
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing lithium storage rate and durability in sphalerite GeP by engineering configurational entropy†
Yanhong
Li
ac,
Jeng-Han
Wang
b,
Tzu-Yu
Liu
b,
Xinwei
Li
e,
Zaiping
Guo
 d,
Meilin
Liu
a and
Wenwu
Li
*ae
d,
Meilin
Liu
a and
Wenwu
Li
*ae
aSchool of Materials Science & Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: wenwuli@skku.edu
bDepartment of Chemistry, National Taiwan Normal University, Taipei, 11677, Taiwan
cFaculty of Materials Science and Engineering/Institute of Technology for Carbon Neutrality, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518060, China
dSchool of Chemical Engineering & Advanced Materials, The University of Adelaide, Adelaide, SA 5005, Australia
eSchool of Chemical Engineering, Sungkyunkwan University (SKKU) 2066, Seoburo, Jangan-gu, Suwon 440-746, Republic of Korea
First published on 8th August 2024
Abstract
Monoclinic GeP with a layered structure, featuring a large capacity, low plateau, and high initial coulombic efficiency (ICE), has been demonstrated as a promising alternative anode material for Li-ion batteries. However, its semiconductor feature and overutilization of expensive Ge pose a significant obstacle to its further advancement. To further improve electronic and Li-ionic conductivity, and reduce the cost, via a mechanochemical method, we synthesize a cubic GeP-based compound, ZnGeSiP3, which possesses a triple cation-mixed sphalerite lattice, affording metallic conductivity and rapid Li-ion diffusion, and thereby outperforms monoclinic GeP due to enhanced conformational entropy, as verified through theoretical calculations and experimental analyses. Various characterization techniques, such as operando X-ray diffraction (XRD), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS), confirm the reversible storage of Li-ions within ZnGeSiP3via intercalation and subsequent conversion reactions, yielding a notable reversible capacity of 1638 mA h g−1 with an ICE of 92% at 100 mA g−1. The graphite-modified ZnGeSiP3 composite exhibits exceptional long-term cycling stability, retaining 981 mA h g−1 after 1600 cycles at 2000 mA g−1, and ultrahigh rate performance, maintaining 568 mA h g−1 at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA g−1, surpassing most previously studied anodes. Drawing inspiration from the favorable entropic effects, we synthesize high-entropy cation-mixed sphalerite-structured GeP-based compounds, including CuSnAlZnGeSiP6, CuSn (or Al)ZnGeSiP5, and SnAlZnGeSiP5, as well as quaternary cation-disordered sphalerite-structured GeP-based compounds of Cu (or Sn, or Al)ZnGeSiP4. Furthermore, we synthesize high-entropy sphalerite-structured GeP-based compounds ZnGeSiPSSe and ZnGeSiP2Se (or S) with disordered cationic and anionic compositions, effectively addressing the challenge of incompatible multiple anions and cations. The phase formation mechanisms of these sphalerite-structured GeP-based compounds can be attributed to their negative phase formation energies, benefiting from the elevated conformational entropy. Crucially, all the aforementioned sphalerite-structured GeP-based compounds have metallic conductivity and showcase superior electrochemical Li-storage properties, including high capacity, high ICE, small polarization loss, and suitable operating potential. Broadly, the high conformational entropy strategy can serve as a new design paradigm for high-performance and cost-effective anodes for LIBs and beyond.
000 mA g−1, surpassing most previously studied anodes. Drawing inspiration from the favorable entropic effects, we synthesize high-entropy cation-mixed sphalerite-structured GeP-based compounds, including CuSnAlZnGeSiP6, CuSn (or Al)ZnGeSiP5, and SnAlZnGeSiP5, as well as quaternary cation-disordered sphalerite-structured GeP-based compounds of Cu (or Sn, or Al)ZnGeSiP4. Furthermore, we synthesize high-entropy sphalerite-structured GeP-based compounds ZnGeSiPSSe and ZnGeSiP2Se (or S) with disordered cationic and anionic compositions, effectively addressing the challenge of incompatible multiple anions and cations. The phase formation mechanisms of these sphalerite-structured GeP-based compounds can be attributed to their negative phase formation energies, benefiting from the elevated conformational entropy. Crucially, all the aforementioned sphalerite-structured GeP-based compounds have metallic conductivity and showcase superior electrochemical Li-storage properties, including high capacity, high ICE, small polarization loss, and suitable operating potential. Broadly, the high conformational entropy strategy can serve as a new design paradigm for high-performance and cost-effective anodes for LIBs and beyond.
Broader contextHigh-entropy materials have garnered considerable attention across various fields including (electro)catalysis, thermoelectrics, and electrochemical energy storage. High-entropy oxides of different structures, encompassing fluorite, perovskite, rock salt, and spinel, exhibit remarkable functionalities inherent to their unique structures. Notably, high-entropy cathodes demonstrate significantly improved performance and durability. However, oxide-based anodes often encounter challenges linked to higher potentials (>1.0 V), which constrain cell voltage and energy density despite their cycling stability and high rate capability. Concurrently, engineering high-entropy configurations for both cationic and anionic sites proves particularly daunting compared to metallic alloys, impeding the realization of their electrochemical potential for desired properties. In this study, we synthesized high-entropy phosphides with a sphalerite lattice, which demonstrate lower working potentials, faster reaction kinetics, and higher energy efficiency. This research aims to establish a correlation between high entropy and lithium storage capabilities. |
Introduction
Amidst the rapid advancement of electric vehicles, there is a growing focus on exploring new electrode materials with high capacity to meet the escalating energy demands of Li-ion batteries (LIBs).1–6 The conventional graphite anode offers a restricted capacity of merely 372 mA h g−1, falling short of achieving the targeted 500 W h kg−1 for LIBs due to its one-electron intercalation reaction.7–13 In stark contrast, emerging alloying-type materials present significant advantages over graphite due to their substantially higher capacity achieved through multi-electron reactions.14–25 Among these, monoclinic GeP with a distinctive layered structure provides a remarkable theoretical capacity of 1913 mA h g−1, five times greater than that of graphite, positioning it as a promising alternative high-capacity anode for LIBs.26–28 The combination of the large capacity, suitable plateau, and high reversibility positions monoclinic GeP as a promising candidate for achieving high energy density and high-rate capabilities. Beyond monoclinic GeP, cubic GeP with a sphalerite lattice theoretically presents a metallic state, facilitated by phosphorus's contribution of delocalized electrons, thus promising more superior electrochemical Li-storage properties. However, sphalerite-structured GeP is only observed under high-pressure conditions due to the energetic challenges associated with its positive phase formation energy.Recently, compounds characterized by high conformational entropy, wherein multiple principal elements coexist at a specific site within a unified lattice structure, have garnered escalating interest across diverse research domains owing to their notable holistic attributes.29–34 Within disordered multicomponent systems, substantial configurational entropy is commonly acknowledged to confer stability upon crystal structures, while the admixture of multiple cations serves to augment chemical and structural diversity.35–39 Additionally, lattice distortions induced by the substitution of ions of varying sizes can enhance both physicochemical and mechanical properties. Due to the cocktail effect, high entropy compounds exhibit significant improvement in electrochemical properties, especially in the cathode field, while research on the anode field is relatively scarce.40–46 Therefore, establishing high configurational entropy for cubic GeP by introducing more cost-effective elements to lower the phase formation energy is a promising strategy to stabilize sphalerite-structured GeP, and simultaneously achieve performance breakthroughs while avoiding the overuse of the relatively expensive Ge.
Establishing high conformational entropy in sphalerite-structured GeP is exceedingly challenging due to the potential air instability and synergistic coordination with phosphorus among various metal cations with differing atomic radii, electronegativities, and bonding characteristics. Achieving high entropy engineering for both cationic and anionic sites simultaneously proves even more difficult than in metallic alloys due to the intricate charge balance considerations involved, thus remaining unrealized to date. Consequently, engineering high conformational entropy in sphalerite-structured GeP with appropriate constituents to attain superior physicochemical properties for Li-storage, while uncovering underlying fundamentals, represents a task of paramount importance yet considerable challenge.
Herein, we, for the first time, synthesized a triple cation-mixed sphalerite-structured GeP-based compound, ZnGeSiP3, utilizing monoclinic GeP with a layered structure and other precursors as raw materials via a simple ball milling method.47 Both experimental measurements and theoretical computations reveal that the synthesized ZnGeSiP3 has faster Li-ionic and electronic transport capabilities compared to its parent phases of layered Ge (or Si)P, and isomorphic ZnGe2P3 and Ge, facilitated by the heightened conformational entropy. Specifically, the ZnGeSiP3 carbon composite presents exceptional long-term cycling stability, retaining 981 mA h g−1 after 1600 cycles at 2000 mA g−1, and ultrahigh rate performance, maintaining 568 mA h g−1 at 22000 mA g−1, surpassing most anodes studied before. In situ XRD and complementary characterization studies corroborate a reversible Li-storage mechanism encompassing (de)intercalation reactions followed by (de)conversion and (de)alloy reactions within the ZnGeSiP3 compound. Motivated by the advantageous effects mediated by entropy, we further synthesize sphalerite-structured GeP-based compounds with high conformational entropy, including CuSnAlZnGeSiP6, CuSn (or Al)ZnGeSiP5, SnAlZnGeSiP5, ZnGeSiPSSe, and ZnGeSiP2S (or Se) as well as quaternary cation-disordered sphalerite-structured GeP-based compounds of Cu (or Sn, or Al)ZnGeSiP4. The phase formation mechanisms of these sphalerite-structured GeP-based compounds are rationalized in terms of their negative phase formation energies, stemming from the increased configurational entropy. Noteworthy is the metallic conductive conductivity attained by the aforementioned high conformational entropy sphalerite-structured GeP-based compounds. This property underscores their superior electrochemical Li-storage characteristics, including high capacity, high energy efficiency, minimal polarization loss, and favorable anodic working potentials, thus positioning them as promising candidates for near-future applications.
Results and discussion
Silicon (Si) is abundant, eco-friendly, cost-effective, and high-capacity, already partially replacing graphite. It enhances lithium storage in cubic SiP with metallic properties. Zinc (Zn) stabilizes the zinc blende structure, contributes via LiZn alloys, and supports industrialization due to its abundance and mature processing technology. Si and Zn, with similar atomic sizes to phosphorus (P) and germanium (Ge), ensure stable coexistence in new materials, and Zn's lower melting point aids synthesis.48 Introducing both Zn and Si increases configurational entropy, reducing Gibbs free energy and stabilizing new material formation. Therefore, we attempted to introduce both Zn and Si into layered GeP to form a metallic ZnGeSiP3 compound. Fig. 1(a) illustrates the inaugural synthesis of the quaternary compound ZnGeSiP3, utilizing a mechanochemical approach with raw materials comprising mixed powders of layer Ge (or Si) P (Fig. S1–S4, ESI†), amorphous red P, and Zn. XRD Rietveld refinement (Fig. 1(b)) confirms the successful formation of the ZnGeSiP3 compound, which exhibits a cation mixed sphalerite structure (Fig. 1(a), space group 216: F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m). Crystallographic parameters are detailed in Tables S1 and S2 (ESI†). Notably, Zn, Ge, and Si demonstrate analogous atomic coordination environments, competitively occupying cationic sites, while P fully occupies the anionic sites within the face-centered cubic zinc blende crystal structure. Despite the positive phase formation energies and charge balance complexity hindering the preparation of the sphalerite-structured Group IVA–VA compounds GeSiP2, GeP, and SiP (Fig. S5, ESI†), we attributed the phase formation mechanism of the triple cation-mixed sphalerite-structured ZnGeSiP3 compound to the increased configurational entropy. This entropy augmentation endows the triple-cation disordered sphalerite-structured ZnGeSiP3 compound with a more negative phase formation energy, facilitating the phase transition from the monoclinic GeP compound (Fig. 1(a)). Furthermore, experimentation with different raw materials demonstrates the stability of the synthesized phase under ambient conditions of temperature and pressure (Fig. S6, ESI†). Despite initial energy differences among the precursors, ball milling minimizes these disparities, leading to the formation of ZnGeSiP3. Consequently, the final product's attributes, including crystallinity, remain largely unaffected by the initial states of the starting materials (Fig. S7 and S8, ESI†). The ultimate state of the ball-milled product depends on conditions such as the ball mill type, rotational speed, milling medium, and ball-to-powder ratio. During initial mixing, the process breaks raw material bonds and forms new ones. Once stability is achieved, properties like morphology and crystallinity remain unchanged. Thermal analysis shows that the ZnGeSiP3 compound is stable at 450 °C in air (Fig. S9, ESI†).
3m). Crystallographic parameters are detailed in Tables S1 and S2 (ESI†). Notably, Zn, Ge, and Si demonstrate analogous atomic coordination environments, competitively occupying cationic sites, while P fully occupies the anionic sites within the face-centered cubic zinc blende crystal structure. Despite the positive phase formation energies and charge balance complexity hindering the preparation of the sphalerite-structured Group IVA–VA compounds GeSiP2, GeP, and SiP (Fig. S5, ESI†), we attributed the phase formation mechanism of the triple cation-mixed sphalerite-structured ZnGeSiP3 compound to the increased configurational entropy. This entropy augmentation endows the triple-cation disordered sphalerite-structured ZnGeSiP3 compound with a more negative phase formation energy, facilitating the phase transition from the monoclinic GeP compound (Fig. 1(a)). Furthermore, experimentation with different raw materials demonstrates the stability of the synthesized phase under ambient conditions of temperature and pressure (Fig. S6, ESI†). Despite initial energy differences among the precursors, ball milling minimizes these disparities, leading to the formation of ZnGeSiP3. Consequently, the final product's attributes, including crystallinity, remain largely unaffected by the initial states of the starting materials (Fig. S7 and S8, ESI†). The ultimate state of the ball-milled product depends on conditions such as the ball mill type, rotational speed, milling medium, and ball-to-powder ratio. During initial mixing, the process breaks raw material bonds and forms new ones. Once stability is achieved, properties like morphology and crystallinity remain unchanged. Thermal analysis shows that the ZnGeSiP3 compound is stable at 450 °C in air (Fig. S9, ESI†).
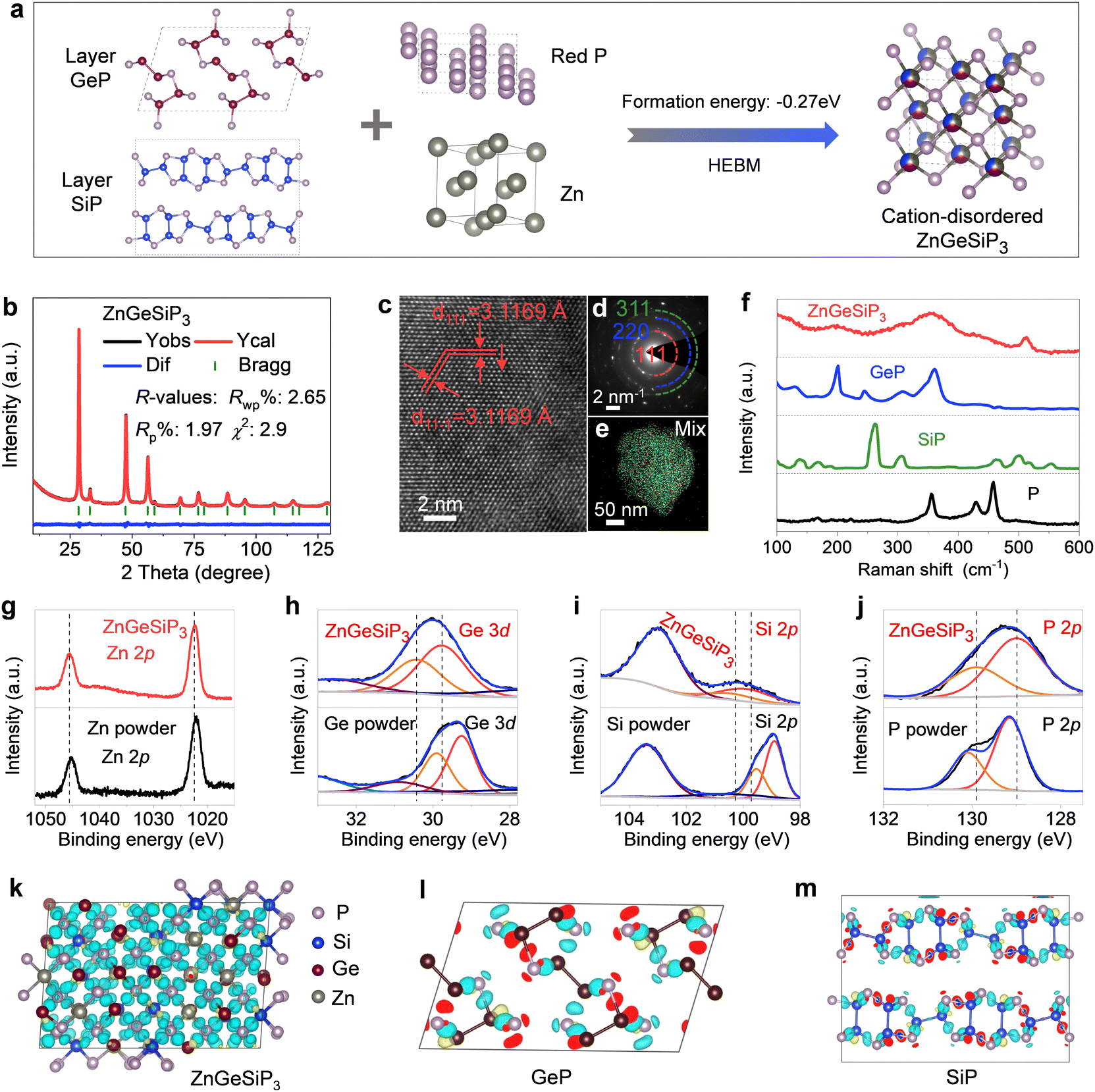 | ||
| Fig. 1 Synthesis, characterization and disordered lattice analysis of ZnGeSiP3. (a) Synthetic scheme. (b) XRD refinement. (c) HRTEM and (d) SAED. (e) Elemental mappings. (f) Raman spectra. (g)–(j) XPS analysis. (k)–(m) Charge density difference. | ||
High-resolution transmission electron microscopy (HRTEM) analysis of the triple-cation mixed sphalerite-structured ZnGeSiP3 compound reveals a distinct d-spacing of 0.312 nm, consistent with the characteristic crystalline plane of (111) within the cubic ZnS lattice, as depicted in Fig. 1(c). Concurrently, the selected area electron diffraction (SAED, inset of Fig. 1(d)) pattern exhibits a halo-like multiple ring feature, indicative of the polycrystalline nature of the ZnGeSiP3 compound. Additionally, the concentric ring pattern observed can be attributed to the overlapping of variously oriented crystallites, denoted by the dashed arcs. This observation aligns well with the crystallographic planes identified through XRD characterization, affirming the absence of additional superstructures and thus confirming the successful formation of the cubic ZnS lattice. Microscopic energy-dispersive X-ray spectroscopy (EDS) elemental mapping offers direct insight into the elemental composition at the individual particle level. Fig. 1(e) and Fig. S10 (ESI†) illustrate that the synthesized sample encompasses all four constituent elements—Zn, Ge, Si, and P—with uniform distribution across the particles, devoid of elemental segregation or the emergence of immiscible phases. This underscores the effective synthesis of the monophasic ZnGeSiP3 compound at the particle level.
We conducted supplementary spectroscopic analyses on the triple-cation mixed sphalerite-structured ZnGeSiP3 compound sample, encompassing Raman spectroscopy and X-ray photoemission spectroscopy (XPS), apart from the aforementioned crystallographic characterization studies. Our investigation into the vibrational properties of the synthesized ZnGeSiP3 compound combines experimental methodologies with computational approaches. Raman vibration modes of the synthesized ZnGeSiP3 compound are depicted in Fig. 1(f) and Fig. S11 (ESI†), demonstrating totally different Raman spectra from its parent phases of layered Ge (or Si)P, and milled red phosphorus, implying a new phase formation. Based on the “special quasi-random structure (SQS)” theory, we built a 144-atom supercell of the triple-cation-disordered sphalerite-structured ZnGeSiP3 and computed its Raman vibrating modes. Leveraging Raman measurements of the triple cation-mixed ZnGeSiP3 compound (Fig. 1(f)), we computationally scrutinized their signature vibrations of Si–P, Zn–P and Ge–P stretching modes located at ca. 500, 410, and 380 cm−1, respectively, as presented in Movie S1 (ESI†). Consistently, high-frequency Si–P stretching modes can be observed in layered SiP, as well as the high-frequency Si–Si stretching modes (≥500 cm−1), as demonstrated in Movie S2 (ESI†); similarly, low-frequency Ge–P stretching modes can be identified in layered GeP, as well as the low-frequency Ge–Ge stretching modes (<400 cm−1), as shown in Movie S3 (ESI†). The mid-frequency Zn–P stretching modes, not surprisingly, are shown in neither layered Ge (or Si)P nor black phosphorus (Movie S4, ESI†).
Next, we analyzed the X-ray photoemission spectroscopy (XPS) spectra of the triple-cation mixed sphalerite-structured ZnGeSiP3 compound by virtue of a joint way of experimental measurements and theoretical calculations. As demonstrated in Fig. 1(g), the high-resolution Zn 2p spectrum exhibited two distinct peaks positioned at around 1045.7 and 1022.4 eV, assigned to Zn 2p1/2 and Zn 2p3/2, respectively. The high-resolution Ge 3d spectrum exhibited two prominent fingerprint peaks centered at approximately 30.45 and 29.75 eV, corresponding to Ge 3d3/2 and Ge 3d5/2, respectively, as presented in Fig. 1(h). Moreover, the high-resolution Si 2p spectra displayed fingerprint peaks at around 100.8 and 100.05 eV, attributed to Si 2p1/2 and Si 2p3/2, respectively (Fig. 2(i)). The high-resolution P 2p spectrum revealed two discernible binding energies at approximately 129.9 and 129.0 eV, corresponding to P 2p1/2 and P 2p3/2, respectively, as shown in Fig. 1(j). In tandem with the experimental findings, computational analysis was conducted to evaluate the induced charge upon bond formation, complementing the XPS results. The charge analysis of ZnGeSiP3 unveiled positive induced charges on all cations (Ge, Zn, and Si), with the highest magnitude observed on Si and the lowest on Ge, while P exhibited a negative charge, as presented in Fig. 1(k). Corresponding to the provided binding energies (Table S3, ESI†), the valences of the constituent elements—Ge, Zn, Si, and P—in the triple cation-disordered ZnGeSiP3 were approximated to be +3, +2, +4, and −3, respectively. These values aligned well with the calculated Bader charge distribution on each constituent element: Ge (+3.0), Zn (+0.6), Si (+3.9), and P (−2.5) (Fig. S12a and b, ESI†). Notably, within the triple cation-disordered ZnGeSiP3 compound, Si demonstrated a more positive charge compared to Ge, attributed probably to the distribution of valence electrons in the outer layer of germanium relative to silicon. The Bader charges for the layered GeP, and SiP are the same that both of the cations (Ge, and Si) are +3.0, and the anion (P) is −3.0. Although the Bader charges for the triple cation-disordered ZnGeSiP3, and the layered Ge (or Si)P are quite similar, their spatial distributions of charges, as shown in Fig. 1(l), (m) and Fig. S12c–f (ESI†), are very different that leads to distinct behaviors of their electrical conductivity. In the triple cation-disordered ZnGeSiP3, charges are evenly distributed, as all the cations (Zn, Ge, and Si) are bonded with anions (P); the homogeneous charge distribution indicates the better electrical conductivity of the ZnGeSiP3, in agreement with the DOS analysis (Fig. 4(e)). On the other hand, the layered Ge (or Si)P has a somewhat inhomogeneous charge distribution, due to the existence of Ge–Ge (or Si–Si) bonds, implying the less conductive Ge (or Si)P (Fig. 4(f)).
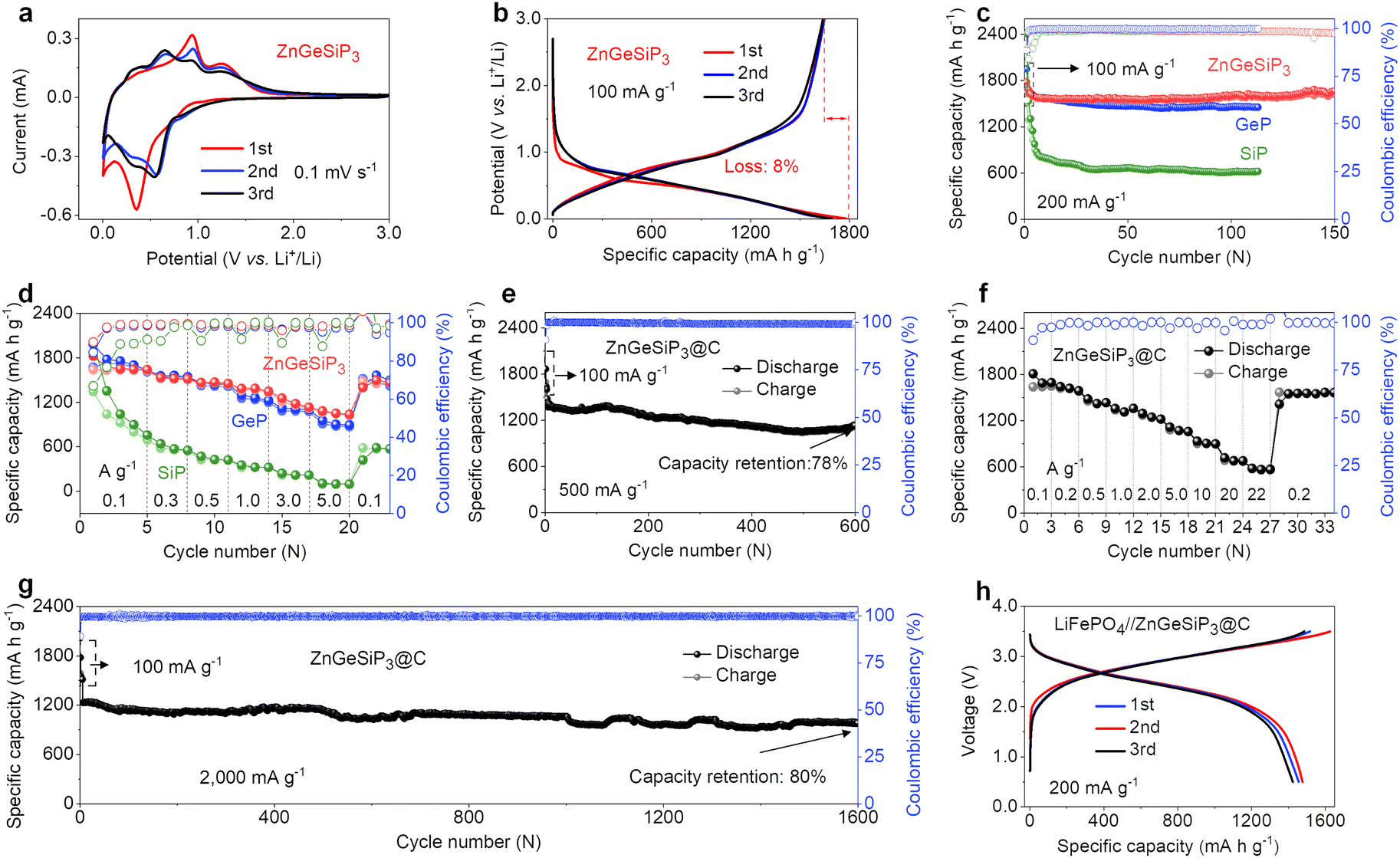 | ||
| Fig. 2 Electrochemical Li-storage properties of ZnGeSiP3, GeP, and SiP. (a) Cyclic voltammetry. (b) GCD profiles. (c) Cycling stability. (d) Rate performance. (e) Cycling stability at 500 mA g−1. (f) Rate performance. (g) Cycling performance at 2000 mA g−1. (h) GCD profiles of the LiFePO4//ZnGeSiP3@carbon full cell. | ||
The distinctive lithium storage characteristics of the ZnGeSiP3 compound synthesized in this study are particularly attractive given its all-lithium-reactive constituents and the cationic mixing lattice structure. To elucidate these unparalleled lithium storage properties, we conducted cyclic voltammetry (CV) and galvanostatic charge and discharge (GCD) analyses. Fig. 2(a) illustrates the initial three CV curves of the ZnGeSiP3 compound obtained at a scan rate of 0.1 mV s−1 within a potential range of 0.005–3.0 V. In the first negative scan, a gradual ramp commences at 1.07 V, indicative of solid electrolyte interface (SEI) formation (Fig. S13a, ESI†). By comparison with the following two, this SEI formation slope is very weak, thus contributing little capacity, which suggests that the as-formed SEI film is very thin and very reversible, leading to a very high ICE, agreeing well with its GCD profile (Fig. 2(b) and Fig. S13b, ESI†). Subsequently, a distinct reduction peak emerges at 0.36 V, attributed to substantial Li-ion absorption by the synthesized ZnGeSiP3 compound, leading to the formation of LixZnGeP3 (x < 3), along with subsequent formation of Li–P and Li–Ge (or Si, or Zn) alloys. Conversely, during the initial positive scan, three oxidation peaks manifest at 0.38, 0.93, and 1.24 V, associated with Li-ionic extraction from Li–Zn (or Ge, or Si) and Li–P alloys, as well as LixZnGeSiP3 (x < 3), respectively. Compared to the first cycle, subsequent cycles exhibit more pronounced peak profiles with slightly reduced polarization, owing to decreased crystallinity due to lattice confinement activation during the initial scanning process. From the second negative sweep onwards, reduction peaks at 0.57 V and 0.39 V indicate the formation of LixZnGeSiP3 (x < 3), Li3P, and other Li alloys of Li–Ge (or Si, or Zn). During the anodic scan, peaks at 0.40 V, 0.67 V, 0.95 V, and 1.24 V indicate Li-ion extraction from Li–Si (or Ge), Li–Zn, Li–P, and LixZnGeSiP3 (x < 3), respectively, based on the working potential of each constituent element. These electrochemical behaviors observed in the CV analysis of the ZnGeSiP3//Li half-cell will be validated through subsequent in situ XRD analysis. The ZnGeSiP3 compound delivers a discharge/charge capacity of 1781/1638 mA h g−1 at a current rate of 100 mA g−1, resulting in an initial coulombic efficiency (ICE) of 92%, comparable to that (>90%) of commercially available graphite electrodes.
Subsequently, we conducted an assessment of the cycling and rate capabilities of the synthesized triple-cation-disordered compound, ZnGeSiP3. As depicted in Fig. 2(c), even after 150 cycles, the ZnGeSiP3-based electrode maintained a capacity of 1619 mA h g−1 at a current density of 200 mA g−1, exhibiting no discernible capacity degradation in comparison to its initial capacity, indicative of its robust cycling stability. Additionally, post-cycling volume change experiments show that ZnGeSiP3 exhibits a volumetric expansion of about 116% (Fig. S14, ESI†), significantly lower than GeP (136%) and SiP (142%). This indicates that ZnGeSiP3 has a considerable advantage in electrode integrity. At elevated current rates, ZnGeSiP3 demonstrates notably rapid rate performance, particularly evident at high rates (Fig. 2(d)). Specifically, across various current rates ranging from 100, 300, 500, 1000, to 3000 mA g−1, the ZnGeSiP3-based electrode yielded capacities of 1634, 1513, 1461, 1372, and 1160 mA h g−1, respectively. Even with a progressive increase in the current density to 5000 mA g−1, the ZnGeSiP3 compound retained a capacity of 1047 mA h g−1. The electrochemical Li-storage characteristics of ZnGeSiP3 outlined above significantly surpass those of layered Ge (or Si) P in terms of high initial Coulombic efficiency, prolonged lifespan, and rapid rate performance (Fig. 2(c), (d) and S15, S16, ESI†).
To further enhance the cycling stability and rate performance, we integrated the pure ZnGeSiP3 compound with graphite, a material commonly used as LIB anodes in practical applications, through a straightforward secondary milling process. The resulting ZnGeSiP3–graphite composite delivered a reversible capacity of 1132 mA h g−1 after 600 cycles at a current density of 500 mA g−1, corresponding to a capacity retention of 78% (Fig. 2(e)). In comparison to the pure ZnGeSiP3 compound, the graphite-modified electrode exhibited enhanced rate performance, maintaining reversible capacities of 1617, 1421, 1245, 1072, 683, and 568 mA h g−1 at current densities of 200, 500, 2000, 5000, 20000, and 22000 mA g−1, respectively. Upon returning to the initial current rate of 200 mA g−1, the electrode restored its original reversible capacity, underscoring its high-rate reversibility (Fig. 2(f) and Fig. S17, ESI†). Following activation with three initial cycles at a low rate of 100 mA g−1, the electrode retained a reversible capacity of 981 mA h g−1 after 1600 cycles at a high rate of 2000 mA g−1, exceedingly twice the theoretical capacity (372 mA h g−1) of commercial graphite (Fig. 2(g)). Compared with recently reported rechargeable anode materials, the newly synthesized multiple mixed-cation phosphides exhibit significant advantages in comprehensive performance, including high ICE, robust cycling stability, and high-rate performance (Table S4, ESI†). To demonstrate practical applicability, we assembled a full battery utilizing the composite as the anode and LiFePO4 as the cathode. As illustrated in Fig. 2(h), the full cell exhibited smooth GCD profiles with an average operating potential of 3.0 V. After 75 cycles (Fig. S18, ESI†), the full cell exhibited negligible capacity degradation, indicating its potential for real-world deployment. Furthermore, when assembled with a high-voltage cathode of LiNi0.5Co0.2Mn0.3O2 (Fig. S19, ESI†), it operated at an average potential of 3.5 V and retained 90% capacity after 60 cycles, demonstrating high practicality.
We conducted operando X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy analyses, along with electrochemical measurements and DFT calculations to elucidate the lithium storage mechanisms inherent to the triple-cation mixed ZnGeSiP3 compound. In Fig. 3(a), we present the in situ XRD pattern evolution depicted through 2D contour plots over the initial two cycles. In the early stages, faint diffraction peaks from conductive carbon black are observed, much weaker than those of ZnGeSiP3 and its lithiation products. Despite the presence of Be13Ca, Be, and BeO in the in situ device, the dynamic transformation of ZnGeSiP3 and its lithiation products is clearly seen throughout two cycles, showing a dynamic evolution of strengthening, weakening, and disappearance. This evolution manifests in three discernible stages: (de)-intercalation, (de)-conversion, and (de)-alloy reactions. During the initial discharge process, the primary peaks at 28.7°, 33.0°, and 47.8° corresponding to ZnGeSiP3 experienced a shift towards lower 2theta angles, concurrently diminishing in intensity (Fig. 3(b)), indicative of a progressive intercalation reaction. Simulations revealed three lithium storage sites at the tetrahedral center within one formula of ZnGeSiP3 (Fig. S20, ESI†), suggesting that the initial intercalation reaction could yield a capacity of 311 mA h g−1 by forming LixZnGeSiP3 (x < 3). Subsequent lithiation reactions rendered the electrode amorphous, devoid of observable XRD peaks. The thermodynamic favorability of these lithium intercalation intermediates (LixZnGeSiP3, x < 3) was confirmed by their negative binding energies (Fig. S21a, ESI†). Notably, the increasingly negative binding energies per lithium ion with higher intercalation levels suggest enhanced affinity for lithium ions in deeply discharged intermediates, thereby facilitating lithium storage kinetics. Accompanying the intercalation of lithium ions, significant crystallographic parameter changes, particularly in volume expansion (Fig. S21b–d, ESI†), led to lattice amorphization. Database references suggest the possible presence of ternary phases such as Li–Zn (or Ge, or Si)–P and Li–Zn–Ge (or Si), among others, within the amorphous electrode. Upon further discharge to 0.005 V, distinctive signals characteristic of binary alloys emerged: LiZn at 24.8° and 41.1° (Fig. 3(c) and Fig. S22b, ESI†), Li7Ge2 at 22.4° and 23.5° (Fig. 3(d) and Fig. S22c, ESI†), Li7Si3 at 23.7° and 40.8° (Fig. 3(e) and Fig. S22d, ESI†), and Li3P at 26.8°, 42.4°, and 43.4° (Fig. 3(f) and Fig. S22e, ESI†), significantly augmenting the lithium storage capacity. Using new Be plates and reducing the carbon content, two additional in situ XRD results confirmed the above conclusions (Fig. S23 and S24, ESI†). Notably, ex situ XPS analyses provided additional evidence of the formation of the aforementioned binary Li-containing alloys, evidenced by their lowered binding energies relative to pristine ZnGeSiP3 powder (Fig. 3(g2)–(j2)).
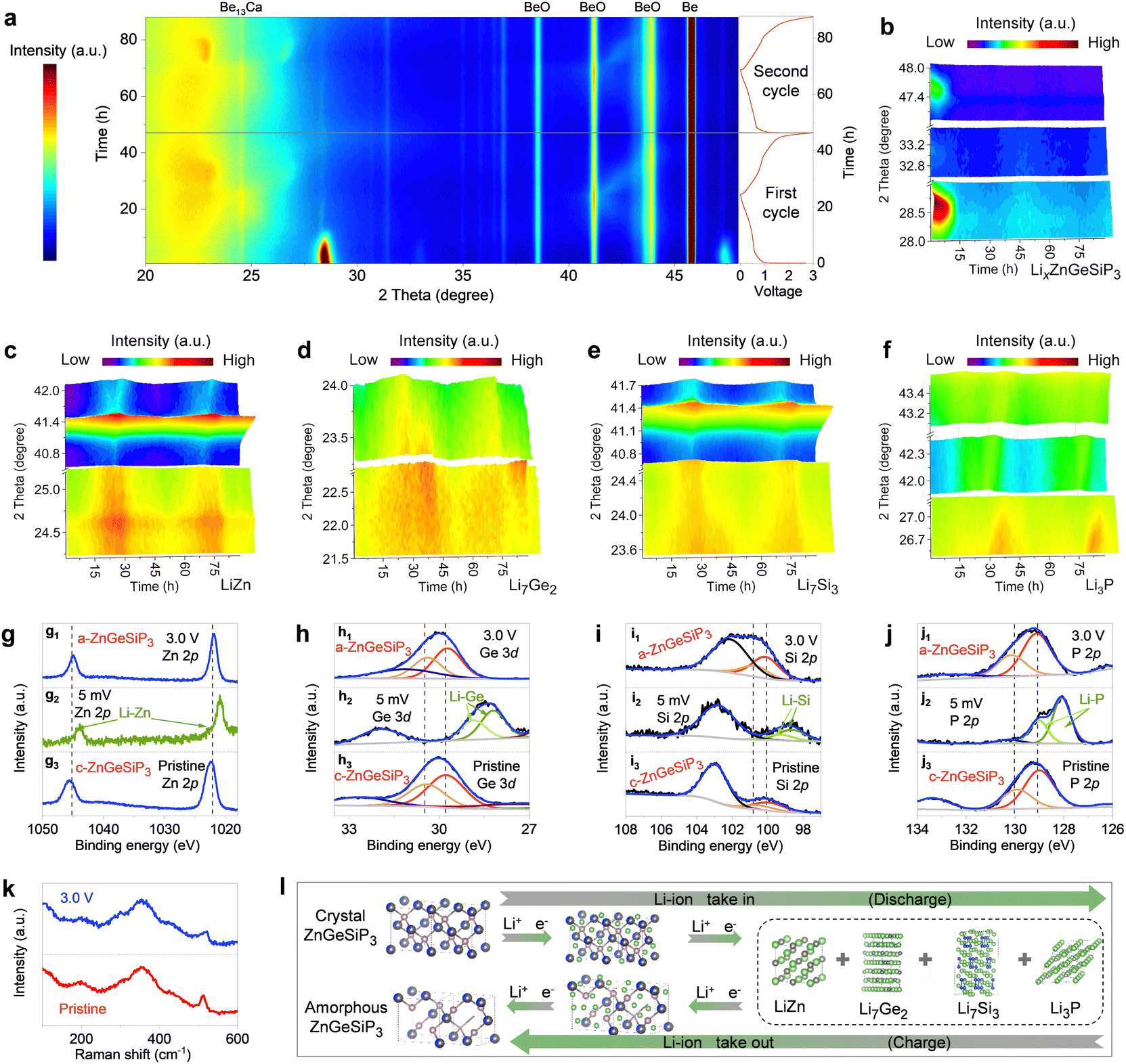 | ||
| Fig. 3 Advanced Li-storage mechanism and characterization of ZnGeSiP3. (a) Operando XRD with 2D contour plots during the initial two cycles. (b)–(f) XRD pattern evolution of the ZnGeSiP3, LiZn, Li7Ge2, Li7Si3, and Li3P. (g)–(j) Ex situ XPS. (k) Ex situ Raman. (l) Schematic diagram of the Li-storage mechanism. | ||
In reverse, throughout the charging phase, the discernible fingerprint XRD peaks associated with Li-containing binary alloys such as Li–Zn, Li–Ge, Li–Si, and Li–P undergo gradual attenuation, indicative of the liberation of Li-ions. Notably, this process closely parallels the discharge phase, as evidenced by the recurrent periodic patterns. Upon reaching full charge, the amorphous state of ZnGeSiP3 is regenerated, corroborated by various operando XRD (Fig. 3(a)–(f)), XPS (Fig. 3(g)–(j)), and Raman (Fig. 3(k)) analyses, which manifest the reappearance of distinctive fingerprint signals compared to the pristine state. Specifically, in comparison with the initial cycle, subsequent in situ XRD observations reveal analogous periodic patterns, thereby substantiating the highly reversible nature of Li-storage processes. Additionally, HRTEM and SAED analyses of the ZnGeSiP3 compound electrodes after the 1st and 10th cycles (Fig. S25, ESI†) revealed distinct sphalerite-like structures, though with a slightly reduced intensity due to decreased crystallinity from cycling. Raman vibration modes of the cycled ZnGeSiP3 remained consistent with pristine powder (Fig. 3(k)), suggesting minimal changes in the coordination environment. Comparatively, the binding energies of lithium-reactive elements associated with the ZnGeSiP3-based electrode remained nearly unchanged upon charging to 3.0 V, except for slight shifts (Fig. 3(g1)–(j1)), attributed to lattice amorphization. The amorphization of the ZnGeSiP3-based electrode post-initial cycling is also reflected in the much smoother discharge profiles of the subsequent cycles compared with the initial one (Fig. 2(b)) and contributes to subsequent improvements in reaction kinetics.
Notably, the subsequent cyclic voltammetry curves, as well as galvanostatic charge and discharge profiles, exhibited analogous lithium storage behaviours to the initial ones (Fig. S13, ESI†), albeit with marginally reduced polarization, likely attributed to the inherently softened crystal lattice, as determined by Young's modulus calculations (Fig. S26, ESI†), and the substantial lattice distortion endowed by high conformational entropy. The nearly identical nature of the initial two discharge curves fully underscores the high reversibility, and superior lithium storage reaction kinetics of ZnGeSiP3 compared to oxygen group compounds previously reported.29,40,49 Moreover, the observed capacity aligned with theoretical predictions, further corroborating proposed reaction mechanisms. Drawing upon the aforementioned characterizations, delineated in Fig. 3(l), the mechanisms governing Li-storage in the ZnGeSiP3 compound can be succinctly summarized as follows:
During discharge:
| ZnGeSiP3 + xLi+ + xe− → LixZnGeSiP3 (x < 3) (intercalation) | (i) |
| LixZnGeSiP3 + (15.833 − x) Li+ + (15.833 − x) e− → LiZn + Li7Ge2 + Li7Si3 + 3Li3P (conversion and alloy reaction) | (ii) |
During charge:
| LiZn + Li7Ge2 + Li7Si3 + 3Li3P − (15.833 − x) Li+ − (15.833 − x) e− → LixZnGeSiP3 (x < 3) (de-alloy and de-conversion reactions) | (iii) |
| LixZnGeSiP3 − xLi+ − xe− → ZnGeSiP3 (amorphous) (x < 3) (de-intercalation) | (iv) |
Thus, the overarching reactions can be expressed as:
| ZnGeSiP3 + (15.833 − x)Li+ + (15.833 − x)e− ↔ LiZn + Li7Ge2 + Li7Si3 + 3Li3P. | (v) |
The above reaction equations illustrate that one mole of the ZnGeSiP3 compound can store 15.833 moles of Li-ions, supporting a reversible capacity of 1638 mA h g−1. The contributions from each Li-reactive constituent of P, Si, Ge, and Zn for the ZnGeSiP3 compound are specified, indicating the specific capacities of 931, 241, 362, and 104 mA h g−1, respectively. As known, the cost of raw materials of P, Si and Zn are much more cost-effective than Ge, thus significantly reducing the battery industry cost compared with the single-component Ge anodes. As for volume change, as compared in Table S5 (ESI†), the ZnGeSiP3 electrode experienced a relatively small volume expansion of 256%, compared with the single-component anodes of P (∼300%), Si (∼392%) or Ge (∼350%) at the expense of sacrificing some capacity, thus contributing to robust cycling stability.50 Moreover, the Li-ionic conductive phase of Li3P and Li7Ge2, and the electronic conductive phase of LiZn and Li7Ge2 are simultaneously produced, well-distributed, and form interfaces within the electrode during cycling, ensuring fast reaction kinetics during the deeply discharged phase. Moreover, the Li3P phase favours the construction of a better solid electrolyte interphase (SEI), promoting cycling stability as reported by Sun's group.51,52
In order to elucidate the superior electrochemical lithium storage characteristics observed in the ZnGeSiP3 compound synthesized herein compared to layered Ge (or Si)P, an investigation was conducted into the lithium ionic diffusion pathways and associated hopping energy barriers utilizing density functional theory (DFT) calculations complemented by experimental validations. Leveraging the cation-mixing attribute of the ZnGeSiP3 compound, we invoked the theory of quasi-disordered structures53 and constructed a 144-atom supercell model for ZnGeSiP3 (Fig. 4(a)). The average energy barrier for lithium ionic diffusion in ZnGeSiP3 was determined to be merely 0.215 eV, markedly lower than that of layered GeP (0.335 eV) and SiP (0.44 eV) (Fig. 4(b) and Fig. S27–S29, ESI†). This enhanced lithium ionic transport property of ZnGeSiP3 is ascribed to its lattice mixing feature, which aligns well with percolation theory.54 Experimentally, galvanostatic intermittent titration technique (GITT, Fig. 4(c), (d) and Fig. S30, ESI†) measurements revealed lithium ionic diffusion coefficients for ZnGeSiP3 ranging from ∼10−11.57 to 10−9.1 cm2 s−1 across the potential window of 3–0.005 V, significantly outpacing those of layered GeP (∼10−12.45 to 10−9.4 cm2 s−1) and SiP (∼10−13.6 to 10−10.36 cm2 s−1), particularly within the voltage range contributing to capacity.
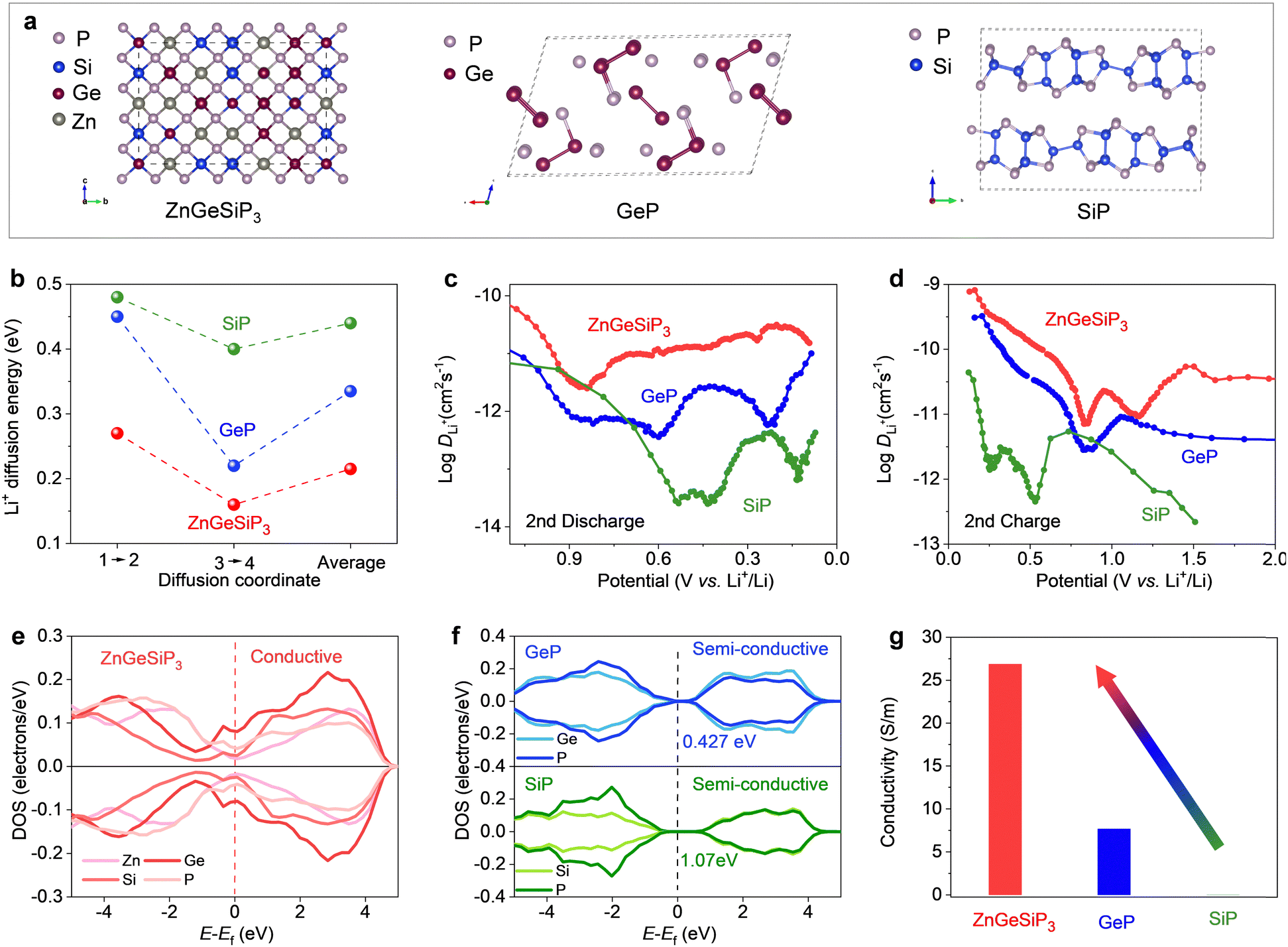 | ||
| Fig. 4 Charge transport properties of ZnGeSiP3, GeP, and SiP. (a) Supercells used for first-principles calculations. (b) Li-ionic hopping energy barriers. (c) and (d) Li-ionic diffusion coefficients derived from GITT measurements. (e) and (f) Projected density of states (PDOS) profiles. (g) Electronic conductivity. | ||
Furthermore, the electronic conductivity of the synthesized sphalerite-structured germanium phosphide-based compound ZnGeSiP3, featuring triple cation mixing, was evaluated and compared to that of layered Ge (or Si)P using a combined approach of DFT calculations and experimental assessments. As illustrated in Fig. 4(e), the total density of states (TDOS) profile intersected the Fermi level, indicating a nonzero value at the Fermi level, indicative of metallic conductivity, a stark contrast to the semiconductor characteristic exhibited by layered Ge (or Si)P (Fig. 4(f)). Analysis of the projected density of states (PDOS) profiles (Fig. S31 and S32, ESI†) revealed nonzero PDOS values for each constituent element—P, Ge, Si, and Zn—at the Fermi level, thereby contributing free electrons at the Fermi level, with phosphorus contributing most significantly due to its tetrahedral coordination geometry, wherein each phosphorus atom is surrounded by four cations and can thus provide a free electron. Experimentally, the electronic conductivity of ZnGeSiP3 was measured to be up to 26.9 S m−1, markedly surpassing that of layered GeP (7.7 S m−1) and SiP (0.001 S m−1) (Fig. 4(g)). Despite the integration of more cost-effective raw materials such as Si and amorphous red P into the cation-mixed zinc blende lattice, the resultant ZnGeSiP3 demonstrated a remarkable breakthrough in electronic conductivity, effectively activating the lithium storage capabilities of the incorporated Si and red P, which exhibit inherently poor electronic conductivity. In summary, a collaborative investigation utilizing first-principles calculations and experimental validations confirms that the triple-cation mixed ZnGeSiP3 compound exhibits higher lithium ionic diffusion coefficients and faster electronic conductivity compared to layered Ge (or Si)P, thus firmly substantiating the superior lithium storage characteristics of the mixed-cation ZnGeSiP3 over layered Ge (or Si)P.
We further design, synthesize and characterize quaternary and high conformational entropy (pentanary and hexanary) cation-disordered germanium phosphide-based compounds on the basis of the sphalerite lattice by introducing additional metals into the cationic site of the triple mixed-cation ZnGeSiP3 compound case and elucidate their electrochemical Li-storage performances. Notably, after the silicon replaces one germanium atom in the binary cation-mixed ZnGe2P3 compound with a sphalerite lattice (Fig. S33 and Tables S6, S7, ESI†), the formed isomorphic triple cation-mixed ZnGeSiP3 compound presented substantially better electrochemical Li-storage properties although the single-component silicon-based anodes generally exhibit significantly inferior electronic conductivity and slower Li-ionic diffusion compared to the Ge counterparts (Fig. S34–S37, ESI†). This improvement can be attributed to the elevated configurational entropy. Motivated by the substantial benefits conferred by high conformational entropy at the cationic sites within the sphalerite lattice, we further incorporated additional metals such as Cu, Sn, and/or Al (Fig. 5(a) and Fig. S38, ESI†), synthesizing high conformational entropy (exceeding 1.5 R) germanium phosphide-based compounds, including CuSnAlZnGeSiP6, CuSn (or Al)ZnGeSiP5, and SnAlZnGeSiP5, as well as quaternary mixed-cation germanium phosphide-based compounds Cu (or Sn, or Al)ZnGeSiP4 as characterized by XRD (Fig. 5(b)–(d)), SAED (inset of Fig. 5(b)–(d)), and elemental mapping (Fig. 5(e)–(g) and Fig. S39–S45, ESI†). The inclusion of metallic elements such as Cu, Sn, and/or Al stems from their well-established industrial infrastructure and wide-ranging applications in daily life. Specifically, metallic Cu enhances both electronic and thermal conductivity, with the latter dissipating heat to prevent battery overheating during operation. Sn exhibits superior electrochemical Li-storage properties, particularly at low temperatures, and contributes to metallic conductivity when tetrahedrally coordinated with phosphorus due to the delocalized electrons provided by phosphorus. Al, on the other hand, is chosen for its low molecular weight and high Li-storage capacity.
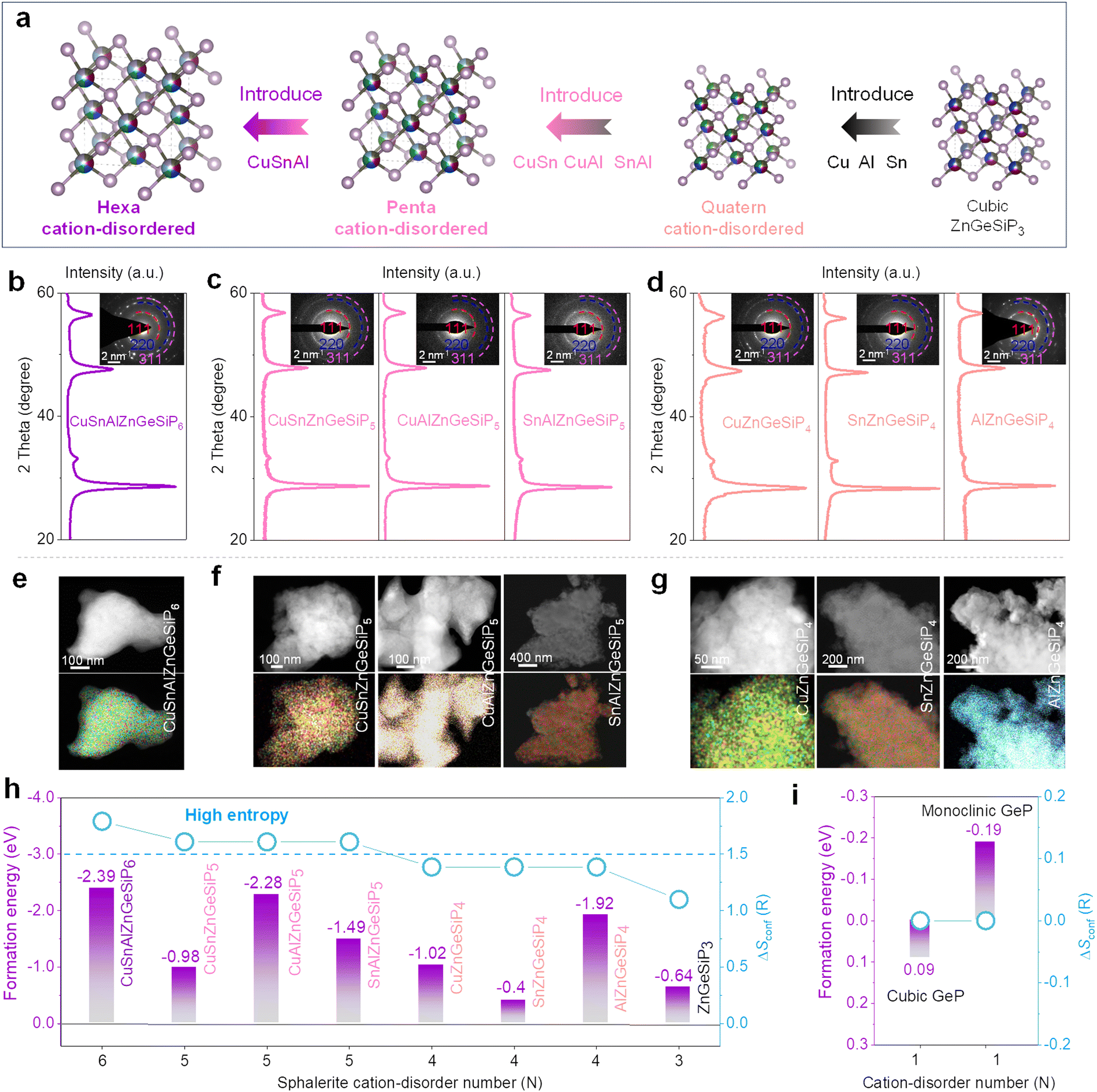 | ||
| Fig. 5 Characterization and calculations of multiple disordered-cation GeP-based compounds of CuSnAlZnGeSiP6, CuSn(or Al)ZnGeSiP5, SnAlZnGeSiP5, and Cu(or Sn, or Al)ZnGeSiP4. (a) Schematic of cationic regulation. (b)–(d) XRD and SAED images. (e)–(g) EDS mappings. (h) and (i) Formation energies along with the configurational entropy. | ||
Subsequently, the mechanisms underlying phase formation were examined through DFT calculations concerning phase formation energies. It is posited that the entropy-mediated stabilization process governs the phase formation mechanism of these diverse germanium phosphide-based compounds, wherein an increasing number of elements occupying the same site in a disordered fashion enhances the configurational entropy. In accordance with the Gibbs free energy equation, ΔG = ΔH − TΔS, the augmentation of conformational entropy results in a reduction of Gibbs free energy, thus facilitating the thermodynamic stabilization of the resultant compounds.55–59 Systems with high entropy can augment the probability of discovering more efficient pathways characterized by lower activation energies, thereby enhancing reaction kinetics. Notably, the phase formation energies of all aforementioned mixed-cation GeP-based compounds with a sphalerite lattice are negative (Fig. 5(h) and (i)), owing to the heightened configurational entropy, thus robustly affirming their stability under ambient conditions.
The electronic structures of the synthesized quaternary and high-conformational-entropy cation-mixed GeP-based compounds with a sphalerite lattice were thoroughly investigated via DFT calculations. As illustrated in Fig. 6, sphalerite-structured mixed-cation GeP-based compounds with high configurational entropies exhibit metallic conductivities, consistent with the calculated non-zero total density of states (TDOS) profiles at their Fermi levels (Fig. 6(a)–(c)). Their projected density of states (PDOS) profiles, depicted in Fig. S46 (ESI†), indicate active participation of all constituent elements in conductivity. The rapid electronic conductivity contributes to achieving superior lithium-storage properties by overcoming the inherent poor electronic conductivity characteristic of high-capacity LIB anode materials such as amorphous red phosphorus, silicon, or germanium-based anodes.
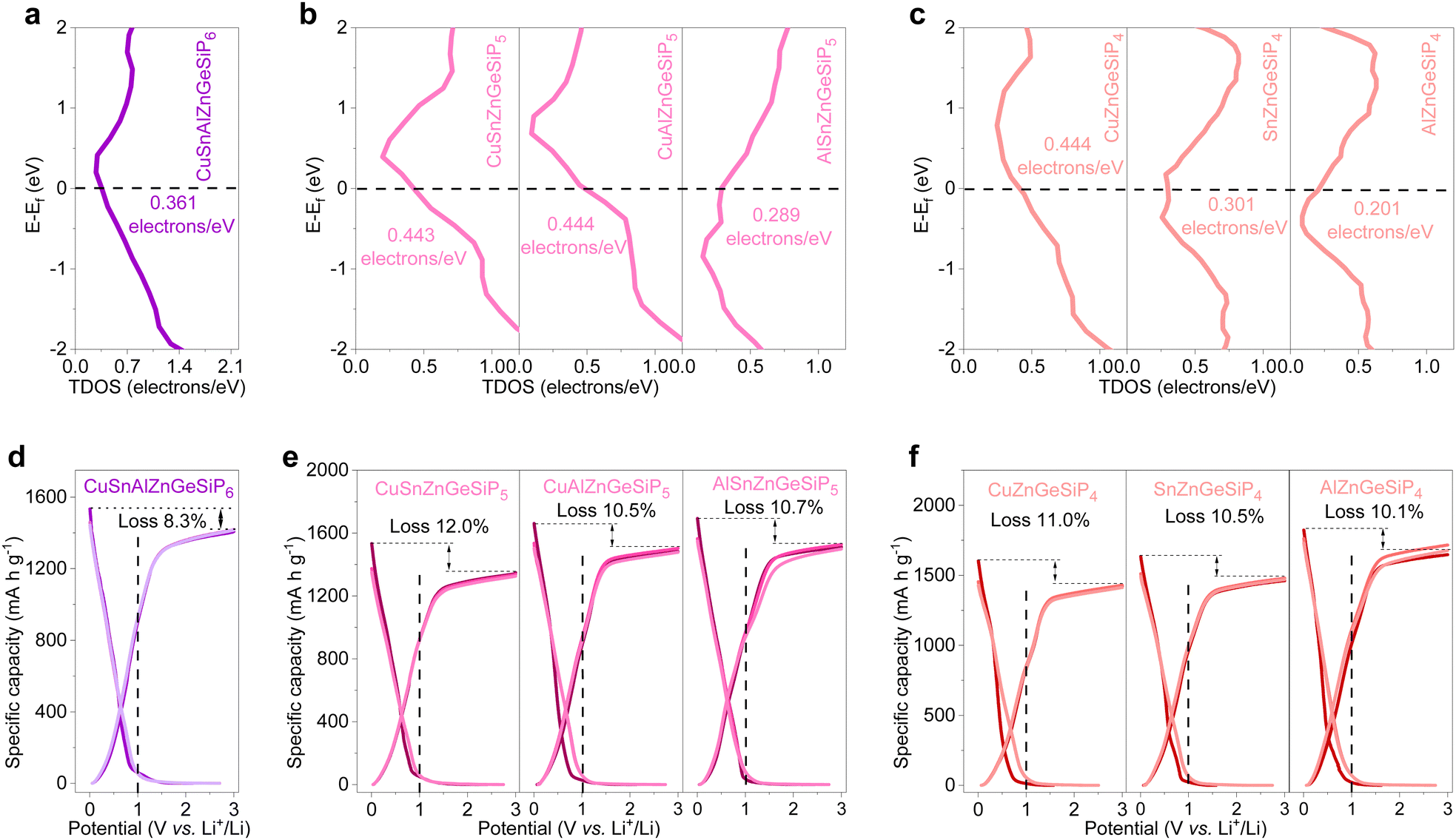 | ||
| Fig. 6 TDOS calculations and GCD profiles of multiple mixed-cation GeP-based compounds of CuSnAlZnGeSiP6, CuSn(or Al)ZnGeSiP5, SnAlZnGeSiP5, and Cu(or Sn, or Al)ZnGeSiP4. (a)–(c) Total density of states (TDOS) profiles. (d)–(f) Initial three charge and discharge profiles. | ||
As anodes for LIBs, the aforementioned multiple, and high conformational entropy germanium phosphide-based compounds demonstrate high reversible capacities ranging from 1349 mA h g−1 for CuSnZnGeSiP5 to 1677 mA h g−1 for AlZnGeSiP4, with a high initial Coulombic efficiency approaching 90% (Fig. 6(d)–(f)). Based on the reversible capacity, all the Li-reactive components actively engage in the Li-storage process and contribute to the high capacity by forming binary lithium alloys. Furthermore, the discharge and charge profiles of these multiple and high conformational entropy germanium phosphide-based compounds, containing various Li-reactive components with substantially different Li-storage working potentials, exhibit relative smoothness without distinct multiple redox reactions (Fig. S47, ESI†). This can be attributed to the modified electrochemical reaction process facilitated by the positive synergy effect resulting from the integration of multiple elements competitively occupying the same site. The constituents of the multiple and high conformational entropy germanium phosphide-based compounds are evenly distributed, and the electrochemical intermediates are abundant and uniformly dispersed, forming rich heterointerfaces between Li-ionic and electronic conductors. This substantially promotes charge and mass transport, as well as reversibility. The smooth discharge and charge profiles facilitate control over discharge depth when applied in a full cell.
Taking into consideration the isomorphic nature of cations and anions, alongside the aforementioned cationic flexibility within a sphalerite lattice, our inquiry broadens its scope to encompass a novel class of high conformational entropy germanium phosphide-based compounds, characterized by mixed cations and anions. This exploration builds upon the framework of the triple disordered-cation ZnGeSiP3 archetype. In order to maximize configurational entropy, we introduce nonmetal elements such as S and/or Se to substitute the phosphorus atom (s) at the anionic site (s), thereby synthesizing compounds ZnGeSiPSSe and ZnGeSiP2Se (or S), which exhibit high degrees of conformational entropy for both cations and anions (Fig. 7(a) and Fig. S48, ESI†). This synthesis is confirmed through various characterization studies, including XRD patterns (Fig. 7(b)), SAED images (inset of Fig. 7(b)), and EDS mappings (Fig. 7(c) and Fig. S49–S51, ESI†). The selection of nonmetal elements S and/or Se is based on their widespread presence in a sphalerite lattice like cubic ZnS (or Se), as well as their ability to enhance electrochemical Li-storage capacity corresponding to the extended working potential window which aids in mitigating electrode structural degradation and enhancing electrochemical intermediate abundance, as well as consequently elevate the heterointerface density, thereby improving the reaction kinetics.
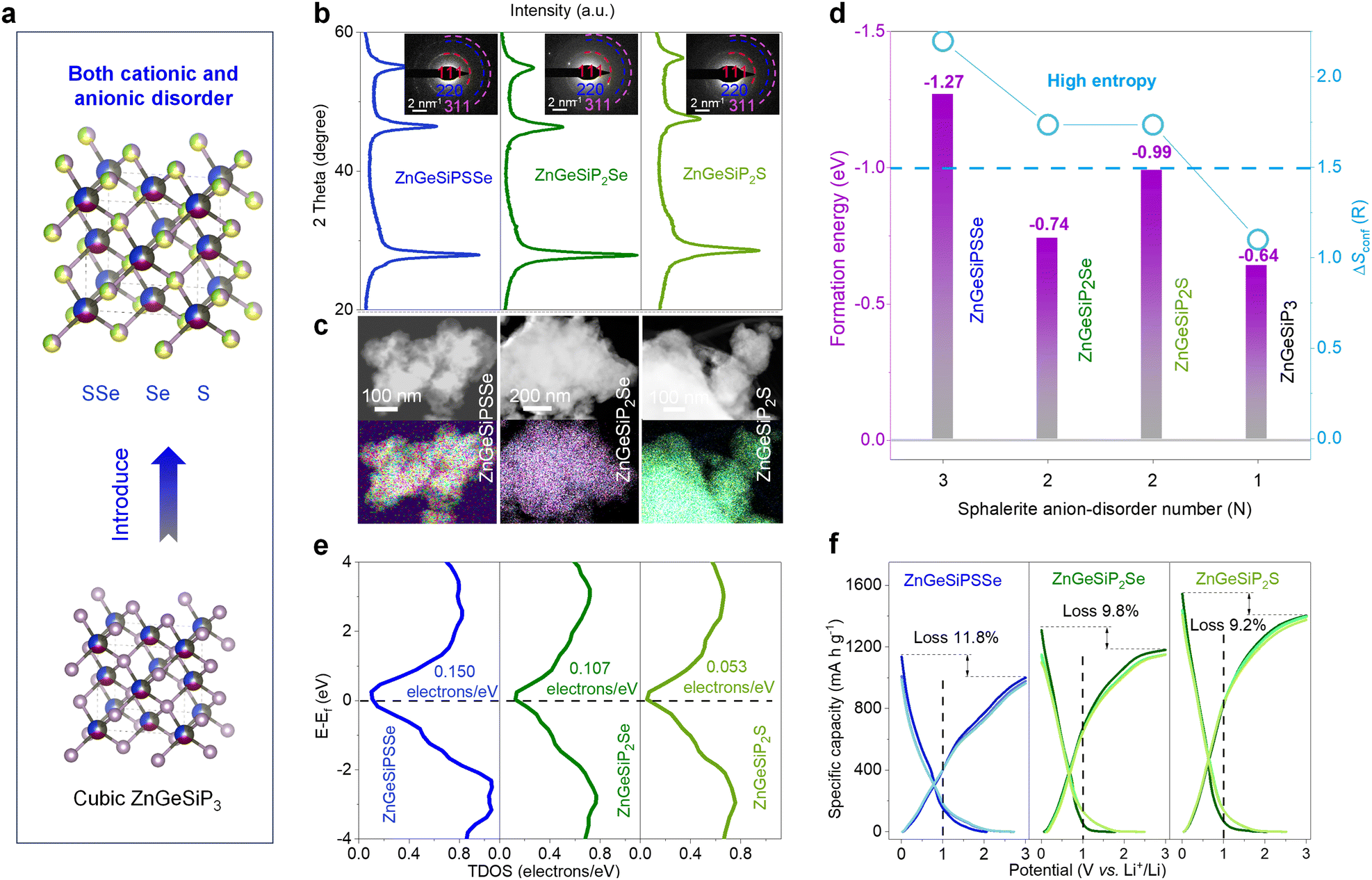 | ||
| Fig. 7 Characterization, calculations and Li-storage performances of high entropy both disordered-cation and -anion compounds ZnGeSiPSSe and ZnGeSiP2Se(or S). (a) Scheme of anionic regulation. (b) XRD and SAED patterns. (c) EDS mappings. (d) Formation energy coupled with the configurational entropy. (e) TDOS profiles. (f) First three charge and discharge profiles. | ||
To elucidate the phase formation mechanisms of these high conformational entropy germanium phosphide-based compounds with mixed cations and anions, we employ first-principles calculations to determine their phase formation energies. Achieving simultaneous engineering of high conformational entropy at both cationic and anionic sites poses greater challenges compared to single-site compounds or metallic alloys, as it necessitates consideration of much more complicated valence balance alongside the atomic radius, electronegativity, and bonding characteristics.60 Despite the inherent challenge posed by the valence balance, typically encountered in cubic phase compounds of MX (M = Ge, or Si; X = S, or, Se), or Zn (or Ge, or Si) P with a sphalerite lattice, the enhanced configurational entropy plays a pivotal role in facilitating phase formation, thus overcoming this obstacle. As illustrated in Fig. 7(d), the negative phase formation energies of ZnGeSiPSSe and ZnGeSiP2Se (or S) indicate their thermodynamic stability under ambient temperature and pressure conditions, a result attributed to their elevated configurational entropy.
The electronic conductivity characteristics of the ZnGeSiPSSe and ZnGeSiP2Se (or S) compounds, which exhibit high conformational entropy and disordered cationic and anionic compositions, are analyzed via density functional theory (DFT) calculations. The total density of states (TDOS) profiles are depicted in Fig. 7(e). The computed electronic structures reveal metallic conductivity in all three aforementioned germanium phosphide-based compounds due to non-zero TDOS profiles at their Fermi levels, attributable to contributions from all constituent elements, as delineated in their respective partial density of states (PDOS) profiles (Fig. S52, ESI†).
Given their metallic conductivity and the presence of all-lithium-reactive components, we further assess the electrochemical lithium-storage properties of these germanium phosphide-based compounds, characterized by high conformational entropy and disordered cationic and anionic compositions, as anodes for LIBs. As illustrated in Fig. 7(f), these high-entropy germanium phosphide-based compounds exhibit superior electrochemical lithium-storage performance, including large capacity, high initial Coulombic efficiency, and broad operating potential, ensuring both high energy density and safety. The synthesized ZnGeSiPSSe and ZnGeSiP2S (or Se) compounds demonstrate discharge/charge capacities of 1135.2/1001.0 and 1544.7/1402.9 (1308.7/1180.4) mA h g−1, respectively, resulting in initial Coulombic efficiencies of 88.2 and 90.8 (90.2)%. Based on their reversible capacity and operating potential characteristics, we infer that both the cationic elements (Zn, Ge, and Si) and anionic constituents (P, S, and/or Se) contribute to their substantial reversible capacity by forming binary Li-containing alloys. In comparison with pristine ZnGeSiP3, the synthesized high conformational entropy germanium phosphide-based compounds ZnGeSiPSSe and ZnGeSiP2S (or Se) exhibit an extended operating potential window, mitigating lithium dendrite growth and ensuring high safety, particularly at elevated rates (Fig. S53, ESI†). The superior electrochemical lithium-storage properties of these high-conformational entropy germanium phosphide-based compounds, featuring concurrent cationic and anionic disorder, can be attributed to their predicted metallic conductivity via first-principles calculations and their rapid Li-ionic diffusion capability.
Element screening in high entropy anodes for LIBs involves navigating a complex phase space with multiple elemental components and varying compositions. Critical in materials science and electrochemistry, selecting elements hinges on assessing stability in structures like the zinc-blende phase, considering atomic properties and bonding characteristics. Theoretical calculations and experimental validation, such as ball milling, are pivotal for advancing these materials. Factors like lithium affinity, conductivity, capacity, abundance, environmental impact, and cost inform element selection. Achieving structural stability often requires high symmetry configurations, though stabilizing high-entropy compositions in low-symmetry structures remains challenging. Evaluating the electrochemical performance encompasses safety, efficiency, and environmental impact considerations, leveraging advanced computational tools like first-principles calculations and machine learning to optimize material performance effectively.
Conclusions
In summary, via a mechanochemical approach, we synthesized, for the first time, a cubic GeP-based compound, ZnGeSiP3, which exhibits a triple mixed-cation sphalerite lattice configuration, with P occupying the anionic site entirely, while Zn, Ge, and Si competitively occupy the cationic sites, as confirmed by XRD structural refinement analysis. Experimental measurements and first-principles calculations corroborate that the ZnGeSiP3 compound displays enhanced reaction kinetics, including accelerated electronic conductivity (metallic conductivity) and expedited lithium-ion diffusion capabilities compared to its parent phases of the layered Ge (or Si)P, as well as isomorphic ZnGe2P3 and Ge, owing to the augmented configurational entropy. Operando characterization studies employing XRD, XPS, and Raman spectroscopy, coupled with electrochemical analyses and theoretical simulations, elucidate the lithium-storage mechanisms of ZnGeSiP3 involving initial intercalation followed by subsequent conversion reactions. The presence of well-dispersed electrochemical intermediates acting as electronic conductors and lithium-ion conductors, alongside enriched heterointerfaces, ensures stable and rapid charge transport throughout the cycling process. The introduction of cost-effective constituent elements, coupled with lattice disordering, facilitates the achievement of a ZnGeSiP3 compound with a capacity of 1638 mA h g−1 and an ICE of 92% at 100 mA g−1. Furthermore, upon modification with graphite, the composite exhibits a capacity of 981 mA h g−1 after 1600 cycles at 2000 mA g−1, and 568 mA h g−1 at a rate of 22000 mA g−1, surpassing those of most previously reported Ge-based anodes while significantly reducing costs. Moreover, by introducing additional elements, high entropy sphalerite-structured GeP-based compounds such as CuSnAlZnGeSiP6, CuSn (or Al)ZnGeSiP5, SnAlZnGeSiP5, ZnGeSiPSSe, and ZnGeSiP2Se (or S) are synthesized due to their negative phase formation energies, attributed to enhanced conformational entropy. These compounds demonstrate superior lithium-storage properties, characterized by high capacity, high ICE, and suitable working potential, facilitated by their metallic conductive conductivity, thereby holding promise for future applications. The high conformational entropy strategy, which involves synergistic interactions among multiple elements to improve performances and mitigate reliance on specific elements to reduce cost, provides a roadmap for the design of next-generation energy-storage devices with enhanced performance.
Experimental section
Materials preparation
Utilizing a straightforward high-energy ball milling (HEBM) approach (Pulverisette 6, FRITSCH), the single-phase sphalerite-structured germanium monophosphide-based compounds, including the triple cation-mixed compound of ZnGeSiP3, quaternary cation-disordered compounds of Cu (or Al, or Sn) ZnGeSiP4, high conformational entropy (pentanary or hexanary) disordered-cation compounds of CuSn (or Al)ZnGeSiP5, SnAlZnGeSiP5, and CuAlSnZnGeSiP6; as well as high conformational entropy both cation- and anion-disordered compounds of ZnGeSiP2S (or Se), and ZnGeSiPSSe were successfully synthesized applying the related mixed elemental powders of Zn, Cu Sn, Al, Ge, Si, P, S, and Se with the right molar ratio as raw materials. To avoid potential oxidation, certain metals underwent additional purification through hydrogen reduction, and purchased red phosphorus underwent further refinement. Following the given stoichiometric ratio, the raw materials were placed in an argon-filled chamber and sealed. The weight ratio of raw materials to steel balls was 1:28, with the ball milling process lasting approximately 8 h at a rotation speed of about 550 rpm. To enhance the ball milling efficiency, a regular program involving alternating forward and reverse rotations was implemented. Furthermore, the graphite-modified ZnGeSiP3 composite was obtained through a second high-energy ball milling process, with a milling time of about 4 h. The weight ratio of ZnGeSiP3 to graphite is 7:2.
Materials characterizations
The crystallographic details of the aforementioned single-phase sphalerite-structural germanium phosphide-based compounds were verified through X-ray diffraction (XRD) utilizing a PANalytical Empyrean instrument with Cu Kα radiation, employing a scanning speed of approximately 1–2° min−1. Field emission scanning electron microscopy (FESEM) with a Hitachi SU8220 instrument was employed to scrutinize the morphology. High-resolution transmission electron microscopy (HRTEM) using a JEM-2100F instrument was employed to elucidate the microstructure. Raman spectra for the aforementioned samples were acquired using a Raman spectrometer (SENTERRA Raman) with a laser excitation wavelength of 532 nm. The surface chemical composition of the samples was investigated through X-ray photoelectron spectroscopy (XPS) using a Thermo Fisher Escalab 250Xi electron spectrometer. Thermogravimetric analysis was conducted in air over the temperature range of room temperature to 1200 °C (TGA/DSC3+, Switzerland).Electrochemical characterizations
For pure phase compound electrodes, the working electrodes were coated with a slurry comprising 70 wt% active materials, 20 wt% carbon black, and 10 wt% Li-PAA on the copper current collector while for carbon composite electrodes, the slurry consisted of a 90 wt% carbon composite and a 10 wt% LiPAA binder without any extra conductive agents. Subsequently, the coated electrodes were dried at 80 °C for over 12 h under vacuum conditions. Li-metal foils were employed as the counter and reference electrodes, and the electrolyte consisted of LiPF6 dissolved in a mixture of EC/DMC/EMC (1:1:1 by volume) with a 2% vinylene carbonate (VC) additive. Polypropylene (PP) (Celgard 2325) was used as the separator. CR2032-type coin cells were assembled within an Ar-filled glovebox with a remaining concentration of H2O and O2 below 0.01 ppm. The loading of materials on the copper foil was approximately 1.2–1.5 mg cm−2. In situ XRD cells with 3–5 mg cm−2 active materials (LIB-LHTXRD-LN, Beijing Scistar Technology Co., Ltd) were assembled using a Be window and fiberglass separator. Electrochemical Li-storage properties were measured using a LAND battery testing system (Wuhan Kingnuo Electronic Co., China) and an electrochemical workstation (Autolab, Pgstat 302N).
Data availability
The data supporting this article have been included in this article or as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22178068). Dr X. Li acknowledges support from the Brain Pool program funded by the Ministry of Science and ICT through the National Research Foundation of Korea (grant no. RS-2023-00304337). We express our thanks to Prof. Hui-Ming Cheng at the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences; Prof. Ho Seok Park at Sungkyunkwan University, for their in-depth comments on the high conformational entropy germanium-phosphide compounds. ML acknowledges the support from the Hightower Endowed Chair and the Georgia Tech Foundation.Notes and references
- S. Tu, B. Zhang, Y. Zhang, Z. Chen, X. Wang, R. Zhan, Y. Ou, W. Wang, X. Liu, X. Duan, L. Wang and Y. Sun, Nat. Energy, 2023, 8, 1365–1374 CrossRef CAS.
- Y. Son, N. Kim, T. Lee, Y. Lee, J. Ma, S. Chae, J. Sung, H. Cha, Y. Yoo and J. Cho, Adv. Mater., 2020, 32, 2003286 CrossRef PubMed.
- J. Zhang, Y. Yan, X. Wang, Y. Cui, Z. Zhang, S. Wang, Z. Xie, P. Yan and W. Chen, Nat. Commun., 2023, 14, 3701 CrossRef CAS PubMed.
- S. Zhang, F. Sun, X. Du, X. Zhang, L. Huang, J. Ma, S. Dong, A. Hilger, I. Manke, L. Li, B. Xie, J. Li, Z. Hu, A. C. Komarek, H.-J. Lin, C.-Y. Kuo, C.-T. Chen, P. Han, G. Xu, Z. Cui and G. Cui, Energy Environ. Sci., 2023, 16, 2591–2602 RSC.
- Y. Huang, C. Wang, H. Lv, Y. Xie, S. Zhou, Y. Ye, E. Zhou, T. Zhu, H. Xie, W. Jiang, X. Wu, X. Kong, H. Jin and H. Ji, Adv. Mater., 2023, 36, 2308675 CrossRef PubMed.
- H. He, J. He, H. Yu, L. Zeng, D. Luo and C. Zhang, Adv. Energy Mater., 2023, 13, 2300357 CrossRef CAS.
- Z. He, Z. Xiao, H. Yue, Y. Jiang, M. Zhao, Y. Zhu, C. Yu, Z. Zhu, F. Lu, H. Jiang, C. Zhang and F. Wei, Adv. Funct. Mater., 2023, 33, 2300094 CrossRef CAS.
- T. Wang, J. Duan, B. Zhang, W. Luo, X. Ji, H. Xu, Y. Huang, L. Huang, Z. Song, J. Wen, C. Wang, Y. Huang and J. B. Goodenough, Energy Environ. Sci., 2022, 15, 1325–1333 RSC.
- S. Ling, X. Li, T. Zhou, R. Yuan, S. Sun, H. He and C. Zhang, Adv. Mater., 2023, 35, e2211201 CrossRef PubMed.
- Y. Shen, J. Zou, M. Zeng and L. Fu, ACS Nano, 2023, 17, 22167–22182 CrossRef CAS PubMed.
- M. Ko, S. Chae, J. Ma, N. Kim, H.-W. Lee, Y. Cui and J. Cho, Nat. Energy, 2016, 1, 16113 CrossRef CAS.
- Z. Liu, X. Wang, J. Hu, J. Meng, C. Niu, F. Liu, L. Cui, R. Yu and L. Mai, Adv. Energy Mater., 2023, 13, 2301715 CrossRef CAS.
- S. Chae, S. H. Choi, N. Kim, J. Sung and J. Cho, Angew. Chem., Int. Ed., 2020, 59, 110–135 CrossRef CAS PubMed.
- S. Chae, S. Park, K. Ahn, G. Nam, T. Lee, J. Sung, N. Kim and J. Cho, Energy Environ. Sci., 2020, 13, 1212–1221 RSC.
- Z. He, C. Zhang, Y. Zhu and F. Wei, Energy Environ. Sci., 2024, 17, 3358–3364 RSC.
- T. Lee, N. Kim, J. Lee, Y. Lee, J. Sung, H. Kim, S. Chae, H. Cha, Y. Son, S. K. Kwak and J. Cho, Adv. Energy Mater., 2023, 13, 2301139 CrossRef CAS.
- Z. Cao, X. Zheng, Q. Qu, Y. Huang and H. Zheng, Adv. Mater., 2021, 33, 2103178 CrossRef CAS PubMed.
- A. M. Li, Z. Wang, T. P. Pollard, W. Zhang, S. Tan, T. Li, C. Jayawardana, S. C. Liou, J. Rao, B. L. Lucht, E. Hu, X. Q. Yang, O. Borodin and C. Wang, Nat. Commun., 2024, 15, 1206 CrossRef CAS PubMed.
- J. Sun, L. Huang, G. Xu, S. Dong, C. Wang and G. Cui, Mater. Today, 2022, 58, 110–118 CrossRef CAS.
- G. Chen, J. Sun, J. Li, X. Du, G. Xu, S. Dong and G. Cui, Angew. Chem., Int. Ed., 2023, 135, 202306141 CrossRef.
- S. Mei, B. Xiang, S. Guo, J. Deng, J. Fu, X. Zhang, Y. Zheng, B. Gao, K. Huo and P. K. Chu, Adv. Funct. Mater., 2023, 34, 2301217 CrossRef.
- Z. Yang, T. Chen, D. Chen, X. Shi, S. Yang, Y. Zhong, Y. Liu, G. Wang, B. Zhong, Y. Song, Z. Wu and X. Guo, Angew. Chem., Int. Ed., 2021, 60, 12539–12546 CrossRef CAS PubMed.
- N. Kim, Y. Kim, J. Sung and J. Cho, Nat. Energy, 2023, 8, 921–933 CrossRef CAS.
- S. Park, J. Sung, S. Chae, J. Hong, T. Lee, Y. Lee, H. Cha, S. Y. Kim and J. Cho, ACS Nano, 2020, 14, 11548–11557 CrossRef CAS PubMed.
- N. Kim, H. Cha, S. Chae, T. Lee, Y. Lee, Y. Kim, J. Sung and J. Cho, Energy Environ. Sci., 2023, 16, 2505–2517 RSC.
- W. Li, X. Li, J. Yu, J. Liao, B. Zhao, L. Huang, A. Ali, H. Zhang, J. H. Wang, Z. Guo and M. Liu, Nano Energy, 2019, 61, 594–603 CrossRef CAS.
- Y. Wei, R. Yao, Y. Zhao, R. Yang, J. Liu, X. Liu and H. Li, Adv. Energy Mater., 2023, 13, 2202884 CrossRef CAS.
- D. T. Ngo, H. T. T. Le, C. Kim, J.-Y. Lee, J. G. Fisher, I.-D. Kim and C.-J. Park, Energy Environ. Sci., 2015, 8, 3577–3588 RSC.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kubel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed.
- L. Su, J. Ren, T. Lu, K. Chen, J. Ouyang, Y. Zhang, X. Zhu, L. Wang, H. Min, W. Luo, Z. Sun, Q. Zhang, Y. Wu, L. Sun, L. Mai and F. Xu, Adv. Mater., 2023, 35, 2205751 CrossRef CAS PubMed.
- S. Zhou, Y. Sun, T. Gao, J. Liao, S. Zhao and G. Cao, Angew. Chem., Int. Ed., 2023, 62, e202311930 CrossRef CAS PubMed.
- Q. Wang, L. Velasco, B. Breitung and V. Presser, Adv. Energy Mater., 2021, 11, 2102355 CrossRef CAS.
- L. Lin, K. Wang, A. Sarkar, C. Njel, G. Karkera, Q. Wang, R. Azmi, M. Fichtner, H. Hahn, S. Schweidler and B. Breitung, Adv. Energy Mater., 2022, 12, 2103090 CrossRef CAS.
- Q. Wang, A. Sarkar, D. Wang, L. Velasco, R. Azmi, S. S. Bhattacharya, T. Bergfeldt, A. Düvel, P. Heitjans, T. Brezesinski, H. Hahn and B. Breitung, Energy Environ. Sci., 2019, 12, 2433–2442 RSC.
- Y. Ding, Z. Wang, Z. Liang, X. Sun, Z. Sun, Y. Zhao, J. Liu, C. Wang, Z. Zeng, L. Fu, M. Zeng and L. Tang, Adv. Mater., 2023, e2302860, DOI:10.1002/adma.202302860.
- C. Yang, J. Xia, C. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan, E. Thimsen, J. Xu, W. Song, E. Hu, X. Ji, S. Hou, X. Zhang, M. S. Ding, S. Hwang, D. Su, Y. Ren, X.-Q. Yang, H. Wang, O. Borodin and C. Wang, Nat. Sustain., 2023, 6, 325–335 CrossRef.
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, Energy Environ. Sci., 2021, 14, 2883–2905 RSC.
- X. Zhao, Z. Fu, X. Zhang, X. Wang, B. Li, D. Zhou and F. Kang, Energy Environ. Sci., 2024, 17, 2406–2430 RSC.
- Y. Li, L. Zhang, H. Y. Yen, Y. Zhou, G. Jang, S. Yuan, J. H. Wang, P. Xiong, M. Liu, H. S. Park and W. Li, Nano-Micro Lett., 2023, 15, 63 CrossRef PubMed.
- K. Wang, W. Hua, X. Huang, D. Stenzel, J. Wang, Z. Ding, Y. Cui, Q. Wang, H. Ehrenberg, B. Breitung, C. Kubel and X. Mu, Nat. Commun., 2023, 14, 1487 CrossRef CAS PubMed.
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y. S. Hu, Angew. Chem., Int. Ed., 2020, 59, 264–269 CrossRef CAS PubMed.
- J. Li, H. Yang, Q. Deng, W. Li, Q. Zhang, Z. Zhang, Y. Chu and C. Yang, Angew. Chem., Int. Ed., 2024, 63, e202318042 CrossRef CAS PubMed.
- M. Chen, W. Hua, J. Xiao, D. Cortie, W. Chen, E. Wang, Z. Hu, Q. Gu, X. Wang, S. Indris, S. L. Chou and S. X. Dou, Nat. Commun., 2019, 10, 1480 CrossRef PubMed.
- X. Liu, Y. Yu, K. Li, Y. Li, X. Li, Z. Yuan, H. Li, H. Zhang, M. Gong, W. Xia, Y. Deng and W. Lei, Adv. Mater., 2024, 36, 2312583 CrossRef CAS PubMed.
- X. Liu, Y. Xing, K. Xu, H. Zhang, M. Gong, Q. Jia, S. Zhang and W. Lei, Small, 2022, 18, e2200524 CrossRef PubMed.
- D. Du, H. He, R. Zheng, L. Zeng, X. Wang, C. Shu and C. Zhang, Adv. Energy Mater., 2024, 14, 2304238 CrossRef CAS.
- W. Li, Y. Li, J.-H. Wang, S. Huang, A. Chen, L. Yang, J. Chen, L. He, W. K. Pang, L. Thomsen, B. Cowie, P. Xiong, Y. Zhou, G. Jang, D. H. Min, J. S. Byun, L. Xu, J.-Q. Huang, K. C. Roh, S. H. Kang, M. Liu, X. Duan and H. S. Park, Energy Environ. Sci., 2024, 17, 5387–5398 RSC.
- J. Liang, Y. Wan, H. Lv, X. Liu, F. Lv, S. Li, J. Xu, Z. Deng, J. Liu, S. Zhang, Y. Sun, M. Luo, G. Lu, J. Han, G. Wang, Y. Huang, S. Guo and Q. Li, Nat. Mater., 2024 DOI:10.1038/s41563-024-01901-4.
- K. Li, G. Yuan, X. Liu, Y. Guo, R. Huang, H. Li, H. Zhang, Q. Jia, Z. Xie, S. Zhang and W. Lei, Adv. Funct. Mater., 2023, 33, 2302348 CrossRef CAS.
- S. Guo, Z. Sun, Y. Liu, X. Guo, H. Feng, S. Luo, C. Wei, Y. Zheng, X. Zhang, K. Kim, H. Liu, P. K. Chu, B. Gao, Q. Zhang and K. Huo, Adv. Energy Mater., 2024, 14, 2303876 CrossRef CAS.
- Y. Cao, S. Zhang, B. Zhang, C. Han, Y. Zhang, X. Wang, S. Liu, H. Gong, X. Liu, S. Fang, F. Pan and J. Sun, Adv. Mater., 2023, 35, 2208514 CrossRef CAS PubMed.
- C. Liu, M. Han, C. L. Chen, J. Yin, L. Zhang and J. Sun, Nano Lett., 2023, 23, 3507–3515 CrossRef CAS PubMed.
- A. Zunger, S. Wei, L. G. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353–356 CrossRef CAS PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed.
- G. Cao, J. Liang, Z. Guo, K. Yang, G. Wang, H. Wang, X. Wan, Z. Li, Y. Bai, Y. Zhang, J. Liu, Y. Feng, Z. Zheng, C. Lu, G. He, Z. Xiong, Z. Liu, S. Chen, Y. Guo, M. Zeng, J. Lin and L. Fu, Nature, 2023, 619, 73–77 CrossRef CAS PubMed.
- T. Li, Y. Yao, Z. Huang, P. Xie, Z. Liu, M. Yang, J. Gao, K. Zeng, A. H. Brozena, G. Pastel, M. Jiao, Q. Dong, J. Dai, S. Li, H. Zong, M. Chi, J. Luo, Y. Mo, G. Wang, C. Wang, R. Shahbazian-Yassar and L. Hu, Nat. Catal., 2021, 4, 62–70 CrossRef CAS.
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed.
- Y. Ye, H. Xie, Y. Yang, Y. Xie, Y. Lu, J. Wang, X. Kong, S. Jin and H. Ji, J. Am. Chem. Soc., 2023, 145, 24775–24784 CAS.
- W. Li, J. H. Wang, L. Yang, Y. Li, H. Y. Yen, J. Chen, L. He, Z. Liu, P. Yang, Z. Guo and M. Liu, Adv. Mater., 2024, 36, e2314054 CrossRef PubMed.
- E. Zhou, X. Luo, H. Jin, C. Wang, Z. Lu, Y. Xie, S. Zhou, Y. Chen, Z. He, R. Ma, W. Zhang, H. Xie, S. Jiao, Y. Lin, D. S. Bin, R. Huang, X. Wu, X. Kong and H. Ji, J. Am. Chem. Soc., 2024, 146, 4752–4761 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental Methods, Fig. S1–S53, Tables S1–S7, and supplementary Movies. See DOI: https://doi.org/10.1039/d4ee01329h |
| This journal is © The Royal Society of Chemistry 2024 |