 powder, as demonstrated by the image in the inset of Fig. 3b and the 4-fold higher fluorescence quantum yield measured (Φf = 0.045 ± 0.003), which essentially matches the Φf value determined for the reference PDI1 solid sample (Φf = 0.045 ± 0.004). As already observed in absorption, the differences between the fluorescent properties of PDI–CB1 and PDI–CB2 powders should be attributed to the distinct arrangement of these molecules in the solid state, thus leading to the formation of dissimilar excimer emitting sites and other non-emitting species.
Despite its relatively low fluorescence quantum yield, PDI–CB1 solid samples exhibit two attractive features for the development of theranostic probes for biological applications: (a) rather bright emission in the red and near-infrared regions (λmaxf ∼700 nm) upon irradiation with low-energy visible light (λabs up to 600 nm, see Fig. 3b), which is often preferred to increase penetration depth and decrease photodegradation and autofluorescence effects in biological tissues; and (b) the high boron content required for efficient boron neutron capture therapy.24 In light of these properties, we explored the preparation of water-dispersible PDI–CB1 nanoparticles (PDI–CB1_NP). With this aim, we applied the precipitation method25 followed by purification through dialysis. Transmission electron microscopy (TEM) and dynamic light scattering (DLS) analysis of the resulting nanoparticles revealed that spherical nanostructures of about 100 nm in diameter and good size uniformity were obtained (Fig. 4a, b and Fig. S5, S6 in the ESI†). In addition, the ζ-potential value measured for PDI–CB1_NP was around −41.2 mV, thus suggesting good colloidal stability without the need of introducing external stabilizing agents. After storage at 4 °C for two weeks, no changes in TEM and DLS measurements were observed for the NP dispersions.
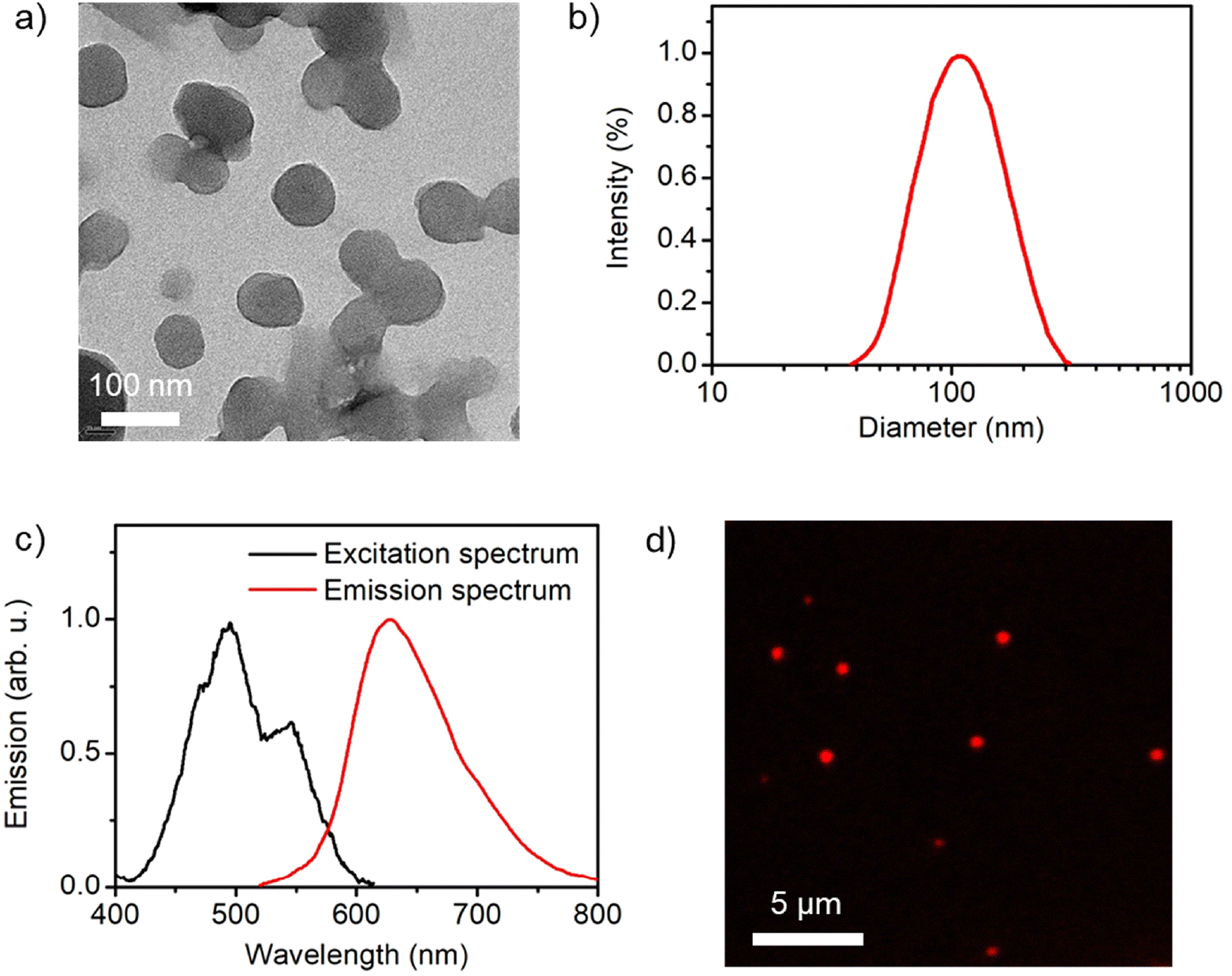 | ||
| Fig. 4 (a) TEM image of PDI–CB1_NP (average diameter = 95 ± 10 nm; see Fig. S6d in the ESI†). (b) Intensity-weighted diameter distributions measured for an aqueous colloidal suspension of PDI–CB1_NP by DLS (average diameter = 112 nm; polydispersity index = 0.12). (c) Fluorescence excitation (λem = 642 nm) and emission (λexc = 500 nm) spectra of an aqueous colloidal suspension of PDI–CB1_NP. (d) Confocal fluorescence microscopy image of PDI–CB1_NP casted on a glass coverslip (λexc = 488 nm, λdetection = 520–780 nm). | ||
More importantly, aqueous colloidal suspensions of PDI–CB_NPs preserved the principal optical properties of PDI–CB bulk powder. Upon visible light excitation, they showed red and near-infrared emission with efficiencies comparable to PDI–CB1 in the solid state (Φf = 0.020, Fig. 4c). In addition, when measuring the fluorescence excitation spectrum of PDI–CB1_NP suspensions, the absorption spectrum of PDI–CB1 bulk powder was reproduced – i.e., a broad absorption spectrum with a red-shifted absorption band extending up to λabs ∼600 nm (Fig. 4c). All these features indicate that excitonic aggregation of the PDI chromophores also occurs in the nanoparticles, where the absorbed excitation energy is funneled towards excimer sites that eventually emit. As a result, PDI–CB1_NP generate sufficient red emission under visible light excitation as to be detected by confocal fluorescence microscopy when dispersed onto a glass coverslip (Fig. 4d). This behavior, combined with their high boron content and aqueous dispersibility, would open the door to the use of PDI–CB1_NP as theranostic platforms for simultaneous fluorescence detection and boron neutron capture therapy.
Conclusions
In this work we have reported the successful synthesis of two perylendiimide (PDI)-based o-carborane derivatives (PDI–CB1 and PDI–CB2) via the insertion reaction of decaborane with alkyne-terminated PDIs (PDI1 and PDI2). The introduction of o-carborane side groups in PDI–CB1 and PDI–CB2 did not affect the optical properties of their PDI units in solution relative to their carborane-free counterparts. Remarkably, the synthesized PDI–carborane conjugates retained the high fluorescence quantum yields (Φf ∼0.9–1) characteristic of PDI fluorophores across a broad range of solvents with varying polarity, which contrasts with most previously reported PDI–o-CB derivatives. Two key structural features contributed to this outcome: the use of a methylene linker to minimize through-bond electronic communication between the PDI and carborane units, and the enhanced solubility provided by the bulky carborane groups positioned at the N-imide sites of the PDI chromophores, thereby preventing aggregation via π–π stacking. Aggregation studies confirmed that PDI–CB1 and PDI–CB2 exhibited significantly higher solubility than their parent PDI compounds, PDI1 and PDI2, in both nonpolar organic solvents (e.g., hexane) and aqueous environments (e.g., dioxane mixtures). Nevertheless, despite the steric hindrance introduced by the o-carborane substituents, fluorescence quenching due to aggregation was observed for PDI–CB1 and PDI–CB2 in the solid state. Yet, PDI–CB1 preserved rather bright red excimer-type emission in its solid form, which was maintained upon formation of water-dispersible nanoparticles that could be detected by confocal fluorescence microscopy. Given their low-energy emission characteristics and high boron content, these nanostructures may have potential applications as theranostic agents for bioimaging and BNCT. In light of the promising results obtained in this work, the development of novel PDI–CB conjugates holds potential. These new conjugates could exhibit enhanced emission efficiencies in the solid state, coupled with valuable optoelectronic properties.Experimental section
Materials
Commercial reagents were used as received from suppliers. The anhydrous MeOH was kept over molecular sieves 3 Å. Decaborane (B10H14) was purchased by Katchem Ltd (Prague) and recrystallized before use. Toluene was purchased from Merck and distilled from sodium benzophenone previously to use. 7-Tridecanone, perylene-3,4,9,10-tetracarboxylic dianhydride, ammonium acetate, sodium cyanoborohydride, NaOH, KOH, imidazole, K2CO3, Et3N, AgNO3 and Et2S were purchased from Sigma-Aldrich and utilized as received. HPLC grade hexane, dioxane, CHCl3, MeOH, CH2Cl2, acetonitrile and DMSO were obtained from Scharlab and used without further purification.All reactions were performed under inert atmosphere employing standard Schlenk techniques. Thin layer chromatography (TLC) was performed on pre-coated aluminum layers of silica gel 60 F254 (0.20 mm thick): MACHEREY-NAGEL Alugram® silica plates SIL G/UV254. Preparative TLC was performed on Pre-coated TLC-plates SIL HD; 0.25 mm thick silica gel 60. Flash column chromatography was performed by using silica gel (SiO2) with 0.04–0.06 mm in particle size and 60 Å in pore size.
Instruments
Infrared spectra were recorded using a Bruker Tensor 27 spectrometer equipped with a Golden Gate Single Reflection Diamond ATR. Nuclear magnetic resonance (NMR) spectra were recorded with Bruker DPX-250 and DPX-360 (1H NMR (250 MHz, 360 MHz, 400 MHz), and DPX-400 spectrometers (11B and 11B{1H} NMR (128 MHz)) and 13C{1H} NMR (100, MHz)). Chemical shifts are reported in ppm using the signal of the residual non-deuterated solvent molecules as a reference for 1H NMR and 13C{1H} NMR. For 11B and 11B{1H} NMR, spectra were measured using quartz tubes and referenced to external BF3·OEt2. All coupling constants are reported in hertz. MALDI-TOF-MS mass spectra were recorded in the negative ion mode using a Bruker Biflex MALDI-TOF spectrometer and dithranol (DIT) as a matrix.UV-vis absorption spectra in solution were recorded in a HP 8452A spectrophotometer (Agilent) with Chemstation software, using 0.1- or 1 cm optical path quartz cuvettes and HPLC-quality solvents. For solid samples, an Agilent Cary 60 spectrophotometer in diffuse reflectance mode and coupled to a remote integrating sphere was used. In this case, the Kubelka–Munk function was used to estimate their absorption spectra. Fluorescence emission spectra were measured in a custom-made spectrofluorometer by using CW laser excitation at λexc = 445 nm and detecting the emitted photons in an Andor ICCD camera coupled to an Andor spectrograph. All the emission spectra registered were corrected by the wavelength dependence of the spectral response of the detection system. Samples were prepared in HPLC grade solvents and adjusted to a response within the linear range. Fluorescence quantum yields in solution were determined using the standard method26 for highly diluted solutions of the compounds of interest to prevent self-absorption processes (absorption <0.1 at the excitation wavelength) and relative to N,N′-bis(1-hexylheptyl)perylene-3,4,9,10-tetracarboxybismide in acetonitrile (Φf = 1).27 For solid samples, absolute fluorescence quantum yields were determined using a Hamamatsu Quantum Yield fluorometer C9920-02G using an integrating sphere.
Scanning electron microscopy images of the nanostructures were obtained in a MERLIN FE-SEM microscope. Dynamic light scattering (DLS) measurements to characterize the nanoparticle diameters and ζ-potentials were measured in a Malvern Zetasizer Nano ZS apparatus. Confocal fluorescence microscopy images were acquired in a Leica TCS SP5 microscope using an air objective (20×, NA = 0.70) and an Ar laser (λ = 488 nm) as excitation source.
Synthetic procedures
The synthesis of the already reported precursors PDI1 and PDI2 is provided in the ESI.†O imide), 1652 (CO imide), 1593, 1578, 1436, 1403, 1337, 1250, 1177, 1108, 1066, 1017, 852, 809, 746, 725, 675, 645 cm−1. MS (MALDI, m/z): calculated for [C40H47B10N2O4]+: 729.5; found 729.9 (M − H).
Author contributions
R. R. – performed the synthesis of the compounds, the characterization of their optical properties and data analysis; S. S. – performed nanostructure preparation and characterization; L. P. – contributed to the synthetic and optical characterization work; J. H. – performed data analysis, supervised the project, prepared the original draft and acquired funding; R. N. – supervised the project, prepared the original draft and acquired funding. All authors have read and approved the final version.Data availability
The authors declare that all data underlying the results are available as part of the main article and the data supporting this article have been included as part of the ESI.† No additional source data are required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants PID2022-141293OB-I00 and PID2022-136892NB-I00 funded by MICIU/AEI/10.13039/501100011033 and by ERDF – “A way of making Europe”. R. Núñez acknowledges financial support from the State Investigation Agency, through the Severo Ochoa Programme for Centers of Excellence in R&D (CEX2023-001263-S). Financial support from the Generalitat de Catalunya (AGAUR) is also acknowledged through 2021 SGR 00064 and 2021 SGR 00442 projects. S. S. acknowledges financial support from DOC-FAM, the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement no 754397. L. P. thanks to the Universitat Autònoma de Barcelona for her predoctoral fellowship. J. H. is a Serra Húnter Fellow.References
- (a) R. N. Grimes, Carboranes, Academic Press, Amsterdam, 3rd edn, 2016 Search PubMed; (b) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS; (c) C. Viñas, Future Med. Chem., 2013, 5, 617–619 CrossRef.
- (a) C. E. Housecroft, Boron: Metallacarbaboranes, Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley, 2011 Search PubMed; (b) P. Farràs, E. J. Juárez-Pérez, M. Lepsik, R. Luque, R. Núñez and F. Teixidor, Chem. Soc. Rev., 2012, 41, 3445–3463 RSC.
- (a) R. Núñez, M. Tarrès, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef; (b) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC; (c) J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed; (d) A. Marfavi, P. Kavianpour and L. M. Rendina, Nat. Rev. Chem., 2022, 6, 486–504 CrossRef PubMed.
- (a) J. N. Hohman, P. Zhang, E. I. Morin, P. Han, M. Kim, A. R. Kurland, P. D. McClanahan, V. P. Balema and P. S. Weiss, ACS Nano, 2009, 3, 527–536 CrossRef CAS; (b) D. Brusselle, P. Bauduin, L. Girard, A. Zaulet, C. Viñas, F. Teixidor, I. Ly and O. Diat, Angew. Chem., Int. Ed., 2013, 52, 12114–12118 CrossRef CAS PubMed.
- (a) C. Viñas and E. Hey-Hawkins, Boron-Based Compounds: Potential and Emerging Applications in Medicine, Wiley, 2018 Search PubMed; (b) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS; (c) C. Viñas, R. Núñez, I. Bennour and F. Teixidor, Curr. Med. Chem., 2019, 26, 5036–5076 CrossRef.
- (a) R. Ziessel, G. Ulrich, J. H. Olivier, T. Bura and A. Sutter, Chem. Commun., 2010, 46, 7978–7980 RSC; (b) A. Harriman, M. A. H. Alamiry, J. P. Hagon, D. Hablot and R. Ziessel, Angew. Chem., Int. Ed., 2013, 52, 6611–6615 CrossRef CAS; (c) D. Hablot, R. Ziessel, M. A. H. Alamiry, E. Bahraidah and A. Harriman, Chem. Sci., 2013, 4, 444–453 RSC; (d) A. V. Safronov, N. I. Shlyakhtina, T. A. Everett, M. R. VanGordon, Y. V. Sevryugina, S. S. Jalisatgi and M. F. Hawthorne, Inorg. Chem., 2014, 53, 10045–10053 CrossRef CAS; (e) X. Wu, J. Guo, J. Zhao, Y. Che, D. Jia and Y. Chen, Dyes Pigm., 2018, 154, 44–51 CrossRef CAS; (f) C. Sang, G. Wang, Y.-C. Wei, Q. Jiang, K. Liu, M. Zhang, Y.-Y. Chen, X. Chang, F. Liu, S. Yin, P.-T. Chou and Y. Fang, CCS Chem., 2022, 4, 1949–1960 CrossRef.
- (a) K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 1418–1420 CrossRef CAS; (b) J. Cabrera-González, C. Viñas, M. Haukka, S. Bhattacharyya, J. Gierschner and R. Núñez, Chem. – Eur. J., 2016, 22, 13588–13598 CrossRef; (c) M. Chaari, Z. Kelemen, J. G. Planas, F. Teixidor, D. Choquesillo-Lazarte, A. Ben Salah, C. Viñas and R. Núñez, J. Mater. Chem. C, 2018, 6, 11336–11347 RSC; (d) Y. Yin, X. Li, S. Yan, H. Yan and C. Lu, Chem. – Asian J., 2018, 13, 3155–3159 CrossRef CAS; (e) X. Wei, M.-J. Zhu, Z. Cheng, M. Lee, H. Yan, C. Lu and J. J. Xu, Angew. Chem., Int. Ed., 2019, 58, 3162–3166 CrossRef CAS.
- (a) K. Liu, C. Shang, Z. Wang, Y. Qi, R. Miao, K. Liu, T. Liu and Y. Fang, Nat. Commun., 2018, 9, 1695 CrossRef PubMed; (b) K. Liu, Z. Wang, C. Shang, X. Li, H. Peng, R. Miao, L. Ding, J. Liu, T. Liu and Y. Fang, Adv. Mater. Technol., 2019, 4, 1800644 CrossRef CAS; (c) N. Ding, K. Liu, Y. Qi, C. Shang, X. Chang and Y. Fang, Sens. Actuators, B, 2021, 340, 129964 CrossRef CAS; (d) Z. Wang, X. Gou, S. K. Liu, X. Chang, G. Wang, W. Xu, S. Lin, T. Liu and Y. Fang, Angew. Chem., Int. Ed., 2022, 61, e202207619 CrossRef CAS PubMed.
- (a) K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS; (b) L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder and M. A. Fox, Chem. – Eur. J., 2012, 18, 8347–8357 CrossRef CAS; (c) K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064–4067 CrossRef CAS PubMed; (d) R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed; (e) D. Tu, P. Leong, Z. Li, R. Hu, C. Shi, K. Y. Zhang, H. Yan and Q. A. Zhao, Chem. Commun., 2016, 52, 12494–12497 RSC; (f) H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2017, 5, 10047–10054 RSC; (g) X. Wu, J. Guo, Y. Quan, W. Jia, D. Jia, Y. Chen and Z. Xie, J. Mater. Chem. C, 2018, 6, 4140–4149 RSC; (h) N. V. Nghia, S. Jana, S. Sujith, J. Y. Ryu, J. Lee, S. U. Lee and M. H. Lee, Angew. Chem., Int. Ed., 2018, 57, 12483–12488 CrossRef CAS; (i) K. L. Martin, J. N. Smith, E. R. Young and K. R. Carter, Macromolecules, 2019, 52, 7951–7960 CrossRef CAS; (j) Y.-J. Cho, S.-Y. Kim, J.-W. Lee, W.-S. Han, C. H. Kim, H.-J. Son and S. O. Kang, Chem. – Eur. J., 2019, 25, 8149–8156 CrossRef CAS; (k) M. S. Mun, C. H. Ryu, H. So, M. Kim, J. H. Lee, H. Hwang and K. M. Lee, J. Mater. Chem. C, 2020, 8, 16896–16906 RSC; (l) S. Sinha, Z. Kelemen, E. Hümpfer, I. Ratera, J.-P. Malval, J. P. Jurado, C. Viñas, F. Teixidor and R. Núñez, Chem. Commun., 2022, 58, 4016–4019 RSC; (m) H. Yang, H. Liu, Y. Shen, S.-T. Zhang, Q. Zhang, Q. Song, C. Lv, C. Zhang, B. Yang, Y. Ma and Y. Zhang, Angew. Chem., 2022, 134, e202115551 CrossRef.
- (a) K. C. Song, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2013, 42, 2351–2354 RSC; (b) N. V. Nghia, J. Oh, S. Sujith, J. Jung and M. H. Lee, Dalton Trans., 2018, 47, 17441–17449 RSC; (c) L. Parejo, M. Chaari, S. Santiago, G. Guirado, F. Teixidor, R. Núñez and J. Hernando, Chem. – Eur. J., 2021, 27, 270–280 CrossRef CAS.
- D. Tu, P. Leong, Z. Li, R. Hu, C. Shi, K. Y. Zhang, H. Yan and Q. Zhao, Chem. Commun., 2016, 52, 12494–12497 RSC.
- (a) M. Chaari, N. Gaztelumendi, J. Cabrera-González, P. Peixoto-Moledo, C. Viñas, E. Xochitiotzi-Flores, N. Farfán, A. Ben Salah, C. Nogués and R. Núñez, Bioconjugate Chem., 2018, 29, 1763–1773 CrossRef CAS; (b) J. Cabrera-González, B. M. Muñoz-Flores, C. Viñas, A. Chávez-Reyes, H. V. R. Dias, V. M. Jiménez-Pérez and R. Núñez, Chem. – Eur. J., 2018, 24, 5601–5612 CrossRef PubMed; (c) C. Bellomo, M. Chaari, J. Cabrera-González, M. Blangetti, C. Lombardi, A. Deagostino, C. Viñas, N. Gaztelumendi, C. Nogués, R. Núñez and C. Prandi, Chem. – Eur. J., 2018, 24, 15622–15630 CrossRef CAS PubMed.
- W. Ma, Y. Wang, Y. Xue, M. Wang, C. Lu, W. Guo, Y.-H. Liu, D. Shu, G. Shao, Q. Xu, D. Tu and H. Yan, Chem. Sci., 2024, 15, 4019–4030 RSC.
- (a) F. Würthner, Chem. Commun., 2004, 1564–1579 RSC; (b) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2015, 116, 962–1052 CrossRef; (c) A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- (a) C. Hua, K. Liu, Y. Wu, W. Xu, J. Zhang, Z. Wang, K. Liu and Y. Fang, ACS Appl. Mater. Interfaces, 2021, 13, 49500–49508 CrossRef CAS; (b) W. Feng, K. Liu, J. Zang, J. Xu, H. Peng, L. Ding, T. Liu and Y. Fang, J. Phys. Chem. B, 2021, 125, 11540–11547 CrossRef CAS PubMed; (c) W. Feng, Q. Jiang, Z. Wang, J. Zang, G. Wang, K. Liu, H. Peng, T. Liu, L. Ding and Y. Fang, J. Phys. Chem. B, 2022, 126, 4939–4947 CrossRef CAS PubMed.
- (a) T. L. Heying, J. W. Ager, S. L. Clark, D. J. Mangold, H. L. Goldstein, M. Hillman, R. J. Polak and J. W. Szymanski, Inorg. Chem., 1963, 2, 1089–1092 CrossRef CAS; (b) M. Thornton-Pett, M. A. Beckett and J. D. Kennedy, J. Chem. Soc., Dalton Trans., 1986, 303–308 RSC.
- (a) H. Langhals and A. Obermeier, Eur. J. Org. Chem., 2008, 6144–6151 CrossRef CAS; (b) D. Shanks, S. Preus, K. Qvortrup, T. Hassenkam, M. B. Nielsen and K. Kilså, New J. Chem., 2009, 33, 507–516 RSC.
- (a) K.-R. Wang, H.-W. An, F. Qian, Y.-Q. Wang, J.-C. Zhang and X.-L. Li, RSC Adv., 2013, 3, 23190–23196 RSC; (b) K. Sun, C. Xiao, C. Liu, W. Fu, Z. Wang and Z. Li, Langmuir, 2014, 30, 11040–11045 CrossRef CAS PubMed.
- A. Toppino, A. R. Genady, M. E. El-Zaria, J. Reeve, F. Mostofian, J. Kent and J. F. Valliant, Inorg. Chem., 2013, 52, 8743–8749 CrossRef CAS PubMed.
- N. Shin, S. Yu, J. H. Lee, H. Hwang and K. M. Lee, Organometallics, 2017, 36, 1522–1529 CrossRef CAS.
- L. D. Wescott and D. L. Mattern, J. Org. Chem., 2003, 68, 10058–10066 CrossRef CAS.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- (a) C. Viñas and E. Hey-Hawkins, Boron-Based Compounds: Potential and Emerging Applications in Medicine, Wiley, 2018 Search PubMed; (b) C. Viñas, R. Núñez, I. Bennour and F. Teixidor, Curr. Med. Chem., 2019, 26, 5036–5076 CrossRef.
- S. Sinha, Z. Kelemen, E. Hümpfner, I. Ratera, J. P. Marval, J. P. Jurado, C. Viñas, F. Teixidor and R. Núñez, Chem. Commun., 2022, 58, 4016–4019 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006.
- T. Kircher and H.-G. Löhmannsröben, Phys. Chem. Chem. Phys., 1999, 1, 3987–3992 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02477j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |