
N-Methylene-C-linked nitropyrazoles and 1,2,4-triazol-3-one: thermally stable energetic materials with reduced sensitivity†
Krishna
Pandey
,
Priyanka
Das
,
Meera
Khatri
and
Dheeraj
Kumar
 *
*
Energetic Materials Laboratory, Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee-247667, Uttarakhand, India. E-mail: dheeraj.kumar@cy.iitr.ac.in
First published on 8th October 2024
Abstract
Recently, there has been a surge in research focusing on triazolone-based energetic materials, propelled by their remarkable properties such as good detonation performance as well as acceptable thermal and physical stability. In this work, a novel combination of the triazolone framework with dinitropyrazoles has been attained using the N-methylene-C-linked approach. Different substituents (NH2, NO2, N3, OH) were utilized on the dinitropyrazole moiety to obtain neutral energetic compounds 3–5 and 8. Furthermore, the hydroxy derivative (compound 8) facilitates the formation of energetic salts 9–13 to fine-tune the overall properties further. All the novel compounds 3–13 were thoroughly characterized by IR, multinuclear NMR spectroscopy, high-resolution mass spectrometry (HRMS), and elemental analysis. Compounds 3, 4, 8, and 10 were further confirmed via15N NMR spectroscopy. The structure of compounds 3 and 8 was also confirmed through single-crystal X-ray diffraction studies. The majority of synthesized compounds showed good thermal stability as well as insensitivity toward external stimuli. Computational studies, including analyses such as Hirshfeld surface, non-covalent interaction, electrostatic potential surface, and HOMO–LUMO analysis, were conducted to examine the influence of substitution at the 4th position on the overall stability of compounds 3, 4, and 8.
1. Introduction
Energetic materials (EMs), including explosives, propellants, and pyrotechnics, represent an emerging class of substances pivotal in accelerating advancements across various sectors such as mining engineering, aerospace exploration, national defense, etc.1–4 In this context, the contemporary utilization of carbon-rich traditional energetic compounds like TNT (2,4,6-trinitrotoluene), RDX (1,3,5-trinitro-1,3,5-triazinane), and HNS [(E)-1,2-bis(2,4,6-trinitrophenyl)ethene] raises concerns because of their drawbacks, including inadequate overall performance (in terms of energy and stability), toxicity, and a lack of environmental compatibility.5–7 In modern days, research into nitrogen-rich azole-based energetic materials has intensified, driven by their remarkable attributes like superior performance, environmental safety, and high physical stability when exposed to external stimuli.8–13 Since various nitrogen-rich heterocycles possess distinct properties, blending them appropriately can aid in mitigating the contradiction between energy and safety.14–18Apart from its applications across sectors like pharmaceuticals, agrochemicals, and coordination chemistry, research on triazolone-based compounds is one of the hotspots in the energetic materials field.19–23 Incorporation of the triazolone ring not only boosts the aromaticity, stability, and overall performance of the entire molecule but also facilitates environmentally friendly combustion products (N2) due to its low carbon-to-hydrogen ratio, potentially reducing environmental damage.24 The milestone energetic compound 5-nitro-1,2,4-triazol-3-one (NTO), which is based on the triazolone framework, demonstrates distinguished properties such as high thermal and physical stability, high density, and the potential to replace traditional explosives RDX and TATB (2,4,6-triamino-1,3,5-trinitrobenzene). The remarkable performance shown by NTO has spurred researchers to delve deeper into triazolone-based energetic compounds, and various modification methods have been employed for this purpose.25,26 In 2015, Shreeve and coworkers investigated the C-functionalization of the triazolone ring with dinitro methyl explosophore, which, upon hydroxyammonium salt formation, resulted in an outstanding energetic molecule (I).27 Furthermore, triazolone-based energetic compounds, both symmetrically (II) and asymmetrically (III) connected, have been explored using different bridges like Azo (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) and –NH– bridging, respectively, with compound III showing promising properties for practical application.20,25 Also, the triazolone ring has been combined with other nitrogen-rich heterocycles (tetrazole, triazole, and furoxan) through C–C and C–N connections, resulting in the formation of energetic compounds (IV–VII), among which V and VII displayed good energetic performance with appropriate overall stability (Fig. 1a–c).25,26,28 The careful examination of the compounds mentioned above indicates that only a select few energetic compounds based on triazolone meet all the high standards and stringent requirements. Therefore, it is still necessary to investigate further strategies for the development of potential triazolone-based energetic compounds.
N–) and –NH– bridging, respectively, with compound III showing promising properties for practical application.20,25 Also, the triazolone ring has been combined with other nitrogen-rich heterocycles (tetrazole, triazole, and furoxan) through C–C and C–N connections, resulting in the formation of energetic compounds (IV–VII), among which V and VII displayed good energetic performance with appropriate overall stability (Fig. 1a–c).25,26,28 The careful examination of the compounds mentioned above indicates that only a select few energetic compounds based on triazolone meet all the high standards and stringent requirements. Therefore, it is still necessary to investigate further strategies for the development of potential triazolone-based energetic compounds.
 | ||
| Fig. 1 (a) Functionlised 1,2,4-triazol-3-one based compounds, (b) symmetrically and asymmetrically azo and NH bridged 1,2,4-triazol-3-one based compounds, (c) asymmetrically C–C and C–N connected 1,2,4-triazol-3-one based compounds, (d) this work. | ||
In the last few decades, employing the N-methylene-C linked approach to connect different energetic frameworks is one of the efficient techniques for achieving energetic compounds with optimal properties.29–34 The most prevalent skeletons used in this method are the dinitropyrazoles, owing to their intrinsic properties such as high density, favorable oxygen balance, and elevated heat of formation. Among the various dinitropyrazoles, 4-amino-3,5-dinitropyrazole (LLM-116) and 4-hydroxy-3,5-dinitropyrazole (OHDNP) are two attractive building blocks that are easily accessible through various synthetic routes and have been increasingly used for the synthesis of new energetic materials. The presence of alternating electron-withdrawing and electron-donating groups in these compounds lead to effective hydrogen bonding and impressive density (ρ = 1.90 g cm−3 for LLM-116, ρ = 1.86 g cm−3 for OHDNP), resulting in enhanced stability and energetic performance.11,31,35–37 In addition, the synthesis of precursors, i.e., N-acetonitrile functionalized derivative, used in the development of N-methylene-C linked energetic compounds, is safer, easily accessible, and straightforward in the case of dinitropyrazoles, particularly in LLM-116 and OHDNP compared to other nitrogen-rich rings. To date, many N-acetonitrile functionalized dinitropyrazole derivatives have been used, where the cyano functionality has been converted into tetrazole and amino/nitramino-1,2,4-oxadiazole, resulting in various N-methylene-C linked energetic compounds with excellent performance.29,38 Despite the remarkable properties of the individual rings, there are no such reports in the literature on dinitropyrazole-triazolone-based energetic compounds. Thus, to seek novel triazolone-based energetic derivatives, this study employs N-acetonitrile functionalized dinitropyrazoles as precursors to synthesize a range of N-methylene-C linked dinitropyrazole and 1,2,4-triazol-3-one derivatives 3–13. Different substituents (NH2, NO2, N3, OH) were used on the 3,5/3,4-dinitropyrazole to study their impact on energetic and stability properties. Moreover, the presence of a hydroxy functionality in compound 8 enables the formation of energetic salts, aiding in the further fine-tuning of physicochemical properties. Most of the compounds synthesized in this study demonstrated excellent physical (IS >40 J, FS >360 N) and thermal stability (Tdec = 200 °C–288 °C), and some of them exhibited detonation performance close to that of traditional explosives such as TATB.
2. Results and discussion
2.1. Synthesis
The synthetic pathways to energetic compounds 3–5 and 8–13 are shown in Schemes 1 and 2, respectively. N-Hydroxyacetimidamide derivatives of 4-amino-3,5-dinitropyrazole (compound 2) and 4-hydroxy-3,5-dinitropyrazole (compound 7) were synthesized from the corresponding N-acetonitrile derivatives (compound 1 and 6, respectively) by using the known literature procedures.29,31 Hydroxyacetimidamide group in 2 and 7 was converted to 4-amino-2,4-dihydro-3H-1,2,4-triazol-3-one by reaction with carbohydrazide in an acidic medium to give methylene-bridged compounds 3 and 8 in excellent yields of 90 and 80% respectively. The amino group of the pyrazole ring in 3 was oxidized in the presence of 30% H2O2 and H2SO4 to yield trinitropyrazole derivative, 4 in 85% yield. Further, the reaction of 4 with sodium azide in methanol resulted in 5-azido-3,4-dinitropyazole derivative, 5 in 78% yield. Because compound 8 possesses an acidic –OH site, the reaction of 8 with various nitrogen-rich bases in acetonitrile solution resulted in energetic salts 9–13. | ||
| Scheme 1 Synthesis of compounds 1–5. | ||
 | ||
| Scheme 2 Synthesis of compounds 6–13. | ||
2.2. Spectral analysis of compounds 3–5 and 8–13
All the energetic compounds described in this study were fully characterized via IR, NMR [1H, 13C{1H}, in a few cases, 15N NMR] spectra, mass spectra, and elemental analysis. The methylene bridge in neutral compounds 3–5 and 8 resonated in the range of 5.26–5.79 ppm in 1H NMR and 46.05–51.63 ppm in 13C NMR. Compared to the neutral compound 8 (5.73 ppm), the peaks corresponding to the methylene bridge in energetic salts, 9–13, were shifted to the upfield region and ranged between 5.25–5.24 ppm in 1H NMR. Meanwhile, in the 13C NMR of energetic salts, a slight downshift is observed compared to methylene carbon of 8 (49.23 ppm), with peaks appearing between 49.59 and 49.68 ppm. In 13C{1H} NMR, the peaks corresponding to the carbonyl carbon of triazolone rings in all compounds were observed in the range of 154.37–157.50 ppm. The peaks corresponding to the NH2 functional group of 4-amino-1,2,4-triazol-3-one moieties in 3 and 8–13 were observed between 5.79–6.79 ppm in proton NMR, whereas, peaks due to the NH groups of all 1,2,4-triazol-3-one derivatives appeared in the range of 10.16 to 11.95 ppm.Apart from 1H NMR and 13C NMR spectra, the structures of compounds 3, 4, 8, and 10 were also confirmed via15N NMR spectra, and the chemical shift measurements were done by taking nitromethane as an external standard. The spectra for compounds 3, 8, and 10 were recorded using DMSO-d6 solvent, while for compound 4, CD3CN was used (Fig. 2). The signals corresponding to chemically non-equivalent nitrogens were identified by comparing them with the analogous compounds previously reported in the literature.25,31 The peaks corresponding to the ring nitrogen atoms [(N6, N7, and N8) in case of 3, 4 and (N5, N6, and N7) in case of 8, 10] in the 1,2,4-triazol-3-one ring appeared in the upfield region, ranging from −190.47 to −243.81 ppm. The peaks corresponding to the NH2 functionality present on the 4-amino-1,2,4-triazol-3-one moieties in the case of compounds 3, 8, and 10 were found in the shielded region at −329.58 (N9), −329.58 (N8), and −329.68 (N8) ppm, respectively. The N-substituted nitrogen (N1) in the pyrazole ring was found in the shielded region (−119.37 to −138.26 ppm) compared to another nitrogen (N2) in the pyrazole ring (−74.07 to −83.68 ppm). The signals corresponding to the nitro groups on the pyrazole ring were observed in the range of −25.85 to −41.16 ppm. The peaks due to the amino group (N4) on the pyrazole ring in compound 3 and the peak corresponding to the hydroxylammonium (N9) cation in 10 were observed at −318.48 and −310.53 ppm, respectively.
 | ||
| Fig. 2 15N NMR spectra of compounds 3, 4, 8, and 10. | ||
2.3. Crystal structure
The structures of 3 and 8 were confirmed by single-crystal X-ray analysis. Suitable single crystals for X-ray diffraction analysis were obtained via slow evaporation of their saturated solution in water and acetonitrile/water mixtures at 298 K. The crystal structures and packing diagrams of 3 and 8 are given in Fig. 3 and 4, respectively. Detailed information about the crystallographic data of these crystals is provided in the ESI.† | ||
| Fig. 3 (a) Thermal ellipsoid plot (50%) and labelling scheme for 3. (b) Dihedral angle between the pyrazole and triazolone ring. (c) Ball and stick packing diagram showing planar stacking in the crystal of 3 viewed along axis. (d) Amplification of hydrogen bonding distribution in 3. | ||
 | ||
| Fig. 4 (a) Thermal ellipsoid plot (50%) and labelling scheme for 8. (b) Dihedral angle between the pyrazole and triazolone ring. (c) Ball and stick packing diagram showing planar stacking in the crystal of 8 viewed along b axis. (d) Amplification of hydrogen bonding distribution in 8. | ||
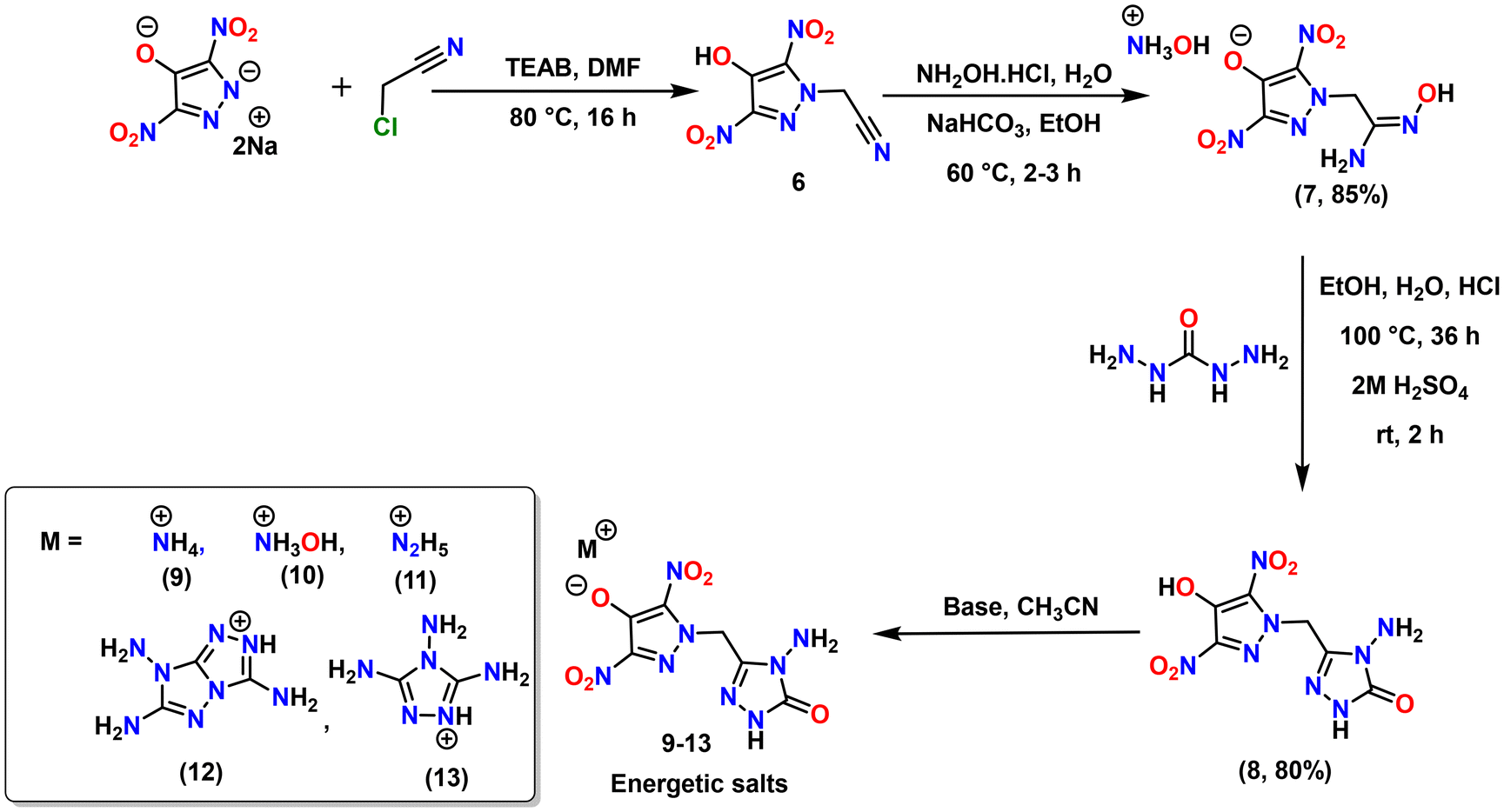
Compound 3 crystallizes in the orthorhombic space group Pbca with a crystal density of 1.81 g cm−3 (108 K) and eight molecules per unit cell (Z = 8). In the single crystal diagram of 3, the methylene bridge carbon shows slight distortion from the tetrahedral geometry (109.5°) with angles N(1)–C(4)–C(5) of 111.3(1)°. The C–C bond length [C(4)–C(5) = 1.49(2) Å] of the N-methylene-C bridges in 3 is slightly longer as compared to the C–N bond length [C(4)–N(1) = 1.46(1) Å]. The dihedral angle between the mean planes through the pyrazole and triazolone ring is 89.14(6)°. The pyrazole moiety is nearly planar because of the strong intra-molecular hydrogen bonding between amino and nitro groups, with the angle between the mean plane through pyrazole and amino/nitro groups being less than 2.16(1)°. Apart from this, the C–N bond length of the C–NH2 group is found to be slightly shorter [C(2)–N(4) = 1.34 (1) Å] compared to the C–N bond lengths of C–NO2 groups [C(1)–N(3) = 1.42(1) Å] (Fig. 3a and b). In the crystal packing of 3, the NH2 group of the triazolone ring has strong hydrogen bonding interactions with the carbonyl of another triazolone ring [N(9)⋯O(5)′ = 2.1(2) Å], and with NO2 group present on the 5th position of pyrazole ring [N(9)⋯O(4) = 2.70 (1) Å]. Triazolone ring nitrogen N(7) also has a strong hydrogen bonding interaction with the carbonyl oxygen of another molecule's triazolone ring [N(7)⋯O(5)′ = 1.86(2) Å]. Compound 3 has layered π–π interactions arranged in a zigzag pattern, giving it excellent thermal stability (Tdec = 288 °C) and insensitivity (IS >40 J) (Fig. 3c and d).
Compound 8 crystallizes in the monoclinic space group P21/n with a crystal density of 1.83 g cm−3 at 100 K and two molecules per unit cell (Z = 2). Similar to 3, the methylene bridge carbon in 8 shows slight distortion from the tetrahedral geometry (109.5°) with angle N(1)–C(4)–C(5) of 110.7(1). The C–C bond length [C(4)–C(5) = 1.49(2) Å] of the N-methylene-C bridges in 8 is somewhat longer as compared to the C–N bond length [C(4)–N(1) = 1.46(2) Å]. The dihedral angles between the mean planes through the pyrazole and triazolone ring in 8 [77.1(7)°]. Unlike 3, the nitro groups in 8 are slightly twisted out of the plane with respect to the pyrazole ring with dihedral angles of 17.2(1)° and 5.44(1)° (Fig. 4a and b). In the packing diagram of 8, the hydroxyl group on the pyrazole ring is participating in strong intra-molecular (with adjacent nitro groups) and inter-molecular hydrogen bonding with NH of second molecule's triazolone ring [O(3)⋯N(6)′ = 2.49(1) Å] as well as with the carbonyl of third molecule's triazolone ring [O(3)⋯O(6)′ = 1.78(3) Å]. There are many other strong intermolecular hydrogen bonding interactions between the carbonyl of one triazolone ring and NH of another molecule's triazolone ring [O(6)⋯N(6)′ = 2.00(1) Å], and between NH2 of one triazolone ring and N of another molecule's triazolone ring [N(8)⋯N(5)′ = 2.22(3) Å]. The abundance of hydrogen bonding in the packing diagram of compound 8 contributes to its high thermal stability (Tdec = 234 °C) and high stability (IS = 33 J) towards external stimuli (Fig. 4c and d).
2.4. Physicochemical and Energetic Properties
Table 1 displays the physicochemical and energetic properties of all the energetic compounds 3–5 and 8–13. Thermal stability and sensitivity measurements were performed to ensure safety during preparation, transportation, and reliable service. Thermal stability was evaluated based on their decomposition temperature, measured using differential scanning calorimetry (DSC) instrument at a heating rate of 5 °C min−1 in a nitrogen atmosphere. Compound 3 has excellent thermal stability, with decomposition temperatures of 288 °C, which is comparable to TNT (Tdec = 295 °C) and better than RDX (Tdec = 204 °C). Meanwhile, other compounds except the azido derivative, 5 (Tdec = 147 °C), display good thermal stability between 174 °C and 234 °C. The impact and friction sensitivity for all the energetic compounds were measured using a standard BAM fall hammer and a BAM friction tester, respectively. Among the neutral compounds, 3 exhibit insensitivity, with an impact sensitivity of >40 J and friction sensitivity of >360 N. In contrast, 4 (IS = 31 J, FS = 360 N), 5 (IS = 26 J, FS = 360 N), and 8 (IS = 33 J, FS = 360 N) demonstrate moderate sensitivities towards impact and friction. Among energetic salts 9–13, all except 10 (IS = 13 J, FS = 240 N) were determined to be insensitive to impact and friction.| Compd. | ρ (g cm−3) | Dv (m s−1) | P (GPa) | ΔHfd (kJ mol−1) | T dec (°C) | ISf (J) | FSg (N) |
|---|---|---|---|---|---|---|---|
| a Density measured using gas pycnometer at 25 °C. b Calculated detonation velocity. c Calculated detonation pressure. d Heat of formation. e Temperature of decomposition (onset). f Impact sensitivity. g Friction sensitivity. | |||||||
| 3 | 1.76 | 7665 | 22 | 99 | 288 | >40 | >360 |
| 4 | 1.79 | 7980 | 26 | 98 | 174 | 31 | 360 |
| 5 | 1.75 | 7803 | 24 | 418 | 147 | 26 | 360 |
| 8 | 1.78 | 7672 | 23 | −43 | 234 | 33 | 360 |
| 9 | 1.74 | 7815 | 24 | 2 | 213 | >40 | >360 |
| 10 | 1.76 | 8083 | 26 | 50 | 181 | 13 | 240 |
| 11 | 1.76 | 8143 | 26 | 150 | 205 | >40 | >360 |
| 12 | 1.71 | 7668 | 22 | 509 | 200 | >40 | >360 |
| 13 | 1.72 | 7740 | 22 | 286 | 202 | >40 | >360 |
| TNT | 1.65 | 6824 | 19.4 | −59.4 | 295 | 15 | >353 |
| RDX | 1.80 | 8795 | 34.9 | 92.6 | 204 | 7.5 | 120 |
| TATB | 1.93 | 8179 | 30.5 | −139.7 | 350 | 50 | >360 |
The density of all compounds was measured using a helium gas pycnometer at room temperature, with values ranging from 1.71 to 1.79 g cm−3. All the compounds were denser than TNT (ρ = 1.65 g cm−3), while the density of 4 (ρ = 1.79 g cm−3) was found to be comparable to that of RDX (ρ = 1.80 g cm−3). The heats of formation were calculated using the Gaussian 09 program suite based on isodesmic reactions. All the prepared energetic compounds except 8 (ΔHf = −43.47 kJ mol−1) show positive heats of formation ranging between 1.97 to 509.46 kJ mol−1. Compared to neutral compound 8, enhancements in heats of formation are observed in all energetic salts 9–13, with the 7H-[1,2,4]triazolo[4,3-b][1,2,4]triazole-3,6,7-triamine (TATOT)+ salt, i.e., 12 (ΔHf = 509.46 kJ mol−1), exhibiting the highest heat of formation. Using heats of formation and density values, the detonation properties were calculated using EXPLO5 (version 7.01.01) software.39 The detonation velocity ranges from 7665 to 8143 m s−1, while the detonation pressure ranges from 21.74 to 26.40 GPa. All the compounds were found to be more energetic than TNT, whereas energetic properties of 4 (Dv = 7980 m s−1 and P = 26.40 GPa), 10 (Dv = 8083 m s−1 and P = 26.04 GPa), and 11 (Dv = 8143 m s−1 and P = 25.53 GPa) were found to be comparable with well-known explosive TATB (Dv = 8179 m s−1 and P = 30.50 GPa).
2.5. Hirsfeld surface analysis and non-covalent interaction (NCI) analysis, electrostatic potential (ESP) and HOMO–LUMO analysis
To acquire a deeper understanding of the structure–property relationship, we did the Hirshfeld surfaces analysis of compounds 3 and 8 using CrystalExplorer17 software, and their Hirshfeld surfaces, 2D fingerprint plots, and the populations contacts are shown in Fig. 5.40 The red spots on Hirshfeld surfaces indicate high close-contact interactions such as O⋯H or N⋯H hydrogen bonding interactions. The high thermal and physical stabilities of compounds 3 and 8 are primarily due to the significant contribution of O⋯H and N⋯H interactions, accounting for more than 48% of the overall interactions (54.8% for 3 and 48.7% for 8). This significant abundance of hydrogen bonding interactions results from the presence of amino and hydroxy functionalities in compounds 3 and 8, respectively.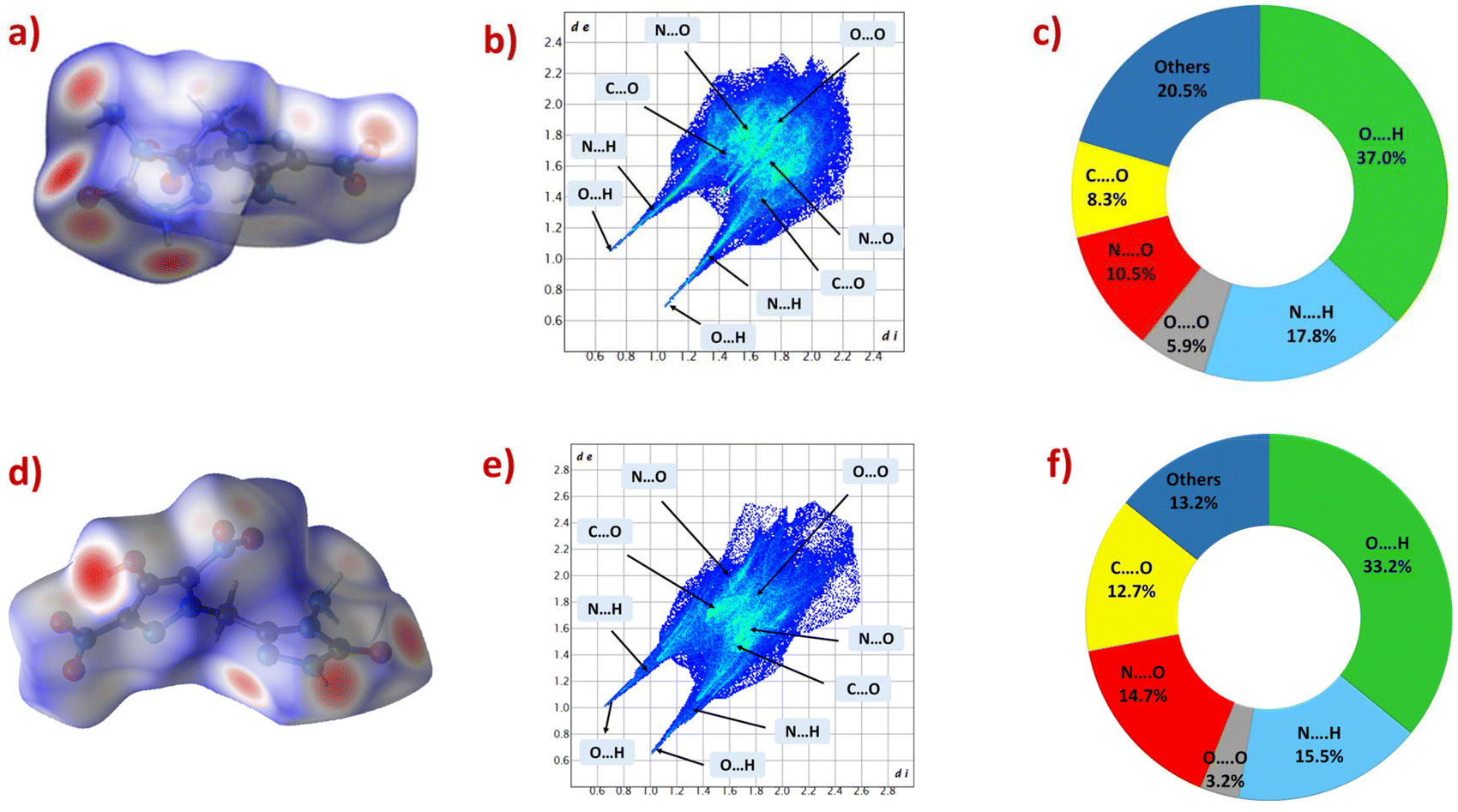 | ||
| Fig. 5 Hirshfeld surfaces (a and d), 2D fingerprint plots (b and e), and populations of molecular interactions (c and f) for 3 and 8, respectively. | ||
Further, electrostatic potential (ESP) and non-covalent interaction (NCI) analysis was done using the Multiwfn software and visualized through VMD software.41,42 The NCI plots of compounds 3 and 8 are shown in Fig. 6, where blue-colored isosurfaces indicate hydrogen bonding interactions and green-colored isosurfaces indicate π–π interactions. In Fig. 6a, a clear depiction of intramolecular hydrogen bonding can be seen between the amino group and two nitro groups of the pyrazole ring. Whereas, in Fig. 6b, hydrogen of hydroxy functionality shows intramolecular hydrogen bonding with one of the nitro groups. Due to this distinction, amino-functionalized 3 may possess better physical and thermal stability than hydroxy-functionalized 8. The superior stability of compound 3 may also be explained by comparing the intensity of the denser green-colored isosurfaces with those of compound 8.
 | ||
| Fig. 6 Non-covalent interaction (NCI) analysis for 3 (a) and 8 (b), respectively. | ||
The ESP-mapped diagrams of synthesized neutral compounds 3, 4, and 8 are shown in Fig. 7(a–c). The geometric optimization of these structures was first realized using the Gaussian 09 program at B3PW91/6-31G(d,p) level. As expected and depicted in Fig. 7a and c, the amino and hydroxy functionalities in compounds 3 and 8 contribute to the even distribution of the electrostatic potential due to the presence of alternating electron withdrawing and donating groups. Apart from that, a more significant electropositive area (red region on ESP surface) indicates higher sensitivity of the energetic compounds. The order of highest electropositive value of the 3,5-dinitropyrazole-based compounds follows 4 (63.40 kJ mol−1) > 8 (54.83 kJ mol−1) > 3 (53.88 kJ mol−1), which supports the order of their sensitivity towards impact and friction.
 | ||
| Fig. 7 Electrostatic potential analysis (ESP) for 3 (a), 4 (b), and 8 (c), respectively; HOMO/LUMO energy levels and energy gaps in 3 (d), 4 (e), and 8 (f), respectively. | ||
To correlate the molecular stability of compounds 3, 4, and 8, the difference (ΔE) between the energies of their highest-occupied molecular orbital (HOMO) and the lowest-unoccupied molecular orbital (LUMO) was computed and shown in Fig. 7(d–f). It is proposed that as ΔE decreases, the molecular stability of the compound decreases because it facilitates electronic excitations, bond cleavage, and decomposition more easily. It is observed that 3 (9.828 eV) and 8 (9.997 eV) show higher energy differences compared to 4 (9.184 eV), indicating better overall stability (physical and thermal stability), which is consistent with the experimental results.
3. Conclusions
A family of N-methylene-C bridged 3,4/3,5-dinitropyrazoles and 1,2,4-triazol-3-ones was synthesized using N-acetonitrile derivatives of nitropyrazoles. Different substituents (NH2, NO2, N3, OH) were employed on the 3,5/3,4-dinitropyrazole moiety to study their effect on energetic and stability properties. Apart from azido (compound 5, Tdec = 147 °C) and trinitropyrazole (compound 4, Tdec = 174 °C) derivatives, other compounds exhibit good thermal stability with decomposition temperature in the range of 180–288 °C. All the compounds were found to be more energetic than traditional explosives TNT, whereas the energetic performance of 4, 10, and 11 was comparable with the well-known secondary explosive TATB. The structure–property relationship was studied for neutral compounds 3, 4, and 8 using the Hirshfeld surface, non-covalent interaction, electrostatic potential surface, and HOMO–LUMO analysis.4. Experimental section
Caution! Although no accidents were observed during the synthesis, handling, and characterization, all the compounds reported in this work are potentially explosive materials and may explode unpredictably under certain conditions. All the compounds must be synthesized only on a small scale (<200 mg). Any mechanical actions involving grinding or scratching must be avoided. In addition, all the manipulations must be strictly carried out in a fume hood behind a polycarbonate safety shield. Eye protection, a face shield, and leather gloves must be worn while handling these compounds.4.1. General methods
All reagents were purchased from Aldrich, TCI, or GLR Innovations in analytical grade and were used as supplied if not stated otherwise. 1H, 13C NMR, and 15N spectra were recorded using a 500 MHz (JEOL ECZ500R/S1) NMR spectrometer operating at 500, 125, and 50.69 MHz, respectively. As external standards, chemical shifts in the 1H and 13C NMR spectra are reported relative to Me4Si and 15N NMR to ammonia. The melting and decomposition temperatures were obtained on a differential scanning calorimeter (S11 6300 EXSTAR) at a scan rate of 5 °C min−1. IR spectra were recorded using KBr pellets for solids on a PerkinElmer FT-IR spectrometer. Densities were measured at room temperature by employing an Anton Par Ultrapyc 5000 gas pycnometer. High-resolution mass spectra quadrupole time-of-flight (HRMS-QTOF) were obtained in ESI mode. Elemental analyses were carried out on an elementar model Vario-EI-III. The impact and friction sensitivity measurements were made using a standard BAM fall hammer (OZM) and a BAM Friction tester (FST ProEX).Suitable crystals of 3 and 8 were obtained by slow evaporation of their saturated solutions in acetonitrile/water and water. The single-crystal diffraction studies were carried out on a Bruker SMART APEX CCD diffractometer with a Mo Kα (λ = 0.710 73 Å) sealed tube. All crystal structures were solved by direct methods. The program SAINT (version 6.22) was used to integrate of the intensity of reflections and scaling. The program SADABS was used for absorption correction. The crystal structures were solved and refined using the SHELXTL (version 6.12) package.43 All hydrogen atoms were included in idealized positions, and a riding model was used. Non-hydrogen atoms were refined with anisotropic displacement parameters.
The heats of formation for energetic compounds 3–5 and 8–13 were obtained using isodesmic reactions (ESI†). The geometric optimization and frequency analyses of the structures were performed by using B3PW91/6-31G(d,p) level without any symmetry restriction as implemented in the Gaussian 09 program.44 All the optimized derivatives are confirmed to be true local energy minima on the potential energy surface without any imaginary frequencies. All calculated gas-phase enthalpies for covalent materials are converted to solid-phase values by subtracting the empirical heat of sublimation obtained based on the molecular surface properties.45 For salts 9–13, the solid phase heats of formation were calculated based on the Born–Haber energy cycle.46,47
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge financial support from Science and Engineering Research Board (SERB/EEQ/2019/000540), New Delhi. We thank DST-FIST [SR/FST/CS-II/2018/72(C)] and IITR for funding the 500 MHz NMR and single-crystal X-ray diffraction facilities at IIT Roorkee. K. P. and P. D. thank DST-Inspire and MHRD for research fellowships, respectively.References
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, Berlin, 2019 Search PubMed.
- D. Kumar and A. J. Elias, Resonance, 2019, 24, 1253–1271 CrossRef.
- K. Pandey, A. Tiwari, J. Singh, P. Bhatia, P. Das, D. Kumar and J. M. Shreeve, Org. Lett., 2024, 26, 1952–1958 CrossRef CAS.
- P. Bhatia, K. Pandey and D. Kumar, Chem. – Asian J., 2024, e202400481 CrossRef CAS.
- I. V. Kuchurov, M. N. Zharkov, L. L. Fershtat, N. N. Makhova and S. G. Zlotin, ChemSusChem, 2017, 10, 3914–3946 CrossRef CAS.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2017, 5, 10437–10441 RSC.
- J. Li, Y. Liu, W. Ma, T. Fei, C. He and S. Pang, Nat. Commun., 2022, 13, 5697 CrossRef CAS.
- T. Zhu, C. Lei, C. Li, H. Yang, C. Xiao and G. Cheng, J. Mater. Chem. A, 2024, 12, 4678–4683 RSC.
- J. Zhang, J. Zhang, J. Singh, W. Wu, R. J. Staples, J. Zhang and J. M. Shreeve, J. Mater. Chem. A, 2024, 12, 5918–5923 RSC.
- P. Bhatia, K. Pandey, P. Das and D. Kumar, Chem. Commun., 2023, 59, 14110–14113 RSC.
- K. Pandey, P. Das, P. Bhatia, V. D. Ghule and D. Kumar, Cryst. Growth Des., 2024, 24, 6790–6799 CrossRef CAS.
- K. Pandey, P. Bhatia, K. Mohammad, V. D. Ghule and D. Kumar, Org. Biomol. Chem., 2023, 21, 6604–6616 RSC.
- X. Yu, J. Tang, C. Lei, C. Xue, G. Cheng, C. Xiao and H. Yang, J. Mater. Chem. A, 2024, 12, 19513–19520 RSC.
- D. Kumar, C. He, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2016, 4, 9220–9228 RSC.
- D. Kumar, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2016, 4, 9931–9940 RSC.
- J. Singh, R. J. Staples, M. Fabin and J. M. Shreeve, J. Mater. Chem. A, 2024, 12, 17501–17509 RSC.
- K. Pandey, P. Bhatia, P. Dolui, V. D. Ghule and D. Kumar, Asian J. Org. Chem., 2022, 11, e202200543 CrossRef CAS.
- P. Bhatia, P. Jangra, V. D. Ghule and D. Kumar, J. Heterocycl. Chem., 2024, 61, 1299–1305 CrossRef CAS.
- L. Wang, Y. Ma, X. H. Liu, Y. H. Li, H. Bin Song and Z. M. Li, Chem. Biol. Drug Des., 2009, 73, 674–681 CrossRef CAS.
- J. Zhu, S. Jin, L. Wan, C. Zhang, L. Li, S. Chen and Q. Shu, Dalton Trans., 2016, 45, 3590–3598 RSC.
- J. Zhang, J. Zhang, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2018, 6, 22705–22712 RSC.
- E. Feng, J. Tang, C. Li, T. Zhu, H. Yang and G. Cheng, Energy Mater. Front., 2024 DOI:10.1016/j.enmf.2024.08.006.
- G. Zhao, D. Kumar, P. Yin, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, Org. Lett., 2019, 21, 1073–7077 CrossRef CAS.
- G. Zhao, D. Kumar, L. Hu and J. M. Shreeve, ACS Appl. Energy Mater., 2019, 2, 6919–6923 CrossRef CAS.
- Y. Liu, Z. Zeng, W. Huang, J. M. Shreeve and Y. Tang, J. Org. Chem., 2022, 87, 4226–4231 CrossRef CAS.
- P. Yang, G. Cheng, J. Tang, W. Hu, G. Zhang and H. Yang, Cryst. Growth Des., 2023, 23, 207–215 CrossRef CAS.
- J. Zhang, Q. Zhang, T. T. Vo, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 1697–1704 CrossRef CAS.
- N. Fischer, K. Hüll, T. M. Klapötke and J. Stierstorfer, J. Heterocycl. Chem., 2014, 51, 85–95 CrossRef CAS.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2017, 23, 7876–7881 CrossRef CAS.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2017, 5, 16767–16775 RSC.
- P. Bhatia, K. Pandey, B. Avasthi, P. Das, V. D. Ghule and D. Kumar, J. Org. Chem., 2023, 88, 15085–15096 CrossRef CAS.
- P. Das, P. Bhatia, K. Pandey and D. Kumar, Mater. Adv., 2024, 5, 171–182 RSC.
- P. Bhatia, V. D. Ghule and D. Kumar, Energy Mater. Front., 2024, 5, 105–111 CrossRef CAS.
- P. Bhatia, P. S. Priya, P. Das and D. Kumar, Energy Mater. Front., 2024, 5, 8–16 CrossRef CAS.
- M. Born, K. Karaghiosoff, T. M. Klapötke and M. Voggenreiter, ChemPlusChem, 2022, 87, e202200049 CrossRef CAS.
- H. Fan, J. Tang, H. Yang and G. Cheng, Cryst. Growth Des., 2024, 24, 6292–6299 CrossRef CAS.
- Y. Liu, J. Li, X. Zhang, C. He and S. Pang, Mater. Chem. Front., 2023, 7, 1046–1057 RSC.
- F. Yang, P. Zhang, X. Zhou, Q. Lin, P. Wang and M. Lu, Cryst. Growth Des., 2020, 20, 3737–3746 CrossRef CAS.
- M. Sucéska, EXPLO5 6.01, Brodarski Institute, Zagreb, Croatia, 2013 Search PubMed.
- M. J. Turner, J. J. Mckinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University ofWestern Australia, 2017 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- SMART: Bruker Molecular Analysis Research Tools, version 5.618, Bruker Analytical X-ray Systems, Madison, WI, 2000 Search PubMed.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem. A, 2006, 110, 1005–1013 CrossRef CAS.
- H. D. B. Jenkins, D. Tudela and L. Glasser, Inorg. Chem., 2002, 41, 2364–2367 CrossRef CAS.
- V. D. Ghule, J. Phys. Chem. C, 2013, 117, 16840–16849 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2381172 and 2381173. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02494j |
| This journal is © The Royal Society of Chemistry 2024 |