
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Additive engineering strategies for improved interfacial stability in lithium metal batteries†
Kun
Ryu‡
 ab,
Kyungbin
Lee‡
a,
Jeonghoon
Lim
c,
Michael J.
Lee
a,
Keun-Hee
Kim
a,
Un Hwan
Lee
d,
Bernardine L. D.
Rinkel
c,
Kyungmo
Kim
a,
Soohyun
Kim
e,
Dayoung
Kim
e,
Dongsek
Shin
e,
Bryan
McCloskey
c,
Joonhee
Kang
*d and
Seung Woo
Lee
*a
ab,
Kyungbin
Lee‡
a,
Jeonghoon
Lim
c,
Michael J.
Lee
a,
Keun-Hee
Kim
a,
Un Hwan
Lee
d,
Bernardine L. D.
Rinkel
c,
Kyungmo
Kim
a,
Soohyun
Kim
e,
Dayoung
Kim
e,
Dongsek
Shin
e,
Bryan
McCloskey
c,
Joonhee
Kang
*d and
Seung Woo
Lee
*a
aGeorge W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: seung.lee@me.gatech.edu
bPritzker School of Molecular Engineering, University of Chicago, Chicago, IL 60637, USA
cDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720, USA
dDepartment of Nanoenergy Engineering, Pusan National University, Busan, 46241, Republic of Korea. E-mail: j.kang@pusan.ac.kr
eLG Energy Solution, Ltd, LG Science Park, Seoul, 07796, Korea
First published on 9th September 2024
Abstract
Electrolyte engineering is crucial for advancing lithium (Li) metal batteries (LMBs). Currently, unstable electrode–electrolyte interfaces limit the stable cycling of LMBs. Here, we introduce an additive engineering approach aimed at strengthening these electrode–electrolyte interfaces by incorporating the ionic additive tetrabutylammonium tetrafluoroborate into a low-concentration tetrahydrofuran ether electrolyte. Our findings reveal that tetrafluoroborate anions minimize corrosion and Li inventory loss. In addition, bulky tetrabutylammonium cations adsorbed onto the anode surface enable uniform and compact Li electrodeposition. This fluorinating and dendrite-suppressing mechanism supports stable high-current and high-capacity operations. Without altering the electrolyte solvation structure, the functional additive forms a robust interface with enhanced charge transport kinetics, specifically a stable solid–electrolyte interphase and cathode–electrolyte interphase. The designed electrolyte demonstrates 150 cycles 82.4% capacity retention in full cells employing 4 mA h cm−2 high-nickel cathodes under practical testing conditions (N/P = 1.75, E/C = 5.1 g A h−1).
Broader contextLithium (Li) metal batteries offer exceptional promise for energy storage devices due to their high energy and power densities. However, the implementation of Li metal anodes is impeded by low Li reversibility, stemming from solid–electrolyte interface (SEI) growth and inactive Li0 formation, and safety concerns due to dendrite penetration. To overcome these challenges, previous research focused heavily on designing electrolytes that maximize Li reversibility and cycle stability. The majority of these improvements were achieved by tailoring the solvation structure, incorporating the anion into the primary solvation sheath of the cation to facilitate the formation of an inorganic-rich SEI. In this work, we implement an additive engineering strategy to form a robust, inorganic-rich SEI and suppress dendrite formation. As a result, high Li reversibility is achieved, along with improved cycle life. Our findings indicate that interfacial instability significantly contributes to the low reversibility observed in conventional low-concentration ether electrolytes. This chemomechanical electrolyte design centered on a simple additive incorporation demonstrates potential for practical Li metal batteries, offering significant cost advantage over more complex electrolyte engineering approaches, marking a step closer to commercialization. |
Introduction
Lithium (Li) metal has been recognized as the ideal anode material for Li secondary batteries, owing to its exceptionally high specific capacity (3860 mA h g−1) and low electrochemical potential (−3.04 V vs. standard hydrogen electrode).1,2 However, its high reactivity, accompanied by continuous Li plating/stripping, triggers a series of detrimental side effects, including dendrite formation, inactive Li0 and solid–electrolyte interface (SEI)-Li+ accumulation, and impeded ion transport, ultimately leading to low reversibility.3,4Electrolyte engineering has played a pivotal role in enhancing Li reversibility and overcoming challenges associated with the Li metal anode (LMA).5 In particular, electrolyte designs with modified solvation structures have been introduced to enable high Li reversibility, including localized high-concentration electrolytes (LHCEs),6–9 fully fluorinated electrolytes (FFEs),10–12 and weakly solvating electrolytes (WSEs).13–15 These systems were engineered to shift the lowest unoccupied molecular orbitals (LUMO) of the electrolyte from solvent molecules towards anions, facilitating anion decomposition.16 Moreover, incorporating anions within the primary solvation sheath induced the formation of inorganic-rich SEI and cathode–electrolyte interphase (CEI), desired for improved Li+ transport kinetics.17,18
Despite these advancements, these electrolyte systems with modified solvation structures suffer from inferior ionic conductivity, manufacturing complexity, and high associated costs, limiting their practicality in commercial Li metal batteries (LMBs).19,20 In this context, developing novel electrolyte chemistry is essential to enhance Li reversibility and enable high-voltage LMB operation, while addressing practical concerns regarding energy density, cycle life, and cost.
Here, we implement an additive engineering strategy to enhance the cycle stability of high-energy LMBs by integrating dendrite and corrosion suppression mechanisms. This involves incorporating tetrabutylammonium tetrafluoroborate (TBATFB), an organometallic compound, into a low-concentration tetrahydrofuran (THF) ether electrolyte (Fig. 1a). THF is selected as a model ether solvent due to its high donor number and dielectric constant, ensuring a structure dominated by solvent-separated ion pairs (SSIP) under low salt concentrations.21 The electrolyte is systematically engineered to optimize Li reversibility and ion transport properties. Although the incorporation of organometallic compounds into conventional electrolytes has been previously reported, the distinguishing feature of the electrolyte presented in this study is its ability to form a stable passivating interface without tailoring the electrolyte solvation structure.22,23 The bulky TBA+ cation was selected to suppress dendrite formation by regulating Li flux.22 Furthermore, the TFB− anion effectively fluorinates the LMA while preserving its metallic lattice structure, which minimizes side reactions and enhanced passivation stability. This synergistic effect leads to compact and uniform Li electrodeposition with high reversibility and enhanced interfacial stability. This improvement addresses the thermodynamic limitations associated with conventional low-salt concentration ether electrolytes at high voltages by forming a stable CEI. Full cells employing 35-μm thick LMA and NCA88 (LiNi0.88Co0.09Al0.03O2, 4 mA h cm−2) cathodes with limited electrolyte (5.1 g A h−1) deliver 82.4% capacity retention after 150 cycles. Moreover, the designed electrolyte presents a notable economic advantage over other contemporary state-of-the-art electrolytes, while delivering comparable electrochemical performances. The additive engineering strategy provides comprehensive insight into improving the anodic and cathodic interfacial stability. The holistic electrolyte engineering addresses longstanding challenges in electrolyte performance through a facile additive approach, offering a promising direction for future electrolyte designs.
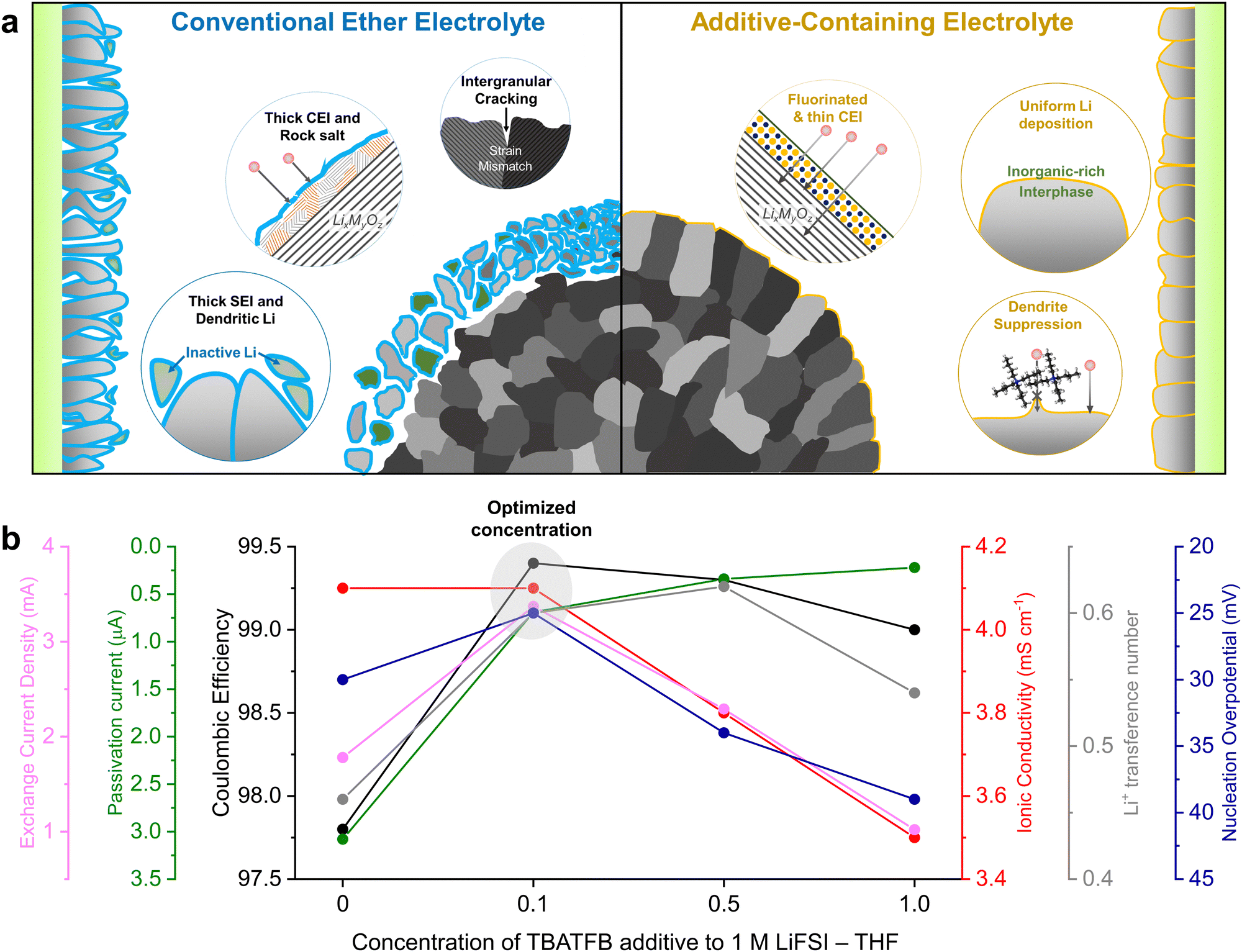 | ||
| Fig. 1 Engineering electrolyte designs for high performance LMBs. (a) Schematic illustrating the interactions between the electrolyte, 4-V class cathode, and Li metal anode (LMA) when utilizing conventional ether electrolyte (CEE, left) and additive-containing electrolyte (ACE, right). (b) Integrated plot for determining the optimized additive concentration by comparing the following categories: exchange current density (light blue), passivation current (green), Coulombic efficiency (black), ionic conductivity (red), Li+ transference number (grey), and nucleation overpotential (blue). | ||
Results and discussion
Electrolyte evaluation and optimization
We initiated the optimization process by adjusting the concentration of the fluorinating additive to enhance compatibility between the electrolyte and the LMA. Li reversibility was evaluated through a Li plating/stripping process on a copper (Cu) substrate, conducted at a current density of 1 mA cm−2 and an areal capacity of 1 mA h cm−2 for 300 cycles (Fig. S1, ESI†). The addition of TBATFB enhanced LMA stability, as evidenced by Coulombic efficiencies (CEs) exceeding 99%, compared to 97.8% efficiency observed in ether electrolytes. The trend remained consistent when electrolytes were tested using the modified Aurbach protocol (Fig. S2, ESI†).24 It is noteworthy that the gradual increase in CEs for 1 M LiFSI + 1 M TBATFB in THF up to the 50th cycle is attributed to the high salt concentration, a trend paralleled with the 3 M LiFSI high-concentration electrolyte (HCE, Fig. S3, ESI†). At 1 mA cm−2, the 0.1 M TBATFB-added electrolyte exhibited the lowest initial Li nucleation overpotential of 25 mV (Fig. S4, ESI†). A lower nucleation overpotential is advantageous as it thermodynamically favors the formation of larger Li nuclei, resulting in a compact Li deposition morphology.25 We further analyzed the ionic conductivity and Li selectivity of these electrolyte systems. The addition of TBATFB generally resulted in reduced ionic conductivity, except at 0.1 M TBATFB, which showed negligible change compared to the ether electrolyte (Fig. S5, ESI†). Electrolyte viscosities increased with increasing TBATFB concentrations, indicating reduced ion mobility within the electrolyte (Fig. S6, ESI†).15,26 An increase in Li+ transference number was observed with the addition of TBATFB (Fig. S7 and S8, ESI†), attributed to attractive Coulomb interactions between TBA+ cations and FSI− and BF4− anions that enhance cationic selectivity.27 Next, we assessed Li passivation stability by measuring leakage current in a Li‖Cu asymmetric cell at 0 V vs. Li/Li+ (Fig. S9, ESI†). Low leakage currents (below 1 μA) underscored the increased stability of the SEI due to TBATFB additives. Exchange current densities of the electrolytes were measured using two methods: extrapolating the Tafel plot and applying the Butler–Volmer equations at low overpotentials (Fig. S10–S12, ESI†), both exhibiting parallel trends. In both methods, the 0.1 M and 0.5 M TBATFB additive demonstrated improved electrochemical activity compared to the ether electrolyte. To optimize the additive-engineered electrolyte, we compared key parameters including exchange current density, passivation stability, CE, ionic conductivity, transference number, and nucleation overpotential across different additive concentrations (Fig. 1b). Our analysis concluded that 1 M LiFSI + 0.1 M TBATFB in THF represents the optimized electrolyte system.Electrolyte solvation structure
The solvation structures of the optimized additive-containing electrolyte (ACE; 1 M LiFSI + 0.1 M TBATFB in THF) were compared with a conventional ether electrolyte (CEE; 1 M LiFSI in THF). Firstly, we applied density functional theory (DFT) computations to verify the binding of TBATFB with THF solvent molecules (Fig. 2a and Fig. S13, ESI†) Next, molecular dynamics (MD) simulations provided further insights into the molecular interactions within the solvation shells (Fig. 2b and c). Both CEE and ACE displayed a distinctive SSIP structure, characterized by Li+ coordination largely dominated by solvent molecules (Fig. 2d).28 The coordination dynamics of Li+ with THF solvents in these electrolyte systems was obtained through Raman spectroscopy (Fig. 2e). The peaks of free THF (948 cm−1) and free FSI− (719 cm−1) remained at identical positions for CEE and ACE. The peak associated with CIP/AGG (742 cm−1) and Li+-coordinated THF peak (955 cm−1) observed in HCE were not present in either CEE or ACE (Fig. S14, ESI†).29 These results indicate that the presence of TBATFB did not alter the electrolyte solvation structure. We proceeded by conducting nuclear magnetic resonance experiments to verify equivalent local environments in CEE and ACE. The 7Li NMR spectra showed a negligible change in the 7Li signal (δ(7Li) = −0.408 ppm for the CEE, compared to δ(7Li) = −0.469 ppm for the ACE), indicating similar Li+ binding energies within CEE and ACE (Fig. 2f). The 19F NMR spectra revealed that the local environment of the FSI− is highly similar in both electrolyte solutions δ(19F) = 51.17 ppm for both the CEE and ACE, while the characteristic BF4− peak is exclusively present in the ACE (δ(19F) = −156.35 ppm; Fig. 2g). This is further supported by the 17O NMR spectra (Fig. 2h). The 17O NMR signal for LiFSI is identical in both electrolyte solutions (δ(17O) = 169.0 ppm). The marginal change in the 17O NMR signal for THF (δ(17O) = 15.5 ppm for CEE, compared to δ(17O) = 15.4 ppm for ACE) suggests that the local coordination environment of the solvent molecules is analogous in both electrolyte solutions.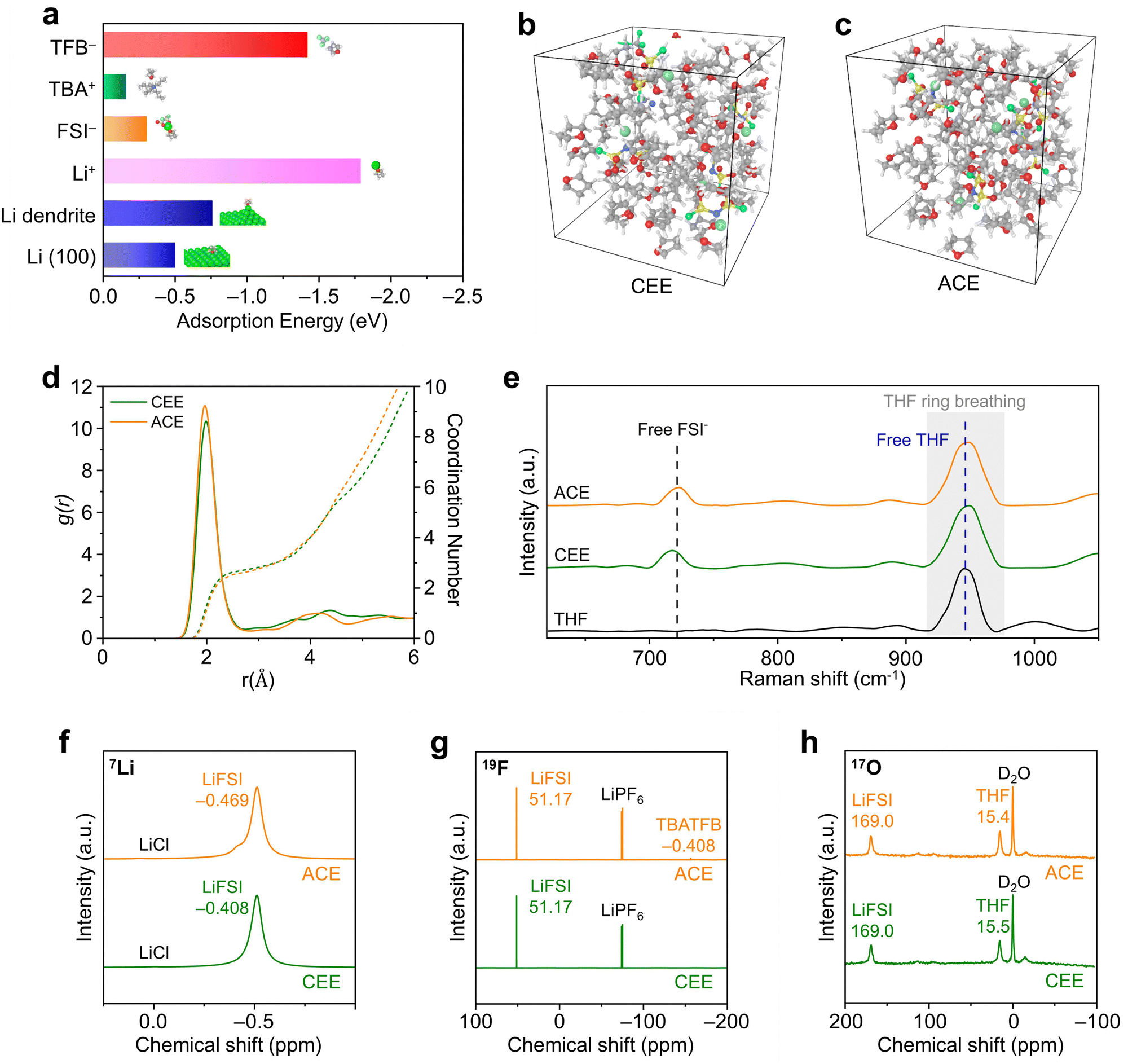 | ||
| Fig. 2 Theoretical and experimental investigation of electrolyte solvation structure. (a) THF adsorption energy against metallic Li and ions within the electrolyte. Snapshots obtained from MD simulations of (b) CEE and (c) ACE and (d) their corresponding Li+ radial distribution function. (e) Raman spectra obtained from pure THF, CEE, and ACE. (f) 7Li, (g) 19F, and (h) 17O NMR spectra of CEE and ACE. | ||
Enhanced Li metal stability
We investigated Li reversibility by evaluating the CEs of Li‖Cu asymmetric cells under heightened current densities and capacities of 3 mA cm−2 and 3 mA h cm−2, respectively (Fig. 3a). The average CEs for CEE and ACE were 97.0% and 99.2%, respectively. LMA corrosion during calendar ageing was assessed after 0, 1, and 5 days of ageing (Fig. 3b and Fig. S15, ESI†). CEE exhibited a relatively linear decrease in CEs with ageing time, while ACE displayed stabilized capacity loss, indicating its self-passivating behavior. To assess LMA stability, Li‖Li symmetric cells were tested under high current densities and areal capacities of 10 mA cm−2 and 4 mA h cm−2 (Fig. 3c) and 20 mA cm−2 and 20 mA h cm−2 (Fig. 3d). Under both conditions, the cells cycled with CEE shorted during early cycles, while ACE showed stable and prolonged cycling performance. ACE also demonstrated stable cycling stability for 1000 cycles at a milder regime of 1 mA cm−2 and 1 mA h cm−2 (Fig. S16, ESI†). To decipher underlying failure mechanisms, Li‖Li symmetric cells were cycled under 1 mA cm−2 and 1 mA h cm−2 for 100 cycles and subjected to analysis. Scanning electron microscopy (SEM) images showed mossy and dendritic Li with porous deposits in CEE, contrasting with dendrite-free densely packed morphology in ACE (Fig. 3e and f). Electrochemical impedance spectroscopy revealed a significant impedance increase from 77 to 141 Ω in CEE (Fig. S17, ESI†). Conversely, ACE showed a modest increase from 75 to 95 Ω, suggesting the formation of a thin and robust SEI.30 The dendrite suppression mechanism of ACE was investigated by measuring the potential energy as a solvated Li+ ion was deposited on the LMA surface (Fig. S18, ESI†). DFT calculations revealed that within one to six THF molecules per Li+, Li[THF]4+ (−1.81 eV) was the most probable solvation structure (Fig. 3g). The potential energy barrier of 0.148 eV for CEE can be attributed to the de-solvation of THF molecules upon Li deposition (Fig. 3h). The sharp decrease in potential energy at the final reaction coordinates indicates thermodynamically favored Li deposition. On the other hand, nudged elastic band (NEB) calculations unveiled an increased energy barrier of approximately 0.199 eV for Li+ for ACE, suggesting a shielding effect by the TBA+ cation layer.31 Typically, dendrite growth is facilitated by three-dimensional (3-D) diffusion of Li+ ions due to increased electric field concentrations at preexisting dendrites, a phenomenon often referred to as the ‘tip effect’.32 As TBA+ cations tend to accumulate near 3-D protrusion, the increased energy barrier in ACE limits Li+ flux on dendrites, effectively mitigate dendrite growth and promoting uniform, compact Li deposition.33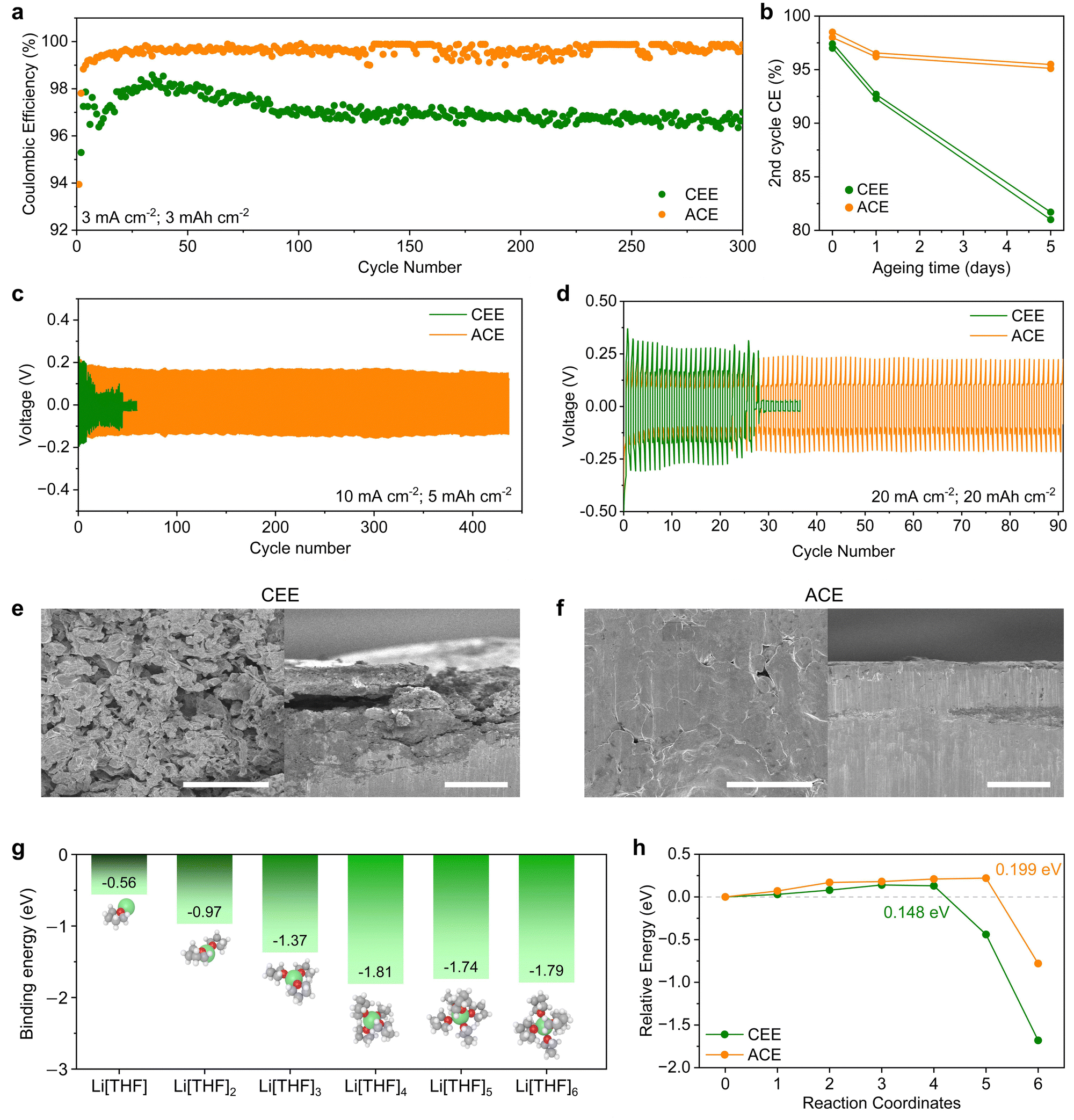 | ||
| Fig. 3 Electrochemical performance, stability, and characterization of LMA. (a) CEs of Li plating/stripping for Li‖Cu asymmetric cells at 3 mA cm−2 and 3 mA h cm−2. (b) Second cycle CEs with varying calendar ageing times. Cycling stability of Li‖Li symmetric cells under (c) 10 mA cm−2 and 4 mA h cm−2 and (d) 20 mA cm−2 and 20 mA h cm−2. SEM images showing surface morphology and cross-sectional views of LMA cycled in (e) CEE and (f) ACE. Scale bars 10 μm. (g) Energies of Li[THF]n+, where n represents the coordination number. (h) Potential energy diagram of Li[THF]4+ calculated using the NEB method at different reaction coordinates. | ||
Characterization of the SEI on cycled Li metal anode
The chemical composition of the SEI was analyzed using X-ray Photoelectron spectroscopy (XPS) complemented by Ar+ sputtering depth profiling (Fig. 4a). XPS elemental analysis revealed a consistent decrease in carbon content in both electrolyte systems upon etching, indicating the formation of SEI with organic-rich outer layer and inorganic-rich inner layer. Specifically, Li2CO3 (290.5 eV) was identified as a predominant carbon species on the LMA cycled in CEE (Fig. 4b), consistent with other low-concentration ether electrolytes.15,34 The significant presence of Li2CO3 is often associated with SEI instability due to its tendency to decompose into gaseous byproducts.35 In the case of the SEI with ACE, participation of BF4− anions during SEI formation were evident, as highlighted by the distinctive B–F (688.1 eV) and Li–F (684.8 eV) peaks in the F 1s spectra (Fig. 4c). The lithium alkyl oxides (RO–Li, 533.0 eV, O 1s) and the C–O (286.0 eV, C 1s) peaks suggests free THF solvent decomposition (Fig. 4d).36 Conversely, the SEI with ACE revealed a pronounced Li2O (529.3 eV) peak, which is known to enhance Li+ diffusion within the SEI.37 The peak intensities in the Li 1s spectra were in good agreement with the peaks shown in the C 1s, O 1s, and F 1s spectra (Fig. 4e). In the S 2p spectra, ACE exhibited lower levels of SO2F (169.6 and 171.2 eV) compared to that with CEE but presented a new SO3 (166.2 eV) peak, indicating more extensive decomposition of FSI− (Fig. 4f).10 The fluorination behavior was verified using MD simulations, wherein we observed rapid decomposition of BF4− at the LMA surface, attributed to its susceptibility to reduction (Fig. S19, ESI†).38 It should be noted that a pronounced distortion in the (100) plane of the Li crystalline lattice was detected upon FSI− anion decomposition (Fig. S20, ESI†). In contrast, BF4− anions successfully fluorinated the LMA while preserving the Li lattice structure due to its stronger bond strength compared to FSI−.39 This promotes the formation of a uniform SEI layer, which helps suppress the growth of Li dendrites.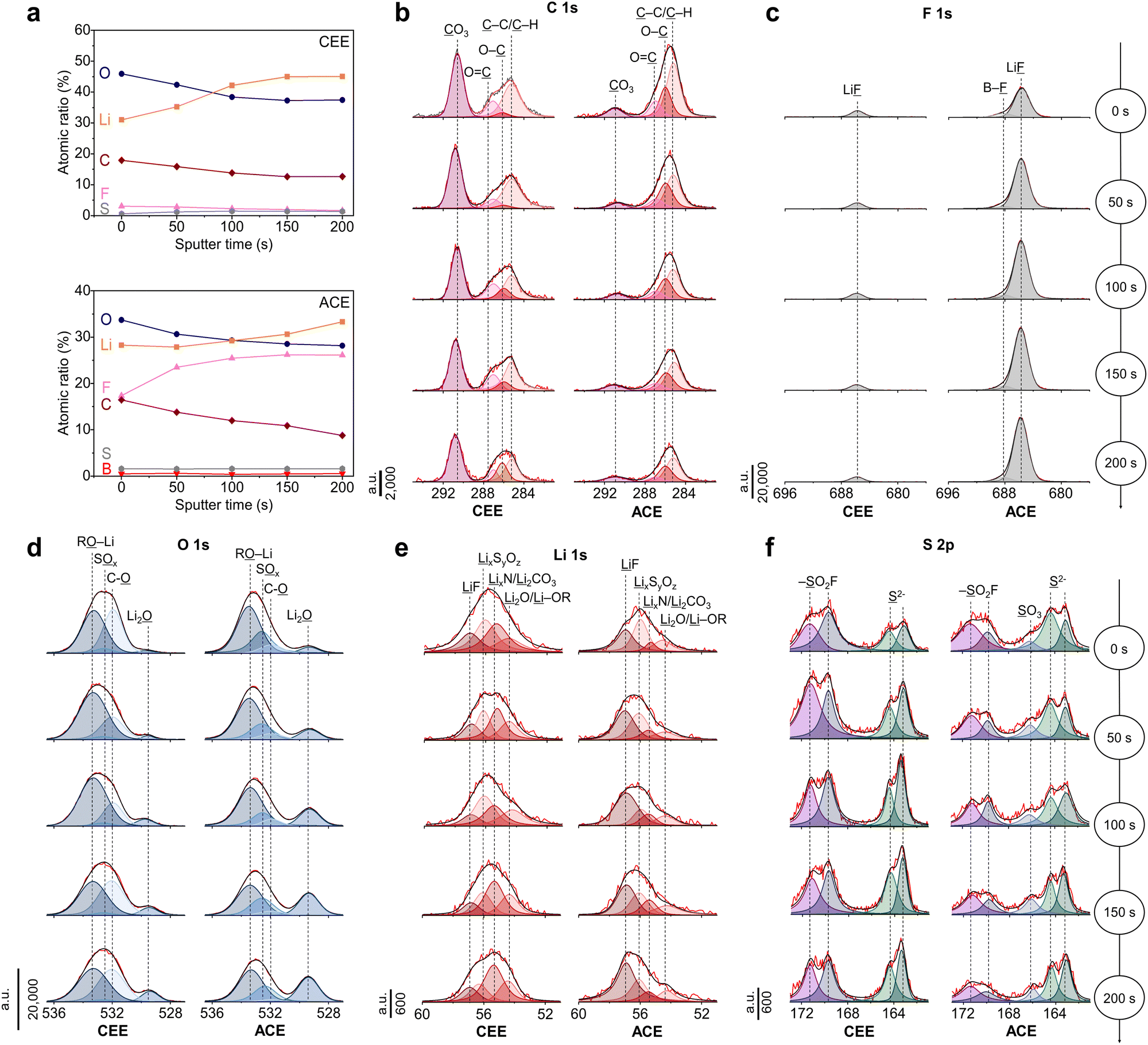 | ||
| Fig. 4 SEI chemical composition. XPS characterization of cycled LMA. (a) Atomic composition ratios at different sputtering times using CEE and ACE. (b) C 1s, (c) F 1s, (d) O 1s, (e) Li 1s, and (f) S 2p spectra of CEE- and ACE-cycled LMAs. XPS spectra are displayed on columns, with each height corresponding to depth profiling results. | ||
Moving towards high energy Li metal batteries
Rate capability tests of the 4 mA h cm−2 loading-LiFePO4 (LFP) cathodes were conducted using CEE and ACE, with current densities varying from 0.8 to 20 mA cm−2 (Fig. S21, ESI†). At a high current of 20 mA cm−2, ACE retained nearly three times the capacity compared to CEE. Upon returning the rate to 2 mA cm−2, both electrolytes showed a recovery of over 99% of their initial capacities. The high capacity recovery in CEE suggests that the reduced capacity observed at higher current densities is predominantly due to the limited charge transport kinetics at the electrode–electrolyte interphases. We further examined the enduring effects of high-rate cycling on LFP capacity retention (Fig. S22, ESI†). After returning to lower current densities, CEE exhibited a progressive capacity decrease, eventually dropping to 50% of its initial capacity within 400 cycles. In contrast, ACE demonstrated 98.5% capacity retention after 500 cycles. Given the excess Li and electrolyte used in half-cell configurations, the observed differences are likely associated with the increased impedance in the CEE system. We then proceeded to evaluate the electrolyte systems under more realistic conditions by fabricating full cells, comprising 35 μm Li with 4 mA h cm−2 loading cathodes. When cycled at 1C, the Li‖LFP cell with ACE exhibited a remarkable 91% capacity retention after 600 cycles, while that with CEE failed within 80 cycles (Fig. S23, ESI†). When we further limited the electrolyte (E/C = 5.1 g A h−1), the Li‖LFP cell with CEE failed within 40 cycles, whereas the cell with ACE showed no capacity loss over 230 cycles at 161.5 mA h g−1 (Fig. S24, ESI†).Employing 4-V class cathodes is essential for achieving higher energy densities. However, in the absence of a stable CEI, free ether solvent molecules react with the metal oxides to generate acidic species, leading to cathode capacity degradation.40 The 4.3 V float test suggested greater oxidative instability in CEE than in ACE (Fig. S25, ESI†). We proceeded to cycling NCA88 half-cells for 50 cycles, where ACE and CEE delivered capacities of 186.7 and 94.7 mA h g−1, respectively (Fig. S26, ESI†). After cycling, CEE exhibited a greater increase in charge transfer impedance compared to ACE (Fig. S27, ESI†). Similarly, galvanostatic intermittent titration technique identified substantial overpotential growth attributed to ohmic loss in the CEE system, while ACE system demonstrated low overpotentials at different states of discharge (Fig. S28, ESI†). Analysis of differential capacity (dQ/dV) of the cycled cathodes highlighted a marked decrease in the H2 to H3 phase transition peak intensities, predominantly attributed to the structural collapse of the layered cathode in the CEE system (Fig. S29, ESI†).41 The structure of NCA cathodes was visualized using high-resolution transmission electron microscopy (TEM, Fig. S30, ESI†). The instability of the layered NCA structure against the electrolyte results in phase transition into a rock-salt structure. ACE-cycled NCA cathodes exhibit a thin and uniform rock-salt layer, while CEE-cycled cathodes have a thick resistive layer. Moreover, polycrystalline domains were observed in CEE-cycled cathode surfaces, as evidenced by numerous arcs with stretched diffraction spots in Fourier transform images, indicative of the rock-salt structure (Fig. S30a, ESI†). These findings suggest that the thick, non-conducting CEI and the collapse of the NCA structure collectively contribute to the reduced capacity in the cycled NCA cathodes with CEE.
We conducted XPS depth profiling analysis on the NCA88 cathodes to better understand the CEI chemistry after cycling (Fig. S31, ESI†). Elemental analysis showed high contents of carbonaceous species originating from PVDF (287.4 and 289.9 eV) in both electrolyte systems (Fig. S32a, ESI†). The C–C and C–O species, likely derived from ether solvents, were more pronounced in the cycled NCA particles with CEE. The O 1s spectra from the cycled cathodes with ACE exhibited consistent compositions at varying depths with minor SOx and NOx peaks (534 eV), attributable to salt decomposition at the CEI (Fig. S32b, ESI†). The higher M–O bond signals (530 eV) in the CEE system indicates its inability to effectively passivate the active cathode surface.6,42 In contrast, ACE provided sufficient protection to the NCA cathode.
Rate capability tests were conducted on NCA88 (4 mA h cm−2) half cells with current densities ranging from 0.8 to 20.0 mA cm−2 (Fig. S33, ESI†). At a high current density of 20 mA cm−2, the NCA cells with CEE failed completely, while the cell with ACE maintained a capacity of 70 mA h g−1. Upon returning to a current density of 2 mA cm−2, the cathode with ACE recovered its original capacity, in contrast to the continued capacity decline in the cell with CEE, which experienced a 36.2% loss post high-rate testing.
Li metal and anode-free full cell performance
We proceeded to fabricate full cells by pairing 50 μm thick LMA with 2 mA h cm−2 loading NCA88 cathodes (Fig. 5a and Fig. S34, ESI†). The full cells with CEE failed to deliver any measurable capacity after 100 cycles, with significant CE fluctuations observed throughout the cycling process (Fig. S35, ESI†). In stark contrast, the cells with ACE maintained an average retention of 80% after 250 cycles. Moreover, a drastic increase in polarization was observed for the cell with CEE compared to stable and steady polarization observed in ACE (Fig. S36, ESI†). The full cells were evaluated under more practical conditions, consisting of 35 μm thick Li and high-loading cathode (4 mA h cm−2, N/P ratio = 1.75) with low E/C ratios (5.1 g A h−1) (Fig. 5b). The full cell with CEE rapidly deteriorated, ultimately failing by the 32nd cycle (Fig. S37, ESI†). Conversely, the ACE-based cell demonstrated robust performance, retaining 82.4% of its initial capacity after 150 cycles (159.3 mA h g−1). We then investigated the cycling performance of the anode-free (Cu‖NCA-88) cells coupled with 2 mA h cm−2 NCA-88 cathode under lean electrolyte conditions (5.1 g A h−1) (Fig. 5c). The anode-free full cell employing CEE experienced a sharp initial decrease in capacity along with significant CE fluctuation (Fig. S38, ESI†). The limited performance of CEE can be ascribed to the linear slope in its voltage profiles, indicative of limited ion transport kinetics within the cell (Fig. S39, ESI†).43 In contrast, the cell with ACE demonstrated enhanced performance by achieving 59% capacity retention after 100 cycles. The LMB performance with ACE aligns with state-of-the-art electrolyte design strategies, including WSEs, LHCEs, and FFEs (Fig. 5d and Tables S3 and S4, ESI†).7,8,11,14,20 In addition to these technical merits, the manufacturing simplicity and cost-effectiveness of ACE offer a substantial economic advantage over alternative electrolyte design strategies (Fig. 5e, f and Tables S1–S4).7–9,11,12,14,15,20,21,44–49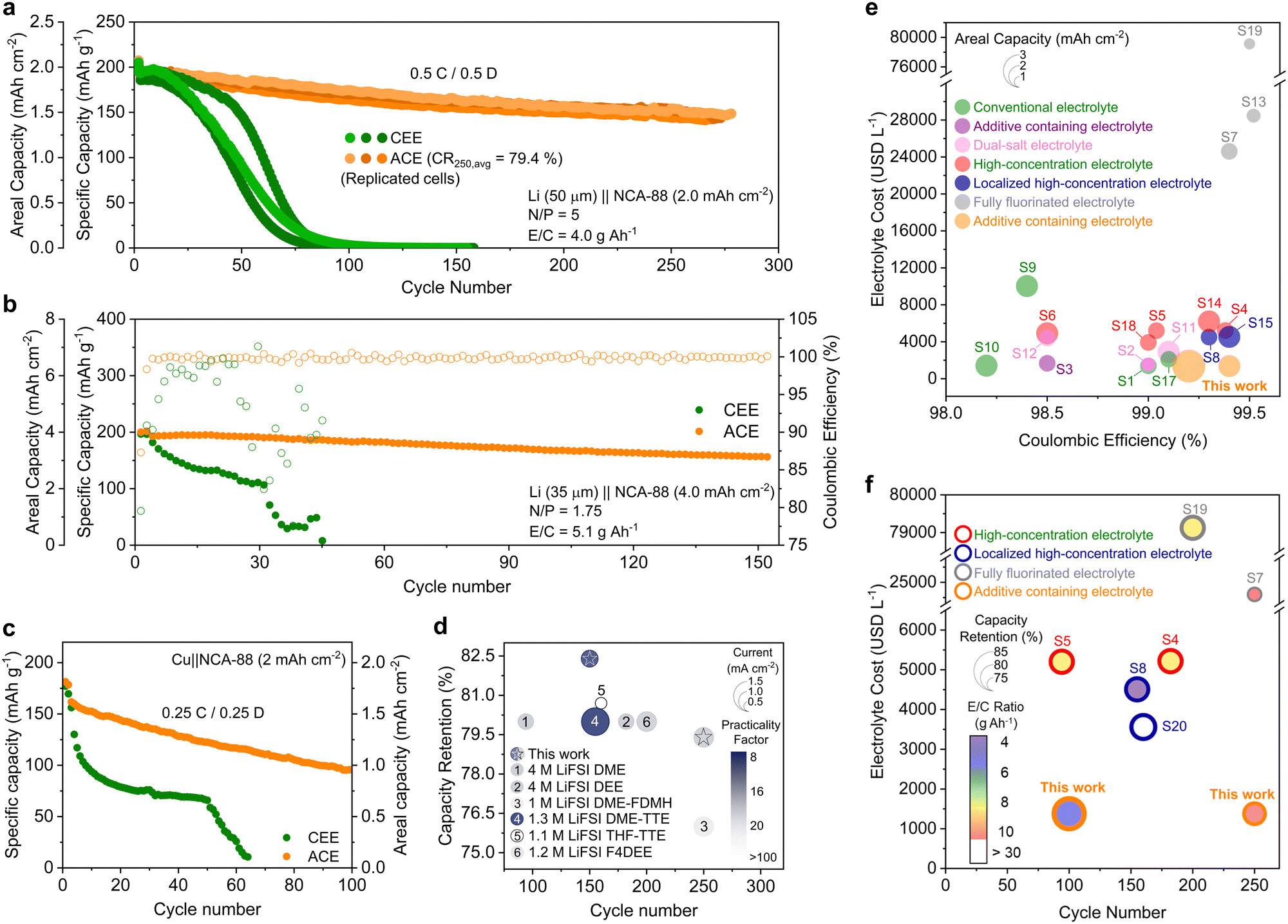 | ||
| Fig. 5 Full-cell performance with designed electrolyte. (a) Long-cycling performance of Li‖NCA88 cells replicated three times. Conditions: 50 μm LMA paired with 2 mA h cm−2 NCA88 cathode, with N/P ratio of 5.0 and E/C ratio of 4.0 g A h−1. The cells cycled at 0.5 C charge and discharge. (b) Cycling performance of Li‖NCA88 cells under practical conditions. Conditions: 35 μm LMA paired with 4 mA h cm−2 NCA88 cathode, with N/P ratio of 1.75 and E/C ratio of 5.1 g A h−1. The cells cycled at 0.1C charge and 1/3C discharge. (c) Cycling performance of anode-free Cu‖NCA-88 cells. Conditions: Cu paired with 2 mA h cm−2 cathode, with E/C ratio of 4.0 g A h−1. (d) Comparison of cycle number, cycle retention, current density, and practicality factor with LMB performance with other ether electrolytes. Practicality factor is defined as the product of the N/P ratio and E/C ratio. Comparison of (e) Li‖Cu Coulombic efficiencies and respective areal capacities and (f) Li‖4-V class cathode cycle number, capacity retention, and E/C ratio plotted against the electrolyte cost for different electrolyte design strategies. | ||
Conclusions
In this work, we have demonstrated the effectiveness of additive engineering in enhancing the stability of electrode–electrolyte interphases within ether-based electrolytes, with a specific focus on additive-containing electrolytes (ACE) as a pivotal proof of concept. Our findings revealed that the incorporation of TBATFB salt into conventional ether electrolytes significantly improves various key parameters, including CE, LMA passivation stability, exchange current density, and Li+ ion selectivity, while preserving the high ionic conductivity innate to ether electrolytes. The endurance and stability conferred by ACE extended the cycle life for high-performance LMBs under practical conditions. Furthermore, our results suggest that the additive engineering approach offers a cost-effective solution for stabilizing electrode–electrolyte interphases without the need to tailor the electrolyte solvation structure. This strategy effectively addresses several fundamental interfacial challenges and performance limitations associated with LMBs. We believe that further research and detailed discussion on the additive engineering strategy will provide fundamental insights into interfacial chemistry as we continue to advance the energy and power density, cost-effectiveness, and cycle life of LMBs.Author contributions
K. R. and S. W. L. conceived the study. K. R. contributed to the project design and conducted the experiments. K. B. L. and M. J. L. contributed to the data analysis. U. H. L and J. K. performed the MD simulations and DFT calculations. J. L., B. L. D. R., B. M. contributed to electrolyte characterization. K.-H. K. and K. K. contributed to cathode characterization. S. K., D. K., D. S. offered insights into the project design. All authors discussed the results and contributed to the manuscript. All authors have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
S. W. L, K. R., K. B. L., S. K., D. K. have filed an invention disclosure (GTRC ID 9365) covering the materials and lithium metal battery applications described in this paper.Acknowledgements
This work was supported by LG Energy Solution. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (ECCS-2025462). We thank Drs. Hasan Celik, Raynald Giovine, and Pines Magnetic Resonance Center's Core NMR Facility (PMRC Core) for spectroscopic assistance. The instrument used in this work was in part supported by NIH S10OD024998.References
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybutin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- W. Deng, X. Yin, W. Bao, X. Zhou, Z. Hu, B. He, B. Qiu, Y. S. Meng and Z. Liu, Nat. Energy, 2022, 7, 1031–1041 CrossRef CAS.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, eabq3750 CrossRef CAS PubMed.
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. M. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy, 2019, 4, 796–805 CrossRef CAS.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. M. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- Y. Lin, Z. Yang, X. Zhang, Y. Liu, G. Hu, S. Chen and Y. Zhang, Energy Storage Mater., 2023, 58, 184–194 CrossRef.
- T. Chen, J. You, R. Li, H. Li, Y. Wang, C. Wu, Y. Sun, L. Yang, Z. Ye, B. Zhong, Z. Wu and X. Guo, Adv. Sci., 2022, 9, 2203216 CrossRef CAS PubMed.
- W. Xue, M. Huang, Y. Li, Y. G. Zhu, R. Gao, X. Xiao, W. Zhang, S. Li, G. Xu, Y. Yu, P. Li, J. Lopez, D. Yu, Y. H. Dong, W. Fan, Z. Shi, R. Xiong, C.-J. Sun, I. Hwang, W.-K. Lee, Y. Shao-Horn, J. A. Johnson and J. Li, Nat. Energy, 2021, 6, 495–505 CrossRef CAS.
- H. Wang, Z. Yu, X. Kong, W. Huang, Z. Zhang, D. G. Mackanic, X. Huang, J. Qin, Z. Bao and Y. Cui, Adv. Mater., 2021, 33, 2008619 CrossRef CAS PubMed.
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. C. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- Y. Chen, Z. Yu, P. Rudnicki, H. Gong, Z. Huang, S. C. Kim, J.-C. Lai, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS PubMed.
- J. Holoubek, H. Liu, Z. Wu, Y. Yin, X. Xing, G. Cai, S. Yu, H. Zhou, T. A. Pascal, Z. Chen and P. Liu, Nat. Energy, 2021, 6, 303–313 CrossRef CAS PubMed.
- F. Wang and J. Cheng, Chem. Sci., 2022, 13, 11570–11576 RSC.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. L. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- S. Weng, X. Zhang, G. Yang, S. Zhang, B. Ma, Q. Liu, Y. Liu, C. Peng, H. Chen, H. Yu, X. Fan, T. Cheng, L. Chen, Y. Li, Z. Wang and X. Wang, Nat. Commun., 2023, 14, 4474 CrossRef PubMed.
- Z. Guo, Z. Cui, R. Sim and A. Manthiram, Small, 2023, 19, 2305055 CrossRef CAS PubMed.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- T. D. Pham, A. Bin Faheem and K. K. Lee, Small, 2021, 17, 2103375 CrossRef CAS PubMed.
- R. Nozu, E. Suzuki, O. Kimura, N. Onagi and T. Ishihara, Electrochim. Acta, 2020, 337, 135711 CrossRef CAS.
- X.-Y. Qin, J.-I. Wang, D.-P. Tang, Y.-J. Mai and L.-Z. Zhang, J. Zhejiang Univ., Sci., 2013, 14, 514–519 CrossRef CAS.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J.-G. Zhang, Adv. Energy Mater., 2018, 8, 1702097 CrossRef.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- Z. Wang, F. Zhang, Y. Sun, L. Zheng, Y. Shen, D. Fu, W. Li, A. Pan, L. Wang, J. Xu, J. Hu and X. Wu, Adv. Energy Mater., 2021, 11, 2003752 CrossRef CAS.
- I. Poli, S. Eslava and P. Cameron, J. Mater. Chem. A, 2017, 5, 22325–22333 RSC.
- Y. Huang, R. Li, S. Weng, H. Zhang, C. Zhu, D. Lu, C. Sun, X. Huang, T. Deng, L. Fan, L. Chen, X. Wang and X. Fan, Energy Environ. Sci., 2022, 15, 4349–4361 RSC.
- X. Liu, Y. Yang, Y. Li, L. Wu, H. Yu, J. Zhang, Y. Liu and Q. Zhao, Energy Adv., 2022, 1, 872–876 RSC.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- T. Wang, Y. Li, J. Zhang, K. Yan, P. Jaumaux, J. Yang, C. Wang, D. Shanmukaraj, B. Sun, M. Armand, Y. Cui and G. Wang, Nat. Commun., 2020, 11, 5429 CrossRef CAS PubMed.
- A. Bayaguud, X. Luo, Y. Fu and C. Zhu, ACS Energy Lett., 2020, 5, 3012–3020 CrossRef CAS.
- Y. Chen, M. Li, Y. Liu, Y. Jie, W. Li, F. Huang, X. Li, Z. He, X. Ren, Y. Chen, X. Meng, T. Cheng, M. Gu, S. Jiao and R. Cao, Nat. Commun., 2023, 14, 2655 CrossRef CAS PubMed.
- B. Han, X. Li, S. Bai, Y. Zou, B. Lu, M. Zhang, X. Ma, Z. Chang, Y. S. Meng and M. Gu, Matter, 2021, 4, 3741–3752 CrossRef CAS.
- B. Y. Lu, W. Li, D. Cheng, B. Bhamwala, M. Ceja, W. Bao, C. Fang and Y. S. Meng, Adv. Energy Mater., 2022, 12, 2202012 CrossRef CAS.
- A. Ramasubramanian, V. Yurkiv, T. Foroozan, M. Ragone, R. Shahbazian-Yassar and F. Mashayek, J. Phys. Chem. C, 2019, 123, 10237–10245 CrossRef CAS.
- H. Kwon, H. Kim, J. Hwang, W. Oh, Y. Roh, D. Shin and H.-T. Kim, Nat. Energy, 2023, 9, 57–69 CrossRef.
- X. Li, J. Zheng, M. H. Engelhard, D. Mei, Q. Li, S. Jiao, N. Liu, W. Zhao, J.-G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 2469–2479 CrossRef CAS PubMed.
- D. H. Jang and S. M. Oh, J. Electrochem. Soc., 1997, 144, 3342–3348 CrossRef CAS.
- K. J. Park, J. Y. Hwang, H. H. Ryu, F. Maglia, S. J. Kim, P. Lamp, C. S. Yoon and Y. K. Sun, ACS Energy Lett., 2019, 4, 1394–1400 CrossRef CAS.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- K. Ryu, M. J. Lee, K. Lee and S. W. Lee, Energy Environ. Mater., 2023, 6, e12662 CrossRef CAS.
- F. Qiu, X. Li, H. Deng, D. Wang, X. Mu, P. He and H. Zhou, Adv. Energy Mater., 2019, 9, 1803372 CrossRef.
- R. Xu, J.-F. Ding, X.-X. Ma, C. Yan, Y.-X. Yao and J.-Q. Huang, Adv. Mater., 2021, 33, 2105962 CrossRef CAS PubMed.
- S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada and Y. Yamada, Nat. Energy, 2022, 7, 1217–1224 CrossRef CAS.
- D. Wang, H. Liu, M. Li, D. Xia, J. Holoubek, Z. Deng, M. Yu, J. Tian, Z. Shan, S. P. Ong, P. Liu and Z. Chen, Nano Energy, 2020, 75, 104889 CrossRef CAS.
- R. Miao, J. Yang, X. Feng, H. Jia, J. Wang and Y. Nuli, J. Power Sources, 2014, 271, 291–297 CrossRef CAS.
- S. Yuan, S. Weng, F. Wang, X. Dong, Y. Wang, Z. Wang, C. Shen, J. L. Bao, X. Wang and Y. Xia, Nano Energy, 2021, 83, 105847 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S38 and Tables S1–S4. See DOI: https://doi.org/10.1039/d4ee02479f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |