
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards the 4 V-class n-type organic lithium-ion positive electrode materials: the case of conjugated triflimides and cyanamides†
Xiaolong
Guo‡
a,
Petru
Apostol‡
 a,
Xuan
Zhou
b,
Jiande
Wang
a,
Xiaodong
Lin
a,
Darsi
Rambabu
a,
Mengyuan
Du
a,
Süleyman
Er
b and
Alexandru
Vlad
*a
a,
Xuan
Zhou
b,
Jiande
Wang
a,
Xiaodong
Lin
a,
Darsi
Rambabu
a,
Mengyuan
Du
a,
Süleyman
Er
b and
Alexandru
Vlad
*a
aInstitute of Condensed Matter and Nanosciences, Molecular Chemistry, Materials and Catalysis, Université catholique de Louvain, Louvain-la-Neuve B-1348, Belgium. E-mail: alexandru.vlad@uclouvain.be
bDIFFER – Dutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ Eindhoven, The Netherlands
First published on 20th November 2023
Abstract
Organic electrode materials have garnered a great deal of interest owing to their sustainability, cost-efficiency, and design flexibility metrics. Despite numerous endeavors to fine-tune their redox potential, the pool of organic positive electrode materials with a redox potential above 3 V versus Li+/Li0, and maintaining air stability in the Li-reservoir configuration remains limited. This study expands the chemical landscape of organic Li-ion positive electrode chemistries towards the 4 V-class through molecular design based on electron density depletion within the redox center via the mesomeric effect of electron-withdrawing groups (EWGs). This results in the development of novel families of conjugated triflimides and cyanamides as high-voltage electrode materials for organic lithium-ion batteries. These are found to exhibit ambient air stability and demonstrate reversible electrochemistry with redox potentials spanning the range of 3.1 V to 3.8 V (versus Li+/Li0), marking the highest reported values so far within the realm of n-type organic chemistries. Through comprehensive structural analysis and extensive electrochemical studies, we elucidate the relationship between the molecular structure and the ability to fine-tune the redox potential. These findings offer promising opportunities to customize the redox properties of organic electrodes, bridging the gap with their inorganic counterparts for application in sustainable and eco-friendly electrochemical energy storage devices.
Broader contextAs the urgency to address climate change escalates, the need for sustainable energy storage solutions is paramount, particularly for integrating renewable energy sources into power grids. Traditional lithium-ion batteries, while instrumental in this energy transition, face challenges including resource scarcity and environmental concerns due to their metal components. Organic electrode materials have emerged as promising alternatives, offering advantages such as sustainability, cost-efficiency, and design flexibility. However, there has been limited availability of organic positive electrodes capable of operating above 3 V versus Li+/Li0 and maintaining stability in ambient air. To address these challenges, this study employs advanced molecular design to introduce a novel class of conjugated triflimides and cyanamides, targeting the 4 V-class n-type organic electrode materials. Specifically, through the mesomeric effect-induced electron density depletion within redox centers, the triflimides demonstrated delithiation potentials between 3.26 V and 3.8 V versus Li+/Li0. On the other hand, Li containing cyanamides exhibited lower delithiation potentials but boasted storage capacity—for instance, the simplest cyanamide displayed a theoretical specific energy level close to 1000 W h kg−1. Comprehensive structural and electrochemical analyses further elucidated the correlation between the molecular structure and the fine-tuning of redox potentials. These groundbreaking findings not only advance the domain of organic electrode materials but also narrow the performance gap between organic and inorganic alternatives, thereby offering new pathways for the development of more sustainable and efficient energy storage systems. |
Introduction
In recent years, lithium-ion batteries (LIBs)1 have been widely used as the primary power source for portable electronic devices as well as in a variety of emerging applications, including electric vehicles and smart grids.2,3 Electric vehicles offer advantages over their conventional thermal engine counterparts, particularly in terms of reduced pollutant emissions and addressing energy crises. Smart grids powered by large stationary batteries are essential enablers of an advanced power system that heavily integrates intermittent renewable energy sources (RESs) such as solar and wind. They are favorable for maintaining global warming below 1.5 °C,4 which will be critical for the future sustainable development of humanity given the frequent extreme worldwide weather events in recent years. However, current LIB technology has limitations in theoretical energy density and practical performance, and the essential elements used, such as lithium, nickel and cobalt, as well as graphite have experienced increasing prices and environmental concerns.5 Consequently, there is a pressing need for alternative materials that align with economic and environmental criteria. Organic electrodes, as opposed to inorganic electrodes, are composed of abundant elements such as C, H, O, N, P, S, etc.6–8 that replace scarce and expensive transition metals, and can be easily synthesized by environmentally friendly processes; with some of these chemicals even being potentially extracted directly from plants,9 fulfilling environmental friendliness and sustainability. Moreover, organic molecule architectures are versatile and straightforward to construct, allowing for better adjustment of theoretical capacity and operational potential, thereby optimizing battery energy density.Electroactive organic materials can be grouped into three main classes: p-type, n-type, and bipolar. The redox mechanism of p-type systems is based on the ingress – removal of anions, evolving at a relatively high working potential.6 Whereas such chemistries are suitable for Li-free batteries10 or commonly known as molecular-ion batteries,11 these are incompatible with commercial Li-ion battery design. Furthermore, a one-electron redox mechanism, and the large mass of the anions inherently limit the energy storage capacity.12 In contrast, n-type molecules have garnered significant attention owing to their compatibility with the characteristics of commercial batteries, as well as their superior molecular flexibility and versatility that enables greater theoretical capacity.7
However, conventional n-type organic battery materials, generally relying on the carbonyl, imine, organosulfur, etc., functionalities, typically display a redox potential lower than 3 V vs. Li+/Li0 (Fig. 1).7,13–15 Consequently, it is imperative to design organic battery materials with a high-working potential, which will offer multiple benefits. Firstly, high-working-potential will result in higher energy per unit charge, resulting in a higher energy density, which is crucial for all chemical energy storage devices. Secondly, given the thermodynamic air stability threshold of about 2.91 V vs. Li+/Li0,16,17 most of the currently available Li-containing organics get oxidized when exposed to ambient atmospheric conditions (i.e., not even possible to handle in a dry room).17,18 Therefore, when n-type organics are used in organic batteries in the lithium-free cathode (oxidized state), lithium metal or its alloy is required as the negative electrode to provide the lithium source. As depicted in Fig. 1A, this poses significant challenges to industrial safety since the perfect solution to lithium dendrites is still indistinct. Thirdly, a high working potential also reduces the occurrence of unwanted electrochemical reactions that can lead to battery degradation and failure over time, when compared to an electrode with a lower working potential at the same power output. This is due to the faster electrochemical reactions required for lower working potential cathodes, considering the same power output.19 Finally, the development of high-working potential positive electrodes presents a promising opportunity to enhance the performance of divalent metal-ion batteries, for which organic materials seem so far to be the best option.20 Therefore, the development of high-working potential n-type organic materials containing Li is of utmost importance, as highlighted in Fig. 1B. Prioritizing the synthesis and optimization of such positive electrode chemistries is crucial to sustainable and clean energy storage technology.
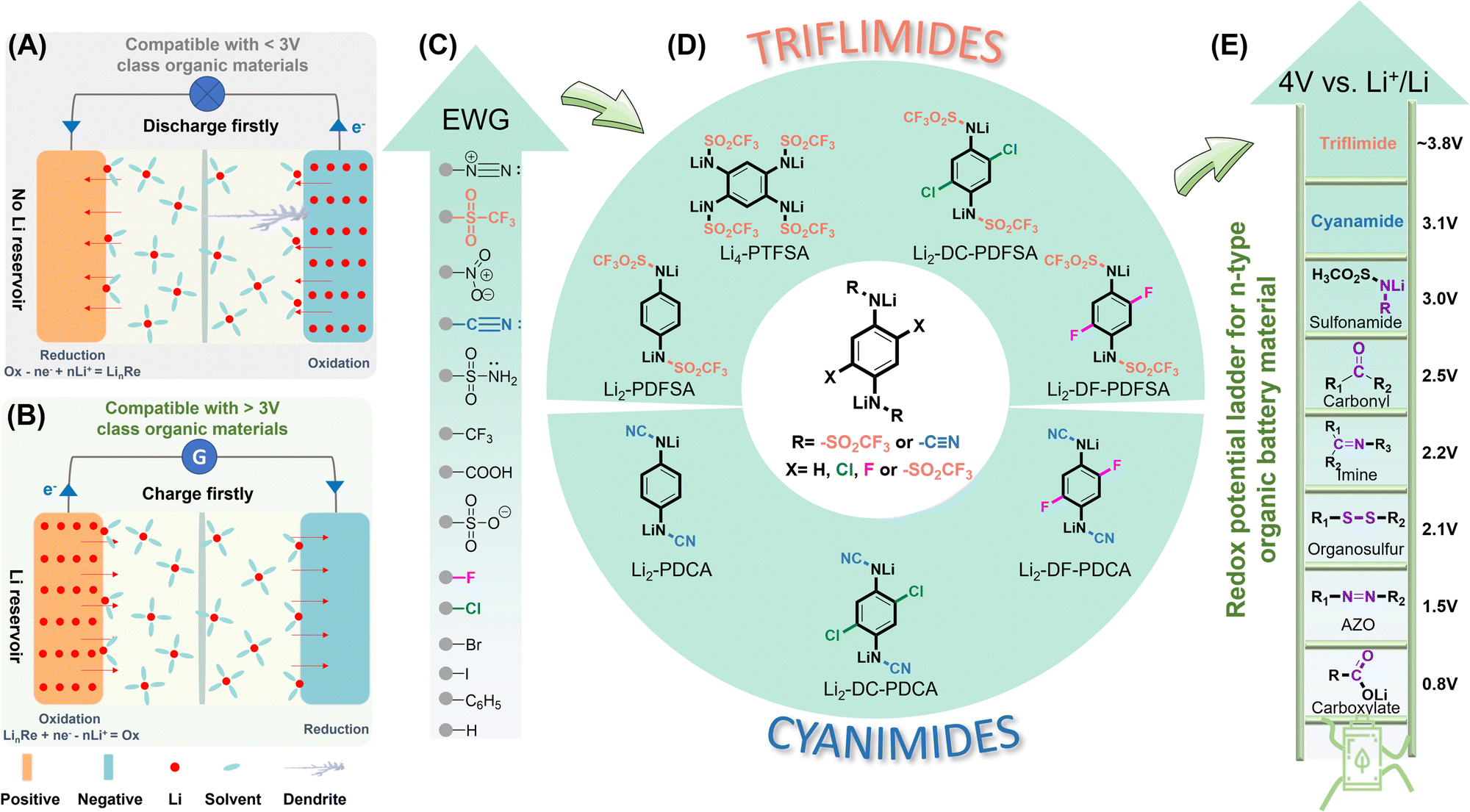 | ||
| Fig. 1 Schematic illustration of a battery (A) with no lithium reservoir and (B) with a lithium reservoir for the positive electrode. (C) The ranking of the electron-withdrawing ability of various functional groups. (D) Rationale behind the molecular design of high-voltage Li-containing triflimides and cyanimides. (E) The redox potential ladder for n-type organic battery materials. EWG, electron withdrawing group. | ||
In recent years, molecular engineering has emerged as a promising strategy to tune the working potential of organic materials. The redox potential of a molecule can be adjusted by fine-tuning the energy levels of its highest occupied molecular orbital (HOMO) and its lowest unoccupied molecular orbital (LUMO), allowing for customization. Based on molecular orbital theory, a molecule with a lower LUMO level exhibits a higher electron affinity, resulting in a higher reduction potential. Lowering the LUMO levels can be achieved through two distinct methods: the through-bond effect and the through-space effect. The former proceeds through inductive21 or mesomeric effects17,22 achieved by introducing electron-donating or -withdrawing groups, as well as the incorporation of spectator cations (Mn+).23–25 The latter includes stereo-electronic chameleonic effects16 and polymorphism.26 The recent development of Li-reservoir organic electrodes has led to a significant increase in working potential, reaching the level of 3.4 V vs. Li+/Li0.17 However, despite these advancements, the number of available organic cathodes satisfying these criteria remains limited.16,17,23,27
The incorporation of electron-withdrawing substituents stands out as an exceptionally efficacious and facile approach to lower the LUMO level, thus raising the redox potential of organic electrode materials in accordance with the rationales of inductive or mesomeric effects.21 For instance, the bistriflimides (F3C–SO2–N–SO2–CF3) and dicyanamides (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–N–CN) demonstrate anodic stability up to 5 V17 and 4.5 V28 (vs. Li+/Li0), respectively, owing to the synergistic effect of the conjugation and the inductive impact of –SO2CF3/–CN groups. Given this perspective and inspired by the recently reported conjugated sulfonamide class,17 herein, we merged a p-phenylenediamine redox core with the triflimides and cyanamides for high voltage positive electrode materials (Fig. 1D). The electron-withdrawing capabilities of the trifuoromethanesulfonyl and cyano groups (Fig. 1C) is anticipated to significantly increase the redox potential, and the intramolecular conjugation to delocalize the negative charges, thereby stabilizing an organic lithium salt in an ambient atmosphere (moisture and air) to prevent hydrolysis (nucleophilic substitution). Therefore, this work discloses two new families of high redox potential Wurster-type redox materials that are air-stable (oxygen and moisture) – conjugated triflimides and cyanamides.
C–N–CN) demonstrate anodic stability up to 5 V17 and 4.5 V28 (vs. Li+/Li0), respectively, owing to the synergistic effect of the conjugation and the inductive impact of –SO2CF3/–CN groups. Given this perspective and inspired by the recently reported conjugated sulfonamide class,17 herein, we merged a p-phenylenediamine redox core with the triflimides and cyanamides for high voltage positive electrode materials (Fig. 1D). The electron-withdrawing capabilities of the trifuoromethanesulfonyl and cyano groups (Fig. 1C) is anticipated to significantly increase the redox potential, and the intramolecular conjugation to delocalize the negative charges, thereby stabilizing an organic lithium salt in an ambient atmosphere (moisture and air) to prevent hydrolysis (nucleophilic substitution). Therefore, this work discloses two new families of high redox potential Wurster-type redox materials that are air-stable (oxygen and moisture) – conjugated triflimides and cyanamides.
Additionally, various EWG substituents (mainly halogen) were also the subject of investigation to determine their regulatory effects on the working redox potential. Specifically, the triflimide family of compounds was explored, including Li2-PDFSA (dilithium 1,4-phenylenebis((trifluoromethylsulfonyl)amide), Li2-DC-PDFSA (dilithium (2,5-dichloro-1,4-phenylene)bis((trifluoromethylsulfonyl)amide))), Li2-DF-PDFSA (dilithium (2,5-fluoro-1,4-phenylene)bis((trifluoromethylsulfonyl)amide)), and Li4-PTFSA (tetralithium benzene-1,2,4,5-tetrayltetrakis((trifluoromethylsulfonyl)amide)). The cyanamide family includes Li2-PDCA (dilithium (1,4-phenylene dicyanamide)), Li2-DC-PDCA (dilithium (1,4-dicyanamido-2,5-dichlorobenzene)), and Li2-DF-PDCA (dilithium (1,4-dicyanamido-2,5-difluorobenzene)) (Fig. 1D).
The triflimide class is found to display a remarkably high average delithiation potential, spanning from 3.26 V to 3.8 V vs. Li+/Li0. In particular, the second delithiation potential of Li2-DF-PDFSA soars at a value as high as 3.91 V vs. Li+/Li0, making this redox couple one of the highest achieved to date for n-type organic chemistries (Fig. 1E). This chemistry approaches the 4 V vs. Li+/Li0 realm, which is a coveted value for n-type organic electrodes. Compared to triflimide, the Li-containing cyanamides display slightly lower delithiation potentials, ranging from 3.1 V to 3.35 V vs. Li+/Li0, yet having a higher storage capacity. As an illustration, Li2-PDCA exhibits a theoretical specific energy level at the active material of nearly 1000 W h kg−1 (3.1 V vs. Li+/Li0, 320 mA h g−1). When used as a battery electrode, the Li2-PDCA electrode retains more than 80% capacity after 100 cycles with one lithium ion exchanging per unit. A fine interplay between the solubility, redox potential, capacity and material utilization is presented, with further development guidelines provided. A full cell employing Li4Ti5O12 as the negative electrode and the cyanamide as the positive electrode material exhibits a specific capacity of approximately 157 mA h gcathode−1 (72 mA h gcathode+anode−1), with a capacity retention of more than 80% after 50 cycles. These promising results offer inspiration for the development of high-voltage and high-energy content Li-ion containing organic positive electrode chemistries that could compete with the current inorganic materials.
Results and discussion
Effect of electron withdrawing groups on molecular conjugation
The impact of electron-withdrawing groups on the electrochemical performances was assessed through the experimental evaluation of various triflimide and cyanamide derivatives (Scheme 1). The triflimides were synthesized using a two-step procedure, involving grafting trifluoromethanesulfonyl groups, followed by lithiation using stoichiometric quantities of lithium hydride in anhydrous diethyl ether, and in an inert environment (Scheme 1A, top, and B route). The protonated versions of cyanamides were synthesized according to a reported procedure;29 thiourea derivatives were synthesized by reacting phenylenediamine with benzoyl chloride and ammonium thiocyanate in acetone. Thiourea was desulfurized with lead(II) acetate to produce cyanamides, then lithiated with lithium methoxide in anhydrous methanol (Fig. 1A, bottom; refer to the experimental part and Fig. S1–S10 in the ESI,† for further details). Notably, the ortho-cyanamide exhibited a propensity for self-condensation,30,31 thus precluding the synthesis of (benzene-1,2,4,5-tetrayl)tetracyanamide.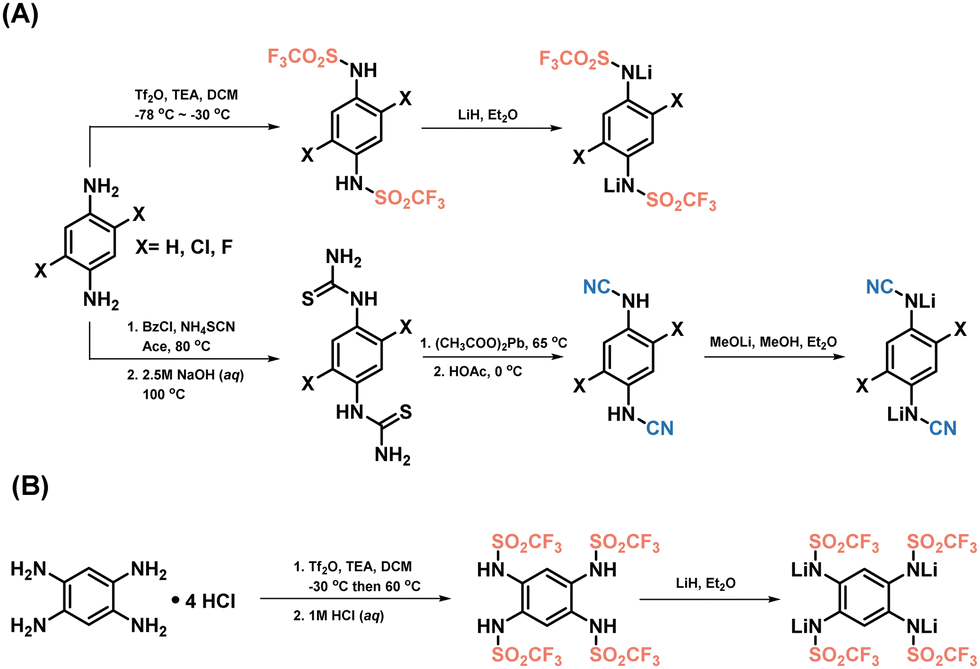 | ||
| Scheme 1 Illustration of the synthetic route for triflimides and cyanamides. The comprehensive experimental procedures can be found in the ESI,† section. | ||
1H NMR and FTIR spectroscopy are reliable methods for investigating the electron density properties, which, in turn, directly impact the electrochemical characteristics.17,23 H2-PDFSA and H2-PDCA were selected as representatives for the spectroscopy study, which is discussed next in detail. Grafting of trifluoromethanesulfonyl and cyano groups onto p-phenylenediamine to form H2-PDFSA and H2-PDCA, respectively, leads to reduced electron density on the nitrogen atom marked by significant deshielding of the amino proton from 4.17 ppm to 11.93 ppm for H2-PDFSA, and 10.05 ppm for H2-PDCA (Fig. 2A). The large chemical shift of N–H indicates the imine proton's acidic nature, rendering it susceptible to exchange by the lithium cation. This is also supported well by the computed electrostatic potential (ESP) map (Table S1, ESI†), wherein the total ESP charge of two amine N atoms in p-phenylenediamine is −1.76e, which turns to −1.38e for H2-PDCA and −1.12e for H2-PDFSA, revealing the strong electron delocalization on both the aromatic ring and the sulfonyl and cyano groups. Simultaneously, compared to the 1H NMR chemical shift of the aromatic peak of p-phenylenediamine, the aromatic protons undergo deshielding in both cases: H2-PDFSA (δ 7.31) and H2-PDCA (δ 6.94) caused by the mesomeric effect (–M) of the grafted trifluoromethanesulfonyl and cyano groups (Fig. 2A).
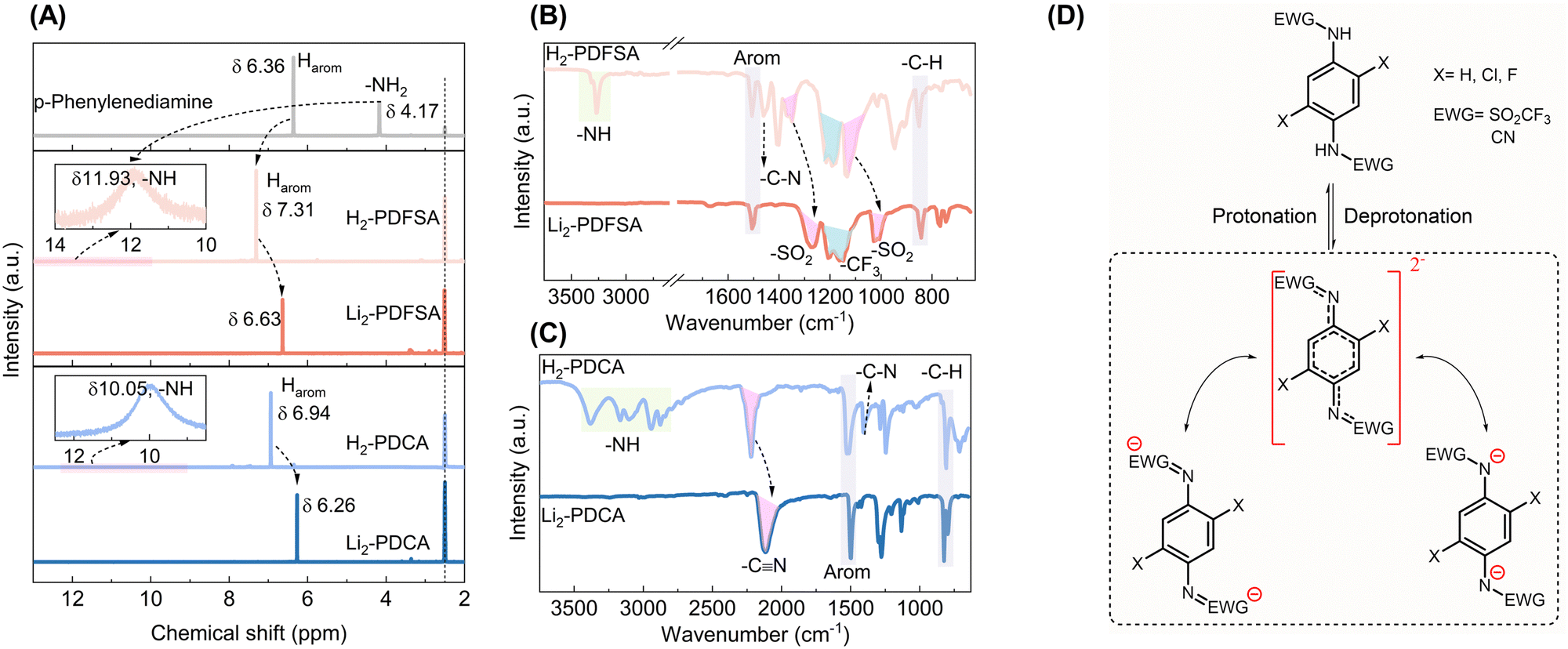 | ||
| Fig. 2 Spectroscopic characterization of H2/Li2-PDFSA and H2/Li2-PDCA as the representatives of triflimide and cyanamide families, respectively. (A) 1H NMR spectra of the starting materials: p-phenylenediamine, protonated form (H2-PDFSA and H2-PDCA), and their lithiated products (Li2-PDFSA and Li2-PDCA). A comparative FTIR analysis of cyanamides (B) and triflimides (C). (D) Schematic representation of the deprotonation and conjugate resonance forms for the triflimides and cyanamides. | ||
Given the higher covalency of the N–H bond, the lithiation process triggers negative overall charge delocalization (yet maintaining the conjugation) of the sulfonamide/cyanamide system via the –C–N– bond. As a result, compared to the protonated forms (H2-PDFSA/PDCA), the pronounced bathochromic shifts of the υ-SO2 (from 1348 cm−1 and 1125 cm−1 to 1274 cm−1 and 1021 cm−1, Fig. 2B) and υ-CN (from 2221 cm−1 to 2117 cm−1, Fig. 2C) vibrations are observed in the anionic forms (Li2-PDFSA/PDCA), attributable to resonance effects (Fig. 2D and Fig. S11, S12, ESI†). The more uniform electron density distribution over the –C–N bridge (between the benzene ring and EWGs) leads to a decrease in its dipole moment and the intensity of the characteristic FTIR band.
Redox mechanism and redox potential tuning through molecular design
The elucidation of the redox mechanism for triflimides and cyanamides was performed through ex situ1H NMR and FTIR spectroscopy analysis. To understand the mechanism, chemically oxidized versions of Li2-DC-PDFSA and Li2-DC-PDCA (denoted as OX-DC-PDFSA and OX-DC-PDCA respectively) were prepared (Fig. S8 and S9, ESI†). In the FTIR analysis, the υ-SO2 bands (1364 cm−1 and 1118 cm−1, OX-DC-PDFSA) and the υ-CN band (2180 cm−1, OX-DC-PDCA) demonstrate a notable hypsochromic shift (Fig. S10, ESI†). This is attributed to the loss of charge delocalization resulting from oxidation, rather than occurring directly on the sulfonyl and cyano groups. The 1H NMR spectrum of the charged Li2-DC-PDFSA phase was found to be similar to that of the chemically oxidized OX-DC-PDFSA (Fig. S13A and B, ESI†). Specifically, the aromatic ring protons (7.12 ppm) were de-shielded to 7.95 ppm upon oxidation, and this process is reversed for the reduced phase. Similar spectroscopic features were observed for Li2-DC-PDCA (Fig. S13C and D, ESI†). Consequently, the redox center in all disclosed triflimides and cyanamides is confirmed to be the para-phenylenediamine building block (Wurster-type redox, Fig. 3A), via a reversible two-electron oxidation to the quinoneimine form, making it the most plausible reaction pathway for these systems.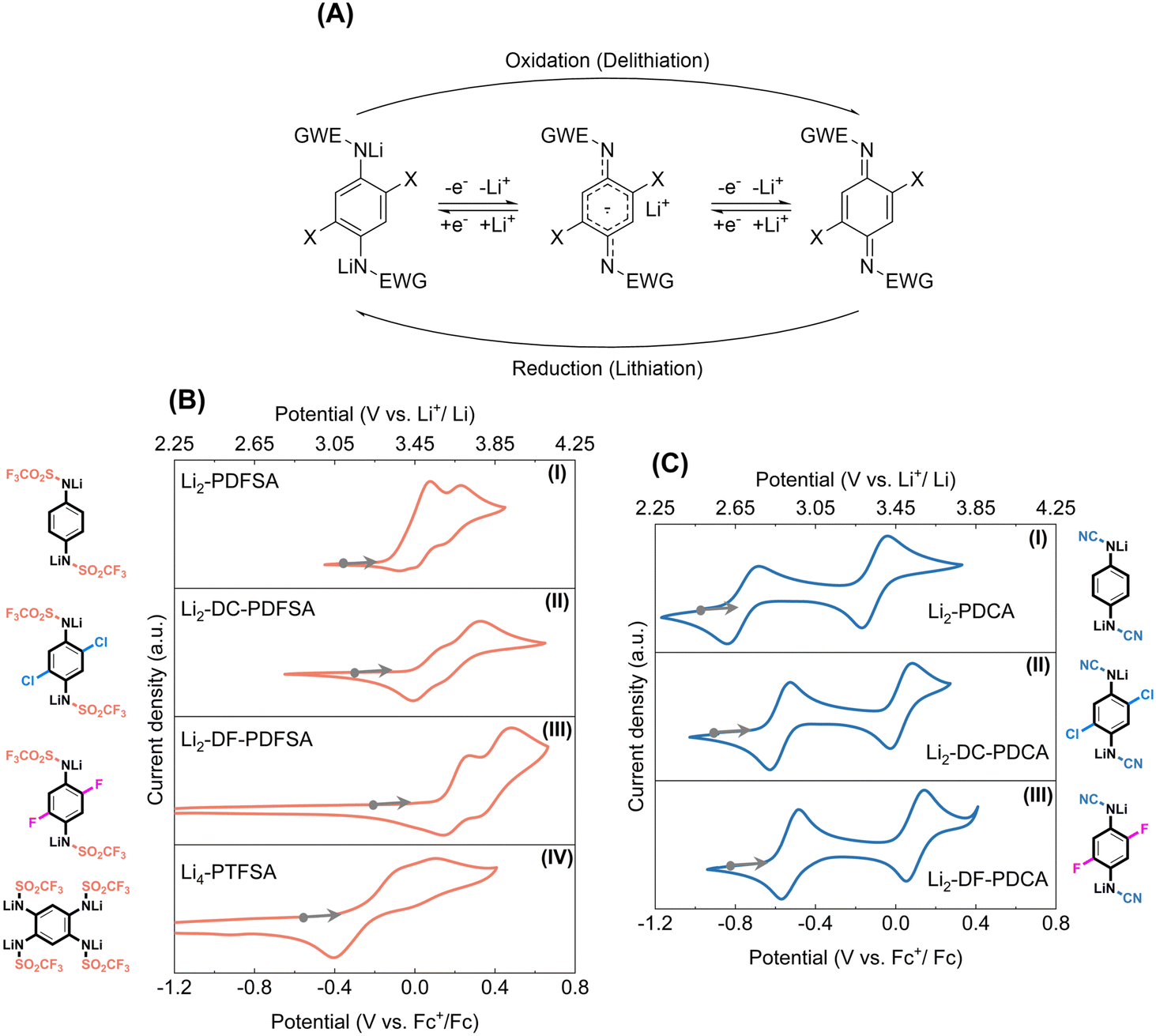 | ||
| Fig. 3 Redox mechanism and electrochemical properties of triflimide and cyanamide materials. (A) Schematic representation of the two-electron redox mechanism that is common to both triflimide and cyanamide materials. The cyclic voltammetry of triflimides (B) and cyanamides (C) measured at 5 mM concentration of active materials, in 0.1 M LiCl DMSO or THF supporting electrolyte, using Pt working and counter electrodes, and an Ag wire as a pseudo reference electrode. Ferrocene was used as an internal reference. | ||
In combination with molecular electrochemistry analysis (liquid-phase cyclic voltammetry, CV) and solid-state electrochemical performance, we deduced that during the charging process (delithiation process), the Li-containing form undergoes the extraction of one electron (−1Li+), leading to the formation of an intermediate radical form. Subsequently, another electron (−1Li+) is extracted, forming a quinonimine moiety (Fig. 3A). In contrast to solid-phase electrochemical processes, which are characterized by a significant number of interfacial reactions, molecular electrochemistry analysis offers a more straightforward approach to evaluate the reversibility and redox potential of materials. The cyclic voltammetry analysis of triflimides and cyanamides is presented in detail in Fig. 3B and C, using Li2-PDFSA and Li2-PDCA as representatives for comparison and discussion. As shown in Fig. 3B(I), Li2-PDFSA exhibits two sequential redox waves at 0.23 V and 0.08 V vs. Fc+/Fc, translating to as high as 3.66 V and 3.51 V vs. Li+/Li0. To the best of our knowledge, these values stand among the highest reported so far for organic n-type redox materials.18 During the first oxidation process the radical anion is generated, while the second oxidation event produces the neutral form, both of which are attributed to the anodic events (delithiation process). The cathodic waves show reversible responses. Notably, the replacement of the methanesulfonyl group with a trifluoromethanesulfonyl moiety, possessing a stronger electron-withdrawing capability, results in a noteworthy increase in the redox potential by 0.5 V as compared to the previously reported analogous material Li2-PDSA.17 The introduction of a cyano group into Li2-PDCA (Fig. 3C(I)) also results in a high redox potential of 3.39 V and 2.75 V vs. Li+/Li0, further confirming the impact of the EWG on the redox potential.
Introducing substituents such as chlorine (–Cl) and fluorine (–F) into molecules has been demonstrated to significantly modulate the electrochemical response of organic and inorganic materials, and this was no exception in this work as well. The order of potential gain with respect to Li+/Li0 followed the expected trend: Li2-PDCA (3.15 V and 3.32 V in solid, 2.75 V and 3.39 V in solution) < Li2-DC-PDCA (3.27 V and 3.45 V in solid, 2.91 V and 3.5 V in solution) < Li2-DF-PDCA (3.35 V in solid, 2.95 V and 3.57 V in solution); and Li4-PTFSA (3.2 V in solid, 3.18 V and 3.35 V in solution) < Li2-PDFSA (3.55 V in solid, 3.51 V and 3.66 V in solution) < Li2-DC-PDFSA (3.62 V in solid, 3.56 V and 3.75 V in solution) < Li2-DF-PDFSA (3.7 V in solid, 3.68 V and 3.91 V in solution) (Fig. 3 and S14, Table 1, ESI†). Importantly, to highlight, the second wave delithiation potential of Li2-DF-PDFSA was found at a value of 3.91 V versus Li+/Li0, making it the sole n-type organic redox material currently available with a potential close to 4 V, a much-coveted characteristic in the n-type organic electrode community.
| Material | E (V vs. Li+/Li0), solution | E (V vs. Li+/Li0), solid phase | Capacity (mA h g−1) |
|---|---|---|---|
| a Note: average delithiation potential for 1st plateau. | |||
| Li2-PDFSA | 3.51 & 3.66 | 3.55 | 140 |
| Li2-DC-PDFSA | 3.56 & 3.75 | 3.62 | 118 |
| Li2-DF-PDFSA | 3.68 & 3.91 | 3.7a | 127 |
| Li4-PTFSA | 3.18 & 3.35 | 3.2 | 78 |
| Li2-PDCA | 2.75 & 3.39 | 3.15 & 3.32 | 320 |
| Li2-DC-PDCA | 2.91 & 3.5 | 3.27 & 3.45 | 225 |
| Li2-DF-PDCA | 2.95 & 3.57 | 3.35a | 276 |
Table 1 summarizes the evaluated values of redox potential for the studied materials. The separation between the two one-electron redox processes for cyanamides is approximately 0.2 V in the solid state and considerably higher, of about 0.6 V in the liquid state; thus, the redox potential gap between the two events is reduced by nearly 0.4 V. It can also be noted that the elevation of the potential for the first electron oxidation process in the solid phase is the main reason for this decrease. Although the exact origin and interpretation of the discrepancy between the molecular liquid phase and solid phase electrochemistry of organic materials remain a matter of debate in the community, our current explanation would reside in the higher activation energy required for the transition from the reduced to the radical intermediate state in the solid state compared to the liquid state. The triflimides in turn exhibit a single plateau in the solid phase due to the larger gap value reduction, which exceeds the gap value (<0.2 V) of triflimides in solution. In addition, voltage hysteresis is observed during discharging only in the Li2-PDFSA case, similarly to Li2-PDSA,17 but not in PDCA and halogen-substituted PDFSA, which suggests that the interaction between sulfonyl substitutions and the free benzene ring site may influence the voltage hysteresis. Finally, from a practical perspective, it should be emphasized that the lithiated organic electrode materials are typically unstable in the ambient environment due to their low working potentials below the oxidation capacity of molecular oxygen and their strong Lewis base properties that capture protons from water. As expected by design, the Li-contained triflimides and cyanamides were found to display very good ambient air condition storage stability (Fig. S15 and S16, ESI†) due to the synergistic effect of resonance and mesomeric effects, exhibiting no oxidation or re-protonation upon exposure to the air moisture environment with an absolute humidity of 10 g m−3 at 25 °C.
The rationale behind the high redox potential regulation of triflimides and cyanamides was further corroborated with 1H NMR analysis, FTIR spectroscopy, and DFT calculations. The increase in the redox potential of the disclosed materials is reflected by a linear increase in the deshielding of aromatic protons in the 1H NMR spectrum (Fig. 4A), as well as a bathochromic shift of aromatic bands in the FTIR spectrum (Fig. 4B). These shifts indicate a reduction in electron density within the redox center (aromatic core) for cyanamides and triflimides. The lithiated molecules (Fig. S17, ESI†) exhibit identical trends, and the overlay of detailed 1H NMR and FTIR spectra is shown in Fig. S18 (ESI†). In contrast, the –SO2– and –CN characteristic FTIR bands exhibit a hypsochromic shift (Fig. S19 and S20, ESI†), indicating an increase in electron density, which suggests π-electron transfer from the aromatic core to EWGs via the mesomeric effect.
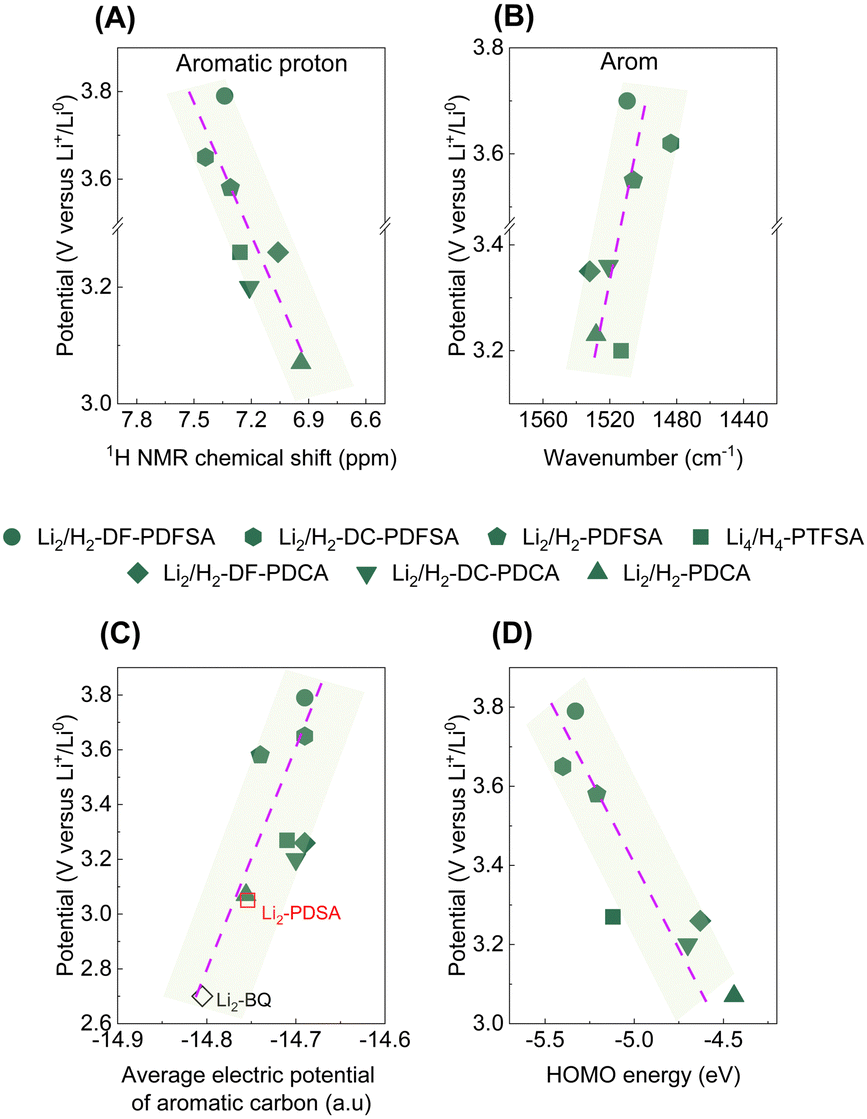 | ||
| Fig. 4 Correlation between the chemical shift of the aromatic proton (A), and the shift of the aromatic band in the FTIR spectra (B) with the delithiation potential of the studied chemistries. Correlation between the average electric potential of aromatic carbon (C; Li2-BQ16 and Li2-PDSA17 were used as benchmark systems), and the HOMO (D) with the delithiation potential of the studied materials. | ||
The computed average electric potentials of the aromatic ring, correlated with the corresponding delithiation potentials, demonstrate that higher overall potentials achieved are associated with higher aromatic electric potentials (lower electron density, Fig. 4C), resulting in a lower HOMO (Fig. 4D). This relationship arises from the thermodynamics of electron transfer reactions between oxidizing and reducing species, which determine the redox potential of a system. Electron density influences this potential by affecting the accessibility of valence electrons for oxidation (HOMO) or reduction (LUMO) reactions. Here, a low electron density (high electric potential) environment restricts the electron donation (oxidation), leading to an increased delithiation potential for the system. In addition, molecules with higher working potentials exhibit smaller average pKa values (Fig. S21, ESI†), attributed to the Hammett constant, which quantifies the influence of substituents on reaction rates and equilibrium constants in aromatic molecules. Importantly, this finding aligns well with a recent study, further reinforcing the association between high redox potentials and the influence of substituents on aromatic systems.32 It is noteworthy that these correlations can be leveraged as guidelines for future developments of aromatic core-based redox chemistries. Furthermore, given that the enhancement of organic electrode potential has proven to be a challenging task, these observations provide valuable insights into how the redox potential of organic electrode materials can be effectively augmented. Particularly, the working potential of organic electrodes can be enhanced by having a depleted electron density in the redox center with the use of electron-withdrawing groups, while maintaining a low molecular weight to gain high theoretical capacity.
Charge storage performances
Galvanostatic cycling was used to investigate the capabilities of the cyanamide and triflimide families as lithium cation storage materials. For this, Li2-PDCA and Li4-PTFSA were used as representative examples, and data are shown in Fig. 5A. The charged-state material (Li0-PDCA, capacity of two electrons, corresponding to 320 mA h g−1, was considered) was observed to dissolve in the electrolyte, resulting in a green coloration of the separator, leading to rapid capacity decay. Comparatively, one-electron cycling (Li2-PDCA ↔ Li1-PDCA) ensured a stable charge–discharge cycling, avoiding the solubility issue – the primary degradation route in organic batteries (Fig. 5A). Therefore, the electrochemical cycling of Li2-PDCA was restricted to one-electron capacity (160 mA h g−1, 2.2–3.3 V vs. Li+/Li0) at a rate of C/10 (equivalent to one Li+ exchange in 5 hours) (Fig. 5B). The cell displays a flat charge/discharge plateau at approximately 2.9 V vs. Li+/Li, slightly above the value of the first redox wave measured in liquid CV (Fig. 3C). This discrepancy may arise from the activation energy difference between solid and liquid phases, as previously discussed. Under these cycling conditions, approximately 80% of the original capacity was retained after 100 cycles with an average Coulombic efficiency of more than 99.5% (Fig. 5C). It should be noted that these are achieved under slow cycling conditions, known to enhance the dissolution–elution effect of organic materials. Furthermore, when the capacity was restricted to 96 mA h g−1, i.e., cycling between Li2-PDCA and Li1.4-PDCA, it facilitated the preservation of additional lithium cation reserves within the cathode. This compensatory effect served to offset the irreversible lithium loss during cycling. Employing this self-sacrifice strategy, a high capacity retention of 84% was achieved after 700 cycles (Fig. 5D) without overpotential fluctuations, preserving the constant operating potential. The rate capability of the Li2-PDCA electrode was also assessed, as illustrated in Fig. S23A and S24B (ESI†). Notably, even with slight polarization, the cell sustains a capacity exceeding 100 mA h g−1 at elevated charge–discharge rates of 1C. The representative Li4-PTFSA was also subjected to galvanostatic charge–discharge cycling in 1 M LiPF6 in EC/DMC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol%). The cycling profile revealed a sloping plateau in the voltage range of 3.1–3.3 V, with the initial discharge capacity approaching 78 mA h g−1, with a two-electron capacity achieved at a rate of C/5 (equivalent to one Li+ exchange in 2.5 hours, Fig. 5E). In the following 50 cycles, the electrode maintained 64 mA h g−1, nearly 80% of its initial capacity (Fig. 5F), with the C-rate performances illustrated in Fig. S23C and D (ESI†).
:1 vol%). The cycling profile revealed a sloping plateau in the voltage range of 3.1–3.3 V, with the initial discharge capacity approaching 78 mA h g−1, with a two-electron capacity achieved at a rate of C/5 (equivalent to one Li+ exchange in 2.5 hours, Fig. 5E). In the following 50 cycles, the electrode maintained 64 mA h g−1, nearly 80% of its initial capacity (Fig. 5F), with the C-rate performances illustrated in Fig. S23C and D (ESI†).
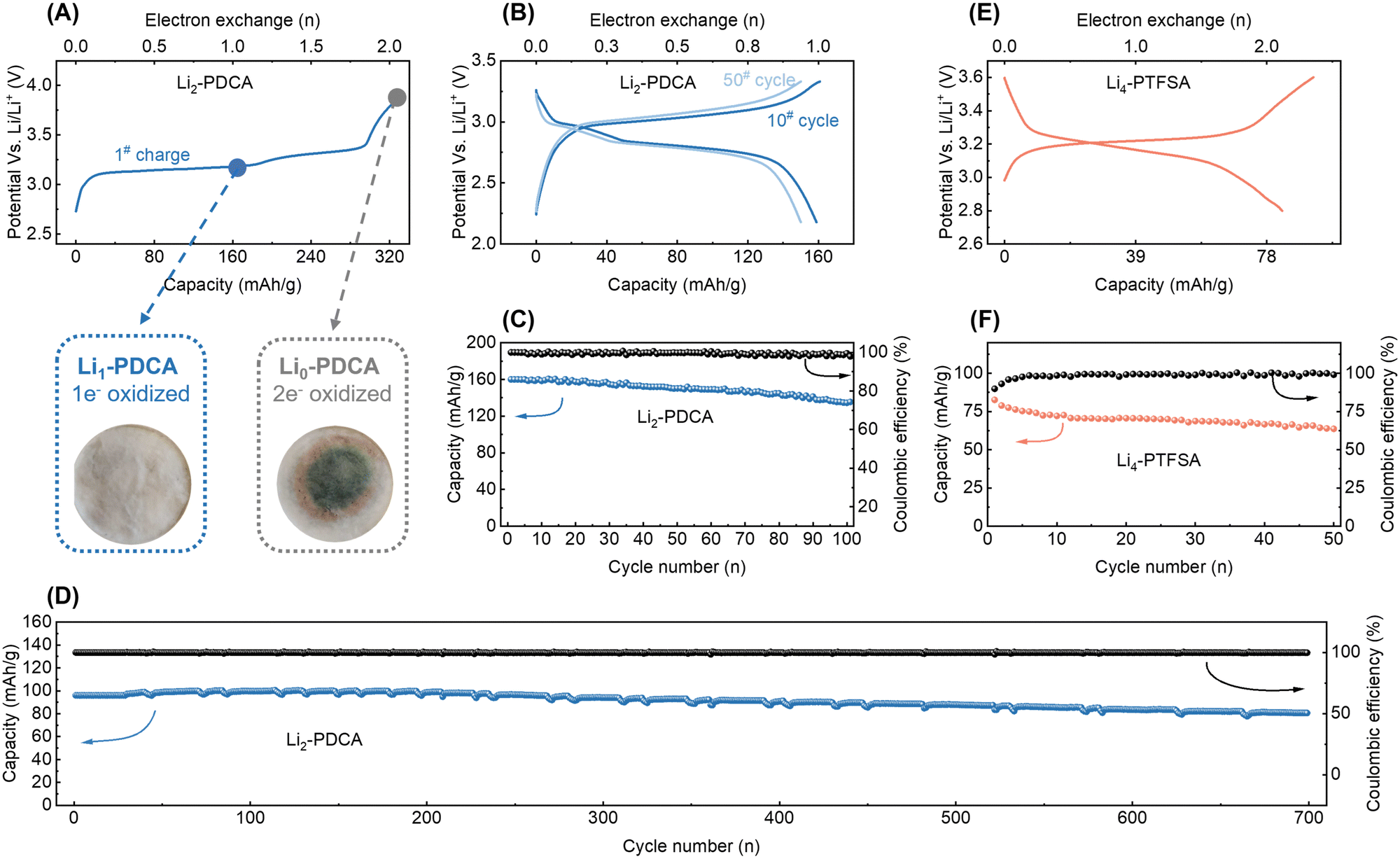 | ||
| Fig. 5 Lithium cation storage performances of cyanamides and triflimides in a half-cell configuration. (A) Potential-capacity galvanostatic charge plot of Li2-PDCA with two-electron capacity, along with visual solubility inspection of a glass fiber separator at various states of charge. (B) Potential-capacity galvanostatic charge curves and the (C) long-term cycling stability of Li2-PDCA restricted to 1-electron capacity at a rate of 0.2C. (D) Long-term cycling stability of Li2-PDCA restricted to 0.6-electron capacity at a rate of 1C. (E) Potential-capacity galvanostatic charge curves and (F) the long-term cycling stability of Li4-PTFSA at a rate of 0.2C. | ||
The studied triflimide and cyanamide materials possess unique attributes distinguishing them from many other available organic n-type battery materials, namely air-stability and lithium-reservoir characteristics. This makes them potentially suitable for use in conventional Li-ion battery design. For instance, a full cell was constructed and evaluated using Li2-PDCA as the positive electrode and Li4Ti5O12 as the negative electrode materials.17 The full cell displayed an output voltage of approximately 1.35 V and a capacity of nearly 157 mA h g[Li2-PDCA]−1 (based on the weight of the positive electrode material), with the capacity retention exceeding 80% after 50 cycles, as shown in Fig. 6A and B.
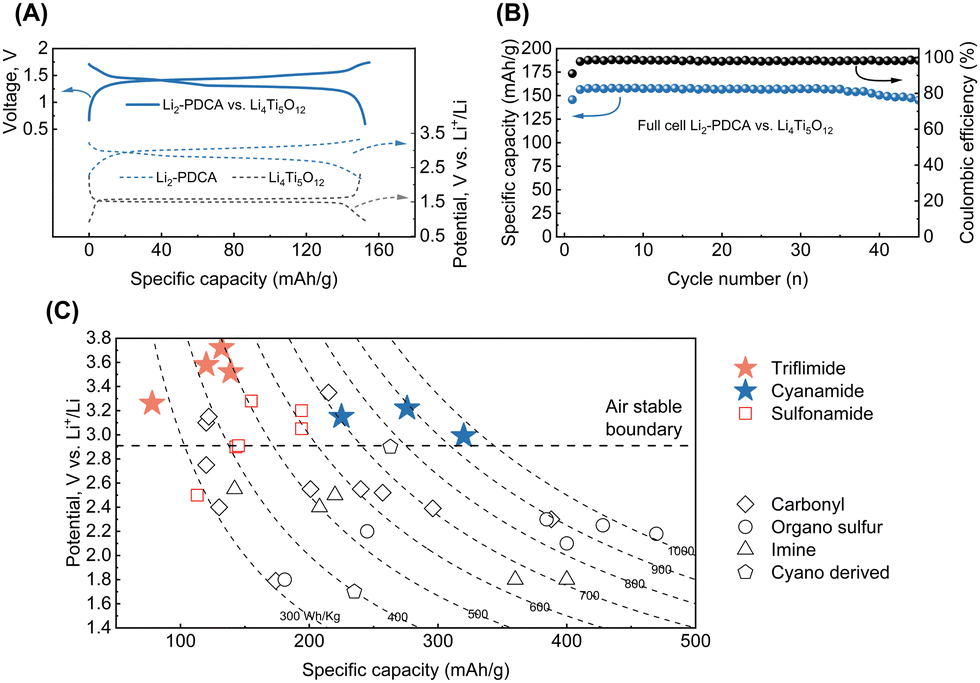 | ||
| Fig. 6 (A) Galvanostatic charge–discharge profiles of Li2-PDCA and Li4Ti5O12 measured in half cells versus Li metal and a full cell cycled at a rate of 0.2C. (B) Specific capacity and coulombic efficiency versus the number of cycles of the full cell achieved at a rate of 0.2C. (C) Comparison of theoretical energy density (performance considered at the material level) of various n-type organic electrode materials (sulfonamides,17,33 carbonyls,16,22,26,34–39 organo sulfur,40–42 imines,14,43,44 and cyano derivatives45) for LIBs, highlighting the performances achieved with triflimides and cyanamides developed in this work. | ||
The energy metrics of the triflimides and cyanamides are next compared with those of n-type organic electrode materials developed thus far, relying on the average working potential, theoretical capacity, and resultant energy density (Fig. 6C). To the best of our knowledge, despite the relatively low capacity given to the high molar mass, the triflimide chemistry disclosed here displays the highest redox potential among n-type organic electrodes, a feature that is highly coveted in energy storage applications. The triflimides' air-stability and high solubility make them prime candidates for use in redox flow batteries (RFBs) that are geared towards large-scale energy storage applications. For example, the Li2/OX-DC-PDFSA redox couple, with a potential of 3.65 V vs. Li+/Li0, exhibits a solubility of over 0.5 M in acetonitrile, a solvent with a wide electrochemical potential stability window.46 In addition, the Li4-PTFSA ↔ Li2-PTFSA redox couple, both of which are organic lithium salts with high solubility in water, is particularly appropriate for aqueous RFBs. Given that mainstream current RFBs are mainly based on toxic and non-sustainable vanadium-based materials,47 these results strongly encourage the use of triflimides with a great deal of potential for future research concerning the establishment of sustainable advanced RFBs. On the other hand, the solid-phase charge storage of triflimides and cyanamides can be optimized by incorporating them in (coordination) polymers,24,48 porous metal or covalent organic frameworks (MOFs or COFs) to suppress their solubility. For instance, delithiated cyanamide based MOFs49 exhibit high conductivity (up to 800 S cm−1) and low solubility in carbonate solvents, indicating that exceptional battery capability could be achieved. Additionally, COFs formed from cyano-group-based molecules are promising candidates for supercapacitors.50 Besides, along with the excellent battery performance exhibited by sulfonamide-based coordination polymers,24 Li4-PTFSA can also benefit from this approach to improve working potential and cycling stability. Finally, solid-state electrolytes present a promising avenue for addressing the solubility issue of electrodes; thus, there is a broad space for continued research to fully explore the practicality of the first-generation triflimides and cyanamides.
Conclusion and outlook
In this study we demonstrate a highly effective method for adjusting the working potential of organic electrode materials for batteries, based on functionalized phenylenediamines with the trifluoromethanesulfonyl and cyano groups, giving rise to seven lithium-containing triflimide and cyanamide compositions. The developed materials display excellent ambient stability and a high-redox potential, expanding the landscape of n-type organic Li-ion positive electrode materials. The triflimides, in particular, exhibit an unprecedented high redox potential of 3.8 V vs. Li+/Li, the highest among those of n-type organic positive electrode materials reported so far. On the other hand, cyanamides possess a theoretical specific energy density of up to 1000 W h g−1 (at the material level), an exceptionally desirable metric. Notably, the Li-containing characteristic empowers cyanamides to be paired with a Li4Ti5O12 negative electrode to form a full cell in a conventional Li-ion battery configuration with a capacity of 157 mA h g[positive]−1 and a capacity retention of over 80% after 50 cycles. The application of the developed materials and methods should not be restricted to conventional lithium-ion batteries alone – these have the potential to contribute substantially to other next-generation energy storage technologies such as redox flow batteries, sacrificial mediators, or for much conveyed multi-valent batteries.Author contributions
X. L. G. and P. A. contributed equally to this work. X. L. G., P. A., and A. V. conceived the project. X. L. G. and P. A. designed and performed the experiments. X. D. L., P. A., J. D. W., X. D. L, D. R, M. Y. D, and A. V. analysed and interpreted the experimental data. X. Z. performed the theoretical calculations. X. Z. and S. E. analysed and interpreted the computational data.Data availability
The datasets generated and analysed in this study are included in the paper and its ESI,† or available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. L. G., J. W., and M. D. acknowledge China Scholarship Council and China-Belgium (Wallonia-Brussels French-speaking Region) Outstanding Young Scholar Scholarships for the funding support. X. L. acknowledges financial support from the Marie Skłodowska-Curie Actions (grant agreement no. 101064286) for his postdoctoral fellowship. D. R. is Chargé de Recherche-FNRS. A. V. is indebted to the European Research Council for support under the European Union's Horizon 2020 research and innovation program (grant agreement no. 770870, MOOiRE), as well as partial support from F.R.S.-FNRS through the F.4552.21-P – MIS – CSA-LION grant. DIFFER is part of the institutes organisation of NWO. This work was sponsored by NWO Exact and Natural Sciences for the use of supercomputer facilities.References
- M. B. Sid, Scrosati. Lithium-ion rechargeable batteries, J. Power Sources, 1994, 51, 79–84, DOI:10.1016/0378-7753(94)01956-8.
- Z. P. Cano, et al., Batteries and fuel cells for emerging electric vehicle markets, Nat. Energy, 2018, 3, 279–289, DOI:10.1038/s41560-018-0108-1.
- Z. Zhu, et al., Rechargeable Batteries for Grid Scale Energy Storage, Chem. Rev., 2022, 122, 16610–16751, DOI:10.1021/acs.chemrev.2c00289.
- J. Rogelj, et al., Paris Agreement climate proposals need a boost to keep warming well below 2 degrees C, Nature, 2016, 534, 631–639, DOI:10.1038/nature18307.
- Time for lithium-ion alternatives, Nat. Energy, 2022, 7, 461 DOI:10.1038/s41560-022-01073-y.
- B. Esser, et al., A perspective on organic electrode materials and technologies for next generation batteries, J. Power Sources, 2021, 482, 228814, DOI:10.1016/j.jpowsour.2020.228814.
- T. B. Schon, B. T. McAllister, P. F. Li and D. S. Seferos, The rise of organic electrode materials for energy storage, Chem. Soc. Rev., 2016, 45, 6345–6404, 10.1039/c6cs00173d.
- Y. Lu and J. Chen, Prospects of organic electrode materials for practical lithium batteries, Nat. Rev. Chem., 2020, 4, 127–142, DOI:10.1038/s41570-020-0160-9.
- X. Zhu and Y. Jing, Natural quinone molecules as effective cathode materials for nonaqueous lithium-ion batteries, J. Power Sources, 2022, 531, 231291, DOI:10.1016/j.jpowsour.2022.231291.
- É. Deunf, et al., Reversible anion intercalation in a layered aromatic amine: a high-voltage host structure for organic batteries, J. Mater. Chem. A, 2016, 4, 6131–6139, 10.1039/c6ta02356h.
- M. Yao, H. Sano, H. Ando and T. Kiyobayashi, Molecular ion battery: a rechargeable system without using any elemental ions as a charge carrier, Sci. Rep., 2015, 5, 10962, DOI:10.1038/srep10962.
- K. Nakahara, et al., Rechargeable batteries with organic radical cathodes, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- J. Kim, et al., Organic batteries for a greener rechargeable world, Nat. Rev. Mater., 2023, 8, 54–70, DOI:10.1038/s41578-022-00478-1.
- C. Peng, et al., Reversible multi-electron redox chemistry of π-conjugated N-containing heteroaromatic molecule-based organic cathodes, Nat. Energy, 2017, 2, 1–9, DOI:10.1038/nenergy.2017.74.
- D. Xu, et al., The Progress and Prospect of Tunable Organic Molecules for Organic Lithium-Ion Batteries, ACS Nano, 2021, 15, 47–80, DOI:10.1021/acsnano.0c05896.
- L. Sieuw, et al., Through-Space Charge Modulation Overriding Substituent Effect: Rise of the Redox Potential at 3.35 V in a Lithium-Phenolate Stereoelectronic Isomer, Chem. Mater., 2020, 32, 9996–10006, DOI:10.1021/acs.chemmater.0c02989.
- J. Wang, et al., Conjugated sulfonamides as a class of organic lithium-ion positive electrodes, Nat. Mater., 2021, 20, 665–673, DOI:10.1038/s41563-020-00869-1.
- Y. Lu, Q. Zhang, F. Li and J. Chen, Emerging Lithiated Organic Cathode Materials for Lithium-Ion Full Batteries, Angew. Chem., Int. Ed., 2023, 135, e202216047, DOI:10.1002/anie.202216047.
- A. Tomaszewska, et al., Lithium-ion battery fast charging: a review, eTransportation, 2019, 1, 100011, DOI:10.1016/j.etran.2019.100011.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Current status and future directions of multivalent metal-ion batteries, Nat. Energy, 2020, 5, 646–656, DOI:10.1038/s41560-020-0655-0.
- H. Kim, et al., High energy organic cathode for sodium rechargeable batteries, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- L. Sieuw, et al., A H-bond stabilized quinone electrode material for Li-organic batteries: the strength of weak bonds, Chem. Sci., 2019, 10, 418–426, 10.1039/c8sc02995d.
- A. Jouhara, et al., Raising the redox potential in carboxyphenolate-based positive organic materials via cation substitution, Nat. Commun., 2018, 9, 4401, DOI:10.1038/s41467-018-06708-x.
- J. Wang, et al., High performance Li-, Na-, and K-ion storage in electrically conducting coordination polymers, Energy Environ. Sci., 2022, 15, 3923–3932, 10.1039/d2ee00566b.
- D. Rambabu, et al., An Electrically Conducting Li-Ion Metal-Organic Framework, J. Am. Chem. Soc., 2021, 143, 11641–11650, DOI:10.1021/jacs.1c04591.
- L. Bernard, et al., Influence of Polymorphism on the Electrochemical Behavior of Dilithium (2,3-Dilithium-oxy)-terephthalate vs. Li, Inorganics, 2022, 10, 62, DOI:10.3390/inorganics10050062.
- A. E. Lakraychi, et al., An air-stable lithiated cathode material based on a 1,4-benzenedisulfonate backbone for organic Li-ion batteries, J. Mater. Chem. A, 2018, 6, 19182–19189, 10.1039/c8ta07097k.
- H. Yoon, et al., Lithium electrochemistry and cycling behaviour of ionic liquids using cyano based anions, Energy Environ. Sci., 2013, 6, 979–986, 10.1039/c3ee23753b.
- M. A. S. Aquino, et al., Superexchange metal-metal coupling in dinuclear pentaammineruthenium complexes incorporating a 1,4-dicyanamidobenzene dianion bridging ligand, J. Am. Chem. Soc., 1992, 114, 5130–5140, DOI:10.1021/ja00039a026.
- R. J. Crutchley, Phenylcyanamide ligands and their metal complexes, Coord. Chem. Rev., 2001, 219, 125–155, DOI:10.1016/S0010-8545(01)00324-1.
- C. A. White, Magneto-structural correlations of novel copper(II) molecular materials, Carleton University, 1999 Search PubMed.
- C. de la Cruz, et al., A Systematic Study on the Redox Potentials of Phenazine-Derivatives in Aqueous Media: A Combined Computational and Experimental Work, ChemSusChem, 2023, 16, e202201984, DOI:10.1002/cssc.202201984.
- J. Wang, et al., A High-Voltage Organic Framework for High-Performance Na- and K-Ion Batteries, ACS Energy Lett., 2022, 7, 668–674, DOI:10.1021/acsenergylett.1c02571.
- W. Wan, et al., Tuning the electrochemical performances of anthraquinone organic cathode materials for Li-ion batteries through the sulfonic sodium functional group, RSC Adv., 2014, 4, 19878–19882 RSC.
- A. Shimizu, et al., Nitrogen-Containing Polycyclic Quinones as Cathode Materials for Lithium-ion Batteries with Increased Voltage, Energy Technol., 2014, 2, 155–158, DOI:10.1002/ente.201300148.
- S. Renault, et al., A green Li–organic battery working as a fuel cell in case of emergency, Energy Environ. Sci., 2013, 6, 2124–2133, 10.1039/c3ee40878g.
- A. Shimizu, et al., Introduction of two lithiooxycarbonyl groups enhances cyclability of lithium batteries with organic cathode materials, J. Power Sources, 2014, 260, 211–217, DOI:10.1016/j.jpowsour.2014.03.027.
- G. S. Vadehra, R. P. Maloney, M. A. Garcia-Garibay and B. Dunn, Naphthalene Diimide Based Materials with Adjustable Redox Potentials: Evaluation for Organic Lithium-Ion Batteries, Chem. Mater., 2014, 26, 7151–7157, DOI:10.1021/cm503800r.
- S. Gottis, A. L. Barres, F. Dolhem and P. Poizot, Voltage gain in lithiated enolate-based organic cathode materials by isomeric effect, ACS Appl. Mater. Interfaces, 2014, 6, 10870–10876, DOI:10.1021/am405470p.
- A. Bhargav, S. V. Patil and Y. Fu, A phenyl disulfide@CNT composite cathode for rechargeable lithium batteries, Sustainable Energy Fuels, 2017, 1, 1007–1012, 10.1039/c7se00135e.
- M. Wu, et al., Highly Reversible Diphenyl Trisulfide Catholyte for Rechargeable Lithium Batteries, ACS Energy Lett., 2016, 1, 1221–1226, DOI:10.1021/acsenergylett.6b00533.
- W. Guo, et al., Bis(aryl) Tetrasulfides as Cathode Materials for Rechargeable Lithium Batteries, Chemistry, 2017, 23, 16941–16947, DOI:10.1002/chem.201703895.
- J. Hong, et al., Biologically inspired pteridine redox centres for rechargeable batteries, Nat. Commun., 2014, 5, 5335, DOI:10.1038/ncomms6335.
- M. Lee, et al., Redox cofactor from biological energy transduction as molecularly tunable energy-storage compound, Angew. Chem., Int. Ed., 2013, 52, 8322–8328, DOI:10.1002/anie.201301850.
- Q. Deng, et al., Exploitation of redox-active 1,4-dicyanobenzene and 9,10-dicyanoanthracene as the organic electrode materials in rechargeable lithium battery, Electrochem. Commun., 2017, 75, 29–32, DOI:10.1016/j.elecom.2016.12.005.
- C. Schotten, et al., Making electrochemistry easily accessible to the synthetic chemist, Green Chem., 2020, 22, 3358–3375, 10.1039/d0gc01247e.
- A. Z. Weber, et al., Redox flow batteries: a review, J. Appl. Electrochem., 2011, 41, 1137–1164, DOI:10.1007/s10800-011-0348-2.
- B. Pan, et al., Polyanthraquinone-Based Organic Cathode for High-Performance Rechargeable Magnesium-Ion Batteries, Adv. Energy Mater., 2016, 6, 1600140, DOI:10.1002/aenm.201600140.
- A. Werner, et al., A Radical Anion Salt of 2,5-Dimethyl-N,N′-dicyanoquinonediimine with Extremely High Electrical Conductivity, Angew. Chem., Int. Ed. Engl., 1986, 25, 740–741, DOI:10.1002/anie.198607401.
- L. Hao, et al., Structural evolution of 2D microporous covalent triazine-based framework toward the study of high-performance supercapacitors, J. Am. Chem. Soc., 2015, 137, 219–225, DOI:10.1021/ja508693y.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, experimental procedures for synthesis and characterization data of compounds, and electrochemical analysis, as well as computation details. See DOI: https://doi.org/10.1039/d3ee02897f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |