
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A homogeneous plating/stripping mode with fine grains for highly reversible Zn anodes†
Zhen
Luo‡
 a,
Yufan
Xia‡
a,
Shuang
Chen
a,
Xingxing
Wu
b,
Esther
Akinlabi
c,
Ben Bin
Xu
*c,
Hongge
Pan
ad,
Mi
Yan
a and
Yinzhu
Jiang
*ab
a,
Yufan
Xia‡
a,
Shuang
Chen
a,
Xingxing
Wu
b,
Esther
Akinlabi
c,
Ben Bin
Xu
*c,
Hongge
Pan
ad,
Mi
Yan
a and
Yinzhu
Jiang
*ab
aSchool of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: yzjiang@zju.edu.cn
bFuture Science Research Institute, ZJU-Hangzhou Global Scientific and Technological Innovation Center, Zhejiang University, Hangzhou 311215, China
cMechanical and Construction Engineering, Faculty of Engineering and Environment, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: ben.xu@northumbria.ac.uk
dInstitute of Science and Technology for New Energy, Xi'an Technological University, Xi'an 710021, China
First published on 22nd August 2024
Abstract
Aqueous zinc metal batteries (AZMBs) have emerged as an attractive energy storage option due to their operational safety, low cost, and environmentally friendly nature. However, the hexagonal close-packed (hcp) Zn anode always exhibits dendritic plating/stripping behavior due to the uncontrollable nucleation and growth of hexagonal platelets, which severely hinders its further development. Herein, we regulate both nucleation and Zn/Zn2+ redox kinetics to enable a homogeneous plating/stripping mode with fine grains, significantly improving the stability and reversibility of the Zn anode. Surface overpotential, which directly determines the nucleation behavior, is prominently amplified by the adsorption of added cordycepin (Cor) on the Zn anode surface. Therefore, a promoted nucleation process with the formation of small-sized Zn grains is achieved. The mitigated Zn growth/dissolution rate caused by the strong steric hindrance effect of Cor further ensures the granular morphology after cycling. Owing to this dual regulation strategy, the Zn anode delivers superior cycling stability of the Zn‖Zn symmetric cell over 900 h at 5 mA cm−2 and an ultra-high cumulative plating capacity of 3 A h cm−2 at 10 mA cm−2. The concept also works in Zn‖Cu asymmetric cells and Zn‖NaV3O8·1.5H2O (NVO) full cells, facilitating excellent reversibility and cycling performance. This work presents a new mode for Zn plating/stripping, which will make highly reversible AZMBs achievable.
Broader contextWhile rechargeable aqueous zinc metal batteries (AZMBs) have been recognized as one of the most promising batteries for scale-up applications, the insane dendritic plating/stripping behavior of the Zn metal anode hinders further development dramatically. As is well known, Zn metal exhibits a hexagonal close-packed (hcp) crystal structure and tends to form large-sized hexagonal platelets. How to completely avoid the formation of dendrites in the long term with Zn hexagonal characteristics still remains under exploration. In this work, we demonstrate a dual regulation strategy that combines fine nuclei and moderate Zn/Zn2+ redox kinetics to ensure granular Zn plating/stripping, which significantly reduces the propensity of dendrite growth. Specifically, the added Cor molecule can preferentially adsorb on the Zn anode surface and amplify the surface overpotential, facilitating to refine the Zn nuclei size. On the other hand, the strong steric hindrance effect of Cor mitigates the Zn growth/dissolution rate, achieving fine Zn grains within the submicron scale. Benefiting from this strategy, a homogeneous Zn plating/stripping mode with fine grains is established, thus prominently enhancing the reversibility and utilization rate of the Zn anode. This work sheds light on the in-depth comprehension of Zn plating/stripping behavior and designing highly reversible Zn anodes. |
Introduction
Aqueous zinc metal batteries (AZMBs) exhibit dominant advantages over traditional lithium-ion batteries (LIBs), primarily due to the high safety and eco-friendliness offered by aqueous electrolytes.1–4 Moreover, metallic zinc exhibits high theoretical capacity (5855 mA h cm−3 or 820 mA h g−1), suitable redox potential (−0.76 V vs. the standard hydrogen electrode), abundant reserves, and relatively high stability in aqueous systems.5–10 The plating/stripping processes of the Zn metal anode play a critical role in AZMBs, since the Zn anode easily displays a dendritic surface morphology with severe volume variation after discharging/charging cycles.11 Numerous efforts focusing on electrolyte additives,12–14 electrode modification,15,16 and artificial solid electrolyte interphase (SEI) fabrication17–20 have been devoted to guiding uniform Zn deposition. However, thorough inhibition of dendritic Zn formation still remains a challenge due to the complex electrochemical reaction at the interface.The Zn deposition process is composed of initial nucleation and subsequent growth of grains.21,22 The manufacturing-inducing defects such as cracks and scratches routinely exist on the Zn anode surface, triggering uneven distribution of local electric fields and nucleation energy barriers.23 Those selected sites with low nucleation energy promote preferential Zn deposition. Zn metal, which exhibits a hexagonal close-packed (hcp) crystal structure, tends to form hexagonal platelets with a (002) plane mostly exposed.24 Beyond the nucleation process, small platelets begin to grow and coarsen with the electrochemical reduction proceeding, resulting in the formation of larger hexagonal plates with sharp edges and corners lying in random directions. These plates with a large tilt angle (∼90°) to the substrate would develop into rampant dendrites, which severely induce local inhomogeneity on the surface and penetrate the separator.25–30 Such dendritic growth is detrimental to the battery in numerous aspects: more intense side reactions (electrode corrosion and hydrogen evolution), higher propensity of internal short circuits, irreversible capacity loss and dramatically reduced Coulombic efficiency (CE).11,23,31–33 During Zn stripping, the intricate surface status will also affect the localized electrochemical environment such as current density, electric field, and charge carrier concentration, which determines different dissolution rates across the electrode surface.34 It is important to note that this kind of reaction rate variation can be exacerbated by uneven deposition.
According to the classical nucleation theory (CNT), the surface overpotential |ηs| acts as a driving force and triggers electrochemical nucleation on the electrode surface, which significantly affects the critical nucleus size (rcrit) and critical nucleation energy barrier (ΔGcrit).8,35,36 The relationships between |ηs|, rcrit and ΔGcrit can be expressed as follows:8
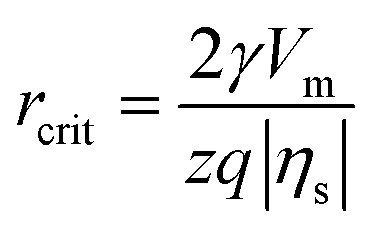 | (1) |
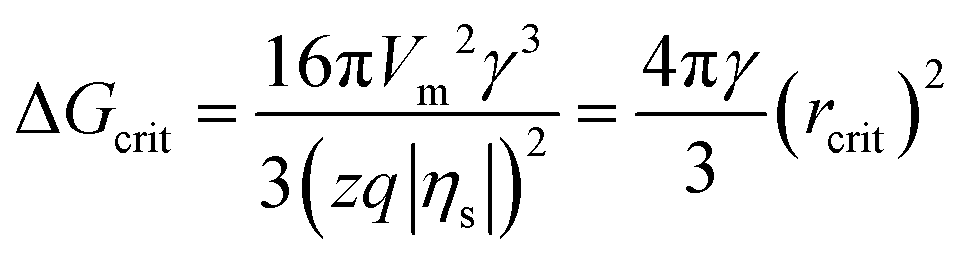 | (2) |
 | ||
| Fig. 1 (a) Illustration of the relationship between the critical nucleation barrier (ΔGcrit) and the critical nucleus size (rcrit) at different surface overpotentials (|ηs|). (b) Schematic illustration of Zn plating/stripping behavior with/without Cor additive. | ||
In this work, we propose a facile strategy to synchronously regulate nucleation and Zn/Zn2+ redox kinetics to realize a homogeneous Zn plating/stripping mode with fine grains, by introducing a trace amount (5 mM, M: mol L−1) of the large molecule cordycepin (Cor) into the 2 M ZnSO4 electrolyte. As shown in Fig. 1b, a smaller |ηs| in a bare ZnSO4 electrolyte induces the formation of large particles at the initial nucleation stage, accompanied by serious side reactions. For further plating, the uncontrollable Zn2+ ion reduction promotes the wild growth of dendrites, simultaneously accelerating side reactions. The dendritic morphology is extremely detrimental to the subsequent stripping process, resulting in disordered dissolution and the formation of “dead Zn”, which eventually leads to the failure of the battery. In contrast, Cor steadily adsorbs on the Zn anode surface and enlarges |ηs|, resulting in the formation of submicron nuclei at the initial plating stage. On account of the strong steric hindrance effect of Cor, the redox kinetics of Zn/Zn2+ is significantly moderated, thus retarding the growth/dissolution of previous deposits and ensuring the homogeneous Zn plating/stripping mode, which prominently enhances the long-term cycling stability and reversibility of the Zn anode under low or high current densities.
Results and discussion
Nucleation regulation by altering the surface overpotential
In order to amplify the surface overpotential (|ηs|) for favorable nucleation, Cor is chosen as an electrolyte additive due to abundant polar groups (amino and hydroxyl) and its large molecular size (Fig. S1, ESI†), which is supposed to spontaneously adsorb on the Zn metal surface and induce a significant steric hindrance effect.40–45 To verify this, we perform first-principles calculations to analyze the adsorption energy (Ead) of Cor and H2O molecules on the Zn metal surface. As shown in Fig. 2a, whether Cor is perpendicular or parallel to Zn (002) facet, it displays much lower Ead (−0.809 eV for vertical state and −1.295 eV for horizontal state) than that of H2O (−0.227 eV), demonstrating the preferential adsorption propensity of Cor. In addition, the horizontally adsorbed Cor on the Zn substrate is under the most stable state (the lowest Ead) due to sufficient surface coverage,42 which may be conducive to increasing the polarization on the Zn anode. The average differential capacitance (![[C with combining macron]](https://www.rsc.org/images/entities/i_char_0043_0304.gif) ) of the Zn anode surface with different concentrations of Cor is identified using cyclic voltammetry (CV) tests (Fig. S2, ESI†), where the 2 M ZnSO4 electrolyte is denoted as BE and the addition of n mM Cor are denoted as BE/nCor (n = 1, 5, 10), respectively. Notably, the Zn anode exhibits reduced with the increase of Cor concentration (Fig. S3 and S4, ESI†), attributed to the shrunken surface area and the formation of a thicker electric double layer (EDL) structure caused by the Cor adsorption.46,47 Furthermore, combined with the scanning electron microscope (SEM) and X-ray diffraction (XRD) analysis, the Zn foil immersed in the BE/5Cor electrolyte displays a flat and neat surface with no irregular flake-like by-products (Fig. S5, ESI†), mainly ascribed to adsorbed Cor suppressing the side reaction of H2O decomposition.
) of the Zn anode surface with different concentrations of Cor is identified using cyclic voltammetry (CV) tests (Fig. S2, ESI†), where the 2 M ZnSO4 electrolyte is denoted as BE and the addition of n mM Cor are denoted as BE/nCor (n = 1, 5, 10), respectively. Notably, the Zn anode exhibits reduced with the increase of Cor concentration (Fig. S3 and S4, ESI†), attributed to the shrunken surface area and the formation of a thicker electric double layer (EDL) structure caused by the Cor adsorption.46,47 Furthermore, combined with the scanning electron microscope (SEM) and X-ray diffraction (XRD) analysis, the Zn foil immersed in the BE/5Cor electrolyte displays a flat and neat surface with no irregular flake-like by-products (Fig. S5, ESI†), mainly ascribed to adsorbed Cor suppressing the side reaction of H2O decomposition.
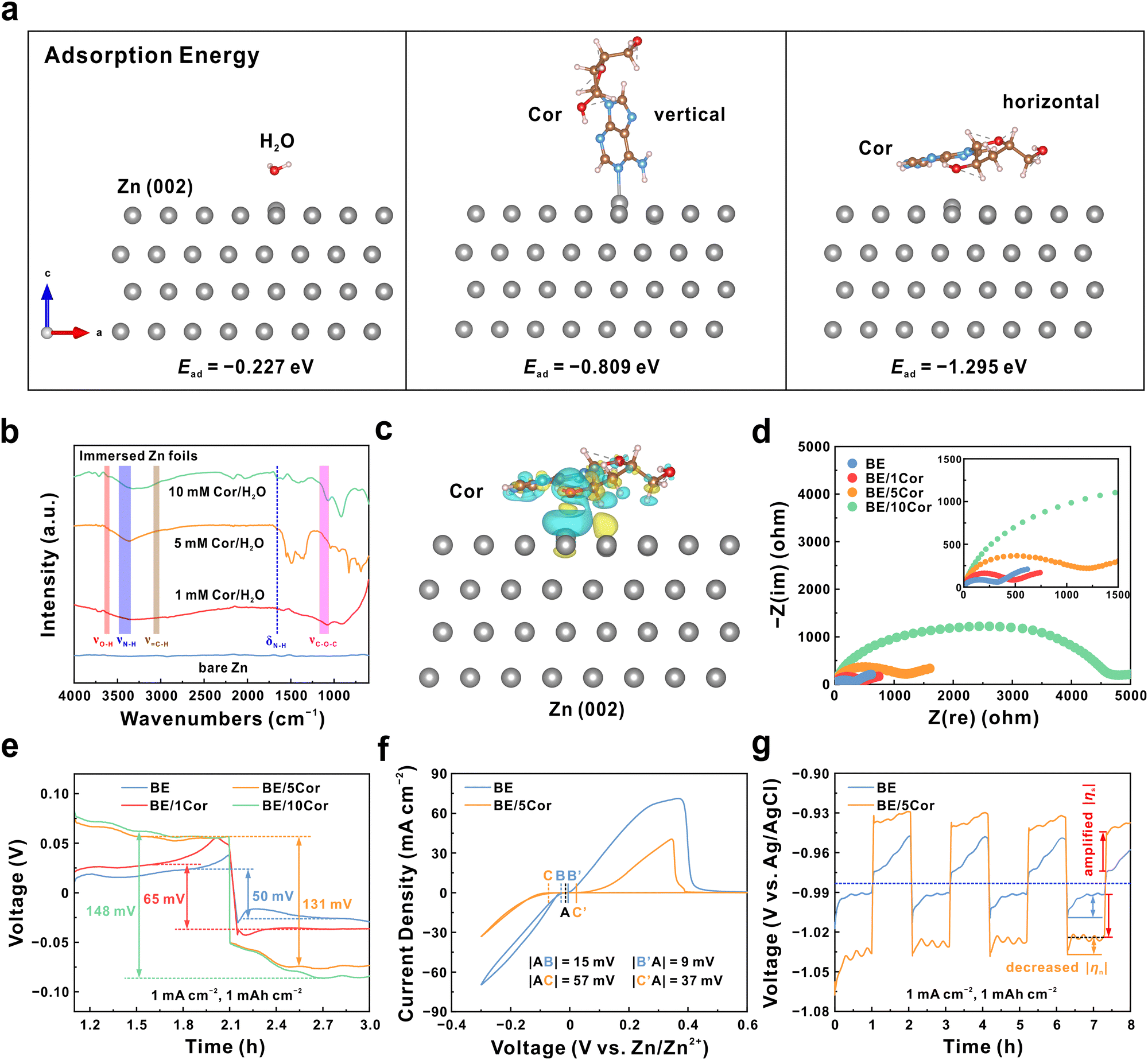 | ||
| Fig. 2 (a) The adsorption energy of H2O, vertical Cor and horizontal Cor molecules on the Zn (002) facet. (b) FTIR spectra of Zn plates immersed in solutions with various concentrations of Cor for 3 days. (c) The charge density difference distribution map of the horizontal Cor molecule on the Zn (002) facet (iso-value = 8 × 10−4 e Bohr−3). The yellow and cyan iso-surfaces represent the increase and decrease in charge density, respectively. (d) EIS spectra of Zn‖Zn symmetric cells. (e) Initial voltage hysteresis of Zn‖Zn symmetric cells at 1 mA cm−2 and 1 mA h cm−2. (f) CV curves of Zn‖Ti asymmetric cells. (g) Voltage profiles of three-electrode beaker cells at 1 mA cm−2 and 1 mA h cm−2. Zn foils were used as the working electrode and counter electrode, and the Ag/AgCl electrode was used as the reference electrode. | ||
We next perform Fourier transform infrared (FTIR) spectroscopy to inspect the interaction between Cor and Zn foil (Fig. 2b). The absorbed signals at 3650–3590, 3500–3300, 3060–3010, 1210–1050, and 1657 cm−1 correspond to the stretching vibrations of O–H, N–H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H from the pyrimidine ring, C–O–C and the bending vibration of N–H in Cor molecule, respectively. These observations explicitly indicate that Cor molecules can spontaneously adhere to the Zn anode in an aqueous system. Moreover, the adsorption type is identified as a chemical interaction according to the distinct charge transfer between Zn and O atoms from the charge density difference distribution map in Fig. 2c. Such a large molecule (10.1 Å, Fig. S1, ESI†) as adsorbate would inevitably incur a strong steric hindrance effect at the interface,44,45 as confirmed by enlarged charge transfer resistance (Rct) in the electrochemical impedance spectroscopy (EIS) shown in Fig. 2d. This result also indicates that the Zn/Zn2+ redox kinetics is moderated, which will be discussed later. It is noteworthy that the significantly increased Rct in the BE/10Cor electrolyte may be attributed to the integrated effect of the natural oxidation layer on the Zn metal surface and excess additive molecules from the supersaturated solution (Fig. S6 and S7, ESI†).48 In addition, the solvation structure of electrolytes with different concentrations of Cor is identified by performing Raman spectroscopy. As shown in Fig. S8 (ESI†), the characteristic peaks present no significant shift with the increase of Cor, suggesting that Cor would not change the coordination environment of Zn2+ ions, which is mainly related to the trace amount and large molecular size of added Cor.
C–H from the pyrimidine ring, C–O–C and the bending vibration of N–H in Cor molecule, respectively. These observations explicitly indicate that Cor molecules can spontaneously adhere to the Zn anode in an aqueous system. Moreover, the adsorption type is identified as a chemical interaction according to the distinct charge transfer between Zn and O atoms from the charge density difference distribution map in Fig. 2c. Such a large molecule (10.1 Å, Fig. S1, ESI†) as adsorbate would inevitably incur a strong steric hindrance effect at the interface,44,45 as confirmed by enlarged charge transfer resistance (Rct) in the electrochemical impedance spectroscopy (EIS) shown in Fig. 2d. This result also indicates that the Zn/Zn2+ redox kinetics is moderated, which will be discussed later. It is noteworthy that the significantly increased Rct in the BE/10Cor electrolyte may be attributed to the integrated effect of the natural oxidation layer on the Zn metal surface and excess additive molecules from the supersaturated solution (Fig. S6 and S7, ESI†).48 In addition, the solvation structure of electrolytes with different concentrations of Cor is identified by performing Raman spectroscopy. As shown in Fig. S8 (ESI†), the characteristic peaks present no significant shift with the increase of Cor, suggesting that Cor would not change the coordination environment of Zn2+ ions, which is mainly related to the trace amount and large molecular size of added Cor.
The chemically adsorbed Cor is supposed to alter the |ηs| due to the reconstructed interface of the Zn anode. According to the galvanostatic charge–discharge (GCD) testing results (1 mA cm−2, 1 mA h cm−2) in Zn‖Zn symmetric cells, the Zn anode presents a voltage hysteresis of 50 mV in BE (Fig. 2e). With the addition of Cor, the voltage hysteresis of symmetric cells is significantly amplified, which demonstrates larger polarization on the Zn anode surface under Cor adsorption. Notably, when the Cor concentration further increases from 5 to 10 mM, the voltage hysteresis of the symmetric cell is mildly increased from 131 to 148 mV, implying the optimum adsorption status of Cor on the Zn anode surface, which is consistent with the results in Fig. S4 (ESI†) and Fig. 2b. This phenomenon is also observed in some reported literature studies using other electrolyte additives.39,49,50 Furthermore, the BE/5Cor solution is determined as the optimum electrolyte for its superior long-term cycling stability (Fig. S9, ESI†) and higher Zn reversibility (Fig. S10, ESI†), which is utilized in the following investigation of the Zn plating/stripping behavior.
From the CV results in Fig. 2f, the onset potentials of both Zn deposition and stripping reaction are postponed in Zn‖Ti asymmetric cells with the BE/5Cor electrolyte. This result reaffirms that the Cor molecule can arouse significant polarization on the Zn anode by impeding charge transfer. Since it is difficult to decouple the Zn plating and stripping process in a symmetric cell, we utilize a three-electrode beaker cell to conduct the GCD test, where Zn foils are used as the working electrode and counter electrode, and the Ag/AgCl electrode is used as the reference electrode (Fig. 2g). The origin of the total overpotential (|ηtotal|) for the Zn plating/stripping process is expressed as follows:
| |ηs| + |ηn| + |ηohm| = |ηtotal| (plating) |
| |ηs| + |ηohm| = |ηtotal| (stripping) |
Moderate Zn/Zn2+ redox kinetics driven by the steric hindrance effect
Except for the initial nucleation stage, the subsequent growth/dissolution behavior of Zn nuclei immensely affects the surface morphology of the Zn anode, which is highly related to Zn/Zn2+ redox kinetics. Therefore, the rate performance of Zn‖Zn symmetric cells is first investigated (Fig. S13, ESI†). Compared with BE, the Zn anode in BE/5Cor shows distinct larger voltage hysteresis at each current density, corresponding to increased |ηs|. The exchange current density (i0) can be derived from the relation between current density (i) and voltage hysteresis (η):56 | (3) |
 | ||
| Fig. 3 (a) The corresponding overpotential versus current density plots from the rate performance of Zn‖Zn symmetric cells revealing the exchange current density (i0) for Zn plating/stripping. CA curves of the Zn anode tested in Zn‖Zn symmetric cells at overpotentials of (b) −150 mV and (c) +150 mV within 200 seconds. Overview of the diffusion pathway for the Zn adatom on (d) the bare Zn (002) surface (IS and FS represent the initial state and final state, respectively); (e) the Zn (002) surface with two diagonally aligned water molecules; and (f) the Zn (002) surface with a horizontal adsorbed Cor molecule. (g) The energy barriers of the three diffusion pathways for the Zn adatom. (h) The calculated transfer numbers of Zn2+ ions in different electrolytes. | ||
To provide an in-depth comprehension of the Cor molecule retarding the Zn/Zn2+ redox kinetics, we perform climbing image nudged elastic band (CI-NEB) calculations and reveal the diffusion behaviors of the Zn adatom under various conditions. The diffusion pathway of the Zn adatom is selected as nine steps with fixed distance along the Zn (002) facet (Fig. 3d). Besides, the diffusion of the Zn adatom beneath two diagonally aligned water molecules (Fig. 3e) and one horizontally adsorbed Cor molecule (Fig. 3f) are also simulated. As illustrated in Fig. 3g, the diffusion energy barriers are relatively low when the adatom migrates along the Zn surface regardless of whether there exist H2O molecules (0.16 or 0.68 eV), corresponding to a mad 2D diffusion in BE. In contrast, a much higher energy barrier of 1.47 eV should be overcome for Zn adatom to migrate through beneath the Cor molecule, indicating the strong steric hindrance effect of Cor.59 Simultaneously, the Zn2+ ion transfer number is also evaluated by a series of EIS and CA analysis. The BE/5Cor electrolyte exhibits a transfer number of 0.16, which is much lower than that of BE (0.31) (Fig. S14, ESI† and Fig. 3h). This anticipative performance further confirms the reduced cation diffusion rate on the Zn anode surface due to the large size of adsorbate, which is highly consistent with the CI-NEB result. Briefly, the adsorbed Cor can induce an intense steric hindrance effect and notably confine the diffusion of the Zn adatom, thus resulting in moderate Zn/Zn2+ redox kinetics and unified plating/stripping behavior.
Highly homogeneous Zn plating/stripping mode with fine grains
To identify the specific surface morphology under dual regulation of nucleation and Zn/Zn2+ redox kinetics, various capacities of Zn are electro-deposited/stripped on copper foil with the surface analyzed by SEM. At 1 mA cm−2 in BE, the deposits primarily consist of anisotropic platelets with minor initial dendrites at the top when the capacity is 0.1 mA h cm−2 in BE (Fig. 4a). When the deposition capacity is increased to 0.5 mA h cm−2, the initial dendrites grow rapidly and develop into moss-like nanowires. Further augmenting the capacity to 1 mA h cm−2, moss-like deposits occupy the entire field of vision. In terms of larger current densities such as 5 and 10 mA cm−2, the ratio of dendrite in deposits contacted with Cu is reduced to a great extent and these deposits seem to be denser and smoother in BE (Fig. S15a and S16a, ESI†), because of the faster kinetics of Zn2+ ions diffusion.60 Nevertheless, due to the difference in atomic activity in each Zn crystal facet, the rapid growth of Zn platelets breaks the flat deposit surface and eventually forms the shape of dendrites. To further investigate the underlying relationship between |ηs| and Zn growth behavior, Zn is deposited at different overpotentials for a certain time in the BE electrolyte (Fig. S17, ESI†). Unlike the large Zn deposits in BE without external overpotential, much finer Zn sheets are observed on the Cu substrate lying in random directions after applying high overpotentials of −200, −220, and −250 mV for 1 min. This result is also consistent with those of CNTs in which higher |ηs| reduces the size of the nucleus. However, as the deposition time increases to 10 min, some erect sheets develop into thick platelets with a width of over 10 μm, which serve as dendrites to pierce the separator or form “dead Zn”, eventually destroying the battery. As a result, reducing the nuclei size without controlling subsequent growth cannot stabilize the Zn plating process. It is worth noting that the difference in electrodeposition morphology with/without external overpotential may be attributed to the influence of surface overpotential on Zn surface diffusion, as confirmed in Fig. S18 (ESI†). With the increase of overpotential, the time of inflection point on the CA curve decreases gradually, indicating that the transition of 2D to 3D diffusion is accelerated.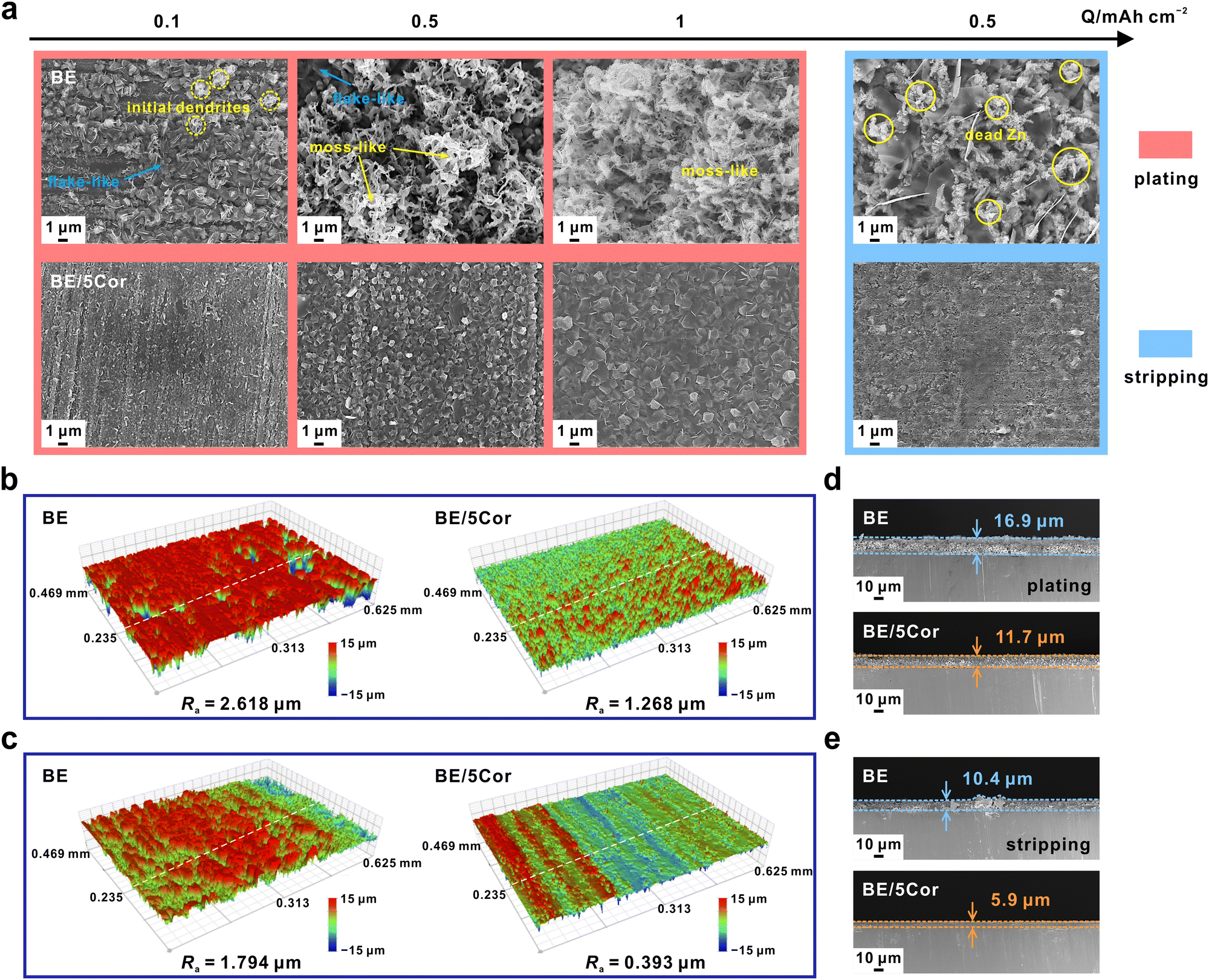 | ||
| Fig. 4 (a) SEM images of plating 0.1, 0.5, and 1 mA h cm−2 of Zn on the Cu substrate, respectively (inside the red rectangle), and stripping half of the pre-deposited Zn with a capacity of 1 mA h cm−2 on the Cu substrate (inside the blue rectangle). The applied current density is 1 mA cm−2. The 3D optical images of (b) deposited Zn with a capacity of 1 mA h cm−2 on the Cu substrate and (c) then stripping 0.5 mA h cm−2 of Zn at 1 mA cm−2. Cross-sectional SEM images of (d) deposited Zn with a capacity of 1 mA h cm−2 on the Cu substrate and (e) then stripping 0.5 mA h cm−2 of Zn at 1 mA cm−2. | ||
In BE/5Cor, benefitting from amplified |ηs|, finer Zn deposits are evenly distributed on the substrate at the initial stage (Fig. 4a and Fig. S15a and S16a, ESI†). As the deposition capacity increases, the surface diffusion is significantly restrained by the huge steric hindrance effect of Cor, thus leading to moderate Zn2+ ion reduction and suppressing distinct Zn grain growth, which achieves uniform electrodeposition with no dendrite or moss-like nanowires. In Fig. S19 (ESI†), it is clearly seen that the initial grain size slightly decreases with the increase of the current density, attributed to the amplified overpotential at higher current density (Fig. S20, ESI†). Even though the capacity increases to 10 times of the original (1 vs. 0.1 mA h cm−2), the expansion of deposit size maintains within the submicron scale (<1 μm), further confirming that the grain growth is suppressed by Cor. Fig. 4b and Fig. S21a (ESI†) present the corresponding 3D optical images of the deposition morphology at 1 mA cm−2 and 1 mA h cm−2. A more uniform surface with a smaller average roughness (Ra) of 1.268 μm is observed in BE/5Cor than that in BE (2.618 μm), which is consistent with the SEM images in Fig. 4a.
Meanwhile, the stripping process is also investigated by pre-electrodepositing Zn with a certain capacity of 1 mA h cm−2 on Cu substrate and then stripping 0.5 mA h cm−2 of Zn with the same current density (Fig. 4a and Fig. S15a and S16a, ESI†). There are two types of Zn stripping behavior identified in BE. At a low current density (1 mA cm−2), an amount of fragmented “dead Zn” remains and intersperses between the adjacent Zn platelets, which cannot participate in the subsequent stripping process. This behavior is highly related to the moss-like deposition under low current density. These moss-like Zn deposits are inherently polycrystalline and mechanically fragile. Although increasing the active area of the Zn anode would reduce the local current density, the reaction inhomogeneity across the whole surface easily fractures the moss-like deposits, therefore exacerbating the uneven stripping. At higher stripping current densities (5 and 10 mA cm−2), the inconsonant dissolution rate of Zn platelets along different directions results in the initially dense deposits becoming loose and separating from each other. As for BE/5Cor, the residual Zn remains compact and interconnecting at either current density, mainly based on the moderate dissolution rate of submicron grains. Remarkably, the residual Zn after stripping at 1 mA cm−2 exhibits high smoothness with a much lower Ra of 0.393 μm than that in BE (1.794 μm) (Fig. 4c and Fig. S21b, ESI†).
In addition, cross-sectional SEM images show that the deposition layers in BE/5Cor are thinner and tightly attached to the Cu substrate before and after stripping, whereas the loosely deposited Zn layers in BE are thicker and rugged, demonstrating the denser plating/stripping behavior under dual regulation of nucleation and Zn/Zn2+ redox kinetics (Fig. 4d, e and Fig. S15b, c and S16b, c, ESI†). The ratio of the deposition layer thickness after stripping (δs) to that before stripping (δp) can reflect the reversibility of Zn. Since the stripping capacity is half of the plating capacity (0.5 vs. 1 mA h cm−2), the ideal value of δs/δp is 0.5. Compared to BE, the value of δs/δp is far closer to 0.5 in BE/5Cor at either current density, which convincingly verifies the homogeneous plating/stripping mode (Fig. S22, ESI†).
On the other hand, side reactions including electrode corrosion and the hydrogen evolution reaction (HER) that occur on the surface of the Zn anode, are considered to disturb the Zn plating/stripping process.11 Since the interfacial chemical environment directly affects the stability of the Zn anode, we measure the pH values of different electrolytes. As shown in Fig. S23 (ESI†), the pH is slightly increased from 3.79 to 4.11 with the addition of Cor, which is conducive to the better anti-corrosion of the Zn anode in a mild acid solution. Subsequently, the linear polarization tests show an increase in corrosion potential and a significant decrease in corrosion current density for the BE/5Cor electrolyte, further demonstrating the anti-corrosion properties of the Zn anode with adsorbed Cor (Fig. S24a, ESI†). Moreover, the linear sweep voltammetry (LSV) testing results show a reduced current density under negative overpotential, which indicates that Cor molecules can slow down the HER rate of the Zn anode and reduce the production of hydrogen (Fig. S24b, ESI†). To further evaluate the resistance of the Zn anode to side reactions during cycling, the Zn foils cycled 10 times at 1 mA cm−2 in different electrolytes with two-electrode beaker cells are characterized by SEM and XRD (Fig. S25, ESI†). After cycling, some light grey fragments are found at the bottom of the beaker cell in BE, which are regarded as “dead Zn” attributed to uneven stripping. The SEM image also shows the mixture of moss-like dendrites and irregular flakes, which are identified as by-products (Zn4SO4(OH)6·5H2O) in XRD patterns (Fig. S25b, ESI†). In sharp contrast, Zn foil cycled in BE/5Cor displays a smoother surface with no discernible by-product formed, implying the uniform Zn plating/stripping process with inhibited side reactions.
Superior electrochemical performance of the Zn anode
We next evaluate the stability of the Zn anode in BE and BE/5Cor electrolytes with Zn‖Zn symmetric cells. As shown in Fig. 5a, the Zn anode displays a much longer stable cycling lifespan in BE/5Cor together with increased voltage hysteresis (over 900 h) than that in BE (∼51 h) at a current density of 5 mA cm−2 and an areal capacity of 5 mA h cm−2. It is worth noting that the increased voltage hysteresis in the Zn‖Zn symmetric cell mainly originated from the amplified |ηs| with the addition of Cor, which has been confirmed in Fig. 2e and g. The stability of the adsorbed Cor upon dynamic plating/stripping is also evaluated by FTIR spectra (Fig. S26, ESI†). Compared with the Zn anode before cycling, the adsorption peaks corresponding to the chemical bonds from the Cor molecule still distinctly exist on the Zn anode surface after 10 cycles at 5 mA cm−2 and 5 mA h cm−2, and are slightly weakened after 30 cycles, indicating that the adsorbed Cor has superior stability during cycling to chronically regulate the plating/stripping mode of the Zn anode. In addition, there is nearly no new adsorption peak appears after cycling, which confirms that the adsorbed Cor would not decompose to form a solid electrolyte interphase (SEI), consistent with the XRD pattern in Fig. S25b (ESI†). Despite the concern that higher |ηs| may do harm to battery performance, the Zn anode in BE/5Cor can even deliver an extended operation time for 600 h under harsher cycling conditions (10 mA cm−2 and 10 mA h cm−2), which is about 15 folds to that in BE (∼41 h, Fig. 5b). The cycling stability of the Zn anode at a high Zn utilization rate of 50% (depth of discharge, DODZn) is further assessed. As shown in Fig. 5c, due to the uneven nucleation and uncontrollable Zn/Zn2+ redox kinetics in the BE electrolyte, the Zn anode is rapidly consumed and perforated, leading to the open circuit of the battery within 30 h. In sharp contrast, the homogeneous plating/stripping mode with fine grains enables the Zn anode to be stably cycled for ∼120 h in the BE/5Cor electrolyte and extends the lifespan of the battery to 220 h with an increase in voltage hysteresis. In fact, the Zn anode undergoes huge volume change under such a high DODZn, which would induce the internal stress accumulating in the bulk electrode.61,62 With the increment of the cycle number (from ∼120 to 220 h), the accumulated internal stress would inevitably destroy the connection of the deposited Zn fine grains and shatter the electrode, thus deteriorating the electric contact and increasing the voltage hysteresis. Impressively, the cumulative plating capacity (CPC) at different current densities (2.25 A h cm−2 for 5 mA cm−2, 3 A h cm−2 for 10 mA cm−2) distinguishes our system with superior performance compared to most of the previous reports with other additives (Fig. 5f and Table S1, ESI†).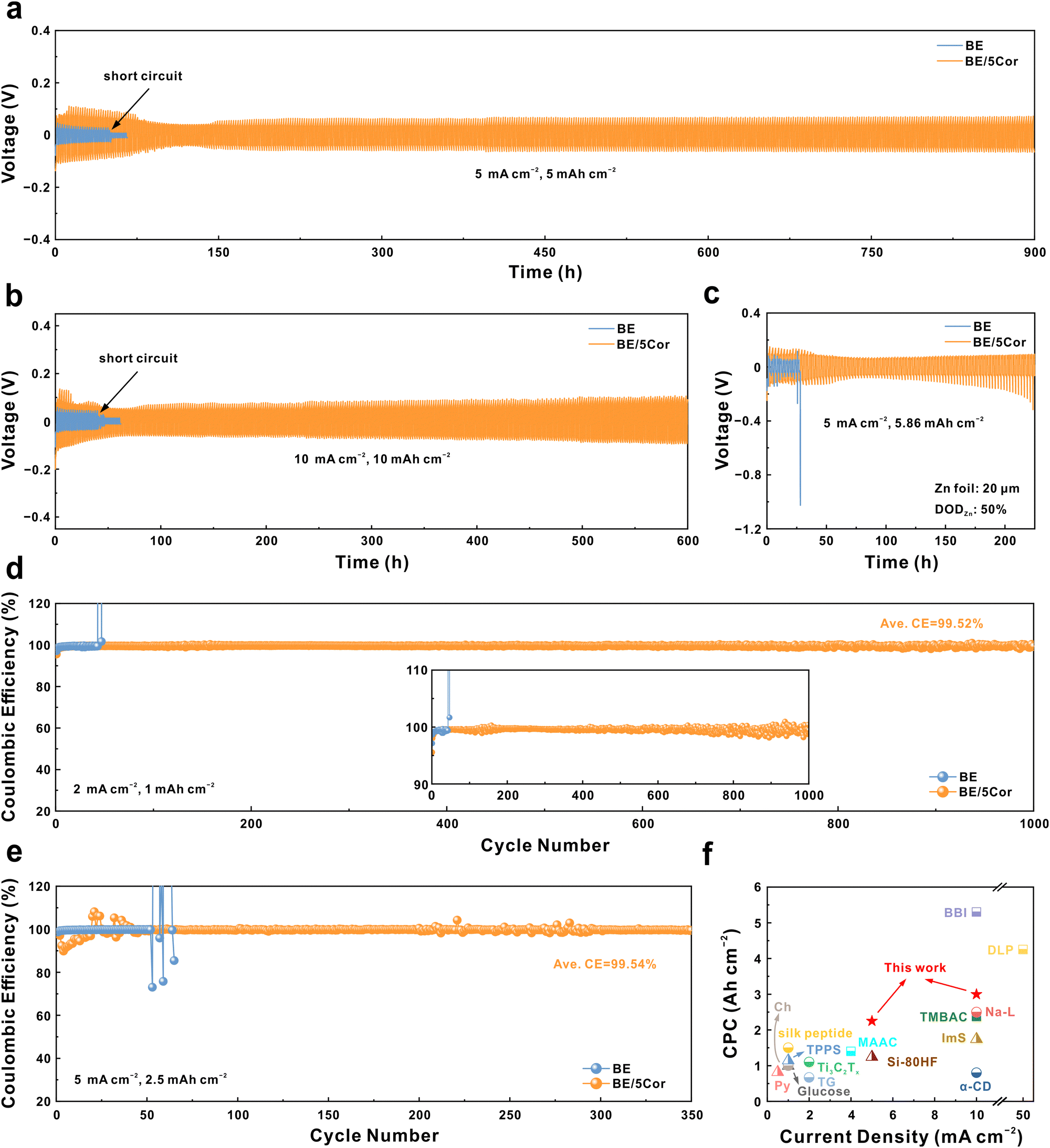 | ||
| Fig. 5 Voltage profiles of Zn‖Zn symmetric cells galvanostatically cycled at (a) 5 mA cm−2, 5 mA h cm−2; (b) 10 mA cm−2, 10 mA h cm−2; and (c) 5 mA cm−2, 5.86 mA h cm−2 (DODZn = 50%), respectively. CE comparisons of Zn‖Cu asymmetric cells at (d) 2 mA cm−2, 1 mA h cm−2 and (e) 5 mA cm−2, 2.5 mA h cm−2. (f) Comparison of the cumulative plating capacity (CPC) in this work with previous reports (see Table S1, ESI†, for details of the data). | ||
We further assess the reversibility of the Zn anode during plating/stripping in Zn‖Cu asymmetric cells, with the Coulombic efficiencies (CEs) recorded in Fig. 5d and e. When cycling at 2 mA cm−2 and 1 mA h cm−2, the Zn‖Cu cell with the BE electrolyte operates for less than 43 h and reaches a short circuit at the 45th cycle, as confirmed by the flat stripping curve with a voltage near 0 V (Fig. S27a, ESI†). In sharp contrast, the cell with the BE/5Cor electrolyte stably cycles for over 1000 times (∼1000 h) with a remarkable average CE of 99.52%, where the homogeneous plating/stripping can be evidenced by the reversible and stable voltage curves (Fig. S27b, ESI†). The larger voltage gap between discharge and charge plateaus in BE/5Cor is also consistent with the phenomenon of amplified |ηs| on the Zn anode surface. The Zn‖Cu cell with the BE/5Cor electrolyte also delivers superior reversibility with an ultra-high average CE of 99.54% for 350 cycles under 5 mA cm−2 and 2.5 mA h cm−2, whereas the cell with BE shows a distinct fluctuation of the CE after 50 cycles due to rampant dendritic growth and formation of “dead Zn”.
To envisage the feasibility of dual regulation for nucleation and Zn/Zn2+ redox kinetics in real-life batteries, Zn‖NVO full cells are assembled using NaV3O8·1.5H2O (NVO) as cathode material. The SEM image and XRD pattern of NVO powders illustrate a nanorod morphology with several micrometers in length and a highly crystalline phase that can be assigned with P21/m space group (JCPDS: 16-0601, Fig. S28 and S29, ESI†). The CV profiles of Zn‖NVO full cells with different electrolytes (Fig. 6a) present similar redox peaks in both curves, which indicates that Cor is prone to work on the Zn anode surface and does not participate in the redox reactions of the NVO cathode during discharging/charging. Fig. 6b and c show the discharge and charge curves of Zn‖NVO cells with/without Cor. With the current density increasing from 0.2 to 5 A g−1, the battery with the BE/5Cor electrolyte still exhibits a superior discharge specific capacity of 140.5 mA h g−1, implying the better rate performance of the Zn anode.
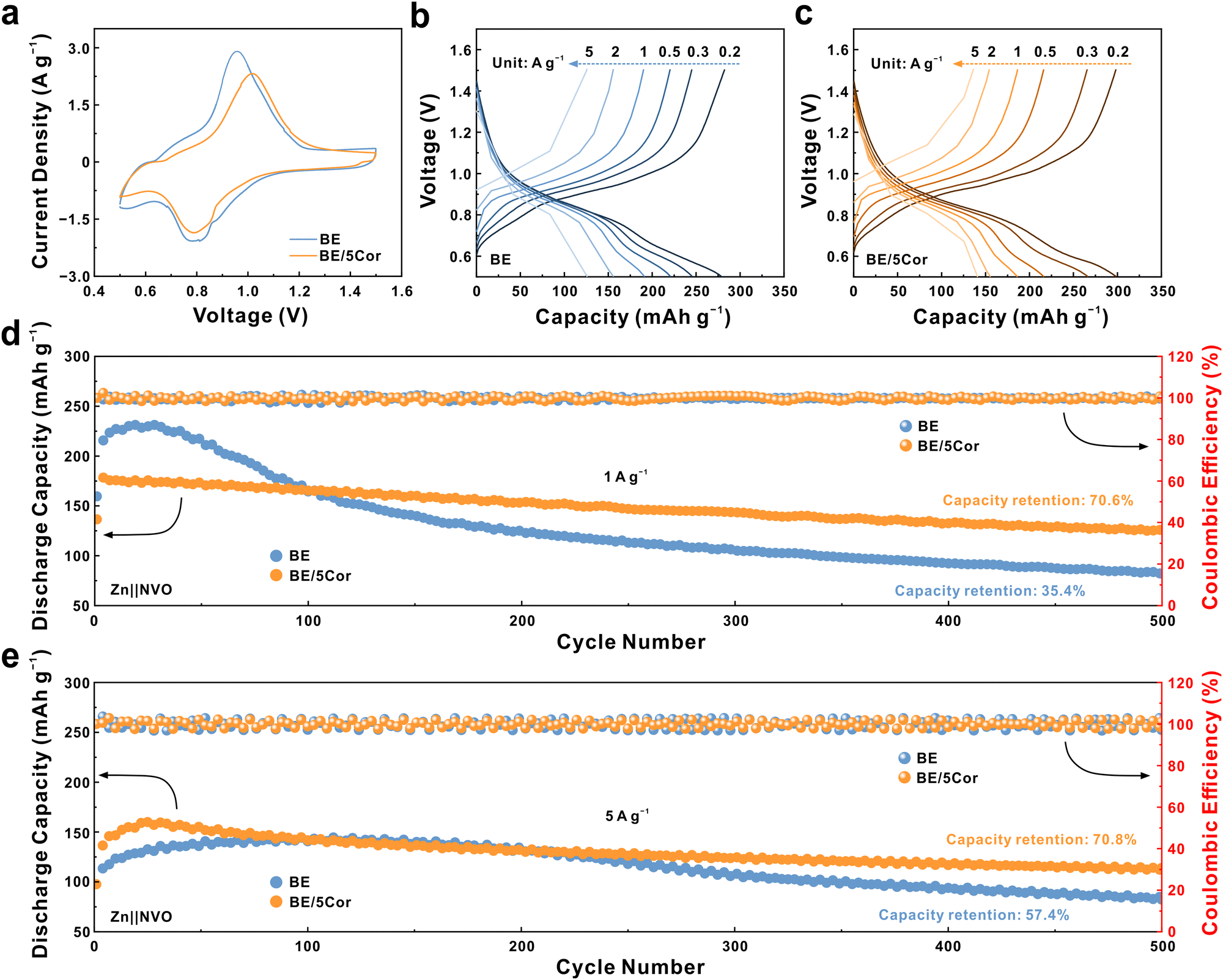 | ||
| Fig. 6 (a) CV curves of Zn‖NVO full cells. The charge/discharge curves of Zn‖NVO full cells at different current densities in (b) BE and (c) BE/5C or electrolytes. Long-term cycling performance of Zn‖NVO full cells at (d) 1 A g−1 and (e) 5 A g−1. | ||
The long-term cycling test of Zn‖NVO batteries (Fig. 6d, at a rate of 1 A g−1) suggests that the full cell with BE undergoes an activated process within 50 cycles to reach a peak specific capacity of 231.8 mA h g−1, then rapidly decays to ∼35.4% of that after 500 cycles due to the severe side reactions under low rate.31,63 Notably, with the adsorbed Cor, the full cell secures 70.6% of its original capacity (125.9 mA h g−1) after 500 cycles. At a higher rate of 5 A g−1, the homogeneous plating/stripping of the Zn anode in the BE/5Cor electrolyte pledges the full cell to achieve an outstanding capacity retention of 70.8% (with a specific capacity of 114 mA h g−1 at the 500th cycle, Fig. 6e). By contrast, the specific capacity in BE is drastically degraded to 57.4% within 500 cycles, suggesting an irreversible capacity loss with dendritic growth/dissolution of the Zn anode. By assembling a Zn‖NVO pouch cell using the BE/5Cor electrolyte and a mass loading of 3.34 mg cm−2 for NVO cathode (Fig. S30a, ESI†), a remarkable capacity retention of 79.3% is achieved at the rate of 0.5 A g−1 over 100 cycles, which demonstrates the feasibility of implementing Cor additive to homogenize Zn plating/stripping in highly reversible AZMBs (Fig. S30b, ESI†).
Conclusions
In summary, we describe a facile strategy to achieve homogeneous Zn plating/stripping with fine grains by regulating both nucleation and Zn/Zn2+ redox kinetics. The added Cor molecules preferentially adsorb on the Zn surface to amplify |ηs| and inhibit side reactions, which significantly reduce the nuclei size and make them uniformly spread over the entire substrate. Meanwhile, the prominent steric hindrance effect caused by Cor retarded Zn surface diffusion alleviates the Zn/Zn2+ redox kinetics for growth/dissolution, limiting the evolution of Zn grains. As such, a homogeneous Zn plating/stripping is facilitated to enable a dendrite-free and high-reversible Zn anode. The Zn‖Zn symmetric cell with BE/5Cor exhibits superior cycling stability under different conditions (900 h at 5 mA cm−2, 5 mA h cm−2 and 600 h at 10 mA cm−2, 10 mA h cm−2). The Zn‖NVO coin cell delivers capacity retention values of 70.6% and 70.8% for 500 cycles at rates of 1 and 5 A g−1, respectively. Remarkably, the Zn‖NVO pouch cell with a mass loading of 3.34 mg cm−2 can even retain 79.3% of its original specific capacity over 100 cycles at 0.5 A g−1. This facile dual regulation strategy opens up another window for the development of future high-performance AZMBs.Author contributions
Y. Jiang and Z. Luo conceived the idea. Z. Luo designed the research. Z. Luo and S. Chen conducted the materials synthesis, electrochemical tests, and materials characterization. Y. Xia carried out the first-principles calculations and analyzed the computational results. Z. Luo and Y. Xia wrote the original manuscript. S. Chen, X. Wu, E. Akinlabi, B. B. Xu, and Y. Jiang reviewed the manuscript and provided the major revision. H. Pan and M. Yan contributed to the data interpretation and mechanism discussion. All authors discussed the results and commented on the manuscript. Z. Luo and Y. Xia contributed equally to this work.Data availability
The raw data supporting this article can be accessed openly by emailing the corresponding authors and through the institutional repository, in line with UKRI and NSFC requirements. The data to evaluate the conclusion have been included in the paper and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (LZ23B030003) and the Fundamental Research Funds for the Central Universities (226-2024-00075). B. B. Xu is grateful for the support from the Engineering and Physical Sciences Research Council (EPSRC, UK) RiR grant – RIR18221018-1.Notes and references
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- M. Qiu, P. Sun, Y. Wang, L. Ma, C. Zhi and W. Mai, Angew. Chem., Int. Ed., 2022, 61, e202210979 CrossRef CAS PubMed.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- Y. Du, Y. Li, B. B. Xu, T. X. Liu, X. Liu, F. Ma, X. Gu and C. Lai, Small, 2022, 18, 2104640 CrossRef CAS PubMed.
- Z. Yi, G. Chen, F. Hou, L. Wang and J. Liang, Adv. Energy Mater., 2021, 11, 2003065 CrossRef CAS.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- H. Yu, Y. Chen, W. Wei, X. Ji and L. Chen, ACS Nano, 2022, 16, 9736–9747 CrossRef CAS PubMed.
- M. Kim, J. Lee, Y. Kim, Y. Park, H. Kim and J. W. Choi, J. Am. Chem. Soc., 2023, 145, 15776–15787 CrossRef CAS PubMed.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- J. Luo, L. Xu, Y. Zhou, T. Yan, Y. Shao, D. Yang, L. Zhang, Z. Xia, T. Wang, L. Zhang, T. Cheng and Y. Shao, Angew. Chem., Int. Ed., 2023, 62, e202302302 CrossRef CAS PubMed.
- J. X. K. Zheng, J. Yin, T. Tang and L. A. Archer, ACS Energy Lett., 2023, 8, 2113–2121 CrossRef CAS.
- H. Ren, S. Li, B. Wang, Y. Zhang, T. Wang, Q. Lv, X. Zhang, L. Wang, X. Han, F. Jin, C. Bao, P. Yan, N. Zhang, D. Wang, T. Cheng, H. Liu and S. Dou, Adv. Mater., 2023, 35, 2208237 CrossRef CAS PubMed.
- R. Zhao, H. Wang, H. Du, Y. Yang, Z. Gao, L. Qie and Y. Huang, Nat. Commun., 2022, 13, 3252 CrossRef CAS PubMed.
- G. Duan, Y. Wang, B. Luo, L. Sun, S. Zheng, J. Huang and Z. Ye, Energy Storage Mater., 2023, 61, 102882 CrossRef.
- Z. Bie, Q. Yang, X. Cai, Z. Chen, Z. Jiao, J. Zhu, Z. Li, J. Liu, W. Song and C. Zhi, Adv. Energy Mater., 2022, 12, 2202683 CrossRef CAS.
- Z. Chen, J. Zhao, Q. He, M. Li, S. Feng, Y. Wang, D. Yuan, J. Chen, H. N. Alshareef and Y. Ma, ACS Energy Lett., 2022, 7, 3564–3571 CrossRef CAS.
- H. Yu, Y. Zeng, N. W. Li, D. Luan, L. Yu and X. W. Lou, Sci. Adv., 2022, 8, eabm5766 CrossRef CAS PubMed.
- Y. Zhu, H. Y. Hoh, S. Qian, C. Sun, Z. Z. Wu, Z. Huang, L. Wang, M. Batmunkh, C. Lai, S. Zhang and Y. L. Zhong, ACS Nano, 2022, 16, 14600–14610 CrossRef CAS PubMed.
- X. Zeng, K. Xie, S. Liu, S. Zhang, J. Hao, J. Liu, W. K. Pang, J. Liu, P. Rao, Q. Wang, J. Mao and Z. Guo, Energy Environ. Sci., 2021, 14, 5947–5957 RSC.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- Y. Zuo, K. Wang, P. Pei, M. Wei, X. Liu, Y. Xiao and P. Zhang, Mater. Today Energy, 2021, 20, 100692 CrossRef CAS.
- J. Wang, Y. Yang, Y. Zhang, Y. Li, R. Sun, Z. Wang and H. Wang, Energy Storage Mater., 2021, 35, 19–46 CrossRef.
- W. Zhang, M. Dong, K. Jiang, D. Yang, X. Tan, S. Zhai, R. Feng, N. Chen, G. King, H. Zhang, H. Zeng, H. Li, M. Antonietti and Z. Li, Nat. Commun., 2022, 13, 5348 CrossRef CAS PubMed.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- C. Li, X. Xie, S. Liang and J. Zhou, Energy Environ. Mater., 2020, 3, 146–159 CrossRef CAS.
- H. Jia, Z. Wang, B. Tawiah, Y. Wang, C.-Y. Chan, B. Fei and F. Pan, Nano Energy, 2020, 70, 104523 CrossRef CAS.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- M. Luo, C. Wang, H. Lu, Y. Lu, B. B. Xu, W. Sun, H. Pan, M. Yan and Y. Jiang, Energy Storage Mater., 2021, 41, 515–521 CrossRef.
- Y. Zhang, X. Han, R. Liu, Z. Yang, S. Zhang, Y. Zhang, H. Wang, Y. Cao, A. Chen and J. Sun, Small, 2022, 18, 2105978 CrossRef CAS PubMed.
- Q. Zou, Z. Liang, W. Wang, D. Dong and Y.-C. Lu, Energy Environ. Sci., 2023, 16, 6026–6034 RSC.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef.
- J. Zhou, F. Wu, Y. Mei, Y. Hao, L. Li, M. Xie and R. Chen, Adv. Mater., 2022, 34, 2200782 CrossRef CAS PubMed.
- R. Qin, Y. Wang, L. Yao, L. Yang, Q. Zhao, S. Ding, L. L. Liu and F. Pan, Nano Energy, 2022, 98, 107333 CrossRef CAS.
- G. Feng, J. Guo, H. Tian, Z. Li, Y. Shi, X. Li, X. Yang, D. Mayerich, Y. Yang and X. Shan, Adv. Energy Mater., 2022, 12, 2103484 CrossRef CAS.
- D. W. Oxtoby, Acc. Chem. Res., 1998, 31, 91–97 CrossRef CAS.
- R. P. Sear, J. Phys. Chem. B, 2006, 110, 21944–21949 CrossRef CAS PubMed.
- Z. Hu, F. Zhang, A. Zhou, X. Hu, Q. Yan, Y. Liu, F. Arshad, Z. Li, R. Chen, F. Wu and L. Li, Nano-Micro Lett., 2023, 15, 171 CrossRef CAS PubMed.
- S.-J. Zhang, J. Hao, D. Luo, P.-F. Zhang, B. Zhang, K. Davey, Z. Lin and S.-Z. Qiao, Adv. Energy Mater., 2021, 11, 2102010 CrossRef CAS.
- Z. Liu, R. Wang, Q. Ma, J. Wan, S. Zhang, L. Zhang, H. Li, Q. Luo, J. Wu, T. Zhou, J. Mao, L. Zhang, C. Zhang and Z. Guo, Adv. Funct. Mater., 2024, 34, 2214538 CrossRef CAS.
- S. Jin, J. Yin, X. Gao, A. Sharma, P. Chen, S. Hong, Q. Zhao, J. Zheng, Y. Deng, Y. L. Joo and L. A. Archer, Nat. Commun., 2022, 13, 2283 CrossRef CAS.
- Z. Luo, Y. Xia, S. Chen, X. Wu, R. Zeng, X. Zhang, H. Pan, M. Yan, T. Shi, K. Tao, B. B. Xu and Y. Jiang, Nano-Micro Lett., 2023, 15, 205 CrossRef CAS PubMed.
- Y. Xia, Z. Hong, L. Zhou, S. Chen, Z. Luo, S. Jin, Y. Huang, Y. Jiang and Y. Wu, J. Energy Chem., 2023, 87, 153–161 CrossRef CAS.
- Y. Zhao, H. Liu, Y. Huyan, D. Lei, N. Li, S. Tian and J. G. Wang, J. Energy Chem., 2023, 79, 450–458 CrossRef CAS.
- S. Yang, A. Chen, Z. Tang, Z. Wu, P. Li, Y. Wang, X. Wang, X. Jin, S. Bai and C. Zhi, Energy Environ. Sci., 2024, 17, 1095–1106 RSC.
- H. Dou, X. Wu, M. Xu, R. Feng, Q. Ma, D. Luo, K. Zong, X. Wang and Z. Chen, Angew. Chem., Int. Ed., 2024, 63, e202401974 CrossRef CAS PubMed.
- C. Huang, X. Zhao, S. Liu, Y. Hao, Q. Tang, A. Hu, Z. Liu and X. Chen, Adv. Mater., 2021, 33, 2100445 CrossRef CAS PubMed.
- K. Bao, M. Wang, Y. Zheng, P. Wang, L. Yang, Y. Jin, H. Wu and B. Sun, Nano Energy, 2024, 120, 109089 CrossRef CAS.
- J. Yin, M. Li, X. Feng, T. Cui, J. Chen, F. Li, M. Wang, Y. Cheng, S. Ding, X. Xu and J. Wang, J. Mater. Chem. A, 2024, 12, 1543–1550 RSC.
- Z. Miao, Q. Liu, W. Wei, X. Zhao, M. Du, H. Li, F. Zhang, M. Hao, Z. Cui, Y. Sang, X. Wang, H. Liu and S. Wang, Nano Energy, 2022, 97, 107145 CrossRef CAS.
- R. Meng, H. Li, Z. Lu, C. Zhang, Z. Wang, Y. Liu, W. Wang, G. Ling, F. Kang and Q. H. Yang, Adv. Mater., 2022, 34, 2200677 CrossRef CAS PubMed.
- Z. Xing, C. Huang and Z. Hu, Coord. Chem. Rev., 2022, 452, 214299 CrossRef CAS.
- X. Yu, Z. Li, X. Wu, H. Zhang, Q. Zhao, H. Liang, H. Wang, D. Chao, F. Wang, Y. Qiao, H. Zhou and S. G. Sun, Joule, 2023, 7, 1145–1175 CrossRef CAS.
- K. Zouhri and S. Y. Lee, Energy Convers. Manage., 2016, 121, 1–12 CrossRef CAS.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- P. Biswal, A. Kludze, J. Rodrigues, Y. Deng, T. Moon, S. Stalin, Q. Zhao, J. Yin, L. F. Kourkoutis and L. A. Archer, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2012071118 CrossRef CAS PubMed.
- P. Cao, X. Zhou, A. Wei, Q. Meng, H. Ye, W. Liu, J. Tang and J. Yang, Adv. Funct. Mater., 2021, 31, 2100398 CrossRef CAS.
- X. Wu, Y. Xia, S. Chen, Z. Luo, X. Zhang, Y. Lu, H. Pan, B. B. Xu, M. Yan and Y. Jiang, Small, 2024, 20, 2306739 CrossRef CAS PubMed.
- L. Zheng, H. Li, X. Wang, Z. Chen, C. Hu, K. Wang, G. Guo, S. Passerini and H. Zhang, ACS Energy Lett., 2023, 8, 2086–2096 CrossRef CAS.
- H. Lu, X. Zhang, M. Luo, K. Cao, Y. Lu, B. B. Xu, H. Pan, K. Tao and Y. Jiang, Adv. Funct. Mater., 2021, 31, 2103514 CrossRef CAS.
- Z. Cai, J. Wang, Z. Lu, R. Zhan, Y. Ou, L. Wang, M. Dahbi, J. Alami, J. Lu, K. Amine and Y. Sun, Angew. Chem., Int. Ed., 2022, 61, e202116560 CrossRef CAS PubMed.
- J. Dong, H. Peng, J. Wang, C. Wang, D. Wang, N. Wang, W. Fan, X. Jiang, J. Yang and Y. Qian, Energy Storage Mater., 2023, 54, 875–884 CrossRef.
- J. Chen, X. Qiao, X. Han, J. Zhang, H. Wu, Q. He, Z. Chen, L. Shi, Y. Wang, Y. Xie, Y. Ma and J. Zhao, Nano Energy, 2022, 103, 107814 CrossRef CAS.
- K. Xu, Commun. Mater., 2022, 3, 31 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02264e |
| ‡ Z. Luo and Y. Xia contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |