
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrong transboundary electron transfer of high-entropy quantum-dots driving rapid hydrogen evolution kinetics†
Hao
Zhao
ab,
Mengyuan
Liu
ab,
Qiansen
Wang
ab,
YuZe
Li
ab,
Yubin
Chen
ac,
Yanping
Zhu
c,
Zhouying
Yue
a,
Jun
Li
a,
Guoliang
Wang
a,
Zhiqing
Zou
a,
Qingqing
Cheng
 *a and
Hui
Yang
*a
*a and
Hui
Yang
*a
aShanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, P. R. China. E-mail: chengqq@sari.ac.cn; yangh@sari.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cNingbo Cotrun New Energy Science & Technology Co., Ltd, Ningbo 315000, P. R. China
First published on 31st July 2024
Abstract
The sluggish Tafel kinetics and high noble metal usage of PtRu-based electrocatalysts in the hydrogen evolution reaction (HER) are the key challenges for practical proton exchange membrane water electrolysis (PEMWE). Here, highly dispersed sub-2 nm PtRuMoFeCoNi high-entropy alloy quantum dots (HEA-QDs) are synthesized through rapid microwave radiation. Advanced spectroscopy methods and theoretical calculations reveal that the strong transboundary electron transfer from Fe/Co/Ni/Mo to Pt/Ru sites weakens the strength of adsorbed hydrogen by enhancing the local electron density around Pt/Ru sites, thus effectively minimizing the Tafel step barrier. The synthesized HEA-QDs demonstrate superior HER activity with an overpotential of 11 mV@10 mA cm−2, obviously outperforming that of Pt/C (18 mV@10 mA cm−2). Strikingly, the PEMWE device using HEA-QDs as a cathodic catalyst with an ultra-low loading of 100 μgPtRu cm−2 exhibits not only excellent performance (1.65 V@1.0 A cm−2), but also outstanding stability, even presenting a reduced cell voltage with a rate of −12.2 μV h−1 over 1000 h of testing at 1.0 A cm−2. Additionally, the HEA-QD cathode cell boasts an energy consumption of 3.98 kWh N m−3 H2 (1.0 A cm−2) and a cost of US$0.88 per kg H2, significantly exceeding the 2030 DOE target. This work offers new guidance for the precise construction of active centers with low kinetic barriers and their potential application in PEMWE devices with low noble metal usage.
Broader contextPEM water electrolysis (PEMWE), capable of directly coupling with intermittent renewable power, is regarded as a promising technology for hydrogen production. However, the sluggish Tafel kinetics of conventional PtRu-based electrocatalysts towards the hydrogen evolution reaction (HER) necessitates massive noble metals to achieve the desired performance, hindering the large-scale commercialization of PEMWE devices. Herein, we construct highly dispersed sub-2 nm PtRuMoFeCoNi high-entropy alloy quantum dots (HEA-QDs) through rapid microwave radiation. The strong transboundary electron transfer from Fe, Co, Ni, and Mo to Pt and Ru weakens the strength of Had on Pt/Ru sites, thereby effectively lowering the Tafel barrier. Consequently, the obtained HEA-QDs exhibit preeminent HER activity and durability. The integrated PEMWE device using HEA-QDs as a cathodic catalyst with an ultra-low loading of 100PtRu μg cm−2 delivers an exceptional activity (1.65 V@1.0 A cm−2), which is superior to that of Pt/C (1.71 V@1.0 A cm−2) with the same loading. Impressively, the PEMWE device with HEA-QDs also exhibits excellent stability even with a negative decay rate of −12.2 μV h−1 at 1.0 A cm−2 over 1000 h. Besides, the energy consumption and cost for H2 production are 3.98 kW h N m−3 H2 and US$0.88 per kg H2, respectively, which surpass the 2030 DOE target. |
Introduction
Proton exchange membrane water electrolysis (PEMWE) is considered one of the most promising technologies for hydrogen production due to its high efficiency, pollution-free operation and rapid response capabilities, which enable direct coupling with intermittent renewable energy sources such as wind and solar.1–5 However, PtRu-based electrocatalysts still represent one of the most effective catalysts for the cathodic hydrogen evolution reaction (HER) in PEMWE devices. Due to the scarcity and expensiveness of Pt and Ru, approaches that can concurrently reduce noble metal usage and enhance the catalytic performance have been the target to pursue, which require significantly improving the catalytic efficiency of active sites. For the HER in PEMWE, the binding strength of hydrogen on the catalyst surface directly affects catalytic activity, following the Sabatier principle.6 Both excessive and insufficient adsorption energy of hydrogen on catalytic sites would result in a high kinetic barrier, and only those with moderate binding strength exhibit high activity.7 Previous studies have demonstrated that Pt/Ru sites in PtRu-based catalysts, which have abundant unoccupied d orbitals, exhibit relatively strong proton adsorption strength (M–H), making it hard to desorb *H2 from the active center, thereby limiting the HER kinetics.8–13 Based on the above considerations, elevating the local electron density around Pt/Ru sites, which can fill the unoccupied d orbitals of active sites, to weaken the M–H energy might be a feasible strategy to promote HER kinetics. Alloying Pt/Ru with other transition metals is a traditional but effective way to realize the modulation of local electron density around the active center through strong ligand or strain effects.14 For example, Hiroshi Kitagawa et al. reported novel PtW solid-solution nanoparticles where electron migration from W to Pt enhanced the negative charge density around Pt sites, achieving record HER performance in acid.15 Furthermore, Tianyi Ma et al. synthesized RuNi alloy nanoparticles that exhibit comparable HER activity to Pt/C due to the enrichment of negative charge on Ru sites.16 Nevertheless, the limited composition and inherent properties of specific elements in the conventional alloys struggle to realize the flexible modulation of the active center, making it difficult to attain the optimal adsorption energy and the best HER kinetics. Therefore, rationally designing catalysts to break such a tailoring limitation of the electronic structure is crucial for improving the HER kinetics and reducing the usage of noble metals in PEMWE devices.High-entropy alloys (HEAs), containing at least five elements with an approximately equal atomic ratio (5–35 at%), have attracted intensive interest as electrocatalysts due to their unique properties, including a high entropy effect, a lattice distortion effect, sluggish diffusion, and a “cocktail” effect.17–19 In particular, the synergistic interactions among multiple metal atoms can induce various ligand and strain effects, thereupon effectively modifying the local charge distribution and d-band structure of active sites in a wide range.20–22 Therefore, the HEA may provide a viable solution to overcome the limitations of traditional Pt/Ru-based alloys in tuning the electronic structure of the active center. For example, Lei Wang et al. reported highly dispersed Pt18Ni26Fe15Co14Cu27 HEA nanoparticles that exhibited excellent HER/MOR (methanol oxidation reaction) activities under alkaline conditions, which was attributed to the appropriate adsorption of intermediates facilitated by the fast site-to-site electron transfer in multi-metal sites.23 Beyond that, HEA also exhibits remarkable thermodynamic and dynamic stabilities owing to the increased atom diffusion barrier and suppressed dislocation motion.24–27 However, the significant differences in standard reduction potentials among various metal ions impede the coupling of different metals into a single nanocrystal using traditional synthesis methods.28–30 Recently, typical HEA particles have been successfully synthesized through a series of novel methods such as carbothermal shock, fast moving bed pyrolysis, arc-discharge, dealloying, etc.31–34 Nevertheless, the particle size of these HEAs is usually large, which leads to the decrease of accessible active sites as well as the HER activity.35,36 Hence, the development of ultra-small PtRu-based HEA particles while optimizing the local electron density around the active center is highly desirable for enhancing the catalytic efficiency and reducing the noble metal usage.
Herein, we propose an electronegativity-driven charge redistribution strategy to weaken the strength of Had on active sites, thereby effectively accelerating the Tafel kinetics. The highly dispersed sub-2 nm ultra-small HEA quantum dots (HEA-QDs) containing Pt, Ru, Mo, Fe, Co, and Ni are synthesized through rapid microwave irradiation. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) confirms the high entropy atomic structure and uniform elemental distribution of ultra-small HEA-QDs. X-ray absorption fine structure (XAFS) spectroscopy and X-ray photoelectron spectroscopy (XPS) reveal the significant electron-donating effect of Fe, Co, Ni, and Mo on enriching the local electron density around Pt/Ru sites. Density functional theory (DFT) calculations and electrochemical characterization elucidate that the strong transboundary electron transfer helps to achieve the optimized adsorption energy of Pt/Ru–H, thereby significantly reducing the energy barrier of the Tafel slope, which is the rate determining step for the HER. As a result, the obtained HEA-QDs exhibit preeminent electrocatalytic activity (11 mV@10 A cm−2) and durability towards the HER compared to the commercial Pt/C (18 mV@10 mA cm−2). Strikingly, the integrated PEMWE device using HEA-QDs as a cathodic catalyst with a low noble metal loading of 100 μgPtRu cm−2 delivers excellent activity (1.65 V@1.0 A cm−2) and surprising stability with even a decreased cell voltage of −12.2 μV h−1 (1.0 A cm−2, over 1000 h). This work not only offers new insights for rationally designing efficient catalysts with rapid reaction kinetics across various fields but also provides potential applications for practical hydrogen production in PEMWEs with low noble metal usage.
Results and discussion
Synthesis and characterization of HEA-QDs
To enhance the local electron density around Pt/Ru active sites, we chose relatively low electronegativity elements (Fig. 1a) including Fe (1.83), Co (1.88), Ni (1.91), and Mo (2.16) as electron donors, which can induce a driving force for directional electron transfer towards the Pt (2.28)/Ru (2.20) sites.30 Additionally, the gradient in electronegativity and spanning multiple periods (Fe, Co, and Ni: 4th period; Mo and Ru: 5th period; and Pt: 6th period) of the selected elements can increase the mixing entropy of the system and further strengthen the sluggish diffusion effect in high-entropy alloys, which is beneficial for improving the stability of the catalyst under reductive or acidic environments. HEA-QDs were synthesized through the microwave-polyol method at 230 °C for 80 s as illustrated in Fig. 1b. The microwave thermal radiation rapidly decomposes the acetylacetone salts, forming seeds, and induces explosive nucleation and uniform growth in triethylene glycol (TEG).37 Meanwhile, TEG as a protecting agent can directly bind to the HEA surface preventing uncontrollable enlargement, thus stabilizing the HEA-QDs.38 Transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) images display that the obtained HEA-QDs are uniformly dispersed with an average diameter of ca. 1.9 nm (Fig. 1c and d and Fig. S1, ESI†). It is noteworthy that this ultrasmall nanostructure can facilitate the overall utilization efficiency of noble metal atoms. As shown in Fig. 1e, the powder X-ray diffraction (PXRD) pattern of HEA-QDs displays broad peaks around 42.0°, owing to the ultra-small particle size and lattice distortion resulting from multiple elements.39 In addition, the atomic-resolution HAADF-STEM image of the HEA-QDs (inset in Fig. S2, ESI†) shows a lattice spacing of 2.16 Å, corresponding to the (111) diffraction plane of a face-center-cubic (fcc) structured nanoscale alloy, which is very different from any pure metals used (Fig. S2, ESI†).34 To probe the atomic structural details of HEA-QDs, aberration-corrected HAADF-STEM was performed. As shown in Fig. 1f and Fig. S3 (ESI†), the various intensities of bright spots elucidate the multielement composition within HEA-QDs due to the different atomic number contrast (Z) while the bright Pt and Ru atoms can be clearly identified compared with other elements. Furthermore, the line profile in Fig. 1g visually indicates that the random distribution of multiple metallic elements results in various atomic distances. Inductively coupled plasma optical emission spectroscopy (ICP-OES) spectra show that the atomic ratio of Pt, Ru, Mo, Fe, Co, and Ni is 17.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17.0:17.4:16.0:5.9:8.1, which is roughly in accordance with the results of energy dispersive spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) in Fig. 1h and Fig. S4 (ESI†). The elemental maps (Fig. 1i) display the homogeneous distribution of elements Pt, Ru, Mo, Fe, Co and Ni in single HEA-QDs. For clear observation, we further analyzed the elemental maps and line scan profiles of HEA-QD aggregates (Fig. S5, ESI†), which also exhibit a uniform distribution of various metal elements over the HEA-QDs. The above results offer concrete evidence regarding the successful synthesis of HEA-QDs. More importantly, using this method and principle, one can successfully synthesize various ultra-small alloys particles from binary to hexinary (Fig. S6–S8, ESI†).
:17.0:17.4:16.0:5.9:8.1, which is roughly in accordance with the results of energy dispersive spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) in Fig. 1h and Fig. S4 (ESI†). The elemental maps (Fig. 1i) display the homogeneous distribution of elements Pt, Ru, Mo, Fe, Co and Ni in single HEA-QDs. For clear observation, we further analyzed the elemental maps and line scan profiles of HEA-QD aggregates (Fig. S5, ESI†), which also exhibit a uniform distribution of various metal elements over the HEA-QDs. The above results offer concrete evidence regarding the successful synthesis of HEA-QDs. More importantly, using this method and principle, one can successfully synthesize various ultra-small alloys particles from binary to hexinary (Fig. S6–S8, ESI†).
 | ||
| Fig. 1 Morphology structure and composition characterization of HEA-QDs. (a) and (b) Preparation procedure. (c) HAADF-STEM image and corresponding (d) size distribution histogram. (e) XRD pattern. (f) AC-STEM image and (g) line profile along the dashed line in (f). (h) TEM-EDS and atomic ratio of different elements from the result of ICP-OES. (i) AC-STEM and corresponding STEM-EDS mapping images. | ||
X-ray absorption fine structure (XAFS) measurements were performed to confirm the electronic structure and coordination environment of active sites in HEA-QDs. Compared with commercial Pt/C and Ru/C, the white line (WL) intensity of the Pt L3-edge significantly decreases (Fig. 2a) and the absorption edge of the Ru K-edge obviously shifts to the left (Fig. 2d) for HEA-QDs, which indicate that excess electrons are transferred into the valence band of Pt and Ru atoms, thus the highly occupied Pt 5d and Ru 4d bands.12 Meanwhile, the linear component fitting analysis in Fig. 2b and e indicates that the average valence states of Pt and Ru elements in HEA-QDs are +0.94 and +0.3, respectively, which are remarkably lower than those of Pt/C (+1.9) and Ru/C (+1.1). The results are probably due to the low electronegativity of Fe, Co, Ni and Mo atoms neighboring Pt/Ru sites, which results in the donation of electron density. Additionally, we further used Fourier transform extended X-ray absorption fine structure (FT-EXAFS) to examine the coordination features of the Pt and Ru sites. As shown in Fig. 2c and f, the average bond lengths of Pt–M (2.1 Å) and Ru–M (2.2 Å) in HEA-QDs significantly differ from the metallic bond lengths in bulk Pt (2.6 Å) and Ru (2.4 Å) references, revealing the remarkable coordination effects between Pt/Ru atoms and other transition metals of Fe, Co, Ni, and Mo. Meanwhile, the presence of Pt–O and Ru–O can be discerned at 1.7 Å and 1.5 Å, respectively, owing to the high oxygen affinity of Pt and Ru elements.40 These features substantiate the increasing electron density around Pt/Ru sites and the formation of high entropy rather than phase segregation structures.
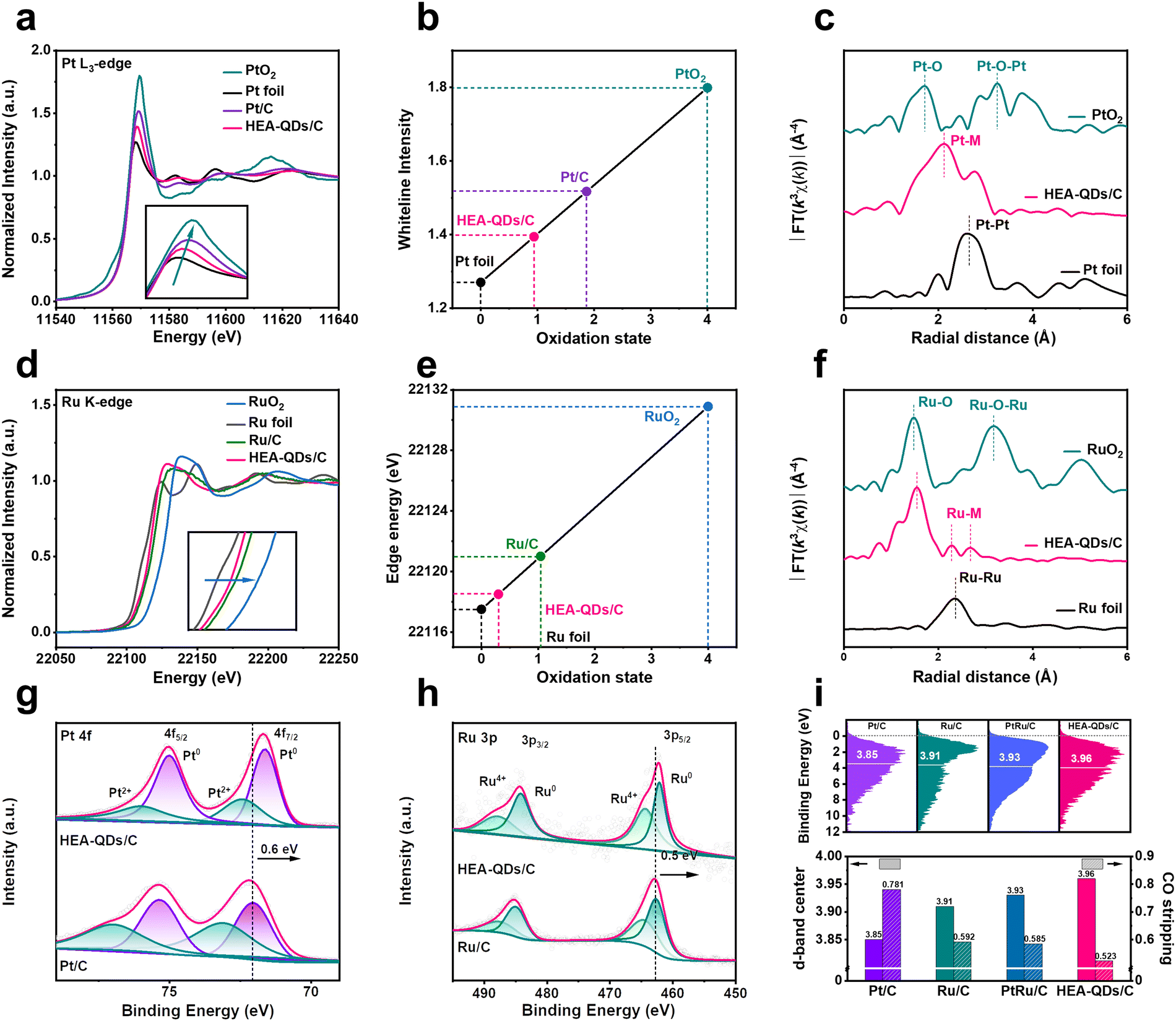 | ||
| Fig. 2 Electronic and geometry structures analysis of HEA-QDs. (a) The XANES spectra of the Pt L3-edge, (b) linear component fitting analysis and (c) FT-EXAFS spectra of Pt. (d) The XANES spectra of the Ru K-edge, (e) linear component fitting analysis and (f) FT-EXAFS spectra of Ru. (g) XPS spectra of Pt and (h) Ru. (i) VBS spectra (top) and CO oxidation peak potentials (down) of different catalysts. | ||
To investigate the surface electronic effects of HEA-QDs, XPS was performed. Compared to commercial Pt/C and Ru/C, the peaks in the Pt0 4f and Ru0 3p XPS spectra of HEA-QDs negatively shift by 0.6 and 0.5 eV, respectively (Fig. 2g and h), suggesting that electrons might be transferred from adjacent transition metal species to Pt and Ru sites. The small fraction of Pt and Ru in +2 and +4 valence states may originate from surface oxidation. On the other hand, the XPS spectra of Fe 2p, Co 2p, Ni 2p, and Mo 3d (Fig. S9, ESI†) indicate that these elements on the surface primarily exist in oxidized states, which could be attributed to the electron-donor effect towards Pt/Ru sites and the oxidation induced by the high surface energy of ultra-small HEA-QDs. Furthermore, XPS-etching was conducted to acquire the interior elemental information. As shown in Fig. S10 (ESI†), the peak of Pt0 4f positively shifts but remains lower than that of Pt/C, while the Ru0 3p negatively shifts with etching deepening from 0.2 nm to 0.6 nm. The metallic state percentage of Fe, Co, and Ni elements increases progressively, confirming the previously mentioned surface oxidation. These results show the complexity of electron interaction among various elements in HEA-QDs. Furthermore, the elemental composition at different etching depths in Fig. S11 (ESI†) demonstrates the high-entropy characteristics of HEA-QDs not only on the surface but also internally.
To further investigate the effects of strong transboundary electron transfer on the d-band structure of surface Pt and Ru sites, which are crucial in determining the adsorption strength of electrocatalytic intermediates, valence band spectra (VBS) were measured.41–43 Compared to the values of Pt/C (−3.85 eV), Ru/C (−3.91 eV), and PtRu/C (−3.93 eV), the d-band center of HEA-QDs/C shifts down to −3.96 eV (Fig. 2i, top), which increases the filling of adsorbate-metal antibonding states, thus the weakened adsorption strength.24 Additionally, the positive shift of the CO stripping peak of HEA-QDs (Fig. 2i, bottom and Fig. S12, ESI†), demonstrates the weakened adsorption of CO on Pt/Ru sites, which is consistent with the results obtained from VBS. The aforementioned results imply that the enhanced local electron density of Pt/Ru sites, which may arise from the strong electron donor effect of Fe, Co, Ni, and Mo elements, can weaken the adsorption strength of H and promote the kinetics of the HER.
Evaluation of electrochemical performance
To evaluate the impact of the strong electron redistribution on electrocatalysis, the HER performance of HEA-QDs was measured in N2-saturated 0.5 M H2SO4 using a three-electrode electrochemical system. Commercial Pt/C, Ru/C, and PtRu/C with small sizes were selected as the references, while the HEA-QDs catalyst was loaded on Vulcan XC-72R carbon support (HEA-QDs/C, Fig. S13 and S14, ESI†). As shown in Fig. 3a, the HEA-QDs exhibit the smallest overpotential (η) of 11 mV/102 mV to achieve the benchmark current density of 10/100 mA cm−2, which are significantly superior compared to that of Pt/C (18 mV/127 mV), Ru/C (45 mV/over 200 mV) and PtRu/C (16 mV/134 mV) under the same noble metal loading. The Tafel slope of HEA-QDs/C (28.7 mV decade−1) is lower than that of Pt/C (29.4 mV decade−1), Ru/C (50.2 mV decade−1) and PtRu/C (37.7 mV decade−1), demonstrating the Volmer–Tafel mechanism on the active sites and the Tafel step as the rate determining step (RDS) during the HER process (Fig. 3b left axis and Fig. S15, ESI†).44,45 It is undeniable that as the applied potential negatively shifts (corresponding to the rise in the Fermi level), the driving force for the formation of H− increases, thereby enhancing the possibility of Heyrovsky step involvement in the HER at high negative potentials.46 Additionally, even comparing the overpotentials at 10 mA cm−2 and Tafel slope values with other advanced electrocatalysts reported recently (Fig. 3c and Table S1, ESI†), HEA-QDs/C can be demonstrated as one of the best electrocatalysts for the HER. In addition, the exchange current density (j0) of HEA-QDs/C calculated using Tafel parameters (Fig. 3b, right axis) is ca. 3.899 mA cm−2, which is 1.53 and 2.56 times higher than that of Pt/C and Ru/C, respectively, confirming the excellent intrinsic HER activity. For a comprehensive evaluation of the HER activity, as shown in Fig. S16 (ESI†), we determined the mass activity (MA) normalized by noble metal loading and the specific activity (SA) normalized by the electrochemically active surface area (ECSA). As shown in Fig. 3d, SA and MA at various overpotentials of HEA-QDs/C exhibit superiority to the references, especially at 200 mV, where the MA & SA of HEA-QDs/C are 1.32 & 1.26 and 2.08 & 2.44 times greater than those obtained from Pt/C & Ru/C, respectively. Additionally, we also synthesized the Pt-based HEA (PtMoFeCoNi, Pt-HEA) and Ru-based HEA (RuMoFeCoNi, Ru-HEA) to explore the synergistic effects between Pt and Ru atoms in HEA-QDs (Fig. S17 and S18, ESI†). Electrochemical HER evaluations (Fig. S19a and b, ESI†) show that the activity of HEA-QDs/C outperforms both Pt-HEA/C and Ru-HEA/C under the same noble metal loading, indicating the existence of synergistic effects between Pt and Ru in HEA-QDs. As shown in Fig. S19c (ESI†), Pt in HEA-QDs/C exhibits a lower oxidation state compared to that in Pt-HEA/C, suggesting that the introduction of Ru atoms can reduce the chemical state of Pt, which can facilitate proton desorption for HEA-QDs/C. Meanwhile, the average oxidation state of Ru in HEA-QDs is analogous to that in Ru-HEA (Fig. S19d, ESI†), possibly due to the broader valence band of Ru element in the HEA, which makes minor adjustments of Pt to its electronic states negligible.47 Besides, as shown in Fig. S20 (ESI†), compared with other small-sized alloys from low entropy to high entropy synthesized by the same methods, the activity of prepared HEA-QDs/C still stands for the top tier.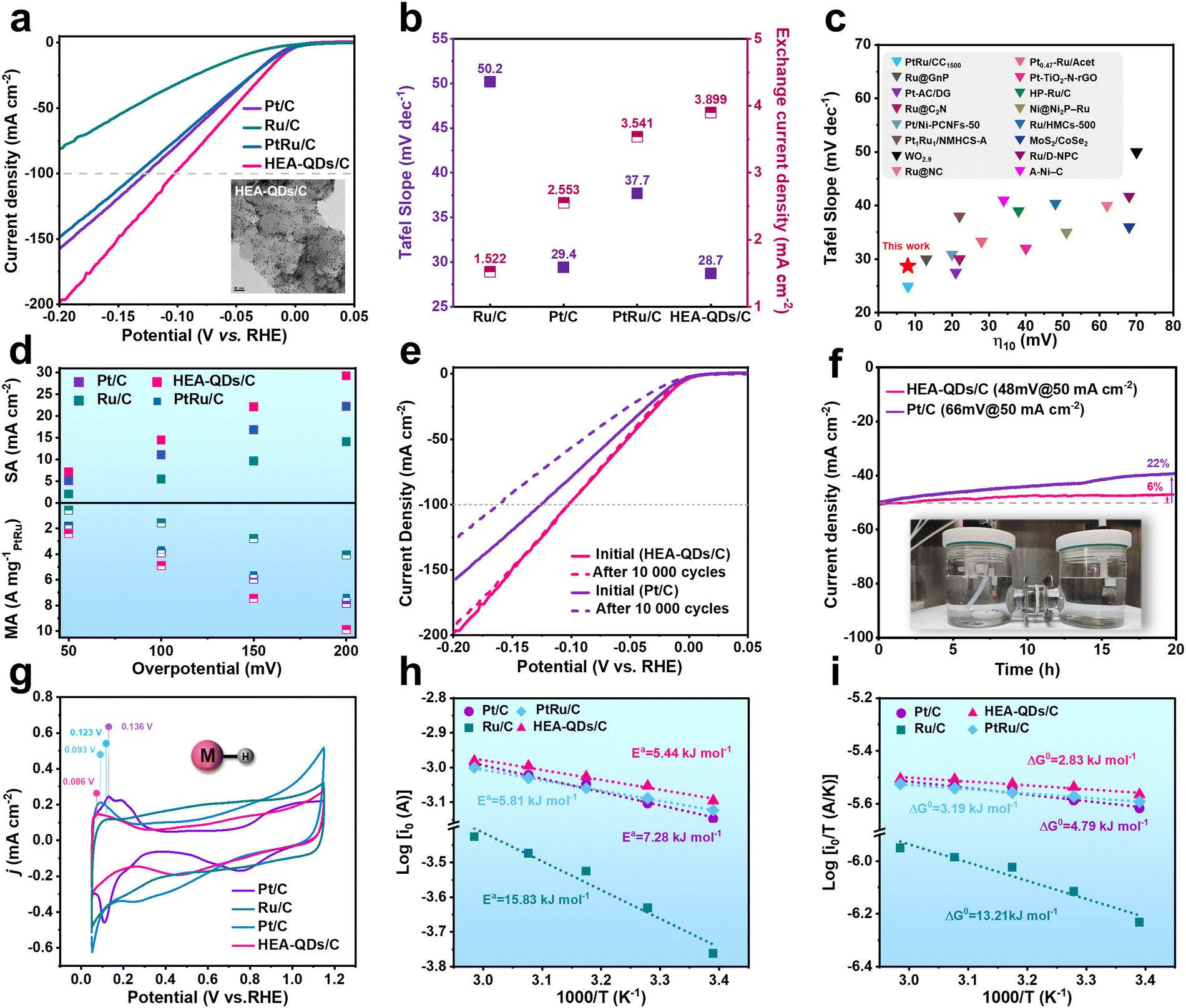 | ||
| Fig. 3 The electrochemistry performance of HEA-QDs toward the HER. (a) Polarization curves (insert is the TEM image of HEA-QDs/C). (b) Tafel slope and corresponding j0. (c) The comparison of η10 values and the Tafel slope between HEA-QDs and recently reported electrocatalysts. (d) Normalized MA and SA at different overpotentials. (e) ADT test and (f) current–time chronoamperometry response. (g) CV curves. (h) Arrhenius plots and (i) Eyring plots at the equilibrium potential in the temperature range of 295–335 K. | ||
Furthermore, accelerated degradation test (ADT) and chronoamperometry were conducted to evaluate the durability of the HEA-QDs/C. As shown in Fig. 3e, the negligible degradation of HEA-QDs/C is observed after 10000 cycles while the Pt/C exhibits obvious deterioration from 66 mV to 90 mV at 50 mA cm−2. Fig. 3f shows a long-term stability test of the HEA-QDs/C at 50 mA cm−2 in 0.5 M H2SO4. After continuous electrolysis for 20 h, HEA-QDs/C demonstrates remarkable stability with only a 6% decay of the initial current density, whereas Pt/C experienced a large decay of over 20%. The superior structural stability of HEA-QDs can be attributed to the high-entropy stabilization effect and a high atomic diffusion barrier. Moreover, as shown in Fig. S21 (ESI†), time-dependent inductively coupled plasma mass spectrometry (ICP-MS) was performed to monitor the dissolution of transition metals during the stability test. The results indicate that the transition metals partially leach under both acidic conditions and reductive potential due to the existence of transition metal oxides on the surface of the HEA-QDs (Fig. S9, ESI†). However, this phenomenon stabilizes after 15 h of testing, which might be attributed to the sluggish diffusion effect of HEA that prevents the transition metals within the lattice of HEA from dissolving, thereby maintaining structural stability. Furthermore, we conducted XRD, TEM, and XPS measurements to explore the degradation mechanism of HEA-QDs. As shown in Fig. S22 (ESI†), the XRD patterns of HEA-QDs/C before and after the stability test confirm the retention of the original fcc structure without the formation of any new phases. TEM images of HEA-QDs/C after a 20 h electrolysis in Fig. S23 (ESI†) show that the small HEA-QDs on XC-72R may undergo migration and form nanowires while still retaining the high-entropy characteristics of individual particles. In addition, the XPS spectra (Fig. S24, ESI†) show that the peak of Pt0 4f exhibits a slightly positive shift while still being lower than that of Pt/C, which can be the primary reason for degradation. On the other hand, the valence states of Mo and Ni elements exhibit a partial reduction from Mo4+ to Mo2+ and Ni2+ to Ni0, respectively, which can be attributed to the prolonged reductive environment. Meanwhile, the Fe and Co elements remain unchanged due to the pinning effects.47 The above results reveal the great improvement in intrinsic activity, atomic utilization efficiency and stability of HEA-QDs, which holds significant potential in the application of low-cost and highly stable PEMWE devices.
To unravel the role of enhanced electron density around active sites in boosting the HER activity, we comprehensively investigated the HER kinetics. The electrochemical impedance spectroscopy (EIS) spectrum at 10 mA cm−2 of HEA-QDs/C displays the smallest semicircle with the charge-transfer resistance (Rct) value of 4.18 Ω, presenting the fastest electron transfer and accelerated HER kinetics (Fig. S25, ESI†). The desorption of the underpotentially deposited hydrogen (Hupd) is an effective way to evaluate the strength of M–H.48 Notably, as shown in Fig. 3g, the oxidation peak of Hupd on HEA-QDs appears at 0.086 V, which is more negative than those of Pt/C (0.136 V), Ru/C (0.123 V), and PtRu/C (0.093 V), implying a weaker M–H bond energy (HBE) for HEA-QDs.49 The decreasing HBE is attributed to the enrichment of electron density around Pt/Ru sites, which can accelerate the coupling and release of protons, thereby promoting the kinetics of RDS (Tafel step).48 In addition, we further analyzed the reaction kinetics based on the Arrhenius and Eyring models. The Arrhenius plots which were tested in H2 saturated 0.5 M H2SO4 are presented in Fig. 3h and Fig. S26 (ESI†). The lower activation energy (Ea) of HEA-QDs/C (5.44 kJ mol−1), compared to that of commercial Pt/C (7.28 kJ mol−1), Ru/C (15.83 kJ mol−1) and PtRu/C (5.81 kJ mol−1), confirms the enhanced reaction kinetics of HEA-QDs.50–52 For a more comprehensive understanding, we also evaluated the Gibbs activation free energy (ΔG0) in the HER process, which considered the impact of temperature and the entropy of activation on the reaction barrier.53 As illustrated in Fig. 3i, the decreased ΔG0 for HEA-QDs/C (2.83 kJ mol−1) further underscores the superior reaction kinetics compared to Pt/C (4.79 kJ mol−1), Ru/C (13.21 kJ mol−1) and PtRu/C (3.19 kJ mol−1). Considering that the Tafel step is the RDS for HEA-QDs during the HER process, the enhanced activity can primarily originate from accelerated Tafel kinetics, i.e., the weakened adsorption of H* on catalytic surface facilitates its coupling and subsequent release. To verify the effect of HEA-QD loading on the Tafel kinetics, polarization curves were obtained and Tafel analysis of HEA-QDs/C with various loadings was performed (Fig. S27a–c, ESI†). The analogous onset potential, overpotential at 10 mA cm−2, and Tafel slope values indicate that the Tafel kinetics of HEA-QDs can be maintained across different loading amounts. Notably, we also analyzed the changes in the Tafel slope values with the average overpotential, where the Tafel slope values are obtained over a small potential region of 10 mV.54 As shown in Fig. S27d (ESI†), the Tafel slope values of HEA-QDs with different loading converge to 29 mV dec−1 in the kinetic region from 0 to 16 mV, further indicating that the Tafel kinetics can be maintained for HEA-QDs with different loading amounts. The continuously increasing Tafel slope in non-kinetic regions can be attributed to the mass transfer effects and bubble formation during the HER.54
Mechanism investigation
DFT calculations were conducted to further investigate the mechanisms for the enhanced HER performance. Through optimizing the structure of different models with a constant metallic atomic ratio based on the results of ICP-OES, we constructed a stable HEA-QD model with the lowest total energy (Fig. 4a and Fig. S28 and S29, ESI†). Notably, despite the subtle distortions after relaxation, the lattice around Pt/Ru sites remains highly stable, implying the structural stability of HEA-QDs during the electrocatalytic process (Fig. 4b). To elucidate the strong transboundary electron transfer mechanisms within HEA-QDs and the influence on adsorbed hydrogen, we conducted local electron density difference and Bader charge analysis. As shown in Fig. 4c, the concentrated electron density around Pt sites with the negative charge of −0.12 |e| can be directly viewed, along with a distinct charge delocalization between Pt and the surrounding metal atoms. Meanwhile, the Ru sites also show a strongly polarized charge redistribution, consistent with abundant electron accumulation on the Ru sites with the value of −0.16 |e| (Fig. 4d). Bader charge analysis in Fig. 4e further indicates the effective electron transfer from Fe, Co, Ni, and Mo to Pt and Ru sites, which aligns well with the XPS and XANE results. To explore the charge distribution in a real electrochemical environment, we analyzed the overall charge density differences between −0.1 V and 0 V (vs. SHE) on HEA-QDs after hydrogen adsorption at Pt/Ru sites.55,56 As shown in Fig. S30 (ESI†), at relatively high Fermi levels, both the catalytic surface and adsorbed proton carry partial negative charges. Concurrently, the depletion of electron density around Pt/Ru sites and the accumulation near the adsorbed proton indicate a partial electron transfer from Pt/Ru sites to the adsorbed hydrogen at −0.1 V. Additionally, we further calculated the charge density differences at −0.1 V (vs. SHE) before and after hydrogen adsorption on Pt/Ru sites (Fig. S31, ESI†). After proton adsorption, the hydrogen atoms adsorbed on Pt and Ru sites of HEA-QDs acquire electronic charges of −0.82 |e| and −0.80 |e|, respectively, which might form an electrostatic repulsion between the adsorbed protons and the catalytic surface, thereby weakening the hydrogen adsorption strength. Furthermore, the projected partial density of states (PDOSs) for various elements have been illustrated in Fig. 4f to demonstrate the detailed electronic structures of HEA-QDs. The significant overlap of d orbitals indicates the strong bonding interactions among various metallic elements, allowing efficient transboundary electron transfer among different atoms. Notably, the Pt 5d orbital occupies the deepest position near −4.7 eV to the Fermi level and the Ru 4d orbital exhibits a broad span ranging from −6.5 to +5.8 eV, which suggest that Pt and Ru atoms in HEA-QDs act as an effective electron reservoir and regulator, respectively, thus accelerating the electron transfer efficiency.47 For the HER process in acid, the adsorption strength of proton on the active sites determines the kinetics of the Tafel step, i.e. two adsorbed protons couple and release hydrogen. As shown in Fig. 4g, the s orbital of adsorbed proton on HEA-QDs shows an evident downshift compared to the free proton, which indicates that the electron-rich Pt/Ru sites transfer abundant active electrons to the adsorbed hydrogen and weaken the adsorption strength effectively. Moreover, the specifical ΔGH* of the HER for Pt/Ru/PtRu sites in HEA-QDs, pure Pt, pure Ru and PtRu alloy (Fig. S32, ESI†) were calculated, which is a reasonable and widely used descriptor for HER kinetics.57,58 As shown in Fig. 4h and Fig. S33 (ESI†), the absolute values of ΔGH* at the Pt (0.04 eV), Ru (0.14 eV) and PtRu (0.16 eV) sites in the HEA-QDs are lower than those in pure Pt (0.22 eV), pure Ru (0.37 eV) and PtRu alloy (0.11 eV for Pt sites, 0.22 eV for Ru sites). These results confirm that the enhanced electron density around Pt/Ru sites weakens the strength of Pt–/Ru–H to an optimum value, thus enabling a rapid H2 release process. | ||
| Fig. 4 Density functional theory calculations for HEA-QDs towards the HER. (a) The top view structural configuration after geometry optimizations. (b) The side view structural configuration and the real spatial contour plots of electronic distributions near the Fermi level. (c) The top and side views of local charge distribution on Pt sites. (d) The top and side views of local charge distribution on Ru sites. (e) The difference in electron amounts on surface atoms between HEA-QDs and pure metals based on Bader charge analysis. (f) The PDOSs of HEA-QDs and (g) the PDOSs for proton adsorption. (h) Calculated hydrogen-adsorption free energy (ΔGH*) profiles for Pt/Ru sites on HEA-QDs, pure Pt and pure Ru. (i) Tafel activation barriers of Pt sites on pure Pt and (j) on HEA-QDs. (k) Tafel activation barriers of Ru sites on pure Ru and (l) on HEA-QDs. | ||
To further understand the origin of the improved HER activity, we investigated the kinetics of the Tafel reaction, which has been proven to be the RDS in the HER process. As shown in Fig. 4i and j, the Pt sites on the Pt(111) surface require an activation barrier (ΔGTS) of 0.84 eV to reach the transition state, which is consistent well with Nørskov's work,59 while the Pt sites on the HEA-QDs(111) surface only need 0.80 eV to reach the same state, indicating accelerated release of two H*. Meanwhile, the desorption of H* at the Ru sites (0.42 eV) on the HEA-QDs(111) surface exhibits an obviously lower ΔGTS than that at Ru sites (0.53 eV) on pure Ru(001) surface (Fig. 4k and l), also suggesting the easier desorption of H*. To further elucidate the underlying mechanisms contributing to the reduced Tafel activation energy on HEA-QDs, we decomposed the Tafel activation barriers of Pt, Ru sites on HEA-QDs(111), Pt sites on Pt(111), and Ru sites on Ru(001) into the geometrical effect (Eg) and local electronic effect (Ee) based on the Hammer decomposition scheme.60,61 On the one hand, as shown in Table S2 and Fig. S34 (ESI†), the Tafel barriers for Pt sites on both Pt(111) and HEA-QDs(111) are primarily dominated by the geometrical effect (Eg), which might be attributed to the bonding competition or direct Pauli repulsion between H*1 and H*2.61 Additionally, the significant decrease in the Ee of Pt sites on HEA-QDs (−0.42 eV) compared to that on pure Pt (0.03 eV) is the primary reason for their lower ETS, which could be attributed to the weakened proton adsorption.61 On the other hand, for Ru sites on HEA-QDs(111) and Ru(001), the Eg is the main contributor to ETS. The reduction in Eg for Ru sites on HEA-QDs (0.53 eV) compared to pure Ru (0.79 eV) results in a lower ETS. The above results reveal that the Pt/Ru sites in HEA-QDs(111) possess faster Tafel kinetics compared to that of pure Pt(111) and pure Ru(001) surfaces, which can be attributed to the enriched electron density around Pt/Ru sites and weakened Pt–/Ru–H strength, resulting in superior HER activity.
Practical application in PEMWE
To evaluate its performance for practical application, the prepared HEA-QDs/C as a cathode catalyst was integrated into an actual PEMWE device with a low noble metal loading (Fig. 5a and Fig. S35, ESI†). The membrane electrode assemblies (MEAs) were prepared through CCM, and integrated with a cathode catalyst (HEA-QDs/C, 0.1 mgPtRu cm−2, commercial Pt/C, 0.1 or 0.3 mgPt cm−2, and commercial PtRu/C, 0.1 mgPtRu cm−2), an anode catalyst (commercial IrO2, 1.5 mg cm−2) and a Nafion 115 membrane (thickness of 125 μm). Field-emission scanning electron microscopy (FESEM) images in Fig. 5b display the cross-sectional morphology of the MEAs, confirming that the cathode/anode catalysts with a thickness of 2.1 μm/4.8 μm, respectively, are the uniform agglomerates attached to the PEM. The corresponding EDS-mappings in Fig. 5c demonstrate the uniform distribution of IrO2 and other multimetallic elements within HEA-QDs/C, ensuring equal access to active sites and efficient charge transfer. The steady-state polarization curve of the PEMWE device using HEA-QDs/C (0.1 mgPtRu cm−2) as the cathode electrocatalyst (Fig. 5d and Fig. S36 and S37, ESI†) reveals a cell voltage of 1.65 V for 1.0 A cm−2, while a high voltage is needed for MEAs with Pt/C (1.71 V) and PtRu/C (1.71 V) cathode catalysts under the same noble metal loading of 0.1 mgPtRu cm−2. Notably, the HEA-QDs/C cathode cell even exhibits a comparable activity to that of Pt/C with a high noble metal loading of 0.3 mgPt cm−2. As shown in Table S3 (ESI†), the HEA-QDs/C demonstrates competitive performance compared with not only commercial Pt/C and PtRu/C but also the other advanced Pt/Ru-based cathodic electrocatalysts reported recently. Moreover, as shown in Fig. S38 (ESI†), the faradaic efficiency of H2 generation for HEA-QDs/C cathode cell is 98.5 ± 0.3%, confirming that the applied current almost contributes to hydrogen production. The obvious superiority of HEA-QDs/C in PEMWE devices can bring out the reduction of noble metal usage and power consumption.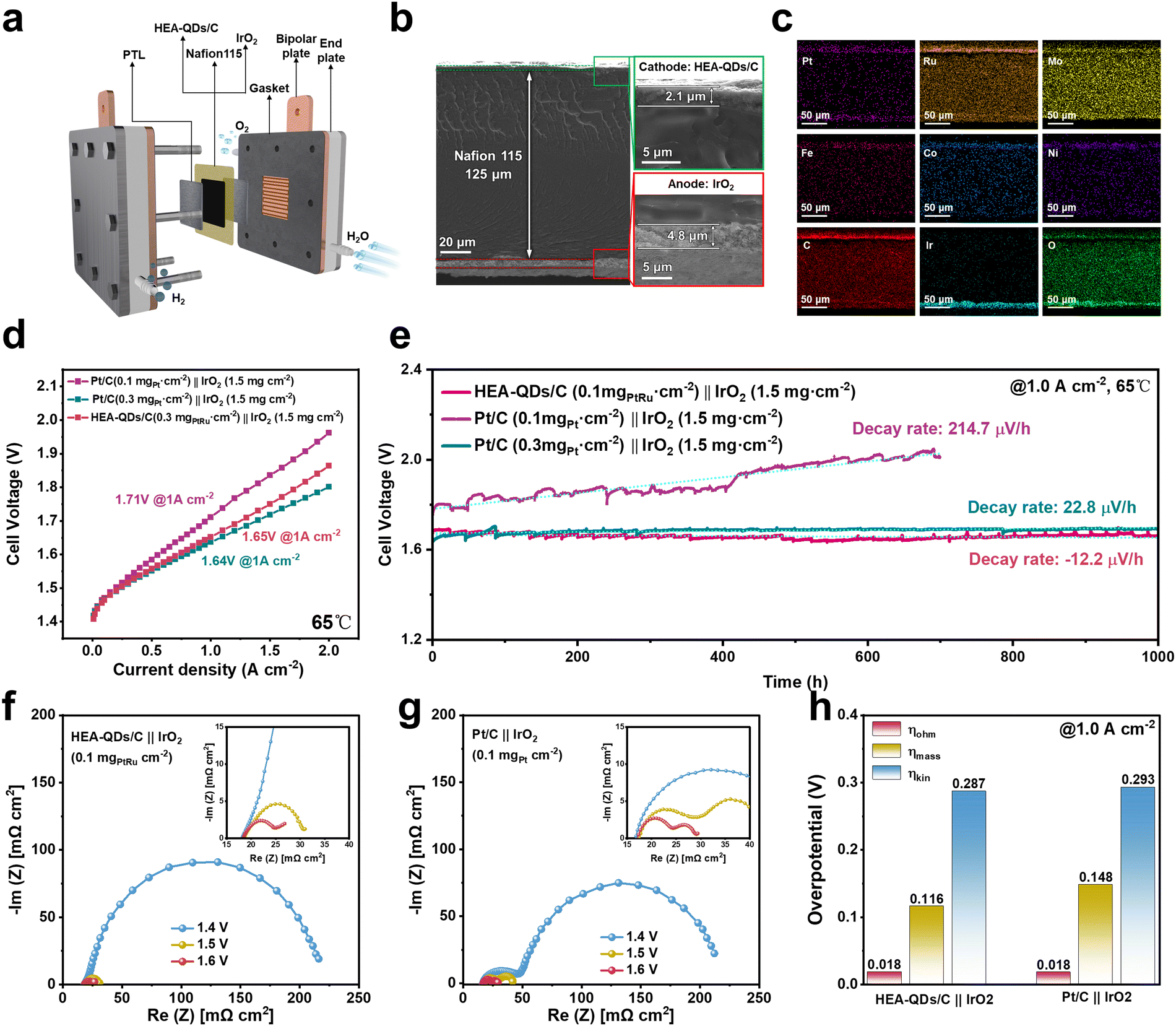 | ||
| Fig. 5 The performance of PEMWE devices. (a) Stack structure and key materials of a PEMWE device. (b) Cross-sectional SEM images of MEAs employing the HEA-QDs/C cathode layer and IrO2 anode layer. (c) SEM-EDS mapping images of MEA. (d) Polarization curves of PEMWE at 65 °C with a Nafion 115 membrane. (e) Chronopotentiometry curve of PEMWE devices operated at 1.0 A cm−2. (f) and (g) EIS curves of PEMWE devices using HEA-QDs/C and Pt/C cathodes. (h) ηkin, ηOhm, and ηmass of PEMWE devices at a current density of 1.0 A cm−2. | ||
Additionally, we also evaluated the performance of PEMWE devices with various loadings of IrO2 (0.1, 1.0, 1.5, and 2.0 mg cm−2) on the anode while maintaining a constant loading of HEA-QDs/C (0.1 mgPtRu cm−2) on the cathode. The results in Fig. S39 (ESI†) indicate that higher IrO2 loadings can effectively reduce the cell voltage of PEMWE devices. However, when the IrO2 loading exceeds 1.0 mg cm−2, the reduction in cell voltage at 1.0 A cm−2 is not as remarkable. Due to the significant impact of IrO2 loading on PEMWE stability, we chose an IrO2 loading of 1.5 mg cm−2 in this work to accurately assess the stability of HEA-QDs/C as a cathode catalyst in PEMWE devices.62
To evaluate the stability of the PEMWE device with different cathode catalysts, stability tests were performed at a constant current density of 1.0 A cm−2. As shown in Fig. 5e, the Pt/C cathode cell with an ultra-low loading of 0.1 mgPt cm−2 exhibits a significant decay with the rate of 214.7 μV h−1, while the cell with high Pt loading of 0.3 mgPt cm−2 shows a much lower decay with the rate of 22.8 μV h−1. Notably, even with an ultra-low total noble loading of 0.1 mgPtRu cm−2, the HEA-QDs/C cathode cell can operate stably at 1.0 A cm−2 for more than 1000 h with a surprising decay rate of −12.2 μV h−1. The electrolysis voltage exhibits a decline during 0–500 h, which results from the partial dissolution of surface transition metals exposing more active sites. During the 500–1000 h testing, the cell voltage slowly increases at a quite low rate. Moreover, the evaluated energy consumption of the HEA-QDs/C cathode cell is 3.98 kWh N m−3 H2 at 1.0 A cm−2, which is lower than that of the reported commercial PEM electrolyzer (4.5–5.0 kWh N m−3 H2).63 According to the calculation method from the US Department of Energy (DOE), the estimated cost of this PEMWE device is US$0.88 per kg H2, which is lower than the 2030 DOE target of US$1.0 per kg of H2.64 The unexceptionable activity and stability demonstrate the promising potential of HEA-QDs as a practical cathode catalyst for real PEMWE applications.
To deduce the origin of enhanced performance, EIS experiments of PEMWE devices using HEA-QDs/C and Pt/C as cathode catalysts were carried out (Fig. 5f and g). Additionally, we fit the EIS Nyquist plots using a typical equivalent electrical circuit model in Fig. S40a (ESI†). The results reveal that both PEMWE devices have the same ohmic resistance (Rs) and anode activation resistance (Raa) while the PEMWE device using the HEA-QDs/C catalyst exhibits a lower cathode activation resistance (Rca) compared to that of the Pt/C catalyst, which confirms the accuracy of performance comparison and the fast cathode reaction kinetics of HEA-QDs/C (Fig. S40b, ESI†). Furthermore, the overvoltage of kinetic (ηkin), ohmic (ηohm) and mass transport (ηmass) at a current density of 1.0 A cm−2 are calculated to clarify the performance difference (Fig. S41, ESI†). As shown in Fig. 5h, the decreasing ηkin and ηohm compared to those of the Pt/C cathode cell demonstrate the superiority of the HEA-QDs/C cathode cell in accelerating the reaction kinetics and mass transfer, respectively.
Conclusion
In conclusion, uniformly ultrasmall PtRuMoFeCoNi HEA-QDs were synthesized by simple rapid microwave radiation. As revealed by XAFS and XPS, the significant transboundary electron transfer from Fe, Co, Ni, and Mo to the Pt/Ru caused by the differences in electronegativity concentrated the local electron density around Pt/Ru sites. Comparative experiments and DFT calculations demonstrated that the enhanced local electron density could optimize the adsorption energy of Pt/Ru–H, thus significantly improving the Tafel kinetics. As a result, the HEA-QDs exhibit superior electrocatalytic activity (11 mV@10 mA cm−2) compared to Pt/C (18 mV@10 mA cm−2) for the HER in acidic solution. Impressively, even with an ultra-low loading of 100 μgPtRu cm−2 in the cathode, the PEMWE device integrated with HEA-QDs as a cathodic catalyst not only exhibits excellent catalytic performance (1.65 V@1.0 A cm−2), but also displays outstanding stability at 1.0 A cm−2, even with a reduced voltage decay rate of −12.2 μV h−1 over 1000 h. This work not only enriches the comprehension of catalytic kinetics in acidic media but also stimulates the practical applications of PEMWE devices with low noble metal usage.Data availability
The data supporting the findings of this study are available within the article and the ESI.† The DFT datasets are available in the ioChem-BD repository with the DOI of DOI: https://doi.org/10.19061/iochem-bd-6-388.65 All other relevant source data are available from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the National Key Research Program (2022YFB4002000), the National Natural Science Foundation of China (22002184, 21533005, 216673275, and 21573107), the Shanghai Science and Technology Innovation Action Plan (20dz1205400, 23ZR1471000 and 22511102500), the Youth Innovation Promotion Association CAS (2021287) and the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF).References
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, L.-W. Wang and D.-J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- L. Deng, S. F. Hung, Z. Y. Lin, Y. Zhang, C. Zhang, Y. Hao, S. Liu, C. H. Kuo, H. Y. Chen, J. Peng, J. Wang and S. Peng, Adv. Mater., 2023, e2305939, DOI:10.1002/adma.202305939.
- J. K. Lee, G. Anderson, A. W. Tricker, F. Babbe, A. Madan, D. A. Cullen, J. D. Arregui-Mena, N. Danilovic, R. Mukundan, A. Z. Weber and X. Peng, Nat. Commun., 2023, 14, 4592 CrossRef CAS PubMed.
- S. Liu, H. Tan, Y. C. Huang, Q. Zhang, H. Lin, L. Li, Z. Hu, W. H. Huang, C. W. Pao, J. F. Lee, Q. Kong, Q. Shao, Y. Xu and X. Huang, Adv. Mater., 2023, e2305659, DOI:10.1002/adma.202305659.
- Y. Wang, M. Zhang, Z. Kang, L. Shi, Y. Shen, B. Tian, Y. Zou, H. Chen and X. Zou, Nat. Commun., 2023, 14, 5119 CrossRef CAS.
- Z. W. Chen, J. Li, P. Ou, J. E. Huang, Z. Wen, L. Chen, X. Yao, G. Cai, C. C. Yang, C. V. Singh and Q. Jiang, Nat. Commun., 2024, 15, 359 CrossRef CAS PubMed.
- I. T. McCrum and M. T. M. Koper, Nat. Energy, 2020, 5, 891–899 CrossRef CAS.
- Y. Li, S. Wang, Y. Hu, X. Zhou, M. Zhang, X. Jia, Y. Yang, B.-L. Lin and G. Chen, J. Mater. Chem. A, 2022, 10, 5273–5279 RSC.
- K. Li, Y. Li, Y. Wang, J. Ge, C. Liu and W. Xing, Energy Environ. Sci., 2018, 11, 1232–1239 RSC.
- S. Wang, X. Gao, X. Hang, X. Zhu, H. Han, W. Liao and W. Chen, J. Am. Chem. Soc., 2016, 138, 16236–16239 CrossRef CAS PubMed.
- F. Zhang, Y. Zhu, Y. Chen, Y. Lu, Q. Lin, L. Zhang, S. Tao, X. Zhang and H. Wang, J. Mater. Chem. A, 2020, 8, 12810–12820 RSC.
- Y. Zhou, Z. Xie, J. Jiang, J. Wang, X. Song, Q. He, W. Ding and Z. Wei, Nat. Catal., 2020, 3, 454–462 CrossRef CAS.
- J. Xu, T. Liu, J. Li, B. Li, Y. Liu, B. Zhang, D. Xiong, I. Amorim, W. Li and L. Liu, Energy Environ. Sci., 2018, 11, 1819–1827 RSC.
- T. Loffler, A. Ludwig, J. Rossmeisl and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 26894–26903 CrossRef CAS PubMed.
- D. Kobayashi, H. Kobayashi, D. Wu, S. Okazoe, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota, S. M. Aspera, H. Nakanishi, S. Arai and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 17250–17254 CrossRef CAS PubMed.
- W. Jin, H. Wu, W. Cai, B. Jia, M. Batmunkh, Z. Wu and T. Ma, Chem. Eng. J., 2021, 426, 130762 CrossRef CAS.
- X. Zuo, R. Yan, L. Zhao, Y. Long, L. Shi, Q. Cheng, D. Liu and C. Hu, J. Mater. Chem. A, 2022, 10, 14857–14865 RSC.
- Y. Zhang, D. Wang and S. Wang, Small, 2022, 18, e2104339 CrossRef PubMed.
- D. B. Miracle and O. N. Senkov, Acta Mater., 2017, 122, 448–511 CrossRef CAS.
- J. Kwon, S. Sun, S. Choi, K. Lee, S. Jo, K. Park, Y. K. Kim, H. B. Park, H. Y. Park, J. H. Jang, H. Han, U. Paik and T. Song, Adv. Mater., 2023, 35, e2300091 CrossRef PubMed.
- J. Hao, Z. Zhuang, K. Cao, G. Gao, C. Wang, F. Lai, S. Lu, P. Ma, W. Dong, T. Liu, M. Du and H. Zhu, Nat. Commun., 2022, 13, 2662 CrossRef CAS PubMed.
- D. Zhang, Y. Shi, H. Zhao, W. Qi, X. Chen, T. Zhan, S. Li, B. Yang, M. Sun, J. Lai, B. Huang and L. Wang, J. Mater. Chem. A, 2021, 9, 889–893 RSC.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- Y. Wang, N. Gong, H. Liu, W. Ma, K. Hippalgaonkar, Z. Liu and Y. Huang, Adv. Mater., 2023, 35, e2302067 CrossRef PubMed.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, I. Gueye, O. Seo, J. Kim, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2020, 11, 12731–12736 RSC.
- R. He, L. Yang, Y. Zhang, D. Jiang, S. Lee, S. Horta, Z. Liang, X. Lu, A. O. Moghaddam, J. Li, M. Ibanez, Y. Xu, Y. Zhou and A. Cabot, Adv. Mater., 2023, e2303719, DOI:10.1002/adma.202303719.
- Y. Chen, X. Zhan, S. L. A. Bueno, I. H. Shafei, H. M. Ashberry, K. Chatterjee, L. Xu, Y. Tang and S. E. Skrabalak, Nanoscale Horiz., 2021, 6, 231–237 RSC.
- H. Minamihara, K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, L. S. R. Kumara, K. Ohara, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2022, 144, 11525–11529 CrossRef CAS PubMed.
- H. Zhu, S. Sun, J. Hao, Z. Zhuang, S. Zhang, T. Wang, Q. Kang, S. Lu, X. Wang, F. Lai, T. Liu, G. Gao, M. Du and D. Wang, Energy Environ. Sci., 2023, 16, 619–628 RSC.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob and H. Xie, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed.
- S. Gao, S. Hao, Z. Huang, Y. Yuan, S. Han, L. Lei, X. Zhang, R. Shahbazian-Yassar and J. Lu, Nat. Commun., 2020, 11, 2016 CrossRef CAS PubMed.
- Y. Li, Y. Liao, L. Ji, C. Hu, Z. Zhang, Z. Zhang, R. Zhao, H. Rong, G. Qin and X. Zhang, Small, 2022, 18, e2107265 CrossRef PubMed.
- Z. Jin, J. Lv, H. Jia, W. Liu, H. Li, Z. Chen, X. Lin, G. Xie, X. Liu, S. Sun and H. J. Qiu, Small, 2019, 15, e1904180 CrossRef PubMed.
- M. W. Glasscott, A. D. Pendergast, S. Goines, A. R. Bishop, A. T. Hoang, C. Renault and J. E. Dick, Nat. Commun., 2019, 10, 2650 CrossRef PubMed.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chemistry, 2005, 11, 440–452 CrossRef CAS PubMed.
- M. Baghbanzadeh, L. Carbone, P. D. Cozzoli and C. O. Kappe, Angew. Chem., Int. Ed., 2011, 50, 11312–11359 CrossRef CAS PubMed.
- X. Chang, M. Zeng, K. Liu and L. Fu, Adv. Mater., 2020, 32, e1907226 CrossRef PubMed.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef CAS PubMed.
- B. Hammer and J. K. Norskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- C. Wei, Y. Sun, G. G. Scherer, A. C. Fisher, M. Sherburne, J. W. Ager and Z. J. Xu, J. Am. Chem. Soc., 2020, 142, 7765–7775 CrossRef CAS PubMed.
- C. Zhang, R. Lu, C. Liu, J. Lu, Y. Zou, L. Yuan, J. Wang, G. Wang, Y. Zhao and C. Yu, Adv. Sci., 2022, 9, e2104768 CrossRef PubMed.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, e1808066 CrossRef PubMed.
- H.-X. Wang, W. L. Toh, B. Y. Tang and Y. Surendranath, Nat. Catal., 2023, 6, 351–362 CrossRef CAS.
- C. Zhan, Y. Xu, L. Bu, H. Zhu, Y. Feng, T. Yang, Y. Zhang, Z. Yang, B. Huang, Q. Shao and X. Huang, Nat. Commun., 2021, 12, 6261 CrossRef CAS PubMed.
- T. Tang, X. Liu, X. Luo, Z. Xue, H. R. Pan, J. Fu, Z. C. Yao, Z. Jiang, Z. H. Lyu, L. Zheng, D. Su, J. N. Zhang, L. Zhang and J. S. Hu, J. Am. Chem. Soc., 2023, 145, 13805–13815 CrossRef CAS PubMed.
- L. Wang, Z. Xu, C. H. Kuo, J. Peng, F. Hu, L. Li, H. Y. Chen, J. Wang and S. Peng, Angew. Chem., Int. Ed., 2023, e202311937, DOI:10.1002/anie.202311937.
- X. Shi, J. Xie, J. Wang, S. Xie, Z. Yang and X. Lu, Nat. Commun., 2024, 15, 302 CrossRef CAS PubMed.
- M. Wu, X. Wang, F. Zhang, Q. Xiang, Y. Li and J. Guo, Energy Environ. Sci., 2024, 17, 619–629 RSC.
- S. Yang, Z. Zhang, A. M. Oliveira, S. Xi, M. Zhiani, J. Zhang, Z. Tu, F. Xiao, S. Wang, Y. Yan and J. Xiao, Adv. Funct. Mater., 2024, 34, 2313275 CrossRef CAS.
- G. Gupta, D. Choudhury, R. Maurya, S. Sharma and M. Neergat, J. Phys. Chem. C, 2023, 127, 23566–23576 CrossRef CAS.
- O. van der Heijden, S. Park, J. J. J. Eggebeen and M. T. M. Koper, Angew. Chem., Int. Ed., 2023, 62, e202216477 CrossRef CAS PubMed.
- K. Chan and J. K. Norskov, J. Phys. Chem. Lett., 2016, 7, 1686–1690 CrossRef CAS PubMed.
- K. Chan and J. K. Norskov, J. Phys. Chem. Lett., 2015, 6, 2663–2668 CrossRef CAS PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- Z. Li, J.-Y. Fu, Y. Feng, C.-K. Dong, H. Liu and X.-W. Du, Nat. Catal., 2019, 2, 1107–1114 CrossRef CAS.
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. D. Gudmundsdo’ttir and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef.
- B. Hammer, Phys. Rev. Lett., 1999, 83, 3681–3684 CrossRef CAS.
- Z.-P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS PubMed.
- Y. Wang, M. Zhang, Z. Kang, L. Shi, Y. Shen, B. Tian, Y. Zou, H. Chen and X. Zou, Nat. Commun., 2023, 14, 5119 CrossRef CAS PubMed.
- J. Xu, H. Jin, T. Lu, J. Li and Y. Liu, Sci. Adv., 2023, 9, eadh1718 CrossRef CAS PubMed.
- L. Yao, F. Zhang, S. Yang, H. Zhang, Y. Li, C. Yang, H. Yang and Q. Cheng, Adv. Mater., 2024, e2314049, DOI:10.1002/adma.202314049.
- M. Alvarez-Moreno, C. de Graaf, N. Lopez, F. Maseras, J. M. Poblet and C. Bo, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee01825g |
| This journal is © The Royal Society of Chemistry 2024 |