
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Avoiding electrochemical indentations: a CNT-cocooned LiCoO2 electrode with ultra-stable high-voltage cycling†
Zhi
Zhu
*ab,
Shuanglong
Xu
a,
Zhenjie
Wang
a,
Xiaohui
Yan
a,
Guiyin
Xu
 b,
Yimeng
Huang
c,
Yuping
Wu
a,
Yin
Zhang
*b and
Ju
Li
*bc
b,
Yimeng
Huang
c,
Yuping
Wu
a,
Yin
Zhang
*b and
Ju
Li
*bc
aConfucius Energy-storage Lab, School of Energy and Environment & Z Energy-storage Center, Southeast University, Nanjing, Jiangsu 211189, China. E-mail: zhizhu@seu.edu.cn
bDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: yinzhang@pku.edu.cn
cDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: liju@mit.edu
First published on 25th July 2024
Abstract
Charging LiCoO2 (LCO) to above 4.5 V induces crystal cracking and seriously deteriorates the battery cycle life. Decreasing the range of the LCO misfit strain during deep de-lithiation is useful for preventing cracks, but this is not always achievable. Here, we demonstrate that the limited electrochemical contact area between electronically conductive carbon and the LCO crystal causes “electrochemical indentations” (ECIs) during charging and discharging. Particularly in fast charging, the high local ΔcLi gradient in LCO would cause a local volume of the surficial lattice to shrink while the rest of the crystal is still under stretching, and hence, drive the ECI to cause cracking. Increasing the electrochemical contact area would reduce the ECI and cracking risk. Therefore, we developed a free-standing CNT-LCO electrode in which all of the LCO particles were intimately wrapped with a dense CNT cocoon to establish a larger true electrical contact area. The simulations demonstrated that the radial ΔcLi and ECI decreased significantly in the cocooned LCO particles. The cocooned LCO electrode maintained good morphology and retained 94% of its energy density after 400 cycles when charged to 4.55 V. By removing the need for a current collector and binder, the volumetric energy density of the CNT-LCO cathode reached 3200 Wh L−1 (electrode).
Broader contextUtilizing the extra capacity of the LiCoO2 (LCO) cathode above 4.5 V is enticing, but it is impeded by crystal cracking and rapid cycling decay for unknown reasons. This work demonstrated that the limited true contact area between LCO and electronic conductive agents is the factor that causes “electrochemical indentations” (ECIs) during high-voltage charging and triggers crystal cracking. We develop a numerical model based on electrochemomechanics and show that avoiding ECI is key to preventing LCO cracking and improving the high-voltage cycling life. Subsequently, a free-standing carbon nanotube-cocooned LCO electrode (CNT-LCO) was developed, where only a 0.5 wt% CNT weaved dense elastic cloth fully wrapped all of the LCO particles, like silkworm cocoons, to maximize the electronic contact area. The CNT-LCO retained 94% of its energy density after 400 cycles when charged to 4.55 V, in contrast to the catastrophic capacity decay using 3 wt% carbon black, with everything else remaining the same. The theory of electrochemical indentation and how a low electronic contact area leads to ECI and cracking are essential for all mixed ionic electronic conductors (MIECs) and even for solid electrolytes, which is a limiting case of MIECs. |
LiCoO2 (LCO) batteries have been used in laptop computers, communication devices and consumer electronic devices for decades because of their high volumetric energy density and good cycling stability.1,2 Commercial LCO cathodes are currently cycled within 4.4 V, 165 mA h g−1, to meet satisfactory cycling life and battery safety; therefore, there is still plenty of extra energy density before getting to its theoretical capacity of 274 mA h g−1.3,4 In recent years, charging LCO beyond 4.5 V to develop the rest of the capacity at high voltage is a hot topic.5–9
In previous works, it has been demonstrated that charging LCO to above 4.5 V involved oxygen redox and triggered particle cracking, which caused the reversible capacity of LCO to decay quickly.8,10,11 Crack formation in LCO was understood from the repeated lattice-volume changes during cycling.12 Thus, many material modifications were conducted on LCO, e.g., doping and coatings,4,8,13,14 to inhibit the phase transformation and decrease the amplitude of the LCO-lattice volume change (ΔV = Vmax − Vmin) during the deep de-lithiation process. However, based on the experimental observation that the crack always originates from the specific sites at the LCO particle surface,6,10 we should be aware that the dynamical spatial gradients of Li-ion concentration in the large LCO single crystals can be a more critical factor for crack formation.
In common cathode electrodes, it was believed that the synergic Li-ion diffusivity and electronic conductivity in the active materials (σ1) and the electronic conductivity (σ2) in carbon black (CB) determined the kinetics of the cathode reaction.15–18 However, as shown in Fig. 1a, the lean CB (<3 wt%) conductive agent that was used in the regular LCO electrode (R-LCO) for high energy density only had a narrow and limited electrical contact area (aA) with the LCO particles, so the local current density (iA ≈ I/aA) would concentrate at the electrical contacting sites (site A), as shown in Fig. 1b. Since the conduction of electrons and Li ions is always coupled in the LCO particles, the high current density resulting in an overpotential (ΔϕA) brings in a high gradient of Li concentration (ΔcLi), causing inhomogeneous lattice-parameter change and dynamic stress inside the single crystal during charging.
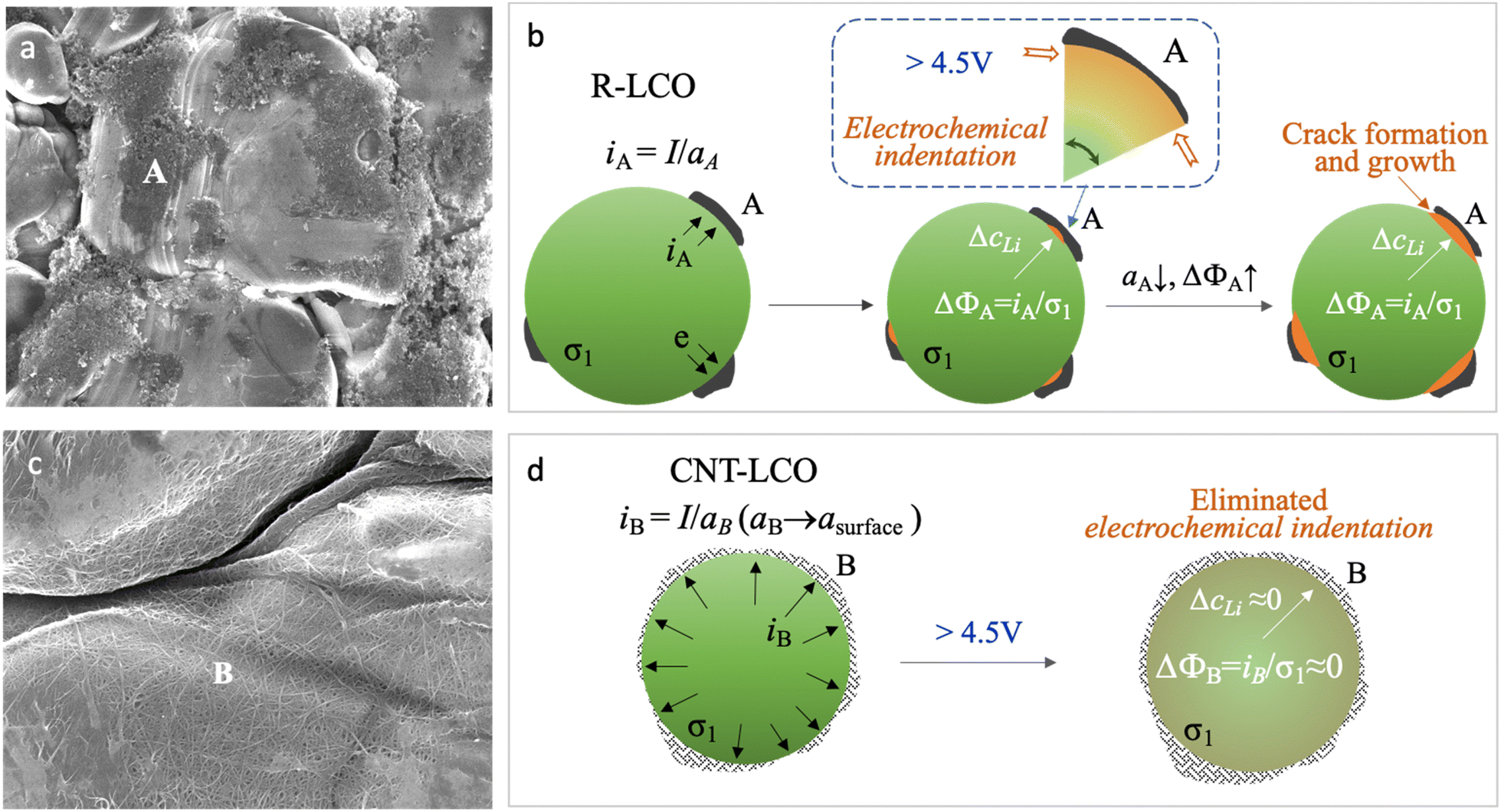 | ||
| Fig. 1 Formation and prevention of electrochemical indentation in LCO particles during high-voltage charging. (a) SEM images of LCO particles and carbon black in R-LCO. (b) Scheme of electrochemical indentation and crack formation in LCO crystals when charging R-LCO to a high voltage. (c) SEM images of CNT-cocooned LCO particles in CNT-LCO. (d) Scheme for preventing electrochemical indentation and crack formation in the LCO crystal when charging CNT-LCO to a high voltage. The fundamental equations for mixed ionic electronic conductor (MIEC) are presented in ref. 27. | ||
This dynamic stress could be bearable when the LCO crystal was charged to an upper voltage of less than 4.5 V.13 However, when charging the LCO beyond 4.5 V at a high rate, the over-delithiated surface (csurface) at site A would first get oxygen redox (O2− → Oα−, α < 2) involved and rapidly tend to shrink the local lattice (Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) csurfaceCoO2), while the less delithiated bulk (Li(csurface + ΔcLi)CoO2) is still under stretching,13 then the high shear stress near site A would act like an electrochemical indentation (ECI) at the crystal surface by the limited-area carbon black, eventually triggering cracking (Fig. 1b). Moreover, the cracks at site A would continue decreasing the electrical contact between the LCO particle and CB (decreasing aA and increasing ΔcLi) and aggravate ECI with continuous crack growth, until the LCO particles lost electrical contact with the CB and eventually terminated the high-voltage battery cycling. Note that we use c in this paper to denote alternatively concentration (unit #Li per m3) or mole fraction (#Li per formula unit, which is dimensionless).
csurfaceCoO2), while the less delithiated bulk (Li(csurface + ΔcLi)CoO2) is still under stretching,13 then the high shear stress near site A would act like an electrochemical indentation (ECI) at the crystal surface by the limited-area carbon black, eventually triggering cracking (Fig. 1b). Moreover, the cracks at site A would continue decreasing the electrical contact between the LCO particle and CB (decreasing aA and increasing ΔcLi) and aggravate ECI with continuous crack growth, until the LCO particles lost electrical contact with the CB and eventually terminated the high-voltage battery cycling. Note that we use c in this paper to denote alternatively concentration (unit #Li per m3) or mole fraction (#Li per formula unit, which is dimensionless).
Eliminating ECI could be the key to preventing LCO crystals from cracking; thus, decreasing the dynamical radial overpotential (Δϕ) during the high voltage de-lithiation process is important to improve the cycling stability of LCO. As the electronic contact between LCO and CB is significant for Δϕ, compared with singly improving σ1 with material doping or singly improving σ2 with a new conductive agent, increasing the electrical contact area for the LCO particles to disperse the current density (i) can be more effective. In this work, we established a CNT cocoon cloth to amply wrap the LCO particles in the electrode, named CNT-LCO (Fig. 1c). Compared to R-LCO, CNT-LCO has the following attributes:
(1) Due to the high aspect ratio of CNT (Fig. S1 and Table S1, ESI†), a small weight percentage of CNT can weave dense elastic cocoon cloth for the LCO particles, like silkworm cocoons, which would not only entwine and cover all LCO particles but also result in thorough electron percolation in the electrode, as shown in Fig. 1c. Such intimate contact between the LCO particles and CNT cocoon will maximize the true electronic contact area (aB → asurface, where asurface is the total surface area of all LCO particles) to decrease iB, ΔϕB and ΔcLi (Fig. 1d). Then, the ECI and cracking in the LCO particles can be greatly prevented during high-voltage cycling, as revealed by our finite-element diffusion-stress modeling of the “contact electromechanics” of mixed ionic electronic conducting (MIEC) LCO with a conductive agent.
(2) CNT can form a complete matrix to establish a robust free-standing electrode without any chemical binder or current collector. Thus, the free-standing CNT-LCO electrode has a high volumetric energy density of 3200 W h L−1 (electrode), which would, in turn, ideally improve the energy density of the LCO full cell from the current 780 W h L−1 (cell) to above 1212 W h L−1 (cell).
Though carbon nanotubes or nanobelts have been used in previous works for LCO electrode preparations,19–21 the mechanisms are not fully understood. Creating a CNT network to enhance the electrical conductivity is far from sufficient to stabilize high-voltage LCO cycling. As we will show, the key is to maximize the true electrical contact area to eliminate the electrochemical indentation. In this work, finite-element simulations and experimental analysis indicated that the plane-to-plane contact between the LCO particles and CNT cocoon efficiently eliminated the electrochemical indentation during high-voltage charging so that CNT-LCO had well-maintained morphologies without cracks and retained 94% of its energy density after 400 cycles when charged to 4.55 V. In addition, the CNT-LCO cathode had a high volumetric energy density of 3200 W h L−1, and the cycled CNT-LCO could be facilely regenerated/recycled without any complicated mechanical or chemical treatment that must be involved in the traditional battery material regeneration/recycling processes.
Results and discussion
We prepared free-standing CNT-LCO electrodes using a vacuum filtration method, which comprised 0.5 wt% CNT and 99.5 wt% commercial LCO particles (Methods). The SEM images of R-LCO and CNT-LCO are compared in Fig. 1a and c, respectively. However, 3 wt% CB in R-LCO only partially covered the LCO particle surfaces, supplying limited electrical contact area (aA) between the LCO particles and CB, the 0.5 wt% CNT in CNT-LCO weaved dense “silkworm cocoons” and fully wrapped all LCO particle surfaces, establishing intimate electrical contacts between them. The TGA and Raman spectra are shown in Fig. S2 and S3 (ESI†), verifying the chemical constituents of the CNT-LCO electrode.The R-LCO and CNT-LCO electrodes were first tested in half cells at 200 mA g−1. The charge/discharge profiles in Fig. 2a indicate that while R-LCO was charged to 198.1 mA h g−1 and discharged to 195.7 mA h g−1 in the 10th cycle between 3.0 and 4.55 V, CNT-LCO was able to be charged to 200.5 mA h g−1 and discharged to 199.1 mA h g−1. After 300 cycles, the charge/discharge profiles of R-LCO were seriously deformed, as shown in Fig. 2a, which could only be charged to 99.3 mA h g−1 and discharged to 97.8 mA h g−1. In contrast, the voltage profile of CNT-LCO remained very stable, and it could still be charged to 192.7 mA h g−1 and discharged to 192.3 mA h g−1. It is quite amazing that without any chemical binder or extra conductive agent and no changes to the active material LCO itself, using just 0.5 wt% CNT (which may be considered both a physical binder and a conductive agent) versus the standard method of electrode preparation can lead to not only better initial performance but also much better cycling durability. This illustrates that the hypothesis of a low electronic contact area leading to ECI is a very important one to check out, and we aim to develop a theory about this later in this paper.
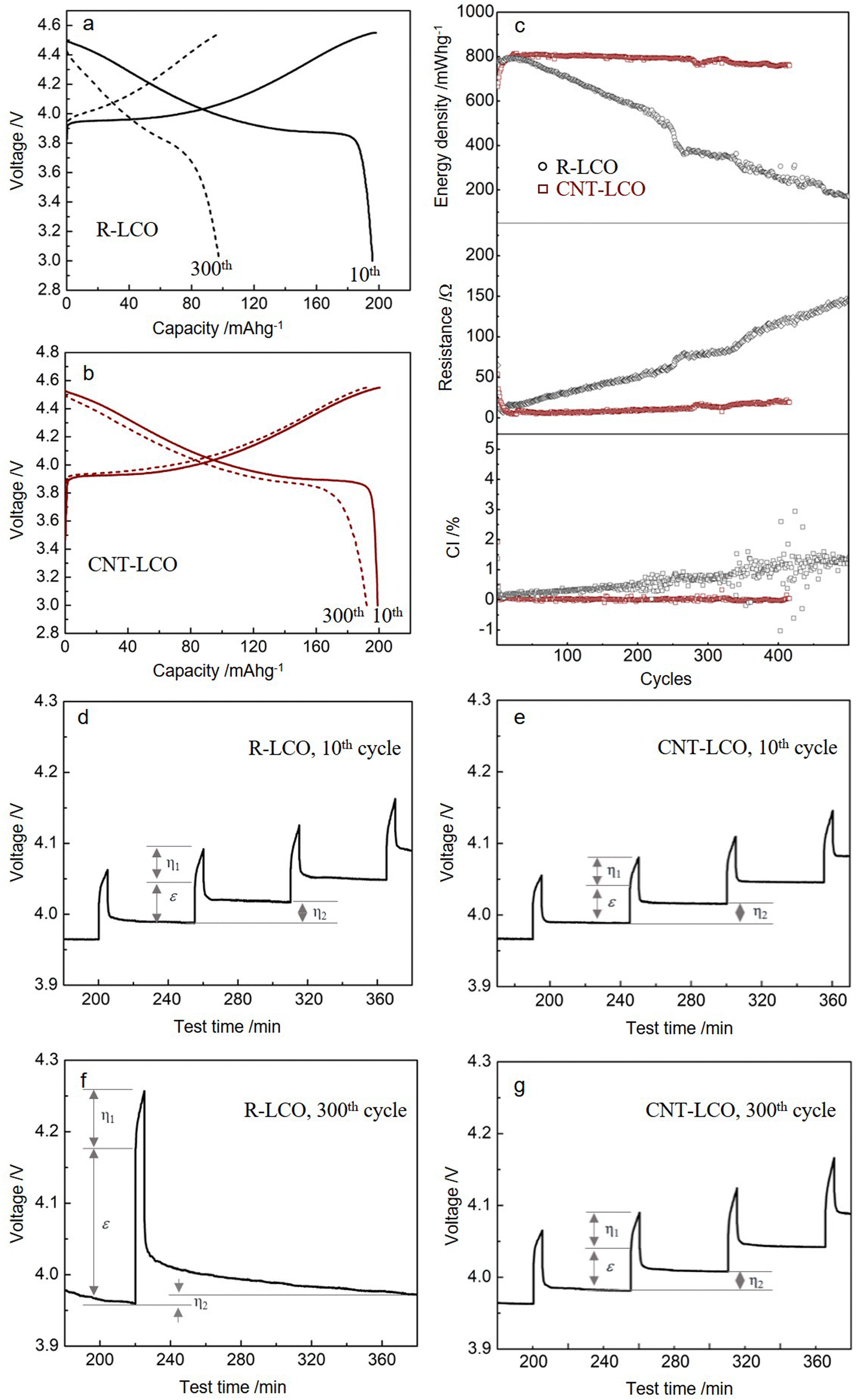 | ||
| Fig. 2 Electrochemical performances of R-LCO and CNT-LCO. (a) and (b) Charge/discharge profiles of R-LCO and CNT-LCO at the 10th and 300th cycles between 3.0 and 4.55 V. (c) Cycling performances of the discharge energy density, Coulombic inefficiency, and interior resistance of R-LCO and CNT-LCO. (d)–(g) GITT profiles of R-LCO and CNT-LCO at the 10th and 300th cycles. | ||
The cycling performances of R-LCO and CNT-LCO at 200 mA g−1 between 3.0 and 4.55 V in half-cells are compared in Fig. 2b. While the discharge energy density of R-LCO decreased to 210 mW h g−1 after 400 cycles, that of CNT-LCO remained as high as 757 mW h g−1, 94.0% of that of the 10th cycle (0.015% decay per cycle). Additionally, while the average polarization resistance (Rp ≡ (Vc − Vd)/2I) of R-LCO increased to above 120 Ω in the 400th cycle, that of CNT-LCO decreased to 21 Ω. The suppressed R indicated that the CNT-LCO cathode had well-maintained electrochemical kinetics during high voltage cycling, which not only promoted the high retention of discharge energy density but also reduced heat generation during fast charging/discharging.
We also note that the Coulombic inefficiency (CI ≡ (100 − CE)%) of CNT-LCO was as low as 0.16%, while that of R-LCO increased to 1.3% after 400 cycles. The CI increase during the R-LCO cycling is likely a result of particle cracking during high-voltage charging, which would not only aggravate the irreversible electrolyte decomposition at the newly generated crack surfaces and create new cathode–electrolyte interphases (CEI) but also impede electronic/polaronic conduction in the following cycles. In contrast, the steady CI during the CNT-LCO cycling indicates that the CNT cocoons greatly suppressed cracking and irreversible side reactions from the electrolyte at high voltage. The cycling performance at higher rates is shown in Fig. S4 (ESI†). Fig. S5 (ESI†) shows that CNT-LCO retained 83.6% and 75.8% of its capacity after 280 cycles when charged to higher upper voltages of 4.60 V and 4.62 V.
To better understand the mechanism for the different electrochemical behaviors during R-LCO and CNT-LCO cycling, GITT tests were performed on the two cathodes. As shown in Fig. 2c and Table S2 (ESI†), from the ε that was mainly contributed by the Ohmic resistance (IRΩ) during the 10th charging, it can be inferred that the electronic resistance in the 0.5 wt% CNT-cocooned CNT-LCO electrode was slightly lower than that of the 3 wt% CB-added R-LCO electrode in the initial cycles. However, while εR-LCO increased by about three times after 300 cycles, εCNT-LCO remained almost the same as in the 10th cycle. This indicates that the matrix for electrical conduction in R-LCO may have been broken during high-voltage cycling, but the CNT network in CNT-LCO maintains long-range electronic percolation throughout the electrode.
More importantly, the GITT results showed that 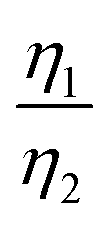 , which was attributed to the polaron diffusion from the interface to the bulk of the LCO crystals, was surprisingly different between R-LCO and CNT-LCO. Fig. 2c and d and Table S2 (ESI†) shows that the
, which was attributed to the polaron diffusion from the interface to the bulk of the LCO crystals, was surprisingly different between R-LCO and CNT-LCO. Fig. 2c and d and Table S2 (ESI†) shows that the 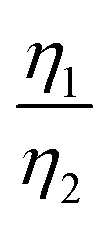 of CNT-LCO (e.g. at an OCV of 4.0 V) was much lower than that of R-LCO in the 10th cycle; moreover, the
of CNT-LCO (e.g. at an OCV of 4.0 V) was much lower than that of R-LCO in the 10th cycle; moreover, the 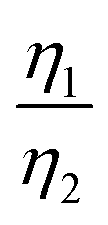 of R-LCO increased by more than 3 times after 300 cycles, whereas that of CNT-LCO changed very little. For cathode crystals, σ1 can be calculated22,23 using eqn (1):
of R-LCO increased by more than 3 times after 300 cycles, whereas that of CNT-LCO changed very little. For cathode crystals, σ1 can be calculated22,23 using eqn (1):
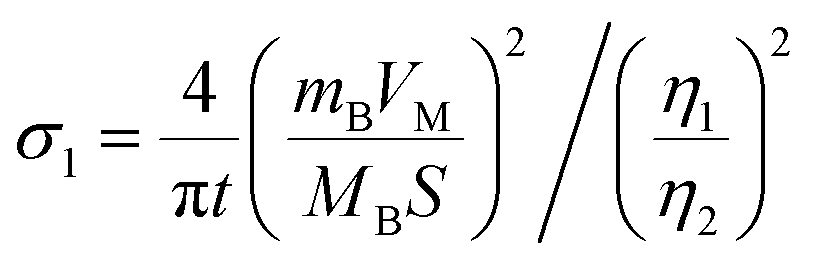 | (1) |
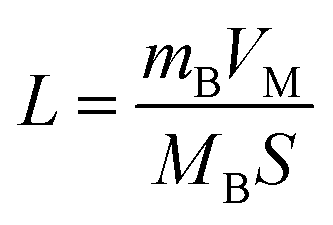 | (2) |
eqn (1) can be rewritten as
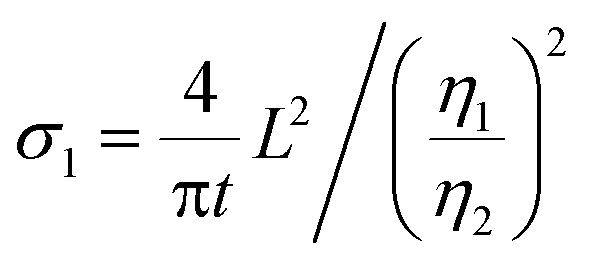 | (3) |
Considering that R-LCO and CNT-LCO have the same LCO particles and should have the same σ1, the value of 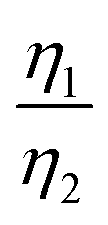 can be proportioned to L in the particle; therefore, the lower
can be proportioned to L in the particle; therefore, the lower 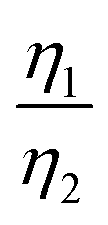 in CNT-LCO indicates that the L in CNT-LCO is much shorter than that in R-LCO. In other words, the full contact between the CNT cocoon and the LCO particles has greatly optimized the polaron diffusion path in LCO and shortened L in the large LCO crystals during cycling. From the GITT results in Fig. 2c–f and Table S2 (ESI†), the L in CNT-LCO can be estimated as 1/1.13 to that in R-LCO in the 10th cycle, while L significantly increased in R-LCO after 300 cycles, it remained much stable in CNT-LCO and could be estimated as less than 1/3.17 of the former.
in CNT-LCO indicates that the L in CNT-LCO is much shorter than that in R-LCO. In other words, the full contact between the CNT cocoon and the LCO particles has greatly optimized the polaron diffusion path in LCO and shortened L in the large LCO crystals during cycling. From the GITT results in Fig. 2c–f and Table S2 (ESI†), the L in CNT-LCO can be estimated as 1/1.13 to that in R-LCO in the 10th cycle, while L significantly increased in R-LCO after 300 cycles, it remained much stable in CNT-LCO and could be estimated as less than 1/3.17 of the former.
The shortened L in CNT-LCO can be a result of the maximized electrical contact interface between the LCO particles and the CNT cocoon (aB → asurface), which would erase the huge Li concentration gradient in LCO and eliminate the ECI. To comprehensively investigate the phenomenon of electrochemical indentation of the MIEC under fast high-voltage charging, we conducted finite-element simulations to accurately model the de-lithiation process in the LCO crystals when charging R-LCO and CNT-LCO. To model the LCO particles in R-LCO, the lithium fluxes were imposed on selected twelve rectangular patches with a size of 9 μm2 each. In contrast, the LCO particles in CNT-LCO experienced a uniform lithium flux over the whole surface. The total current contributed by each LCO particle (I) was maintained at 200 mA g−1 for both electrodes.
As shown in Fig. 3a, the LCO particles in R-LCO exhibit a larger radial lithium concentration gradient compared to that in CNT-LCO during charging. Specifically, when the lithium mole fraction dropped to 0.55 near the electric contact area with CB (site A), the lattice at the LCO surface would trigger the phase transition from a hexagonal structure to a monoclinic structure. This low Li concentration at the surface can lead to an irreversible structural transformation that significantly affects battery life. Since the de-lithiation process introduces a volume change of around 2.5%,13 the nonuniform de-lithiation resulting from a small contact area produces a high Li gradient and nonuniform volume change. This incompatibility would lead to an extremely high local mechanical stress of up to 2000 MPa, as shown in Fig. 3b, which approaches the yield strength of the LCO crystal at around 2700 MPa.
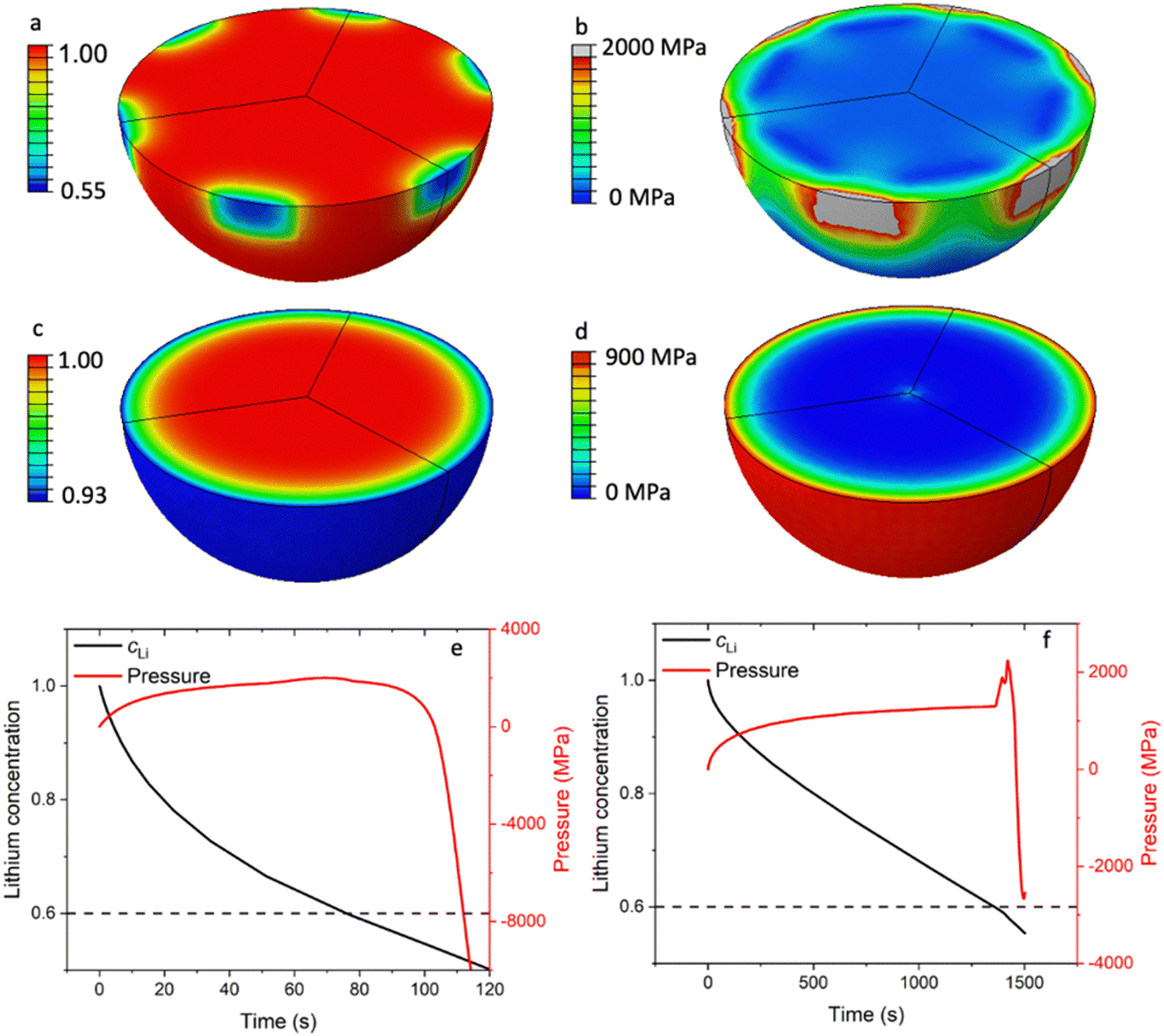 | ||
| Fig. 3 FEM simulations of the electrochemical indentation of R-LCO and CNT-LCO. (a) Li concentration in the LCO particle in R-LCO, (b) the von Mises stress field of LCO in R-LCO, (c) Li concentration in the LCO particle in CNT-LCO, (d) the von Mises stress field of LCO in CNT-LCO, (e) lithium concentration and pressure at a characteristic surface point in R-LCO and (f) the lithium concentration and pressure at a characteristic surface point in CNT-LCO. | ||
ECI is similar to mechanical indentation,24 where a small mechanical load is applied to the materials through a sharp indenter. Here, the partial contact produces a nonuniform Li concentration and volume change, which further leads to a localized stress field near the contact area. Therefore, we can call this phenomenon electrochemical indentation, which is a contact electromechanics problem analogous to the contact problems in mechanics.25 Electrochemical indentation imposes a high local mechanical stress near the surface of LCO particles, which could lead to early cracks and degrade the LCO particles in R-LCO. However, Fig. 3c shows that the LCO particles in CNT-LCO have a much smoother and spherically symmetric Li concentration field because of the fully wrapped CNT cocoon on the LCO particle surface. Note that, under the same charge/discharge current, the ΔcLi between the surface and centroid within the LCO particle in CNT-LCO was only 7% of the initial concentration, while that of R-LCO reached 45%. Due to the relatively low Li concentration gradient, the mechanical interior stress within the LCO particles in CNT-LCO was much lower, as shown in Fig. 3d, and was only around 900 MPa at the outer surface.
More importantly, upon further charging, the deep de-lithiation would involve oxygen redox and cause the lattice at the LCO surface to shrink, which is contrary to the initial volume expansion in bulk. As shown in Fig. 3e, this abnormal shrinkage would apply an additional load to the surface and lead to the inelastic deformation of the LCO particles in R-LCO at around 100s of charging. On the other hand, the LCO particles in CNT-LCO remained elastic and undamaged until 1500s of charging, when the abnormal shrinkage flipped the sign of the pressure field near the surface from positive (compression) to negative (tension). Therefore, the time to the initial damage (td) in the LCO particles was significantly prolonged from 100s in R-LCO to 1500s in CNT-LCO after the introduction of the CNT-cocoon lamination. The finite element simulations demonstrated that an enlarged electronic contact area between the LCO particles and CNT cocoon is greatly favorable for erasing the high local Li concentration gradient and eliminating the electrochemical indentation in the LCO particles during high-voltage cycling.
In Fig. 3f, sharp pressure spikes and transitions from positive to negative pressure are illustrated, corresponding to the non-linear volumetric changes depicted in Fig. S6 (ESI†). During the de-lithiation phase, the unit cell volume of LixCoO2 initially increases from 96.50 Å3 to 98.90 Å3 as the lithium content x approaches 0.6. This volumetric expansion is primarily due to the increased electrostatic repulsion among the CoO2 layers as lithium ions are progressively removed. As de-lithiation continues beyond this point, the unit cell volume subsequently contracts to 95.31 Å3. This contraction is linked to the repositioning of the CoO2 layers in response to the significant lithium removal, resulting in a denser structural configuration. The abrupt change in the partial volume of lithium at the critical x of 0.6 leads to these sharp pressure spikes and the transition from a compressive to a tensile load.
ECI during high-voltage charging would damage the LCO crystal and cause particle cracking; therefore, the elimination of the ECI can be key to maintaining the integrity of LCO particles during high-voltage cycling. Fig. 4a and b compare the morphologies of the LCO particles in R-LCO and CNT-LCO, respectively, after 120 cycles. The SEM images show that in the cycled CNT-LCO electrode, the CNT cocoon still fully covered and tightly wrapped all of the LCO particles, maintaining intimate electrical contact between the LCO particles and the CNT cocoon, whereas much of the LCO particles lost electrical contact with the CB in R-LCO. More importantly, while severe corrosion and deep cracks were found everywhere on the LCO surface in R-LCO, the LCO particles in CNT-LCO still had smooth surfaces.
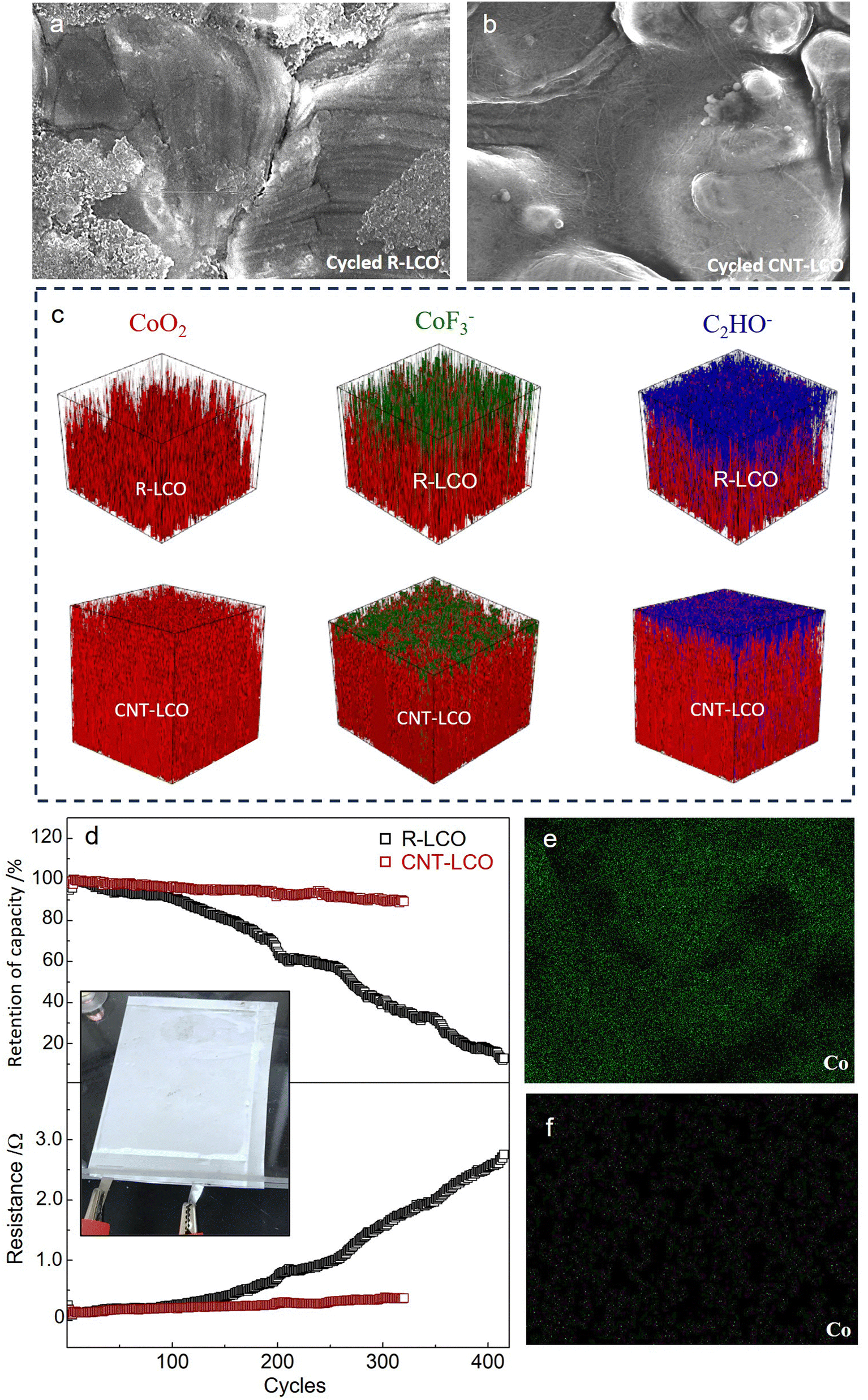 | ||
| Fig. 4 Particles after cycling and full-cell cycling performance. (a) and (b) SEM images of the LCO particles in R-LCO and CNT-LCO after high-voltage cycling. (c) TOF-SIMS analysis of the surface of the cycled R-LCO11 and CNT-LCO electrodes. (d) Cycling performance of discharge energy retention and cell resistances of R-LCO and CNT-LCO full cells. (e) and (f) EDS mapping of Co on the graphite anode from the cycled R-LCO (e) and CNT-LCO (f) full cells. | ||
The cracks at the LCO particle surface would introduce and expose new particle surfaces to the electrolyte and increase the interface area between the cathode and electrolyte. The increase of the free surface area would seriously aggravate the side reactions with liquid electrolytes at high voltages, including the oxygen release and electrolyte decomposition, so that the maintained morphology of LCO particles in CNT-LCO would suppress these side reactions and prolong high-voltage cycling life. To better compare the side reactions that occurred on the surfaces of the two cathodes, we performed TOF-SIMS to analyze the surficial compositions of R-LCO and CNT-LCO after cycling. Fig. 4c and Fig. S7 (ESI†) show the distribution of the CoF3− component at the surface of the two cathodes, which is a representative indicator of cathode corrosion from HF attack in the electrolyte. While the CoF3− layer was thick at the surface of R-LCO and in a penetrating fashion towards the deep,11 it was much thinner at the CNT-LCO surface. In other words, the surface of R-LCO was seriously corroded, and the damage spread into the particle bulk (along the cracks in the crystal) after high-voltage cycling, but such corrosion was very weak and only occurred at the outermost surface of the CNT-LCO.
Fig. 4c and Fig. S7 (ESI†) also compare the distribution of C2HO− species to evaluate electrolyte decomposition. It was quite clear that the C2HO− layer on the CNT-LCO surface was also much thinner than that of R-LCO,11 indicating that the electrolyte decomposition on the CNT-LCO surface was greatly prevented during cycling. The suppressed surface corrosion and electrolyte decomposition at the CNT-LCO surface confirmed the prevention of LCO cracks.
We then tested R-LCO and CNT-LCO in pouch full cells that were paired with commercial graphite anodes and a lean electrolyte (2 g A h−1) at a current density of 200 mA g−1. As shown in Fig. 4d, the discharge capacity retention of the CNT-LCO full cell was 91% after 300 cycles, whereas that of R-LCO degraded to 37%. The interior resistances were also quite different between the two cells: that of R-LCO increased to 1.65 Ω after 300 cycles, while it was only 0.34 Ω for the CNT-LCO cell. The stable cycling stability of the CNT-LCO full cell not only verified the validity of the CNT cocoon in stabilizing high-voltage LCO cycling, but it also indicated that the cocooning technique has practical application potential. After cycling, we also measured the Co distribution at the surface of the graphite anodes that disassembled from the cycled full cells. The EDS mapping in Fig. 4e and f shows that the Co deposition on the graphite anode from the CNT-LCO cell was much less than that from the R-LCO cell. It can be inferred that the prevention of LCO cracks also greatly suppressed Co dissolution from the cathode, which further maintained the interior resistances and stabilized high-voltage full-cell cycling.
In the CNT-LCO electrode, 0.5 wt% CNT was not only fully wrapped around all LCO particles but also self-intertwined to be a free-standing electrode so that no additional binder or metal current collector was necessary for preparing the electrode, as shown in Fig. 5a. In this work, we compressed this free-standing electrode to 4.45 g cm−3 (Fig. 5b), then the volumetric energy density of CNT-LCO reached ∼3200 W h L−1 (electrode) based on the whole free-standing cathode volume, and the volumetric and gravimetric energy densities of the new designed full cell can be expected to be significantly improved to 1212.5 W h L−1 (cell) and 341 W h kg−1 (cell) (Fig. S8 and the Discussion section, ESI†), respectively.
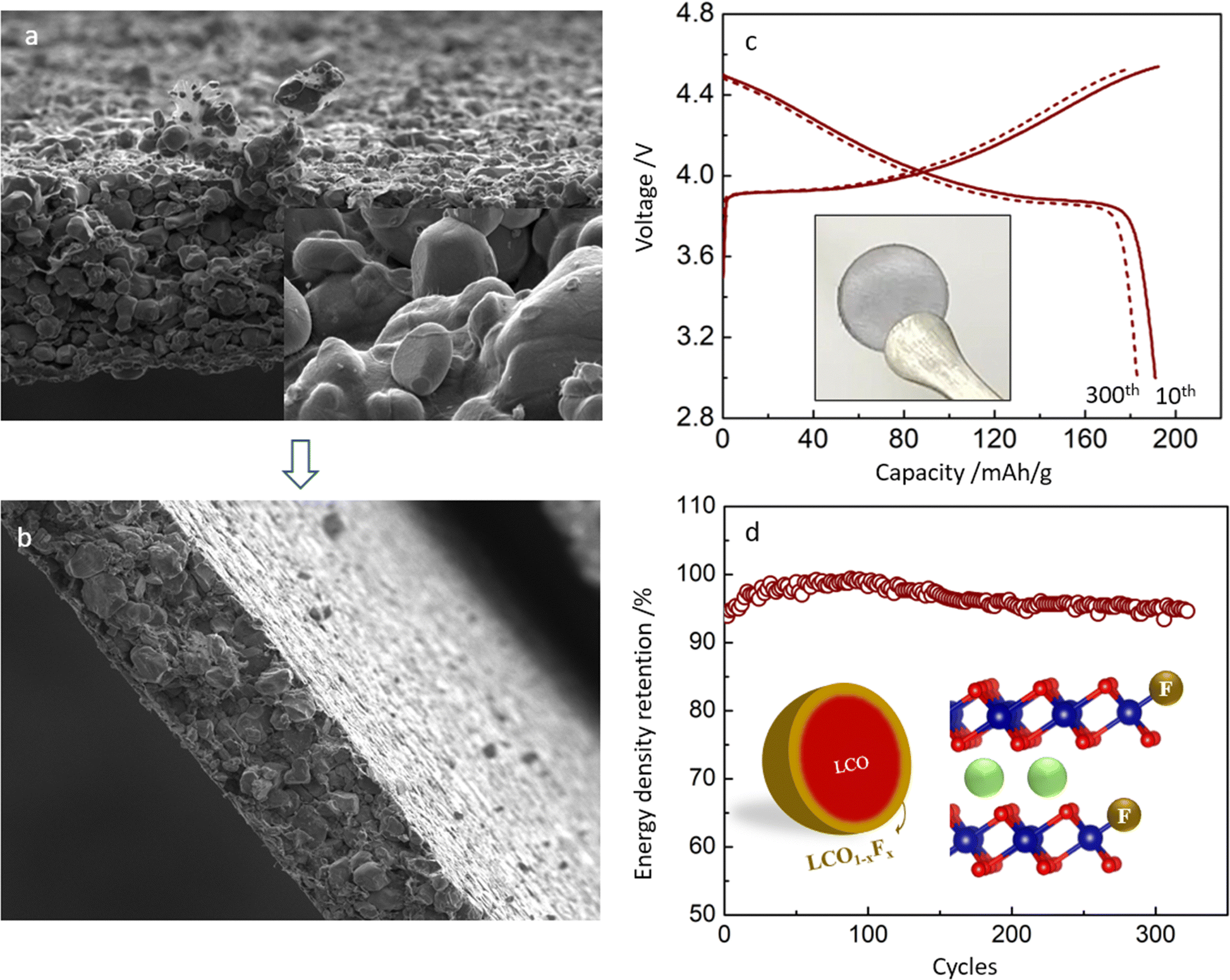 | ||
| Fig. 5 Images of the free-standing CNT-LCO electrode and cycling performance after regeneration. (a) Images of the free-standing CNT-LCO electrode before pressing. The inset shows the LCO particles in CNT-LCO. (b) Images of the free-standing CNT-LCO electrode after rolling pressing to a high density. (c) Charge/discharge profiles of the cycled CNT-LCO after in situ regeneration; the inset is the photograph of a new free-standing CNT-LCO electrode that was made of in situ regenerated materials. (d) Cycling performance of the regenerated CNT-LCO electrode; the inset is the proposed schematic of the F-doped lattice at the surface of the regenerated LCO particles. | ||
Besides the improvements in high-voltage cycling stability and energy density, the free-standing CNT-LCO cathode also makes it easier to recycle at the end of life. Owing to the absence of an aluminum current collector and sticky polymeric binder, the cycled CNT-LCO electrode can be efficiently regenerated and recycled with a facile in situ air annealing treatment at 500–600 °C, which oxidizes the CNT-cocoon and organic species at the surface. This does not require the complicated separation and collection processes of the active materials, which must be involved in the regular battery regeneration and recycling process.7,26 The thermogravimetric (TG) analysis of the regeneration process is shown in Fig. S9 (ESI†). The recycled LCO materials are still robust and can be again directly used for a new free-standing CNT-LCO electrode (inset in Fig. 5c) in the cell.
Fig. 5c and d show the charge/discharge profile and cycling performance of the regenerated CNT-LCO cathode between 3.0 and 4.55 V in half-cells. Though the reversible capacity of the regenerated CNT-LCO was slightly lower than that of the fresh electrode (Fig. 2a), it was surprising that the cycling stability even improved, as the decay of energy density was less than 0.013% per cycle after the 150th cycle. The improved cycling stability of the regenerated CNT-LCO can be understood from the appropriate lattice doping of F at the LCO particle surface during annealing (Fig. 5d), where the F element came from the CEI that formed in the previous long cycling. The F doping at the regenerated LCO surface is demonstrated by the XPS analysis, as shown in Fig. S10 (ESI†). An appropriate amount of F doping at the LCO particle surface could greatly prevent lattice-oxygen escape and HF corrosion from the electrolyte, which both favored high-voltage LCO cycling stability. This result indicates that free-standing CNT-LCO can be a promising green battery candidate.
Conclusions and outlook
In this work, we established a CNT-cocooned LCO electrode that was totally free of a metallic current collector or chemical binder. The plane-to-plane contact between the LCO particles and the CNT cocoon in the free-standing CNT-LCO electrode optimized the electronic and ionic diffusion paths in the large LCO crystals, eliminated the electrochemical indentation, and significantly extended the high-voltage LCO battery cycling life. The free-standing CNT-LCO electrode had an ultra-high volumetric energy density of 3200 W h L−1 (electrode) based on the whole electrode and could be simply recycled/regenerated with an economic in situ air annealing process.This CNT-cocooned free-standing electrode technology can also be generalized to other electrode materials, such as high-Ni NMC cathodes, LiFePO4 cathodes, graphite anodes, and Si anodes. The CNT cocoon will not only improve the cycling stability of the active materials (especially under fast charging) but also improve the energy density of the cell. In addition to the simple, environmentally friendly and cost-friendly recycling and regeneration process, the CNT-cocooned free-standing electrodes provide important guidance for new sustainable electrode design and battery fabrication. To promote this free-standing electrode technology in larger pouch cells, new electrode structures and fabrication technologies are being developed.
Lastly, we believe that the theory of electrochemical indentation (ECI) and how a low electronic contact area leads to ECI-induced cracking is fundamentally important for all mixed ionic electronic conductors (MIEC) and even for solid electrolytes (SE), which is just a limiting case of the MIEC (electronic transference number telectronic < 10−4).27 With no changes to the active material, using just 0.5 wt% CNT (serving both as a physical binder and a conductive agent) instead of the standard electrode preparation led to better battery performance and durability. This very surprising demonstration and the underlying mechanism should be realized by the broader electrochemical material community.
Methods
Electrode preparation
The pristine commercial LiCoO2 particles (C-LCO) were ordered from Targray Corp Ltd (https://www.targray.com/li-ion-battery/cathode-materials/cathode-active-materials). The R-LCO electrode was prepared using a regular slurry coating process on an Al foil, which comprised 95 wt% commercial LCO particles, 3 wt% carbon black and 2 wt% PVDF binder, and then vacuum-dried at 110 °C for 3 h. The CNT-LCO electrode was prepared using a vacuum filtration method. A commercial CNT solution was used for the CNT-LCO preparation, which was ordered from OCSIAL (https://ocsial.com/nanotubes/), the details of which are listed in Table S1 (ESI†). A slurry with 99.5 wt% of commercial LCO particles and 0.5 wt% CNT was ultrasonically mixed in an aqueous solution and then filtered on a round PVDF filter at room temperature. The deposition was washed with water, filtered 3 times, vacuum-filtered in air for 12 h, and vacuum-dried at 110 °C for 3 h. The mass loadings of LCO in R-LCO and CNT-LCO were both controlled at around 20 mg cm−2, and both of the electrodes were deeply pressed using a rolling press.Material characterization
Scanning electron microscopy was performed using a Zeiss Merlin high-resolution scanning electron microscope with X-ray spectroscopy (EDS). The thermogravimetric (TG) analysis was performed on a NETZSCH STA449F3 TG-DSC instrument in air at a heating rate of 5 °C per min. The Raman spectra were obtained using a Horiba Jobin-Yvon HR800 Raman spectrometer with an 830nm laser. For the time-of-flight secondary-ion mass spectrometry (TOF-SIMS) studies, ION-TOF TOF-SIMS 5 was used under the pressure of the analysis chamber below 1.1 × 10−9 mbar. Organic imaging in a delay extraction mode with pulsed 30 keV Bi3+ (pulsed current: 0.16–0.28 pA) ion beam was applied for high lateral resolution mapping (<800 nm) analysis; the typical analysis area was 100 × 100 μm2, with 1 keV Cs+ ion beam sputtering at the same time (69.27–82.74 nA, 300 × 300 μm2 sputter raster).
Electrochemical tests
R2032 coin cells were fabricated with the above cathode, a Li metal anode, a Celgard 2400 polymeric separator, and a commercial electrolyte solution of 1.2 M LiPF6 was dissolved in a mixture of EC and DEC in a volume ratio of 1:1. Pouch full cells were fabricated with the above cathodes, a commercial graphite anode (double-side coated on Cu foil), a separator of Celgard 2400 polymer and a commercial electrolyte solution of 1.2 M LiPF6 was dissolved in a mixture of EC and DEC in a volume ratio of 1:1, and 2wt% vinylene carbonate additive (ordered from Soulbrain MI). The commercial graphite anode had a loading density of ∼15 mg cm−2 with ∼4.5 mA g cm−2 (tested in half-cell at 0.2C). The pouch full cells of R-LCO were fabricated with double layers of electrodes (one graphite anode foil with a double-sided coating and two R-LCO cathode foils with a single-sided coating). The pouch full cells of CNT-LCO were fabricated with double layers of electrodes (two graphite anode foils with single-sided coating and one free-standing CNT-LCO cathode). The amount of electrolyte added in the pouch cell was about 2 g A−1 h−1. A LAND CT2001A 8-channel automatic battery test system (Wuhan Lanhe Electronics) was used to charge/discharge the cells. An electrochemical workstation (Gamry Instr, Reference 3000) was used for the galvanostatic intermittent titration technique (GITT) with a constant current for 200 seconds followed by 50 min relaxation until 4.6 V (specifically, the relaxation time for R-LCO in the 300th cycle was 150 min). All the electrochemical tests were carried out at room temperature.
Finite element simulations
Detailed justification of the physical model for mixed conductor is presented in ref. 27. We simulated the de-lithiation processes of the R-LCO and CNT-LCO particles using the ABAQUS finite element package. The diameter of each spherical particle was 15 μm. The concentration field of LicLi was determined by solving the standard diffusion equation. The concentration of Li was normalized by the Li concentration at the LiCoO2 composition, such that cLi varied between 0 and 1. The diffusivity of Li, DLi, was taken as 6.5 × 10−11 cm2 s−1.13 The total current per particle for R-LCO and CNT-LCO was 0.19 nA, corresponding to 200 mA g−1. The particles were initially pristine and subjected to a constant Li flux at the surface contact. We adopted a chemomechanical model to describe lithium-induced volume change. The total strain was decomposed into three parts, εij = εcij + εeij + εpij, where εcij is the volumetric strain due to the change in the Li concentration, εeij is the elastic strain, and εpij is the plastic strain. The volumetric strain obeys a bi-linear relation ![[small epsi, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a1.gif) cij = αċLi, where α = 0.126 if cLi < 0.6 and α = −0.0625 if cLi ≧ 0.6. The elastic strain follows the Hooke's law,
cij = αċLi, where α = 0.126 if cLi < 0.6 and α = −0.0625 if cLi ≧ 0.6. The elastic strain follows the Hooke's law, 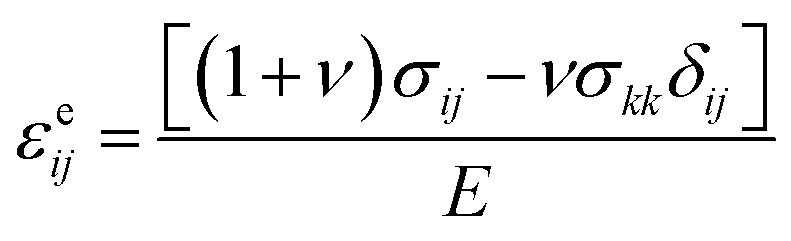 , where E is Young's modulus, ν is the Poisson ratio, σij is the mechanical stress, and δij is the Kronecker delta. The plastic strain εpij obeys the classical J2 flow rule, where the material yields when the von Mises stress
, where E is Young's modulus, ν is the Poisson ratio, σij is the mechanical stress, and δij is the Kronecker delta. The plastic strain εpij obeys the classical J2 flow rule, where the material yields when the von Mises stress  is equal to the yield strength σY. For the LCO particle, we choose E = 191 GPa, ν = 0.24, and σY = 2.6 GPa.28 When the R-LCO particle was charged for 90 s, the mechanical stress from the electrochemical indentation started to trigger plasticity near the contact. For the particles in CNT-LCO, the stress remained below the yield strength after 600 s of charging.
is equal to the yield strength σY. For the LCO particle, we choose E = 191 GPa, ν = 0.24, and σY = 2.6 GPa.28 When the R-LCO particle was charged for 90 s, the mechanical stress from the electrochemical indentation started to trigger plasticity near the contact. For the particles in CNT-LCO, the stress remained below the yield strength after 600 s of charging.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.References
- Y. C. Lyu, et al., An Overview on the Advances of LiCoO(2)Cathodes for Lithium-Ion Batteries, Adv. Energy Mater., 2021, 11, 2000982, DOI:10.1002/aenm.202000982.
- M. G. Kim and J. Cho, Reversible and High-Capacity Nanostructured Electrode Materials for Li-Ion Batteries, Adv. Funct. Mater., 2009, 19, 1497–1514, DOI:10.1002/adfm.200801095.
- Y. Y. Huang, et al., Mg-Pillared LiCoO2: Towards Stable Cycling at 4.6 V, Angew. Chem., Int. Ed., 2021, 60, 4682–4688, DOI:10.1002/anie.202014226.
- L. L. Wang, et al., A Novel Bifunctional Self-Stabilized Strategy Enabling 4.6 V LiCoO2 with Excellent Long-Term Cyclability and High-Rate Capability, Adv. Sci., 2019, 6, 1900355, DOI:10.1002/advs.201900355.
- J. N. Zhang, et al., Dynamic evolution of cathode electrolyte interphase (CEI) on high voltage LiCoO2 cathode and its interaction with Li anode, Energy Storage Mater., 2018, 14, 1–7, DOI:10.1016/j.ensm.2018.02.016.
- X. R. Yang, et al., Pushing Lithium Cobalt Oxides to 4.7 V by Lattice-Matched Interfacial Engineering, Adv. Energy Mater., 2022, 12, 2200197, DOI:10.1002/aenm.202200197.
- J. X. Wang, et al., Direct and green repairing of degraded LiCoO2 for reuse in lithium-ion batteries, Nat. Sci. Rev., 2022, 9, nwac097, DOI:10.1093/nsr/nwac097.
- J. N. Zhang, et al., Trace doping of multiple elements enables stable battery cycling of LiCoO2 at 4.6 V, Nat. Energy, 2019, 4, 594–603, DOI:10.1038/s41560-019-0409-z.
- W. J. Xue, et al., Stabilizing electrode-electrolyte interfaces to realize high-voltage Li‖LiCoO2 batteries by a sulfonamide-based electrolyte, Energy Environ. Sci., 2021, 14, 6030–6040, 10.1039/d1ee01265g.
- Z. Zhu, et al., Gradient-morph LiCoO 2 single crystals with stabilized energy density above 3400 W h L−1, Energy Environ. Sci., 2020, 13, 1865–1878 RSC.
- Z. Zhu, et al., A Surface Se-Substituted LiCo [O2–δSeδ] Cathode with Ultrastable High-Voltage Cycling in Pouch Full-Cells, Adv. Mater., 2020, 32, 2005182 CrossRef CAS PubMed.
- J. X. Zhang, et al., Interfacial Design for a 4.6 V High-Voltage Single-Crystalline LiCoO2 Cathode, Adv. Mater., 2022, 34, 210835, DOI:10.1002/adma.202108353.
- Q. Liu, et al., Approaching the capacity limit of lithium cobalt oxide in lithium ion batteries via lanthanum and aluminium doping, Nat. Energy, 2018, 3, 936–943, DOI:10.1038/s41560-018-0180-6.
- S. L. Mao, et al., Outside-In Nanostructure Fabricated on LiCoO2 Surface for High-Voltage Lithium-Ion Batteries, Adv. Sci., 2022, 9, 2104841, DOI:10.1002/advs.202104841.
- X. M. Fan, et al., In situ inorganic conductive network formation in high-voltage single-crystal Ni-rich cathodes, Nat. Commun., 2021, 12, 5320, DOI:10.1038/s41467-021-25611-6.
- R. Rojaee and R. Shahbazian-Yassar, Two-Dimensional Materials to Address the Lithium Battery Challenges, ACS Nano, 2020, 14, 2628–2658, DOI:10.1021/acsnano.9b08396.
- X. F. Wang, et al., Fluorine doped carbon coating of LiFePO4 as a cathode material for lithium-ion batteries, Chem. Eng. J., 2020, 379, 122371, DOI:10.1016/j.cej.2019.122371.
- W. Lee, et al., Advances in the Cathode Materials for Lithium Rechargeable Batteries, Angew. Chem., Int. Ed., 2020, 59, 2578–2605, DOI:10.1002/anie.201902359.
- D. L. Chao, et al., A V2O5/Conductive-Polymer Core/Shell Nanobelt Array on Three-Dimensional Graphite Foam: A High-Rate, Ultrastable, and Freestanding Cathode for Lithium-Ion Batteries, Adv. Mater., 2014, 26, 5794–5800, DOI:10.1002/adma.201400719.
- S. Luo, et al., Binder-Free LiCoO2/Carbon Nanotube Cathodes for High-Performance Lithium Ion Batteries, Adv. Mater., 2012, 24, 2294–2298, DOI:10.1002/adma.201104720.
- M. Xie, B. Li and Y. Zhou, Free-standing high-voltage LiCoO 2/multi-wall carbon nanotube paper electrodes with extremely high areal mass loading for lithium ion batteries, J. Mater. Chem. A, 2015, 3, 23180–23184 RSC.
- Y.-C. Chien, et al., Rapid determination of solid-state diffusion coefficients in Li-based batteries via intermittent current interruption method, Nat. Commun., 2023, 14, 2289, DOI:10.1038/s41467-023-37989-6.
- Z. Shen, L. Cao, C. D. Rahn and C.-Y. Wang, Least Squares Galvanostatic Intermittent Titration Technique (LS-GITT) for Accurate Solid Phase Diffusivity Measurement, J. Electrochem. Soc., 2013, 160, A1842, DOI:10.1149/2.084310jes.
- G. H. Lee, et al., High-Strength Chemical-Vapor Deposited Graphene and Grain Boundaries, Science, 2013, 340, 1073–1076, DOI:10.1126/science.1235126.
- K. L. Johnson and K. L. Johnson, Contact mechanics, Cambridge university press, 1987 Search PubMed.
- J. X. Wang, et al., Direct conversion of degraded LiCoO2 cathode materials into high-performance LiCoO2: A closed-loop green recycling strategy for spent lithium-ion batteries, Energy Storage Mater., 2022, 45, 768–776, DOI:10.1016/j.ensm.2021.12.013.
- Y. Zhang, Y. Dong and J. Li, Electrochemical shock and transverse cracking in solid electrolytes, Acta Mater., 2024, 265, 119620, DOI:10.1016/j.actamat.2023.119620.
- E. J. Cheng, N. J. Taylor, J. Wolfenstine and J. Sakamoto, Elastic properties of lithium cobalt oxide (LiCoO2), J. Asian Ceram. Soc., 2017, 5, 113–117 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00722k |
| This journal is © The Royal Society of Chemistry 2024 |