
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModification of cobalt bis(dicarbollide) ions with nitrile groups on carbon atoms: a unique low-temperature skeletal rearrangement due to the specific electron-donor character of CN substitution†
Ece Zeynep
Tüzün
 ab,
Drahomír
Hnyk
a,
Dmytro
Bavol
a,
Miroslava
Litecká
a,
Jindřich
Fanfrlík
c and
Bohumír
Grűner
*a
ab,
Drahomír
Hnyk
a,
Dmytro
Bavol
a,
Miroslava
Litecká
a,
Jindřich
Fanfrlík
c and
Bohumír
Grűner
*a
aInstitute of Inorganic Chemistry, Czech Academy of Sciences, 25068 Řež, Czech Republic. E-mail: gruner@iic.cas.cz
bDpt. of Inorganic Chemistry, Charles University, Hlavova 2030, 12843 Prague 2, Czech Republic
cInstitute of Organic Chemistry and Biochemistry, Czech Adademy of Sciences, 166 10, Prague 6, Czech Republic
First published on 1st November 2024
Abstract
Herein, we report on the synthesis and stereochemistry of mono- and isomeric dinitrile derivatives of [(1,2-C2B9H11)2-3,3′-Co]− ions. The shape and electronic properties of CN groups can apparently surmount the strain associated with the substitution of two vicinal carbon positions. Owing to electron donation to the cage, this results in a substituent-induced rearrangement of one of the carbon atoms to the upper pentagonal ring. The molecular structure of this isomer was confirmed using sc-XRD and DFT chemical computations.
A bis-icosahedral cobalt bis(1,2-dicarbollide)(1-) anion,1–3 [(1,2-C2B9H11)2-3,3′-Co]− (1−), is characterized by fully occupied orbitals of the metal with 18 e−, aromaticity,4 very robust chemical, thermal and radiation stability, low-nucleophilic properties,5 and its behaviour like stealth amphiphile6 or superchaotrope7 in aqueous solutions. It has attracted considerable attention as an anionic scaffold for various emerging applications.
Considering medicinal chemistry8 on which we currently focus, the parent ion has been proven to have relatively low toxicity and can easily penetrate cells.9,10 The ion forms supramolecular assemblies and11 vesicles,12 interacts strongly with proteins,13 can serve as a delivery agent for BNCT,14,15 transport cationic peptides through cell membranes,7 enhance the antibiotic action of some drugs16 and can specifically inhibit the HIV-Protease enzyme with an IC50 value of 1 μmol.9
The anion 1− can be readily modified on the B(8) vertex through electrophile-induced nucleophilic substitution (EINS) pathways.3,17 With this advantage, the nucleophile is a cyclic ether that produces species containing an oxonium atom.17 Such rings can be easily cleaved with a broad range of nucleophilic reagents.18 This method has shown high potential in the medicinal chemistry of ion 1− and for the development of polymers,19 dendrimers20,21 and materials,22 although at the expense of limited possibilities of modifying the length of the linker and stereochemistry.1
Considering the experimental difficulties in the initial stages of development, functionalization of the ion 1− on carbon atoms has only recently emerged as a versatile method for creating anionic building blocks applicable in the rational design of biologically active boron-cluster compounds for various therapeutic targets, such as enzyme inhibitors, antibacterials, and cytotoxic species, or in the construction of advanced molecular architectures and materials.2,8 The use of the latter approach allows for the introduction of different dipole moments and possibilities of fine-tuning the proton affinity of protoble groups (OH, NH3, COOH, etc.) as well as the distance between the functional group and the cage by a one atom increment. These may be the critical factors in attaining optimal interactions with the active site of enzymes such as carbonic anhydrase IX.23 Considering disubstitutions, carbon functionalization offers, in principle, three isomers with different space orientations of groups, denoted previously as chiral racemic-, and symmetric meso- and vicinal-forms. Racemic isomers, which are characterized by the largest separation between substituents, are usually the most abundant species resulting from reactions of the lithiated anion 1− with electrophiles. In contrast, vicinal isomers have not been isolated from direct reactions yet, although their presence in low ratios has been observed through HPLC in several reaction mixtures.2
Compounds substituted with a nitrile group belong to the most valuable reagents and building blocks in organic chemistry because of their smooth interconversions to carboxylates, amines, and heterocycles via cycloaddition and other reactions. Considering synthesis, it is known that the direct introduction of functional groups on the C-atoms of ion 1− (with no linker) may present certain difficulties connected with shielding of the reaction sites due to the steric and electronic effects of the bulky anionic cage. Also, the physicochemical properties of the resulting compounds can be significantly altered, as was recently exemplified by the substitution with –NH2 and –N3 groups.24 Thus, although nitriles attached via an alkane linker could be easily prepared starting from the respective alkyl methylsulfonyl esters,25 synthesis of the first member of the series was unsuccessful. Indeed, our previous attempts to prepare the target nitriles using cyanogen bromide or trimethylsilyl cyanide resulted in high-yield bromination25 or silylation of the cage carbon(s) instead, due to the ambiguous behaviour of the bromine or silylium ion under the applied reaction conditions. However, we show here that low-temperature reaction of the lithiated cage with toluene sulfonyl cyanide (TsCN) led to the smooth formation of the respective nitriles with the general formulae [(1-CN-1,2-C2B9H10)(1′,2′-C2B9H11)-3,3′-Co(III)]− (2−) and [(CN-C2B9H10)2-Co(III)]− (3−). As evidenced by HPLC analyses and mass spectroscopy of the reaction mixtures, the reaction did not go to full completion and produced equilibrium mixtures of the parent anion with the corresponding nitriles, even when high ratios of ion 1− to the base and TsCN of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.0 were used. Starting from a ratio of 1:1.50, also several isomeric trisubstituted products were formed in low quantities; however, these could not be isolated in sufficient purity due to their limited hydrolytic stability during RP chromatography in an aqueous mobile phase, which was essential for separation of the mixture of anionic products.
:3.0 were used. Starting from a ratio of 1:1.50, also several isomeric trisubstituted products were formed in low quantities; however, these could not be isolated in sufficient purity due to their limited hydrolytic stability during RP chromatography in an aqueous mobile phase, which was essential for separation of the mixture of anionic products.
The main products were isolated using extraction into diethyl ether followed by separation with flash chromatography on C18 RP Bűchi columns. The monosubstituted compound 2− was isolated as the main product from the reactions carried out in ratios up to 1:1.75 and was fully characterized by a combination of HPLC, HRMS, and NMR techniques (see in ESI†). Concerning the disubstituted compounds 3−, the presence of several isomers was observed in the reaction mixture. However, only two main products, stable towards air and moisture, could be isolated. The first one, characterized using spectral methods and XRD corresponded to the red rac-isomer with the formula [(1,1′-CN-1,2-C2B9H10)2-3,3′-Co(III)]− (3a−) (see Fig. 1 and Scheme 1), which was apparently the most thermodynamically stable species out of the three “conventional” isomers. This formed as the main product when the cooling and stirring of the reaction mixture to −82 °C was continued for at least for 3 h. This compound was separated and characterized using analytical HPLC, HRMS, NMR, and sc-XRD (see Fig. 1 and the ESI†). The ratio of the second species increased upon faster warming up the reaction mixture and prevailed when the cooling bath was completely removed after 30 min after the addition of TsCN. However, the patterns observed in the 11B NMR spectrum of this isomer did not match with the expected meso-isomer. Instead, the 11B NMR spectrum contained 13 distinguishable boron signals, of which one part corresponding to the unsubstituted dicarbollide ligand matched closely with the pattern for the parent ion 1−, showing boron signals with intensities of 1:1:2:2:2:1. The pattern of the second ligand was completely asymmetric, corresponding to 9 signals of intensity 1, among which, however, five peaks overlapped by coincidence. Two distinguishable CH signals could be seen in the 1H NMR spectrum; however, only one signal could be observed for the CH and Ccage carbons in the 13C NMR analysis and two signals for CN groups (see the ESI†).
 | ||
| Fig. 1 Crystal structure of the anion rac-3a−; the Me4N+ cation is omitted for clarity (ORTEP view, ellipsoids are drawn with 40% probability). The structural parameters and selected interatomic distances and angles are presented and discussed in the Crystallographic part in the ESI.† | ||
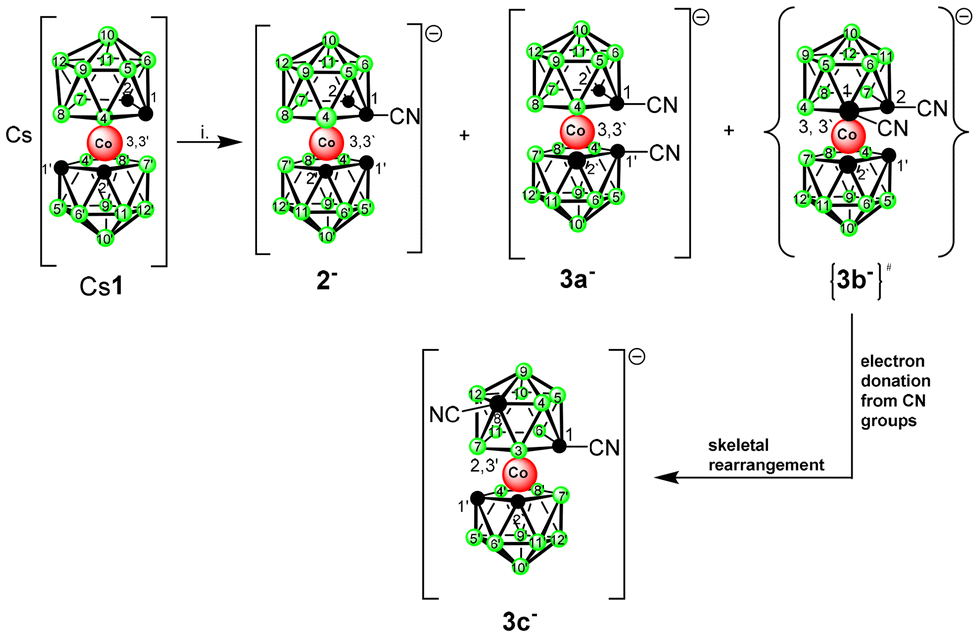 | ||
| Scheme 1 Synthesis of nitriles; conditions: i. BuLi, DME, −82 °C, then TsCN in DME at −82 °C, ratios of 1−:reagents, from 1:1.5 to 1:3.0, 12 h stirring from 30 min to 3 h, −82 °C to 20 °C during 12 h, extraction, RP chromatography. #Assumed intermediate isomer. | ||
The structure of this species was unambiguously confirmed by X-ray diffraction analysis performed on crystals isolated from two independent runs, for certainty (Fig. 2). Curiously, the sc-XRD structure showed the migration of one of the substituted skeletal carbon atoms to the upper pentagon of the dicarbollide ion, which was non-adjacent to Co(III). Only one rearranged isomer corresponding to the formula [(1,8-CN2-1,2-C2B9H9)(1′,2′-C2B9H11)-2,3′-Co(III)]− (3c−) could be seen; therefore the rearrangement was highly stereospecific. According to HPLC analysis, the rearrangement proceeded during warming up the reaction mixture from −82 °C up to room temperature. Concerning the ionic full-sandwich metal bis(dicarbollides), such a low-temperature pathway has no precedent. Only the 1,7-isomer of cobalt bis(dicarbollide) is known, in which both carbons remain adjacent to the Co(III) atom. This is typically prepared by cobalt insertion into the [7,9-C2B9H12]− ion and eventually is also accessible via thermal rearrangement in a high-temperature interval starting at 346 °C.3,26 The closely related thermal isomerization of the mixed [3-CpCo-1,2-C2B9H11] sandwich was studied in more detail in the range 400–700 °C. Depending on the temperature, variable ratios of several isomers were formed, inclusive of the 1,8-species.27,28 Therefore, the introduction of two linear-shaped and electron-rich nitrile groups on one ligand of the cage had unexpectedly important consequences. Our considerations, supported by theoretical computations performed at the SMD(Et2O)/BP86/AE1//BP86/AE1 level of theory, assume that the nucleophilic reaction with CN as the electrophile occurs initially at two adjacent carbon atoms, forming a kinetic transient vicinal-isomer with the assumed formulation {(1,2-CN2-1,2-C2B9H9)(1′,2′-C2B9H11)-3,3′-Co(III)}− {3b−} (Scheme 1 and Fig. 3). The spontaneous rearrangement proceeded relatively rapidly; therefore, the presence of the transient isomer could not be reliably identified using HPLC or NMR methods. Although the CN group is an electron-withdrawing substituent towards 2D aromatics, it acts in a different way with respect to the current substitution on the CoC2B9 “icosahedron”. Namely, the IBO charge on carbons bearing the CN groups was computed to be −0.22, a number which is not different from that detected in the parent ion by the same computational approach. The IBO HOMO/LUMO gap was computed to be only 0.11 eV, which indicates an extreme instability of the starting 1,2-isomer of the dinitrile derivative, where the latter seemed to exist below time scaling of the NMR approach. Note that such a carbon charge in the case of its azido-substitution was 0.00, which was ascribed to the electron-withdrawing abilities of the N3 group.24 On that basis, transfer of electron density from the CN groups to the boron cage is apparent and causes an extreme elongation of the C(1)–C(2) nearest-neighbour separation from 1.68 Å in Cs+ salt of the parent ion29 to 2.27 Å in the BP86/AE1 space of {3b−}. Such an elongation of this distance beyond bonding limits is known from the chemistry of mixed sandwich complexes of aryl-, alkyl-, and alkane-substituted dicarbollide ligands and fragments containing transition metals of the second and third row, such as RuCp, RhCOD, and IrCOD, where the metal central unit is electron-poor, containing 16 valence electrons only, which is compensated with semi-open structures called iso-closo-, hyper-closo-, or pseudo-closo.30–33 Interestingly, exceptionally long C–C nearest-neighbour separations were observed in C,C-disubstituted-o-carboranes; the longest for NH2 groups reached almost 2 Å.34–36
 | ||
| Fig. 2 Crystal structure of the anion 3c−; the cation is omitted for clarity (ORTEP view, ellipsoids are drawn with 40% probability). The structural parameters and selected interatomic distances and angles are given and discussed in the Crystallographic part in the ESI.† | ||
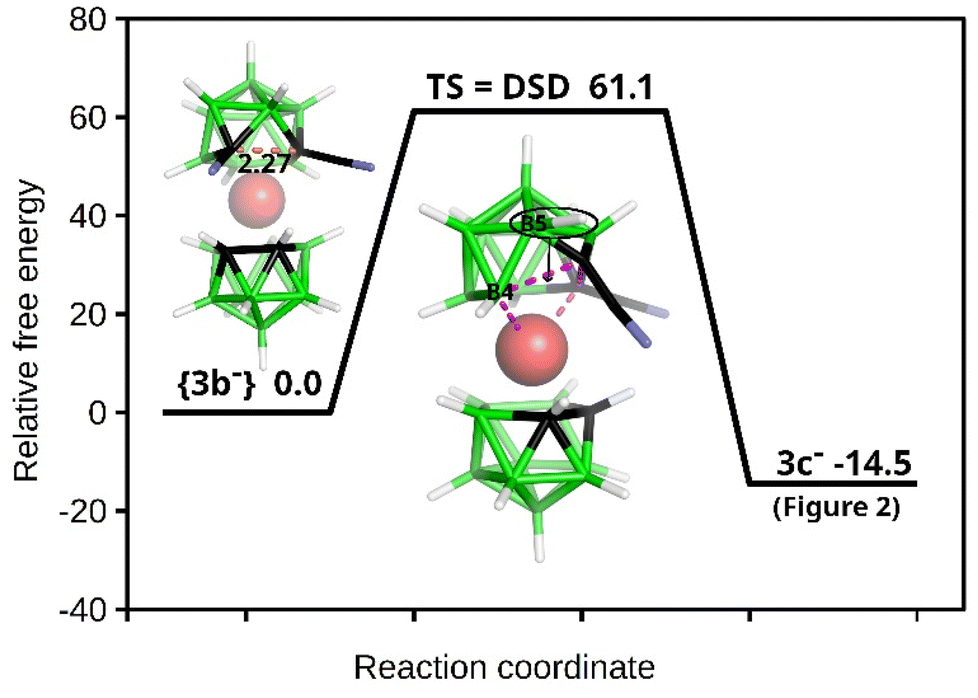 | ||
| Fig. 3 Schematic potential energy surface of the DSD-based rearrangement with the relative Gibbs free energies (kcal mol−1) as computed at the SMD(Et2O)/BP86/AE1//BP86/AE1 level of theory (for the methodology description and the Cartesian geometries of the stationary points, see Computational Details in the ESI†). The C(1)–C(2) separation in {3b−} is given in Å. | ||
In the case of our dinitrile motif, the rearrangement process occurred in the Co–C(1)–C(2)–B(4) diamond (see Fig. 3), and the resulting isomer had an IBO HOMO/LUMO gap of 4.44 eV, which seemed to be a driving force for the rearrangement. The DSD-like transition state was characterized by a strong imaginary frequency at −541 cm−1. Consequently, the B(5)–H vertex, as a part of a six-membered ring, moved to the upper four-membered CB3 girdle, which was facilitated by the positive region dictated by cobalt. In other words, C(1) and B(5) interchanged their positions after the rearrangement and two pentagonal rings were present in the resulting 3c− structure. Note that this reaction profile corresponds to the classical DSD 1,2-closo-C2B10H10 to 1,7-closo-C2B10H10 rearrangement computed e.g. in ref. 37 Note that the reaction profiles in boron-cluster chemistry can be very complex because there exist relatively small energy differences between many intermediate and transition states. Consequently, the boron-cluster reaction may involve many competing pathways.38
Another diverse mechanism consisting in 1e− reductive opening and oxidative closure of the cage, observed previously39 for the rearrangement of the mixed sandwich [3-CpCo-1,2-C2B9H11] to a mixture of isomers inclusive of the 1,8-species apparently, does not apply in the current case, due to fact that the isomerisation to 3c proceeded under an argon atmosphere, without the presence of any oxidation agent and in a stereospecific way.
Some analogy can be also seen with rearrangements observed previously in the case of neutral mixed Mo, W, Ru, Rh, and Ir complexes containing a dicarbollide ligand substituted with sterically demanding aryl/alkyl,40 MeS, or Me2S groups41 and COD or norbornadiene as the second ligand. Typically, this interconversion occurs during or inherently after metal fragment insertion into the substituted dicarbollide ligand at temperatures between 20 °C and 60 °C. Such complexes containing 2nd and 3rd-row transition metals with more diffuse f and d orbitals are inherently less stable and more prone to cage opening and isomerisation. In addition, the crowding from sterically demanding substituents on carbon atoms is an essential condition for elongating the carbon–carbon bond that gives rise to this pathway. This is, however, dissimilar to the case of the full cobalt(III) sandwich and rod-like CN groups as substituents described herein. In addition, no example of rearrangement due to post-modification of an icosahedral metallacarborane cage has yet been described, even in the mixed sandwich series.
Conclusion
Synthetic ways to obtain carbon-substituted nitriles of the cobalt bis(dicarbollide) ion were developed. Mono- and disubstituted compounds are now available. This in turn may lead to an expanding family of various new derivatives using approaches adapted from organic chemistry, exoskeletal metal complexes, polymers, etc. The main achievement, however, consists in attaining a unique stereochemistry of the substitution via direct modification of the cage. It has been shown that the introduction of a particular class of substituents, in our case nitrile groups, can enable low-temperature skeletal rearrangement of carbon atoms in one dicarbollide ligand of the cobalt bis(dicarbollide) ion to the C(8) position of the cage. This was proven feasible even though the structure of ion 1− is very robust and invulnerable towards rearrangement at temperatures up to 300 °C or higher. Based on computational support, the skeletal interconversion was determined to occur as a consequence of the entry of electron-donating CN groups sitting in vicinal carbon positions followed by a DSD mechanism, in which the Co⋯C(1)⋯C(2)⋯B(4) belt represents the “square” with individual next-nearest separations spanning the interval 2.1–2.7 Å (BP86/AE1). As opposed to 2D aromatics, the CN group behaves here as an electron donor. The rearrangement proceeds in a regioselective way, and may thus offer direct access to a broad selection of other groups on ion 1− in a geometrically unique arrangement. This may be useful for fine-tuning the interactions of the cobalt bis(dicarbollide) ion with biological targets, for the design of protein inhibitors, anticancer, antimicrobial agents, etc.Data availability
The data supporting of this article, consisting of the 11B, 1H, 13C NMR spectra and HRMS results, have been included as part of the ESI.† Further, details relating to yields and signal assignments are included.The crystallographic data for the compounds Me4N3a and Me4N3c have been deposited with the Cambridge Crystallographic Data Centre with the CCDC 2370282 and 2370281, respectively. Moreover, CheckCif files were uploaded during submission.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for funding from the Czech Science Foundation Grants No. 2114409S to BG (synthesis and crystallography) and No. 23-05083S to JF (DFT computations).References
- B. P. Dash, R. Satapathy, B. R. Swain, C. S. Mahanta, B. B. Jena and N. S. Hosmane, J. Organomet. Chem., 2017, 849–850, 170–194 CrossRef CAS.
- L. Pazderová, E. Z. Tüzün, D. Bavol, M. Litecká, L. Fojt and B. Grűner, Molecules, 2023, 28, 67 CrossRef PubMed.
- R. N. Grimes and R. N. Grimes, Metallacarboranes of the Transition and Lanthanide Elements, 2016 Search PubMed.
- J. Poater, M. Sola, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- V. Dorďovič, Z. Tošner, M. Uchman, A. Zhigunov, M. Reza, J. Ruokolainen, G. Pramanik, P. Cígler, K. Kalikova, M. Gradzielski and P. Matějíček, Langmuir, 2016, 32, 6713–6722 CrossRef PubMed.
- Y. Chen, A. Barba-Bon, B. Grüner, M. Winterhalter, M. A. Aksoyoglu, S. Pangeni, M. Ashjari, K. Brix, G. Salluce, Y. Folgar-Cameán, J. Montenegro and W. M. Nau, J. Am. Chem. Soc., 2023, 145, 13089–13098 CrossRef CAS PubMed.
- M. Gos, J. Cebula and T. M. Goszczyński, J. Med. Chem., 2024, 67, 8481–8501 CrossRef CAS PubMed.
- P. Cígler, M. Kožísek, P. Rezáčová, J. Brynda, Z. Otwinowski, J. Pokorná, J. Plešek, B. Grűner, L. Dolečková-Marešová, M. Máša, J. Sedláček, J. Bodem, H. G. Krausslich, V. Král and J. Konvalinka, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15394–15399 CrossRef PubMed.
- M. Tarres, E. Canetta, C. Viñas, F. Teixidor and A. J. Harwood, Chem. Commun., 2014, 50, 3370–3372 RSC.
- J. Cebula, K. Fink, J. Boratynski and T. M. Goszczynski, Coord. Chem. Rev., 2023, 477, 19 CrossRef.
- C. Teixidor-Viñas, F. Teixidor and A. J. Harwood, in Boron-Based Compounds: Potential and Emerging Applications in Medicine, ed. E. HeyHawkins, C. Teixidor-Viñas, John Wiley & Sons Ltd, Chichester, 2018, pp. 159–173 Search PubMed.
- K. Fink, J. Boratynski, M. Paprocka and T. M. Goszczynski, Ann. N. Y. Acad. Sci., 2019, 1457, 128–141 CrossRef CAS PubMed.
- M. Nuez-Martinez, C. I. G. Pinto, J. F. Guerreiro, F. Mendes, F. Marques, A. Munoz-Juan, J. A. M. Xavier, A. Laromaine, V. Bitonto, N. Protti, S. G. Crich, F. Teixidor and C. Viñas, Cancers, 2021, 13(24), 6367 CrossRef CAS PubMed.
- A. G. Beck-Sickinger, D. P. Becker, O. Chepurna, B. Das, S. Flieger, E. Hey-Hawkins, N. Hosmane, S. S. Jalisatgi, H. Nakamura, R. Patil, M. D. H. Vicente and C. Viñas, Cancer Biother.Radiopharm., 2023, 38, 160–172 CrossRef PubMed.
- E. Vaňková, K. Lokočová, P. Kašparová, R. Hadravová, I. Křížová, O. Mat'atková, J. Masák and V. Šícha, Pharmaceuticals, 2022, 15, 534 CrossRef PubMed.
- J. Plešek, S. Heřmánek, A. Franken, I. Císařová and C. Nachtigal, Collect. Czech. Chem. Commun., 1997, 62, 47–56 CrossRef.
- A. A. Druzina, A. V. Shmalko, I. B. Sivaev and V. I. Bregadze, Russ. Chem. Rev., 2021, 90, 785–830 CrossRef.
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC.
- R. Núñez, E. J. Juarez-Perez, F. Teixidor, R. Santillan, N. Farfan, A. Abreu, R. Yepez and C. Viñas, Inorg. Chem., 2010, 49, 9993–10000 CrossRef PubMed.
- J. Cabrera-Gonzalez, V. Sanchez-Arderiu, C. Viñas, T. Parella, F. Teixidor and R. Núñez, Inorg. Chem., 2016, 55, 11630–11634 CrossRef CAS PubMed.
- F. Teixidor, R. Núñez and C. Viñas, Molecules, 2023, 28, 24 CrossRef PubMed.
- M. Kugler, J. Nekvinda, J. Holub, S. El Anwar, V. Das, V. Šícha, K. Pospíšilová, M. Fábry, V. Král, J. Brynda, V. Kašička, M. Hajdůch, P. Řezáčová and B. Grűner, ChemBioChem, 2021, 22, 2741–2761 CrossRef CAS PubMed.
- Z. E. Tüzün, L. Pazderová, D. Bavol, M. Litecká, D. Hnyk, Z. Růžičková, O. Horáček, R. Kučera and B. Grűner, Inorg. Chem., 2024, 4c03257, DOI:10.1021/acs.inorgchem.
- S. El Anwar, Z. Ružičková, D. Bavol, L. Fojt and B. Grűner, Inorg. Chem., 2020, 59, 17430–17442 CrossRef CAS PubMed.
- V. V. Volkov, G. S. Yurev, S. G. Vasileva, G. S. Voronina and K. G. Myakishev, Bull. Acad. Sci. USSR, Chem. Sci., 1990, 39, 1355–1359 CrossRef.
- M. K. Kaloustian, R. J. Wiersema and M. F. Hawthorne, J. Am. Chem. Soc., 1972, 94, 6679–6686 CrossRef CAS.
- R. B. King, I. Silaghi-Dumitrescu and I. Şovago, Inorg. Chem., 2009, 48, 5088–5095 CrossRef CAS PubMed.
- A. Zalkin, T. E. Hopkins and D. H. Templeton, Inorg. Chem., 1967, 6, 1911–1915 CrossRef CAS.
- J. Bould and J. D. Kennedy, J. Organomet. Chem., 2014, 749, 163–173 CrossRef CAS.
- L. S. Alekseev, A. V. Safronov, F. M. Dolgushin, A. A. Korlyukov, I. A. Godovikov and I. T. Chizhevsky, J. Organomet. Chem., 2009, 694, 1727–1735 CrossRef CAS.
- R. M. Garrioch, G. M. Rosair and A. J. Welch, J. Organomet. Chem., 2000, 614, 153–157 CrossRef.
- R. T. Baker, Inorg. Chem., 1986, 25, 109–111 CrossRef CAS.
- J. M. Oliva, N. L. Allan, P. V. Schleyer, C. Viñas and F. Teixidor, J. Am. Chem. Soc., 2005, 127, 13538–13547 CrossRef CAS PubMed.
- J. X. Li, R. L. Pang, Z. F. Li, G. Q. Lai, X. Q. Xiao and T. Müller, Angew. Chem., Int. Ed., 2019, 58, 1397–1401 CrossRef CAS PubMed.
- J. Llop, C. Viñas, J. M. Oliva, F. Teixidor, M. A. Flores, R. Kivekas and R. Sillanpää, J. Organomet. Chem., 2002, 657, 232–238 CrossRef CAS.
- B. M. Gimarc, D. S. Warren, J. J. Ott and C. Brown, Inorg. Chem., 1991, 30, 1598–1605 CrossRef CAS.
- D. Hnyk and D. A. Wann, in Boron: The Fifth Element, Springer, Dordrecht, 2015, vol. 20, ch. 5, pp. 17–48 Search PubMed.
- W. Y. Man, G. M. Rosair and A. J. Welch, Dalton Trans., 2015, 44, 15417–15419 RSC.
- A. V. Safronov, F. M. Dolgushin, P. V. Petrovskii and I. T. Chizhevsky, Organometallics, 2005, 24, 2964–2970 CrossRef CAS.
- M. M. Vinogradov, Y. V. Nelyubina, A. A. Pavlov, V. V. Novikov, N. V. Shvydkiy and A. R. Kudinov, Organometallics, 2017, 36, 791–800 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, crystallographic tables, and additional supporting figures. CCDC 2370281 and 2370282. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02588a |
| This journal is © The Royal Society of Chemistry 2024 |