
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExponentially enhanced photocatalytic alkane production from biomass-derived fatty acid decarboxylation via self-heating-induced conformational inversion†
Qiang
Huang‡
ad,
Chunlin
Hao‡
b,
Guibao
Guo
*b,
Hongwei
Ji
ad,
Shengli
An
*c,
Wanhong
Ma
 *ad and
Jincai
Zhao
ad
*ad and
Jincai
Zhao
ad
aKey Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: whma@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, Inner Mongolia University of Science & Technology, Baotou, 014010, P. R. China
cInner Mongolia Key Laboratory of Advanced Ceramic Materials and Devices, Inner Mongolia University of Science and Technology, Baotou, 014010, P. R. China
dUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 28th August 2024
Abstract
Heating is the most convenient means to enable unfavourable chemical reactions to upscale their application, but it is rarely used in photocatalysis because its driving force has always been identified as the intensity of excitable light and charge-separation efficiency, both of which are temperature-independent and negatively correlated. Herein, we report that the heat from relaxed incident light could significantly promote the conversion of long-chain fatty acids to alkanes with one less carbon atom (Cn−1 alkanes) mediated by TiO2 photocatalysis for bio-fuel preparation. Employing high-boiling-point solvents could maximize the reaction temperature and allow the relaxed heat to accumulate within substrate molecules with amphiphilic groups as bond strain is made available by deforming the long C-chain onto the surface of TiO2. In this way, photocatalysis could run at high temperatures (∼254 °C) and under very concentrated sunlight (∼26 W cm−2), with its scale upgraded from the traditional far less than mmol L−1 level to the unprecedented mol L−1 level for a single output.
Broader contextImproving the efficiency of photocatalytic reactions has been a long-sought goal, but it is generally considered that it is weakly temperature-dependent and therefore cannot be significantly improved by heating. At present, improvement methods focus on modified catalysts. Therefore, the present study reports a simple system that allows the output concentration of the photocatalytic long-chain fatty acid decarboxylation reaction to be upgraded beyond the mM limit to the M level by the self-heating effect of the photocatalyst with unmodified TiO2, which can also be performed under focused outdoor sunlight. Owing to the conformational change in the fatty acid adsorbed on the catalyst at high temperature, photoinduced electron–hole pairs changed the direction of attacking fatty acid molecules, which led to an increase in reaction temperature dependence. |
Introduction
Decarboxylation is a basic reaction in organic synthesis, which can be used to produce fine chemicals and for other applications, especially the conversion of waste biomass-derived fatty acids into high-value-added biofuels.1,2 In China, about ten million tons of biodiesel are produced from fatty acids via the catalytic decarboxylation process.3,4 Although fatty acids stem from non-fossil biomass, decarboxylation operation commonly requires a lot of fossil energy to power a temperature of 300–400 °C, ∼20 MPa H2 and transition metal-based catalysts.5 Instead, solar energy-powered photocatalysis has the potential to allow this conversion of fatty acids into bio-fuel via a safer method with lower environmental and production costs. Photocatalytic technologies can utilize sunlight as a driving force to produce electron–hole pairs with redox capability and then catalyse the transformation of many important chemical reactions.6–14 However, after decades of research, photocatalysis examples involving solid/liquid heterogenous reactions that can replace existing traditional processes are very rare because of their generalized poor single output concentration (1–102 μM). One of the main reasons for this is that for almost all photocatalytic transformations that are available to date, their turnovers cannot be increased linearly with the increase in incident light intensity due to the commonly shared severe self-quenching effect of excited states under high-intensity irradiation.15,16 Significantly differing from thermal catalysis that enables mass production via the input of considerable heat energy, photocatalysis generally fails to directly store and re-use relaxed photons. Even for downhill decarboxylation reactions in solutions,17 the situation has not been fundamentally transformed. In the early 1970s, Bard and co-workers found that short-chain fatty acids could be decarboxylated into Cn−1 alkanes over TiO2 powder under ultraviolet light (≤380 nm),18 the so-called photo-Kolbe reaction, but the yields of alkanes were at a trace concentration level (nM). Since then, some work has been conducted to improve this, and although an algal photoenzyme with quantum efficiencies of up to 80% has been reported19–23 and related industrialization strategies have been attempted,24 breakthrough progress in decarboxylation efficiency targeted at practical scale production is still lacking.20–22,24–30For a photocatalytic system that relies solely on solar energy to achieve large-scale production, in theory, it at least requires that the catalyst must cover most of the solar broadband spectrum and is able to be effectively excited. However, for any real photocatalyst, only a limited range of sunlight with frequencies equal to or beyond the energy of the band gap (Eg) is available for reactions. The majority of the incident sunlight below Eg has to be converted to heat dissipation through vibrational–rotational excitation of the solvent, reactant, catalysts and even the reactor's wall. Unfortunately, the heat formed from excitation–relaxation, inner conversion (IC) of vibrational–rotational excitation, or separated electron–hole pairs recombination is hardly reused in the photocatalysis itself, since there is a lack of the reverse IC or S1 → S0 mechanism for common photocatalytic materials, unlike thermal activation delayed fluorescence (TADF), where reverse intersystem crossing (rISC) from long-life triplet excitation states is available.31 This is generally true for most photocatalytic reactions no matter how high the temperature is raised, where the efficiency is even lowered, since high temperatures accelerate the recombination of separated electron–hole pairs too.15,32,33 Plasma catalysis was revealed to be more efficient under high light intensity and temperature,34,35 but it is limited to the use of a lot of noble metals. Until recently, there have been few works on the up-hill reactions reconsidering the elusive effect of heating on the heterogeneous photocatalytic redox and separated electron–hole recombination. For example, Zhou et al. reported that the solar-to-hydrogen (STH) process in solar water-splitting could be significantly enhanced when the water temperature was raised from room temperature to 60–70 °C.36Also, our group work revealed the photocatalytic hydrogenated dehalogenation of polybrominated diphenyl ethers (PBDEs) on In2O3 by H2 could be smoothly carried out only when the system temperature was at more than 140 °C,37 whereas when alcohol was used as a hole sacrificial agent the dehalogenation at room temperature was the nearly same rate as that at 140 °C.38 From these examples, we intuitively deem that these photocatalytic conversions initiated by photoinduced holes at the valence band (hvb+), once they become the rate-determining step, would exhibit a different dependence between the reaction efficiency and temperature. We previously reported some hvb+-initiated reactions that were indeed able to become the rate-determining step.39 However, what types of photocatalytic reactions could use the relaxed light energies for lifting the reaction efficiency and what universal mechanism it depends on are little understood at present. More specifically, whether the manufacture of biofuels from fatty acids by TiO2-based photocatalytic decarboxylation could significantly accelerate and achieve a high substrate-concentration conversion through self-heating from incident sunlight remains unknown.
In this work, through detailed investigation of the TiO2-based photocatalytic conversion of long-chain fatty acids into Cn−1 alkanes in solvents with different boiling points only by using the relaxed heat from incident 410 nm light to boil the system, we aimed to show that such a decarboxylation of long-chain fatty acids involves such a thermally activated photocatalytic transformation. The fatty acid decarboxylation output per single operation reached the mol L−1 level and the output efficiency at high concentration increased linearly with the intensity of the incident light. Long-chain fatty acids from C14 to C20 could all be smoothly converted into the corresponding Cn−1 alkanes in a range of 80–95% yields under the irradiation of a 768 W LED and a strong focus on the outside sunlight (∼26 W cm−2), respectively. Detailed mechanism investigation indicated that, through forcing the upright long-chains of the fatty acids down on the TiO2 surface with the self-heating means from the null photon energy for energy storage, could strain the C–COOH bonds of the fatty acid to become close to react with the separated electrons–holes more effective to realize decarboxylation. Our results indicated that the exhaust incident light energy was able to be used again to further assist the promotion of decarboxylation promotion.
Results and discussion
Photocatalytic stearic acid decarboxylation enabled by a photothermal conversion effect
We selected stearic acid as a model substrate to investigate photocatalytic decarboxylation in a photoreactor equipped with an air-cooled return pipe, in which n-tetradecane as the solvent, N2 atmosphere at normal pressure, and the well-known P-25 TiO2 as the photocatalyst made up our typical acid reaction system (Fig. S1, ESI†). Significantly different from small molecular carboxyl acids, such as acetic acid and propionic acid, the stearic acid adsorbed on the TiO2 surface extended the TiO2 catalyst absorption edge from ∼380 nm to ∼420 nm, close to the visible region (Fig. 1a). This is mainly because the long fatty carbon chain hanging from the polar carboxyl is more favourable for delocalization of the conduction band electrons of TiO2 coordinated with carboxylate than that of short-chain acids, which results in lowering the potential of the conduction band of the TiO2, thereby leading to a redshift of the absorption band edge of TiO2.40 Also, the diffuse reflectance FTIR (DRIFT) spectra (Fig. 1b) indicated that stearic acid was connected to the surface of TiO2via an O,O′–bidentate coordination bond at room temperature (referring to the latter, see Fig. 5). Accordingly, a blue LED array light source (410 nm; the wavelength distribution is shown in Fig. S1c, ESI†) with high power (total power, 768 W, intensity, 10.25 W cm−2) was selected as an excitation light source to excite the TiO2–C18H36O2 complex for photocatalytic decarboxylation. Upon the irradiation, the 0.05 M stearic acid in n-tetradecane solvent was rapidly converted into the main product n-heptadecane in a 95.1% yield and 95.4% selectivity in 0.5 h (Fig. 2a). In control experiments, the stearic acid was thermally stable and was not converted under light irradiation alone without a catalyst or in the case with a catalyst but heated up to 254 °C by an external heat source without light irradiation. The main Cn−1 product formation was consistent with previously reported photocatalytic transformations of small molecular fatty acids and even long-chain ones under specially controlled room temperature conditions,27 but the rate of decarboxylation was increased by dozens of times. As expected, without any insulation measures for our photoreactor, the temperature of the reaction suspending solution (25 mL) rose very rapidly and even reached the boiling point (254 °C) of the n-tetradecane solvent to drive the reflux reaction after 6 min light irradiation (Fig. 2b).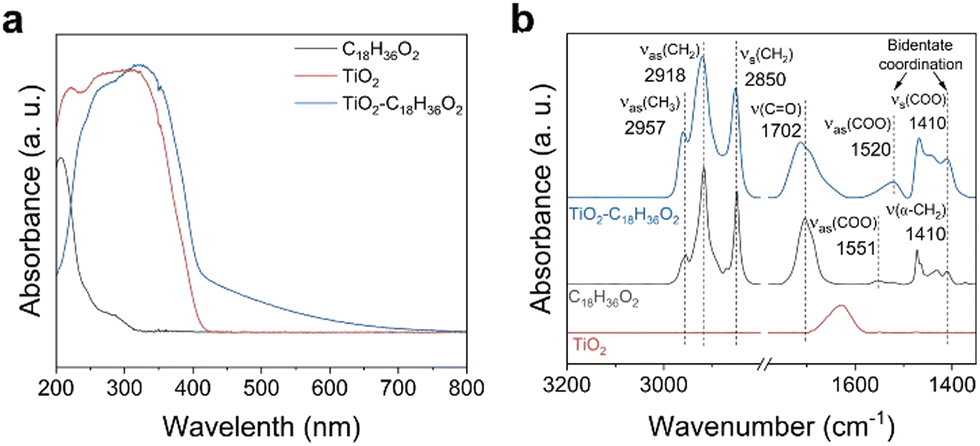 | ||
| Fig. 1 Adsorption of stearic acid on TiO2. (a) UV-visible absorption spectra. (b) Diffuse reflectance FTIR spectra obtained at room temperature. | ||
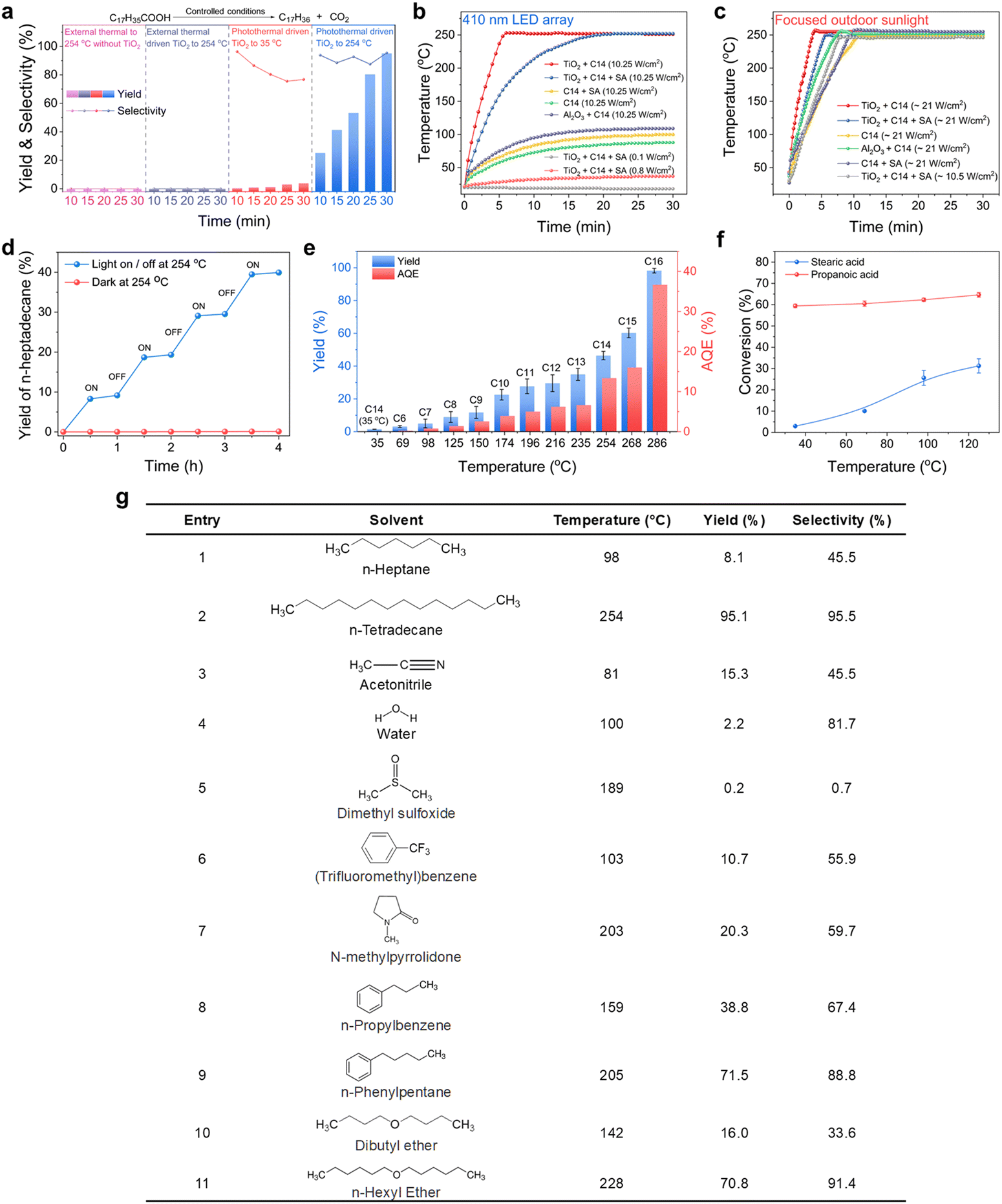 | ||
| Fig. 2 Photocatalytic decarboxylation of stearic acid over TiO2 at solvent-locked boiling temperature. (a) Comparison of the yield and selectivity for the decarboxylation of stearic acid under different conditions. (b) Temperature increment for different reaction media under the same LED light irradiation and (c) under focused outdoor sunlight. (d) Alternating on and off light experiments for the decarboxylation of stearic acid at solvent-locked temperature (n-tetradecane, 254 °C) and full-course dark reaction at 254 °C maintained by an external heat source without light irradiation. (e) Yields and AQEs for the photocatalytic decarboxylation of stearic acid at different boiling points with a range of n-alkanes acting as the solvent. (f) Promotion difference by the self-heating effect between stearic acid and propanoic acids (from 35 °C to 125 °C). (g) TiO2-based photothermal catalytic decarboxylation of stearic acid in some solvents with different polarities and boiling points. General reaction conditions: reaction solution volume (25 mL), TiO2 (125 mg), stearic acid (0.05 M), n-octadecane (internal standard, 0.05 M), reaction pressure (0.1 MPa), solvents locked reaction temperature, N2 atmosphere, LED (410 nm, 10.25 W cm−2) irradiation for 15 min in (e) and for 30 min in (g). All the catalytic performance data in the figures are averages of at least three experiments. | ||
Obviously, this was a typical photo-to-heat event since TiO2 is an excellent material with a good photothermal conversion effect.41 The photothermal conversion efficiency η under solid circumstances was 42.99% under sunlight and we measured η to be about 3.89% under the solution conditions with 410 nm light irradiation (for the detailed measurement method see the ESI†). As a control, an Al2O3/C14 solvent suspending system under identical irradiation conditions was tested and showed a slightly higher temperature rise (up to about 100 °C) than the pure solvent case, but significantly lower than that of the TiO2/C14 solvent case (∼254 °C) (Fig. 2b). This proved that TiO2 had a superior photothermal conversion effect than Al2O3 under incident light with a wavelength greater than 410 nm. All the systems were allowed to reach boiling reflux while exposed to highly focused outdoor sunlight (Fig. 2c). Furthermore, the photo-to-heat efficiency in this system exponentially decayed with the intensity of incident light in n-tetradecane solvent (Fig. S2c, ESI†). This is one of the main reasons why the TiO2-based photothermal conversion effect was often overlooked in the past while the intensity of incident light was controlled as very low. The decarboxylation system was discontinuously externally heated to boiling at 254 °C. A significant increase in n-heptadecane yield was detected only under light exposure, suggesting that the reaction involved thermal-assisted photocatalysis rather than photo-driven thermocatalysis and that no induction period existed (Fig. 2d). The slight increase in n-heptadecane yield after switching off the light was attributed to the continued conversion of the intermediates accumulated during the light period (Fig. 2d). Initially, we followed the conventional theory and believed that the heat created from incident light irradiation would not provide an additional benefit to this target transformation. To prove the point, we carried out the same photocatalytic reaction in n-tetradecane solvent but controlled the reaction temperature close to ambient temperature (35 °C) through strong wind-cooling. However, there was very little conversion of stearic acid and little product n-heptadecane formation under the same high-power irradiation (see Fig. 2a, 3rd column). This seemed to be a serious violation of the previous photocatalytic basis of the dependency relationship between the temperature and the reaction rate.33 We speculated that this anomalous relationship was due to accelerated mass transfer due to the boiling behaviours of the solvent accelerating the mass transfer of the substrate into the catalyst surface. Therefore, other n-alkanes with boiling points below and above that of n-tetradecane were tested for the same photocatalytic conversion of stearic acid. Fig. 2e clearly shows that although these n-alkane solvents all could keep the reaction system boiling and refluxing, the yield of n-heptadecane, without exception, increased with the increase in the boiling point of the solvents. That is, without changing the intensity of incident light at all, the higher the boiling point of the solvent, the greater the resultant yield. This adequately evidenced that it was the high reaction temperature rather than the boiling itself that accelerated the decarboxylation of stearic acid. The light excitation in our system should be divided into two roles: (i) successful excitation used to produce ecb−/hvb+ pairs for launching redox events; (ii) invalid excitation that goes to generating heat for in situ flattening the upright long-chains of stearic acid to moved them close to the TiO2 surface so as to drive ecb−/hvb+ decarboxylation (see the later description). Their respective contributions to the overall reaction efficiency were furthermore quantified. We determined the corresponding apparent quantum efficiency (AQE) under different solvent conditions (Fig. 2e) and found the AQE that was ∼0.08% at 35 °C was increased to 36.67% at 286 °C with the n-hexadecane solvent boiling. Considering the decarboxylation reaction at forced locking near room temperature (35 °C) as a conventional photocatalytic reaction, the AQE value means that 0.08% of the light absorbed by the system was exploited in TiO2 to generate reactive ecb−/hvb+ pairs. When the reaction temperature was locked by the solvent to 286 °C, the 36.7% AQE means that 36.6% (i.e. 36.7% – 0.08%) of the light absorbed by the system was converted to effective heat (to force the C–COOH bond of stearic acid to come close to the hvb+ sites for oxidation), by which the AQE had increased by nearly 500 times. All of the results demonstrated the decarboxylation of stearic acid was photocatalyzed and could be significantly accelerated by the self-heating originating from incident light irradiation. Instead, when small molecule acids (e.g. propionic acid) were used as a substrate to examine this self-heating effect on promoting photocatalyzed decarboxylation, no obvious acceleration occurred with control of the temperature from 35 °C to 125 °C (ensuring that propionic acid would not escape into the gas phase, from where it could not completely return) (Fig. 2f), which was highly consistent with previous observations. That is, the photocatalytic decarboxylation of long-chain stearic acid is exceptionally beyond our general understanding of the temperature sensitivity of photocatalytic reactions. Next, after determining the influence of other external factors, we focused on uncovering and confirming the novel mechanism behind the temperature effect of long-chain fatty acid photocatalytic decarboxylation with great practical potential.
We first screened other reaction conditions, including the wavelength of incident irradiation (Fig. S3, ESI†), and found that the yield increased with the decrease in the wavelength of incident light under external heating to boiling (due to the insufficient photothermal effect of TiO2 under high wavelength irradiation), indicating the absorbed light by TiO2 was able to play dual roles for photocatalysis and photo-to-heat conversion. To further determine that it was not solely n-alkanes as solvents that can cause the self-heating effect to enhance the reaction efficiency, more varieties of solvents were used to confirm the self-heating to boiling and the temperature sensitivity of the photocatalytic decarboxylation of stearic acid. As depicted in Fig. 2g, all the reaction solutions were self-heated to boiling point under cold 410 nm light irradiation with high power in spite of the polarity of the solvent. However, the reactivity of the substrates with increasing temperature was completely different for the non-polar and polar solvent systems. For the former, the higher the boiling point of the non-polar solvent, the faster the reaction; whereas for the latter, the conversion of the substrate did not increase even when the polar solvents reached a high boiling temperature (see entries 5 and 7), suggesting that polar solvents competitively occupy and even deplete the catalyst's active sites,42–45 thereby reducing the possibility of both adsorption and reaction of the stearic acid on the polarized TiO2 surface. Such an adverse solvent effect in conventional photocatalysis is common, which is also what has long constrained the large-scale conversion of photocatalytic reactions. Under such circumstances, even rising temperatures will not significantly change the competitive situation. However, when there is a solvent and substrate with a completely different polarity, like our stearic acid/TiO2/n-alkane solvent system, it is possible for the solvent effect to enable only the substrate to experience greater reactivity through a heat-forced change of the adsorption conformation on TiO2 active sites. This favourable solvent effect can be summarized as the synergistic effect of two reasons: (i) n-alkane solvents do not compete with substrates in adsorption on TiO2-active sites and, help the decarboxylated products, n-alkanes too, to rapidly escape from the active sites by their excellent compatibility at a similar polarity, especially at high temperatures, thereby keeping the catalyst operating at a high activity state; (ii) n-alkane solvents with a high boiling point can accumulate the heat from the TiO2-photothermal effect, and efficiently transfer this energy to the long-chain moiety of the fatty acid substrate adsorbed on TiO2 sites, facilitating decarboxylation of the substrates through changes of the conformation of the carbon chain closer to the TiO2 active sites. Thus, it is more conducive for the non-diffusible hvb+ (into solution) to attack nearby sites. In addition, we quantified the thermodynamic parameters for the photocatalytic decarboxylation of stearic acid at various temperatures ranging from 35 °C to 286 °C. The photocatalytic decarboxylation of stearic acid followed first-order reaction dynamics (Fig. S4, ESI†). The rate constant k at 286 °C was up to 4.19 × 10−3 s−1, which was more than 410 times that at 35 °C (1.0 × 10−5 s−1).
Scope of fatty acids and performances under outdoor sunlight irradiation
To confirm whether most of the biomass-derived linear chain fatty acids were able to take advantage of this photo-to-heat effect for enhancing photocatalytic decarboxylation, a series of linear chain saturated fatty acids were studied under the optimized reaction conditions (Table 1). In n-heptadecane solvent (boiling point 292 °C), the fatty acids with a carbon number more than C12 could be converted to their corresponding n-alkanes less-one carbon in high yields (72–96%) in 15 min (Table 1, entries 1–11). In addition, we explored the suitability of branched-chain fatty acids for this system, and found that isostearic acid decarboxylates were similar to long straight-chain fatty acids in enabling a relaxed heat-accelerated conversion (Table 1, entry 19). However, their selectivity was a little less than that of the straight-chain ones (∼60.7% yield). We believe that there could be some side reactions, such as elimination reactions into diverse olefins, for the branched-chain intermediates. The average output rates of Cn–1n-alkane products were 14.37–40.73 mmol gcat−1 h−1 in our designed reaction system, which was much higher than those previously reported for photocatalysis and thermal catalytic transformations operated under harsh processes conditions (Table S1, ESI†). When we scaled up the concentration of the substrate stearic acid from 0.05 M to 0.5 M, the once-through output of n-heptadecane product was obtained with very high concentrations (∼0.43 M) after 6 h irradiation (Table 1, entry 5), which was 5 orders of magnitude higher than the previous semiconductor photocatalytic and algal photoenzyme transformations, which were limited in the range of 1–102 μM order of magnitudes in a once-through operation.21 This is the closest to the practicality of high-flux fatty acids conversion so far. More importantly, in a concentration-scale reaction of mixtures of fatty acids, various fatty acids could be converted independently at the same time, affording the corresponding Cn–1n-alkanes in outstanding yields (∼85%, entry 18 in Table 1). This showed the practicality of this operation for industrially crude fatty acids conversion (because there were mixed raw fatty acids for most occasions). Moreover, the TiO2 catalyst presented similar Cn–1n-alkane yields across five cycles (Fig. S5a, ESI†), and the XRD patterns of the catalyst after the fifth catalytic run showed little change in its crystal structure (Fig. S5b, ESI†). The BET surface area even increased after the fifth catalytic run, presumably due to the depletion of residue hydroxyl groups or H2O on the TiO2 surface (Fig. S5d, ESI†) and the well-known self-cleaning action created by UV light irradiation on the TiO2 surface.46 The cryogenic ESR feature of the TiO2 sample did not change fundamentally before and after the photothermal reaction, and no significant peaks of carbon-free radicals and trapped/accumulated electrons (i.e. Ti3+) were observed (Fig. S5c, ESI†). This indicated there was little of the carbon deposition and Ti4+/3+redox on the used TiO2 surface that traditional thermal- and photocatalysis systems are often faced with. Even extended to 30 photocatalytic cycles, the TiO2 photocatalyst still showed no significant loss of activity or change in catalyst properties (Fig. S6, ESI†), demonstrating its potential for the industrial transformation of long-chain fatty acids upon solar exposure. All the results demonstrated the outstanding stability of this “star” catalyst in such photothermal catalysis.|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Boiling point (°C) | Product | Conv. (%) | Yield (%) |
| General reaction conditions: reaction solution (25 mL), substrate concentration (0.05 M), n-octadecane (internal standard, 0.05 M), TiO2 (125 mg), n-heptadecane solvent locked reaction temperature (292 °C), reaction pressure (0.1 MPa), LED (410 nm, 10.25 W cm−2) irradiation for 15 min. All the catalytic performance data in the table are averages from at least three experiments.a n-Eicosane as an internal standard (0.05 M).b n-Tetradecane solvent locked reaction temperature (254 °C), irradiation for 30 min.c Stearic acid concentration (0.5 M), 6 h.d Stearic acid (0.25 M) and palmitic acid (0.25 M), n-tetradecane solvent locked reaction temperature (254 °C), irradiation for 6 h. | |||||
| 1 | C22H44O2 (behenic acid) | 306 (8 kPa) | C21H44 (n-heneicosane) | 100 | 95.6 |
| 2 | C21H42O2 (heneicosanoic acid) | 292 (5.3 kPa) | C20H42 (n-eicosane) | 100 | 96.7 |
| 3 | C20H40O2 (arachidic acid) | 328 | C19H40 (n-nonadecane) | 100.0 | 96.6 |
| 4a | C19H38O2 (nonadecanoic acid) | 300 | C18H38 (n-octadecene) | 100.0 | 93.3 |
| 5b | C18H36O2 (stearic acid) | 383 | C17H36 (n-heptadecane) | 99.7 | 95.1 |
| 100.0 | 86.2c | ||||
| 6 | C17H34O2 (heptadecanoic acid) | 363 | C16H34 (n-hexadecane) | 100.0 | 85.6 |
| 7 | C16H32O2 (palmitic acid) | 351 | C15H32 (n-pentadecane) | 100.0 | 84.5 |
| 8 | C15H30O2 (pentadecanoic acid) | 339 | C14H30 (n-tetradecane) | 100.0 | 85.3 |
| 9 | C14H28O2 (myristic acid) | 326 | C13H28 (n-tridecane) | 100.0 | 79.1 |
| 10 | C13H26O2 (tridecanoic acid) | 312 | C12H26 (n-dodecane) | 100.0 | 76.1 |
| 11 | C12H24O2 (lauric acid) | 298 | C11H24 (n-undecane) | 100.0 | 72.3 |
| 12 | C11H22O2 (undecanoic Acid) | 280 | C10H22 (n-decane) | 100.0 | 66.8 |
| 13 | C10H20O2 (capric acid) | 268 | C9H20 (n-nonane) | 100.0 | 64.7 |
| 14 | C9H18O2 (nonanoic Acid) | 254 | C8H18 (n-octane) | 88.0 | 57.8 |
| 15 | C8H16O2 (caprylic acid) | 239 | C7H16 (n-heptane) | 100.0 | 45.8 |
| 16 | C7H14O2 (caprylic acid) | 223 | C6H14 (n-hexane) | 100.0 | 38.6 |
| 17 | C6H12O2 (hexanoic acid) | 205 | C5H12 (n-undecane) | 100.0 | 36.7 |
| 18d | Stearic acid & palmitic acid (mol ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
— | n-Heptadecane | 97.4 | 86.0 |
| n-Pentadecane | 89.5 | 85.4 | |||
| 19 |
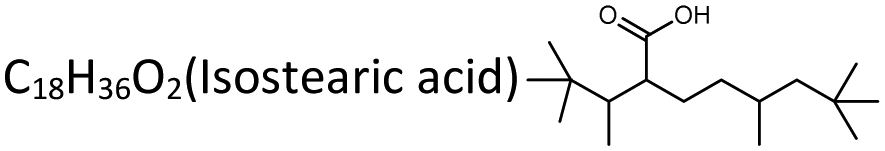
|
382 |
|
100.0 | 60.7 |
It is worth noting that for the straight-chain fatty acids with a carbon number below C12, their yields decreased significantly with the decrease in the C-number of fatty chains at the boiling point of n-tetradecane (see entries 12–17 in Table 1). We initially believed this was due to the shorter contact times between these low boiling-point substrates and TiO2 compared to that of the long-chain ones, because the majority of the low boiling-point substrates escaped from the suspending solution in to the gas phase. The results in Fig. 2f show the very close rates in the photocatalytic conversion of propionic acid between 125 °C and 35 °C, differing from the stearic acid case under identical conditions, clearly indicating it was the size of the fatty chain of the fatty acids that decided the temperature sensitivity rather than the simple evaporation degree of the fatty acids. Now, it could be summarized that the short-chain fatty acids could be transformed at room temperature but there was no temperature-dependent yield; while the long-chain ones showed just the opposite case. We anticipated this photocatalytic conversion of long-chain fatty acids into value-added alkanes by means of self-heating could be carried out on a large scale under outdoor sunlight irradiation. Thus, we scaled up the experiment from 25 mL to 500 mL (0.05 M stearic acid). For this purpose, the outdoor sunlight was concentrated by a collecting mirror (equipped with a dish light concentrator with automatic solar tracking, with a collecting mirror area of ∼3.3 m2), and the focused incident irradiation area was 132 cm2 (Fig. 3 and Movie S1, ESI†). The light intensity was enhanced from ∼0.1 W cm−2 to 21 W cm−2, which could rapidly heat the reaction solution to boiling (note, the reactor was without any insulation measure mainly for the sake of observation. In practical application though, the size of the collecting mirror could be significantly reduced if the photoreactor was well isolated). The decarboxylation reaction was nearly finished within 120 min, and the yield of n-heptadecane reached ∼89% (Fig. 3b). We further expanded this photocatalysis to be more scaled up to 1500 mL with an initial 0.1 M of stearic acid. Therefore, we used a larger collecting mirror (area ∼9.5 m2) to provide the larger light area (314 cm2, 26 W cm−2) required for the larger-scale reaction (Movie S2, ESI†). As expected, although the light receiving area was not increased by 6 times (should be 6 × 132 = 792 cm2), the decarboxylation reaction still finished in 120 min under nearly similar light intensity (Fig. 3c). The almost constant output rate as in expanding the reaction volume (see Fig. 3c) showed a different kinetics feature than that of traditional photocatalysis. Thanks to the product's rapid removal from the TiO2 surface and always constant occupancy of the substrate on TiO2 active sites, the output rate per unit volume concentration of TiO2 in our system should be exclusively decided by the reactivity of conformation of the substrate's molecular strain induced on the TiO2 active sites. Once the temperature of the system remained the same at the boiling point, the conformational change was consistent, and the photocatalytic output rate per unit volume concentration of TiO2 should be the same regardless of the increase in volume. We obtained a 78.9% yield of product isolation by reduced pressure distillation (see ESI† sunlight collection device section). NMR and MS analyses proved that the isolated product was n-heptadecane (Fig. S7, ESI†).
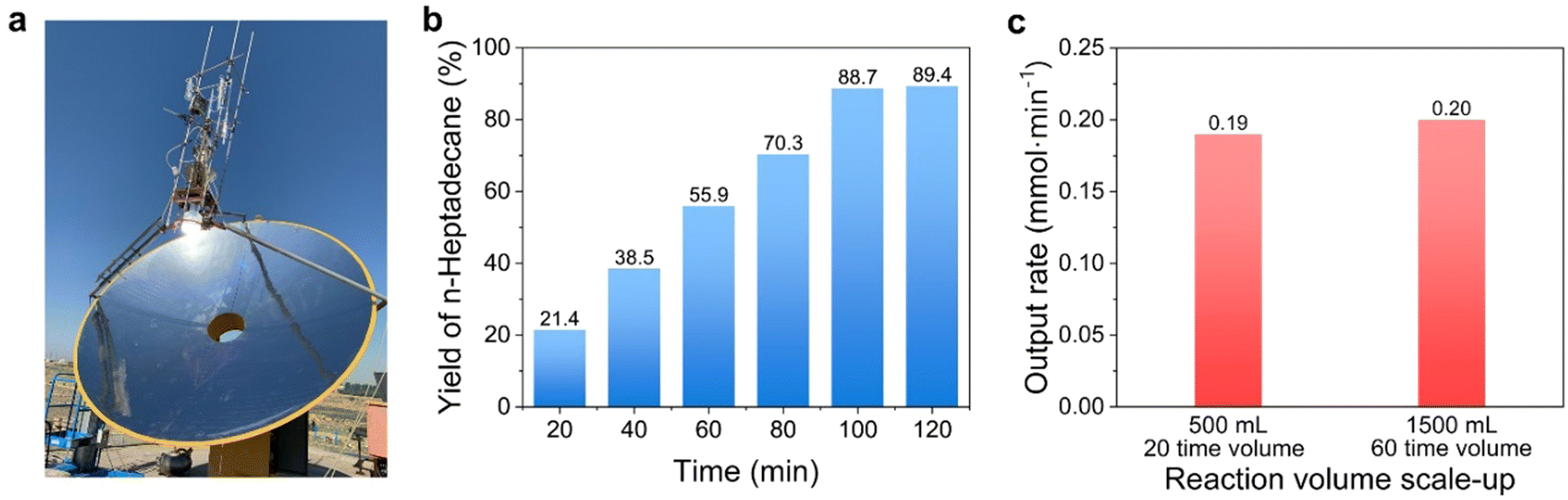 | ||
| Fig. 3 Stearic acid photocatalytic decarboxylation scaled-up in volume over TiO2 under outdoor solar irradiation. (a) Device for focusing sunlight for the photocatalytic reaction. The detailed parameters of the device are shown in the ESI.† (b) Yield of n-heptadecane for the scaled-up test (0.5 M–500 mL). The test was performed under solar radiation in Baotou, China (11/6/2017, 14–30 °C). The starting concentration and volume of stearic acid were 0.05 M and 500 mL, respectively. The power of focused sunlight was about 21 W cm−2. The catalyst dosage was 2.5 g (5 mg mL−1). (c) Output rates at different scaled-up levels. The scaled-up test (0.1 M–1500 mL) was performed under solar radiation in Baotou, China (29/6/2023, 14–35 °C and 30/6/2023, 16–36 °C). The starting concentration of stearic acid was 0.1 M but the volume of suspending solution was expanded up to 1500 mL. The power of focused sunlight was about 26 W cm−2. The catalyst loading was 7.5 g (5 mg mL−1). | ||
In situ observations of the decarboxylation reaction by ESR, FTIR, and XPS
Why does the temperature-rate dependency in the TiO2-based photocatalytic decarboxylation at the n-tetradecane solvent boiling point only work for long-chain fatty acids of more than twelve carbons? And why do these short-chain ones, such as propionic acid, not work? According to the above temperature-dependent transformation results, namely, the shorter the chain of the fatty acids is, the less their transformation depends on the temperature. The typical photo-Kolbe decarboxylation process39,47 should be reconsidered from its oxidation step of photoinduced hvb+ predominating over the reduction step of the conduction band electrons (ecb−), which would have a prerequisite for cleavage of the C–COO− bonds of fatty acids. In situ ESR determinations of the photocatalytic conversion of several linear fatty acids were carried out at a much lower temperature (∼100 K) for direct observation of whether photoinduced hvb+ could still react with short-chain fatty acids (Fig. 4a). Generally, signals with g = 2.007, g = 2.014, and the shoulder at about g = 2.020–2.028 arose from a typical photoinduced hvb+ centre of P-25 TiO2, while a sharp signal at g⊥ = 1.990 and shoulder at g|| = 1.957 were also observed, assigned to lattice electron trapping sites in anatase TiO2. Previous studies have suggested the location of the hvb+ traps to be either surface OH sites48–50 or subsurface oxygen anions.51,52 Upon irradiation with a xenon lamp, the features of trapped ecb− did not show much difference between the C18 and C3 cases. However, the characteristic peaks of photoinduced hvb+ were significantly suppressed from the C5 case onwards, while they nearly completely disappeared for the C3 acid case. Instead, the C-centred radical was formed, indicating that the C3 acid was still able to be oxidized by the photoinduced hvb+ even at 100 K. On the contrary, very distinct hvb+ characteristic peaks were observed for both the C12 acid and C18 acid, which indicated that the photoinduced hvb+ really did not react with them at 100 K at all.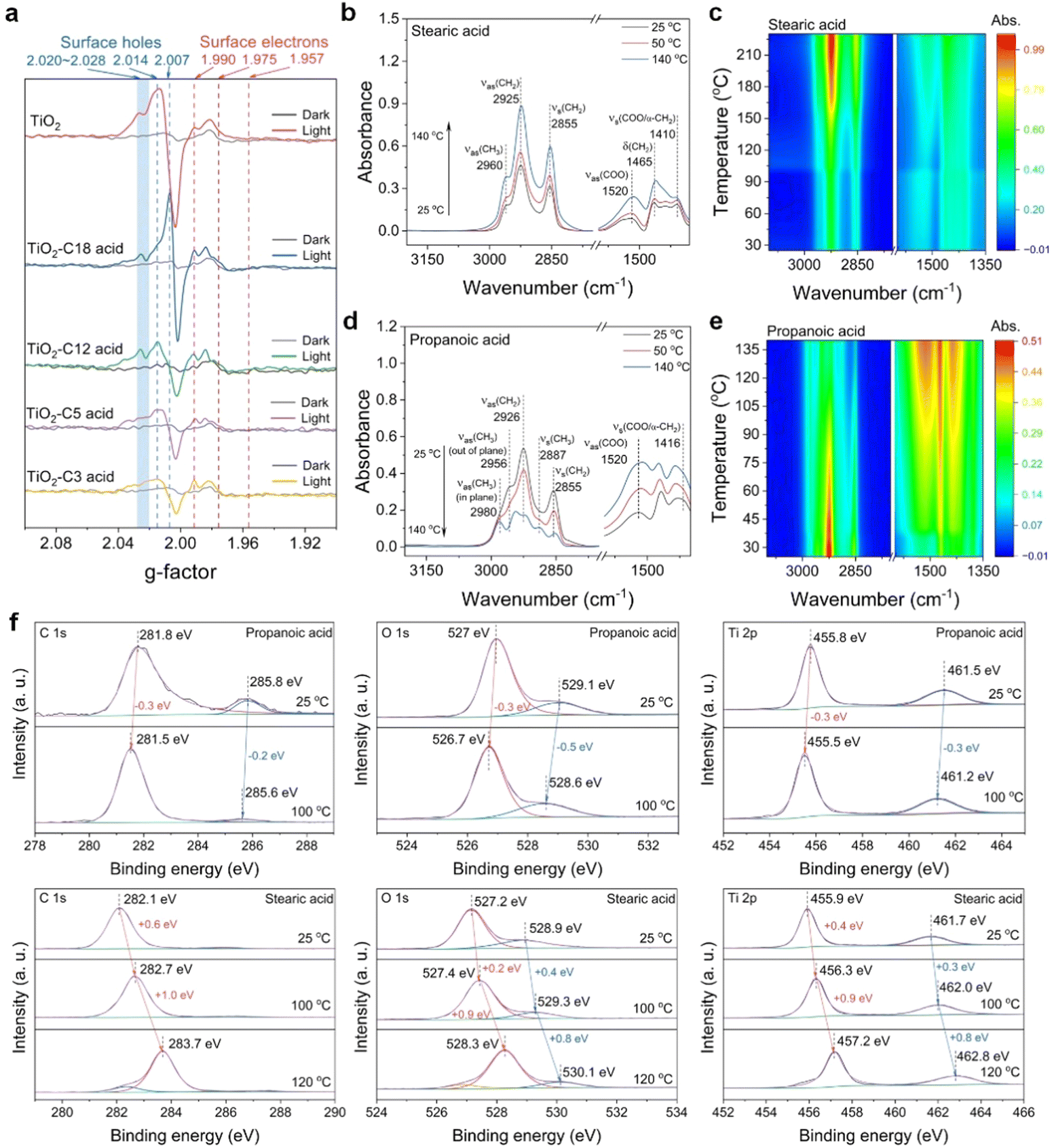 | ||
| Fig. 4 In situ observations of the decarboxylation reaction. (a) ESR observation of the photocatalytic decarboxylation reaction by TiO2 at low temperature (100 K). (b) In situ DRIFT spectra and 2D plot (c) of stearic acid over TiO2 with a programmed temperature process (from 25 °C to 140 °C). (d) In situ DRIFT spectra and 2D plot (e) of propionic acid over TiO2 with a programmed temperature process (25–140 °C). (f) In situ XPS of propionic acid and stearic acid over TiO2, respectively, at high temperatures. ESR sample preparation: 10 mg of 20 wt% TiO2-fatty acids were placed in NMR tubes; then, the air in the tubes was replaced with Ar in a glove box. | ||
Given that photoinduced hvb+ plays a key role, what principle makes the size of the fatty chain of acids initiate hvb+ oxidation; in other words, what properties of the long-chain moiety of fatty acid decide the exhausted photons converted heat available for photocatalytic redox? To reveal the truth, we used in situ DRIFT spectroscopy with programmed temperature means to observe the situation of stearic acid on the TiO2 photocatalyst surface (Fig. 4b). The absorbance of the symmetric and antisymmetric stretching vibration peaks of –CH2– (2925 and 2855 cm−1) in the non-polar long-chain moiety of stearic acid gradually increased with the temperature increase (For the additional high-temperature data, see Fig. S13, ESI†). Also, the antisymmetric stretching vibration absorbance of the end group –CH3 rose, both of which indicated the whole process of the fatty chain from standing up to setting down on the surface of TiO2 with the temperature increase. A typical wavenumber difference between the antisymmetric stretching vibration absorbance of –COO−, Δν (νas(COO) − νs(COO) = 1520 cm−1 − 1410 cm−1 = 110 cm−1)53,54 was obtained from the adsorption of stearic acid to TiO2. Comparison with the spectrum of the dissolved stearic acid (Δν (νas(COO) − νs(COO)) = 1551 cm−1 − 1410 cm−1 = 141 cm−1) indicated the formation of bidentate carboxylate linkages (see Fig. S13c and d, ESI†). However, the antisymmetric stretching vibration absorbance of –COO− also increased (1520 cm−1), while the symmetrical stretching vibration absorbance increased insignificantly (1410 cm−1). This indicated that the fatty chain portion of stearic acid was gradually attached to the surface of TiO2 driven by heat, and one of the bidentate coordination bonds between the carboxyl and TiO2 was gradually destroyed, resulting in a decrease in the stability of the complex relative to the vertical bidentate coordination (1520 and 1410 cm−1). On the contrary for propionic acid, the absorbance of the symmetric and antisymmetric stretching vibrational peaks of CH2 and CH3 (2980, 2926, 2887, 2855 cm−1) decreased with increasing the temperature (Fig. 4d), and the bidentate coordination remained stable.55,56
We expected further support from both angle-resolved XPS at room temperature and in situ heating XPS determination for stearic acid and propionic acid loaded on TiO2, respectively. The angle-resolved XPS features (Fig. S14, ESI†) demonstrate that the stearic acid and propionic acid differed in their carbon chain arranging patterns on the TiO2 surface at room temperature. The former's anchoring was far away from the TiO2 surface and its long carbon chain was upright, while the latter was closer to the TiO2 surface and the whole molecule lay flat without distinction at room temperature. Furthermore, with raising the temperature, new anchoring and arranging situations with bigger binding energies were developed in the stearic acid/TiO2 case, but no longer in the propionic acid case (Fig. 4f). This suggested that temperature has a stronger effect on stearic acid anchoring on TiO2 owing to the length of the carbon chain.
Inspired by Le Chatelier's principle,57,58 we believe that the adsorption stable state of stearic acid, which is achieved at room temperature with the carbon chains upright on TiO2, is broken when it reaches high temperature. Both in situ heating DRIFT-FTIR and XPS analyses demonstrated that such a change from an upright to a flat C–COOH bond of stearic acid on the surface of TiO2 (see Fig. 5a) had indeed taken place with the rising temperature. It is very possible that the flattened long-carbon chain on the TiO2 surface at the high temperature we observed is a prototype of how Chatelier's principle is obeyed. Here, the increasing interaction between the non-polar chain and polar TiO2 surface with the rising temperature makes the delocalization of the polarity of carboxylate spread to the entire long-chain, just as high temperature can break the pattern of a micelle formed by amphiphilic molecules in an oil/water interface and cause the upright arrangement to collapse. The change provides a new possible pathway for hvb+ oxidizing the C–COOH bond, which once seemed too remote to work at room temperature. In this way, hvb+-dominated oxidation of the strained C–COOH bond followed by proton/electron coupling to the tiled C-terminal is easier than to the remote upright C–COOH bond at room temperature. The more important consequence of this is that bending the straight carbon chain of stearic acid at high temperature results in a C–COOH bond with more strain, i.e. invalid excitation relaxed into heat is converted to internal energy and transferred to the α-C–COOH bond. Such energy storage is very beneficial for hvb+ subsequently cutting off the C–COOH bond. However, short-chain carboxylic acids, such as propionic acid, where the whole molecule including its ethyl group is polar due to the little polarization isolation, are prone to be close to the photoinduced hvb+ sites and oxidized whether they are upright or prostrate to the polar TiO2 surface (Fig. 5a). In this condition, hvb+ oxidation is no longer controlled by the α-C–COOH bond absence (Fig. 5a), but it does not use the heat energy from the relaxed light irradiation anymore because of the typically negative temperature-dependent efficiency of general photocatalysis. Namely, the positive effect of temperature accelerating the reaction collision probability is totally cancelled out by the increase in recombination of separated ecb−/hvb+ pairs with the temperature. Thus, the short-chain acids show no significant effect from the temperature.
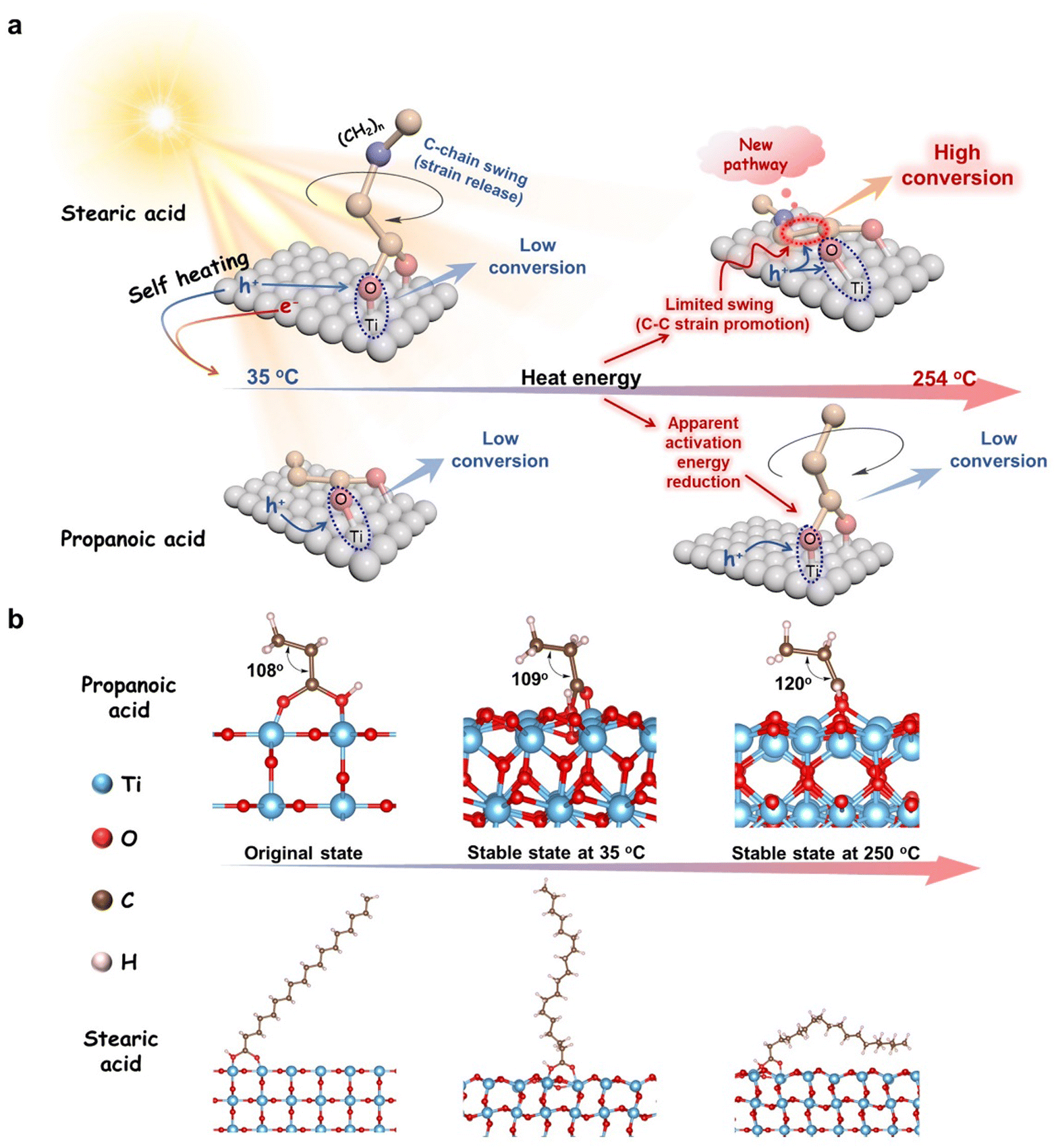 | ||
| Fig. 5 Plausible reaction pathway for the photocatalytic decarboxylation of fatty acids over TiO2 with different temperature-independent AQEs. (a) Long-chain fatty acids accumulate heat energy through shifting to attach to the surface of TiO2 at high temperatures to enable hvb+ activation and the short-chain fatty acids undergo an upright flip at high-temperature that counteracts the enhanced high-temperature hvb+ activation over TiO2. (b) Theoretical calculation results showing different adsorption models of propionic acid and stearic acid on the TiO2 surface between 35 °C and 254 °C. | ||
We further verified this proposal with theoretical calculations. Straight-chain fatty acids were modelled to act with the TiO2 surface at an original angle of 45° and molecular dynamics (MD) simulations were carried out at 35 °C and 250 °C, respectively, to identify their stable conformations after adsorption. After a 100 ps MD simulation, the carbon chains of stearic acid at 250 °C showed some folding of the carbon chains from the initial state towards the surface of TiO2, whereas they were almost completely upright on the surface of TiO2 at 35 °C (Fig. 5b). That is, the change of conformation on the TiO2 surface with the temperature increases forced the entire stearic acid molecule, including its carboxyl head, significantly closer to the TiO2 surface than the case at 35 °C (refer to Fig. S14 (ESI†)). This suggests the oxidative decarboxylation of photogenerated hvb+ becomes easier. The total energy changes accompanied by the anchoring transition are shown in Fig. S15 (ESI†). The carbon chain of propionic acid was too short to allow visualizing the conformational changes on the model, but the trend of adsorption changes could be reflected in the change of C–C–C bond angles. The bond angle increased from 108° in the initial state to 120° with the increase in temperature, indicating that the propionic acid molecule tended towards a little more chaotic arrangement at higher temperatures. The related MD movies are shown in Movie S3 (ESI†). In addition, the bond energy of the α-C–COOH bond changes calculated based on the model provide additional support for the findings (Fig. S16, ESI†). The results indicated that as the temperature rose from 35 °C to 250 °C, the upright of the long-chain of stearic acid on the TiO2 surface adopted a flat down conformation. In addition, compared to the short-chain propionic acid case, this event of forcing the entire stearic acid molecule closer to the TiO2 surface also weakened the α-C–COO bond more (ΔBE ∼ 47 kJ mol−1) and made it more prone to be oxidized by photogenerated hvb+, i.e. the conformational shift brought about a difference in reactivity. The results of the theoretical calculations were basically consistent with our proposal of why only long-carbon chain fatty acids enable the relaxed heat from failure excitation to accelerate the main photocatalytic decarboxylation process.
Conclusions
We presented a combination of TiO2-based photocatalysis and a photothermal conversion effect synergistic route to achieve the high-flux conversion of long-chain fatty acids to Cn–1n-alkane biofuels under strong light irradiation. With a high boiling point of n-alkane as the solvent, the high-power irradiation was sufficient to enable the formed heat from invalid excitation light to boil the reaction system above 250 °C and to accumulate the strain of the target C–COO− bond by forcing the standing-up long-chain to become flat on the surface of TiO2, which allowed the product to be obtained with high concentrations (∼0.5 M). This work demonstrates that the inevitable photo-to-heat effect could play a significant role in photocatalytic industrialization using small-sized reactors and high-incident light intensity.Author contributions
Q. H. and W. M. conceived and designed the experiments. Q. H. and C. H. conceived the figures and tables. G. G. and W. M. supervised the research and revised the manuscript. Q. H. and C. H. carried out the experiments and characterizations. H. Q., W. M. and S. A. analysed the data. All authors discussed and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (grant no. 2022YFA1205603, no. 2018YFA0209302), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB3600000), the National Natural Science Foundation of China (grants no. 22188102, no. 22176192, no. 21827809).References
- D. J. Liu, J. Sun, B. A. Simmons and S. Singh, ACS Sustainable Chem. Eng., 2018, 6, 7232–7238 CrossRef CAS
.
- L. Q. Yang, K. L. Tate, J. B. Jasinski and M. A. Carreon, ACS Catal., 2015, 5, 6497–6502 CrossRef CAS
- Y. Li, Y. F. Wen, B. K. Chen, X. Fu and Y. Wu, Energy Sustainable Dev., 2023, 75, 60–71 CrossRef
- P. M. Yeletsky, R. G. Kukushkin, V. A. Yakovlev and B. H. Chen, Fuel, 2020, 278, 118255 CrossRef CAS
- X. C. Wang, M. Arai, Q. F. Wu, C. Zhang and F. Y. Zhao, Green Chem., 2020, 22, 8140–8168 RSC
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu, M. Heggen, L. Lin, C. Ma, M. Wang, F. Fan, S. Jin and F. Wang, Nat. Energy, 2019, 4, 575–584 CrossRef CAS
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- M. R. Becker, E. R. Wearing and C. S. Schindler, Nat. Chem., 2020, 12, 898–905 CrossRef CAS PubMed
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed
- T. Ju, Y. Q. Zhou, K. G. Cao, Q. Fu, J. H. Ye, G. Q. Sun, X. F. Liu, L. Chen, L. L. Liao and D. G. Yu, Nat. Catal., 2021, 4, 304–311 CrossRef CAS
- X. D. Li, Y. F. Sun, J. Q. Xu, Y. J. Shao, J. Wu, X. L. Xu, Y. Pan, H. X. Ju, J. F. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS
- H. Yan, Y. B. Tang, X. Y. Sui, Y. C. Liu, B. W. Gao, X. F. Liu, S. F. Liu, J. H. Hou and W. Ma, ACS Energy Lett., 2019, 4, 1356–1363 CrossRef CAS
- Z. Geng, Y. Yu, A. J. Offen and J. Liu, Nat. Catal., 2023, 6, 1241–1247 CrossRef CAS
- Ł. Jęczmionek and K. Porzycka-Semczuk, Fuel, 2014, 131, 1–5 CrossRef
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS
- P. P. Samire, B. Zhuang, B. Légeret, A. Baca-Porcel, G. Peltier, D. Sorigué, A. Aleksandrov, F. Beisson and P. Müller, Sci. Adv., 2023, 9, EADG3881 CrossRef CAS PubMed
- N. S. Scrutton, Science, 2017, 357, 872–873 CrossRef CAS PubMed
- D. Sorigue, B. Legeret, S. Cuine, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugiere, Y. Coute, D. Nurizzo, P. Muller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier and F. Beisson, Science, 2017, 357, 903–907 CrossRef CAS PubMed
- D. Sorigue, B. Legeret, S. Cuine, P. Morales, B. Mirabella, G. Guedeney, Y. Li-Beisson, R. Jetter, G. Peltier and F. Beisson, Plant Physiol., 2016, 171, 2393–2405 CrossRef CAS PubMed
- D. Sorigué, K. Hadjidemetriou, S. Blangy, G. Gotthard, A. Bonvalet, N. Coquelle, P. Samire, A. Aleksandrov, L. Antonucci, A. Benachir, S. Boutet, M. Byrdin, M. Cammarata, S. Carbajo, S. Cuiné, R. B. Doak, L. Foucar, A. Gorel, M. Grünbein, E. Hartmann, R. Hienerwadel, M. Hilpert, M. Kloos, T. J. Lane, B. Légeret, P. Legrand, Y. Li-Beisson, S. L. Y. Moulin, D. Nurizzo, G. Peltier, G. Schirò, R. L. Shoeman, M. Sliwa, X. Solinas, B. Zhuang, T. R. M. Barends, J. P. Colletier, M. Joffre, A. Royant, C. Berthomieu, M. Weik, T. Domratcheva, K. Brettel, M. H. Vos, I. Schlichting, P. Arnoux, P. Müller and F. Beisson, Science, 2021, 372, EABD5687 CrossRef PubMed
- S. Simic, M. Jakstaite, W. T. S. Huck, C. K. Winkler and W. Kroutil, ACS Catal., 2022, 12, 14040–14049 CrossRef CAS PubMed
- I. Izumi, F. R. F. Fan and A. J. Bard, J. Phys. Chem., 1981, 85, 218–223 CrossRef CAS
- K. Liu, A. Litke, Y. Su, B. G. van Campenhout, E. A. Pidko and E. J. Hensen, Chem. Commun., 2016, 52, 11634–11637 RSC
- Z. P. Huang, Z. T. Zhao, C. F. Zhang, J. M. Lu, H. F. Liu, N. C. Luo, J. Zhang and F. Wang, Nat. Catal., 2020, 3, 170–178 CrossRef CAS
- M. M. E. Huijbers, W. Zhang, F. Tonin and F. Hollmann, Angew. Chem., Int. Ed., 2018, 57, 13648–13651 CrossRef CAS PubMed
- W. Zhang, M. Ma, M. M. E. Huijbers, G. A. Filonenko, E. A. Pidko, M. van Schie, S. de Boer, B. O. Burek, J. Z. Bloh, W. J. H. van Berkel, W. A. Smith and F. Hollmann, J. Am. Chem. Soc., 2019, 141, 3116–3120 CrossRef CAS
- H. T. Duong, Y. Wu, A. Sutor, B. O. Burek, F. Hollmann and J. Z. Bloh, ChemSusChem, 2021, 14, 1053–1056 CrossRef CAS
- A. Al Mousawi, D. M. Lara, G. Noirbent, F. Dumur, J. Toufaily, T. Hamieh, T. T. Buo, F. Goubard, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2017, 50, 4913–4926 CrossRef CAS
- R. R. Ishiki, H. M. Ishiki and K. Takashima, Chemosphere, 2005, 58, 1461–1469 CrossRef PubMed
- L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS PubMed
- Y. C. Zhang, S. He, W. X. Guo, Y. Hu, J. W. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed
- L. A. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Q. Zhao, L. Henderson, L. L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS
- Z. Wan, C. Wang, C. Yang, D. Ma, H. Ji, C. Chen, W. Ma and J. Zhao, Appl. Catal., B, 2023, 324, 122237 CrossRef CAS
- W. Chang, C. Y. Sun, X. B. Pang, H. Sheng, Y. Li, H. W. Ji, W. J. Song, C. C. Chen, W. H. Ma and J. C. Zhao, Angew. Chem., Int. Ed., 2015, 54, 2052–2056 CrossRef CAS
- Y. Yan, W. Shi, Z. Yuan, S. He, D. Li, Q. Meng, H. Ji, C. Chen, W. Ma and J. Zhao, J. Am. Chem. Soc., 2017, 139, 2083–2089 CrossRef CAS
- L. L. Qu, D. L. Huang, H. F. Shi, M. B. Gu, J. L. Li, F. Dong and Z. J. Luo, J. Phys. Chem. Solids, 2015, 85, 173–179 CrossRef CAS
- L. L. Wang, M. Wang, Z. P. Xu, W. Yu and H. Q. Xie, Sol. Energy Mater. Sol. Cells, 2020, 212, 110575 CrossRef CAS
- S. Jung and S. Park, ACS Catal., 2017, 7, 438–442 CrossRef CAS
- T. D. Pham and B. K. Lee, Chem. Eng. J., 2017, 307, 63–73 CrossRef CAS
- W. Wu, G. Q. Shan, Q. Xiang, Y. Q. Zhang, S. J. Yi and L. Y. Zhu, Water Res., 2017, 122, 78–85 CrossRef CAS
- P. Maki-Arvela, M. Snare, K. Eranen, J. Myllyoja and D. Y. Murzin, Fuel, 2008, 87, 3543–3549 CrossRef CAS
- J. Y. Park, Science, 2018, 361, 753 CrossRef CAS PubMed
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS
- C. D. Jaeger and A. J. Bard, J. Phys. Chem., 1979, 83, 3146–3152 CrossRef CAS
- J. R. Harbour and M. L. Hair, J. Phys. Chem., 1979, 83, 652–656 CrossRef CAS
- V. Brezova, A. Stasko and L. Lapcik, J. Photochem. Photobiol., A, 1991, 59, 115–121 CrossRef CAS
- R. F. Howe and M. Gratzel, J. Phys. Chem., 1987, 91, 3906–3909 CrossRef CAS
- D. Bahnemann, A. Henglein and L. Spanhel, Faraday Discuss. Chem. Soc., 1984, 78, 151–163 RSC
- K. D. Dobson and A. J. McQuillan, Spectrochim. Acta, Part A, 1999, 55, 1395–1405 CrossRef
- I. Dolamic and T. Bürgi, J. Phys. Chem. B, 2006, 110, 14898–14904 CrossRef CAS PubMed
- H. P. Lin, Y. F. Liu, Y. X. Liu, Z. X. Yang and J. L. Lin, J. Phys. Chem. C, 2021, 125, 4567–4579 CrossRef CAS
- Y. K. Chen, Y. F. Lin, Z. W. Peng and J. L. Lin, J. Phys. Chem. C, 2010, 114, 17720–17727 CrossRef CAS
- H. Wang, L. Wang, D. Lin, X. Feng, Y. M. Niu, B. S. Zhang and F. S. Xiao, Nat. Catal., 2021, 4, 418–424 CrossRef CAS
- W. Lin and C. J. Murphy, ACS Cent. Sci., 2017, 3, 1096–1102 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: [DETAILS]. See DOI: https://doi.org/10.1039/d4ee02026j |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |