
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoupling light- and oxygen-induced degradation mechanisms of Sn–Pb perovskites in all perovskite tandem solar cells†
Yang
Bai‡
ab,
Ruijia
Tian‡
a,
Kexuan
Sun‡
a,
Chang
Liu
*ab,
Xiting
Lang
a,
Ming
Yang
a,
Yuanyuan
Meng
a,
Chuanxiao
Xiao
a,
Yaohua
Wang
a,
Xiaoyi
Lu
a,
Jingnan
Wang
a,
Haibin
Pan
a,
Zhenhua
Song
a,
Shujing
Zhou
a and
Ziyi
Ge
 *ab
*ab
aZhejiang Provincial Engineering Research Center of Energy Optoelectronic Materials and Devices, Ningbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, Ningbo 315201, China. E-mail: liuchang1@nimte.ac.cn; geziyi@nimte.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 2nd August 2024
Abstract
Efficiencies of all-perovskite tandem solar cells are dominantly constrained by the challenges pertaining to defects and stability within tin–lead (Sn–Pb) perovskite sub-cells. On top of the well-studied oxygen oxidation, defects related to iodide and the consequent generation of I2 upon light illumination pose significant degradation risks, leading to Sn2+ → Sn4+ oxidation. To address this, we screen phenylhydrazine cation (PEH+)-based additives of varying polarities, which strongly coordinate with Sn for reinforcing the Sn–I bond, and interacting electrostatically with negatively charged defects (VSn, VFA, ISn, and I−i). The synergistic effects suppress the photo-induced formation of I2 and the subsequent oxidation of Sn–Pb perovskites, circumventing the stability concerns of narrow bandgap (NBG) perovskite solar cells (PSCs) under operational conditions. The reducing agent 2-mercaptobenzimidazole (MBI) was further introduced into the precursor solution, which not only demonstrates strong resistance to oxygen erosion, but also reduces the deep-level defect density of the Sn–Pb perovskites. Consequently, single-junction Sn–Pb cells achieve a champion efficiency of 23.0%. The enhanced light stability allows these cells to retain 89.4% of their initial efficiency after 400 hours of continuous operation, as assessed by tracking the maximum power point (MPP). We further integrated the Sn–Pb perovskite into a two-terminal (2T) monolithic all-perovskite tandem cell and achieved a PCE of 27.9% (27.2% certified). Meanwhile, the encapsulated tandem device maintained 90.3% of its initial PCE after 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h through MPP tracking. The work offers new ideas for tackling the stability issues related to light-triggered oxidation.
h through MPP tracking. The work offers new ideas for tackling the stability issues related to light-triggered oxidation.
Broader contextAll-perovskite tandem cells have the potential to surpass the Shockley–Queisser (SQ) limit of single-junction solar cells, relying on high-performance tin-lead (Sn–Pb) perovskite solar cells (PSCs). Herein, our research specifically addresses previously unexplored and inadequately decoupled degradation pathways related to light-induced and oxygen-induced mechanisms within Sn–Pb perovskites. A series of phenylhydrazine cationic additives of different polarities were designed to enhance the Sn–I bond strength by forming strong coordination bonds with tin (Sn) while also electrostatically interacting with intrinsic charged defects. The synergistic effects suppress the photo-induced formation of I2 and the subsequent oxidation of Sn–Pb perovskite, circumventing the stability issues of narrow bandgap (NBG) PSCs under continuous light illuminations. Further bolstering the stability in air, we introduced 2-mercaptobenzimidazole (MBI). By suppressing defects and extending carrier lifetime, the single-junction NBG cell delivers an impressive PCE of 23.0%. We then integrated a 2T monolithic all-perovskite tandem cell and achieved a PCE of 27.9% (certified 27.2%) with excellent long-term stability. The knowledge gained from this study lays a solid foundation for the commercial application of perovskite technology and provides new insights into perovskite research. |
Introduction
Organometallic halide perovskites have attracted widespread attention due to their remarkable light-absorbing and electronic properties, which have promoted the PCE of PSCs by over 26.1% to date.1–6 To further break the Shockley–Queisser (SQ) limits of single-junction cells, two-terminal (2T) all-perovskite tandem solar cells have been developed, typically with the combination of a narrow bandgap (NBG, ∼1.26 eV) mixed tin–Pb (Sn–Pb) perovskite as the bottom cell and a wide bandgap (WBG, ∼1.78 eV) bromide–iodide (Br–I) based perovskite as the top cell.7–10 Despite theoretical predictions suggesting efficiencies over 40%, current real-world performance remains below 28%,5,11–17 with only a few reports exceeding 29%.18The fact that the NBG mixed Sn–Pb perovskite is highly sensitive to oxidants would introduce numerous traps or defects into the polycrystalline film, severely limiting the photovoltaic efficiency and stability of the bottom sub-cells as well as the tandem cells.8,19–22 Specifically, Sn4+/Sn2+ has a significantly smaller standard reduction potential (+0.15 V) than that of Pb4+/Pb2+ (+1.67 V), causing easy oxidation of Sn2+ → Sn4+ even in the presence of a low level of oxygen. Sn4+ ions not only act as non-radiative recombination centers for considerable voltage loss of NBG sub-cells, but also engender severe p-type self-doping of the Sn–Pb perovskite film due to the spontaneous reaction of Sn4+ → Sn2+ + 2h+, where two holes are released to the valence band, leading to unmatched energy level alignment.23,24
To weaken the oxidation of Sn2+, numerous reducing agents and antioxidants, such as tin fluoride (SnF2),25 formamidine sulfinic acid (FSA),26 hydrazine sulfate (HS),27 and 4-hydrazine benzoic acid (HBA),28 were introduced into Sn–Pb perovskite precursors. These efforts effectively mitigate the oxygen induced Sn2+ → Sn4+ during solution preparation, film deposition and film storage processes, suppressing Sn4+-related defects and elongating the shelf-stability of devices under ambient conditions.
It is proposed that the exposure of a Sn-containing perovskite film to long-term light illumination under operational conditions would account for Sn2+ oxidation as well, which has been scarcely studied thus far.29–31 In the Pb-based perovskite, light-accelerated ion migration causes the destruction of the perovskite lattice, which induces the generation of VI and interstitial iodine (I−i/I+i), subsequently producing oxidized iodine (I2).32–36 Huang proposed that such effects would be exacerbated in the Sn–Pb perovskite considering that a weaker Sn–I bond than Pb–I readily leads to the migration and oxidation of I− (as schematically illustrated in Fig. S1, ESI†).29 Wang et al. further elaborated this by revealing the Sn–I octahedral framework distortion upon photon injection, when MA+/FA+ organic cations in the lattice transfer energy to the Sn–I octahedral framework through coupling.37 The continuous accumulation of energy causes the Sn–I bond to break and release free I−, subsequently generating elemental iodine.38 Afterwards I2 could further initiate Sn2+ → Sn4+ in view of its higher standard redox potential φ (I2/I−) of +0.54 V than that of φ (Sn4+/Sn2+), which is fatal to device efficiency and stability. Besides, Saif A. et al. unveiled that the oxidized degradation product SnI4 rapidly evolved into I2 under the combined stresses of light and oxygen, generating more SnI4 and establishing a recycling degradation mechanism.39 Such a decomposition pathway severely limits the operational stability of NBG PSCs and 2T all perovskite tandem solar cells under light and thermal stresses, despite previous strategies including use of antioxidant additives and rigorous encapsulations having been attempted.29,32,40,41
Towards the end, the fundamental concept to mitigate light-induced I− migration is to concomitantly strengthen Sn–I bonding interaction and suppress the iodine-related defects (including VI, VSn, VFA, ISn, and I−i). Phenylhydrazine-based cations (PEH+) have thus been screened as additives in this work thanks to their strong coordination with tin for reinforcing the Sn–I bond, as well as the intense electrostatic interactions with negatively charged defects, in comparison with the widely utilized phenylammonium cations (details are discussed in the main context). From the characterization studies including X-ray photoelectron spectroscopy (XPS), liquid-state 1H nuclear magnetic resonance (1H NMR) and Fourier transform infrared spectroscopy (FTIR), it is revealed that the coordination with tin and passivation effects on various defects successively incremented with higher electrostatic potentials of PEH+ based additives (PEHCl-OC, PEHCl-F, and PEHCl-CN). It resultantly affords the strongest resistance to light illumination of the Sn–Pb perovskites with PEHCl-CN (largest dipole moment) incorporation, delivering NBG PSCs with a PCE of 21.7%. As an effective passivation agent to charged vacancies, the PEH+ cation shows substantially sacrificed reducing properties to oxygen as compared to the phenylhydrazine (PE) analogue, due to the salification process. Therefore, the reducing agent (2-mercaptobenzimidazole, MBI) has been further introduced into the Sn–Pb precursor along with PEHCl-CN, to inhibit the degradation pathway from the ambient air. With suppressed defects and an elongated charge carrier lifetime, the single-junction NBG cell affords an impressive PCE of 23.0%. Subsequently, we fabricated monolithic all-perovskite tandem solar cells using optimized NBG perovskite layers, achieving an exceptional PCE of 27.9%, with a certified value of 27.2%. The encapsulated tandem device maintained 90.3% of its initial PCE after 300h through maximum power point (MPP) tracking. Besides, the tandem device retained 90.7% of its original PCE after storing it in an N2-filled atmosphere for 2400 hours.
Results and discussion
Defect passivation by the PEHCl-X (-OC, -F, -CN) system
PEHCl was utilized as the research system in this work. Compared with the commonly used phenylammonium cations (PA+),5 which has been proven to be effective in passivating Sn–Pb perovskites, PEH+ shows more negative adsorption energy on the perovskite surface (−5.73 eV vs. −4.64 eV in Fig. S2 and S3, ESI†). It suggests a stronger passivation effect of PE+ than that of PA+ counterparts. This is possibly due to the electronegative N for increasing the electropositivity on the –NH to NH3+ cation, leading to stronger binding with negatively charged vacancies. Substituting groups with different electron-donating/withdrawing characteristics at the para-position of the hydrazine were further introduced to regulate the polarities of the molecules. The calculated electrostatic potential distribution is shown in Fig. 1a–c. The proposed working mechanisms of PEHCl-based agents on the Pb–Sn perovskite are schematically illustrated in Fig. 1d. The hydrogen in two different chemical environments in –NH to NH3+ would form N–H–I hydrogen bonds with iodine in the perovskite, preventing the formation of shallow energy level defects. Furthermore, the strong interaction between PEH+ with tin will enhance the oxidation barrier of Sn2+. By regulating the molecular dipole moment, the electrostatic interactions with defects would also be improved, thereby enhancing the formation energy of various defects. The following series of theoretical calculations and experimental characterization will verify these conjectures.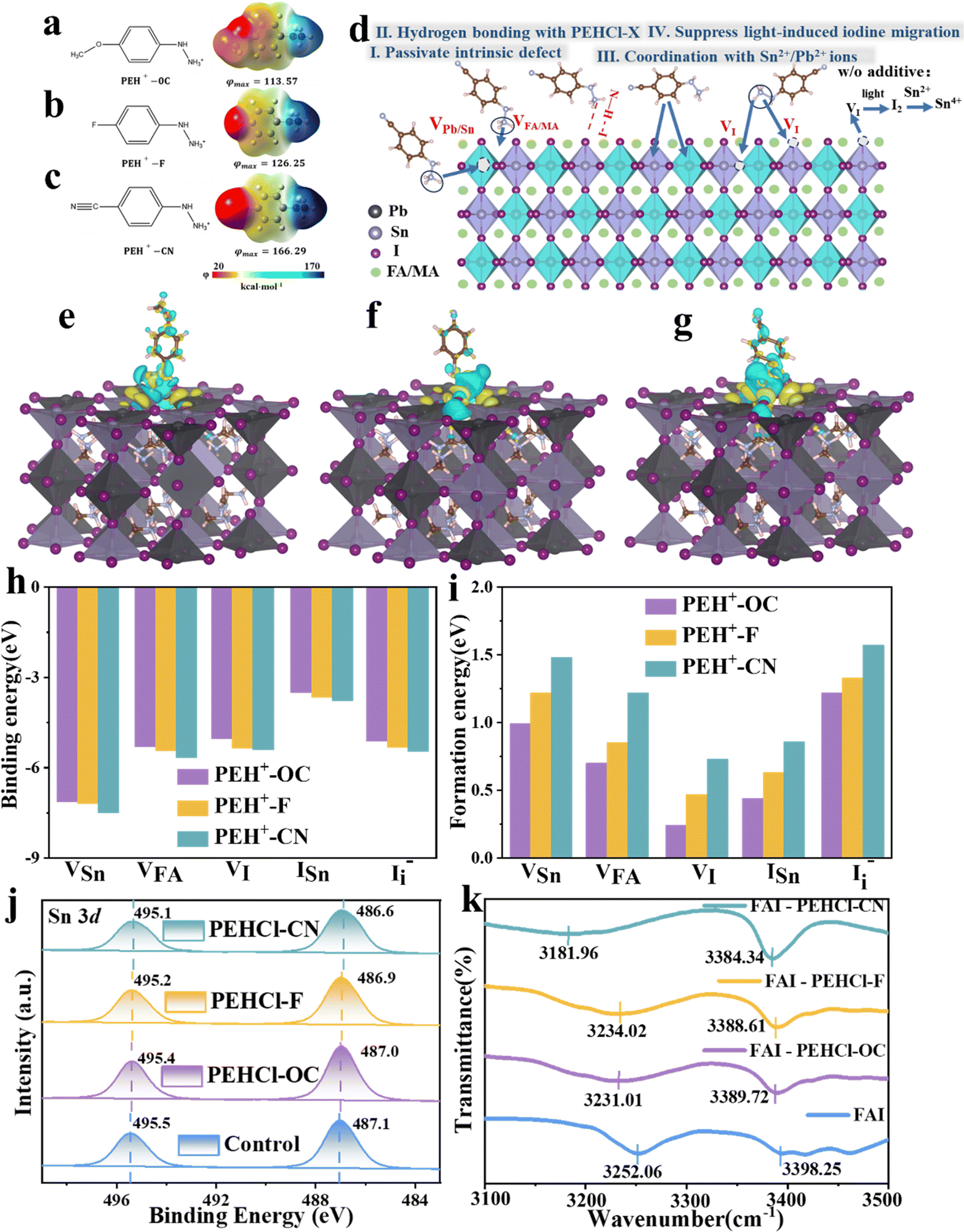 | ||
| Fig. 1 PEHCl-X (-OC, -F, -CN) passivation mechanism. (a)–(c) The structure and calculated electrostatic potentials (φ) of three passivators (PEHCl-OC, PEHCl-F and PEHCl-CN). The right colour bar from red to blue marks the increase of electropositivity. (d) Schematic diagram of the passivation mechanism of PEHCl-X. The electron density distribution of (e) PEHCl-OC, (f) PEHCl-F, and (g) PEHCl-CN after absorption on the perovskite. Yellow represents areas of increased charge, while light blue indicates areas of decreased charge. (h) The binding energy (Eb) of surface Sn, FA and I vacancy defects (VSn, VFA and VI), the I interstitial defect (I−i), I substituted at the Sn site (ISn) adsorbed by FA0.5MA0.5Sn0.5Pb0.5I3 and FA0.5MA0.5Sn0.5Pb0.5I3 on PEHCl-X. (i) The formation energy (Ef) between the passivators and various defects (define the Ef value of the original film as zero). (j) Sn 3d XPS spectra of control, PEHCl-OC-modified, PEHCl-F-modified, PEHCl-CN-modified films. (j) FTIR spectra of FAI, FAI/PEHCl-OC, FAI/PEHCl-F and FAI/PEHCl-CN film complex. | ||
The electrostatic potential (φmax) differs on the –NH to NH3+ side in an order of φ(PEHCl-F) < φ(PEHCl-OC) < φ(PEHCl-CN) in Fig. 1a–c. The effects of three molecules on passivating the perovskite surfaces (through both ionic bonding and hydrogen bonding) were investigated through density functional theory (DFT). Calculated from the three different surface adsorption orientations (horizontal, vertical, and oblique adsorption) of additives on the Sn–Pb perovskite surface, the results are shown in Fig. S3 and S5 (ESI†). It has been demonstrated that PEHCl-OC and PEHCl-F tend to adsorb vertically on the surface, while PEHCl-CN tends to adsorb obliquely on the surface. From Fig. S3 (ESI†), the additive with the highest electrostatic potential (PEHCl-CN) exhibits a more negative adsorption energy of −6.48 eV than that of PEHCl-OC (−5.73 eV) and PEHCl-F (−6.06 eV), suggesting the strongest interaction between PEHCl-CN and the perovskite. Furthermore, by evaluating the differential charge density and plotting the cross-sectional distribution of charge density (Fig. 1e–g and Fig. S4, ESI†), charge accumulation is observed between the perovskite surface and additives. Notably, in the case of PEHCl-CN, a more pronounced accumulation of electrons is evident, elucidating the stronger and more stable adsorption of perovskites, as compared to those of PEHCl-OC and PEHCl-F.
Previous studies have evinced that surface defects greatly affects the degradation pathway of the Sn–Pb perovskites.22,42,43 To understand the influence of PEHCl-X adsorption on the surface defects of perovskites, the binding and formation energies of various defects on its surface were calculated. Binding energy (Eb) summarized in Fig. 1h demonstrates that PEHCl-CN with the highest φmax has the strongest binding to acceptor-type defects (VFA, VSn, ISn, and I−i) on the perovskite surface.5 It is explained that the stronger electron-withdrawing cyano group in PEHCl-CN strongly absorbs the electron density of adjacent atoms, leaving a higher electron positivity on the –NH to NH3+ side, thereby enhancing the electrostatic interaction with negatively charged defects. The formation energies (Efs) of various types of defects were further calculated (Fig. 1i), where the Ef value of the original film was defined as zero. It can be clearly seen that with the incorporation of the three additives, the Ef value of acceptor-type defects (VFA, VSn, ISn, and I−i) moves to a positive value. This is due to the enhanced ionic bond strength between PEHCl-X and the charged defects, resulting in a larger potential barrier in the formation of acceptor defects.44,45
For the donor-type defect VI, which could have originated from the presence of uncoordinated metal elements on the perovskite surface,46,47 the passivation might have arisen from the strong interaction between Sn and the PEHCl-CN additive. To verify this, XPS measurements were carried out. After adding PEHCl-X, the XPS peak of Sn 3d5/2 shifts from 495.5 to 495.4, 495.2, and 495.1 eV, and that of Sn 3d3/2 shifts from 487.1 to 487.0, 486.9, and 486.6 eV, respectively, for PEHCl-OC, PEHCl-F and PEHCl-CN, as illustrated in Fig. 1j. The XPS peak shifted downward to a lower binding energy, indicating that the electron density around Sn increased and its electron-deficient under-coordinated state was improved. This is due to the coordination between PEHCl-X with higher electronegativity and Sn, which also indirectly passivates the VI defect. In addition, by increasing the electron density around Sn, the oxidation of Sn2+ to Sn4+ can be effectively prevented. Meanwhile, the results in Fig. S6 (ESI†) testify to the presence of Cl 2p with the incorporation of hydrochloride, which would also directly passivate the iodine vacancies in perovskite films.48,49 These are consistent with the increase in the VI formation energy (Fig. 1i). It verifies our conjecture that the incorporation of PEHCl-X strengthened the Sn–I bond in the perovskite lattice, preventing it from breaking and causing the migration of free iodide ions. Additionally, the hydrogen bonding between –NH to NH3+ and I− could also contribute to suppressing the migration of I−, thus enhancing the VI formation energy.
To further testify the N–H–I hydrogen bonding between PEHCl-X and perovskite, 1H NMR was employed (Fig. S7, ESI†). We used FAI instead of SnI2 and PbI2 to rule out the interaction between the additives and Sn–Pb perovskite. After the addition of FAI, the N–H bonds of the hydrazine group in two different chemical environments of the PEHCl-series molecules all shift to a higher field. Among them, the proton resonance signals of –NH and –NH3+ moved from 9.15 to 8.76 ppm (–0.39 ppm) and 10.62 to 10.17 ppm (–0.45 ppm) respectively, upon the addition of PEHCl-CN. This is because the I− atom is too large, and the ability of the nucleus to bind the electron cloud becomes weaker, which enhances the shielding effect. At the same time, the hydrogen atoms on the hydrazine group in PEHCl-X act as proton donors. After interacting with iodine, the density of the hydrazine electron cloud on PEHCl-X increases, further enhancing the shielding effect of the electron cloud on hydrogen protons. Therefore, the chemical shift of the hydrogen spectrum moves toward the high-field direction, and the chemical shift becomes smaller. The above verifies the N–H–I hydrogen bond between the hydrazine group and perovskite. Meanwhile, the increment in the electron cloud density on the hydrazine group would also weaken the electron-donating ability of the Sn2+, subsequently inhibiting its oxidation. The PEHCl-OC and PEHCl-F incorporation shows similar results, but with fewer peak shifts as compared to that with PEHCl-CN treatment.
Fourier transform infrared (FTIR) spectroscopy was also recorded to assess the hydrogen bonds and coordination interactions. From Fig. 1k, it is seen that with the addition of PEHCl-X to FAI, the N–H vibrational peaks located at 3252.06 cm−1 experienced downshifts by −21.05 cm−1, −18.04 cm−1 and −70.1 cm−1 for PEHCl-OC, PEHCl-F and PEHCl-CN, respectively, and the other peaks located at 3398.25 cm−1 downshifts by −8.53 cm−1, −9.64 cm−1 and −13.91 cm−1. The reason for this shift is that the proton acceptor iodine transfers electrons into the antibonding orbital of the proton donor N–H, resulting in the elongation of the N–H bond and a red shift in the vibration frequency. This is consistent with the 1H NMR results, once again proving the hydrogen bonding interaction between the PEHCl-X molecules and perovskite.50 The coordination interaction was also investigated by mixing PEHCl-X with SnI2, the FTIR spectra are shown in Fig. S8 (ESI†). For PEHCl-OC, PEHCl-F and PEHCl-CN, N–H vibrational bonds of the amino groups located at 3214.40, 3208.96 and 3207.87 cm−1 move to a lower wavenumber direction due to coordination with Sn2+ ions.49 Such a shift indicates that the N with a lone pair of electrons on the PEHCl-X molecule and the Sn2+ in the perovskite may form an N→ Sn coordination bond, which together with XPS proves the interaction (Fig. 1j).
Inhibiting the formation of light-induced iodine
Effects of PEHCl-X passivation on the photo-generated I2 and its subsequent oxidation of Sn2+ → Sn4+ were investigated. Firstly, the generation of I2 in the perovskite film under light illumination was quantitatively evaluated by ultraviolet (UV) absorption spectra in Fig. S9 (ESI†).29,51 The perovskite films were immersed in toluene, followed by illumination for a gradient period from 10 to 40 h. As can be seen from Fig. S9e (ESI†), the generation of I2 quantity (absorbance at 500 nm) decreases sequentially in control, PEHCl-OC-treated, PEHCl-F-treated and PEHCl-CN-treated films. This suggests that the lower density of defects (including VI, VSn, VFA, ISn, and I−i) suppresses the halide ion migration under illumination, thus stabilizing the Sn–Pb perovskites.Scanning electron microscopy (SEM) measurements were then performed to study the evolution of perovskite film morphologies under light illuminations. By comparing the fresh films in Fig. 2a–d and Fig. S10, S11 (ESI†), it is seen that the addition of PEHCl-X imposes marginal effects on grain sizes, while suppressing the formation of residual PbI2 on grain boundaries.13,52 It might be explained that the intermediates formed by the coordination between PEHCl-X and SnI2 lead to a complete reaction between FAI and SnI2. Upon continuous illumination for 20, 30, and 40 hours, the control film shows severe degradation of the perovskite grains with the emergence of large amounts of PbI2 areas, as shown in Fig. 2a–d and Fig. S10, S11 (ESI†). Contrastingly, the incorporation of PEHCl-X additives shows good resistance of the film morphologies to light illumination, among which, the PEHCl-CN addition reserves an intact film morphology with suppressed precipitation of PbI2. It is beneficial for improving the device performance, reducing hysteresis behavior and improving device stability. Consistent with the SEM results, the X-ray diffraction (XRD) analysis of the four films reveals that the control film exhibits an additional peak at 12.7° (Fig. S12, ESI†),22 assigning to the (001) lattice plane of PbI2. The intensity of the peak decreases sequentially in the order of PEHCl-OC, PEHCl-F, and PEHCl-CN.
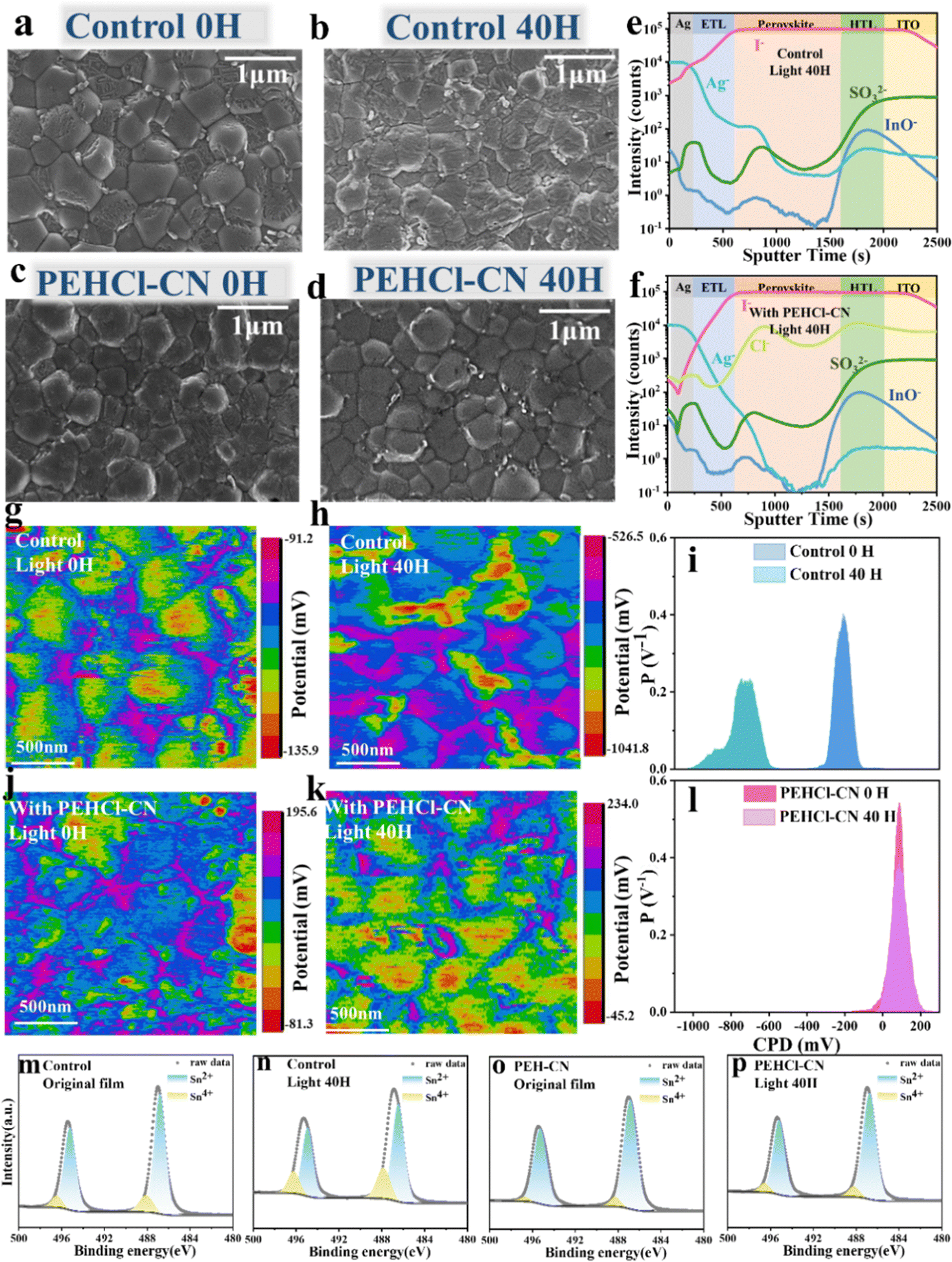 | ||
| Fig. 2 Stability of NBG Sn–Pb perovskite films under light illumination. Scanning electron microscopy (SEM) images of perovskite films without illumination and after illumination for 40 hours (a) and (b) pristine perovskite films and (c) and (d) perovskite films with PEHCl-CN. Time-of-flight secondary ion mass spectrometry (ToF-SIMS) profiles of the control film (e) and PEHCl-CN modified (f) PSC (both after 40 hours of illumination treatment). Kelvin probe force microscopy (KPFM) images under illumination for 0 hours and 40 hours: (g) and (h) pristine perovskite films and (j) and (k) perovskite films with PEHCl-CN, the corresponding CPD potential distributions in (i) and (l). The Sn 3d XPS of control (m) and (n), and PEHCl-CN (o) and (p) perovskite films under the same conditions as above. The specific content of Sn4+ is in Table S1 (ESI†). | ||
The cross-sectional morphological evolution and associated iodide distributions in PSCs were also analyzed by cross-sectional SEM images and energy dispersive spectroscopy (EDS).53,54 To simulate real device scenarios, stacked layers of ITO/PEDOT:PSS/perovskite/C60/BCP/Ag were examined. Obviously, the control film shows massive pinholes and grain boundaries that could stimulate non-radiative recombination and structural degradation. The corresponding EDS analysis for iodide in the perovskite films (Fig. S13, ESI†) reveals increased iodide diffusion from the perovskite to the C60 layer over prolonged light illumination periods, confirming significant halide ion migration across the layers. The migration of iodide ions ultimately gives rise to the precipitation of I2, which would directly oxidize Sn2+ to Sn4+. Additionally, I2 will escape from the perovskite, penetrating into the adjacent transmission layer, or even corroding the electrode, which would further accelerate device degradation. Conversely, in PEHCl-CN-treated samples, the structural integrity could be well maintained after illumination, and the corresponding EDS spectrum as well demonstrates suppressed iodide migration to the adjacent layers after 40 hours of illumination (Fig. S13, ESI†).
Time-of-flight secondary ion mass spectrometry (ToF-SIMs) depth profiling was then conducted to analyze the diffusion of iodine in the device layers after 40 hours of illumination. As shown in Fig. 2e and f, our ToF-SIM measurements have identified several ions: Ag−, originating from the silver electrode; I−, indicative of the perovskite layer itself; SO3−, associated with the hole transporting layer PEDOT:PSS; and InO− from the ITO substrate and the additional anions detected were chloride ions (Cl−). Obviously, the concentration of iodine in the electron transporting layer (ETL) and the Ag electrode of the control device is significantly reduced in the PEHCl-CN-treated device over that in the control device. It evidences that PEHCl-CN could effectively prevent the diffusion of iodine to other functional layers, thus inhibiting the corrosion of the electrode and the subsequent device degradation.33 Meanwhile, the distribution of Cl− reflects the overall distribution of additives in the perovskite film. It can be seen from Fig. 2f that Cl− ions are mainly distributed on the upper surface and lower interface of the perovskite, while are less distributed in the middle of the perovskite. Furthermore, the 3D distribution image of chloride ions was further drawn to demonstrate such a phenomenon (Fig. S14, ESI†). This could be explained that the majority of the grain boundaries are formed at the bottom and upper surface of the perovskite films, rather than in the bulk film. The additives reside on these grain boundaries.
XPS measurements were performed to quantitatively characterize the Sn2+ → Sn4+ process initiated by photo-induced I2 in the perovskite film (Fig. 2m–p and Fig. S15, ESI†). The films were aged at gradient light illumination periods of 0 h, 20 h and 40 h. In line with the changing trend of the I2 content (Fig. S9e, ESI†), the PEHCl-OC, PEHCl-F and PEHCl-CN treated perovskite films show successively decreased Sn4+ under light aging, which is all significantly lower than that in the pristine film. Among PEH-X series additives, PEHCl-CN shows the strongest oxidation resistance to I2, with the marginal Sn4+ XPS peak presented (Fig. 2o). It could be explained that the migration of iodine ions induced by light is inhibited, thereby fundamentally inhibiting the oxidation of Sn2+ with the incorporation of PEHCl-CN. The results are consistent with the previous 1H NMR discussions (Fig. S7, ESI†).
To dig into the effects of additives on the electrical charge distribution of the perovskite films, Kelvin probe force microscopy (KPFM) measurements were conducted to inspect the surface potential of perovskite films (Fig. 2g, h, j, k and Fig. S16, S17, ESI†), each with an area of 2 μm × 2 μm. Contact potential difference (CPD) was recorded by detecting the electrostatic force between the scanning tip and film surface, i.e. Vtip − Vsample.55 According to Fig. 2i and l, the control and PEHCl-CN-treated fresh films exhibit CPD peaks at −210.2 mV and 98.7 mV. The more positive CPD in the PEHCl-CN treated film compared to that in the control film is indicative of a lower work function and thus more n-type characteristics. This can be attributed to the fact that the addition of PEHCl-CN brings a dipole moment and dielectric environment to the perovskite grains, compensating for the excess holes in the Sn–Pb perovskite.23 It could subsequently reduce the degree of p-type self-doping.56 Combining the ultraviolet photoelectron spectroscopy (UPS) energy spectrum and the optical band gap of ≈1.26 eV (Fig. S18, ESI†), the energy level diagram can be deduced as shown in Fig. S19 (ESI†). It is seen that the Fermi level of the perovskite film shifts upward by 0.09 eV after PEHCl-CN incorporation, which is consistent with the above KPFM results. As polarity increases, the surface properties (Fig. S17a and d, ESI†) gradually transit to more n-type doping (82.1 mV for PEHCl-F and 17.6 mV for PEHCl-OC). Meanwhile, films treated with the PEHCl-X series of additives exhibit a narrower distribution of CPD values, indicating improved uniformity of perovskite grains.
Under 20 hours of continuous illumination, a CPD peak at around −900 mV appears in the control film (Fig. S16a, ESI†). This could be assigned to the formation of a high work function PbI2 region (∼5.9 eV),57,58 which originated from the light-induced ion migration for degrading perovskite into I2 and PbI2. Moreover, the decomposed perovskite film exhibits a more negative surface potential with the peak shifting from −210.2 mV to −760.5 mV (Fig. 2i). This might explain that photo-induced I2 accelerates the formation of Sn4+, making severe p-type doping in the Pb–Sn perovskite film.59 These are consistent with the UV and XPS results discussed previously. An elongated light duration (up to 40 hours) (Fig. 2k and l) further expands the CPD distribution, shifting the surface potential more negatively. In clear contrast, the PEHCl-CN-treated perovskite film demonstrates strong resistance to photo-induced p-type self-doping or degradation, without noticeable CPD shifts or the emergence of a high work function PbI2 region.
The above results and discussions strongly support the view that the incorporation of PEHCl-CN effectively inhibits light-induced I2-related Sn–Pb degradation. This could be ascribed to the suppressed formation of iodine-related defects (including VI, VSn, VFA, ISn, and I−i) and strengthened Sn–I bonding interactions, which increases the potential barrier for iodine ion migration. It has important implications for solving the significant stability issues of Sn–Pb PSCs under practical operational conditions.
Resistance of Pb–Sn perovskites to air oxidation
In addition to intrinsic defects and the related I2-triggered degradation pathway of the Sn–Pb perovskites, Sn2+ could be directly oxidized by ambient air. The above results confirmed that the PEHCl-CN cation could form strong electrostatic interactions with the intrinsic defects, preventing the generation of I2 and the Sn2+ oxidation. It is noted that the salification of PEHCl into PEHCl significantly weakens the intrinsic reducing property of PEHCl, as can be confirmed by the color change experiment of the precursor solution. From Fig. 3e, PEHCl-CN shows limited ability to inhibit the oxidation of Sn2+ → Sn4+ in the precursor solution under exposure to air. Thus, we introduced the 2-mercaptobenzimidazole (MBI) molecule (structure shown in Fig. 3c) The thiol group (–SH) has been shown to significantly improve the stability of Pb-based perovskites and has strong reducing properties.31 Due to the effect of the electron-donating group imidazole, the hydrogen atom at the end of the thiol group is easier to fall off. In addition, MBI can also form hydrogen bonds with O2 in the air through the thiol group to inhibit the oxidation of Sn2+. This process was verified by subsequent DFT, and to understand the mechanism of the enhanced stability of SnI2 complexes, we performed cyclic voltammetry characterization and analyzed the redox potentials of SnI2, SnI2–PEHCl-CN and SnI2–MBI complexes. As shown in Fig. S20 (ESI†), the calculated highest occupied molecular orbital (HOMO) energy level is −4.736 (0.064–4.8) eV for the SnI2–MBI complex, which is lower than −4.71 (4.8–0.09) eV for SnI2. The −4.711 (4.8–0.089) eV for the SnI2–PEHCl-CN complex is similar to that for SnI2. The deeper the HOMO energy level, the more difficult it is to oxidize the SnI2–MBI complex, indicating that MBI can significantly increase the oxidation barrier, which is consistent with the phenomenon of reduced oxidation in solution discussed above.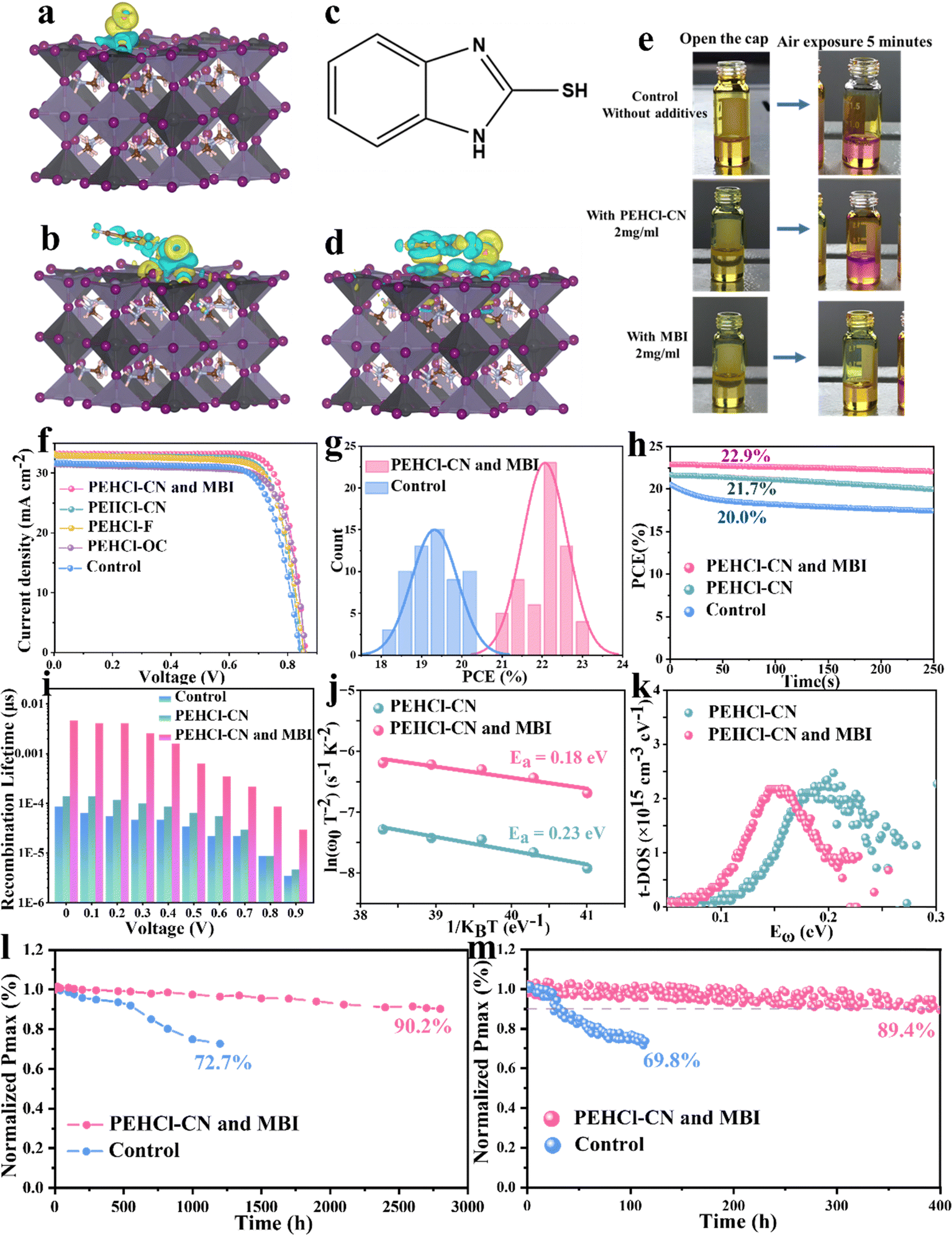 | ||
| Fig. 3 Characterization of air resistance and performance of single-junction Sn–Pb PSCs. The electron density distribution of (a) O2 and (b) O2-PEH+-CN after absorption on perovskite. (c) Molecular structural formula of MBI. (d) The electron density distribution of O2–MBI after absorption on perovskite. (e) Color changes of the FA0.7MA0.2Cs0.1Sn0.5Pb0.5I3 precursor solution under different conditions. (f) J–V curves of the best-performing solar cells. (g) Histogram of PCEs for 60 single-junction Sn–Pb PSCs. (h) SPO efficiency of the single-junction PSCs. (i) Recombination lifetime of devices under different biases derived from the Nyquist plots in Figure S. (j) The Arrhenius plots of the attempt-to-escape frequencies to extract the defect activation energy (Ea) for PSCs. (k) Trap density of states (t-DOS) of PSCs. Maximum power point (MPP) tracking of control devices and devices with PEHCl-CN and MBI under 1 sun illumination in the N2 environment. (l) Long-term stability test of control devices and devices with PEHCl-CN and the MBI Sn–Pb cell stored in an N2-filled glovebox. (h) MPP tracking of control devices and devices with PEHCl-CN and MBI under 1 sun illumination in the N2 environment. | ||
Previous research indicates that Sn2+ oxidation typically results from the adsorption of oxygen molecules (O2) on the Sn–Pb perovskite surface.60 Our investigation into the charge density distribution affected by O2, O2–MBI, and O2–PEHCl-CN adsorption (Fig. 3a, b and d) using DFT shows that MBI and PEHCl-CN alter the charge distribution on the perovskite surface. MBI forms hydrogen bonds with O2, facilitating O2–MBI co-adsorption on the perovskite, a phenomenon also observed with O2–PEHCl-CN. Cross-sectional charge density analysis (Fig. S21, ESI†) reveals a significant reduction in charge transfer between O2 and Sn in the presence of MBI and PEHCl-CN, indicating their protective effect against O2 oxidation. Notably, the MBI system exhibits a markedly reduced charge transfer, highlighting its superior efficacy in inhibiting O2-induced oxidation of perovskite crystals compared to PEHCl-CN.
After incorporating PEHCl-CN and MBI, negligible Sn4+ could be detected from the XPS spectrum of the fresh film, which is a significant reduction compared to 4.98% of Sn4+ in the PEHCl-CN-treated perovskite and 12.66% of Sn4+ in the control film, respectively (Fig. S22, ESI†). Under air exposure for 20 and 40 h, the integration of the XPS peak area defines the Sn4+ content to be 26.29% and 36.58%, respectively, in the control film, which is similar to those in only the PEHCl-CN-treated film. In sharp contrast, the content of Sn4+ shows to be as low as 9.49% in the PEHCl-CN and MBI incorporated films after 40 h of exposure to air (Fig. S23 and Table S1, ESI†), demonstrating the strong antioxidant properties of MBI, thereby contributing to higher quality perovskite films and highly stable devices.
To further understand the effect of PEHCl-X series and MBI additives on the crystallinity of the Sn–Pb perovskites, as well as their structural stability, XRD measurements were performed (Fig. S24, ESI†). For the control film, the diffraction peak of ∼14° corresponding to the (100) crystal plane almost disappears after 30 hours of exposure to air, indicating complete degradation of the perovskite crystals. With the incorporation of PEHCl-X additives, the intensity of the (100) diffraction peak increases with the full width at half maximum (FWHM) narrowed from 0.10483° to 0.09409°, 0.0895° and 0.0927°, respectively, for PEHCl-OC, PEHCl-F, and PEHCl-CN treatments, indicative of the improved crystallinity.61 This might be due to the stronger rapid crystallization interaction of higher polarity additives with the perovskite that stimulates rapid nucleation and fine crystallization processes. While for PEHCl-X incorporation alone, the diffraction peak of (100) gradually disappears after 40 h of aging in air (Fig. S25, ESI†), further confirming the absence of the reducing property of PEHCl-X. For binary treatment with PEHCl-CN and MBI, the perovskite crystal is well maintained even after 40 h of exposure, in line with the XPS results under air aging, indicating that the existence of MBI is the main reason for resisting oxygen corrosion to the perovskite lattice.
Depth-resolved grazing incident XRD (GIXRD) patterns were then conducted to quantitatively analyze the residual stress in the perovskite films, which were recorded at different Ψ values ranging from 10° to 50°. Based on the following equation: 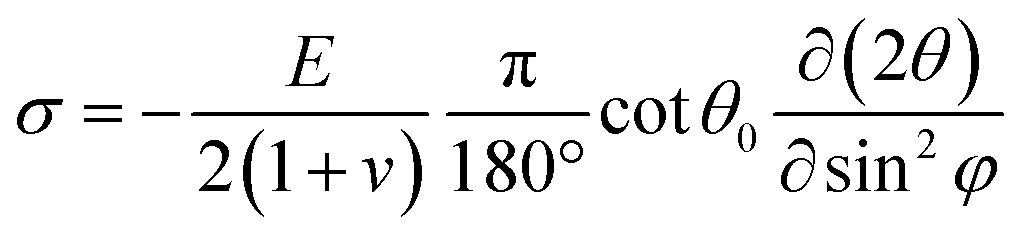 (where φ and E are the angles of the diffraction vector corresponding to the sample surface normal and the perovskite modulus),62 2θ − sin2Ψ plots (derived from the GIXRD patterns in Fig. S26, ESI†) shown in Fig. S27 (ESI†) reveal tensile strains of 54 MPa, 50 MPa, 35 MPa, 34 Mpa, and 9 Mpa for pristine, PEHCl-OC-treated, PEHCl-F-treated, PEHCl-CN-treated and PEHCl-CN and MBI-treated films, respectively. The release of residual stress in the perovskite film is beneficial for the photovoltaic performance and stability of PSCs. For the control film, the lattice is severely deformed due to the presence of tensile stress. After modification by PEHCl-CN and MBI, residual stress can be released and the perovskite grain boundaries can be healed, thereby obtaining a high-quality perovskite film.
(where φ and E are the angles of the diffraction vector corresponding to the sample surface normal and the perovskite modulus),62 2θ − sin2Ψ plots (derived from the GIXRD patterns in Fig. S26, ESI†) shown in Fig. S27 (ESI†) reveal tensile strains of 54 MPa, 50 MPa, 35 MPa, 34 Mpa, and 9 Mpa for pristine, PEHCl-OC-treated, PEHCl-F-treated, PEHCl-CN-treated and PEHCl-CN and MBI-treated films, respectively. The release of residual stress in the perovskite film is beneficial for the photovoltaic performance and stability of PSCs. For the control film, the lattice is severely deformed due to the presence of tensile stress. After modification by PEHCl-CN and MBI, residual stress can be released and the perovskite grain boundaries can be healed, thereby obtaining a high-quality perovskite film.
Charge carrier dynamics were further explored using steady-state and time-resolved photoluminescence (PL and TRPL) measurements. From the PL spectra in Fig. S28 (ESI†), PEHCl-X treatment improves the PL quenching of perovskite, and the degree of enhancement also follows the same trend as the dipole moment increment, where the most polar PEHCl-CN shows the highest PL intensity. The incorporation of MBI further enhances the fluorescence intensity, and the peak position was blue-shifted, suggesting fewer surface defects and lower non-radiative recombination losses. Correspondingly, bi-exponentially fitting the TRPL spectra (Fig. S29, ESI†) affords a notably elongated carrier recombination lifetime from 5.96 μs (control) to 8.00 μs (PEHCl-OC), 8.12 μs (PEHCl-F), 8.15 μs (PEHCl-CN), and 8.46 μs (PEHCl-CN and MBI), respectively, consistent with the PL spectra. Fitting details are illustrated in Table S2 (ESI†).63
To evaluate the charge recombination rate in Sn–Pb based PSC devices, transient photovoltage (TPV) measurements were performed. Under light illumination, voltage attenuation is observed, which is closely related to the separation of carriers. Faster signal decay indicates more internal defects in the film, which intensifies the recombination of carriers. As shown in Fig. S30a (ESI†), the decay time of the device with PEHCl-CN and MBI incorporation was found to be 0.42 ms, which is longer than that of the control device (0.36 ms) and the PEHCl-CN incorporated device (0.40 ms), implying a lower charge recombination rate and thus fewer internal defects.29 Then the transient photocurrent (TPC) was measured. A shorter carrier lifetime of TPC from 1.5 μs (control) to 0.85 μs and 0.68 μs could be extracted for PEHCl-CN and PEHCl-CN and MBI treatment (Fig. S30b, ESI†), respectively, indicating substantially improved charge extraction efficiency from perovskite to the adjacent transporting layers.64
Performance and stability of NBG single-junction PSCs
Motivated by the improved film properties observed with the addition of PEHCl-CN and MBI, planar single-junction Sn–Pb PSCs were fabricated employing an inverted p–i–n configuration of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS)/NBG perovskite/C60/bathocuproine (BCP)/Ag.65 As shown in Fig. S31 (ESI†), we optimized the optimal concentrations of the three additives PEHCl-X (-OC, -F, and -CN) and MBI, the former were all 2.0 mg ml−1, while the optimal concentration of MBI was 4.0 mg ml−1. Following the modification of the perovskite film with PEHCl-CN and MBI, significant enhancements in photovoltaic parameters were observed: the open circuit voltage (VOC) increases from 0.83 V to 0.86 V, the short circuit current density (JSC) increases from 31.66 mA cm−2 to 33.2 mA cm−2, and the fill factor (FF) improves from 75.4% to 80.3%, as shown in Fig. 3f and Table S5 (ESI†). Prior studies have indicated that such an increment in JSC is crucial, as it directly influences the photocurrent density matching in all-perovskite tandem solar cells. External quantum efficiency (EQE) measurements corroborate the high JSC values observed in PEHCl-CN and MBI modified PSCs (Fig. S32, ESI†). The highest-performing device records a PCE of 23.0%, significantly surpassing the 20.1% of the control PSCs. Moreover, the Sn–Pb cell demonstrated a stabilized PCE of 22.9%, showcasing its substantial stability potential (Fig. 3h). Furthermore, 60 single-junction NBG solar cells with or without PEHCl-CN and MBI were fabricated, and the average PCE of the additives increases to 22.1% (relative to 19.3% of the control device), as shown in Fig. 3g.Space-charge limited-current (SCLC) measurements were performed to quantitatively investigate the trap densities (Nt) in the perovskite films based on hole-only devices and electron-only devices (Fig. S34a and c, ESI†).66 From the I–V results in Fig. S34 b and d (ESI†), the trap field limit voltage (VTFL) and Nt values of the perovskite film could be extracted and are summarized in Tables S3 and S4 (ESI†).67 Upon PEHCl-CN and MBI treatment, the VTFL decreased from 0.55 to 0.25 V (hole-only device) and from 0.52 to 0.22 V (electron-only device). Meanwhile the perovskite film shows reduced Nt values from 8.51 × 1016 cm−3 (hole-only device) and 8.05 × 1016 cm−3 (electron-only device) to 3.57 × 1016 cm−3 and 3.40 × 1016 cm−3 respectively, accrediting to inhibition of iodine ion migration as well as the passivation of halide defects and under-coordinated Sn2+ and Pb2+.
To dig into the charge recombination dynamics at the device level, electrochemical impedance spectroscopy (EIS) spectra of the PSCs at external voltages of 0–0.9 V were recorded and are shown in Fig. S35 (ESI†). Fig. S36 (ESI†) shows a comparison of the EIS spectra at 0.4 V, which reveals the significantly incremented recombination resistance (Rrec) values of PEHCl-CN (955 Ω) and MBI-modified (1527 Ω) Sn–Pb perovskite devices as compared to that of the control device (202 Ω). The larger Rrec values of PEHCl-CN and MBI integrated devices indicate suppressed charge recombination losses. The carrier recombination lifetimes of the perovskite films can be extracted from Fig. S33 (ESI†) and ar summarized in Fig. 3i.68 The elongated carrier recombination lifetimes further support the effective defect passivation and reduced carrier recombination in devices with PEHCl-CN and MBI treatment. As can be seen from Fig. S37 (ESI†), the built-in potential (Vbi) of the PSC increases from 0.44 V for the control device to 0.47 V for the device with the incorporation of PEHCl-CN. This parameter increases to 0.61 V with the incorporation of MBI, which is due to the reduction of trap-assisted recombination losses, which corresponds to a higher open circuit voltage (VOC) of the device.
To quantitatively analyze the density of deep-level trap states of the perovskite films, thermal admittance spectroscopy was further conducted. From Fig. 3j, the defect activation energy (Edefecta) is reduced from 0.23 eV for the PEHCl-CN film to 0.18 eV for PEHCl-CN and MBI-treated films, respectively, indicative of a lower probability of defect formation. The energetic defect distribution (t-DOS) of the films can be derived, as can be shown in Fig. 3k.67 It is seen that the density of trap states is greatly reduced with PEHCl-CN and MBI incorporation. These results, together with the previous PL and TRPL and EIS spectra, indicate that non-radiative recombination is significantly suppressed. Additionally, previous studies have demonstrated that energy disorder will broaden the t-DOS distribution and cause additional electronic states in the band tail to extend into the forbidden gap. It can be observed that with the incorporation of PEHCl-CN and MBI, the t-DOS distribution (Fig. 3k) becomes narrower, which will further strengthen the splitting of the quasi-Fermi level, thus obtaining a larger VOC.23,69
We also measured the stability of the devices, and the results are provided in Fig. 3l and m. For the analysis of the long-term storage stability, the optimized NBG device retained 90.2% of its original PCE after an extended long-term test lasting for 2800 h, while the control ones degrade rapidly, retaining only 72.7% after 1200 h of shelf-storage in a N2-filled glovebox. Subsequently, the operational stability was evaluated by tracking the MPP under AM1.5 G. The PEHCl-CN and MBI treated cells maintain over 89.4% of the initial PCE for 400 h, while the control devices show only 69.8% of the original PCE after only 115 h. This significant improvement is likely due to the mitigation of light-induced ion migration and iodine defect-related degradation.
Performance and stability of tandem devices
Finally, the monolithic 2T all-perovskite tandem solar cells were fabricated by stacking the optimized NBG perovskite layers described above (see details in the Methods) as the bottom sub-cell and the 1.78 eV WBG perovskite as the top sub-cell.20,70 The WBG solar cells show a PCE of 18.7%, with a VOC of 1.27 V, a JSC of 17.95mAcm−2 and a FF of 82.3% (Fig. S38, ESI†). The configuration of the tandem cells is ITO/NiOx/Me-4PACz/WBG-PSCs/C60/SnO2/Au/PEDOT:PSS/NBG-PSCs/C60/BCP/Ag (Fig. 4a). A representative cross-sectional SEM image of the tandem cells is displayed in Fig. 4b. The champion device delivered an impressive PCE of 27.9%, with a VOC value of 2.08 V, a JSC value of 16.48 mA cm−2 and a FF of 81.4% (Fig. 4c), and PCE was stabilized at 27.6% (Fig. 4f). Meanwhile, a reputable third-party organization gives certified efficiencies of 27.2% and 27.1% with forward and reverse voltage scanning, respectively (Fig. S39, ESI†), exhibiting negligible hysteresis. JSC values of the bottom NBG and top WBG sub-cells integrated from EQE spectra (Fig. 4e) were in agreement with the JSC values obtained from J–V measurements and certification results. 60 all-perovskite tandem solar cells with and without PEHCl-CN and MBI were fabricated (in Fig. 4d), which demonstrates the superior reproducibility of the target device.
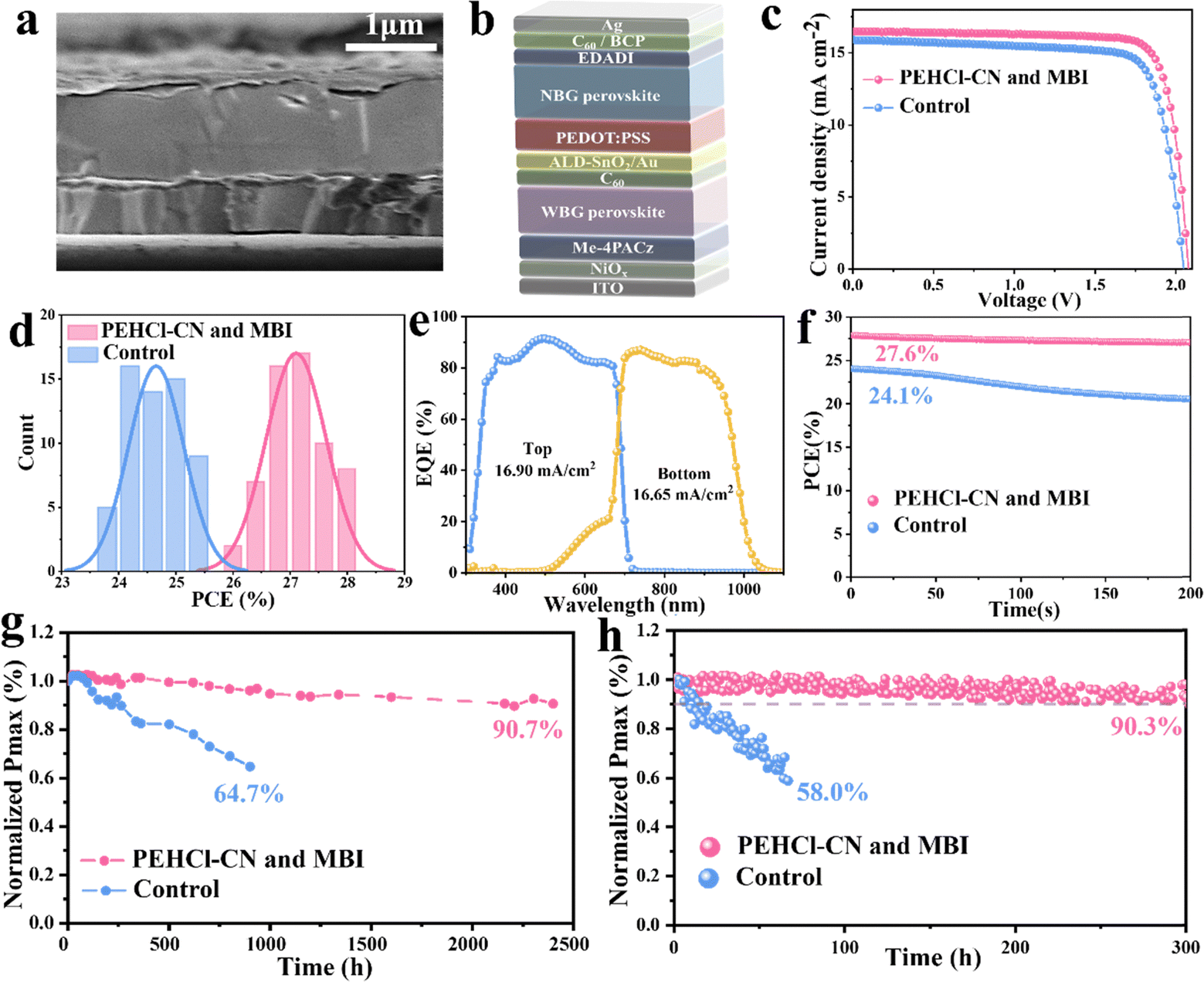 | ||
| Fig. 4 Performance of 2T all-perovskite tandem solar cells. (a) Cross-sectional SEM image. (b) Schematic picture of all-perovskite 2T device with PEHCl-CN and MBI modifications. (c) J–V curves of the best-performing control, PEHCl-CN and MBI-modified solar cells. (d) Histogram of PCEs for 60 tandem cells. (e) EQE curves of the tandem cell. (f) SPO efficiency of the best-performing tandem solar cell. (g) Long-term stability test of control devices and devices with PEHCl-CN and MBI tandem cells stored in a N2-filled glovebox. (h) MPP tracking of control devices and devices with PEHCl-CN and MBI under 1 sun illumination in the N2 environment. | ||
Long-term shelf-storage and operational stabilities were also evaluated,71 as presented in Fig. 4g and h. Impressively, the encapsulated optimized tandem devices maintain 90.3% of the initial PCE after 300h of MPP tracking under 1 sun illumination, while that of the control device shows rapid degradation with only 58.0% of the original PCE retained after 67 h. Besides, the PCE of the control devices drops to 64.7% of the initial value after 900 h of shelf storage in a N2-filled glovebox. Conversely, the optimized tandem devices retain 90.7% of the original PCE after an extended long-term test lasting for 2400 h, implying a significant improvement in stability through PEHCl-CN and MBI incorporation.
Conclusions
This study decouples the degradation mechanisms of Sn–Pb NBG perovskites induced by light and ambient air, respectively. To mitigate the oxidized degradation pathway induced by photo-generated I2, polarity engineering on the PEHCl series additives was conducted. Specifically, PEHCl-CN with the largest dipole moment not only affords strong coordination with Sn for strengthening the Sn–I bond, but also effectively passivates the defects by forming electrostatic interactions with negatively charged vacancies and interstitials. The synergistic effects prevent the photo-induced formation of I2 and the subsequent oxidation of the Sn–Pb perovskites. By further incorporating the reducing agent MBI into the precursor solution, the degradation pathway in the ambient air has also been circumvented. This binary modification leads to an extended carrier lifetime and a decrease in the trap density, culminating in a PCE of 23.0% for champion single-junction NBG PSCs. Moreover, the PCE of 2T all-perovskite tandem solar cells, employing PEHCl-CN and MBI-modified bottom Sn–Pb sub-cells, achieved an exceptional PCE of 27.9%. The tandem devices demonstrate remarkable stability during both MPP tracking and shelf-storage.Author contributions
C. L. conceived the idea and guided the work. Y. B. and R. T. designed the experiments. K. S. performed DFT simulation calculations; X. L. and C. X. characterized the KPFM test. M. Y., Y. M., Y. W., X. L., J. W., H. P., Z. S. and S. Z. fabricated the perovskite films and devices and measured their performance. Y. B. and R. T. analyzed the measured results. C. L. and Z. G. helped to revise the manuscript. All authors commented on this paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Science Fund for Distinguished Young Scholars (21925506) and the National Natural Science Foundation of China (U21A20331, 81903743, 22279151, and 22275004).References
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim, M. G. Kim, T. J. Shin and S. Il Seok, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef CAS PubMed.
- D. Luo, W. Yang, Z. Wang, A. Sadhanala, Q. Hu, R. Su, R. Shivanna, G. F. Trindade, J. F. Watts, Z. Xu, T. Liu, K. Chen, F. Ye, P. Wu, L. Zhao, J. Wu, Y. Tu, Y. Zhang, X. Yang, W. Zhang, R. H. Friend, Q. Gong, H. J. Snaith and R. Zhu, Science, 2018, 360, 1442–1446 CrossRef CAS PubMed.
- R. Lin, J. Xu, M. Wei, Y. Wang, Z. Qin, Z. Liu, J. Wu, K. Xiao, B. Chen, S. M. Park, G. Chen, H. R. Atapattu, K. R. Graham, J. Xu, J. Zhu, L. Li, C. Zhang, E. H. Sargent and H. Tan, Nature, 2022, 603, 73–78 CrossRef CAS PubMed.
- J. Zhou, L. Tan, Y. Liu, H. Li, X. Liu, M. Li, S. Wang, Y. Zhang, C. Jiang and R. Hua, Joule, 2024, 8, 1691–1706 CrossRef CAS.
- K. Leng, W. Fu, Y. Liu, M. Chhowalla and K. P. Loh, Nat. Rev. Mater., 2020, 5, 482–500 CrossRef CAS.
- S. Gu, R. Lin, Q. Han, Y. Gao, H. Tan and J. Zhu, Adv. Mater., 2020, 32, 1907392 CrossRef CAS PubMed.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang, M. Wei, M. I. Saidaminov, Y. Gao, J. Xu and M. Xiao, Nat. Energy, 2019, 4, 864–873 CrossRef CAS.
- G. E. Eperon, T. Leijtens, K. A. Bush, R. Prasanna, T. Green, J. T.-W. Wang, D. P. McMeekin, G. Volonakis, R. L. Milot and R. May, Science, 2016, 354, 861–865 CrossRef CAS PubMed.
- C. Wang, Y. Zhao, T. Ma, Y. An, R. He, J. Zhu, C. Chen, S. Ren, F. Fu, D. Zhao and X. Li, Nat. Energy, 2022, 7, 744–753 CrossRef CAS.
- R. Lin, Y. Wang, Q. Lu, B. Tang, J. Li, H. Gao, Y. Gao, H. Li, C. Ding and J. Wen, Nature, 2023, 620, 994–1000 CrossRef CAS PubMed.
- S. Zhou, S. Fu, C. Wang, W. Meng, J. Zhou, Y. Zou, Q. Lin, L. Huang, W. Zhang and G. Zeng, Nature, 2023, 624, 69–73 CrossRef CAS PubMed.
- D. Yu, M. Pan, G. Liu, X. Jiang, X. Wen, W. Li, S. Chen, W. Zhou, H. Wang and Y. Lu, Nat. Energy, 2024, 9, 298–307 CrossRef CAS.
- J. Zhou, S. Fu, S. Zhou, L. Huang, C. Wang, H. Guan, D. Pu, H. Cui, C. Wang and T. Wang, Nat. Commun., 2024, 15, 2324 CrossRef CAS PubMed.
- H. Chen, A. Maxwell, C. Li, S. Teale, B. Chen, T. Zhu, E. Ugur, G. Harrison, L. Grater, J. Wang, Z. Wang, L. Zeng, S. M. Park, L. Chen, P. Serles, R. A. Awni, B. Subedi, X. Zheng, C. Xiao, N. J. Podraza, T. Filleter, C. Liu, Y. Yang, J. M. Luther, S. De Wolf, M. G. Kanatzidis, Y. Yan and E. H. Sargent, Nature, 2023, 613, 676–681 CrossRef CAS PubMed.
- C. Liu, R. Lin, Y. Wang, H. Gao, P. Wu, H. Luo, X. Zheng, B. Tang, Z. Huang and H. Sun, Angew. Chem., 2023, 135, e202313374 CrossRef.
- National Renewable Energy Laboratory (NREL), Photovoltaic Research, Best Research-Cell Efficiency Chart, 2023, https://www.nrel.gov/pv/cell-efficiency.html.
- L. Lanzetta, T. Webb, N. Zibouche, X. Liang, D. Ding, G. Min, R. J. E. Westbrook, B. Gaggio, T. J. Macdonald, M. S. Islam and S. A. Haque, Nat. Commun., 2021, 12, 2853 CrossRef CAS PubMed.
- J. Tong, Z. Song, D. H. Kim, X. Chen, C. Chen, A. F. Palmstrom, P. F. Ndione, M. O. Reese, S. P. Dunfield and O. G. Reid, Science, 2019, 364, 475–479 CrossRef CAS PubMed.
- R. K. Gunasekaran, J. Jung, S. W. Yang, D. Im, W. C. Choi, Y. Yun and S. Lee, ACS Energy Lett., 2023, 9, 102–109 CrossRef.
- S. Hu, K. Otsuka, R. Murdey, T. Nakamura, M. A. Truong, T. Yamada, T. Handa, K. Matsuda, K. Nakano and A. Sato, Energy Environ. Sci., 2022, 15, 2096–2107 RSC.
- Q. Sun, Z. Zhang, H. Yu, J. Huang, X. Li, L. Dai, Q. Wang, Y. Shen and M. Wang, Energy Environ. Sci., 2024, 17, 2512–2520 RSC.
- D. Ricciarelli, D. Meggiolaro, F. Ambrosio and F. De Angelis, ACS Energy Lett., 2020, 5, 2787–2795 CrossRef CAS.
- Q. Chen, J. Luo, R. He, H. Lai, S. Ren, Y. Jiang, Z. Wan, W. Wang, X. Hao and Y. Wang, Adv. Energy Mater., 2021, 11, 2101045 CrossRef CAS.
- K. Xiao, R. Lin, Q. Han, Y. Hou, Z. Qin, H. T. Nguyen, J. Wen, M. Wei, V. Yeddu and M. I. Saidaminov, Nat. Energy, 2020, 5, 870–880 CrossRef CAS.
- W. Zhang, H. Yuan, X. Li, X. Guo, C. Lu, A. Liu, H. Yang, L. Xu, X. Shi and Z. Fang, Adv. Mater., 2023, 35, 2303674 CrossRef CAS PubMed.
- J. Cao, H. Loi, Y. Xu, X. Guo, N. Wang, C. Liu, T. Wang, H. Cheng, Y. Zhu and M. G. Li, Adv. Mater., 2022, 34, 2107729 CrossRef CAS PubMed.
- J. Wang, M. A. Uddin, B. Chen, X. Ying, Z. Ni, Y. Zhou, M. Li, M. Wang, Z. Yu and J. Huang, Adv. Energy Mater., 2023, 13, 2204115 CrossRef CAS.
- Z. Zhang, X. Tian, C. Wang, J. Jin, Y. Jiang, Q. Zhou, J. Zhu, J. Xu, R. He and Y. Huang, Energy Environ. Sci., 2022, 15, 5274–5283 RSC.
- X. Liu, T. Wu, C. Zhang, Y. Zhang, H. Segawa and L. Han, Adv. Funct. Mater., 2021, 31, 2106560 CrossRef CAS.
- T. Webb and S. A. Haque, Energy Environ. Sci., 2024, 17, 3244–3269 RSC.
- X. Lu, K. Sun, Y. Wang, C. Liu, Y. Meng, X. Lang, C. Xiao, R. Tian, Z. Song and Z. Zhu, Adv. Mater., 2024, 2400852 CrossRef CAS PubMed.
- Y. Zhang, Q. Song, G. Liu, Y. Chen, Z. Guo, N. Li, X. Niu, Z. Qiu, W. Zhou and Z. Huang, Nat. Photonics, 2023, 17, 1066–1073 CrossRef CAS.
- X. Li, H. Yang, A. Liu, C. Lu, H. Yuan, W. Zhang and J. Fang, Energy Environ. Sci., 2023, 16, 6071–6077 RSC.
- X. Ren, J. Wang, Y. Lin, Y. Wang, H. Xie, H. Huang, B. Yang, Y. Yan, Y. Gao and J. He, Nat. Mater., 2024, 23, 810–817 CrossRef CAS PubMed.
- Y. Zhang, B. Zhao, L. Liu, J. Zhou, X. Ma and N. Wang, Small, 2024, 20, 2306115 CrossRef CAS PubMed.
- H. Zhang, L. Pfeifer, S. M. Zakeeruddin, J. Chu and M. Grätzel, Nat. Rev. Chem., 2023, 7, 632–652 CrossRef CAS PubMed.
- L. Lanzetta, T. Webb, N. Zibouche, X. Liang, D. Ding, G. Min, R. J. E. Westbrook, B. Gaggio, T. J. Macdonald, M. S. Islam and S. A. Haque, Nat. Commun., 2021, 12, 2853 CrossRef CAS PubMed.
- W. Tang, T. Liu, M. Zhang, F. Yuan, K. Zhou, R. Lai, Y. Lian, S. Xing, W. Xiong and M. Zhang, Matter, 2023, 6, 3782–3802 CrossRef CAS.
- D. He, P. Chen, M. Hao, M. Lyu, Z. Wang, S. Ding, T. Lin, C. Zhang, X. Wu and E. Moore, Angew. Chem., Int. Ed., 2024, 63, e202317446 CrossRef CAS PubMed.
- M. Hu, Y. Zhang, J. Gong, H. Zhou, X. Huang, M. Liu, Y. Zhou and S. Yang, ACS Energy Lett., 2023, 8, 1035–1041 CrossRef CAS.
- J. Tong, Q. Jiang, A. J. Ferguson, A. F. Palmstrom, X. Wang, J. Hao, S. P. Dunfield, A. E. Louks, S. P. Harvey, C. Li, H. Lu, R. M. France, S. A. Johnson, F. Zhang, M. Yang, J. F. Geisz, M. D. McGehee, M. C. Beard, Y. Yan, D. Kuciauskas, J. J. Berry and K. Zhu, Nat. Energy, 2022, 7, 642–651 CrossRef CAS.
- H. Su, J. Zhang, Y. Hu, Y. Yao, X. Zheng, Y. She, B. Jia, L. Gao and S. (Frank) Liu, Adv. Funct. Mater., 2023, 33, 2213123 CrossRef CAS.
- X. Guo, J. Su, Z. Lin, X. Wang, Q. Wang, Z. Zeng, J. Chang and Y. Hao, iScience, 2021, 24, 102276 CrossRef CAS PubMed.
- C. Li, X. Wang, E. Bi, F. Jiang, S. M. Park, Y. Li, L. Chen, Z. Wang, L. Zeng, H. Chen, Y. Liu, C. R. Grice, A. Abudulimu, J. Chung, Y. Xian, T. Zhu, H. Lai, B. Chen, R. J. Ellingson, F. Fu, D. S. Ginger, Z. Song, E. H. Sargent and Y. Yan, Science, 2023, 379, 690–694 CrossRef CAS PubMed.
- K. Zhang, Y. Wang, M. Tao, L. Guo, Y. Yang, J. Shao, Y. Zhang, F. Wang and Y. Song, Adv. Mater., 2023, 35, 2211593 CrossRef CAS PubMed.
- J. Wu, M.-H. Li, J.-T. Fan, Z. Li, X.-H. Fan, D.-J. Xue and J.-S. Hu, J. Am. Chem. Soc., 2023, 145, 5872–5879 CrossRef CAS PubMed.
- B. Liu, H. Chen, J. Cao, X. Chen, J. Xie, Y. Shu, F. Yan, W. Huang and T. Qin, Adv. Funct. Mater., 2024, 34, 2310828 CrossRef CAS.
- W. Zhao, J. Xu, K. He, Y. Cai, Y. Han, S. Yang, S. Zhan, D. Wang, Z. Liu and S. Liu, Nano-Micro Lett., 2021, 13, 169 CrossRef CAS PubMed.
- F. Fu, S. Pisoni, Q. Jeangros, J. Sastre-Pellicer, M. Kawecki, A. Paracchino, T. Moser, J. Werner, C. Andres and L. Duchêne, Energy Environ. Sci., 2019, 12, 3074–3088 RSC.
- F. Liu, Q. Dong, M. K. Wong, A. B. Djurišić, A. Ng, Z. Ren, Q. Shen, C. Surya, W. K. Chan and J. Wang, Adv. Energy Mater., 2016, 6, 1502206 CrossRef.
- X. Li, W. Zhang, X. Guo, C. Lu, J. Wei and J. Fang, Science, 2022, 375, 434–437 CrossRef CAS PubMed.
- T. Zhang, F. Wang, H.-B. Kim, I.-W. Choi, C. Wang, E. Cho, R. Konefal, Y. Puttisong, K. Terado, L. Kobera, M. Chen, M. Yang, S. Bai, B. Yang, J. Suo, S.-C. Yang, X. Liu, F. Fu, H. Yoshida, W. M. Chen, J. Brus, V. Coropceanu, A. Hagfeldt, J.-L. Brédas, M. Fahlman, D. S. Kim, Z. Hu and F. Gao, Science, 2022, 377, 495–501 CrossRef CAS PubMed.
- S. Tan, T. Huang, I. Yavuz, R. Wang, T. W. Yoon, M. Xu, Q. Xing, K. Park, D.-K. Lee, C.-H. Chen, R. Zheng, T. Yoon, Y. Zhao, H.-C. Wang, D. Meng, J. Xue, Y. J. Song, X. Pan, N.-G. Park, J.-W. Lee and Y. Yang, Nature, 2022, 605, 268–273 CrossRef CAS PubMed.
- Z. Zhang, Y. Huang, C. Wang, Y. Jiang, J. Jin, J. Xu, Z. Li, Z. Su, Q. Zhou and J. Zhu, Energy Environ. Sci., 2023, 16, 3430–3440 RSC.
- H. Chen, H. Yan and Y. Cai, Chem. Mater., 2022, 34, 1020–1029 CrossRef CAS.
- C. Ma, B. Kim, D.-H. Kang, S.-W. Kim and N.-G. Park, ACS Energy Lett., 2021, 6, 2817–2824 CrossRef CAS.
- H. Li, B. Chang, L. Wang, Y. Wu, Z. Liu, L. Pan and L. Yin, ACS Energy Lett., 2024, 9, 400–409 CrossRef CAS.
- B. Li, H. Di, B. Chang, R. Yin, L. Fu, Y. Zhang and L. Yin, Adv. Funct. Mater., 2021, 31, 2007447 CrossRef CAS.
- J. Luo, R. He, H. Lai, C. Chen, J. Zhu, Y. Xu, F. Yao, T. Ma, Y. Luo and Z. Yi, Adv. Mater., 2023, 35, 2300352 CrossRef CAS PubMed.
- C. Zhu, X. Niu, Y. Fu, N. Li, C. Hu, Y. Chen, X. He, G. Na, P. Liu, H. Zai, Y. Ge, Y. Lu, X. Ke, Y. Bai, S. Yang, P. Chen, Y. Li, M. Sui, L. Zhang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 815 CrossRef CAS PubMed.
- T. Zhao, C.-C. Chueh, Q. Chen, A. Rajagopal and A. K.-Y. Jen, ACS Energy Lett., 2016, 1, 757–763 CrossRef CAS.
- H. Chen, S. Teale, B. Chen, Y. Hou, L. Grater, T. Zhu, K. Bertens, S. M. Park, H. R. Atapattu and Y. Gao, Nat. Photonics, 2022, 16, 352–358 CrossRef CAS.
- Z. Zhang, R. Zhu, Y. Tang, Z. Su, S. Hu, X. Zhang, J. Zhang, J. Zhao, Y. Xue and X. Gao, Adv. Mater., 2024, 36, 2312264 CrossRef CAS PubMed.
- J. Song, T. Kong, Y. Zhang, X. Liu, M. Saliba and D. Bi, Adv. Funct. Mater., 2023, 33, 2304201 CrossRef CAS.
- R. Tian, C. Liu, Y. Meng, Y. Wang, R. Cao, B. Tang, D. Walsh, H. Do, H. Wu and K. Wang, Adv. Mater., 2023, 2309998 Search PubMed.
- Y. Wang, Y. Meng, C. Liu, R. Cao, B. Han, L. Xie, R. Tian, X. Lu, Z. Song and J. Li, Joule, 2024, 8, 1120–1141 CrossRef CAS.
- Y. Jiang, J. Wang, H. Zai, D. Ni, J. Wang, P. Xue, N. Li, B. Jia, H. Lu and Y. Zhang, J. Am. Chem. Soc., 2022, 144, 5400–5410 CrossRef CAS PubMed.
- H. Gao, K. Xiao, R. Lin, S. Zhao, W. Wang, S. Dayneko, C. Duan, C. Ji, H. Sun, A. D. Bui, C. Liu, J. Wen, W. Kong, H. Luo, X. Zheng, Z. Liu, H. Nguyen, J. Xie, L. Li, M. I. Saidaminov and H. Tan, Science, 2024, 383, 855–859 CrossRef CAS PubMed.
- S. Tan, C. Li, C. Peng, W. Yan, H. Bu, H. Jiang, F. Yue, L. Zhang, H. Gao and Z. Zhou, Nat. Commun., 2024, 15, 4136 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02427c |
| ‡ Yang Bai, Ruijia Tian and Kexuan Sun contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |