
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaser-induced high-entropy alloys as long-duration bifunctional electrocatalysts for seawater splitting†
Yunchao
Xie‡
 ab,
Shichen
Xu‡
c,
Andrew C.
Meng
d,
Bujingda
Zheng
a,
Zhenru
Chen
a,
James M.
Tour
*cefg and
Jian
Lin
*a
ab,
Shichen
Xu‡
c,
Andrew C.
Meng
d,
Bujingda
Zheng
a,
Zhenru
Chen
a,
James M.
Tour
*cefg and
Jian
Lin
*a
aDepartment of Mechanical and Aerospace Engineering, University of Missouri, Columbia, MO 65201, USA. E-mail: LinJian@missouri.edu
bDepartment of Mechanical and Manufacturing Engineering, Miami University, Oxford, OH 45056, USA
cDepartment of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005, USA. E-mail: tour@rice.edu
dDepartment of Physics and Astronomy, University of Missouri, Columbia, MO 65201, USA
eDepartment of Materials Science and NanoEngineering, Rice University, 6100 Main Street, Houston, Texas 77005, USA
fDepartment of Computer Science Engineering, Rice University, 6100 Main Street, Houston, Texas 77005, USA
gNanoCarbon Center and the Rice Advanced Materials Institute, Rice University, 6100 Main Street, Houston, Texas 77005, USA
First published on 7th October 2024
Abstract
Electrocatalytic seawater splitting has garnered significant attention as a promising approach for eco-friendly, large-scale green hydrogen production. Development of high-efficiency and cost-effective electrocatalysts remains a frontier in this field. Herein, we report a rapid in situ synthesis of FeNiCoCrRu high-entropy alloy nanoparticles (HEA NPs) by direct CO2 laser induction of metal precursors on carbon paper under ambient conditions. Due to the induced ultrahigh temperature and ultrafast heating/quenching rates, FeNiCoCrRu HEA NPs with sizes ranging from 5 to 40 nm possess uniform phase homogeneity. FeNiCoCrRu HEA NPs exhibit exceptional bifunctional electrocatalytic activities, delivering overpotentials of 0.148 V at 600 mA cm−2 for the hydrogen evolution reaction and 0.353 V at 300 mA cm−2 for the oxygen evolution reaction in alkaline seawater. When assembled FeNiCoCrRu HEA NPs to an electrolyzer, it shows a negligible voltage increase at 250 mA cm−2 even after over 3000-hour operation. This superior performance can be attributed to the high-entropy design, large electrochemical specific area, and excellent chemical and structural stability. An operando Raman spectroscopy study discloses that the Ni and Ru sites serve as active sites for hydrogen evolution, while the Ni site acts as an active site for oxygen evolution. This work demonstrates a laser-induced eco-friendly nanomaterial synthesis. The systematic studies offer an in-depth understanding of HEA design and its correlation with high-efficiency seawater splitting.
Broader contextHigh-entropy materials have attracted much attention as prospective electrocatalysts due to their unprecedented physicochemical properties. Despite these advances, many challenges remain impeding the practical applications of high-entropy materials. This work reports an in situ, rapid synthesis of FeNiCoCrRu high-entropy alloy nanoparticles by direct CO2 laser induction of metal complexes under ambient conditions. The laser induced ultrahigh temperature (>3200 K), ultrafast heating and quenching rates (105 K s−1), high entropy effect and sluggish diffusion effect render the formation of single-phase solid solution FeNiCoCrRu without phase separation and segregation. When explored for seawater electrolysis, FeNiCoCrRu showed high-efficiency bifunctional electrocatalytic performance and outstanding long duration of >3000 hours. The superior performance can be attributed to the high-entropy design, large electrochemical specific area, chemical and structural stabilization. An operando Raman spectroscopy study discloses the Ni and Ru sites serve as active sites for hydrogen evolution, while Ni sites acts as active sites for oxygen evolution. |
1. Introduction
Hydrogen is a promising and eco-friendly energy source, offering significant potential to reduce greenhouse gas emissions and mitigate climate change. Unlike grey and blue hydrogen, green hydrogen is produced by water electrolysis using renewable energy sources such as wind or solar power, which has gained significant momentum.1 However, relying solely on freshwater electrolysis for large-scale green hydrogen production is impractical, especially in renewable energy-rich but arid regions. Therefore, developing seawater electrolysis as a complementary technology is essential to balance water use between human consumption and hydrogen production, ensuring sustainability in water-scarce areas.2–4 To date, a wide range of electrocatalysts, encompassing (oxy)hydroxides, nitrides, phosphides, and sulfides, have been engineered for seawater electrolysis.5 Nonetheless, seawater electrolysis encounters three primary challenges: the generation of hypochlorite byproducts, severe corrosion at the anode, and the formation of scale and biofouling at the cathode.3 These issues render the active sites inert, thus leading to significant deterioration in electrolyzer performance. Consequently, it is imperative to rationally design highly efficient and robust electrocatalysts capable of addressing these obstacles in the context of high-efficiency seawater electrolysis.Recently, high-entropy materials (HEMs), containing five or more equimolar metal elements, have attracted much attention as prospective electrocatalysts for seawater electrolysis.6–8 This interest arises from the unique properties of HEMs, including inherent thermodynamic and kinetic stability, outstanding corrosion resistance, and unprecedented physicochemical properties.9 For example, TiNbTaCrMo high-entropy alloy nanoparticles (HEA NPs) were prepared through a confinement-assisted arc and plasma shock method within a vacuum chamber.6 It exhibited a requisite overpotential of 0.97 V at 50 mA cm−2 for the hydrogen evolution reaction (HER) in alkaline seawater. In another study, Ni–Fe–Cr–Mn–Co high-entropy (oxy)hydroxides with a flower-like structure were synthesized on nickel foam via a solvothermal process, displaying an overpotential (η10) of 0.223 V at 10 mA cm−2 when used for the oxygen evolution reaction (OER) in alkaline seawater.7 Wang et al. prepared FeCoNiMnMo HEA NPs through molten salt reduction of mixed metal oxides for simulated seawater electrolysis.8 FeCoNiMnMo HEA NPs delivered remarkable OER activity with an η10 value of 0.237 V and showed stability of 200 h at a current density of 100 mA cm−2. While many of these synthesis methods have proven to be effective and productive, they often suffer from several drawbacks. First, many of these processes prove to be time-consuming, spanning from hours to days, and demand large amounts of chemicals and solvents. Additionally, they typically require specialized and sophisticated facilities to maintain high temperatures and/or an inert gas environment. Obviously, these approaches fall short in swiftly screening high efficiency electrocatalysts, given the vast combinatorial libraries for HEM design. Another limitation is that these HEMs are free-standing powders, which require mixture with a binder like Nafion and a subsequent spray-coating step onto conductive substrates. This process diminishes the exposure of active sites, and hinders mass and charge transfer, not to mention the increase in the electrode weight and capital investment. Moreover, vigorous bubble formation during electrolysis imposes significant mechanical stress, leading to a high chance of electrocatalyst detachment from the electrode. To mitigate these issues, a straightforward and effective approach to directly fabricate self-supported electrocatalysts on conductive substrates is quite desirable.
Recently, laser induction has garnered much attention to producing self-supported HEMs using nanosecond (ns) or femtosecond (fs) lasers.10–13 Yang and coworkers implemented ns laser synthesis of CoCrFeNiAl high-entropy oxides on diverse substrates under low pressure.10 Huang et al. synthesized FeCoNiCuPtIr HEA by irradiating metal precursors loaded on multiwall carbon nanotube paper with an ns laser in an inert environment.11 In another work, a set of HEAs and high-entropy ceramics were prepared through ns laser ablation of metal precursors in organic solvents.12 Hegde and his collaborators reported the fabrication of HEAs and multi-component metal oxides on conductive substrates using an fs laser.13 Despite these advances, the requirement for a vacuum or an inert environment, coupled with the high capital cost of ns or fs lasers, impedes their practical applications for scalable HEM synthesis. As an alternative, CO2 lasers have been explored for the fabrication of graphene, metals, alloys, and oxides/sulfides, owing to its simplicity, scalability, cost-effectiveness, and robustness.14–16 To the best of our knowledge, there are no reports exploring the use of CO2 lasers for HEM fabrication.
Herein, we demonstrated the in situ, rapid synthesis of FeNiCoCrRu HEA NPs with porous structures on a conductive substrate via direct CO2 laser induction under ambient conditions. The laser-induced ultrahigh temperature (>3200 K), ultrafast heating and quenching rates (105 K s−1), and high entropy design rendered FeNiCoCrRu HEA NPs with uniformly distributed metal elements and no phase separation. When explored for seawater electrolysis, FeNiCoCrRu HEA NPs showed high-efficiency and stable bifunctional electrocatalytic performance, with overpotentials of 0.148 V at 600 mA cm−2 for the HER and 0.353 V at 300 mA cm−2 for the OER, and a superior long duration of >3000 hours operated at 250 mA cm−2. In situ Raman spectra indicated that for HER, the Ni sites facilitate H2O adsorption and dissociation, while the Ru sites simultaneously accelerate the combination of H* to form H2. For OER, the Ni sites facilitate the deprotonation of *OH to *O intermediate, thereby accelerating O2 production. The advances of this work in the field can be summarized as follows. First, a laser induction method for the in situ, rapid synthesis of single-phase HEAs under ambient conditions has been demonstrated. It features improved output efficiency, minimum energy consumption, and a shortened production cycle. Second, a systematic study on the correlation of HEA design with electrocatalytic performance has been performed, which would accelerate the discovery of new HEA electrocatalysts with distinctive and tailorable properties for energy conversion, water treatment, biomedical, and sensing applications. Third, the development of long-lasting, high-efficiency, and bifunctional HEA electrocatalysts holds potential for cost-effective electrolyzers for direct seawater splitting, making large-scale green hydrogen production more economical.
2. Results and discussion
2.1 Synthesis procedure
Fig. 1a illustrates the synthesis of quinary FeNiCoCrRu HEA NPs via direct CO2 laser induction of metal precursors on carbon paper under ambient conditions. In a typical process, five metal chlorides of FeCl3, NiCl2, CoCl2, CrCl3 and RuCl3 as well as sodium citrate were mixed in aqueous solution, and then drop-cast on pre-cleaned carbon paper (Table S1, ESI†). After drying, the carbon paper was irradiated directly with a CO2 laser under ambient conditions. Compared to other methods, this direct laser induction technique offers several advantages.17–19 The laser induces an ultrahigh localized temperature while maintaining ultrafast heating and quenching rates. To elucidate the temperature profile, an infrared pyrometer was deployed for non-contact temperature measurement during the CO2 laser induction process (Fig. 1b). The surface temperature reached a peak temperature of 3283 K at a heating rate of 1.23 × 105 K s−1. In the subsequent cooling process, the temperature first dropped to 3107 K in 30 ms, featuring a quenching rate of 5.9 × 103 K s−1. This initial cooling rate was slower than the subsequent stage, during which the temperature cooled to 823 K in 10 ms, showing a cooling rate of 2.28 × 105 K s−1. These two distinguished cooling stages are beneficial to alloying reactions at a high-temperature range of 3283–3107 K and then phase fixation by rapid cooling in the range of 3107–823 K. Hence, the ultrafast heating and quenching rates during the short laser induction duration create far-from-equilibrium conditions, enabling the synthesis of HEA NPs without phase separation.20 Laser induction enables the rapid production of NPs, offering a highly efficient method for synthesizing them with precise control over size and composition. As a demo, FeNiCoCrRu HEA NPs were prepared on carbon paper with areas ranging from 1.2 cm2 to 16 cm2 (Fig. S1 and Video S1, ESI†). By deploying spray-coating, a line laser, and a roll-to-roll apparatus,21 it is expected that this process can achieve continuous, scalable production. Moreover, laser induction enables the direct growth of HEA NPs on conductive substrates, eliminating the need for transferring or post-processing steps. This facilitates the exposure of active sites, promotes mass and charge transfer, and boosts stability.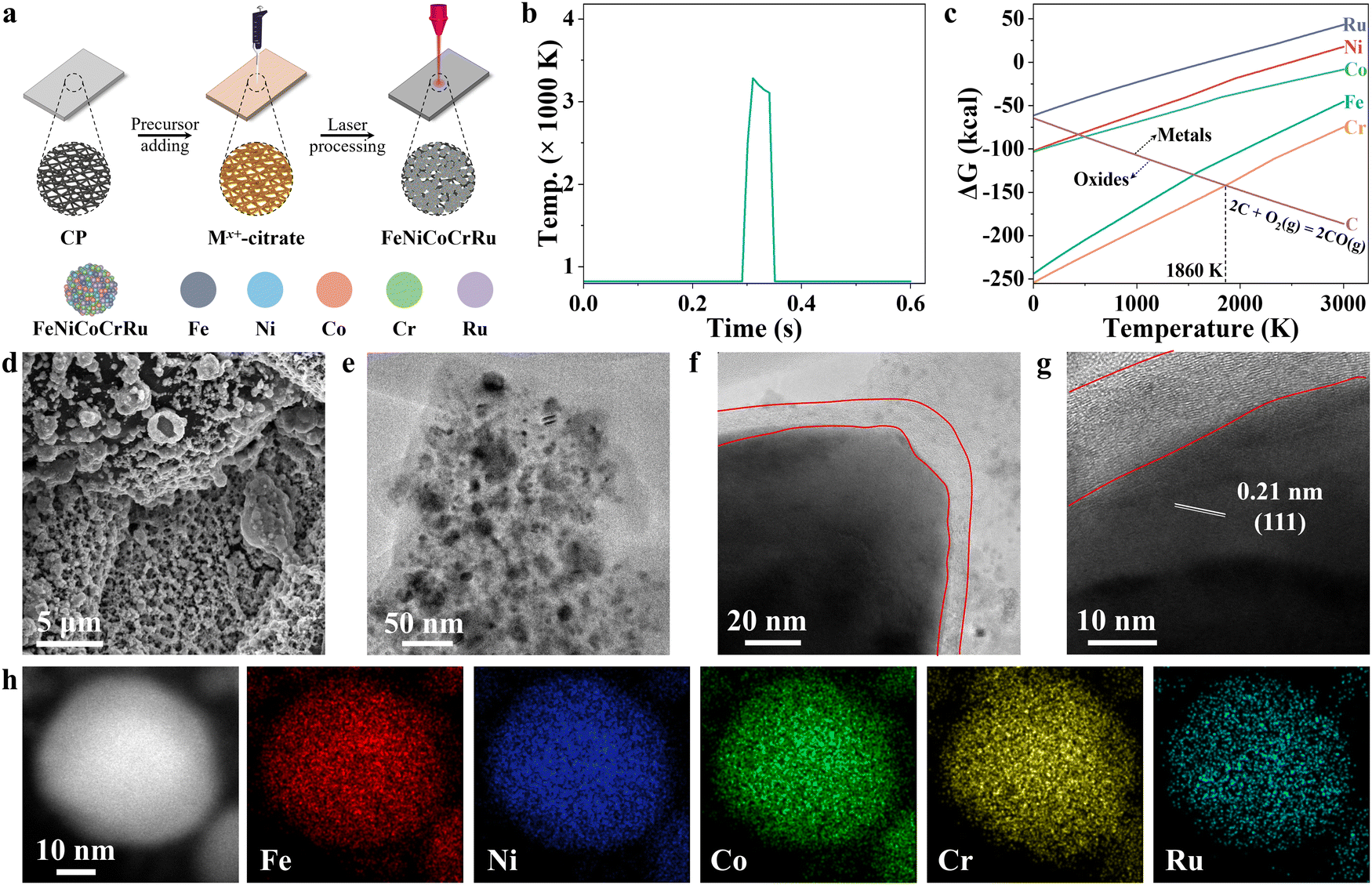 | ||
| Fig. 1 (a) Schematic showing the synthesis steps of FeNiCoCrRu HEA NPs using a CO2 laser. (b) A temperature profile during CO2 laser irradiation. (c) An Ellingham diagram showing the Gibbs free energies for the five metal elements as a function of temperature. (d) SEM, (e) TEM, (f)–(g) high-resolution TEM (HRTEM), (h) high-angle annular dark-field STEM (HAADF-STEM) and energy dispersive X-ray (STEM-EDX) elemental mapping images of HEA NPs. | ||
In the realm of electrocatalyst design, it is crucial to consider the roles of different elements. In this context, Fe, Ni, and Co emerge as primary active sites due to their superior affinity for OH* and H2O,22 while Cr effectively promotes the charge transfer and provides corrosion resistance, and Ru effectively lowers the energy barrier.23 These distinct contributions underscore the importance of a well-balanced composition for achieving optimal electrocatalytic performance. Previous studies have indicated that the atomic size difference (δ), the mixing enthalpy (ΔHmix), the mixing entropy (ΔSmix), and the mixing valence electron concentration (VECmix) should each fall within specific ranges to facilitate the formation of single-phase solid solution: 0 ≤ δ ≤ 8.5, −22 ≤ ΔHmix ≤ 7, 11 ≤ ΔSmix ≤ 19.5, and VECmix ≥ 8.24 Regarding FeNiCoCrRu, these four parameters were theoretically calculated as 2.90, −5.28, 13.38, and 8.2, respectively, indicating that it can form a single-phase solid solution HEA (Note S1 and Tables S2–S4, ESI†).
The following study shows that sodium citrate is a vital component in the laser-induced synthesis of FeNiCoCrRu HEA NPs. It facilitates the formation of a uniform and smooth layer on the carbon paper, benefiting the formation of small NPs. During laser irradiation, sodium citrate thermally decomposes into solid carbon, carbon dioxide, and water.25 The presence of solid carbon helps mitigate the oxidation tendency, a critical concern in a nanoscale HEA alloying process.26 An Ellingham diagram is presented to illustrate the oxidation potential of five metal elements and their relative ease of oxide reduction (Fig. 1c). It is evident that all five metal elements tend to form metallic elements in a carbothermal reduction environment when the temperature exceeds 1860 K. A control experiment without sodium citrate in the precursor solution results in the readily release of metal ions into the water, as indicated by an observable color change (Fig. S2, ESI†). Moreover, sodium citrate acts as a chelating agent to stabilize metal ions by forming negatively charged metal citrate complexes, while the carbon paper is positively charged under weakly acidic conditions.27 This induces strong electrostatic interactions between metal salts and carbon paper, facilitating the formation of a uniform coating layer. Overall, sodium citrate is crucial for ensuring the stability and uniformity of the synthesized HEA NPs.
2.2 Characterization analysis
To investigate the structures, electron microscopy studies were conducted. The scanning electron microscopy (SEM) image shows that FeNiCoCrRu HEA NPs with porous structures are well distributed and anchored to the surface of carbon paper (Fig. 1d). FeNiCo, FeNiCoRu and FeNiCoCr NPs also exhibited similar distributions (Fig. S3, ESI†). The porous structures with preferentially exposed edges offer advantages in promoting electrocatalytic activity by increasing mass transfer and reaction kinetics. According to the solid–liquid–gas interface theory,28,29 microscale roughness not only induces a strong capillary force for effective electrolyte pumping, but also reduces the interfacial adhesion to promote the release of gas bubbles from the active sites. To investigate the surface wettability, contact angle measurements were carried out (Fig. S4 and Video S2, ESI†). The carbon paper showed a contact angle of 78.5°, demonstrating its relatively hydrophobic characteristics. Upon loading with FeNiCoCrRu HEA NPs, the surface becomes hydrophilic, indicating the hydrophilic nature of FeNiCoCrRu HEA NPs.The transmission electron microscopy (TEM) image shows that FeNiCoCrRu HEA NPs have sizes ranging from 5 to 40 nm (Fig. 1e). High-resolution TEM (HRTEM) image displays FeNiCoCrRu NPs wrapped by a carbon layer averaging 10 nm (Fig. 1f–g). This carbon layer can serve as a chloride ion barrier and a corrosion-resistant layer, allowing the diffusion of water, hydroxide ions, and gas bubbles while preventing chloride ion penetration.30 Moreover, it can optimize the absorption behavior of intermediates, effectively improve the conductivity of the electrocatalysts, and promote charge transfer.31 As shown in Fig. 1g, a lattice fringe with an interplanar distance of 0.21 nm is visible, corresponding to the (111) plane of the face-centered cubic (FCC) structure.32 High-angle annular dark-field STEM (HAADF-STEM) and energy dispersive X-ray (EDX) elemental mapping on a FeNiCoCrRu HEA NP demonstrate a uniform distribution of Fe, Ni, Co, Cr, and Ru without obvious phase separation (Fig. 1h), which is further confirmed by EDX line-scan signal intensities (Fig. S5, ESI†). Inductively coupled plasma-mass spectrometry (ICP-MS) was used to determine the compositions, which shows Fe0.17Ni0.19Co0.34Cr0.1Ru0.2 (Note S2 and Table S5, ESI†). From the ICP-MS results, the calculated ΔSmix value of 12.8 falls in the range of 11 to 19.5, confirming that FeNiCoCrRu should be classified as a HEA rather than a medium entropy alloy.24
X-ray diffraction (XRD) patterns were collected to investigate the crystalline structure and phase composition of FeNiCoCrRu HEA NPs. Five transition metals (Fe, Ni, Co, Cr, and Ru) have dissimilar physiochemical properties, including atomic size (ranging from 0.124 nm to 0.134 nm), electronegativity (1.66 to 2.20), and crystal structures (FCC, BCC, and HCP) (Table S2, ESI†). In addition, it is hard to form miscible alloys of Ru with Fe and Ni due to substantial thermodynamic energy barriers (Fig. S6, ESI†).33 Therefore, it is a daunting task to alloy them into a nanoscale single-phase solid solution without phase separation and segregation. The XRD spectra of single metal samples (Fe, Ni, Co, Cr, and Ru) were first collected (Fig. S7a, ESI†). The Ru and Ni samples exhibited intensive diffraction peaks corresponding to their metallic forms, whereas the Fe, Co, and Cr samples showed weak diffraction peaks indicative of metal oxides. Specifically, the Ru sample displayed five intensive diffraction peaks at 38.5°, 42.3°, 44.2°, 58.5° and 69.5°, corresponding to the (100), (002), (101), (102), and (110) planes of HCP Ru phase (JCPDS 06-0663).34 The Ni sample exhibited two intensive diffraction peaks at 44.6 and 51.9, corresponding to the (111) and (200) planes of FCC Ni phase (JCPDS 04-0850).32 The Fe sample had a weak diffraction peak at 35.7°, corresponding to the (311) plane of Fe3O4 (JCPDS 65-3107).35 The Co sample displayed two weak peaks at 36.5° and 42.6°, corresponding to the (311) and (400) planes of Co2O3 (JCPDS 07-3333).36 The Cr sample showed two weak diffraction peaks at 33.8° and 36.4°, corresponding to the planes (104) and (110) of Cr2O3 (JCPDS 38-1479).37 When these five metal elements were mixed in an equimolar ratio (Fig. 2a), FeNiCoCrRu HEA NPs exhibited two diffraction peaks at 42.8° and 49.7°, corresponding to the (111) and (200) planes of an FCC structure (JCPDS 04-0850), respectively.32 Additionally, the other two peaks at 44.4° and 53.7° are attributed to graphitic carbon paper.38 The absence of peaks related to individual metals or their oxides implies the successful synthesis of single-phase solid solution FeNiCoCrRu HEA NPs via direct CO2 laser induction. The successful formation of the single-phase solid solution HEA can be attributed to both thermodynamics and kinetic factors.26,39 From a thermodynamic perspective, the high configurational entropy (1.61R) of the quinary FeNiCoCrRu (Fig. S8, ESI†) and ultrahigh temperature (>3200 K) can enhance the solubility limit of each element in the alloy system, regardless of their dissimilar physiochemical properties.26 This promotes further dissolution of the element-rich phases, ultimately resulting in the formation of a stable, homogeneously mixed single-phase solid solution. Moreover, ultrafast heating/quenching rates (105 K s−1) effectively inhibit phase segregation and the coarsening of HEA NPs that happen during the cooling process. From a kinetic perspective, the quinary FeNiCoCrRu exhibits diverse local energy at each atomic site due to the different types of bonding arising from various local atomic configurations, greatly hindering the kinetics of diffusion and phase transformation.39 Hence, both thermodynamic and kinetic factors contribute to the successful formation of single-phase solid solution FeNiCoCrRu HEA NPs.
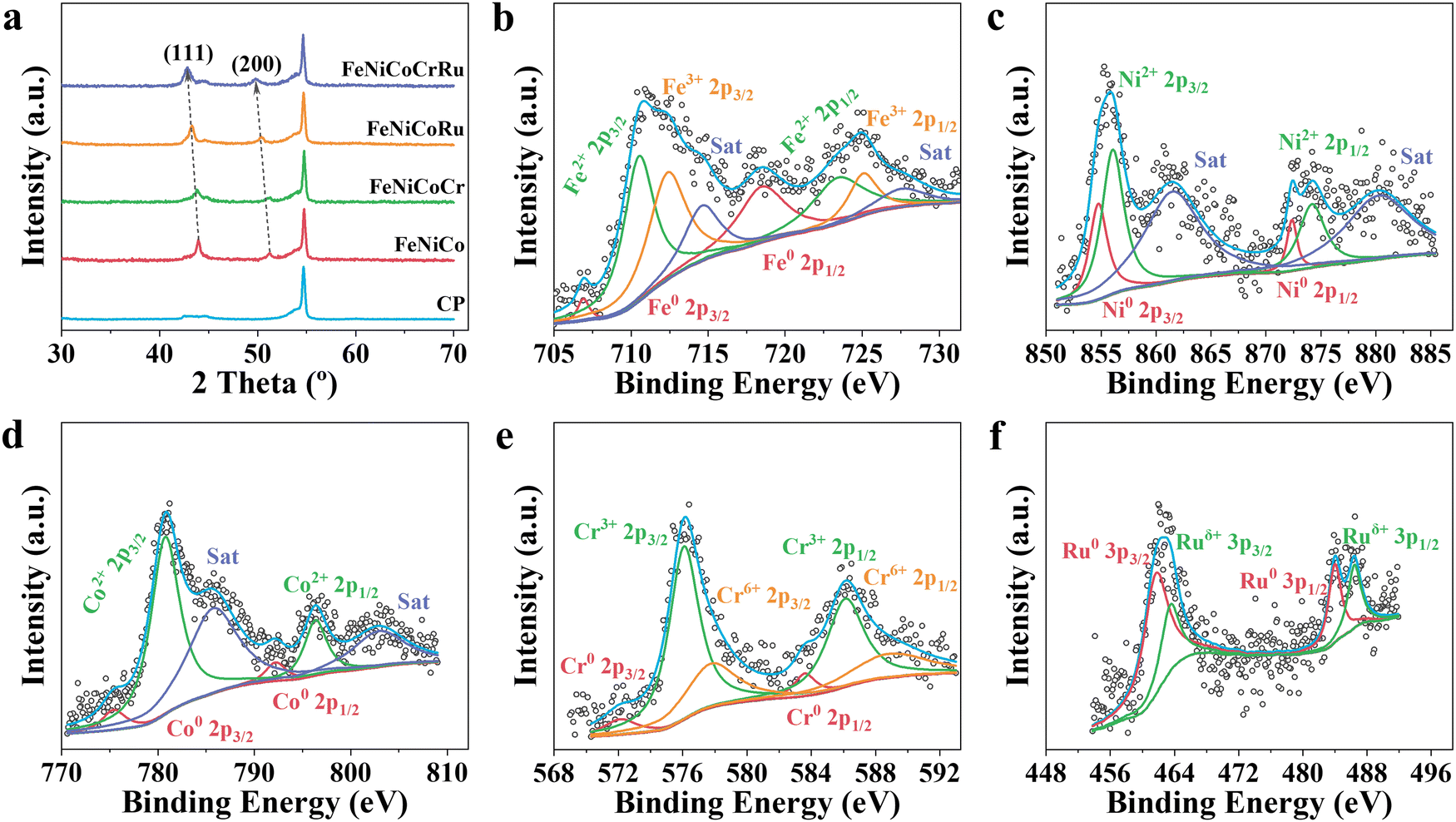 | ||
| Fig. 2 (a) XRD spectra of FeNiCoCrRu, FeNiCoRu, FeNiCoCr, FeNiCo, and carbon paper (CP). High-resolution (b) Fe 2p, (c) Co 2p, (d) Ni 2p, (e) Cr 2p, and (f) Ru 3p spectra of FeNiCoCrRu HEA NPs. | ||
Compared to the XRD spectra of FeNiCoRu, FeNiCoCr, FeNiCo, and FeNi (Fig. 2a and Fig. S7a, ESI†), the diffraction peaks associated with the (111) and (200) planes of FeNiCoCrRu HEA NPs shift towards smaller 2 theta angles, indicating significant lattice distortion in FeNiCoCrRu.32 The formation of FeNiCoCrRu by partially substituting Ni sites with Fe, Co, Cr, and Ru introduces a lattice mismatch and alters bond distances, thereby inducing lattice distortion.32 Peak broadening, as is reflected in the increased full width at half maximum, is correlated with the increased number of incorporated metal elements. This trend indicates a reduction in the crystallite size of the FeNiCoCrRu HEA NPs. Raman spectra (Fig. S7b, ESI†) of FeNiCoCrRu HEA NPs further confirm the absence of metal oxides. The three vibrational peaks appearing at 1328, 1570, and 2680 cm−1 can be assigned to D, G and 2D bands of the graphitic carbon from the carbon paper.
X-ray photoelectron spectroscopy (XPS) was conducted to elucidate the chemical composition and oxidation states of FeNiCoCrRu HEA NPs. The survey spectrum (Fig. S9a, ESI†) clearly shows the presence of five metal elements (Fe, Ni, Co, Cr, and Ru) and two non-metallic elements (C and O), consistent with the EDS mapping results. High-resolution Fe 2p, Ni 2p, Co 2p, Cr 2p, and Ru 3p spectra are shown in Fig. 2b–f, indicating that all metal elements exist in both metallic and oxidized forms. The synergistic effect between metallic and oxidized states of metal elements in FeNiCoCrRu HEA NPs could promote the electron transfer, leading to an enhanced electrocatalytic activity.40 In the Fe 2p spectrum (Fig. 2b), two minor peaks centered at 706.9 and 718.5 eV correspond to metallic Fe0. The presence of the oxidized Fe2+ and Fe3+ species are revealed by four main peaks at 710.5, 723.4, 712.4, 725.1 eV and two satellite peaks at 714.6 and 727.5 eV, respectively.32 Further observation showed that the oxidized Fe2+/Fe3+ species are dominant, which can be ascribed to the high chemical activity and low redox potential of Fe. The Ni 2p spectrum (Fig. 2c) shows two peaks at 854.8 and 872.4 eV, which are attributed to the metallic Ni0. Another two peaks at 856.1 and 874.2 eV accompanied by satellite peaks at 861.5 and 880.2 eV show the existence of oxidized Ni2+.40 As seen in the Co 2p spectrum (Fig. 2d), most Co species are the oxidized Co2+ (780.7 and 796.3 eV), while minor metallic Co0 also exists (775.4 and 792.2 eV).40 The Cr 2p spectrum, as shown in Fig. 2e, is deconvoluted into six peaks with the first two ones (576.1 and 586.1 eV) assigned to Cr3+ and the other two ones (577.8 and 588.9 eV) attributed to Cr6+, respectively.41 Two minor peaks at 572.1 and 583.6 eV correspond to Cr0. The Ru 3p spectrum reveals the existence of both metallic Ru (461.6 and 463.4 eV) and Ruδ+ (483.9 and 486.4 eV) (Fig. 2f).42 As shown in the C 1s and Ru 3d spectra (Fig. S9b, ESI†), the two broad peaks at 280.8 and 284.8 eV are assigned to Ru0, while the other two peaks at 282.3 and 286.0 eV correspond to Ruδ+ 3d5/2 and Ruδ+ 3d3/2, respectively.43 The C 1s spectrum shows four characteristic peaks including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (sp2, 284.4 eV), C–C (sp3, 284.9 eV), C–O (286.9 eV) and CO (288.7 eV).32 The O 1s spectrum (Fig. S9c, ESI†) exhibits three distinctive peaks, which can be assigned to the lattice oxygen (529.5 eV), vacancy oxygen (530.4 eV) and adsorbed oxygen (531.6 eV), respectively.32 The lattice oxygen and adsorbed oxygen may originate from amorphous metal oxides and H2O on the surface. The presence of the oxygen vacancies typically promotes the adsorption of oxygen intermediates (e.g., O*, OH*, and OOH*), thus enhancing the electrocatalytic performance.44
C (sp2, 284.4 eV), C–C (sp3, 284.9 eV), C–O (286.9 eV) and CO (288.7 eV).32 The O 1s spectrum (Fig. S9c, ESI†) exhibits three distinctive peaks, which can be assigned to the lattice oxygen (529.5 eV), vacancy oxygen (530.4 eV) and adsorbed oxygen (531.6 eV), respectively.32 The lattice oxygen and adsorbed oxygen may originate from amorphous metal oxides and H2O on the surface. The presence of the oxygen vacancies typically promotes the adsorption of oxygen intermediates (e.g., O*, OH*, and OOH*), thus enhancing the electrocatalytic performance.44
To deepen the understanding of the electron interaction among the metal elements, high-resolution spectral analysis of FeNiCoRu, FeNiCoCr, and FeNiCo was conducted (Fig. S10, ESI†). It is evident that as the number of alloying elements increases, the Ru 3p shifts towards lower binding energies. This shift indicates an increase in the electron density of Ru, suggesting electron transfers from the 3d transition metal atoms, particularly Cr, to Ru atoms within FeNiCoCrRu. This electron redistribution phenomenon can be attributed to the higher electronegativity of Ru (2.20) compared to those of the 3d transition metals, such as Cr (1.66), Fe (1.83), Co (1.88), and Ni (1.91).32 Consequently, the 3d transition metals become more electron-deficient while Ru is more electron-efficient. The electronic structure of Ru strongly influences adsorption/desorption energies of the reaction intermediates, thus promoting the electrocatalytic activity.32
2.3 Electrochemical performance
The electrocatalytic performance of FeNiCoCrRu HEA NPs for the HER and OER was first evaluated using a typical three-electrode configuration in an alkaline freshwater electrolyte (1 M KOH). All potentials were calibrated to the reversible hydrogen electrode (RHE) with 95% iR-correction. Cyclic voltammetry (CV) was conducted to electrochemically activate and stabilize the electrocatalysts followed by linear sweep voltammetry (LSV). As derived from Fig. 3a, FeNiCoCrRu HEA NPs show overpotentials of 0.002 V and 0.051 V at 10 and 100 mA cm−2, respectively, for the HER. These values are the lowest among those of FeNiCoRu (0.004 V and 0.071 V), FeNiCoCr (0.112 V and 0.238 V), FeNiCo (0.176 V and 0.286 V), FeNi (0.165 V and 0.292 V), Ni (0.126 V and 0.247 V), and Fe (0.127 V and 0.351 V) (Fig. S11a and Table S6, ESI†). Impressively, FeNiCoCrRu HEA NPs need overpotentials of only 0.078 V, 0.113 V, and 0.150 V to attain 200, 400, and 600 mA cm−2, respectively. The improved HER activity with the increased number of alloying elements further validates the synergistic functions of these metals. Notably, FeNiCoCrRu HEA NPs outperform many recently reported HER electrocatalysts (Table S7, ESI†). It is noted that the substantial fluctuation observed at high current densities in the LSV curves is caused by the bursting of H2 gas bubbles from the catalyst surface (Fig. 3a).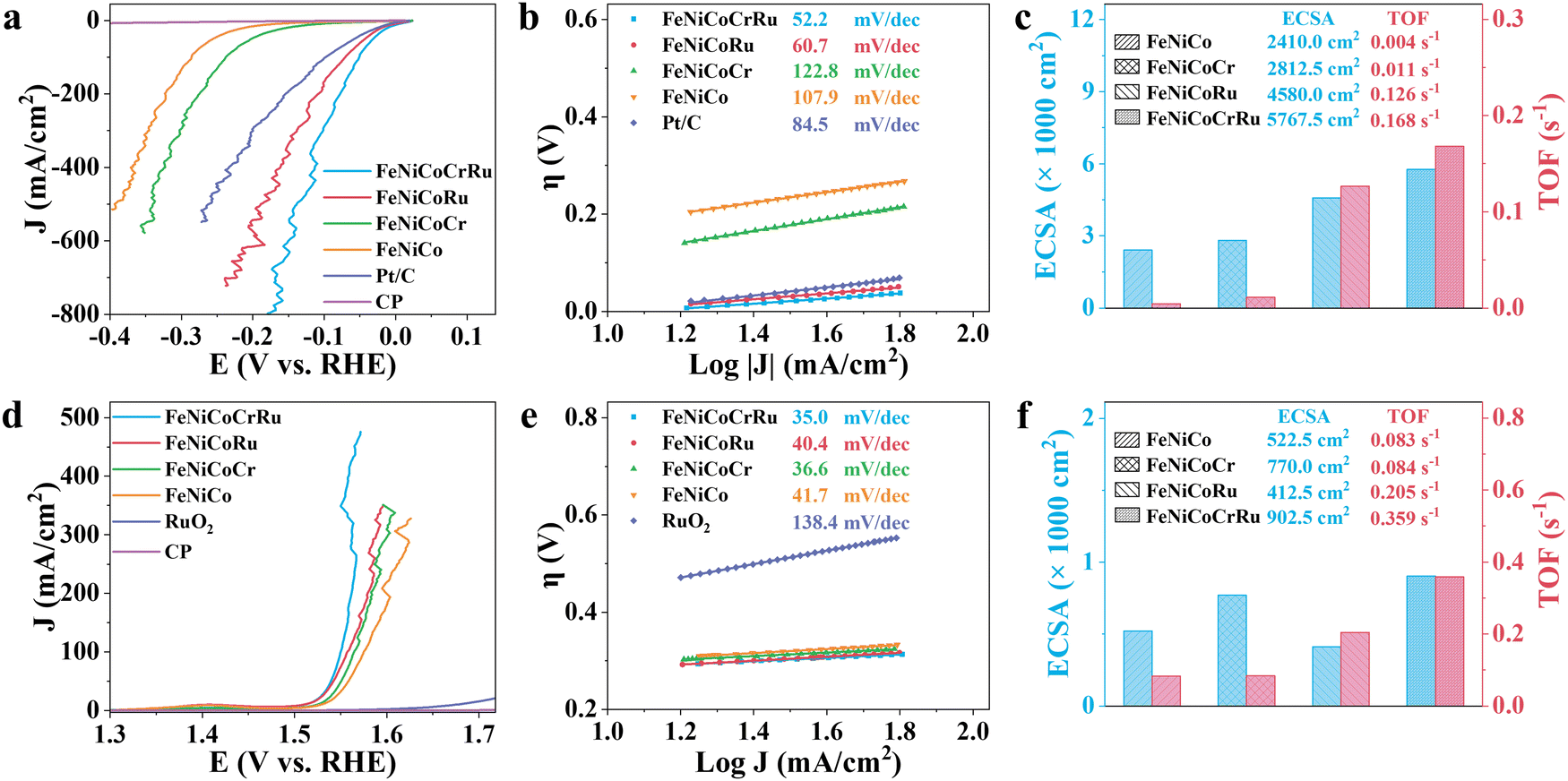 | ||
| Fig. 3 Electrocatalytic water splitting performance of FeNiCoCrRu in 1 M KOH. LSV curves of (a) HER and (d) OER at a scan rate of 10 mV s−1 with 95% iR compensation. Tafel slope plots of (b) HER and (e) OER. ECSA and TOF plots of (c) HER and (f) OER. | ||
Tafel plots were subsequently derived from the LSV curves to evaluate the HER reaction mechanism (Fig. 3b and Fig. S11b, ESI†). The remarkably enhanced HER kinetics of FeNiCoCrRu HEA NPs is attested by the lowest Tafel slope of 52.2 mV dec−1, revealing that the hydrogen generation process likely follows the Volmer–Heyrovsky pathway in alkaline electrolytes (Note S3, ESI†).45 This value is lower than those of FeNiCoRu (60.7 mV dec−1), FeNiCoCr (122.8 mV dec−1), FeNiCo (107.9 mV dec−1), and Pt/C (84.5 mV dec−1), indicating that the high-entropy design and the associated synergistic effect facilitate faster HER kinetics. Electrochemical impedance spectroscopy (EIS) was conducted to further investigate the charge transfer kinetics and mass transfer capacities (Fig. S12 and Table S6, ESI†). The Nyquist plots exhibit distinctive semicircular patterns in all EIS spectra, from which the charge transfer resistance (Rct) and the inherent resistance of the electrode and electrolyte (Rs) can be derived. Among these electrocatalysts, FeNiCoCrRu HEA NPs exhibits the smallest Rct of 0.477 Ω, showing a fast charge transfer rate during the HER process.
In general, electrocatalytic efficiency arises from the interplay of both the quantity and quality of exposed active sites on electrocatalysts, which can be quantified as electrochemical active surface area (ECSA) and turnover frequency (TOF).46 These parameters represent comprehensive outcomes influenced by various factors such as composition, size, defects, conductivity, and interparticle resistances. The ECSA was determined based on the double-layer capacitance (Cdl) in the non-faradaic region (Fig. 3c and Fig. S13 and Table S6, ESI†). FeNiCoCrRu HEA NPs exhibit the largest ECSA value of 5767.5 cm−2, much larger than those of FeNiCoRu (4580 cm−2), FeNiCoCr (2812.5 cm−2), and FeNiCo (2410 cm−2). A larger ECSA means more active sites are available for electrocatalytic reactions, enabling greater adsorption of water molecules and thereby enhancing HER activity. In terms of TOF (Fig. 3c and Note S4 and Table S6, ESI†), FeNiCoCrRu HEA NPs possess a rate of 0.168 H2 s−1 at an overpotential of 0.15 V, which is significantly superior to those of FeNiCoRu (0.126 H2 s−1), FeNiCoCr (0.011 H2 s−1), and FeNiCo (0.004 H2 s−1). Moreover, the mass activity of FeNiCoCrRu HEA NPs (474.39 A g−1) at an overpotential of 0.1 V is 2.1, 24.7, and 29.1 times higher than those of FeNiCoRu (221.76 A g−1), FeNiCoCr (19.23 A g−1), and FeNiCo (16.29 A g−1), respectively. These results clearly demonstrate the highest intrinsic electrocatalytic activity of FeNiCoCrRu HEA NPs toward the HER. The presence of Ru is crucial in this enhanced HER performance, which can be primarily attributed to its ability to effectively adsorb reaction intermediates like OH* and H*.47
Next, the OER performance was investigated in the same alkaline freshwater electrolyte. As shown in Fig. 3d, the carbon paper showed negligible OER activity, while the benchmark RuO2 electrocatalyst required an overpotential of 0.446 V to achieve a current density of 10 mA cm−2. In comparison, FeNiCoCrRu HEA NPs demonstrate lower overpotentials of 0.321 V, 0.331 V, and 0.332 V at current densities of 100, 200 and 300 mA cm−2, respectively. The activity improvement can be distinctly concluded compared to FeNiCoRu (0.327 V, 0.349 V, and 0.359 V), FeNiCoCr (0.336V, 0.353 V, and 0.372 V), and FeNiCo (0.346 V, 0.369 V, and 0.379 V) (Fig. S14a and Table S6, ESI†). Furthermore, FeNiCoCrRu HEA NPs achieve 400 mA cm−2 at an overpotential of 0.329 V, notably lower than that required for hypochlorite generation (0.479 V, Note S5, and Fig. S15, ESI†). This indicates that hypochlorite generation is not thermodynamically preferrable, suggesting FeNiCoCrRu HEA NPs are promising candicates for alkaline seawater electrolysis.48 As shown in Fig. 3e and Fig. S14b (ESI†), FeNiCoCrRu HEA NPs exhibit the smallest Tafel slope of 35 mV dec−1 among FeNiCoRu (40.4 mV dec−1), FeNiCoCr (36.6 mV dec−1), and FeNiCo (41.7 mV dec−1), indicating faster OER reaction kinetics. Additionally, FeNiCoCrRu HEA NPs possesses the lowest Rct of 1.325 Ω, the largest ECSA of 902.5 cm−2, and the highest TOF of 0.359 s−1 (Fig. 3f and Fig. S16, S17, and Table S6, ESI†). These results collectively indicate that FeNiCoCrRu HEA NPs are also highly effective electrocatalysts for the OER.
The effects of reaction conditions on the electrocatalytic performance were also investigated (Table S1, ESI†). Eight additional samples were prepared by tuning laser processing parameters (power and rate) and varying the ratios of metal salts to sodium citrate. Notably, FeNiCoCrRu HEA NPs synthesized with a power of 18 W, a rate of 10 cm s−1 and a metal-to-citrate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 exhibited the best HER and OER activities (Fig. S18, ESI†). It is obvious that power, rate, and metal-to-citrate ratio significantly affect the morphology and size of HEA NPs, which in turn influence their electrocatalytic performance (Fig. S19, ESI†). In addition, three other quinary HEA NPs including FeNiCoCuRu, FeNiCoMnRu, and FeNiCoMoRu were prepared under the same conditions, except that Cr was replaced with Cu, Mn, and Mo, respectively (Table S1, ESI†). As shown in Fig. S20 (ESI†), FeNiCoCrRu HEA NPs exhibited superior HER and OER activities compared to these other quinary HEA NPs, further confirming the critical role of the Cr in promoting HER and OER activities. It is deduced that introducing Cr into the HEA design changes the oxidation states of the catalytically active Fe, Ni, and Co, facilitates electron redistribution, reduces the deprotonation energy,49 and induces lattice strain.50
:2 exhibited the best HER and OER activities (Fig. S18, ESI†). It is obvious that power, rate, and metal-to-citrate ratio significantly affect the morphology and size of HEA NPs, which in turn influence their electrocatalytic performance (Fig. S19, ESI†). In addition, three other quinary HEA NPs including FeNiCoCuRu, FeNiCoMnRu, and FeNiCoMoRu were prepared under the same conditions, except that Cr was replaced with Cu, Mn, and Mo, respectively (Table S1, ESI†). As shown in Fig. S20 (ESI†), FeNiCoCrRu HEA NPs exhibited superior HER and OER activities compared to these other quinary HEA NPs, further confirming the critical role of the Cr in promoting HER and OER activities. It is deduced that introducing Cr into the HEA design changes the oxidation states of the catalytically active Fe, Ni, and Co, facilitates electron redistribution, reduces the deprotonation energy,49 and induces lattice strain.50
Although hydrogen production through freshwater electrolysis is a technologically mature process, scaling it up for large-scale production requires substantial amounts of ultrapure water. This presents a significant hurdle, particularly in renewable energy-rich yet water-scarce regions, underscoring the need for more sustainable alternatives.51 In this context, seawater electrolysis emerges as a more attractive solution since seawater is considered an unlimited water resource on our planet.2–4 However, seawater electrolysis faces additional challenges, including electrode side reactions, corrosion and scaling caused by the complex components of seawater. To mitigate these issues, several research groups have made pioneering advancements.2–4,52 Xie et al. developed a direct seawater electrolysis system that incorporates hydrophobic porous waterproof breathable membrane and a self-dampening electrolyte.2 This innovative system features a higher self-driven water migration rate, 100% ion-blocking efficiency, and excellent antifouling capability. Qiao and coworkers manipulated the local reaction environment for seawater electrolysis by introducing a Lewis acid layer of CrOx over transition metal oxides.3 The Lewis acid layer significantly enhance water dissociation and captures hydroxyl anions, thus facilitating HER kinetics while also preventing unwanted precipitation. Detailed discussions on the feasibility and advancements of using seawater for hydrogen production are provided in Note S6 (ESI†). Hence, to extend the practicability of FeNiCoCrRu HEA NPs for future industrial applications, it is necessary to evaluate the electrocatalytic activities in an alkaline seawater electrolyte (1 M KOH + 0.5 M NaCl). As expected, FeNiCoCrRu HEA NPs exhibit outstanding performance towards both HER and OER in 1 M KOH + 0.5 M NaCl electrolyte (Fig. 4a–c and Table S8, ESI†). In detail, FeNiCoCrRu HEA NPs show overpotentials of 0.052 V at 100 mA cm−2 for HER and 0.320 V at100 mA cm−2 for OER. Meanwhile, the corresponding Tafel slopes are 52.4 mV dec−1 for HER and 42.7 mV dec−1 for OER, indicating superior reaction kinetics in 1 M KOH + 0.5 M NaCl electrolyte (Fig. S21, ESI†). The Rct for HER and OER are 0.322 Ω and 2.609 Ω, respectively (Fig. S22, ESI†). These results are very close to those in 1 M KOH.
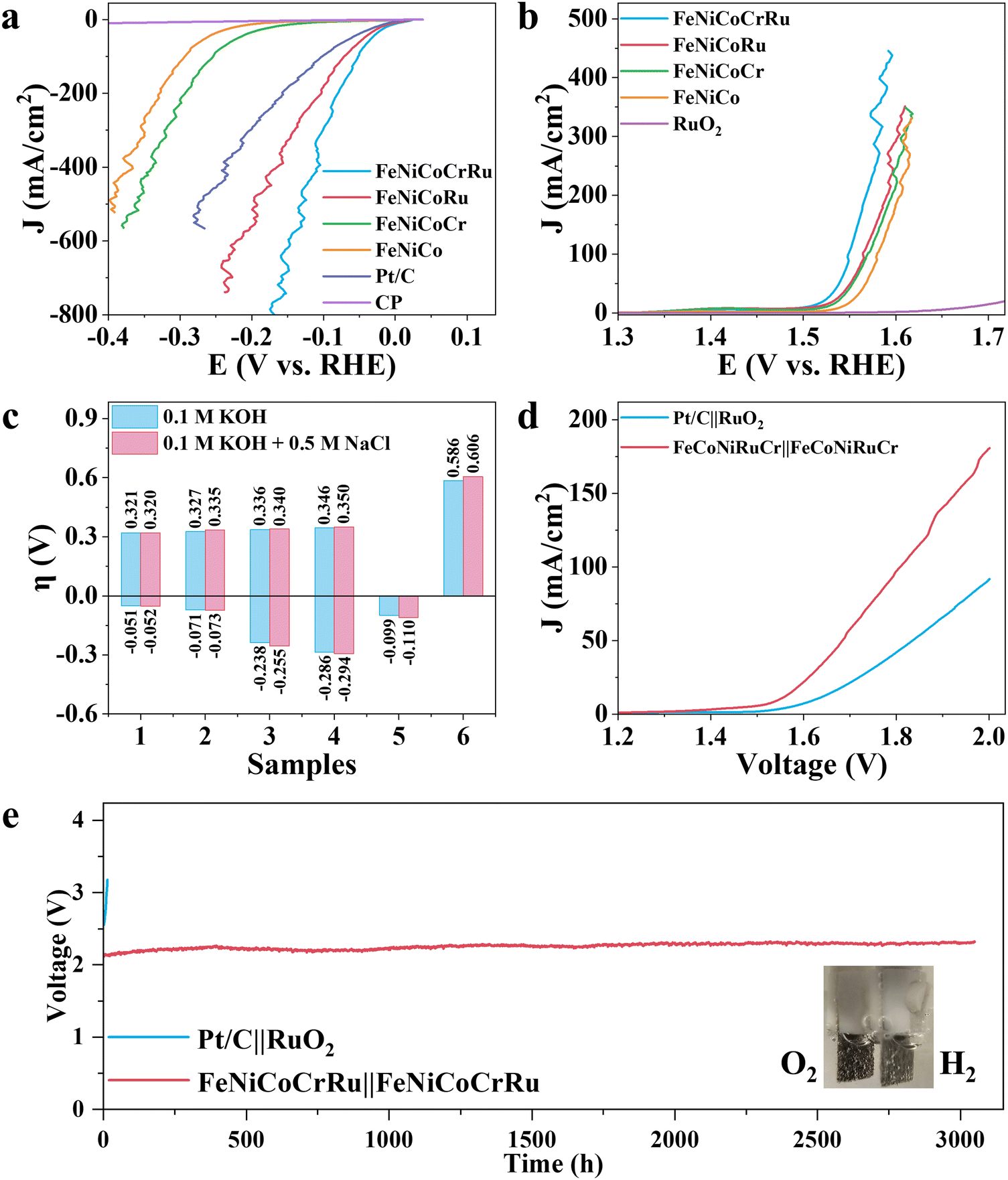 | ||
| Fig. 4 LSV curves of (a) HER and (b) OER in alkaline seawater electrolyte (1 M KOH + 0.5 M NaCl) with 95% iR compensation. (c) Comparison of overpotentials at current density of 100 mA for both HER and OER over various electrocatalysts. 1: FeNiCoCrRu, 2: FeNiCoRu, 3: FeNiCoCr, 4: FeNiCo, 5: Pt/C, and 6: RuO2. (d) LSV curves of FeNiCoCrRu||FeNiCoCrRu and Pt/C||RuO2 electrolyzers without iR-correction. (e) Chronopotentiometry curves of FeNiCoCrRu||FeNiCoCrRu and Pt/C||RuO2 electrolyzers at 250 mA cm−2. | ||
Given the outstanding HER and OER performance in alkaline seawater, it is reasonable to propose that FeNiCoCrRu HEA NPs can serve as a highly efficient and stable bifunctional electrocatalyst for seawater electrolysis. Consequently, two electrolyzers, i.e., FeNiCoCrRu||FeNiCoCrRu and Pt/C||RuO2 were assembled to investigate the overall seawater electrolysis performance. As shown in Fig. 4d, the FeNiCoCrRu||FeNiCoCrRu electrolyzer exhibits superior overall seawater electrolysis performance, which can drive 10, 50, and 100 mA cm−2 at cell voltages of 1.594, 1.683, and 1.808 V, respectively. These values are significantly lower than those of the Pt/C||RuO2 electrolyzer and are either lower than or comparable to those reported in recent studies (Table S9, ESI†). We further tested the full-cell electrochemical performance of the FeNiCoCrRu||FeNiCoCrRu electrolyzer across different voltage ranges (Fig. S23, ESI†). It is obvious that three LSV curves overlap well, and current density exhibits a linear relationship with applied voltage starting at 1.6 V. It means that hydrogen and oxygen gas bubbles are released quickly without blocking active sites, further confirming the advantages of the porous structures. To evaluate the faradaic efficiency of the FeNiCoCrRu||FeNiCoCrRu electrolyzer, the generated gas was collected and compared with theoretical values (Note S7 and Fig. S24a, ESI†). The experimental gas volume closely matches the theoretical values, resulting in Faradaic efficiency of 99.6% and 97.7% for hydrogen and oxygen, respectively. From a practical perspective, industrial water splitting is usually performed in highly alkaline electrolytes (3–6 M KOH) at elevated temperatures (60–80 °C).53 Hence, it is of great importance to evaluate the seawater splitting performances of the FeNiCoCrRu||FeNiCoCrRu electrolyzer under such working conditions (Fig. S24b, ESI†). The electrolyzer delivered a current density of 100 mA cm−2 at applied voltages of 1.848 V, 1.748 V, 1.753 V and 1.734 V in 1 M KOH + 1 M NaCl, 3 M KOH + 0.5 M NaCl, 6 M KOH + 0.5 M NaCl, and 1 M KOH + 0.5 M NaCl (60 °C), respectively. In addition, the FeNiCoCrRu||FeNiCoCrRu electrolyzer was easily actuated by a 1.5 V AA battery (Fig. S25 and Video S3, ESI†).
To assess the suitability of the electrocatalyst for industrial applications, long-term stability tests were conducted on both FeNiCoCrRu||FeNiCoCrRu and Pt/C||RuO2 electrolyzers using chronopotentiometry at 250 mA cm−2 (Fig. 4e).2 Notably, the applied voltage across the Pt/C||RuO2 electrolyzer experienced a significant surge after only 10 hours, indicating poor stability. In contrast, the FeNiCoCrRu||FeNiCoCrRu electrolyzer demonstrated exceptionally stability, with a voltage increase rate of only 0.05 mV h−1. In detail, the voltage increased by just 0.153 V after 3050 hours of continuous operation, which is comparable to recent reports.2,4,52,54 Stable gas bubbles were consistently observed emanating from the electrode surfaces (inset of Fig. 4e and Video S4, ESI†). After the stability test, the electrolyte underwent active chlorine species and ICP-MS tests to investigate hypochlorite concentrations and the dissolution of metal species, respectively. Minimal hypochlorite formation was observed during the long-term test, as indicated by negligible changes in absorbance (Note S8 and Fig. S26, ESI†). This means that the applied voltage was predominantly utilized to generate oxygen rather than activating chlorine species. The concentrations of Fe, Ni, Co, Cr, and Ru dissolved in the electrolyte were all less than 0.5 mg L−1 (Table S10, ESI†), suggesting excellent corrosion resistance of FeNiCoCrRu at high current densities in 1 M KOH + 0.5 M NaCl. The metal composition of both the anode and cathode was also investigated using ICP-MS (Table S11, ESI†). The cathode showed a slight decrease in Fe and Co species, whereas the anode exhibited a slight decrease in Fe, Cr and Ru species. Furthermore, the morphology, crystal structure, and chemical composition of FeNiCoCrRu were investigated after the stability test using SEM, TEM, Raman, XRD, and XPS. SEM images (Fig. S27, ESI†) revealed that both the cathode and the anode retained their porous structures with 3D interconnected networks. HRTEM images (Fig. S28, ESI†) of both the anode and cathode clearly displayed lattice spacing of 0.18 nm, corresponding to the (200) planes of the FCC phase. HAADF-STEM images, EDX elemental mapping images, and line-scan spectra (Fig. S29, ESI†) showed a uniform distribution of all five metal elements without phase segregation in both the anode and cathode. XRD spectra (Fig. S30, ESI†) indicated that the cathode maintained a good crystalline structure, while the anode exhibited slight crystal deterioration. XPS (Fig. S31, ESI†) confirmed the constancy of elemental composition for the cathode, whereas the anode showed a deficiency of Cr on the surface, likely due to Cr leaching into the electrolyte.55 These experimental results not only demonstrate the superior stability of FeNiCoCrRu HEA NPs but also validate the catalyst design strategy.
2.4 Catalytic mechanism
To further investigate the reaction mechanism, an in situ Raman spectroscopy study was performed to reflect the surficial-active species of FeNiCoCrRu HEA NPs during alkaline seawater electrolysis. As illustrated in Fig. 5a, a home-made testing cell consists of FeNiCoCrRu HEA NPs, Pt wire, and Ag/AgCl as the working, counter, and reference electrode, respectively. The electrolyte (1 M KOH + 0.5 M NaCl) flows over the surface of FeNiCoCrRu HEA NPs to carry away generated gas bubble. A laser beam passes through a quartz window and is focused on the surface of FeNiCoCrRu HEA NPs to identify changes in active species during HER and OER. As shown in Fig. 5b, in the absence of applied potential, the spectrum exhibits three dominant peaks at 1310, 1589 and 2620 cm−1 corresponding to D, G, and 2D bands of carbon fibers, and a peak at 500 cm−1 can be assigned to Ni–O bonds.32 Upon applying a potential of 50 mV, a new Raman peak corresponding to the Ru–H bond appears at ∼2050 cm−1, revealing the presence of Ru–H intermediates.32 Close observation shows that the peak intensities of Ni–O and Ru–H continuously increase as the applied potential increases, indicating enhanced HER activity. At an open circuit potential, FeNiCoCrRu HEA NPs give rise to a new Raman peak at ∼500 cm−1 (Fig. 5c), which is attributed to the bending mode of Ni–O bonds, suggesting that the Ni sites serve as the binding sites for the OH species. As the potential increases from 1.5 V to 1.6 V, the peak intensity slightly decreases, which may be due to the consumption of the local concentration of OH species. Based on these experimental results, the following HER and OER reaction mechanisms are proposed (Fig. 5d and e). The Tafel slope (52.2 mV s−1) of FeNiCoCrRu HEA NPs reveals that the hydrogen generation process follows the Volmer–Heyrovsky pathway.45 The water dissociation into H* and subsequent adsorption of H* are the rate-determining steps, which dictate the overall water dissociation rate.32 The in situ HER Raman of FeNiCoCrRu HEA NPs reveals that Ni and Ru active sites stabilize different intermediates, with Ni facilitating H2O adsorption and dissociation while Ru simultaneously accelerates the combination of H* and H2.32 As shown in Fig. 5d, the H2O* molecule is first absorbed onto the surface of FeNiCoCrRu HEA NPs, destabilizing the H–OH bond. This is followed by the dissociation of the H–OH bond, leading to the co-adsorption of OH* and H* intermediates. The H* intermediate then reacts with a proton that is provided by another H2O molecule dissociation. Finally, the formed H2 molecule is released from the surface of FeNiCoCrRu HEA NPs. Regarding the OER reaction pathway, the deprotonation of OH* to form the *O intermediate is the rate-determining step.56 As shown in Fig. 5e, the OER process begins with the adsorption of OH− from the electrolyte onto the surface of FeNiCoCrRu HEA NPs, forming an adsorbed OH*. The adsorbed OH* undergoes deprotonation via reacting with another OH− to form an adsorbed O* and an H2O molecule. Then, another OH− is adsorbed, which reacts with the adsorbed O* to form a superoxol intermediate (OOH*). Finally, the OOH* undergoes a deprotonation step via reaction with OH−, leading to the formation of O2 and H2O molecules.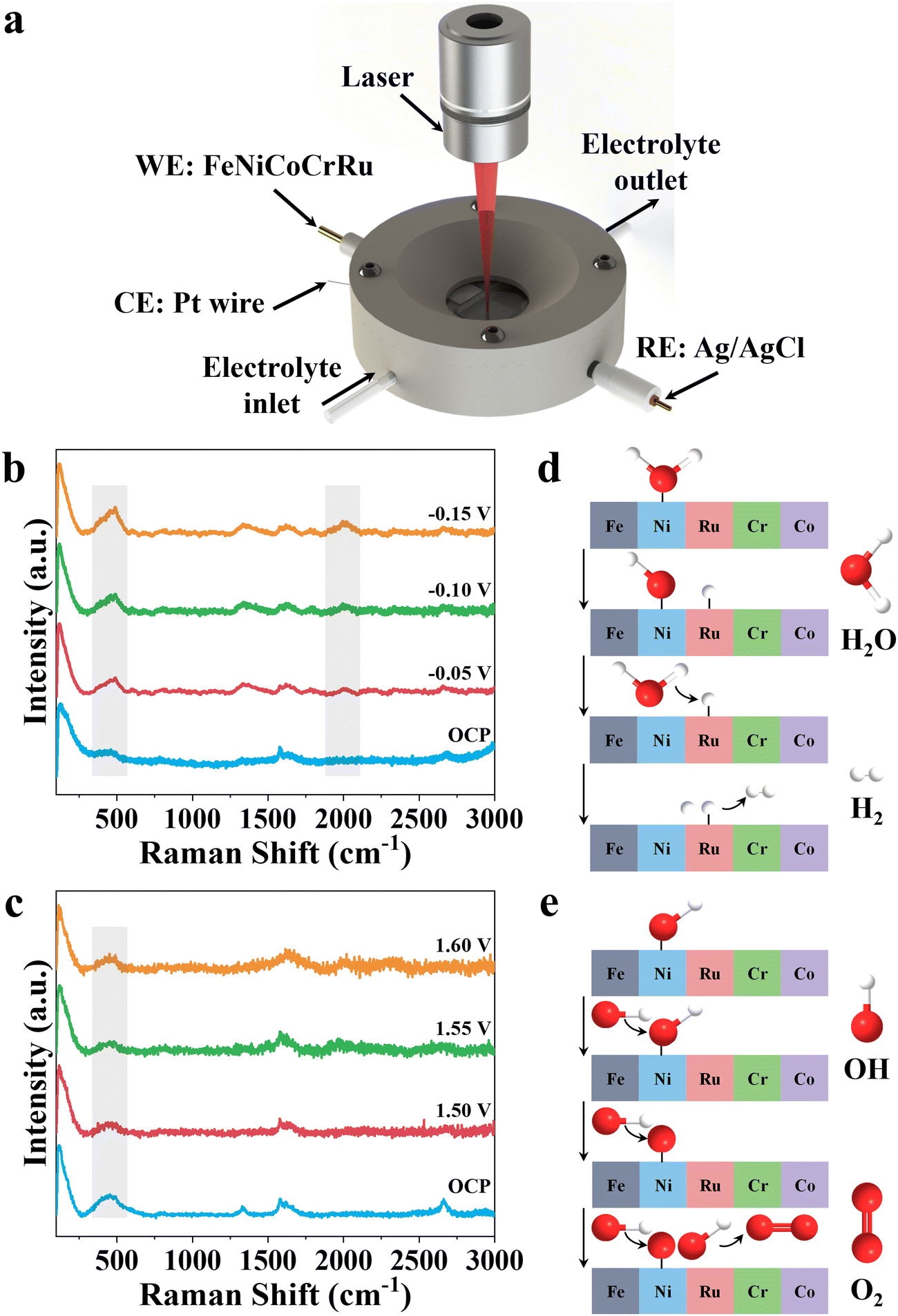 | ||
| Fig. 5 (a) In situ Raman spectroscopy setup. In situ Raman spectra of the FeNiCoCrRu HEA at various potentials for (b) HER and (c) OER in 1 M KOH + 0.5 M NaCl electrolyte. Proposed catalytic mechanism of (d) HER and (e) OER in the alkaline seawater electrolyte. | ||
3. Conclusions
We have successfully demonstrated the in situ, rapid synthesis of quinary FeNiCoCrRu HEA NPs anchored onto carbon paper by direct CO2 laser induction under ambient conditions. The ultrahigh temperature, high entropy effect, ultrafast heating/quenching rates, and sluggish diffusion effects enabled the formation of FeNiCoCrRu HEA NPs with uniform distribution of all five metal elements, without phase separation or segregation. FeNiCoCrRu HEA NPs exhibit remarkable performance as a high-efficiency and stable bifunctional electrocatalyst in both alkaline freshwater and seawater electrolytes. For instance, FeNiCoCrRu HEA NPs delivered overpotentials of 0.148 V at 600 mA cm−2 for the HER and 0.353 V at 300 mA cm−2 for the OER in alkaline seawater electrolyte. The assembled FeNiCoCrRu||FeNiCoCrRu electrolyzer exhibited negligible voltage increase at 250 mA cm−2 even after >3000 hours of continuous operation. This outstanding bifunctional electrocatalytic activities and long duration can be attributed to high-entropy design, electron redistribution, large electrochemical active surface area, and excellent chemical and structural stability. In situ Raman spectra indicated that, regarding the HER mechanism, the Ni sites facilitate H2O adsorption and dissociation, while the Ru sites simultaneously accelerate the combination of H* to form H2. Regarding the OER mechanism, the Ni sites facilitate the deprotonation of *OH to form *O intermediate, thereby accelerating O2 production. This work not only demonstrates the advantages of laser induction for eco-friendly, high-throughput, and scalable catalysts synthesis, but also provides deeper insights into the design of HEA NPs and their correlation with seawater electrolysis.4. Materials
4.1 Chemicals
Iron(III) chloride hexahydrate (FeCl3·6H2O, Sigma), nickel(II) chloride hexahydrate (NiCl2·6H2O, Fisher), cobalt(II) chloride hexahydrate (CoCl2·6H2O, Sigma), chromium(III) chloride hexahydrate (CrCl3·6H2O, Aldrich), ruthenium(III) chloride hydrate (RuCl3·xH2O, Aldrich), copper(II) chloride dihydrate (CuCl2·2H2O, Fisher), manganese(II) chloride tetrahydrate (MnCl2·4H2O, Fisher), sodium molybdate dihydrate (Na2MoO4·2H2O, Fisher), trisodium citrate dihydrate (Na3C6H5O7·2H2O, Sigma), Pt/C (20 wt%, Alfa Aesar), ruthenium oxide (RuO2, Alfa Aesar), potassium hydroxide (KOH, Fisher), sodium chloride (NaCl, Sigma Aldrich), Nafion (D520, 5 w/w in water and 1-propanol, ThermoScientific), ethanol (200 proof, Decon Labs), and carbon paper (AvCarb MGL370, FuelCell) were used as received without any purification. Deionized water (DI H2O) was used to prepare aqueous solutions.4.2 Preparation of FeNiCoCrRu HEA NPs
CP was first heated at 500 °C for 10 min to increase the wettability and then cut into small pieces with a size of 1 × 1.2 cm2. CP was then washed subsequently with deionized water and ethanol at least three times and dried in an oven. After that, a precursor solution of FeNiCoCrRu was prepared by dissolving 0.096 mmol FeCl3·6H2O, 0.096 mmol NiCl2·6H2O, 0.096 mmol CoCl2·6H2O, 0.096 mmol CrCl3·6H2O, 0.096 mmol RuCl3·xH2O and 0.960 mmol Na3C6H5O7·2H2O in a mixture of DI H2O (4.8 mL) and ethanol (1.2 mL). Then, 48 μL of FeNiCoCrRu precursor solution was drop-cast onto CP and dried at 90 °C on a hotplate. Then, CP was irradiated using a commercial CO2 laser (Universal Laser Systems VLS3.50/60DT) and was washed with deionized water at least three times. An optimum recipe to produce FeNiCoCrRu NPs was obtained via repeating the above-mentioned steps sixteen times. The spot size and focal length of CO2 laser are 0.03 mm and 50.8 mm, respectively. The optimal laser processing parameters were determined to be a laser power of 18 W and a scanning rate of 10 cm s−1. For comparison, FeNiCoRu, FeNiCoCr, FeNiCo, FeNi, Ru, Cr, Co, Ni, and Fe were also synthesized under identical reaction conditions except the composition of the metal precursor solutions. More details can be found in Table S1 (ESI†).4.3 Preparation of Pt/C and RuO2
For comparison with conventional benchmarks, commercial Pt/C and RuO2 electrocatalysts were also prepared. First, 4 mg of a commercial electrocatalyst (Pt/C or RuO2) was ultrasonicated in 1 mL of the mixture solution containing deionized water, ethanol and Nafion (v/v/v = 19/5/1) for 4 h. Then, 150 μL of the dispersion mixture was drop-cast onto CP and dried at 90 °C on a hotplate. The loading mass of Pt/C and RuO2 was calculated to be 0.5 mg cm−2.4.4 Material characterization
The crystallinity and phase purity of FeNiCoCrRu HEA NPs were obtained using an X-ray diffractometer (Rigaku SmartLab) with Cu Kα radiation (λ = 0.15406 nm). The surface morphology of FeNiCoCrRu HEA NPs was observed on a FEI Quanta 600F environmental scanning electron microscope (SEM). Scanning/transition electron microscopy (S/TEM) characterization was performed using a ThermoScientific Spectra 300 STEM equipped with a Super-X X-ray energy dispersive spectroscopy (EDS) silicon drift detector (0.7 sr). Elemental mapping experiments were conducted at an accelerating voltage of 300 kV and a beam current of ∼200 pA to quantify the composition of FeNiCoCrRu HEA NPs. The chemical composition and oxidation state of FeNiCoCrRu HEA NPs were obtained using X-ray photoelectron spectroscopy (315 PHI Quantera) under a pressure of 5 × 10−9 Torr.4.5 Electrochemical measurements
All electrochemical measurements were conducted on a Biologic SP-200 electrochemical workstation with a three-electrode configuration. The as-prepared FeNiCoCrRu HEA NPs, graphite rod and Ag/AgCl (filled with 1 M KCl) were used as the working, counter, and reference electrodes, respectively. There were two types of electrolytes including alkaline freshwater (1 M KOH) and alkaline saline (1 M KOH + 0.5 M NaCl). All measured potentials were calibrated to RHE according to the equation: ERHE = E(Ag/AgCl) + 0.222 + 0.059 × pH. CV was conducted at a scan rate of 100 mV s−1 to activate and stabilize the electrocatalysts. LSV data were collected at a scan rate of 10 mV s−1 with 95% iR-correction. Electrochemical impedance spectroscopy (EIS) tests were carried out at an overpotential of 0.3 V in a frequency range of 100000 to 0.1 Hz at an amplitude of 5 mV. Long-term stability was evaluated using a chronopotentiometry technique at 250 mA cm−2. To estimate the double layer capacitance (Cdl), CV measurements in a non-faradaic region (0–0.1 V for HER and 1.1–1.2 V for OER) were collected at different scanning rates. Cdl was calculated by plotting ΔJ = Ja − Jc at 0.05 V (HER) and 1.15 V (OER) as a function of the scan rate. The electrochemical active surface area (ECSA) was obtained using the equation ECSA = Cdl/(Cs × A), where Cs is the capacitance of an atomically smooth planar surface (0.04 mF cm−2 in the alkaline electrolyte), and A is the electrode area (1 cm2). Turnover frequency (TOF) is calculated using the equation TOF = (total number of hydrogen turnover × J)/(total number of active sites × ECSA). More calculation details can be found in Note S4 (ESI†). In situ Data for Raman analysis were recorded using a home-made 3-electrode electrochemical cell using Raman spectroscopy with a 633 nm laser beam. The in situ Raman spectra characterization under different applied potentials was performed. The acquisition time for each spectrum was set to be 20 s with 2 sweeps.
Author contributions
Y. X prepared the electrocatalysts and conducted electrochemical tests. S. X. conducted XRD, XPS, and ICP-MS characterizations under the guidance of J. M. T. SEM, TEM, HRTEM, and EDS characterizations were done by A. M. B. Z. printed the in situ Raman cell and Z. C. drew the schematic. Both B. Z. and Z. C. made code for data analysis. J. L. conceived the idea, managed the research progress, and provided regular guidance. Y. X. drafted the first manuscript which was revised by J. L., S. X., A. M., and J. M. T. All authors commented and agreed on the final version.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
All authors claim there are no competing interests.Acknowledgements
J. L. thanks the financial support from U.S. National Science Foundation (award numbers: 1825352 and 1933861) and U.S. Department of Interior (grant number: R21AC10073-00). J. L. and J. M. T. acknowledge the financial support from U.S. Army Corps of Engineers, ERDC (grant number: W912HZ-21-2-0050).Notes and references
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef
.
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef PubMed
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 Search PubMed
- R. Fan, C. Liu, Z. Li, H. Huang, J. Feng, Z. Li and Z. Zou, Nat. Sustainability, 2024, 7, 158–167 CrossRef
- X. Liu, J. Chi, H. Mao and L. Wang, Adv. Energy Mater., 2023, 13, 2301438 CrossRef CAS
- X. Wang, Q. Peng, X. Zhang, X. Lv, X. Wang and Y. Fu, J. Colloid Interface Sci., 2022, 607, 1580–1588 CrossRef CAS PubMed
- M. Shi, T. Tang, L. Xiao, J. Han, X. Bai, Y. Sun, S. Chen, J. Sun, Y. Ma and J. Guan, Chem. Commun., 2023, 59, 11971–11974 RSC
- P. Li, Y. Yao, W. Ouyang, Z. Liu, H. Yin and D. Wang, J. Mater. Sci. Technol., 2023, 138, 29–35 CrossRef CAS
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, Energy Environ. Sci., 2021, 14, 2883–2905 RSC
- J. X. Yang, B.-H. Dai, C.-Y. Chiang, I. C. Chiu, C.-W. Pao, S.-Y. Lu, I. Y. Tsao, S.-T. Lin, C.-T. Chiu, J.-W. Yeh, P.-C. Chang and W.-H. Hung, ACS Nano, 2021, 15, 12324–12333 CrossRef CAS PubMed
- Y. Lu, K. Huang, X. Cao, L. Zhang, T. Wang, D. Peng, B. Zhang, Z. Liu, J. Wu, Y. Zhang, C. Chen and Y. Huang, Adv. Funct. Mater., 2022, 32, 2110645 CrossRef CAS
- B. Wang, C. Wang, X. Yu, Y. Cao, L. Gao, C. Wu, Y. Yao, Z. Lin and Z. Zou, Nat. Synth., 2022, 1, 138–146 CrossRef
- C. Hegde, C. H. J. Lim, T. H. Teng, D. Liu, Y.-J. Kim, Q. Yan and H. Li, Small, 2022, 18, 2203126 CrossRef CAS
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. G. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS
- H. Deng, K. Sattari, Y. Xie, P. Liao, Z. Yan and J. Lin, Nat. Commun., 2020, 11, 6325 CrossRef
- B. Zheng, G. Zhao, Z. Yan, Y. Xie and J. Lin, Adv. Funct. Mater., 2023, 33, 2210084 CrossRef
- R. Jiang, Y. Da, X. Han, Y. Chen, Y. Deng and W. Hu, Cell Rep. Phys. Sci., 2021, 2, 100302 CrossRef
- Q. Liu and S. W. Chen, Trends Chem., 2022, 4, 918–934 CrossRef
- X. Hu, D. Zuo, S. Cheng, S. Chen, Y. Liu, W. Bao, S. Deng, S. J. Harris and J. Wan, Chem. Soc. Rev., 2023, 52, 1103–1128 RSC
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef PubMed
- M. Yamada, N. Soma, M. Tsuta, S. Nakamura, N. Ando and F. Matsumoto, Int. J. Extreme Manuf., 2023, 5, 035004 CrossRef
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef
- A. Takeuchi and A. Inoue, Mater. Trans., 2005, 46, 2817–2829 CrossRef
- X. Wang, Q. Dong, H. Qiao, Z. Huang, M. T. Saray, G. Zhong, Z. Lin, M. Cui, A. Brozena, M. Hong, Q. Xia, J. Gao, G. Chen, R. Shahbazian-Yassar, D. Wang and L. Hu, Adv. Mater., 2020, 32, 2002853 CrossRef
- Y. Yao, Z. Huang, L. A. Hughes, J. Gao, T. Li, D. Morris, S. E. Zeltmann, B. H. Savitzky, C. Ophus, Y. Z. Finfrock, Q. Dong, M. Jiao, Y. Mao, M. Chi, P. Zhang, J. Li, A. M. Minor, R. Shahbazian-Yassar and L. Hu, Matter, 2021, 4, 2340–2353 CrossRef
- S. Li, H. Xie, Q. Dong, S. Jing, T. Li, L. Xu and L. Hu, Small Struct., 2023, 4, 2200176 CrossRef
- L. Wu, F. Zhang, S. Song, M. Ning, Q. Zhu, J. Zhou, G. Gao, Z. Chen, Q. Zhou, X. Xing, T. Tong, Y. Yao, J. Bao, L. Yu, S. Chen and Z. Ren, Adv. Mater., 2022, 34, 2201774 CrossRef
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu, S. Hu, F. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC
- Y.-Y. Ma, C.-X. Wu, X.-J. Feng, H.-Q. Tan, L.-K. Yan, Y. Liu, Z.-H. Kang, E.-B. Wang and Y.-G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC
- B. Wang, M. Lu, D. Chen, Q. Zhang, W. Wang, Y. Kang, Z. Fang, G. Pang and S. Feng, J. Mater. Chem. A, 2021, 9, 13562–13569 RSC
- J. Hao, Z. Zhuang, K. Cao, G. Gao, C. Wang, F. Lai, S. Lu, P. Ma, W. Dong, T. Liu, M. Du and H. Zhu, Nat. Commun., 2022, 13, 2662 CrossRef PubMed
- R. F. Zhang, X. F. Kong, H. T. Wang, S. H. Zhang, D. Legut, S. H. Sheng, S. Srinivasan, K. Rajan and T. C. Germann, Sci. Rep., 2017, 7, 9577 CrossRef PubMed
- T. Zhu, J. Huang, B. Huang, N. Zhang, S. Liu, Q. Yao, S.-C. Haw, Y.-C. Chang, C.-W. Pao, J.-M. Chen, Q. Shao, Z. Hu, Y. Ma and X. Huang, Adv. Energy Mater., 2020, 10, 2002860 CrossRef
- K. Lellala, Energy Fuels, 2021, 35, 8263–8274 CrossRef
- S. S. Kanakkillam, B. Krishnan, D. A. Avellaneda and S. Shaji, Colloids Surf., A, 2020, 594, 124657 CrossRef
- X. Liu and J. Wu, Electrochim. Acta, 2019, 320, 134577 CrossRef
- Q. Si, M. Matsui, T. Horiba, O. Yamamoto, Y. Takeda, N. Seki and N. Imanishi, J. Power Sources, 2013, 241, 744–750 CrossRef CAS
- Y. Sun and S. Dai, Sci. Adv., 2021, 7, eabg1600 CrossRef CAS PubMed
- Q. Wang, Z. Jia, J. Li, Y. He, Y. Yang, Y. Li, L. Sun and B. Shen, Small, 2022, 18, 2204135 CrossRef CAS
- T. X. Nguyen, J. Patra, J.-K. Chang and J.-M. Ting, J. Mater. Chem. A, 2020, 8, 18963–18973 RSC
- Z. Zhang, J. Cai, H. Zhu, Z. Zhuang, F. Xu, J. Hao, S. Lu, H. Li, F. Duan and M. Du, Chem. Eng. J., 2020, 392, 123655 CrossRef
- Q. He, Y. Zhou, H. Shou, X. Wang, P. Zhang, W. Xu, S. Qiao, C. Wu, H. Liu, D. Liu, S. Chen, R. Long, Z. Qi, X. Wu and L. Song, Adv. Mater., 2022, 34, 2110604 CrossRef
- F. Wang, P. Zou, Y. Zhang, W. Pan, Y. Li, L. Liang, C. Chen, H. Liu and S. Zheng, Nat. Commun., 2023, 14, 6019 CrossRef
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef
- J. Kibsgaard and T. F. Jaramillo, Angew. Chem., Int. Ed., 2014, 53, 14433–14437 CrossRef PubMed
- F. S. M. Ali, R. L. Arevalo, M. Vandichel, F. Speck, E.-L. Rautama, H. Jiang, O. Sorsa, K. Mustonen, S. Cherevko and T. Kallio, Appl. Catal., B, 2022, 315, 121541 CrossRef CAS
- W. Tong, M. Förster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS
- A. Abdelhafiz, B. Wang, A. R. Harutyunyan and J. Li, Adv. Energy Mater., 2022, 12, 2200742 CrossRef
- T. X. Nguyen, Y.-H. Su, C.-C. Lin, J. Ruan and J.-M. Ting, Adv. Sci., 2021, 8, 2002446 CrossRef
- H. Jin, J. Xu, H. Liu, H. Shen, H. Yu, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Sci. Adv., 2023, 9, eadi7755 CrossRef PubMed
- T. Li, B. Wang, Y. Cao, Z. Liu, S. Wang, Q. Zhang, J. Sun and G. Zhou, Nat. Commun., 2024, 15, 6173 CrossRef
- M. Ning, F. Zhang, L. Wu, X. Xing, D. Wang, S. Song, Q. Zhou, L. Yu, J. Bao, S. Chen and Z. Ren, Energy Environ. Sci., 2022, 15, 3945–3957 RSC
- Z. Cai, J. Liang, Z. Li, T. Yan, C. Yang, S. Sun, M. Yue, X. Liu, T. Xie, Y. Wang, T. Li, Y. Luo, D. Zheng, Q. Liu, J. Zhao, X. Sun and B. Tang, Nat. Commun., 2024, 15, 6624 CrossRef PubMed
- M. Zhang, X. He, K. Dong, H. Zhang, Y. Yao, C. Yang, M. Yue, S. Sun, Y. Sun, D. Zheng, Y. Luo, Q. Liu, N. Li, B. Tang, J. Liu and X. Sun, Chem. Commun., 2023, 59, 9750–9753 RSC
- H. Zhu, S. Sun, J. Hao, Z. Zhuang, S. Zhang, T. Wang, Q. Kang, S. Lu, X. Wang, F. Lai, T. Liu, G. Gao, M. Du and D. Wang, Energy Environ. Sci., 2023, 16, 619–628 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee01093k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |