
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Distinguishing bulk redox from near-surface degradation in lithium nickel oxide cathodes†
Lijin
An
a,
Jack E. N.
Swallow
a,
Peixi
Cong
a,
Ruomu
Zhang
a,
Andrey D.
Poletayev
 ab,
Erik
Björklund
ab,
Pravin N.
Didwal
ab,
Michael W.
Fraser
ab,
Leanne A. H.
Jones
a,
Conor M. E.
Phelan
a,
Namrata
Ramesh
a,
Grant
Harris
c,
Christoph J.
Sahle
d,
Pilar
Ferrer
e,
David C.
Grinter
e,
Peter
Bencok
e,
Shusaku
Hayama
e,
M. Saiful
Islam
ab,
Robert
House
ab,
Peter D.
Nellist
ab,
Robert J.
Green
cf,
Rebecca J.
Nicholls
a and
Robert S.
Weatherup
*abe
ab,
Erik
Björklund
ab,
Pravin N.
Didwal
ab,
Michael W.
Fraser
ab,
Leanne A. H.
Jones
a,
Conor M. E.
Phelan
a,
Namrata
Ramesh
a,
Grant
Harris
c,
Christoph J.
Sahle
d,
Pilar
Ferrer
e,
David C.
Grinter
e,
Peter
Bencok
e,
Shusaku
Hayama
e,
M. Saiful
Islam
ab,
Robert
House
ab,
Peter D.
Nellist
ab,
Robert J.
Green
cf,
Rebecca J.
Nicholls
a and
Robert S.
Weatherup
*abe
aDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK. E-mail: robert.weatherup@materials.ox.ac.uk
bThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot OX11 0RA, UK
cDepartment of Physics and Engineering Physics, University of Saskatchewan, Saskatoon, Canada S7N 5E2
dESRF – The European Synchrotron, 71 Avenue des Martyrs, 38000 Grenoble, France
eDiamond Light Source, Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK
fStewart Blusson Quantum Matter Institute, University of British Columbia, Vancouver, Canada V6T 1Z1
First published on 13th September 2024
Abstract
Ni-rich layered oxide cathodes can deliver higher energy density batteries, but uncertainties remain over their charge compensation mechanisms and the degradation processes that limit cycle life. Trapped molecular O2 has been identified within LiNiO2 at high states of charge, as seen for Li-rich cathodes where excess capacity is associated with reversible oxygen redox. Here we show that bulk redox in LiNiO2 occurs by Ni–O rehybridization, lowering the electron density on O sites, but importantly without the involvement of molecular O2. Instead, trapped O2 is related to degradation at surfaces in contact with the electrolyte, and is accompanied by Ni reduction. O2 is removed on discharge, but excess Ni2+ persists forming a reduced surface layer, associated with impeded Li transport. This implicates the instability of delithiated LiNiO2 in contact with the electrolyte in surface degradation through O2 formation and Ni reduction, highlighting the importance of surface stabilisation strategies in suppressing LiNiO2 degradation.
Broader contextIncreasing the capacity of Li-ion batteries requires cathodes which can reversibly deintercalate more lithium without leading to structural instability and severe capacity fade. To this end, Ni-rich layered cathodes are under development for next-generation batteries, with LiNiO2 the archetypal system for investigating their charging mechanisms. However, the role played by different redox centres in LiNiO2 is still debated, and the connections with structural instabilities and associated degradation are not yet fully established. Recent reports have suggested the involvement of molecular O2 in the bulk redox process at high states of charge, with direct experimental detection of O2 based on techniques that probe 100–200 nm into the surface of the few μm-sized cathode particles. Here, we combine a broad suite of X-ray spectroscopies with varying information depths (10 nm to 10 μm) to separate the bulk redox from surface degradation. We reveal that trapped O2 formation in LiNiO2 is primarily associated with degradation at surfaces in contact with the electrolyte, rather than contributing to the bulk redox process. Interpretation of experimental spectra using theoretical calculations shows that bulk charge compensation proceeds by Ni–O rehybridization. These findings highlight the importance of using bulk sensitive techniques to understand redox, and suggests design strategies for stabilising high energy density Ni-rich cathodes. |
Introduction
Layered transition metal (TM) oxides, LiTMO2 (TM = Co, Ni, Mn, etc.), are the cathode materials of choice for commercial high-energy density Li-ion batteries, reversibly intercalating Li over thousands of cycles.1,2 Ni-rich stoichiometries are increasingly favoured to increase capacity, and lower Co content, which is expensive and has ethical concerns around its mining.3 In the traditional picture of charge compensation, Li+ removal is compensated by an increase in the formal oxidation state of the redox-active TM centres via a single electron transfer. However, it is well-established that there are accompanying changes in TM–O bond covalency meaning both TM and O sites are involved.4–9The archetypal Ni-rich cathode material, LiNiO2, undergoes several first-order structural phase transitions upon delithiation, with significant degradation observed at high potentials that has been associated with severe lattice changes,10,11 structural degradation,12,13 gas evolution,14,15 and parasitic reactions with the electrolyte.16,17 However, the causality and connections between these different modes of degradation are not yet fully established. Oxygen loss from the cathode surface and the associated formation of a reduced surface layer have been widely observed for Ni-rich cathode materials particularly above the H2–H3 transition,15,18–20 and are found to depend critically on the upper cut off voltage (UCV) and electrolyte formulation.17,21–23 Recently, studies of Ni-rich cathodes at high potentials (≥4.3 V during charge) have also shown the emergence of a distinct signature in O K-edge resonant inelastic X-ray scattering (RIXS) spectra at an excitation energy of ∼531.5 eV.24–26 For Li-rich materials that show excess capacity beyond TM cation redox, this signature is typically taken as evidence of the formation of molecular O2 trapped in pores throughout the cathode bulk, as a result of charge compensation by non-bonding O orbitals.27,28 However, the TM vacancies necessary to accommodate this are not expected in LiNiO2, with experimental samples typically containing excess Ni. Furthermore, LiNiO2 does not exhibit excess capacity that might be associated with molecular O2 redox. Nevertheless, bulk sensitive Ni K-edge X-ray absorption Near Edge Structure (XANES) measurements of LiNiO2 have indicated a plateauing of the main edge half-height position at similarly high states of charge, which has been taken as evidence of the formal Ni oxidation state no longer changing in the bulk, and thus a change in the redox mechanism.29
Although O K-edge RIXS and the related fluorescence yield X-ray absorption spectroscopy (FY-XAS) are widely referred to as bulk-sensitive (∼200 nm information depth), in the context of Li-ion cathode materials where typical secondary particle diameters are >5 μm, these methods probe <10% of the particle volume nearest to the surface. Attribution of bulk molecular O2 redox based on these methods alone is therefore ambiguous. Similar concerns have been raised around identifying oxygen redox with hard X-ray photoelectron spectroscopy (HAXPES), where typical probing depths are tens of nm.30 Solid-state 17O magic-angle-spinning nuclear magnetic resonance (NMR) spectroscopy provides an alternative bulk-averaged approach to estimate the amount of O2 present in the lattice.31 However, it does not resolve the spatial distribution of O2 molecules, nor has it been reported for LiNiO2 to date. Whereas, online electrochemical mass spectrometry (OEMS) can quantify the gas release associated with O-loss from the cathode surface, it does not probe molecular O2 that remains trapped within the cathode.15,20,32 The extent to which oxygen redox is involved in charge compensation in the LiNiO2 bulk thus remains unclear, motivating approaches that can provide comparable information with surface- and bulk-sensitivity.
Here we combine complementary core-loss spectroscopies to obtain a depth-resolved (10 nm to >10 μm) account of the redox processes in LiNiO2 and distinguish reversible bulk redox processes from near-surface degradation. X-ray Raman Spectroscopy (XRS, >10 μm information depth) reveals that in the bulk of LiNiO2 secondary particles there is a continuous change in both the Ni L3,2-edge and O K-edge spectra with state of charge (SoC) up to 4.8 V. This is consistent with charge compensation proceeding by rehybridization between the Ni and O centres, lowering the electron density on O sites but with Ni–O coordination still preserved. Features of trapped molecular O2 appear at potentials of ≳4.2 V in O K-edge FY-XAS, accompanied by increased Ni2+ contributions in the Ni L3,2-edge. Importantly, these changes are less pronounced in bulk-averaged XRS measurements indicating that formation of molecular O2 is a predominantly surface process. Total Electron Yield (TEY)-XAS measurements (∼10 nm information depth) confirm that a densified NiO-like layer forms in direct contact with electrolyte, whilst FY-XAS measurements are consistent with an extended cation mixing layer in which Ni2+ ions have migrated to occupy Li sites. Scanning transmission electron microscopy–electron energy loss spectroscopy (STEM–EELS) further confirms this picture of a reduced surface layer (RSL) that extends ∼200 nm into the surface for LiNiO2 which has been cycled to 4.8 V vs. Li/Li+. This understanding emphasises the importance of strategies to stabilise the interfaces of Ni-rich cathode materials in contact with electrolyte (e.g. cathode coatings/gradients, electrolyte formulation), rather than bulk stabilisation approaches (e.g. pillaring) that might sacrifice capacity.
Results and discussion
Trends in bulk chemical state
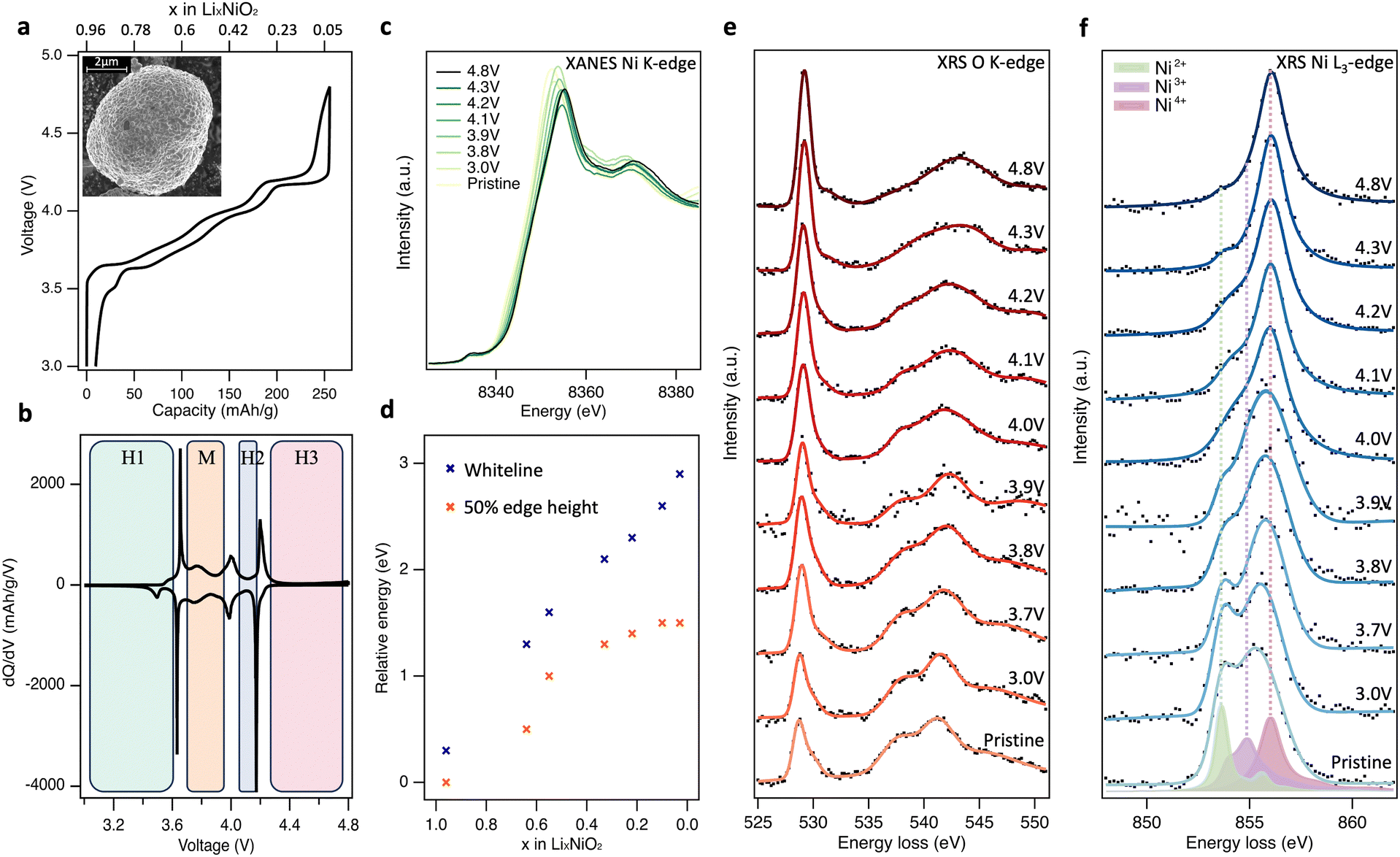
The charge–discharge profile for the 2nd cycle of the composite polycrystalline LiNiO2 electrode is shown in Fig. 1(a), together with an inset showing a scanning electron micrograph (SEM) of the pristine LiNiO2 active material (see ESI,† Fig. S1 for further characterisation). The ∼5 μm diameter spheroidal LiNiO2 secondary particles are composed of sub-μm primary particles. The voltage profiles show distinct plateaus associated with the first-order structural phase transitions of LiNiO2 on delithiation, apparent as maxima in the dQ/dV plots (Fig. 1(b)), at potentials consistent with prior literature.15,33,34 Powder X-ray diffraction (XRD) of the pristine material (see ESI,† Fig. S1c) closely resembles the calculated pattern for LiNiO2 with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. This hexagonal H1 phase transitions to the monoclinic M phase at ∼3.67 V, then to the H2 phase at ∼4.0 V, followed by the H3 phase at ∼4.2 V. The voltage profile shows noticeable hysteresis above ∼4.3 V, with the voltage rapidly dropping from 4.8 V to ∼4.2 V on discharge. However, the capacity reached at 4.8 V is 256 mA h g−1 which compares with a maximum theoretical capacity of 264 mA h g−1, based on the pristine material having ∼4% Ni excess as determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES).26 This provides an initial indication that the full capacity of the electrode can be accounted for by formal Ni redox alone, without obvious excess capacity associated with molecular O2 redox.
m space group. This hexagonal H1 phase transitions to the monoclinic M phase at ∼3.67 V, then to the H2 phase at ∼4.0 V, followed by the H3 phase at ∼4.2 V. The voltage profile shows noticeable hysteresis above ∼4.3 V, with the voltage rapidly dropping from 4.8 V to ∼4.2 V on discharge. However, the capacity reached at 4.8 V is 256 mA h g−1 which compares with a maximum theoretical capacity of 264 mA h g−1, based on the pristine material having ∼4% Ni excess as determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES).26 This provides an initial indication that the full capacity of the electrode can be accounted for by formal Ni redox alone, without obvious excess capacity associated with molecular O2 redox.
 | ||
| Fig. 1 Bulk-sensitive probing of LiNiO2 redox processes. (a) 2nd cycle charge–discharge profile of LiNiO2 electrode cycled at a rate of C/20 between 3.0 and 4.8 V vs. Li/Li+. Inset: Scanning electron microscope (SEM) image of pristine LiNiO2 particles. (b) Corresponding differential capacity plots (dQ/dV). (c), Normalised Ni K-edge XANES spectra (transmission mode) of LiNiO2 at different SoC. (d) Plot of the energy shift in normalised Ni K-edge whiteline and 50% edge height positions relative to pristine LiNiO2. (e) and (f) XRS (∼10 μm information depth) of the O K-edge and Ni L3-edge core-loss spectra for LiNiO2 electrodes at different SoC during the 2nd charge cycle. Experimental XRS data is marked as black dots and represented with smooth solid trace lines. Charge transfer multiplet (CTM) calculations of formally Ni2+ (green), Ni3+ (purple), and Ni4+ (pink) environments. See ESI,† Fig. S2 for fitted XRS Ni L3,2-edges. | ||
Fig. 1(c) shows normalised transmission Ni K-edge XANES spectra for the LiNiO2 electrodes at different SoC (x in LixNiO2) during the 2nd charge cycle. As expected, the Ni K-edge shifts to higher energies as the formal Ni oxidation state increases, with the removal of valence electrons leaving the Ni nucleus less-shielded such that it has a higher effective charge, and the core-level becomes more strongly bound. Both the energy of the fractional (normalised) edge height and the position of the whiteline (intensity maximum) are routinely used as indirect measures of average oxidation state.35,36 A continuous shift to higher energy in both the edge half-height and whiteline is observed up to 4.2 V, x = 0.22 (Fig. 1(d)). The two trends diverge with further delithiation, with the whiteline monotonically shifting to higher energy up to the furthest measured extent of delithiation (4.8 V, x = 0.03), while the half-height position plateaus with little variation between x = 0.10 and x = 0.03. The plateau of half-height position has previously been taken as an indication that Ni is no longer involved in the redox mechanism at high SoC,24,25 however the continuing shift in whiteline position would suggest otherwise. Indeed, the edge-position is known to be sensitive to other factors including bond length and ligand covalency.37
To resolve this ambiguity without introducing surface sensitivity as a confounding factor, bulk-sensitive XRS was performed to collect O K-edge (Fig. 1(e)) and Ni L3-edge (Fig. 1(f)) spectra at the same SoC as the XANES. XRS probes lower-energy O 1s → 2p and Ni 2p → 3d transitions using hard X-rays (10 keV), achieving an information depth of ∼10 μm which is similar to Ni K-edge XANES. In Fig. 1(e), pristine LiNiO2 exhibits a prominent asymmetric O K pre-edge feature centred at 528.8 eV associated with transitions from O 1s → O 2p-Ni 3d hybridised states, and main edge features above 535.0 eV associated with transitions from O 1s → O 2p-Ni 4s,p hybridised states. On delithiation, the pre-edge peak is seen to continuously increase in relative intensity, whilst losing its asymmetry and shifting by 0.4 eV to a higher peak energy of 529.2 eV. There is also an accompanying shift in the main edge half-height position from ∼536.0 eV for pristine LiNiO2 up to 539.5 eV at 4.8 V, and the shape of the main edge changes indicating a change in the O2p and Ni4s,p orbital hybridisation. Importantly, across the potentials probed, the feature arising at ∼531.5 eV associated with the formation of molecular O2 is not strongly pronounced.24–26
The corresponding Ni L3-edge XRS (Fig. 1(f)) for pristine LiNiO2 shows a broad line shape composed of three main features at 853.6 eV, 854.9 eV, and 856.1 eV. There remains debate over the ground state of LiNiO2 (see Supplementary Note 1, ESI†) and a variety of models based on alternating layers of NiO6 octahedra and Li have been proposed. The simplest model, in which all NiO6 octahedra are equivalent with a formal oxidation state of Ni3+, is compatible with XRD data but not with measurements using more local probes.38,39 As a result, more complex models involving time or spatially varying distortions of the octahedra have been proposed. These include structures with Jahn–Teller (J–T) distortions, where two different Ni–O bond lengths are present and the formal oxidation state remains Ni3+,40 and spin disproportionated structures, where Ni2+ (S = 1), Ni3+ (S = ½), and Ni4+ (S = 0) octahedra coexist and interconvert dynamically at room temperature.41,42 Recent temperature-dependent XAS and X-ray magnetic circular dichroism (XMCD) shows strong evidence for such disproportionation in LiNiO2,41 which is consistent with other correlated nickelate compounds, including AgNiO2, which show disproportionation and strong covalency between frontier O 2p and Ni 3d states.43–47
Charge transfer multiplet (CTM) calculated L3-edges for the three Ni environments with formal oxidation states of +2, +3 and +4 are overlaid on the pristine LiNiO2 spectra in Fig. 1(f), corresponding to the three main features at 853.6 eV, 854.9 eV, and 856.1 eV seen in Ni L3-edge XRS. Simulation parameters have been optimised based on experimental data (Supplementary Note 3, ESI†). Each simulated spectra can be thought of as a superposition of metal–ligand hole configurations,47 with the formally Ni2+, Ni3+, and Ni4+ octahedra having ground-state configurations of 0.80|3d8〉 + 0.19|3d9![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) 〉 + 0.01|3d102〉, 0.25|3d7〉 + 0.58|3d8〉 + 0.16|3d92〉 + 0.01|3d103〉, 0.04|3d6〉 + 0.33|3d7〉 + 0.48|3d82〉 + 0.14|3d93〉 + 0.01|3d104〉 respectively. In CTM calculations, increasing ligand hole contributions indicate an increasing degree of Ni–O covalency for higher formal oxidation states. Linear combinations of the simulated spectra match closely to the Ni L3-edge spectra from XRS, FY-XAS and TEY-XAS at all SoC (ESI,† Fig. S2–S5), indicating that the simulated spectra for the Ni2+, Ni3+ and Ni4+ environments are suitable descriptions despite the small changes in octahedral environment expected for different phases.
〉 + 0.01|3d102〉, 0.25|3d7〉 + 0.58|3d8〉 + 0.16|3d92〉 + 0.01|3d103〉, 0.04|3d6〉 + 0.33|3d7〉 + 0.48|3d82〉 + 0.14|3d93〉 + 0.01|3d104〉 respectively. In CTM calculations, increasing ligand hole contributions indicate an increasing degree of Ni–O covalency for higher formal oxidation states. Linear combinations of the simulated spectra match closely to the Ni L3-edge spectra from XRS, FY-XAS and TEY-XAS at all SoC (ESI,† Fig. S2–S5), indicating that the simulated spectra for the Ni2+, Ni3+ and Ni4+ environments are suitable descriptions despite the small changes in octahedral environment expected for different phases.
On cycling to higher potentials, the XRS shows a continuous growth in the intensity of the Ni4+ feature (see Fig. 2(d)), initially at the expense of Ni2+ up to 3.9 V, x = 0.55, and then Ni3+ up to 4.8 V, x = 0.03. This evolution of Ni species upon delithiation matches that expected from disproportionation.41 At 4.8 V, the spectrum closely matches Ni L3-edge simulations of Ni4+ (ESI,† Fig. S6) with 4–5% Ni2+. This is consistent with the excess Ni detected with ICP-OES occupying Li sites, as similarly sized Ni2+, and thus preventing all sites reaching Ni4+.26,48 The bulk sensitivity of XRS suppresses contributions from surface layers which are otherwise seen even for inverse partial fluorescence yield (IPFY) measurements (ESI,† Fig. S7), including for reference Ni4+ compounds.49,50 Importantly this shows that charge compensation in the LiNiO2 bulk proceeds predominantly through Ni–O rehybridization across the whole cycling range, lowering the electron density on O sites, but without a significant role for molecular O2 redox. This contrasts with several reports of oxygen redox in this potential range for LiNiO2 and Ni-rich layered cathode materials, based on detection of the molecular O2 feature with less bulk-sensitive O K-edge RIXS.24–26,51
 | ||
| Fig. 2 Near-surface probing of LiNiO2 redox processes. (a) and (b) FY-XAS (∼200 nm information depth) of the O K-edge and Ni L3-edge, and (c) TEY-XAS (∼10 nm information depth) of the Ni L3-edge for LiNiO2 at different SoC. (d) Relative intensities of Ni2+, Ni3+, and Ni4+ components based on fitting CTM calculated spectra to XRS, FY-XAS, and TEY-XAS spectra (see fitting results in ESI,† Fig. S2–S5). | ||
Near-surface degradation
To further investigate the origins of molecular O2 reported at high SoC, the same core levels were measured using soft XAS in FY mode (Fig. 2(a) and (b)). The spectra for pristine LiNiO2 closely resemble those obtained with XRS, however an additional feature is apparent at 532.3 eV in the O K-edge, and the Ni2+ feature in the Ni L3-edge is more intense. These same features are seen for NiO (see ESI,† Fig. S8 for O K-edge), and correspond to a NiO-like RSL,52 whose contribution is not detected in the more bulk-sensitive XRS. On delithiation, the XAS data show similar trends to the XRS until 4.1 V, x = 0.33, with the Ni L3-edge showing the Ni4+ feature increasing at the expense of Ni2+ and then Ni3+, and some growth in the O K pre-edge. At higher SoC there are significant deviations between FY-XAS and XRS spectra. Most notably a feature at ∼531.5 eV is seen to emerge in the O K-edge, which although initially weak at 4.2 V, x = 0.22, shows significant intensity at 4.8 V, x = 0.03 (see integrated peak areas in ESI,† Fig. S9). This feature corresponds to the same absorption energy as molecular O2, whose vibrational structure has been detected in LiNiO2 and other conventional Ni-rich layered oxides in several recent reports.25,26 Whereas this molecular O2 signature and the Ni2+ feature grow in FY-XAS, the O K pre-edge peak and the Ni4+ feature in the Ni L3-edge are supressed in FY-XAS compared to the XRS. This suggests a near-surface molecular O2 redox process associated with RSL growth, in which Ni is reduced toward Ni2+ and molecular O2 forms i.e., NiO2 → NiO2−x + ½xO2. Similar trends are observed with the more surface-sensitive TEY-XAS (fits shown in ESI,† Fig. S4) consistent with RSL formation proceeding from electrolyte-exposed surfaces.21,52 Importantly, the bulk-sensitive XRS (Fig. 1(e) and (f)) does not detect such Ni reduction or O2 formation, even after charging to 4.8 V, highlighting the key connection between the formation of trapped molecular O2 and the increase in Ni2+ species close to the cathode surface.We now investigate the reversibility of this near-surface molecular O2 redox process and how its extent changes with upper cutoff voltage (UCV). Fig. 3(a) shows that after discharging from a UCV of 4.8 V to 4.0 V, the molecular O2 feature at ∼531.5 eV disappears from the O K-edge, but a prominent RSL feature at 532.6 eV remains. On discharge to 3.0 V, the RSL feature further grows in intensity relative to the pre-edge feature, with accompanying increases in the Ni2+ feature for the Ni L3-edge spectra (Fig. 3(b) and (c)). This is even more prominent in the surface-sensitive TEY-XAS (Fig. 3(c)), indicating the RSL is more densified near to the surface. Comparison to an electrode where the UCV is 4.2 V confirms that the extent of RSL formation is much greater for the UCV of 4.8 V, consistent with previous studies where significant RSL formation occurs at SoC above the H2-H3 transition in Ni-rich cathodes.20,21,52 Longer-term cycling (150 cycles, ESI,† Fig. S10) further shows that the UCV of 4.8V leads to greater voltage hysteresis and charge transfer impedance reflecting this more extensive RSL formation.
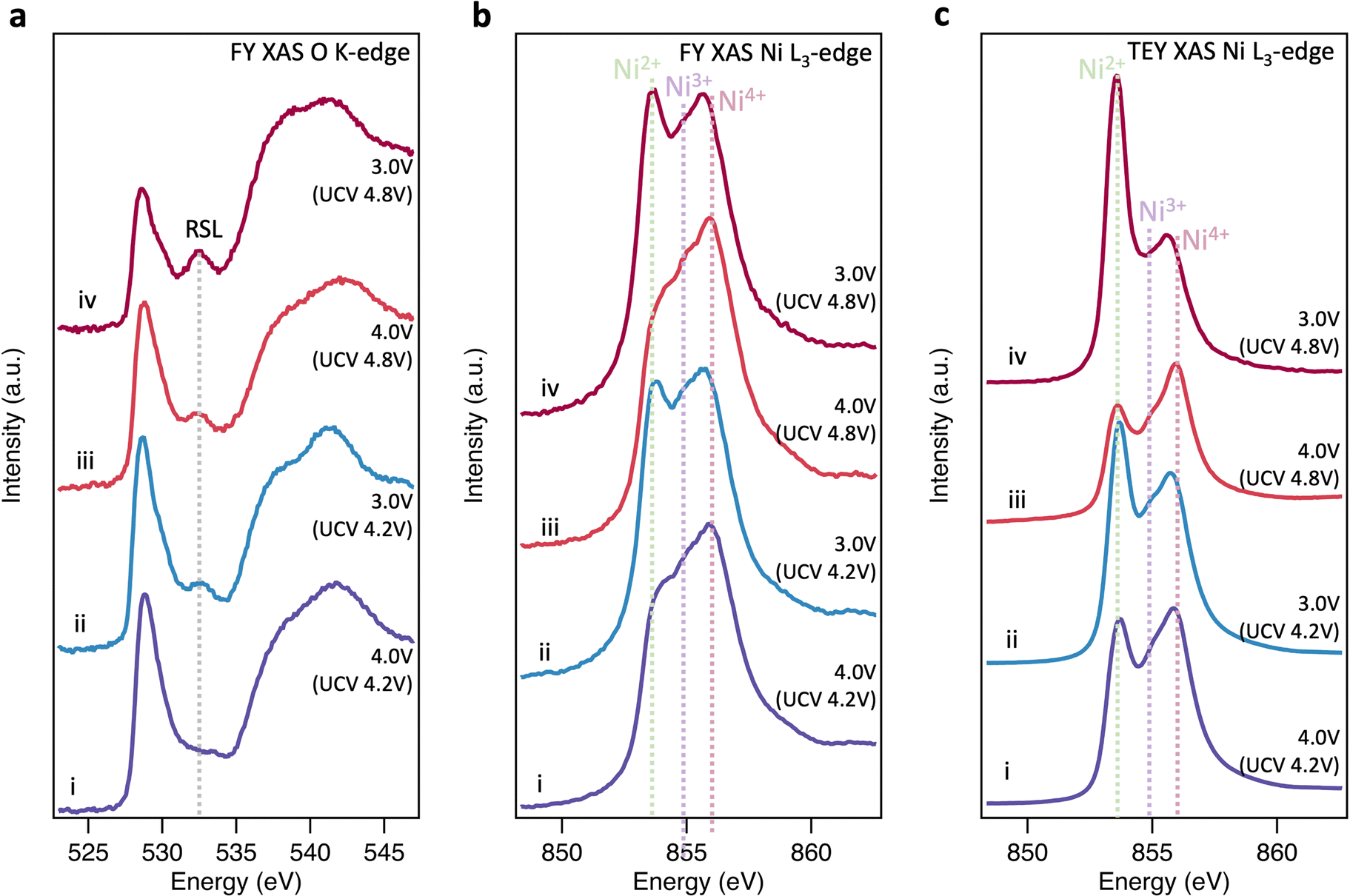 | ||
| Fig. 3 Discharge behaviour of LiNiO2. (a), (b) FY-XAS (∼200 nm information depth) of the O K-edge and Ni L3-edge, and (c) TEY-XAS (∼10 nm information depth) of the Ni L3-edge for LiNiO2 cycled to a UCV of 4.2 V before being discharged to (i) 4.0 V and (ii) 3.0 V, with parallel samples cycled to a higher UCV of 4.8 V and then back to (iii) 4.0 V and (iv) 3.0 V. | ||
Although comparison of TEY and FY mode XAS confirms the RSL is found predominantly near the sample surface, it provides only limited insight into the depth over which it is distributed. To spatially resolve the extent of the RSL at high SoCs, STEM-EELS was performed for LiNiO2 charged to 4.8 V (Fig. 4). Depth-resolved Ni L3-spectra show a decreasing proportion of the lower energy (more reduced) component (peak A1) on moving towards the bulk of the particle, stabilising at ∼200 nm from surface, consistent with more Ni2+ species at the surface and more Ni4+ in the bulk. Similarly, the O K-edge shows a higher pre-edge intensity (peak B1) towards the bulk of the particle correlating with higher Ni oxidation state and Ni–O covalency. This extended RSL region where the Ni oxidation state is seen to vary over ∼200 nm is attributable to a cation mixing layer in which Ni2+ ions have migrated to occupy Li sites, and is consistent with the differences seen between TEY-XAS, FY-XAS and XRS observations. Notably, a similar extent of RSL formation is not observed at intergranular cracks away from the LiNiO2 surface, presumably as electrolyte does not fully penetrate these cracks for the low cycle numbers considered here. This indicates a key role of the electrolyte in promoting RSL formation, with electrolyte infiltration into internal cracks likely proceeding over multiple cycles.
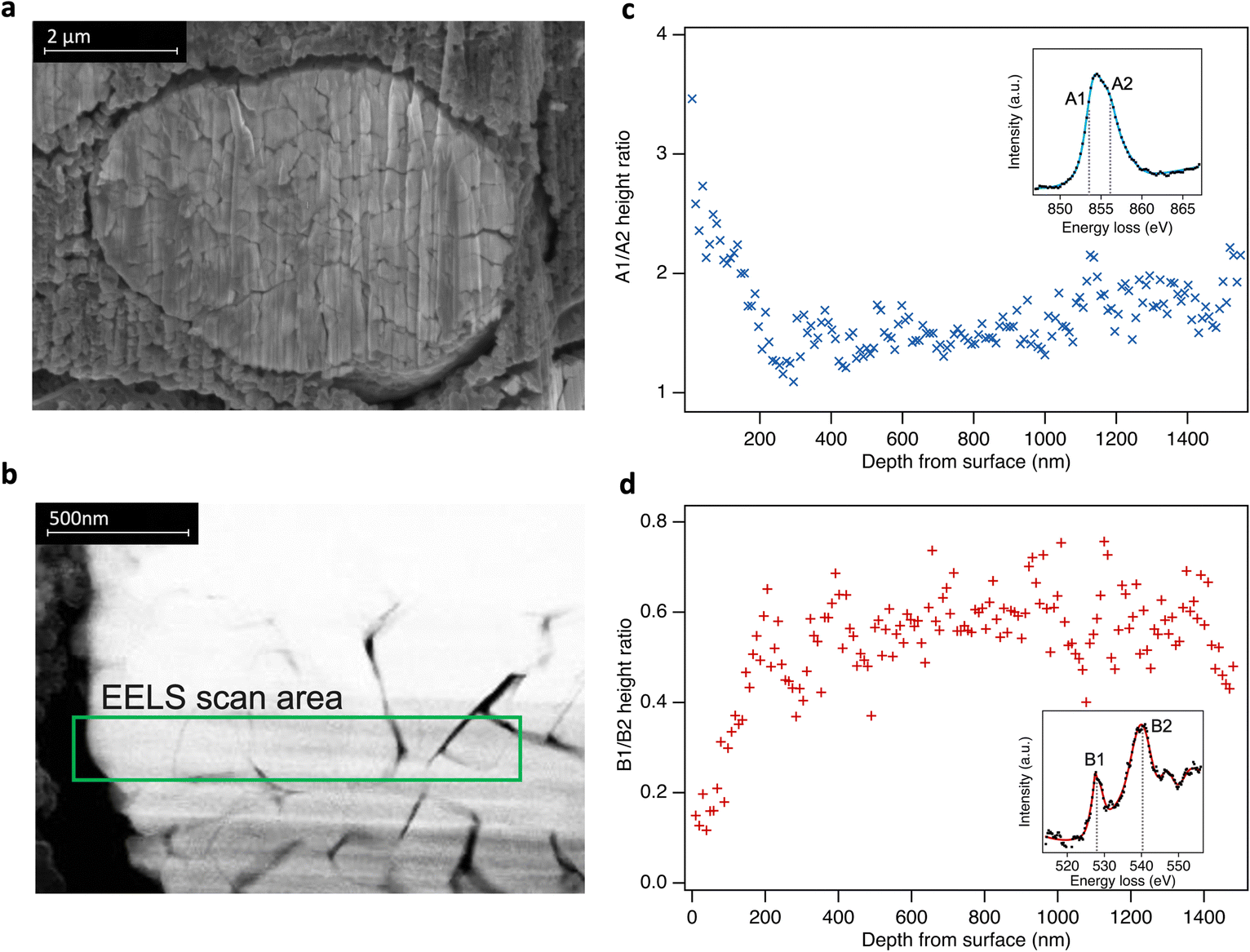 | ||
| Fig. 4 (a) Cross-sectional scanning electron microscopy (secondary electron detection) of LiNiO2 particle from an electrode charged to 4.8 V. (b) Selected STEM-EELS scan area of 1.5 μm from surface to bulk (left to right) of the particle. (c) and (d) Fitted peak ratios of depth-resolved Ni L3- and O K-edge EELS spectra using a simplified two peak fit in each case (see ESI,† Fig. S11). Insets: Examples of EELS spectra. | ||
Bulk electronic and geometric structural evolution of LiNiO2
Having shown that bulk redox in LiNiO2 occurs by Ni–O rehybridization, we now consider further the associated changes in electronic and geometric structure. Fig. 5(a) shows high-energy-resolution fluorescence detection (HERFD-)XANES Ni K-edge spectra of pristine LiNiO2 and after cycling to 4.8 V. Notably the main edge half-height position is shifted ∼2.1 eV higher compared to LiNiO2, a more distinct change than seen in the transmission mode measurements of Fig. 1(c) (∼1.5 eV), as a result of the fine-structure features along the rising edge now being better resolved. Fig. 5(c) compares the similarly bulk-sensitive O K-edge XRS spectra of the same samples. Since we anticipate that differing Ni–O bond lengths yield distinct signatures in the O K pre-edge in any model of the material, we chose the zigzag J–T P21/c structure for LiNiO2 spectral calculations. More complex time-varying LiNiO2 model structures are computationally prohibitive for spectral calculations and although the P21/c structure has a formal oxidation state of Ni3+, this will predominantly affect the Ni 3d states, which only weakly influence the main features of the Ni and O K-edge spectra. Density functional theory (DFT) calculated Ni and O K-edge spectra for LiNiO2 and NiO2 (Fig. 5(b) and (d)), reproduce the features of the experimental spectra extremely well, showing the same pre-edge peaks, number of fine structure features, and similar trends in intensity and linewidths across the whole spectral range. The relative energy shifts are also captured well, giving confidence in the sufficiency of the chosen structure models (P21/c for LiNiO2, and Rm for NiO2).
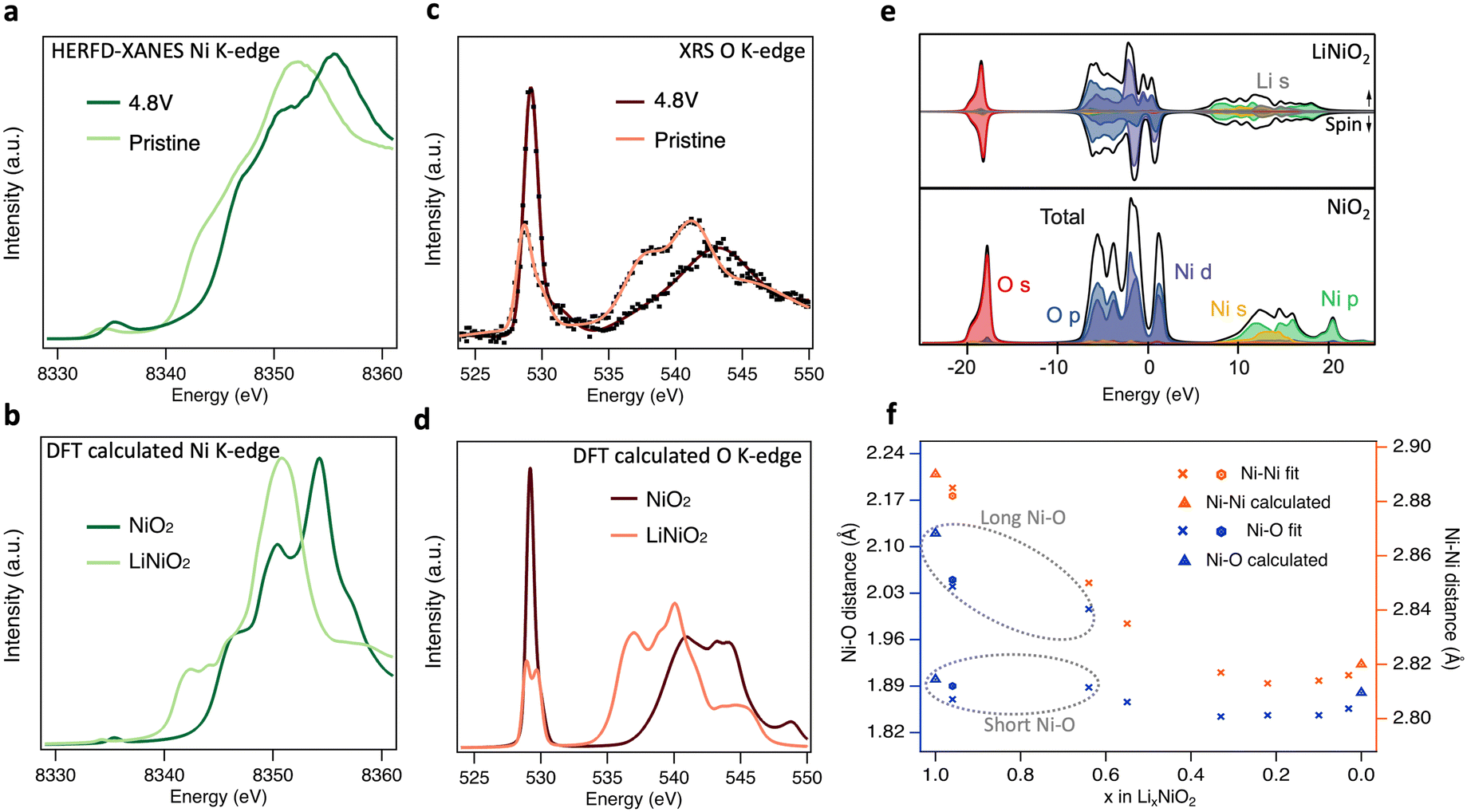 | ||
| Fig. 5 Electronic and geometric structural changes of LiNiO2 upon delithiation. (a) Experimental HERFD-XANES and b, core–hole calculated Ni K-edge spectra of pristine and charged LiNiO2. (c) Experimental XRS with smooth trace lines and (d) core–hole calculated O K-edge spectra of pristine and charged LiNiO2. (e) Ground-state partial and total density of states for LiNiO2 (top) and NiO2 (bottom). Fermi energies are set to zero. (f) Ni–Ni and Ni–O distances determined from the Fourier-transformed EXAFS spectra (details in ESI,† Table S1 and Fig. S12, S13). Note that the short/long Ni–O lengths of pristine (hexagons), 3.0 V and 3.8 V (crosses) LiNiO2 are related to the disproportionated model applied for EXAFS fitting. Bond lengths for the geometry optimised structures from DFT calculations used in (b), (d), (e) are shown as triangles in f. | ||
The origin of the spectral features can be understood by comparison to ground-state partial density-of-states (pDOS) shown in Fig. 5(e), and consideration of the allowed spectroscopic transitions. The first unoccupied states in both LiNiO2 and NiO2 lie just above 0 eV, showing mixed O 2p and Ni 3d orbital character and giving rise to the pre-edge peaks in the experimental Ni (∼8335 eV) and O (∼529 eV) K-edges. A sizable gap separates the next set of unoccupied states which give rise to the main edges in the Ni (≳8340 eV) and O (≳535 eV) K-edges, and have Ni 4s,p character, with some Li 2s contribution also seen in this region for LiNiO2. This gap widens by ∼2.9 eV from LiNiO2 to NiO2 which can be related to a decrease in average Ni–O bond length associated with the change in geometric structure.53,54 We note that the DFT calculated Ni K-edge spectra show weaker pre-edge features than experiment, attributable to quadrupolar transitions not being considered in the calculations.55
A clear splitting of the calculated O K pre-edge peak in Fig. 5(d) for LiNiO2 resembles the asymmetric pre-edge in the XRS experimental data. The O K pre-edge becomes far more intense in the 4.8 V sample and the peak splitting seen in the calculated spectrum of LiNiO2 is lost. This corresponds with the change from D4h site symmetry for the J–T distorted Ni3+ octahedra used in the LiNiO2 calculation, where d orbital splitting arises from the elongation of two Ni–O bonds, to Oh site symmetry for the Ni4+ octahedra of NiO2, where this d orbital splitting is lost. The growth in intensity of the O K pre-edge feature is also consistent with the CTM calculations, where the increased ligand hole contributions for the Ni4+ octahedra indicate an increasing degree of Ni–O covalency on delithiation. The increase of O K pre-edge intensity by a factor of ∼2 on full delithiation (see ESI,† Fig. S9) corresponds closely to the factor of ∼1.8 obtained based on the proportions of Ni species fitted to the Ni L3,2-edge XRS spectra (Fig. 2(d)) and their respective electron configurations. Further evidence for increased Ni–O covalency is apparent from the emergence of more distinct fine-structure features (∼8347 eV and 8351 eV) in the Ni K-edge, attributable to ligand-to-metal charge transfer shakedown transitions,56 as well as satellite peaks in the Ni L3,2-edge that are most clearly seen in FY-XAS measurements (see ESI,† Fig. S14b) and are well-reproduced in the CTM calculated Ni4+ spectrum. In addition, Bader charge analysis57 based on the ground-state DFT calculations shows the ionic charge of the Ni only modestly changes from +1.41 to +1.56 e− between LiNiO2 and NiO2, whilst a more significant change from −1.15 to −0.78 e− is seen for the O charges.
Fig. 5(f) shows the nearest Ni–O and Ni–Ni distances extracted by fitting to EXAFS spectra for LiNiO2 at different SoC. Since fitting with the single Ni–O bond length model showed significantly higher Debye–Waller factors at low SoC, and disproportionation is expected to persist up to 3.9 V based on Fig. 2(d), a model with two Ni–O bond lengths (ratio of short:long Ni–O bond based on the disproportionated model and associated XRS fittings) was instead used to fit pristine, 3.0 V and 3.8 V LiNiO2 (see ESI,† Fig. S15 and Table S2). The Ni–O and Ni–Ni bond distances obtained show good agreement with both the J–T P21/c LiNiO2 and the disproportionated structure, however, the short:long bond ratios of the disproportioned model show lower Debye–Waller factors, supporting assignment of this structure.
Similar trends in weighted average Ni–O bond lengths are seen to operando neutron diffraction measurements,34 with Ni–O bond length gradually shrinking in line with the change in structure, increased oxidation state and increased covalency at high SoC. Notably, above the H2–H3 transition (x ≤ 0.22) a modest increase in the Ni–O bond length is observed. This has been associated with a loss of the stabilising effect of Li–O covalency at high SoC, leading to Ni–O bond elongation alongside the sudden c-lattice collapse related to the H2–H3 transition, and increased charge transfer from the O to Ni sites.11,58,59 This changing covalency, seen as continuous spectral changes in Fig. 1(c) and (d), can account for the plateauing in half-height position of the Ni K main-edge at high SoC in transmission XANES (Fig. 1(d)). As well as highlighting the limitations of applying a single metric to assess changes in oxidation state, the limited sensitivity of the Ni K-edge fractional-edge height reflects that it arises from transitions to Ni 4s,p states, in contrast to the O K- and Ni L3,2-edges which probe transitions to O 2p-Ni 3d hybridised states.
Conclusion
In summary, bulk sensitive XRS measurements reveal that in the bulk of LiNiO2, charge compensation occurs by Ni–O rehybridization without the involvement of molecular O2. From an initially disproportionated structure where formally Ni2+, Ni3+, and Ni4+ octahedra coexist, the Ni4+ features of the Ni L3,2-edge continuously grow on delithiation, initially at the expense of Ni2+ and subsequently Ni3+ features (see Fig. 6). There is a concomitant increase in O K pre-edge intensity, consistent with significant lowering of electron density on the O sites at potentials where O loss is expected.9,50 However, significant signatures of molecular O2 formation are not detected throughout the bulk suggesting its formation remains kinetically hindered.60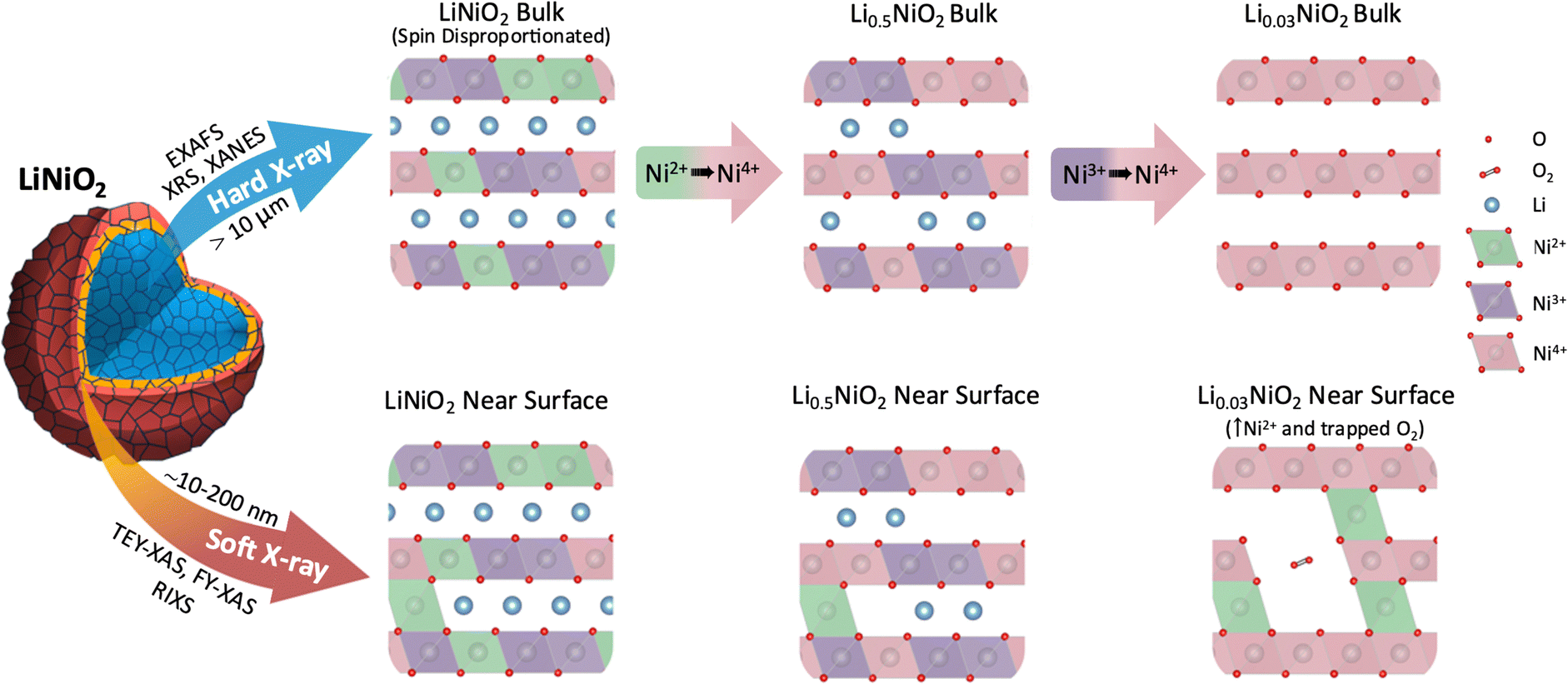 | ||
| Fig. 6 Schematic representation of LiNiO2 bulk charge compensation mechanism and surface degradation processes probed by different X-ray spectroscopy techniques. The differences in LiNiO2 delithiation occurring at the surface and in the bulk, including the accommodation of trapped O2 in pores formed near the surface by Ni2+ species migrating to the Li layer. Continuous oxidation of Ni in bulk LiNiO2 at high SoC is distinguished from the near surface degradation. | ||
FY-XAS measurements reveal evidence of molecular O2 formation in the outer ∼200 nm of the cathode surface, with the growth in intensity of the Ni4+ feature plateauing above 4.2 V, and features of trapped molecular O2 emerging alongside increased Ni2+ contributions. This is consistent with molecular O2 trapped in voids formed by Ni2+ entering the Li layers (see Fig. 6). STEM-EELS reveals a RSL that extends ∼200 nm into the LiNiO2 surface following cycling to 4.8 V, showing a gradual change in oxidation state across its thickness. The absence of this extended RSL at internal surfaces of the secondary cathode particles, e.g. interparticle cracks, suggests its formation is driven by contact with the electrolyte.
The trapped molecular O2 feature disappears on discharging to 4.0 V, but a significantly increased near-surface Ni2+ contribution is retained. Although our results do not fully exclude some reversible molecular O2 redox, online electrochemical mass spectroscopy studies have reported O2 evolution occurring on discharge.15 We therefore suggest that this may arise from the release of trapped O2 associated with structural changes, including abrupt c-lattice expansion and particle cracking (ESI,† Fig. S16).
Our findings highlight the importance of combining bulk- and surface-sensitive techniques to fully confirm the extent to which molecular O2 redox processes in cathode materials are bulk phenomena contributing to reversible charge compensation, rather than involved in surface degradation as revealed here for LiNiO2. The understanding developed of the surface instability of LiNiO2 associated with rehybridisation at high SoC, emphasises the importance of strategies such as cathode coatings, composition gradients, and electrolyte formulation to stabilise Ni-rich cathode surfaces in contact with electrolyte, rather than bulk stabilisation approaches (e.g. pillaring) that might unduly sacrifice capacity. This study thus provides a solid basis for future exploration of molecular O2 formation and Ni–O rehybridisation in Ni-rich cathodes in different electrolyte environments, and for further investigations to separate bulk redox and near-surface degradation processes in a broad range of cathode materials.
Experimental
Sample preparation
Commercial grade LiNiO2 powder was obtained from BASF, without any deliberate doping or coating added. This was characterised by SEM (Zeiss Merlin, 2 kV, Inlens detector), XRD (Rigaku Miniflex, Cu Kα source), and X-ray Photoelectron Spectroscopy (XPS, PHI Versaprobe III, Al Kα source), see ESI,† Fig. S1. Free-standing electrodes were prepared by calendaring a mixture of 80 wt% LiNiO2 powder, 10 wt% conductive acetylene black and 10 wt% polytetrafluoroethylene (PTFE) binder. Electrochemical tests of the LiNiO2 cathodes were performed in 2032 coin cells (316 stainless steel, Cambridge Energy Solutions) using Li metal disks as negative electrodes and borosilicate glass fibre separators (borosilicate, GF/A, Whatman) soaked with 120 μL of LP57 electrolyte (1 M LiPF6 in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 of EC:EMC). The assembled cells were charged up to 4.2 V at C/20 (calculated based on a theoretical capacity of LiNiO2 of 275 mA h g−1), held for 30 min and then cycled back to 3.0 V at the same rate. This was immediately followed by a second charge at C/20 (ESI,† Fig. S1), with a voltage hold for 10 hours at the desired endpoint. The SoC of delithiated LixNiO2 is calculated based on the charge–discharge capacity curve presented in Fig. 1(a), starting from x = 0.96 for pristine and 3.0 V LiNiO2 (based on the ∼4% Ni excess). All potentials mentioned in this work are referenced to Li/Li+. For ex situ measurements, cathodes were recovered from the cycled coin cells by disassembly in a glove box under Ar atmosphere (O2 < 1 ppm, H2O < 1 ppm). Recovered cathodes were washed in dimethyl carbonate (anhydrous, ≥99%, Sigma Aldrich) solvent and dried before heat-sealing in aluminised mylar (Fresherpack, 130 μm) pouches for XANES, EXAFS, and XRS, or transferring in a vacuum suitcase for XAS, XPS.
:7 of EC:EMC). The assembled cells were charged up to 4.2 V at C/20 (calculated based on a theoretical capacity of LiNiO2 of 275 mA h g−1), held for 30 min and then cycled back to 3.0 V at the same rate. This was immediately followed by a second charge at C/20 (ESI,† Fig. S1), with a voltage hold for 10 hours at the desired endpoint. The SoC of delithiated LixNiO2 is calculated based on the charge–discharge capacity curve presented in Fig. 1(a), starting from x = 0.96 for pristine and 3.0 V LiNiO2 (based on the ∼4% Ni excess). All potentials mentioned in this work are referenced to Li/Li+. For ex situ measurements, cathodes were recovered from the cycled coin cells by disassembly in a glove box under Ar atmosphere (O2 < 1 ppm, H2O < 1 ppm). Recovered cathodes were washed in dimethyl carbonate (anhydrous, ≥99%, Sigma Aldrich) solvent and dried before heat-sealing in aluminised mylar (Fresherpack, 130 μm) pouches for XANES, EXAFS, and XRS, or transferring in a vacuum suitcase for XAS, XPS.
Electron microscopy
A Thermo Scientific Helios G4 CXe Plasma FIB (PFIB) was used to prepare the STEM lamellae. For lift-out, a thin Pt layer was deposited onto the surface region of interest (ROI), trenches were patterned around the ROI to make 4 μm-thick lamellae. A W needle was then used to lift each lamella and place them on a FIB lift-out grid (Cu, Agar Scientific). Each lamellae was then thinned down to around 50 nm thick for STEM-EELS and polished with a low dose, low energy beam (<0.3 nA, 5 keV) to minimise ion beam damage. Inert transfer between PFIB and an Ar glovebox was achieved using a Gatan iLoad system.Spatially resolved EELS of the lamellae was performed using a JEOL ARM200F equipped with cold field emission gun operated at 200 keV and spherical aberration probe corrector. Dual EELS was acquired using a Gatan GIF Quantum 965 ER with energy resolution of around 1 eV at 0.25 eV per channel dispersion. Inert transfer between glovebox and STEM was achieved using a JEOL double-tilt vacuum transfer holder.
Given LiNiO2 is less stable when highly delithiated, radiation damage should be considered when evaluating the oxidation state of Ni in EELS. The energised Xe ion beam in PFIB and electron beam in STEM can both induce reduction of LiNiO2 and NiO2 towards NiO.61 The absolute A2/A1, and B2/B1 ratios seen in the LiNiO2 bulk reflect some degree of ion/electron beam induced reduction. Nevertheless, equal acquisition time and constant electron beam current during the EELS scans ensure a consistent radiation dose (3 × 103 e− Å−2) such that the trends in EELS spectra and the spatial variations seen near their surface are still representative.
X-ray spectroscopy
Transmission Ni K-edge XANES and EXAFS spectra were collected with a laboratory-based easyXAFS300+ spectrometer (easyXAFS, WA, US). X-rays are generated with a liquid-cooled Ag anode X-ray tube, before monochromation by a Si (551) spherically bent crystal analyser. A helium-filled box with polyimide windows is placed in the beam path for better X-ray transparency while a steel plate with a 9 × 3 mm slot is placed after each sample to lower the background. The transmitted intensity is measured with a silicon drift detector (KETEK, Munich, Germany) placed behind the sample. Each acquisition was performed over 45 min and 30 scans were collected for each sample to obtain good statistics. NiO reference spectra were also collected for each batch of measurements for energy calibration. Data pre-processing was performed with the EasyXANES package to convert the measured intensity into linear attenuation coefficient, μ. Data reduction and analysis were performed using the Demeter package (version: 0.9.26).
In situ HERFD-XANES was performed at Diamond Light Source's beamline I20 with aluminised mylar (Fresherpack, 130 μm) pouch cells containing free-standing LiNiO2 cathodes, with Li metal disks as negative electrodes and a Celgard 2325 separator soaked with 80 μl of LP57 electrolyte (1 M LiPF6 in 3:7 of EC:EMC). The cells were held at the desired potentials, and measured through the cathode side of the pouches using an X-ray emission spectrometer equipped with three Si(444) analyser crystals.62 The spectrometer was set to the maximum of the Ni Kβ1,3 line (8266 eV), and the incident energy was scanned using the four-bounce Si(111) monochromator. The spectrometer was calibrated using a Ni foil, measuring the Kβ line with the incident energy tuned +300 eV from the Ni K-edge.
TEY- and FY-XAS measurements were performed at ES-2 of beamline B07-B at Diamond Light Source, with the exit slits set to 50 μm in the dispersive direction, yielding a flux of between 1 × 1011 (O K-edge) and 2 × 1011 (Ni L3,2-edge) photons s−1. All samples were measured with the incident beam normal to the electrode surface, yielding a beam footprint of 150 × 100 μm. FY-XAS measurements were acquired using an Al coated Si photodiode directed at the sample with its surface normal at ∼45° to incident beam direction. Simultaneous TEY-XAS measurements were obtained using a SR570 low-noise current amplifier (Stanford Research Systems) to collect the current between the sample plate and an isolated steel washer in front of the sample biased to +90 V. Separate IPFY-XAS measurements of the Ni L3,2-edge were acquired using a Vortex silicon drift detector (Hitachi) at the I10 beamline at Diamond Light Source, with FY and TEY mode measurements simultaneously acquired. All spectra are divided by the drain current measured from the last X-ray mirror, to correct for variations in incident photon flux. The photon energy scale is calibrated using a NiO sample.63 O K-edge spectra are background-subtracted using a straight line fitted to the pre-edge region, followed by intensity normalization to the post-edge region at 550 eV. Ni L3,2-edge spectra are normalized to the intensity at 867 eV after removal of a linear background.
XRS measurements were performed at the European Synchrotron Radiation Facility at the ID20 beamline.64 X-rays are generated from three U26 revolver undulators, before being collimated, and then monochromated by a liquid–nitrogen cooled double-crystal Si(111) pre-monochromator. The beam from a second Si(311) channel-cut post-monochromator is focussed onto a ∼20 × 20 μm2 spot at the sample position by a mirror in Kirkpatrick–Baez geometry. The sample surface was positioned at a grazing angle of ∼1° relative to the incident beam direction, to maximise the illuminated area and the sample was scanned over a region of ∼10 mm during the 4-hour measurement to minimise beam-induced changes. Inelastically scattered photons were recorded using 72 spherically bent Si(660) crystal analysers with energy loss events in the vicinity of both O K-edge and Ni L3,2-edge. O K- and Ni L3,2-edges were recorded at momentum transfers of q = 6.9 ± 0.5, and all data extraction and treatment were performed as described in ref. 65.
Charge-transfer multiplet calculations
Ni L3,2-edge multiplet simulations were performed at the ligand field level of theory using the many-body code, Quanty.66 This was implemented using the same single-cluster NiO6 Hamiltonian as Green et al.,47 where Ni 2p, Ni 3d ligand shells are explicitly included (see Supplementary Note S3, ESI†). Parameters used in Ni2+ calculation (eV): Δ = 5.5, 10Dq = 0.71, Veg = 2.627, Vt2g = 1.524. Parameters used in Ni3+ calculation (eV): Δ = −0.5, 10Dq = 0.93 with Jahn–Teller splitting of Δeg = 0.15 and Δt2g = 0.10 where Δeg is the difference between the x2 − y2 and 3x2 − r2 onsite energies and Δt2g is the difference between the xy and xz/yz onsite energies, V3x2 − r2 = 2.43, Vx2 − y2 = 3.33, Vxz/yz = 1.41, Vxy = 1.93. Parameters used in Ni4+ calculation (eV): Δ = −6.5, 10Dq = 0.78, Veg = 3.456, Vt2g = 2.004.DFT spectral calculations
Density functional theory (DFT) calculations were carried out using the plane wave pseudopotential code CASTEP67 and the Perdew–Burke–Ernzerhof (PBE) form of the generalized gradient approximation functional,68 with the addition of the G06 semi-empirical dispersion correction69 to better account for van der Waals forces. The zig-zag J–T P21/c structure for LiNiO2, and the Rm structure for NiO2 were used for pristine LiNiO2 and fully delithiated materials respectively.33 Each structure was initially geometry optimised using appropriate plane wave cut-off energies (900 eV) and k-points (0.03 Å−1k-point spacing) determined via convergence of the total energy. The geometry of the system was considered optimized when the maximum forces on the ions were below 0.001eV Å−1 for NiO2 and 0.01 eV Å−1 for LiNiO2 consistent with other studies.33,70 Calculations of the pDOS and core–hole spectra were subsequently performed. The energy scale of the ground-state pDOS assumes the material is an insulator and sets the Fermi energy, Ef, to zero. Since core orbitals are not treated explicitly in the pseudopotential method, a unique pseudopotential is generated for an excited atom possessing a core–hole. For O and Ni K-edges, a core–hole is placed on the O 1s or Ni 1s orbitals respectively. A supercell is generated to prevent interactions between neighbouring core–holes. For spectral calculations, the plane wave energy cut-off, k-point sampling and cell size were increased until no visible effect on the spectrum was seen. Spectral calculations were handled using the OptaDOS programme.71 Lorentzian broadening was performed using full widths at half maximum of 0.14 and 0.8 eV for the O and Ni K-edges respectively, which should reflect the lifetimes of radiative and non-radiative transitions.72,73 The Gaussian component was then adjusted as a free parameter to match the experimental data, but remained fixed for the same edges to allow for comparison. The Lorentzian component is given energy dependence to account for the energy dependence of the lifetime. This was done by summing the set width with a factor that varies linearly with energy as implemented in Optados. The calculated spectra were rigidly shifted to align with the first absorption peaks of the experimental data to allow better comparison. In cases where the system under investigation possessed more than one inequivalent excitation site, separate spectra were generated, energy aligned74 and combined before rigidly shifting.
Author contributions
LA, JENS and RSW conceived the study. LA performed electrode preparation and electrochemical testing, with assistance from RAH. MWF performed SEM. LA, JENS, CJS and RSW performed the XRS. LA, AP, EB, PND, MWF, LAHJ, CMEP and RSW performed the soft XAS with support from DCG, PF and PB. LA, JENS, AP, CJS, and RSW performed XAS and XRS analysis. LA and PC performed the lab-based XANES and EXAFS, with analysis performed by PC. LA, JENS, EB, PND and RSW performed HERFD-XANES with support from SH. JENS, NR and RJN performed DFT calculations. RZ and PDN performed PFIB, STEM-EELS and related analysis. GH and RJG performed CTM calculations. LA, JENS, PC and RSW wrote the paper with contributions from all authors.Data availibility
The data supporting this article have been included as part of the ESI.† The corresponding data sets are available from the ORA repository, https://doi.org/10.5287/ora-yxpnqgero.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors acknowledge funding from the Faraday Institution (Faraday.ac.uk; EP/S003053/1, FIRG001, FIRG007, FIRG008, FIRG016, FIRG024) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (EXISTAR, grant agreement No. 950598) and under the Marie Sklodowska-Curie Actions (ISOBEL, grant agreement No. 101032281). We acknowledge support from the Engineering and Physical Science Research Council (EPSRC) through grants EP/K040375/1, EP/L022907/1, EP/T001038/1, and EP/R010145/1 (Henry Royce Institute). R. J. G. acknowledges funding from the Natural Sciences and Engineering Research Council of Canada (NSERC). R. A. H. acknowledges funding from the Royal Academy of Engineering under the Research Fellowship scheme. R. S. W. acknowledges a CAMS-UK Fellowship through the Analytical Chemistry Trust Fund and a UKRI Future Leaders Fellowship (MR/V024558/1). We acknowledge access to the David Cockayne Centre for Electron Microscopy. We thank Diamond Light Source (DLS) for beamtime on beamlines B07B, I10, and I20 under proposals SI33283, MM33062, and SP32010. We acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities under proposal MA-5753 and thank Blanka Detlefs for assistance and support in using beamline ID20.References
- E. Wikner, E. Björklund, J. Fridner, D. Brandell and T. Thiringer, How the utilised SOC window in commercial Li-ion pouch cells influence battery ageing, J. Power Sources Adv., 2021, 8, 100054 CrossRef CAS.
- J. E. Harlow, et al., A Wide Range of Testing Results on an Excellent Lithium-Ion Cell Chemistry to be used as Benchmarks for New Battery Technologies, J. Electrochem. Soc., 2019, 166, A3031–A3044 CrossRef CAS.
- “This Is What We Die For”: Human Rights Abuses in the Democratic Republic of the Congo Power the Global Trade in Cobalt, Amnesty International, 2016, Index: AFR 62/3183/2016.
- P. Kuiper, G. Kruizinga, J. Ghijsen, G. A. Sawatzky and H. Verweij, Character of Holes in LixNi1−xO and Their Magnetic Behavior, Phys. Rev. Lett., 1989, 62, 1214 CrossRef CAS.
- T. Mizokawa, et al., Role of oxygen holes in LixCoO2 revealed by soft X-ray spectroscopy, Phys. Rev. Lett., 2013, 111, 1–5 CrossRef.
- J. M. Tarascon, et al., In Situ Structural and Electrochemical Study of Ni1−xCoxO2 Metastable Oxides Prepared by Soft Chemistry, J. Solid State Chem., 1999, 147, 410–420 CrossRef CAS.
- K. Kleiner, et al., On the Origin of Reversible and Irreversible Reactions in LiNixCo(1 − x)/2 Mn(1 − x)/2O2, J. Electrochem. Soc., 2021, 168, 120533 CrossRef CAS.
- Z. Wu, et al., Unveiling the Evolution of LiCoO2 beyond 4.6 V, ACS Energy Lett., 2023, 8, 4806–4817 CrossRef CAS.
- A. R. Genreith-Schriever, et al., Oxygen hole formation controls stability in LiNiO2 cathodes, Joule, 2023, 7, 1623–1640 CrossRef CAS.
- H. Li, N. Zhang, J. Li and J. R. Dahn, Updating the Structure and Electrochemistry of LixNiO2 for 0 ≤ x ≤ 1, J. Electrochem. Soc., 2018, 165, A2985–A2993 CrossRef CAS.
- S. Lee, L. Su, A. Mesnier, Z. Cui and A. Manthiram, Cracking vs. surface reactivity in high-nickel cathodes for lithium-ion batteries, Joule, 2023, 7, 2430–2444 CrossRef CAS.
- S. Ahmed, et al., Visualization of Light Elements using 4D STEM: The Layered-to-Rock Salt Phase Transition in LiNiO2 Cathode Material, Adv. Energy Mater., 2020, 10, 2001026 CrossRef CAS.
- C. S. Yoon, D. W. Jun, S. T. Myung and Y. K. Sun, Structural Stability of LiNiO2 Cycled above 4.2 v, ACS Energy Lett., 2017, 2, 1150–1155 CrossRef CAS.
- J. K. Papp, et al., A comparison of high voltage outgassing of LiCoO2, LiNiO2, and Li2MnO3 layered Li-ion cathode materials, Electrochim. Acta, 2021, 368, 137505 CrossRef CAS.
- L. de Biasi, et al., Phase Transformation Behavior and Stability of LiNiO2 Cathode Material for Li-Ion Batteries Obtained from In Situ Gas Analysis and Operando X-Ray Diffraction, ChemSusChem, 2019, 12, 2240–2250 CrossRef CAS.
- D. Aurbach, et al., The Study of Surface Phenomena Related to Electrochemical Lithium Intercalation into LixMOy Host Materials (M = Ni, Mn), J. Electrochem. Soc., 2000, 147, 1322 CrossRef CAS.
- R. Pan, E. Jo, Z. Cui and A. Manthiram, Degradation Pathways of Cobalt-Free LiNiO2 Cathode in Lithium Batteries, Adv. Funct. Mater., 2023, 33, 1–11 Search PubMed.
- F. Lin, et al., Surface reconstruction and chemical evolution of stoichiometric layered cathode materials for lithium-ion batteries, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, Oxygen Release and Its Effect on the Cycling Stability of LiNixMnyCozO2 (NMC) Cathode Materials for Li-Ion Batteries, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- D. Streich, et al., Operando Monitoring of Early Ni-mediated Surface Reconstruction in Layered Lithiated Ni–Co–Mn Oxides, J. Phys. Chem. C, 2017, 121, 13481–13486 CrossRef CAS.
- W. M. Dose, et al., Electrolyte Reactivity at the Charged Ni-Rich Cathode Interface and Degradation in Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2022, 14, 13206–13222 CrossRef CAS.
- C. M. E. Phelan, et al., Role of Salt Concentration in Stabilizing Charged Ni-Rich Cathode Interfaces in Li-Ion Batteries, Chem. Mater., 2024, 36, 3334–3344 CrossRef CAS.
- F. Kong, et al., Kinetic Stability of Bulk LiNiO2 and Surface Degradation by Oxygen Evolution in LiNiO2-Based Cathode Materials, Adv. Energy Mater., 2019, 9, 1–12 Search PubMed.
- N. Li, et al., Unraveling the Cationic and Anionic Redox Reactions in a Conventional Layered Oxide Cathode, ACS Energy Lett., 2019, 4, 2836–2842 CrossRef CAS.
- A. S. Menon, et al., Oxygen-Redox Activity in Non-Lithium-Excess Tungsten-Doped LiNiO2 Cathode, PRX Energy, 2023, 2, 1 CrossRef.
- M. Juelsholt, et al., Does trapped O2 form in the bulk of LiNiO2 during charging?, Energy Environ. Sci., 2024, 17, 2530 RSC.
- M. Zhang, et al., Pushing the limit of 3d transition metal-based layered oxides that use both cation and anion redox for energy storage, Nat. Rev. Mater., 2022, 7, 522–540 CrossRef.
- R. A. House, et al., The role of O2 in O-redox cathodes for Li-ion batteries, Nat. Energy, 2021, 6, 781–789, DOI:10.1038/s41560-021-00780-2 Preprint at.
- N. Li, et al., Unraveling the Cationic and Anionic Redox Reactions in a Conventional Layered Oxide Cathode, ACS Energy Lett., 2019, 4, 2836–2842 CrossRef CAS.
- Z. W. Lebens-Higgins, et al., How Bulk Sensitive is Hard X-ray Photoelectron Spectroscopy: Accounting for the Cathode-Electrolyte Interface when Addressing Oxygen Redox, J. Phys. Chem. Lett., 2020, 11, 2106–2112 CrossRef CAS.
- R. A. House, et al., First-cycle voltage hysteresis in Li-rich 3d cathodes associated with molecular O2 trapped in the bulk, Nat. Energy, 2020, 5, 777–785 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, Chemical versus electrochemical electrolyte oxidation on NMC111, NMC622, NMC811, LNMO, and conductive carbon, J. Phys. Chem. Lett., 2017, 8, 4820–4825 CrossRef CAS.
- M. Mock, M. Bianchini, F. Fauth, K. Albe and S. Sicolo, Atomistic understanding of the LiNiO2–NiO2 phase diagram from experimentally guided lattice models, J. Mater. Chem. A, 2021, 9, 14928–14940 RSC.
- P. H. Chien, et al., New Insights into Structural Evolution of LiNiO2 Revealed by Operando Neutron Diffraction, Batteries Supercaps, 2021, 4, 1701–1707 CrossRef CAS.
- R. J. Woolley, B. N. Illy, M. P. Ryan and S. J. Skinner, In situ determination of the nickel oxidation state in La2NiO4+δ and La4Ni3O10−δ using X-ray absorption near-edge structure, J Mater Chem, 2011, 21, 18592–18596 RSC.
- W. E. O’Grady, K. I. Pandya, K. E. Swider and D. A. Corrigan, In Situ X-Ray Absorption Near-Edge Structure Evidence for Quadrivalent Nickel in Nickel Battery Electrodes, J. Electrochem. Soc., 1996, 143, 1613–1617 CrossRef.
- M. L. Baker, et al., K- and L-edge X-ray absorption spectroscopy (XAS) and resonant inelastic X-ray scattering (RIXS) determination of differential orbital covalency (DOC) of transition metal sites, Coord. Chem. Rev., 2017, 345, 182–208 CrossRef CAS PubMed.
- A. Rougier, C. Delmas and A. V. Chadwick, Non-cooperative Jahn-Teller effect in LiNiO2: An EXAFS study, Solid State Commun., 1995, 94, 123–127 CrossRef CAS.
- J. H. Chung, et al., Local structure of LiNiO2 studied by neutron diffraction, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 8–11 Search PubMed.
- S. Sicolo, M. Mock, M. Bianchini and K. Albe, And Yet It Moves: LiNiO2, a Dynamic Jahn-Teller System, Chem. Mater., 2020, 32, 10096–10103 CrossRef CAS.
- A. D. Poletayev, R. J. Green, J. E. N. Swallow, L. An, L. Jones, G. Harris, P. Bencok, R. Sutarto, J. P. Cottom, B. J. Morgan, R. A. House, R. S. Weatherup and M. S. Islam, Temperature-Dependent Dynamic Disproportionation in LiNiO2, arXiv, 2024, preprint, arXiv:2211.09047 DOI:10.48550/arXiv.2211.09047.
- K. Foyevtsova, I. Elfimov, J. Rottler and G. A. Sawatzky, LiNiO2 as a high-entropy charge- and bond-disproportionated glass, Phys. Rev. B, 2019, 100, 1–7 CrossRef.
- T. Mizokawa, D. Khomskii and G. Sawatzky, Spin and charge ordering in self-doped Mott insulators, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 11263–11266 CrossRef CAS.
- V. Bisogni, et al., Ground-state oxygen holes and the metal-insulator transition in the negative charge-transfer rare-earth nickelates, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- S. Johnston, A. Mukherjee, I. Elfimov, M. Berciu and G. A. Sawatzky, Charge disproportionation without charge transfer in the rare-earth-element nickelates as a possible mechanism for the metal-insulator transition, Phys. Rev. Lett., 2014, 112, 1–5 CrossRef PubMed.
- E. Wawrzyńska, et al., Charge disproportionation and collinear magnetic order in the frustrated triangular antiferromagnet AgNiO2, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 094439 CrossRef.
- R. J. Green, M. W. Haverkort and G. A. Sawatzky, Bond disproportionation and dynamical charge fluctuations in the perovskite rare-earth nickelates, Phys. Rev. B, 2016, 94, 1–5 CrossRef.
- J. Zheng, et al., Ni/Li Disordering in Layered Transition Metal Oxide: Electrochemical Impact, Origin, and Control, Acc. Chem. Res., 2019, 52, 2201–2209 CrossRef CAS PubMed.
- L. Jin, et al., Hidden Hydroxides in KOH-Grown BaNiO3 Crystals: A Potential Link to Their Catalytic Behavior, Chem. Mater., 2023, 35, 9434–9443 CrossRef CAS.
- H. Huang, et al., Unusual double ligand holes as catalytic active sites in LiNiO2, Nat. Commun., 2023, 14, 1–14 Search PubMed.
- Z. W. Lebens-Higgins, et al., Revisiting the charge compensation mechanisms in LiNi0.8Co0.2-: YAlyO2 systems, Mater. Horiz., 2019, 6, 2112–2123 RSC.
- E. Björklund, et al., Cycle-Induced Interfacial Degradation and Transition-Metal Cross-Over in LiNi0.8Mn0.1Co0.1O2-Graphite Cells, Chem. Mater., 2022, 34, 2034–2048 CrossRef.
- A. H. De Vries, L. Hozoi and R. Broer, Origin of the chemical shift in X-ray absorption near-edge spectroscopy at the Mn K-edge in manganese oxide compounds, Int. J. Quantum Chem., 2002, 91, 57–61 CrossRef.
- A. Miglio, C. P. Heinrich, W. Tremel, G. Hautier and W. G. Zeier, Local Bonding Influence on the Band Edge and Band Gap Formation in Quaternary Chalcopyrites, Adv. Sci., 2017, 4, 1700080 CrossRef PubMed.
- F. M. F. De Groot, et al., 1s2p Resonant inelastic X-ray scattering of iron oxides, J. Phys. Chem. B, 2005, 109, 20751–20762 CrossRef CAS PubMed.
- S. DeBeer, et al., X-ray absorption edge and EXAFS studies of the blue copper site in stellacyanin: Effects of axial amide coordination, J. Phys. Chem. B, 2000, 104, 10814–10819 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- C. Delmas, et al., On the behavior of the LixNiO2 system: An electrochemical and structural overview, J. Power Sources, 1997, 68, 120–125 CrossRef CAS.
- A. O. Kondrakov, et al., Charge-transfer-induced lattice collapse in Ni-rich NCM cathode materials during delithiation, J. Phys. Chem. C, 2017, 121, 24381–24388 CrossRef CAS.
- F. Kong, et al., Kinetic Stability of Bulk LiNiO2 and Surface Degradation by Oxygen Evolution in LiNiO2-Based Cathode Materials, Adv. Energy Mater., 2019, 9, 1–12 Search PubMed.
- Y. Koyama, T. Mizoguchi, H. Ikeno and I. Tanaka, Electronic structure of lithium nickel oxides by electron energy loss spectroscopy, J. Phys. Chem. B, 2005, 109, 10749–10755 CrossRef CAS.
- S. Diaz-Moreno, et al., The Spectroscopy Village at Diamond Light Source, J. Synchrotron Radiat., 2018, 25, 998–1009 CrossRef PubMed.
- G. Van Der Laan, J. Zaanen, G. A. Sawatzky, R. Karnatak and J. M. Esteva, Comparison of x-ray absorption with x-ray photoemission of nickel dihalides and NiO, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 4253–4263 CrossRef CAS.
- S. Huotari, et al., A large-solid-angle X-ray Raman scattering spectrometer at ID20 of the European Synchrotron Radiation Facility, J. Synchrotron Radiat., 2017, 24, 521–530 CrossRef CAS.
- C. J. Sahle, et al., Planning, performing and analyzing X-ray Raman scattering experiments, J. Synchrotron Radiat., 2015, 22, 400–409 CrossRef CAS PubMed.
- M. W. Haverkort, Quanty for core level spectroscopy - Excitons, resonances and band excitations in time and frequency domain, J. Phys.: Conf. Ser., 2016, 712, 012001 CrossRef.
- S. J. Clark, et al., First principles methods using CASTEP, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. Chen, C. L. Freeman and J. H. Harding, Charge disproportionation and Jahn–Teller distortion in LiNiO2 and NaNiO2: A density functional theory study, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 85108 CrossRef.
- A. J. Morris, R. J. Nicholls, C. J. Pickard and J. R. Yates, OptaDOS: A tool for obtaining density of states, core-level and optical spectra from electronic structure codes, Comput. Phys. Commun., 2014, 185, 1477–1485 CrossRef CAS.
- S. T. Perkins, et al., Tables and Graphs of Atomic Subshell and Relaxation Data Derived from the LLNL Evaluated Atomic Data Library (EADL), Z = 1–100. UCRL-50400-30, 1991.
- J. L. Campbell and T. Papp, Widths of the atomic K-N7 levels, At. Data Nucl. Data Tables, 2001, 77, 1–56 CrossRef CAS.
- D. A. Eustace, et al., First-principles calculation of spectral features, chemical shift and absolute threshold of ELNES and XANES using a plane wave pseudopotential method, J. Phys.: Condens. Matter, 2009, 21, 6 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02398f |
| This journal is © The Royal Society of Chemistry 2024 |