
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe evolution of photocatalytic H2O2 generation: from pure water to natural systems and beyond
Yuyang
Tang
 abc,
Wuming
Wang
abc,
Jiaqi
Ran
abc,
Cheng
Peng
*d,
Zuxin
Xu
abc and
Wenhai
Chu
*abc
abc,
Wuming
Wang
abc,
Jiaqi
Ran
abc,
Cheng
Peng
*d,
Zuxin
Xu
abc and
Wenhai
Chu
*abc
aState Key Laboratory of Pollution Control and Resources Reuse, College of Environmental Science and Engineering, Tongji University, Shanghai 200092, China. E-mail: 1world1water@tongji.edu.cn
bMinistry of Education Key Laboratory of Yangtze River Water Environment, Tongji University, Shanghai 200092, China
cShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China
dKey Laboratory of Interfacial Physics and Technology, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: pengcheng@sinap.ac.cn
First published on 13th August 2024
Abstract
Photocatalytic hydrogen peroxide (PHP) production represents a sustainable approach that mimics photosynthesis using sunlight to generate H2O2 from water and oxygen. This review explores the intricate mechanisms of PHP and provides a critical assessment on the development of PHP in different systems, including pure water, natural water, microdroplets, and coupling systems. Furthermore, their potential applications in environmental remediation, biomedicine and the production of high-value chemicals are discussed. Moreover, the current limitations of PHP efficiency and stability, particularly in the pure water system, are summarized, and possible solutions to overcome these challenges are proposed as well. The objective of this paper is to present perspectives on improving the PHP efficiency and to advance the sustainable development of PHP technology.
Broader contextHydrogen peroxide (H2O2) presents the potential of green chemistry, playing a crucial role in sustainable development across applications in biomedical therapy, environmental remediation, and chemical synthesis. This review offers a comprehensive overview of the critical advancements in photocatalytic H2O2 production, emphasizing the significance of these methods in alignment with global sustainability objectives. The diverse systems under investigation, including pure water, natural water, innovative coupling systems, and microdroplets, present unique approaches to the efficient and environmentally friendly production of H2O2. The pure water system sets high-efficiency standards, while natural water systems highlight the scalability of the technology and its environmental compatibility. Coupling systems showcase the synergistic potential of integrated processes, enhancing the value of solar energy utilization. Microdroplet systems introduce an innovative approach, providing controlled and intensified reaction environments that optimize resource utilization. These advancements represent significant scientific achievements and establish the foundation for a more sustainable and environmentally conscious future. |
Introduction
Hydrogen peroxide (H2O2) is recognized as a versatile and sustainable chemical reagent with widespread applications in chemical synthesis,1–3 bleaching,4,5 food processing,6 biomedical therapy7 and environmental remediation.8–10 H2O2 exhibits potent oxidative properties across a broad pH range, and its only byproduct is the clean, non-toxic water. Upon activation, its derivatives like hydroxyl radicals (˙OH), can degrade virtually all forms of organic pollutants or disrupt key components within microbial cells, thereby achieving the removal of organic contaminants or the eradication of pathogenic microorganisms.11,12The anthraquinone method is the predominant industrial process for H2O2 production.13,14 Initially, anthraquinone undergoes hydrogenation with a metallic catalyst, such as palladium or platinum, resulting in the formation of hydrogenated anthraquinone. Subsequently, the hydrogenated anthraquinone is then subjected to catalytic oxidation in an oxygen-enriched environment, resulting in the production of H2O2.15 This method allows for the continuous and efficient production of H2O2 by subsequently regenerating anthraquinone through the recycling of hydrogenated anthraquinone back to its original form. However, the anthraquinone method for H2O2 production is constrained by considerable energy consumption, the reliance on precious metal catalysts, the necessity for careful handling due to potential hazards, rigorous control demands for recycling and regeneration processes, and safety concerns on H2O2 transport and storage. Therefore, it is urgent to develop more sustainable methods for H2O2 production.
Photocatalytic H2O2 production (PHP) represents a promising approach that mimics photosynthesis by leveraging sunlight as the energy source to enable the generation of H2O2 from pure water and oxygen (eqn (1)).16–20 This process involves the use of a semiconductor photocatalyst to harness sunlight, driving the reduction of oxygen and the oxidation of water to yield H2O2. This artificial photosynthesis process presents substantial potential for sustainable H2O2 production with minimized environmental impact and energy consumption. Moreover, the use of PHP process has significant potential for a wide range of applications, especially for on-site preparation in environmental remediation and biomedical applications contexts. Currently, commercial H2O2 has a concentration of approximately 30–70 wt%, whereas low concentrations of H2O2 are commonly used in practical applications.10 The PHP operates solely on solar energy, allowing for the on-site production of low-concentration H2O2 solutions using water and oxygen (air) as raw materials, thereby circumventing the transportation, storage costs, and safety hazards associated with high-concentration H2O2 solutions.
 | (1) |
Currently, extensive research is focused on optimizing the PHP process in pure water, with the exploration of a variety of photocatalysts such as metal oxides,21,22 sulfides23,24 and innovative polymers including graphitic carbon nitride (g-C3N4),25–28 resorcinol-formaldehyde (RF) resins,29,30 metal–organic frameworks (MOFs)31,32 and covalent organic frameworks (COFs).33–35 PHP can be implemented in various reaction systems. Sacrificial agent systems, as the earliest developed, typically involve the addition of sacrificial agents to rapidly consume holes, providing excess electrons, thus enhancing and accelerating the generation of H2O2. However, with a deeper understanding of the PHP process mechanism and the pursuit of absolutely green technology, the pure water system has become mainstream. The C5N2 with piezoelectric effect reported by Ma et al.36 achieved a solar-to-chemical conversion (SCC) efficiency of up to 2.6% under low light (0.1 sun) in pure water, marking a significant milestone in the field of PHP. In recent years, coupling systems capable of achieving synergistic catalysis or dual-phase reactions have emerged, differing from traditional sacrificial agent systems. These innovations ensure an ultra-high yield of H2O2 compared to pure water systems while generating high-value by-products or reducing separation difficulties, thereby promoting the advancement of PHP from an application value perspective. Additionally, the emergence of natural water systems and microdroplet systems has further expanded the research and prospects of PHP technology (Fig. 1). Given that the emergence of these advancement, a timely review of the progress and potential outlook in different PHP systems is necessary.
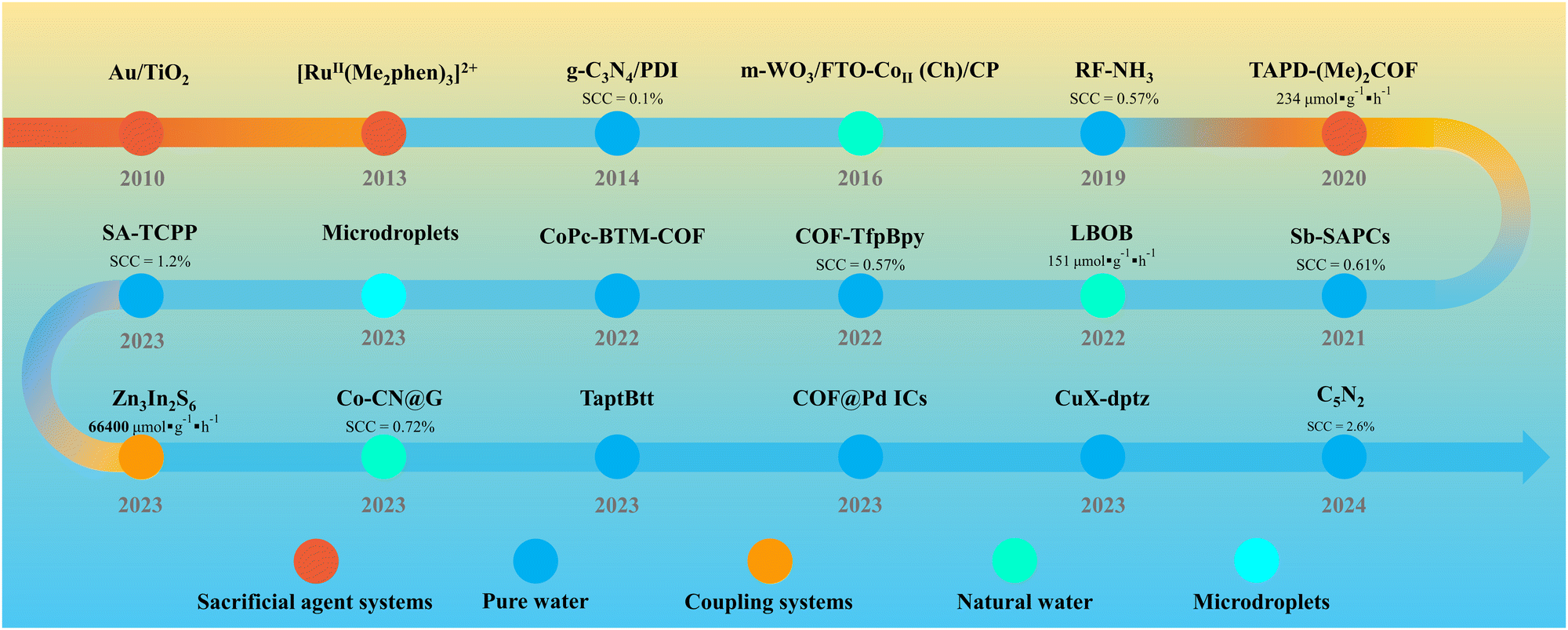 | ||
| Fig. 1 The development of PHP photocatalysts in various systems. | ||
In this review, we provide an in-depth exploration of the advancements in PHP production, with a specific emphasis on analyzing innovative strategies and technological breakthroughs achieved in different systems. A systematical review of the evolutionary progression of PHP, ranging from its application in pure water, which can serve as a simple model system, to its adaptability in natural water and microdroplets, is conducted. We also discuss the integration of PHP with high-value chemical photosynthesis in coupling systems, highlighting the strong correlation between sustainability and industrial application. Additionally, we provide a summary of the primary challenges that impede the large-scale implementation of PHP and propose possible solutions to address these obstacles. We expect to promote environmental protection and the development of sustainable technologies through our research efforts aimed at enhancing the efficiency and applicability of PHP.
Mechanisms of the PHP process
Fig. 2a illustrates the potential reactions during the PHP process. Under sunlight illumination, the photons with energy higher than the band gap are absorbed, photogenerated electron–hole pairs are induced, leading to the excitation of photogenerated electrons from the valence band (VB) to the conduction band (CB), whilst leaving photogenerated holes in the VB. Subsequently, these photogenerated carriers migrate to the surface of photocatalyst, photogenerated electrons engage in reduction reactions while photogenerated holes are involved in oxidation reactions. However, part of the photogenerated electrons and holes recombine either within the bulk or on the surface of the photocatalyst, they have no contribution to the PHP process. | ||
| Fig. 2 (a) Photoexcitation and charge transfer pathways in a photocatalyst. The Scheme of H2O2 producing via (b) 2e− ORR and 4e− WOR pathway, (c) 2e− ORR and 2e− WOR pathway and (d) 4e− ORR and 2e− WOR pathway. (e) Classical combinations of ORR and WOR pathways. | ||
During the production of H2O2, the choice between oxygen reduction reaction (ORR) or water oxidation reaction (WOR) is predominantly governed by the energy band levels, where the 4e− ORR (Pathway III) and 4e− WOR (Pathway VI) do not produce H2O2, competing against 2e− ORR and 2e− WOR. When the edge potential of CB is below 0.68 V (vs. NHE at pH = 0), H2O2 can be synthesized via the indirect two-step 1e− ORR (Pathways Ia and Ib) or the direct one-step 2e− ORR (Pathway II). The indirect one possesses a kinetic advantage, producing superoxide radicals (O2˙−) as intermediates, requiring only one electron per step. However, the direct pathway is more favorable in thermodynamics due to −0.33 V is more negative than 0.68 V (vs. NHE at pH = 0). Therefore, the precise regulation of H2O2 production through direct or indirect ORR pathways, and the elucidation of its generation mechanism in direct correlation with the catalyst structure, remains a focal point in the field of PHP research. When the edge potential of VB is above 1.76 V (vs. NHE at pH = 0), H2O2 can be generated through the 2e− WOR. Similarly, it can be categorized into the direct on-step 2e− WOR (Pathway V) and the indirect two-step 1e− WOR with free ˙OH as intermediates (Pathway IV). Nevertheless, this criterion is rigorous for the VB, leading most photocatalysts to predominantly follow the 2e− ORR and 4e− WOR mechanisms (Fig. 2b). Intriguingly, recent studies have identified cases where H2O2 is generated concurrently through both the 2e− ORR and 2e− WOR pathways (Fig. 2c), and have even explored the exclusive generation of H2O2 solely through the 2e− WOR pathway (Fig. 2d). It is crucial to emphasize that the intermediate ˙OH detected in studies may originate from free OH− (E(OH−/˙OH) = 1.99 V) rather than directly from H2O, as the potential of E(H2O/˙OH) = 2.73 V is excessively high. This detail is often overlooked in most research related to 2e− WOR. In summary, the pathways of overall photosynthesis via different ORR and WOR pathways are illustrated in Fig. 2e.37–39 The 4e− ORR & one-step 2e− WOR process is not included as no studies have reported its occurrence.
The PHP process in pure water
The ideal H2O2 photosynthesis system leverages the abundant water and oxygen available on Earth as feedstock, accomplished through ORR and WOR as two integral half-reactions.40–42 However, the light-driven WOR is challenging to accomplish, and the sluggish kinetics of the WOR half-reaction significantly limit the efficiency of H2O2 generation. In the early sacrificial agent systems, common organic solvents such as isopropanol (IPA), ethanol (EtOH), and formic acid (HCOOH) can consume holes and provide additional electrons,40,43–45 thereby driving the 2e− ORR process. However, in 2014, Shiraishi et al.46 firstly reported the photocatalyst (g-C3N4/PDI), perylene diimide (PDI) modified graphite-like carbon nitride (g-C3N4), which exhibited the capability to achieve a 90% selectivity for the 2e− ORR pathway in pure water. This remarkable result has drawn attention to the PHP process in pure water, leading to the development of numerous high-performance catalysts and the exploration of various PHP mechanisms, including the 2e− ORR pathway, the 2e− ORR and 2e− WOR dual-channel pathway and the 2e− WOR pathway (Table 1). Today, pure water serves as the optimal model system for studying PHP mechanisms. It obviates the concern of sacrificial agent residue in situ applications and demonstrates significant potential for environmental remediation,47–53 biomedical therapy7 and chemical synthesis.54–56| Photocatalysts | Reaction pathway | Energy band/eV | Atmosphere | Light source | Reaction conditions (∼cat., ∼H2O, temperature) | H2O2 yield/μmol g−1 h−1 (h) | AQEa | SCCb efficiency | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Apparent quantum yield. b Solar to chemical conversion. | |||||||||
| TPE-AQ | 2e− ORR | 1.61 | Air | λ > 400 nm, 100 mW cm−2 | 10 mg, 20 ml | 909 (1) | 57 | ||
| 1 sun, AM 1.5G | 100 mg, 80 ml | 0.26% (1) | |||||||
| Nano-BZT | Two-step 1e− ORR | 2.22 | O2 | λ > 420 nm, 300 W | 5 mg, 50 ml, 285 K | 1456 (12) | 58 | ||
| DE7-M | Two-step 1e− ORR | 2.34 | O2 | 1 sun, AM 1.5G | 5 mg, 3 ml, 313 K | 2200 (5) | 59 | ||
| A white LED | — | 8.7% (420 nm) | |||||||
| 1 sun, AM 1.5G | 200 mg, 50 ml, 313 K | 0.23% (5) | |||||||
| PC-MB-3 | One-step 2e− ORR | 1.88 | Air | λ > 420 nm | 40 mg, 20 ml | 1385 (12) | 1.44% (630 nm) | 60 | |
| CNP-s | 2e− ORR | 2.80 | O2 | 1 sun, AM 1.5G | 0.5 mg, 5 ml | 3200 (1) | 61 | ||
| Furan-BILP | Two-step 1e− ORR | 2.19 | O2 | λ > 420 nm, 300 W | 0.5 mg, 1.0 ml, R.T. | 2200 (2) | 62 | ||
| BBTz | Two-step 1e− ORR | 2.2 | Air | 1 sun, AM 1.5G | 5 mg, 25 ml | 7274 (1) | 7.14% (475 nm) | 63 | |
| QAP2 | Two-step 1e− ORR | 1.82 | Air | λ > 420 nm, 300 W | 10 mg, 50 ml, 298 K | 380 (1) | 64 | ||
| — | 1 sun, AM 1.5G | — | ∼0.17% | ||||||
| CuBr-dptz | Two-step 1e− ORR | 1.22 | Air | λ > 400 nm, 300 W | 5 mg, 80 ml H2O | 1874 (1) | 0.4% (400 nm) | 0.08% | 65 |
| Sb-SAPC | Two-step 1e− ORR | 2.63 | O2 | λ > 420 nm, 300 W | 100 mg, 50 ml, 298 K | 324 (1) | 66 | ||
| λ > 420 nm, 300 W | 60 mg, 30 ml, 298 K | 17.6% (420 nm) | |||||||
| 1 sun, AM 1.5G | 500 mg, 100 ml | 0.61% (8.33) | |||||||
Nv–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–CN N–CN |
Two-step 1e− ORR | 1.88 | O2 | λ > 420 nm, 300 W | 20 mg, 20 ml, 298 K | 137 (1) | 36.2% (400 nm) | 67 | |
| 22.1% (420 nm) | |||||||||
| 1 sun, AM 1.5G | 20 mg, 20 ml, 298 K | 0.23% | |||||||
| DMCR-1NH | Two-step 1e− ORR | 2.60 | O2 | λ > 420 nm, 300 W | 5 mg, 11 ml, 298 K | 2588 (3) | 68 | ||
| FS-COFs | One-step 2e− ORR | 2.17 | O2 | λ > 420 nm, 300 W | 5 mg, 20 ml, 298 K | 3904.2 (1) | 6.21% (420 nm) | 69 | |
| HEP-TAPT-COF | One-step 2e− ORR | 2.30 | O2 | 1 sun, AM 1.5G | 300 mg, 60 ml | 1750 | 15.35% (420 nm) | 0.65% | 70 |
| TpDz COF | One-step 2e− ORR | 2.20 | O2 | λ> 420 nm | 3 mg, 18 ml, 293 K | 7327 (1) | 71 | ||
| 1 sun, AM 1.5G | — | 11.9% (420 nm) | 0.62% | ||||||
| TZ-COF-1.5 | Two-step 1e− ORR | 1.98 | Air | λ > 420 nm, 300 W | 15 mg, 30 ml, 298 K | 268 (1.5) | 72 | ||
| λ > 420 nm, 300 W | 45 mg, 30 ml | 0.6% (475 nm) | |||||||
| 1 sun, AM 1.5G | 45 mg, 30 ml | 0.036% (1.5) | |||||||
| RF-GQDs-0.4 | Two-step 1e− ORR | O2 | λ > 420 nm, 300 W | 10 mg, 50 ml, 323 K | 2450 (1) | 73 | |||
| 1 sun, AM 1.5G | 400 mg, 150 ml, 323 K | ∼1.1% (1) | |||||||
| P5R95F | One-step 2e− ORR | 1.54 | O2 | λ > 420 nm, 300 W | 50 mg, 30 ml, 298 K | 176 (6) | 74 | ||
| 1 sun, AM 1.5G | 150 mg, 50 ml, 333 K | ∼0.9% (5) | |||||||
| RF@Nf-1.0 | 2e− ORR | 1.78 | O2 | λ > 420 nm, 11.8 mW cm−2 | 50 mg, 30 ml, 298 K | 190 (6) | 75 | ||
| 1 sun, AM 1.5G | 150 mg, 50 ml, 298 K | ∼0.35% (24) | |||||||
| APFac | Two-step 1e− ORR | 1.54 | O2 | λ > 420 nm, 300 W | 10 mg, 50 ml | — | 76 | ||
| 1 sun, AM 1.5G | 400 mg, 150 ml, 323 K | ∼0.54% (2) | |||||||
| rGO@MRF-0.5 | Two-step 1e− ORR | 1.90 | O2 | λ > 420 nm, 300 W | 25 mg, 100 ml, 298 K | 861 (2) | 77 | ||
| 1 sun, AM 1.5G | 400 mg, 150 ml, 323 K | ∼1.23% (2) | |||||||
| CHF-DPDA | 2e− ORR & 2e− WOR | 2.35 | O2 | λ > 420 nm, 100 mW cm−2, 300 W | 40 mg, 20 ml | 1725 | 16.0% (420 nm) | 78 | |
| 1 sun, AM 1.5G | 375 mg, 75 ml | ∼0.78% | |||||||
| COF-TfpBpy | One-step 2e− ORR & 2e− WOR | 2.58 | Air | λ > 420 nm, 40.8 mW cm−2 | 15 mg, 10 ml, 298 K | 695 (0.66) | 79 | ||
| λ > 300 nm, 40.8 mW cm−2 | 600 mg, 400 ml, 298 K | 8.1% (420 nm) | 0.57% | ||||||
| TTF-BT-COF | Two-step 1e− ORR & 2e− WOR | 1.64 | O2 | λ > 420 nm, 20.3 mW cm−2 | 5 mg, 10 ml, 298 K | 2760 (1) | 80 | ||
| λ > 420 nm, 20.3 mW cm−2 | 50 mg, 100 ml | 11.9% (420 nm) | 0.49% (1) | ||||||
| COF-N32 | Two-step 1e− ORR & 2e− WOR | 2.43 | O2 | λ > 420 nm, 100 mW cm−2, 300 W | 25 mg, 50 ml, 298 K | 605 (12) | 6.2% (459 nm) | 81 | |
| λ > 420 nm, 100 mW cm−2, 300 W | 3 mg, 2 ml | 0.31% | |||||||
| TD-COF | Two-step 1e− ORR & 2e− WOR | 2.05 | O2 | 400 < λ < 700 nm, 100 mW cm−2 | 1 mg, 4 ml | 4620 (4) | (1) | 69 | |
| TT-COF | Two-step 1e− ORR & 2e− WOR | 2.06 | O2 | 400 < λ < 700 nm, 100 mW cm−2 | 1 mg, 4 ml | 4245 (4) | (1) | 69 | |
| TaptBtt | Two-step 1e− ORR & 2e− WOR | 2.29 | Air | λ > 420 nm, 300 W | 15 mg, 10 ml | 1407 (1.5) | 82 | ||
| λ > 420 nm, 12.5 mW cm−2, 300 W | 75 mg, 60 ml | 4.6% (450 nm) | |||||||
| 1 sun, AM 1.5G | 75 mg, 60 ml | 0.30% | |||||||
| RF-DHAQ-2 | Two-step 1e− ORR & 2e− WOR | 2.17 | O2 | λ > 420 nm, 300 W | 10 mg, 50 ml | 1820 | 11.6% (420 nm) | 83 | |
| 1 sun, AM 1.5G | 400 mg, 150 ml, 323 K | ∼1.2% (1) | |||||||
| SA-TCPP | 2e− ORR & 2e− WOR | 1.57 | O2 | λ > 420 nm, 300 W | 25 mg, 50 ml, 353 K | 1255 (4) | 84 | ||
| λ > 420 nm, 300 W | 45 mg, 30 ml, 353 K | 14.9% (420 nm) 1.1% (940 nm) | |||||||
| 1 sun, AM 1.5G | 150 mg, 50 ml, 328 K | ∼1.2% (3) | |||||||
| Co14-(L-CH3)24 | Two-step 1e− ORR & 2e− WOR | 1.95 | O2 | 300 < λ < 1100 nm, 300 W | 5 mg, 10 ml, 298 K | 146.6 (0.66) | 85 | ||
| Sv-ZIS | Two-step 1e− ORR & 2e− WOR | 2.15 | O2 | λ ≥ 400 nm | 20 mg, 30 ml, 288 K | 1706.4 (2) | 9.9% (420 nm) | 86 | |
| 1 sun, AM 1.5G | 40 mg, 30 ml | 0.81% (0.5) | |||||||
| Mn/AB-C3N4 | 2e− WOR | 2.60 | O2 | λ > 427 nm, 40 W | 40 mg, 22 ml water with 0.55 M KOH | 255 (7) | 0.43% (427 nm) | 87 | |
2e− ORR single-channel pathway
N group and nitrogen vacancy (Nv) into g-C3N4 (Nv–CN–CN) to form dual-defect sites. Such electron-rich structure localized the charge density distribution, leading to not only enhanced light absorption and charge carrier separation, but also improved the selectivity and activity of H2O2 generation. Li et al.89 synthesized nitrogen-rich C3N5 with an asymmetric structure via the polymerization of triazole and triazine frameworks (Fig. 3a). This structure exhibited non-overlapping positive and negative charge centers, resulting in a dipole moment. The inherent self-induced polarization field facilitated directional bulk-phase photogenerated charge separation, ultimately enhancing charge migration (Fig. 3b).
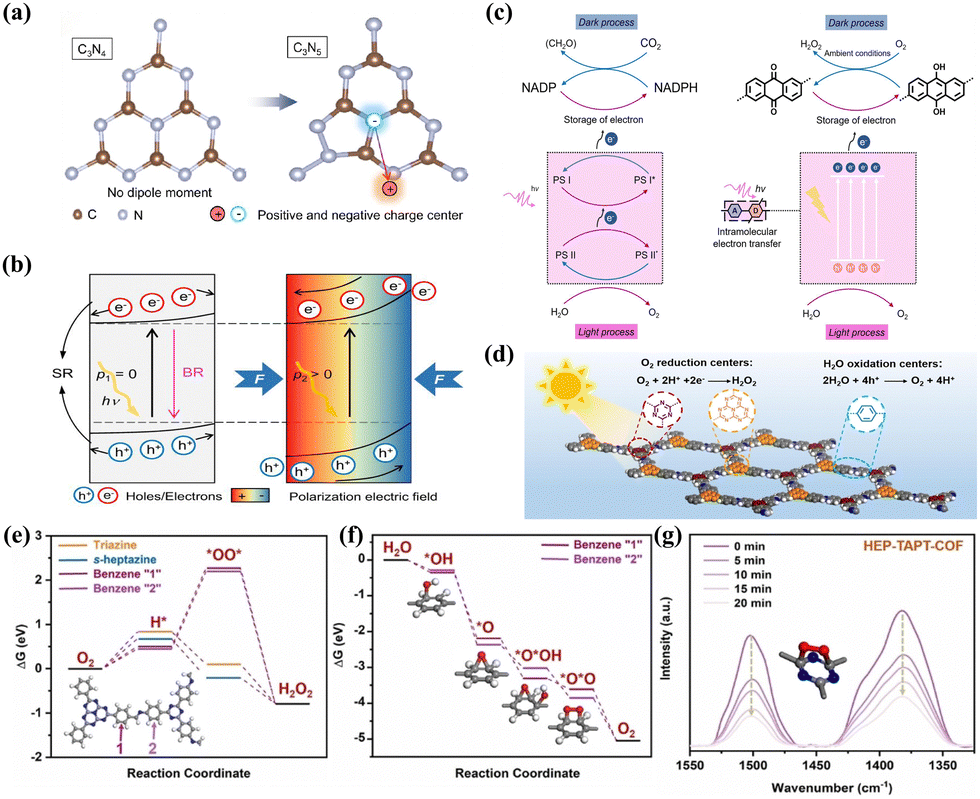 | ||
| Fig. 3 (a) Structural unit and dipole moments of C3N4 and C3N5 with positive and negative charge centers.89 (b) Mechanism of the field effect to promote the separation of photocatalytic carriers.89 (c) The mechanism behind photosynthesis of H2O2 by TPE-AQ.57 (d) The schematic diagram of photocatalytic H2O2 production by HEP-TAPT-COF.70 (e) Free energy of 2e− ORR pathway on different active sites in HEP-TAPT-COF.70 (f) Free energy of 4e− WOR pathway on different active sites in HEP-TAPT-COF.70 (g) In situ DRIFT spectra of HEP-COFs during photocatalytic H2O2 production.70 | ||
Optimizing the structure of electron acceptor or donor units presents a viable approach to promoting the charge carrier separation. Ye et al.57 developed a D–A conjugated polymer TPE-AQ with redox-active groups that function as electron acceptors for electron storage (Fig. 3c). The improved electron storage properties enhanced the charge carrier separation efficiency, increased the selectivity of 2e− ORR, and reduced the exciton binding energy.
Designing hetero-structured photocatalysts is an alternative for efficient PHP in pure water. Zhao et al.73 enhanced photocatalytic H2O2 activity to 2450 μmol g−1 h−1 by employing a freeze-drying technique to synthesize graphene quantum dots (GQDs) modified with resin (RF-GQDs). The additional electron acceptors, facilitating the electrons transfer from resin to GQDs. Similarly, Tian et al.77 created a sandwich-structured polymer photocatalyst (rGO@MRF) by integrating mesoporous formaldehyde resin onto reduced graphene oxide through an interfacial self-assembly method, facilitating an efficient chain of electron transfer processes.
Separating redox centers is another effective strategy. Mou et al.72 synthesized a TZ-COF photocatalyst, in which the pyrene and imidazole group served as the donor and acceptor unit, respectively. By establishing a D–π–A structure between the imidazole group and pyrene as a charge transfer channel, they were able to promote directed electron transfer from pyrene to the imidazole, effectively inhibiting the recombination of photoexcited charges. Chen et al.70 synthesized a crystalline COF-based photocatalyst with separated redox centers (HEP-TAPT-COF) (Fig. 3d). By calculating the Gibbs free energy (ΔG) of the intermediate products, it was determined that the carbon atoms of the s-heptazine and triazine moieties were the active sites for ORR, while WOR occurred on the benzene ring “1” and “2” (Fig. 3e and f). The structural evolution of the triazine group in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) further confirmed this point (Fig. 3g).
Moreover, it is also necessary to explore new synthesis methods to prepare new materials with excellent photoelectric properties. Zhang et al.65 synthesized zig-zag two-dimensional coordination polymers (CuX-dptz) by directly combining CuX (X = Cl, Br, I) with 3,6-di(pyridin-4-yl)-1,2,4,5-tetrazine (dptz) ligand in acetonitrile solution under ultrasonic conditions at room temperature. The layered structure of CuX-dptz conferred high charge transfer capabilities, while the halogen atoms modulated the coordination environment of the Cu sites.
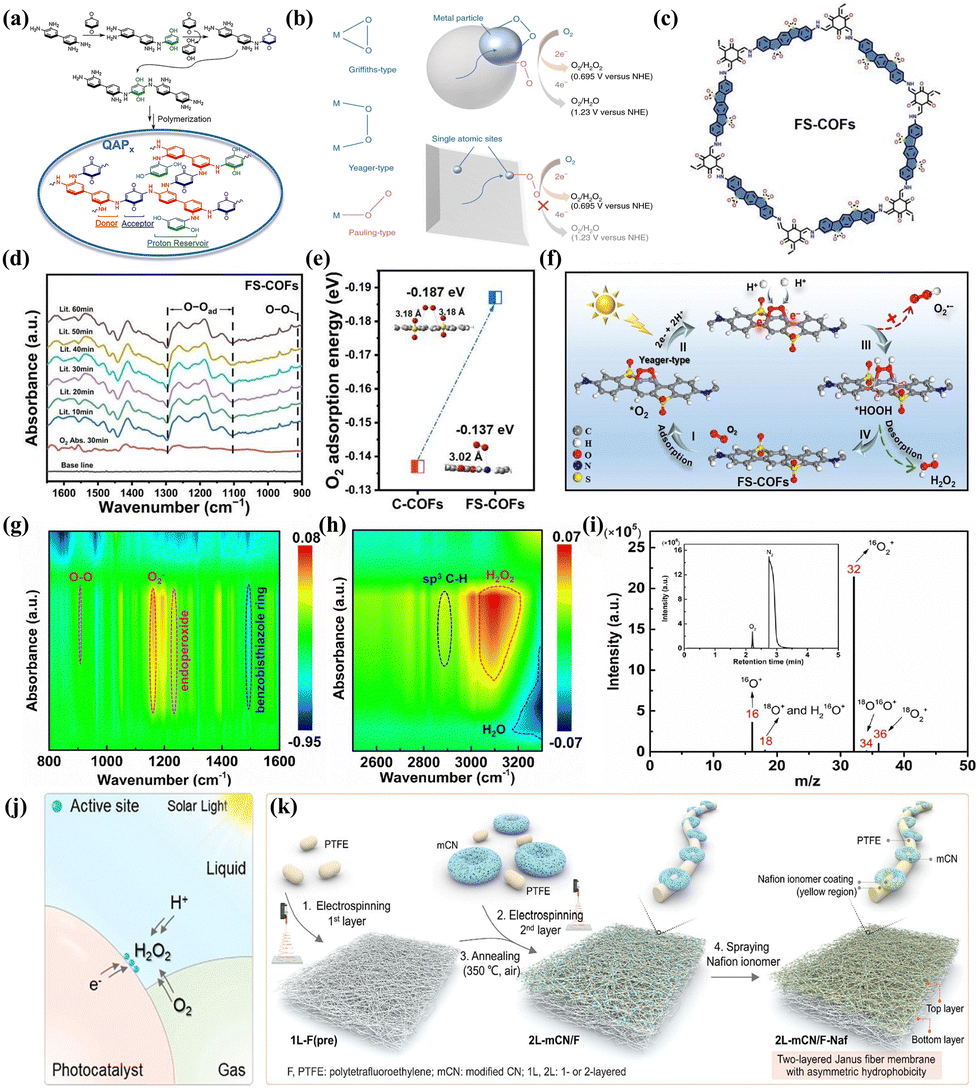 | ||
| Fig. 4 (a) The mechanism of proton reservoir within QAP2.64 (b) The schematic diagram of O2 adsorption on metal surface and ORR on a metal particle (top) and an isolated atomic site (bottom).66 (c) The structure of FS-COFs.90 (d) In situ DRIFTS spectra of FS-COFs over time during photocatalysis.90 (e) The adsorption energy of O2 on optimum site of FS-COFs.90 (f) The mechanism of H2O2 production by FS-COFs.90 The in situ DRIFT spectra of BBTz under illumination for 60 min at 10-min intervals from (g) 800 to 1600 cm−1 and (h) 3300 to 2500 cm−1.63 (i) MS spectra of gas-phase products and TIC (inset) for BBTz in the O2 to 1O2 conversion pathways during the ORR.63 (j) The schematic diagram of gas–liquid–solid triple-phase interfaces.91 (k) The synthesis route for the Janus 2L-mCN/F-Naf fiber membrane.91 | ||
On the other side, the type of O2 adsorption on the active sites of the photocatalyst significantly influences the selectivity between 2e− ORR/4e− ORR and the one-step 2e− ORR/two-step 1e− ORR. Specifically, the O2 molecular adsorption on active sites are primarily categorized into three typical configurations: Pauling-type (end-on), Griffiths-type (side-on), and Yeager-type (side-on).92,93 Teng et al.66 prepared an antimony single-atom photocatalyst (Sb-SAPC) with remarkable selectivity for 2e− ORR (Fig. 4b). In contrast to conventional metal particles, where both end-on and side-on adsorption of O2 molecules occurred simultaneously, Sb-SAPC demonstrated exclusive end-on adsorption of O2 on isolated Sb atomic sites. This distinctive O2 adsorption behavior promoted the formation of Sb-μ-peroxide (Sb-OOH), attenuated the dissociation of the O–O bond, and consequently established an effective pathway for 2e− ORR to produce H2O2. Luo et al.90 synthesized FS-COFs using a Schiff base condensation reaction (Fig. 4c). The vibrational signals of –O–O– and 1,4-endoperoxide intermediates indicated that the FS-COFs possessed a more robust adsorption capacity for O2, which was predominantly of the Yeager type in the initial configuration (Fig. 4d). DFT calculations also demonstrated that the introduction of sulfone groups altered the O2 adsorption energy (Fig. 4e). As a result, O2 adsorbed on FS-COFs can be simultaneously provided with two electrons and two protons to form *HOOH instead of *OOH by direct one-step 2e− ORR (Fig. 4f).
Optimizing the formation pathway between active sites and intermediates can also promote the 2e− ORR pathway. Cheng et al.63 reported that singlet oxygen (1O2) could be converted into endoperoxides through a [4+2] cycloaddition on the thiazole-based conjugated polymer (BBTz), thereby lowering the energy barrier for H2O2 formation. In situ DRIFT spectroscopy revealed that the infrared vibration intensity of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH+ and the π-skeleton in benzobisthiazole progressively increased and decreased, respectively (Fig. 4g). Furthermore, a new sp3 C–H vibration signal confirmed this process (Fig. 4h). Concurrently, isotope labeling experiments vividly illustrated the photogenerated hole-induced water oxidation for O2 evolution on BBTz (Fig. 4i). Yang et al.62 described a photocatalytic covalent furan-benzimidazole polymer film (Furan-BILP) containing O and N heterocycles with O–C–CN bonding. The C atom within the Furan ring was involved in binding to the adsorbed *OOH, while the H atom of *OOH engages in hydrogen bonding with the N atom located within benzimidazole ring. This interaction resulted in the stabilization of the *OOH intermediate and promoted 2e− ORR through the formation of a stable six-membered ring structure. By adjusting the relative nitrogen locations of diazine rings in COF photocatalysts, Liao et al.71 reported that pyridazine can selectively stabilize the endoperoxide intermediate, leading to a more effective direct 2e− ORR pathway compared to pyrimidine and pyrazine.
NH+ and the π-skeleton in benzobisthiazole progressively increased and decreased, respectively (Fig. 4g). Furthermore, a new sp3 C–H vibration signal confirmed this process (Fig. 4h). Concurrently, isotope labeling experiments vividly illustrated the photogenerated hole-induced water oxidation for O2 evolution on BBTz (Fig. 4i). Yang et al.62 described a photocatalytic covalent furan-benzimidazole polymer film (Furan-BILP) containing O and N heterocycles with O–C–CN bonding. The C atom within the Furan ring was involved in binding to the adsorbed *OOH, while the H atom of *OOH engages in hydrogen bonding with the N atom located within benzimidazole ring. This interaction resulted in the stabilization of the *OOH intermediate and promoted 2e− ORR through the formation of a stable six-membered ring structure. By adjusting the relative nitrogen locations of diazine rings in COF photocatalysts, Liao et al.71 reported that pyridazine can selectively stabilize the endoperoxide intermediate, leading to a more effective direct 2e− ORR pathway compared to pyrimidine and pyrazine.
Directly enhancing the contact between O2 and the photocatalyst can also accelerate the redox reaction kinetics. In a typical PHP process, only oxygen molecules dissolved in water are reduced to H2O2 on the surface of the photocatalyst. However, the relatively low solubility of O2 in water (0.9 mM at 298 K, 1 atm pressure) and its low diffusion coefficient (2.1 × 10−5 cm2 s−1) significantly impede the transport of O2 molecules to the active sites on the surface of the photocatalyst.94,95 Constructing a gas–liquid–solid triphase interface (TPI) is an effective strategy to overcome this limitation (Fig. 4j).94–96 For instance, Li et al.91 prepared an asymmetric hydrophobic bilayer Janus fiber membrane photocatalyst (Fig. 4k) by spray-coating with amphiphilic Nafion monomers possessing hydrophobic –CF2 and hydrophilic –SO3H groups. By tailoring the hydrophilic–hydrophobic characteristic and microenvironment, this photocatalyst demonstrated moderate hydrophobicity, ensuring a sufficient supply of O2 for the mCN active sites. Upon immersion in pure water, a 1.5 cm piece of the photocatalyst exhibited a noteworthy H2O2 production rate of 5.38 mmol g−1 h−1.
2e− ORR and 2e− WOR dual-channel pathway
The PHP process with dual-channel pathway involves both the 2e− ORR and 2e− WOR mechanisms. However, the 2e− WOR pathway requires the VB edge potential of the photocatalyst exceeds 1.76 V (vs. NHE at pH = 0), leading to very limited photocatalysts success in achieving dual-mode H2O2 production thus far. The realization of dual-channel pathways has been associated with the presence of acetylenic, pyridine, pyrimidine, thiophene, and thiazole functional groups. These groups possess a strong electron-donating capability and feature conjugated structures. Cheng et al.78 synthesized a photocatalyst (CHF-DPDA) through the incorporation of acetylenic functional groups into coupled covalent heptazine frameworks (CHFs) using the Friedel–Crafts reaction. The X-ray absorption near-edge structure (XANES) spectroscopy, in situ DRIFT spectra, and DFT calculations demonstrated that the 2e− ORR occurred on the s-heptazine moiety, while the 2e− WOR occurred on the benzene ring and the acetylene or diacetylene bond in CHFs. Similarly, Chang et al.80 synthesized a redox-active molecular structure COF (TTF-BT-COF) by covalently coupling tetrathiafulvalene (oxidative site) with benzothiazole (reductive site) (Fig. 5a). Liu et al.81 utilized triamines coated with triazine and featuring varying numbers of phenyl groups as exciton modulation precursors, successfully synthesizing COF-N32 with optimal intramolecular polarity. This configuration promoted exciton formation and dissociation, consequently boosted the π-conjugation of COFs to enhance the photostability. Yue et al.69 engineered two thienyl-containing COFs (TD-COF and TT-COF) by modulating N-heterocycle units (pyridine and triazine). The thiophene unit served for ORR, while the benzene ring (linked to the thiophene by the imine bond) served for WOR. Both COFs synthesized H2O2 efficiently via indirect 2e− ORR and direct 2e− WOR pathways, with TD-COF achieving a H2O2 yield of 4060 μmol g−1 h−1 in pure water. Qin et al.82 synthesized three imine-linked donor–acceptor COFs (TpaBtt, TapbBtt, and TaptBtt) by combining benzotrithiophene-trialdehyde (Btt) with various monomers possessing different electron-accepting capacities, including 4,4′,4′′-triphenylamine (Tpa), 1,3,5-tris(4-aminophenyl) benzene (Tapb), and 1,3,5-triazine (Tapt) (Fig. 5b). The band structure of TaptBtt is thermodynamically suitable for the dual-channel pathways of 2e− ORR and 2e− WOR (Fig. 5c). The number of transferred electrons in TaptBtt is close to 2 (Fig. 5d), indicating the advantage of linearly bound interactions between imine bonds and TaptBtt/Btt linkers in the 2e− ORR pathway. The results of the H218O photocatalytic experiments (Fig. 5e) indicate that TaptBtt exhibits a dual process of 2e− ORR and 2e− WOR. This research offers profound understanding of the dynamic interplay between intramolecular patterns and the chemistry of connections in polymers, furnishes an exemplary conception for the design of COFs that engender H2O2via dual-channel pathways.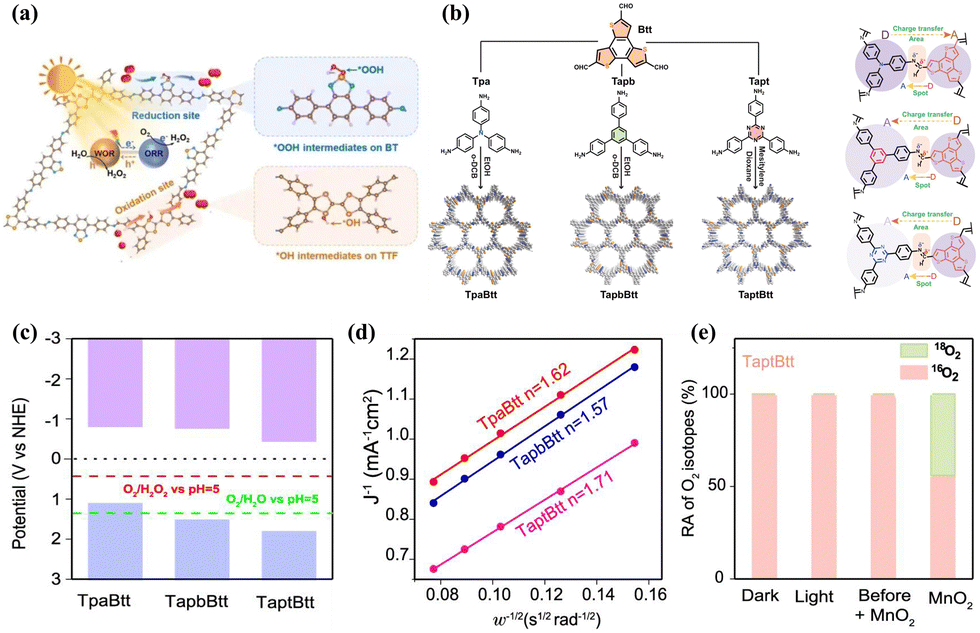 | ||
| Fig. 5 (a) The mechanism behind photosynthesis of H2O2 by TTF-BT-COF.80 (b) Synthesis of TpaBtt, TapbBtt and TaptBtt and directionality of electron transfer between functional motifs and imine linkage in TpaBtt, TapbBtt, and TaptBtt.82 (c) Energy band values of TpaBtt, TapbBtt, and TaptBtt.82 (d) Koutecky–Levich plots of TpaBtt, TapbBtt and TaptBtt obtained by RDE measurements.82 (e) ˙O2− yields of TpaBtt, TapbBtt and TaptBtt detected by NBT method under light conditions.82 | ||
Other types of photocatalyst without above functional groups are capable of facilitating PHP processes through both 2e− ORR and 2e− WOR pathways as well. Zhao et al.83 utilized the Stöber method to integrate electron-deficient 1,4-dihydroxyanthraquinone (DHAQ) into the RF resin. By adjusting the donor–acceptor ratio, the energy band structure was modified, thereby enabling the realization of the PHP process through a dual-channel pathway. Zhang et al.84 reported on a self-assembled tetrakis(4-carboxyphenyl) porphyrin supramolecule (SA-TCPP) that corroborated the participation of carboxyl groups in 2e− WOR pathway whereas photo-generated electrons promoted the formation of H2O2 at the active pyridinic N–H sites. High performance liquid chromatographytime of flight-mass spectrometry (HPLC-TOF-MS) spectra demonstrated that the hole-induced H2O2 formation process includes a photo-induced transformation of –COOH to –CO3H groups (Fig. 6a and b). Isotope experiments also confirmed the active participation of 18O from 18O-labelled water in the formation of H2O2 through the thermolytic break down of –COO18OH (Fig. 6c). Hou et al.97 decreased the energy barrier for 2e− WOR in COFs through cyano-functionalization. Liu et al.85 constructed a metal–organic cage photocatalyst, Co14-(L-CH3)24, where metal sites and imidazole sites of the ligands facilitated for the 2e− ORR and 2e− WOR, respectively. Peng et al.86 synthesized ultrathin ZnIn2S4 nanosheets with sulfur vacancies (Sv-ZIS). The presence of sulfur vacancies strongly altered the coordination structure of ZnIn2S4, thereby modulating the adsorption abilities to intermediates and preventing the overoxidation of H2O to O2. XANES spectra of the Zn K-edge and In K-edge were gathered to examine the structure of Sv-ZIS. The reduction in the valence states of Zn and In corroborated the formation of Sv in Sv-ZIS (Fig. 6d and e). Extended X-ray absorption fine structure (EXAFS) spectra clearly revealed the coordination peaks of Zn–S, In–S, Zn–Zn, and In–In in both pristine ZIS and Sv-ZIS (Fig. 6f). The planar-averaged charge density difference (CDD) along the c-axis from the DFT calculation results demonstrated that Sv not only altered the environment of the surrounding atoms but also profoundly affected the distribution of surface electron density (Fig. 6g). Sv on the surface and in the inner layer significantly reduced the adsorption energy of H+ and O2, thereby enhancing their adsorption strength (Fig. 6h).
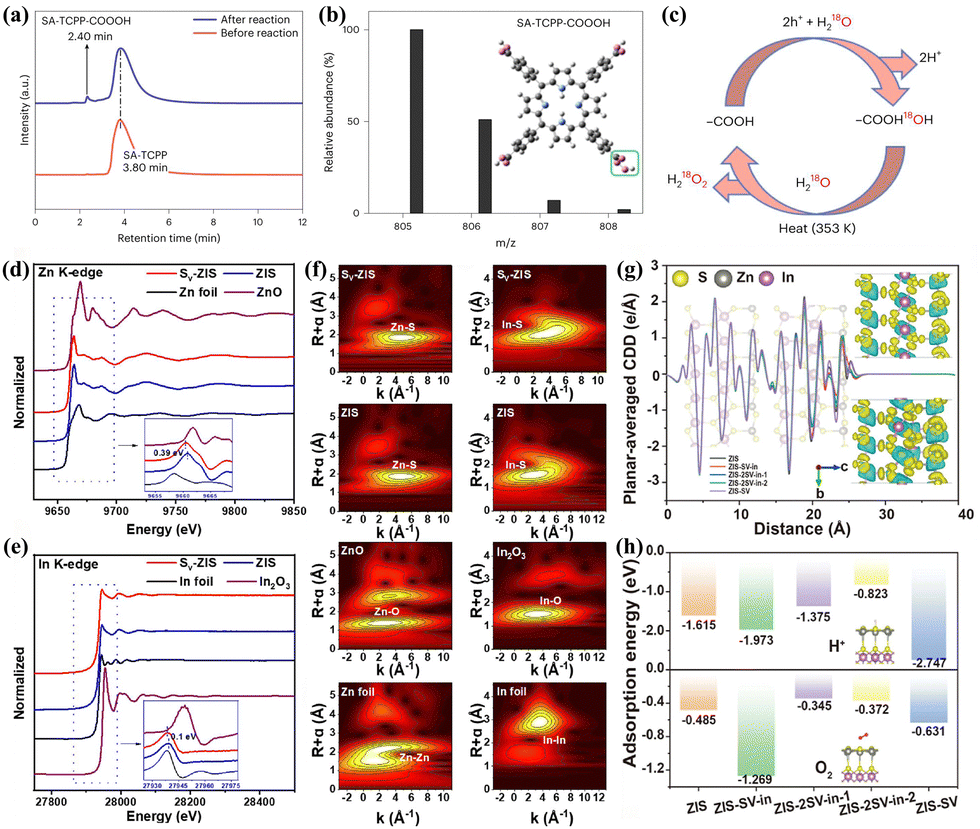 | ||
Fig. 6 (a) HPLC for SA-TCPP before and after reaction.84 (b) The molecular ion peak obtained from the ESI(−)-TOF-MS spectrum for the 2.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) min HPLC peak. Inset is the molecular structure of SA-TCPP-COOOH.84 (c) The mechanism for H2O2 production on SA-TCPP by holes according to the isotopic experiments.84 Normalized XANES spectra of Sv-ZIS and ZIS at (d) the Zn K-edge and (e) the In K-edge. The inset is the first derivative of XANES spectra.86 (f) Wavelet transform analysis of the k3-weighted EXAFS for SV-ZIS and ZIS at the Zn K-edge and the In K-edge.86 (g) Planar-averaged CDD of ZIS, ZIS-SV-In, ZIS-2SV-In, ZIS-2SV-In-2, and ZIS-SV.86 (h) Adsorption energy of H+ and O2 on the S site of the different models.86 min HPLC peak. Inset is the molecular structure of SA-TCPP-COOOH.84 (c) The mechanism for H2O2 production on SA-TCPP by holes according to the isotopic experiments.84 Normalized XANES spectra of Sv-ZIS and ZIS at (d) the Zn K-edge and (e) the In K-edge. The inset is the first derivative of XANES spectra.86 (f) Wavelet transform analysis of the k3-weighted EXAFS for SV-ZIS and ZIS at the Zn K-edge and the In K-edge.86 (g) Planar-averaged CDD of ZIS, ZIS-SV-In, ZIS-2SV-In, ZIS-2SV-In-2, and ZIS-SV.86 (h) Adsorption energy of H+ and O2 on the S site of the different models.86 | ||
2e− WOR single-channel pathway
The single-channel pathway discussed herein refers to 2e− WOR concurrently taking place with 4e− ORR. In 2023, Ren et al.87 reported a photocatalyst (Mn–AB–C3N4) featuring coordinatively unsaturated Mn–Nx sites dispersed on aryl amino-substituted g-C3N4 nanosheets. Continuous-wave W-band and X-band EPR spectra were employed to investigate the electron spin and local environment of Mn atoms in the photocatalyst (Fig. 7a). This photocatalyst operates via a single 2e− WOR pathway, with Mn–OOH acting as an intermediate in the 4e− ORR process.98 DFT calculations indicated that, photogenerated holes in the VB of Mn–AB–C3N4 facilitate H2O2 formation through two primary mechanisms: a direct 2e− WOR process that oxidizes H2O to H2O2, and an indirect process in which OH− is initially oxidized by photogenerated holes to generate ˙OH and subsequently combines to form H2O2 (Fig. 7b).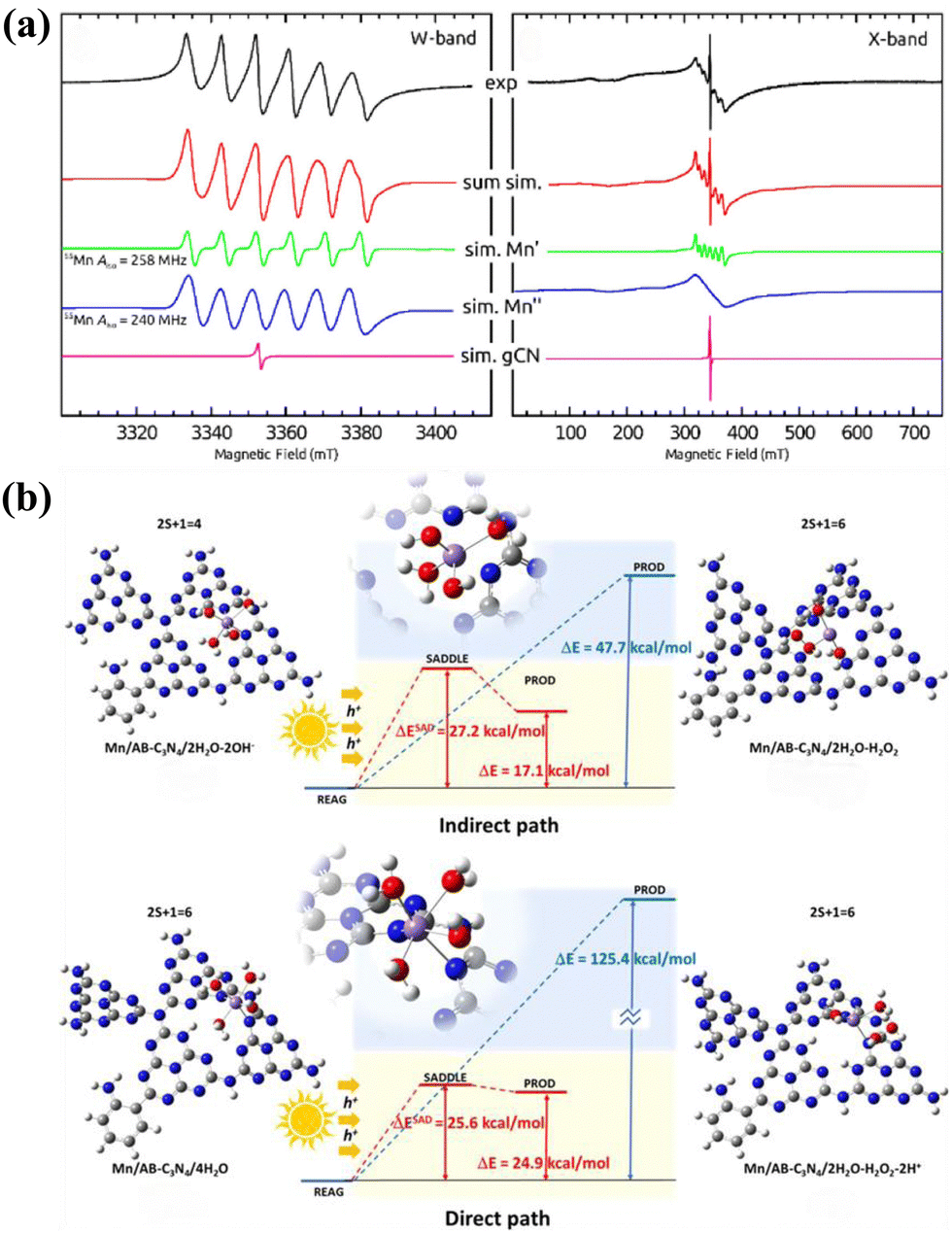 | ||
| Fig. 7 (a) cw W-band and cw X-band EPR spectra of the Mn/AB-C3N4 catalyst and the respective EPR signal deconvolution.87 (b) The mechanism behind photosynthesis of H2O2 by Mn/AB-C3N4.87 | ||
The PHP process in coupling systems
The mentioned coupling systems refer to two distinct types of sacrificial agent systems, encompassing the integration of PHP processes with other photosynthetic processes or involving interactions between different phases. The development of such coupled systems has the advantage of maintaining ultra-high H2O2 generation rates while producing high-value added products or reducing separation difficulties. This expands the application value of sacrificial agent systems from different perspectives and can be divided into single-phase systems with bifunctional photocatalysts and dual-phase systems (Table 2).| Photocatalysts | Reaction pathway | Energy band/eV | Atmosphere | Light source | Reaction conditions | H2O2 yield /μmol g−1 h−1 (h) | AQE | SCC efficiency | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| ZrS1−yS2−x (15/100) NBs | Two-step 1e− ORR | 1.98 | O2 | 1 sun, AM 1.5G | 50 mg, 30 ml H2O (1 mmol benzylamine) | 1562 (5) | 11.4% (500 nm) | 99 | |
| 10.8% (600 nm) | |||||||||
| TA-Por-sp2-COF | Two-step 1e− ORR | 1.70 | Air | λ > 600 nm, 15 W | 5 mg, 20 ml acetonitrile solvent (1 mmol benzylamine, 1 mmol p-xylene), RT | 55.6 (2) | 100 | ||
| Zn3In2S6 | Two-step 1e− ORR & one-step 2e− ORR | 2.80 | O2 | λ > 400 nm, 300 W | 5 mg, 5 ml H2O (25 mM THIQs acetonitrile), 298 K | 66400 (0.33) | 101 | ||
| ASCN-3 | Two-step 1e− ORR | 2.35 | O2 | 1 sun, AM 1.5G | 10 mg, 20 ml acetonitrile /water solution (40/60 vol%) with 0.2 mmol 4-MBA | 12912 (4) | 11.7% (420 nm) | 102 | |
| Cu3-BT-COF | Two-step 1e− ORR | 1.92 | O2 | λ > 420 nm, 300 W | 5 mg, 11.6 mM FAA aqueous solution, 298 K | 468 (4) | 7.98% (420 nm) | 0.62% | 103 |
| Py-Da-COF | Two-step 1e− ORR | 2.53 | O2 | λ > 420 nm, 300 W | 5 mg, 5 ml H2O | 461 (1) | 104 | ||
| λ > 420 nm, 300 W | 5 mg, 5 ml H2O (90 vol% EtOH) | 682 (1) | 2.4% (420 nm) | ||||||
| λ > 420 nm, 300 W | 5 mg, 5 ml H2O (90 vol% BA) | 1242 (1) | 4.5% (420 nm) | ||||||
| 1 sun, AM 1.5G | — | 0.09% | |||||||
| sonoCOF-F2 | Two-step 1e− ORR | 2.86 | O2 | λ > 420 nm, 300 W | 3 mg, 5 ml H2O, 293 K | 1889 (1.5) | 105 | ||
| λ > 420 nm, 300 W | 3 mg, 5 ml H2O, (10 vol% BA), 293 K | 2422 (1.5) | |||||||
| λ > 420 nm, 300 W | 50 mg, 60 ml H2O, (90 vol% BA), 293 K | 4125 (168) | |||||||
| — | — | 4.8% (420 nm) | |||||||
| PMCR-1 | Two-step 1e− ORR | 0.71 | Air | λ > 420 nm, 300 W | 10 mg, 22 ml H2O, 298 K | 1294 (1) | 106 | ||
| O2 | λ > 420 nm, 300 W | 10 mg, 22 ml H2O, 298 K | 1445 (1) | ||||||
| λ > 420 nm, 300 W | 10 mg, 22 ml 10:1 water:EtOH mixtures, 298 K |
1941 (1) | |||||||
| λ > 420 nm, 300 W | 10 mg, 22 ml 10:1 water:IPA mixtures, 298 K |
2265 (1) | |||||||
| λ > 420 nm, 300 W | 10 mg, 22 ml 10:1 water:BA mixtures, 298 K |
5500 (1) | 14% (420 nm) |
Single-phase systems with bifunctional photocatalysts
The role of traditional sacrificial agents in reaction systems is deliberate; they are generally not the desired end products but are consumed to enhance the overall reaction process. H2O2 is produced solely through the 2e− ORR pathway. This inevitably increases the total cost, as most high-value sacrificial agents, such as alcohols, are converted into low-value aldehydes or acids, thereby wasting the oxidative potential of the holes. Nevertheless, the bifunctional single-phase system involves the integration of the PHP process with another photo-oxidation process within a single phase, a photocatalyst is utilized to facilitate both processes simultaneously. Tian et al.99 utilized zirconium trisulfide (ZrS3) nanoribbons with abundant disulfide (S22−) and sulfide anion (S2−) vacancies to enable dual-function photocatalysis, allowing for simultaneous generation of H2O2 and benzonitrile in benzylamine solutions. Liu et al.100 employed cyano-terminated porphyrin as a precursor to synthesize the highly conjugated TA-Por-sp2-COF through cyano self-polymerization. The as-prepared bifunctional photocatalyst exhibited remarkable efficiency in the selective oxidation of benzylamine or thioanisole, as well as in the production of H2O2. Chang et al.103 reported that the Cu3-BT-COF photocatalyst can effectively promote the PHP and the furfuryl alcohol (FFA) photo-oxidation (Fig. 8a). The generation of O2 and HOOH is closely related to the two successive hydrogenation steps during the dehydrogenation process of FFA. The charge density difference analysis results indicated that oxidation and reduction reactions occurred on Cu3 and BT, respectively (Fig. 8b). Additionally, adsorption energy calculations demonstrated that FFA had a greater propensity to adsorb onto Cu3, while BT was more inclined to adsorb O2 compared to FFA (Fig. 8c). Li et al.102 coupled the selective oxidation reaction of 4-methoxybenzyl alcohol (4-MBA) with ORR. As the consumption of 4-MBA continued, the H2O2 yield gradually decreased, but surged upon replenishment (Fig. 8d), illustrating the simultaneous consumption of photogenerated electrons and holes. In the 2D isotope-labeling control experiment, the observation of 1H-labeled H2O2 confirmed that the H atoms in H2O2 primarily originated from 4-MBA rather than H2O, further illustrating the synergistic effect (Fig. 8e). Luo et al.101 utilized tetrahydroisoquinolines (THIQs) as unique proton donors, promoting the formation of H2O2 while conducting highly selective semi-dehydrogenation reactions to produce economically valuable dihydroisoquinolines (DHIQs) under the influence of a bifunctional Zn3In2S6 photocatalyst. Under visible light (400 > nm), unprecedented rates approaching stoichiometric levels of 66.4 and 62.1 mmol g−1 h−1 can be achieved (Fig. 8f). The enhancement of the O–O stretching vibrations of *OOH and HO–OH, and the OOH bending vibrations of *HOOH in the in situ FTIR spectra with prolonged irradiation intervals indicated that H2O2 on the surface of Zn3In2S6 was formed via a superoxide intermediate pathway (Fig. 8g).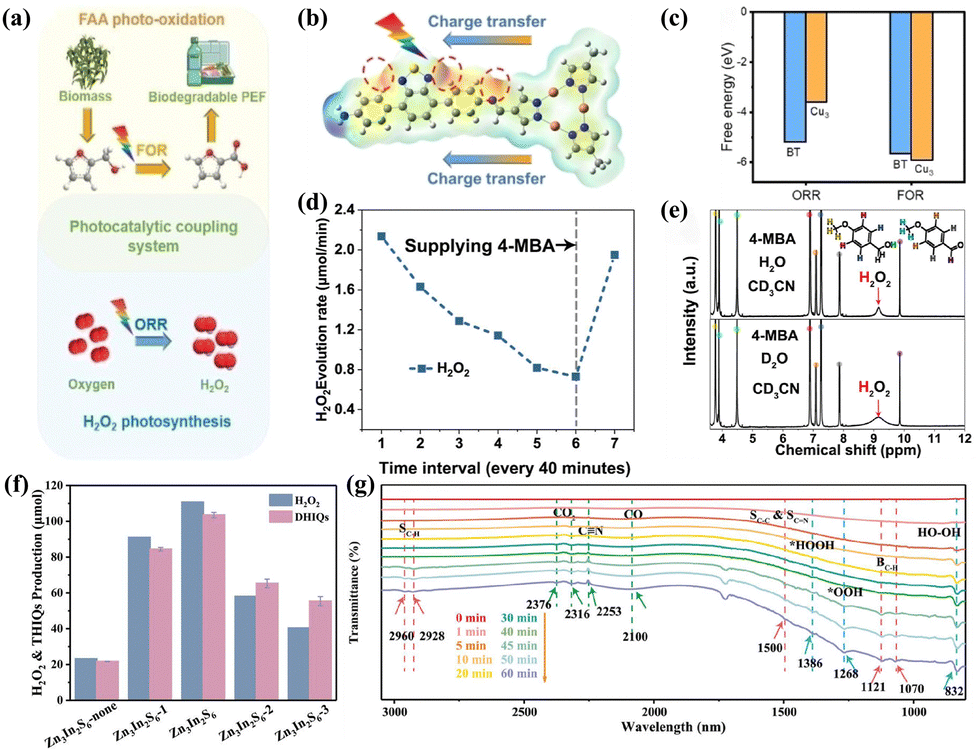 | ||
| Fig. 8 (a) The schematic representation of H2O2 photosynthesis coupled with FFA photo-oxidation.103 (b) Differential charge density diagram of Cu3-BT-COF.103 (c) Adsorption energy of Cu3 and BT units in Cu3-BT-COF for FFA and O2.103 (d) The H2O2 production rate of ASCN-3.102 (e) 1H NMR spectra of the different photocatalytic solutions after photocatalysis.102 (f) The yields of DHIQs and H2O2.101 (g) Time-dependent in situ FTIR spectra of Zn3In2S6 in THIQ/CH3CN solution.101 | ||
Dual-phase systems
The dual-phase system offers increased cost-effectiveness attributed to the straightforward separation process. Isaka et al.107 reported on the application of a biphasic benzyl alcohol/water (BA/water) system in the PHP process. This biphasic system utilized a spontaneous separation mechanism, wherein benzyl alcohol (BA) was oxidized to benzaldehyde in the organic phase, while H2O2 was generated in the aqueous phase, facilitating efficient photogenerated carrier separation and enhanced catalytic efficiency (Fig. 9a and c). The use of hydrophobic alkylated MOF photocatalysts ensured the separation between the photocatalyst and the produced H2O2, preventing the reduction of H2O2 to OH− and –OH radicals (Fig. 9b).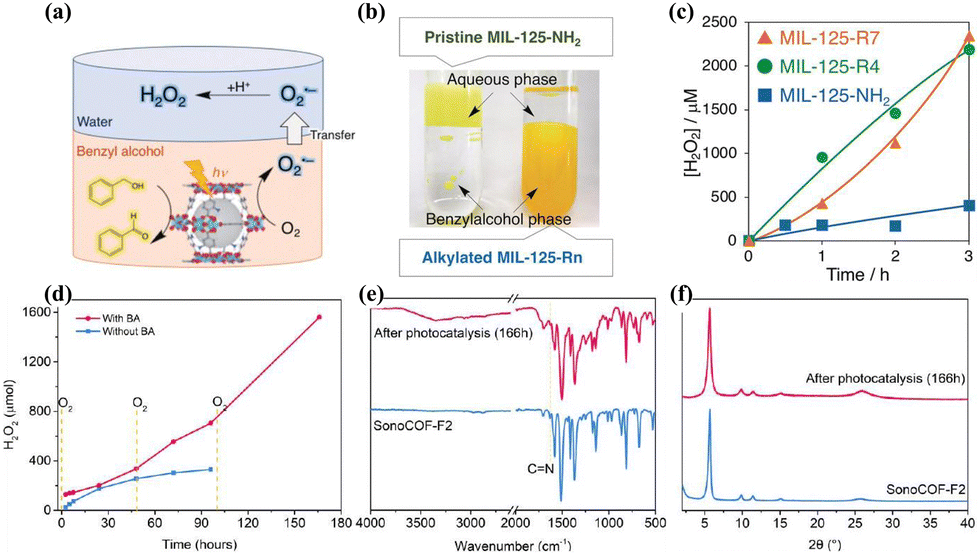 | ||
| Fig. 9 (a) Photocatalytic H2O2 production utilizing the two-phase system.107 (b) Digital photographs of two-phase systems.107 (c) The rate of H2O2 production.107 (d) Comparison of long-term photocatalytic H2O2 production using sonoCOF-F2 with and without two-phase systems.106 (e) FTIR and (f) PXRD spectra of sonoCOF-F2 before and after long-term photocatalytic testing using two-phase systems.106 | ||
The separation of the photocatalyst from H2O2 in a biphasic system plays a crucial role in preserving the structural integrity of the photocatalyst for long-term stability. Zhao et al.105 observed that the photocatalyst sonoCOF-F2 transformed into a low-crystallinity acylamide-linked COF after 96 hours of continuous photocatalytic testing, resulting in decreased electron coupling efficiency and diminished photocatalytic activity. Nevertheless, the reaction rate exhibited nearly linear kinetics throughout a continuous PHP process lasting up to 166 hours in the water/BA biphasic system, validating the efficacy of the biphasic system in preserving the stability of the photocatalyst (Fig. 9d–f). Similarly, Sun et al.104 illustrated that the pyrene-containing Py-Da-COF not only displayed notable stability of the photocatalyst but also effectively inhibited H2O2 decomposition in the biphasic system.
The PHP process in natural water
Recent studies have shown a growing interest in exploring the viability of utilizing natural water, such as rivers, lakes, and oceans, as alternatives to pure water in the PHP process. This innovative approach seeks to diminish the operational costs associated with the PHP process and improve the adaptability and ease of deployment of the systems. Nevertheless, the alternative to utilizing natural water sources presents a range of challenges, primarily stemming from the differences in photocatalytic performance observed between natural water and pure water mediums. Various factors, including dissolved salts, organic compounds, and microorganisms presented in natural water sources, can diminish the intensity of light on the surfaces of photocatalytic materials. Moreover, these constituents might compete with the desired reactants for available active sites or modify the charge distribution on the surface of the photocatalyst due to their complex composition.108–111 Hence, it is necessary to conduct in-depth research on the implementation of the PHP process in natural water.Some photocatalysts demonstrate adaptability to complex water environments and even exhibit catalytic efficiency superior to that in pure water. For example, Wu et al.112 successfully synthesized an PHP photocatalyst, PM-CDs-x, through the phenol condensation reaction in seawater using carbon dots, organic dye molecule cyanidin, and 4-methoxybenzaldehyde as precursors. It was found that the presence of Na+ in seawater had promoted the ionization of carboxyl groups, enhanced the electronegativity of carbonyl oxygen atoms, thereby increasing the charge and electron trapping barrier of carbon dots, and prolonged the lifetime of electrons (Fig. 10a). Additionally, the functional groups on the surface of carbon dots, such as hydroxyl (–OH), carbonyl (CO), and carboxyl (–COOH), effectively captured electrons (Fig. 10b), accelerating electron transfer and suppressing electron–hole recombination, significantly enhancing the photocatalytic activity in seawater (Fig. 10c). Gopakumar et al.113 developed a multiphase photocatalyst based on hydrolysis lignin (LBOB) capable of directly producing H2O2 from seawater. The deprotonation action of Na+ on the –OH groups within the lignin component of the LBOB rendered lignin an effective electron trap. Moreover, the impact of the intrinsic ions Ca2+, Mg2+, K+, and SO42− found in seawater, in conjunction with traditional acids and bases like H2SO4 and NaOH for PHP process by LBOB, was expounded upon (Fig. 10d and e), with experiments demonstrating a stable increase in H2O2 concentration concomitant with salt concentration, and the addition of extraneous acids and bases in seawater also substantially augmenting the photogenerated yield of H2O2. Calculations on the transformation of O2 at the oxide/lignin interface revealed that the proton loss from the oxidized surface during the redox process could be compensated by the reintroduction of protons and H atoms derived from lignin. In all configurations, sodium ions were found to be adsorbed above the oxide surface, in close proximity to the oxygen molecule. After simulating the departure of the ions, the HO2 complex demonstrated stability, indicating that following proton transfer to the oxygen molecule, the cations could stably accumulate on the negatively charged surface of the oxide (Fig. 10f). Wang et al.114 synthesized an efficient photocatalyst for PHP in natural seawater by anchoring cobalt (Co) single atoms onto a two-dimensional sulfur-doped graphitic carbon nitride/reduced graphene oxide (Co-CN@G) heterostructure. This unique single-atom heterostructure leveraged the photothermal effect to suppress the recombination of photogenerated charge carriers and accelerate reaction kinetics. Photogenerated electrons on Co-CN@G were transferred to the antibonding orbitals of O2 through robust electronic coupling with Co atoms, promoting O2 activation and protonation, thereby reducing the energy barrier for H2O2 generation. Zhang et al.115 proposed utilizing silver quantum dots for the photocatalytic synthesis of H2O2 in seawater. The decomposition of the resulting ˙OH radicals enhanced the inactivation efficiency of marine microorganisms in seawater from 72.3% to 99.4%, thereby expanding the application of photocatalytic technology in the field of marine antifouling. Similarly, Zhang et al.116 developed TiO2 quantum dot loaded g-C3N4 nanosheets, which, leveraging the positive role of salt ions, demonstrated efficient H2O2 generation in a seawater environment. Wang et al.117 anchored Au covalently onto Ni5P4 to create an active and durable photocatalyst for H2O2 synthesis from real seawater. The modification with plasma Au significantly suppressed charge carrier recombination, and halide ions in seawater facilitated the generation of H2O2.
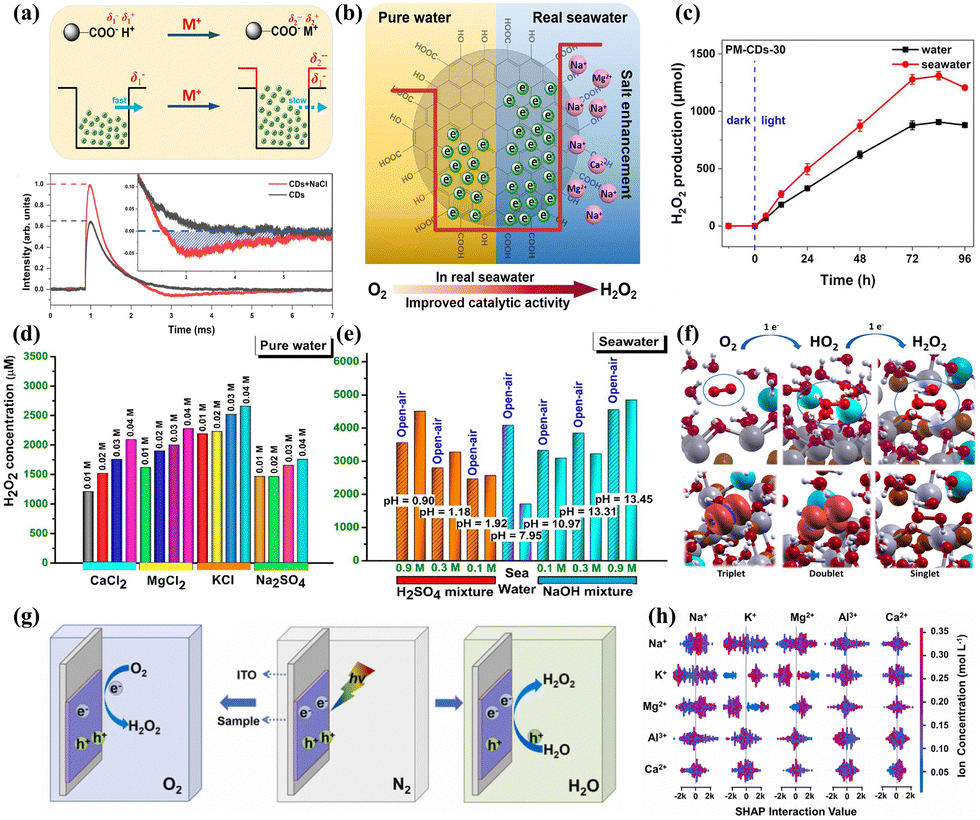 | ||
| Fig. 10 (a) The schematic diagram of the electron sink model for CDs and TPV curves of CDs powders before and after adding NaCl.112 (b) The mechanism of H2O2 photoproduction by PM-CDs-30 in seawater.112 (c) The H2O2 photoproduction by PM-CDs-30.112 (d) Photocatalytic H2O2 production using the LBOB catalyst using various acids, NaOH, and ethanol.113 (e) Photocatalytic H2O2 production using the LBOB catalyst in real seawater containing different concentrations of H2SO4 and NaOH.113 (f) The mechanism through which O2 is evolved into H2O2 by adding two electrons to the BiOBr interface in the presence of Na+ ions (blue spheres).113 (g) The schematic of in situ TPV test under different atmospheres.118 (h) Interaction diagram of Mg2+, Ca2+, K+, Na+ and Al3+.118 | ||
In the intricate natural water systems, researching photocatalytic mechanisms presents a formidable challenge. Li et al.118 were the first to employ transient photovoltage (TPV) technology (Fig. 10g) to investigate the effects of metal ion types and concentrations on the H2O2 yield in photocatalysts prepared from quercetin and methylene blue, known as CQM. By analyzing the TPV curves, which reveal the surface effective electron count (the higher the photovoltage intensity, the greater the number of electrons collected by the ITO electrode), they determined the contribution of different ions to the hydrogen peroxide yield as follows: Mg2+ > Al3+ > Ca2+ > K+. SHapley Additive exPlanation (SHAP) analysis further indicated that interactions between metal ions could enhance the yield of H2O2 (Fig. 10h). Guided by predictions from machine learning (ML), the H2O2 yield in actual seawater systems could reach 11306 μmol g−1 h−1 with CQM. This offers a promising strategy for investigating the mechanisms of photocatalytic hydrogen peroxide production in natural water systems and guiding the design, selection, and optimization of photocatalysts.
The PHP process in microdroplets
Microdroplets, tiny liquid droplets ranging in size from micrometers to millimeters, have garnered attention due to their high surface-to-volume ratio.119 The air–water interface of microdroplets exhibits unique physicochemical properties, including enhanced electric fields, localized solvation effects, and reduced reaction activation energies,120–123 which are considered pivotal factors in accelerating chemical reaction rates within microdroplets and promoting spontaneous reactions.124–126Li et al.127 leveraged the properties of microdroplets to significantly enhance the separation efficiency of limited electron–hole pairs and the slow charge transfer efficiency at the semiconductor–solution interface during the photocatalytic process. They employed a sonochemical precipitation method to synthesize various photocatalysts and generated microdroplets with diameters ranging from 100 to 500 micrometers on superhydrophobic quartz chips (Fig. 11a–c). The study revealed a substantial increase in H2O2 production within the microdroplets compared to the bulk-phase solution, attributed to the enhanced O2 availability in the microdroplets (Fig. 11d). Micro-Raman spectroscopy analysis unveiled the reinforcement of the interfacial electric field in the microdroplets facilitates the separation of photogenerated charges and promoting the PHP process (Fig. 11e). Moreover, the size of the microdroplets is a principal factor affecting the PHP process. As microdroplet size decreases, the photocatalytic decomposition of H2O2 intensifies. Microdroplets exceeding 300 μm in diameter show a positive cumulative H2O2 concentration following 1 hour of light irradiation, in contrast to smaller microdroplets, which exhibit a negative cumulative concentration (Fig. 11f). Additionally, the enhanced solvation at the interface reduced the reaction energy barrier, further facilitating the production of H2O2. Moreover, Feng et al.128 achieved synergistic hollow reactions (internal) and microdroplets (external) enrichment of O2 by combining the confined catalysis of ZnCdS@PDA with microdroplets, thereby enhancing the effectiveness of O2. Similarly, the exceptionally high interfacial electric field within the microdroplets further improved the efficiency of electron–hole separation. This concept offers a highly effective approach to accelerate chemically confined gas diffusion in liquids.
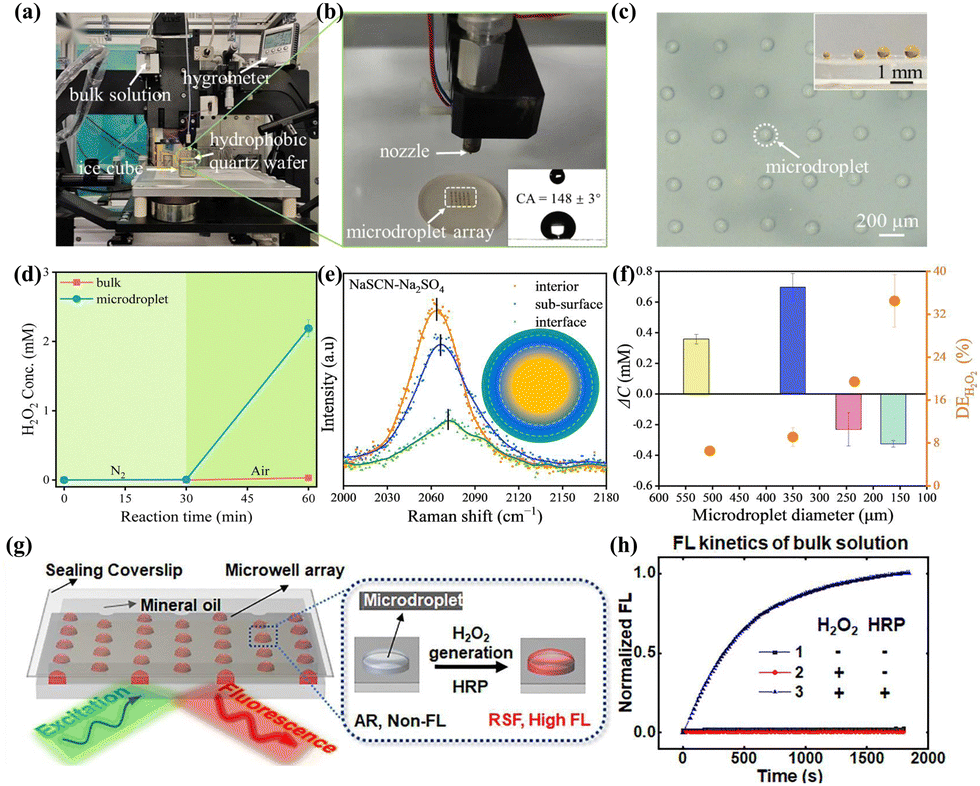 | ||
| Fig. 11 (a) Photos of ink-jet printing system for generating microdroplets with a specific diameter. The inset in (b) is the water contact angle (CA) of the superhydrophobic quartz wafer.127 (c) The image of an microdroplet array with a uniform diameter.127 (d) Photocatalytic H2O2 production in microdroplets and bulk solution under different reaction atmospheres.127 (e) Raman spectra of v(CN) measured in different regions of microdroplet. The inset is the detected regions.127 (f) Photocatalytic H2O2 decomposition efficiency and the concentration changes in microdroplets of different sizes under an air atmosphere.127 (g) The schematic illustration of the fabrication of an array of stabilized water microdroplets and time-lapsed FL imaging of spontaneous H2O2 generation.129 (h) Proof of the essential role of H2O2 and HRP in AR to RSF fluorescence conversion.129 | ||
Employing microdroplet systems presents a very direct method for augmenting the yield of H2O2. To broadly apply the PHP in microdroplets on an industrial scale, an innovative continuous gas spray technique is considered.127 This method involves a top-down approach, where a stream of air or oxygen, after undergoing humidification, is used to atomize the photocatalyst solution into uniform microdroplets. These microdroplets are then evenly dispersed within the reactor, capturing ambient light to enhance the PHP process. The resulting mixture can either be processed further or directly filtered for immediate use, presenting an effective method for wastewater treatment and disinfection. This approach is particularly advantageous in areas with abundant sunlight and limited rainfall.
Moreover, Zhou et al.129 developed a powerful tool for deciphering the microscopic chemical mechanisms within microdroplets. They fabricated stable and geometrically customizable water microdroplets utilizing micropore confinement and non-volatile oil encapsulation techniques (Fig. 8g). By innovatively employing a horseradish peroxidase (HRP) red fluorescent probe system sensitive to H2O2, along with delayed in situ fluorescence imaging technology, the continuous, real-time monitoring of H2O2 molecules within singular water microdroplets was achieved (Fig. 8h). Introducing such technique into the PHP process in microdroplets facilitates a profound comprehension of the mechanisms, laying a foundation for future applications.
Summary and outlook
In recent years, significant advancements have been achieved in the study of the PHP process in pure water, the coupling systems, natural water, and microdroplets. Nonetheless, PHP research is still in its early phases, with the current H2O2 yield remaining relatively limited, highlighting a substantial gap in readiness for large-scale applications. Consequently, further efforts are imperative to realize the practical deployment of the PHP process. Below, we summarize the key challenges for future research in this field and proposed potential solutions.Photocatalysts play a pivotal role in the PHP process. Enhancing the efficiency of photocatalysts and extending their lifespan are critical issues. Currently, under pure water conditions, photocatalysts exhibit a maximum SCC efficiency of approximately 1%, and most evaluations are of short duration, offering limited insight into long-term stability. Testing multiple indices such as H2O2 yield, AQY, and SCC efficiency under uniform conditions (including light intensity, light area, testing time, temperature, photocatalyst mass and reaction liquid volume) over a long period will be more conducive to evaluating the performance of the photocatalyst. Combining short-term repeated tests with long-term real-time tests will also be more beneficial for assessing the stability of the photocatalyst. In terms of photocatalyst design, strategies such as morphology control, functional group modification, doping, the construction of D–A structures, and heterojunctions have the potential to effectively promoting the generation of H2O2. Moreover, in the pathway of H2O2 generation, electrocatalytic methods resemble photocatalytic ones, with the primary distinction being the source of electrons. During photocatalysis, semiconductor materials are excited by light, leading to the separation of electron–hole pairs, which then engage in redox reactions upon contact with water and oxygen. In contrast, during electrocatalysis, an external power source supplies the electrons, with electrolytes and oxygen undergoing redox reactions near the electrodes. Thus, the insights garnered from catalyst design in electrocatalysis can be intertwined with those from photocatalysis to develop more efficacious photocatalysts. Photocatalysts capable of effectively integrating the direct 2e− ORR and 2e− WOR are anticipated to exhibit exceptional performance and stability.
Further investigation on the mechanism of the PHP process requires more sophisticated characterization and computational methods. Current studies employ photoluminescence spectroscopy (PL), time-resolved photoluminescence (TRPL) to evaluate carrier dynamics, electron spin resonance (ESR) to confirm the presence of active intermediates, and X-ray photoelectron spectroscopy (XPS) to detect surface active sites. However, these methods do not provide direct evidence of charge separation and migration. Most research is limited to analyzing the relationship between charge migration and design structure through DFT calculations. Introducing in situ techniques (i.e. in situ electron microscopy and in situ X-ray absorption fine structure), quantitative isotope labeling, and spatiotemporal resolution techniques can more accurately elucidate the charge transfer dynamics in H2O2 generation. Combining DFT with the Hubbard correction (DFT+U) and other theoretical calculation methods to study structure and electronic properties can facilitate the development of efficient photocatalysts.
Simplifying the preparation process and reducing the cost of photocatalysts represent critical research directions. The synthesis of photocatalysts often necessitates high temperatures, long reaction periods, or the use of toxic organic solvents, and lacks a scalable approach for large-scale fabrication. Therefore, identifying a scalable, economical, and environmentally friendly synthesis pathway is imperative. Methods such as sol–gel and microwave-assisted synthesis, which are characterized by their mildness or rapidity, should be extensively explored. Concurrently, efforts should be devoted to minimizing the cost of raw materials while ensuring the photocatalysts maintain high performance.
In the development of environmentally friendly, energy-efficient, and sustainable PHP processes, a key focus should be on harnessing the potential of natural water sources and atmospheric oxygen. Additionally, the innovation and optimization of efficient reactor systems play a crucial role in advancing the efficacy and sustainability of the PHP process. Integrating these elements can pave the way for a more environmentally conscious and energy-efficient approach to the PHP process, ensuring its long-term viability and impact in various applications.
Author contributions
Y. T. wrote the manuscript. W. C. and C. P. supervised and revised the manuscript. W. W., J. R. and Z. X. participated discussed on the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 52325001 and 52170009), the Program of Shanghai Academic Research Leader, China (no. 21XD1424000), the International Cooperation Project of Shanghai Science and Technology Commission (no. 20230714100), and the Fundamental Research Funds for the Central Universities.References
- W. Zhan, L. Ji, Z.-M. Ge, X. Wang and R.-T. Li, Tetrahedron, 2018, 74, 1527–1532 CrossRef CAS.
- D. H. Bremner, A. E. Burgess and F. B. Li, Appl. Catal., A, 2000, 203, 111–120 CrossRef CAS.
- J. J. Peng, F. Shi, Y. L. Gu and Y. Q. Deng, Green Chem., 2003, 5, 224–226 RSC.
- R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2006, 45, 206–222 CrossRef CAS PubMed.
- R. N. Gurram, M. Al-Shannag, N. J. Lecher, S. M. Duncan, E. L. Singsaas and M. Alkasrawi, Bioresour. Technol., 2015, 192, 529–539 CrossRef CAS PubMed.
- L. Xing, W. Zhang, L. Fu, J. M. Lorenzo and Y. Hao, Food Chem., 2022, 385, 132555 CrossRef CAS PubMed.
- J. Ma, X. Peng, Z. Zhou, H. Yang, K. Wu, Z. Fang, D. Han, Y. Fang, S. Liu, Y. Shen and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210856 CrossRef CAS PubMed.
- M. Ksibi, Chem. Eng. J., 2006, 119, 161–165 CrossRef CAS.
- M.-h Zhang, H. Dong, L. Zhao, D.-x Wang and D. Meng, Sci. Total Environ., 2019, 670, 110–121 CrossRef CAS PubMed.
- L. Pi, J. Cai, L. Xiong, J. Cui, H. Hua, D. Tang and X. Mao, Chem. Eng. J., 2020, 389, 123420 CrossRef CAS.
- A. Mirzaei, Z. Chen, F. Haghighat and L. Yerushalmi, Chemosphere, 2017, 174, 665–688 CrossRef CAS PubMed.
- J. P. Ribeiro and M. I. Nunes, Environ. Res., 2021, 197, 111129 CrossRef PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvio, V. Colic, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- A. T. Murray, S. Voskian, M. Schreier, T. A. Hatton and Y. Surendranath, Joule, 2019, 3, 2942–2954 CrossRef CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- C. Chu, Q. Zhu, Z. Pan, S. Gupta, D. Huang, Y. Du, S. Weon, Y. Wu, C. Muhich, E. Stavitski, K. Domen and J.-H. Kim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6376–6382 CrossRef CAS PubMed.
- Z. Teng, N. Yang, H. Lv, S. Wang, M. Hu, C. Wang, D. Wang and G. Wang, Chem, 2019, 5, 664–680 CAS.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- B. He, Z. Wang, P. Xiao, T. Chen, J. Yu and L. Zhang, Adv. Mater., 2022, 34, 2107480 CrossRef PubMed.
- T. Liu, Z. Pan, J. J. M. Vequizo, K. Kato, B. Wu, A. Yamakata, K. Katayama, B. Chen, C. Chu and K. Domen, Nat. Commun., 2022, 13, 1034 CrossRef CAS PubMed.
- H. Zhuang, L. Yang, J. Xu, F. Li, Z. Zhang, H. Lin, J. Long and X. Wang, Sci. Rep., 2015, 5, 16947 CrossRef CAS PubMed.
- J. Xu, Z. Chen, H. Zhang, G. Lin, H. Lin, X. Wang and J. Long, Sci. Bull., 2017, 62, 610–618 CrossRef CAS PubMed.
- M. A. Mohamed, M. F. M. Zain, L. J. Minggu, M. B. Kassim, N. A. S. Amin, W. N. W. Salleh, M. N. I. Salehmin, M. F. M. Nasir and Z. A. M. Hir, Appl. Catal., B, 2018, 236, 265–279 CrossRef CAS.
- Y. Shan, Y. Guo, Y. Wang, X. Du, J. Yu, H. Luo, H. Wu, B. Boury, H. Xiao, L. Huang and L. Chen, J. Colloid Interface Sci., 2021, 599, 507–518 CrossRef CAS PubMed.
- Y. Yang, C. Zhang, D. Huang, G. Zeng, J. Huang, C. Lai, C. Zhou, W. Wang, H. Guo, W. Xue, R. Deng, M. Cheng and W. Xiong, Appl. Catal., B, 2019, 245, 87–99 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- Y. Shiraishi, M. Matsumoto, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2021, 143, 12590–12599 CrossRef CAS PubMed.
- Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2018, 54, 9270–9273 RSC.
- N. Kolobov, M. G. Goesten and J. Gascon, Angew. Chem., Int. Ed., 2021, 60, 26038–26052 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef CAS PubMed.
- X. Yu, B. Viengkeo, Q. He, X. Zhao, Q. Huang, P. Li, W. Huang and Y. Li, Adv. Sustainable Syst., 2021, 5, 2100184 CrossRef CAS.
- J. Ma, C. Peng, X. Peng, S. Liang, Z. Zhou, K. Wu, R. Chen, S. Liu, Y. Shen, H. Ma and Y. Zhang, J. Am. Chem. Soc., 2024, 146, 21147–21159 CrossRef CAS PubMed.
- J. Ma, X. Peng, Z. Zhou, Y. Shen and Y. Zhang, Chin. Chem. Lett., 2023, 34, 108784 CrossRef CAS.
- L. Sun, P. Li, Z. Shen, Y. Pang, X. Ma, D. Qu, L. An and Z. Sun, Adv. Energy Sustainable Res., 2023, 4, 2300090 CrossRef CAS.
- H. Cheng, J. Cheng, L. Wang and H. Xu, Chem. Mater., 2022, 34, 4259–4273 CrossRef CAS.
- M. Teranishi, S.-I. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850–7851 CrossRef CAS PubMed.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 RSC.
- G.-h Moon, W. Kim, A. D. Bokare, N.-e Sung and W. Choi, Energy Environ. Sci., 2014, 7, 4023–4028 RSC.
- V. Maurino, C. Minero, G. Mariella and E. Pelizzetti, Chem. Commun., 2005, 2627–2629 RSC.
- X. Zeng, Z. Wang, N. Meng, D. T. McCarthy, A. Deletic, J.-H. Pan and X. Zhang, Appl. Catal., B, 2017, 202, 33–41 CrossRef CAS.
- M. Wen, K. Mori, Y. Kuwahara, T. An and H. Yamashita, Appl. Catal., B, 2017, 218, 555–569 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- S. K. Loeb, P. J. J. Alvarez, J. A. Brame, E. L. Cates, W. Choi, J. Crittenden, D. D. Dionysiou, Q. Li, G. Li-Puma, X. Quan, D. L. Sedlak, T. D. Waite, P. Westerhoff and J.-H. Kim, Environ. Sci. Technol., 2019, 53, 2937–2947 CrossRef CAS PubMed.
- A. Torres-Pinto, M. J. Sampaio, C. G. Silva, J. L. Faria and A. M. T. Silva, Appl. Catal., B, 2019, 252, 128–137 CrossRef CAS.
- C. Chu, Q. Li, W. Miao, H. Qin, X. Liu, D. Yao and S. Mao, Appl. Catal., B, 2022, 314, 121485 CrossRef CAS.
- Y. Ju, H. Li, Z. Wang, H. Liu, S. Huo, S. Jiang, S. Duan, Y. Yao, X. Lu and F. Chen, Chem. Eng. J., 2022, 430, 133168 CrossRef CAS.
- M. Zhang, Y. Jiang, X. Xu, X. Yu, W. Shen, M. Luo, L. Ding and H. Chen, J. Alloys Compd., 2022, 925, 166604 CrossRef CAS.
- Y. Lu, Y. Huang, Y. Zhang, T. Huang, H. Li, J.-J. Cao and W. Ho, Chem. Eng. J., 2019, 363, 374–382 CrossRef CAS.
- Y. Zhu, Y. Sun, J. Khan, H. Liu, G. He, X. Liu, J. Xiao, H. Xie and L. Han, Chem. Eng. J., 2022, 443, 136501 CrossRef CAS.
- C. Feng, J. Luo, C. Chen, S. Zuo, Y. Ren, Z.-P. Wu, M. Hu, S. Ould-Chikh, J. Ruiz-Martinez, Y. Han and H. Zhang, Energy Environ. Sci., 2024, 17, 1520–1530 RSC.
- H. He, Z. Wang, J. Zhang, C. Shao, K. Dai and K. Fan, Adv. Funct. Mater., 2024, 34, 2315426 CrossRef CAS.
- X. Xu, Y. Sui, W. Chen, W. Huang, X. Li, Y. Li, D. Liu, S. Gao, W. Wu, C. Pan, H. Zhong, H.-R. Wen and M. Wen, Appl. Catal., B, 2024, 341, 123271 CrossRef CAS.
- Y. Ye, J. Pan, Y. Shen, M. Shen, H. Yan, J. He, X. Yang, F. Zhu, J. Xu, J. He and G. Ouyang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2115666118 CrossRef CAS PubMed.
- B. Liu, W. Zhang, Q. Zhang, Y. Guan and Z. Lu, Small, 2023, e2303907 CrossRef PubMed.
- L. Liu, M. Y. Gao, H. Yang, X. Wang, X. Li and A. I. Cooper, J. Am. Chem. Soc., 2021, 143, 19287–19293 CrossRef CAS PubMed.
- Q. Wu, Y. Liu, J. Cao, Y. Sun, F. Liao, Y. Liu, H. Huang, M. Shao and Z. Kang, J. Mater. Chem. A, 2020, 8, 11773–11780 RSC.
- H. Yang, C. Li, T. Liu, T. Fellowes, S. Y. Chong, L. Catalano, M. Bahri, W. Zhang, Y. Xu, L. Liu, W. Zhao, A. M. Gardner, R. Clowes, N. D. Browning, X. Li, A. J. Cowan and A. I. Cooper, Nat. Nanotechnol., 2023, 18, 307–315 CrossRef CAS PubMed.
- T. Yang, Y. Jin, Y. Wang, A. Kong, Y. Chen, Y. Zou, C. Liu, G. Wei and C. Yu, Adv. Funct. Mater., 2023, 33, 2300714 CrossRef CAS.
- J. Cheng, S. Wan and S. Cao, Angew. Chem., Int. Ed., 2023, 62, e202310476 CrossRef CAS PubMed.
- B. Sheng, Y. Xie, Q. Zhao, H. Sheng and J. Zhao, Energy Environ. Sci., 2023, 16, 4612–4619 RSC.
- J. Zhang, H. Lei, Z. Li, F. Jiang, L. Chen and M. Hong, Angew. Chem., Int. Ed., 2023, e202316998 Search PubMed.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata, C. Su, B. Liu and T. Ohno, Nat. Catal., 2021, 4, 374–384 CrossRef CAS.
- X. Zhang, P. Ma, C. Wang, L. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang, X. Zhou and K. Zheng, Energy Environ. Sci., 2022, 15, 830–842 RSC.
- P. Das, G. Chakraborty, J. Roeser, S. Vogl, J. Rabeah and A. Thomas, J. Am. Chem. Soc., 2023, 145, 2975–2984 CrossRef CAS PubMed.
- J. Yue, L. Song, Y. Fan, Z. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef PubMed.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202217479 CrossRef CAS PubMed.
- Q. Liao, Q. Sun, H. Xu, Y. Wang, Y. Xu, Z. Li, J. Hu, D. Wang, H. Li and K. Xi, Angew. Chem., Int. Ed., 2023, 62, e202310556 CrossRef CAS PubMed.
- Y. Mou, X. Wu, C. Qin, J. Chen, Y. Zhao, L. Jiang, C. Zhang, X. Yuan, E. Huixiang Ang and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309480 CrossRef CAS PubMed.
- C. Zhao, X. Wang, S. Ye and J. Liu, Solar RRL, 2022, 6, 2200427 CrossRef CAS.
- Y. Shiraishi, K. Miura, M. Jio, S. Tanaka, S. Ichikawa and T. Hirai, ACS Mater. Au, 2022, 2, 709–718 CrossRef CAS PubMed.
- Y. Shiraishi, M. Jio, K. Yoshida, Y. Nishiyama, S. Ichikawa, S. Tanaka and T. Hirai, JACS Au, 2023, 3, 2237–2246 CrossRef CAS PubMed.
- X. Wang, X. Yang, C. Zhao, Y. Pi, X. Li, Z. Jia, S. Zhou, J. Zhao, L. Wu and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202302829 CrossRef CAS PubMed.
- Q. Tian, X. K. Zeng, C. Zhao, L. Y. Jing, X. W. Zhang and J. Liu, Adv. Funct. Mater., 2023, 33, 2213173 CrossRef CAS.
- H. Cheng, H. Lv, J. Cheng, L. Wang, X. Wu and H. Xu, Adv. Mater., 2022, 34, e2107480 CrossRef PubMed.
- M. Kou, Y. Wang, Y. Xu, L. Ye, Y. Huang, B. Jia, H. Li, J. Ren, Y. Deng, J. Chen, Y. Zhou, K. Lei, L. Wang, W. Liu, H. Huang and T. Ma, Angew. Chem., Int. Ed., 2022, 61, e202200413 CrossRef CAS PubMed.
- J. N. Chang, Q. Li, J. W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- F. Liu, P. Zhou, Y. Hou, H. Tan, Y. Liang, J. Liang, Q. Zhang, S. Guo, M. Tong and J. Ni, Nat. Commun., 2023, 14, 4344 CrossRef CAS PubMed.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- C. Zhao, X. Wang, Y. Yin, W. Tian, G. Zeng, H. Li, S. Ye, L. Wu and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218318 CrossRef CAS PubMed.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- J. Lu, J. Liu, L. Dong, J. Lin, F. Yu, J. Liu and Y. Lan, Angew. Chem., Int. Ed., 2023, 62, e2115666118 Search PubMed.
- H. Peng, H. Yang, J. Han, X. Liu, D. Su, T. Yang, S. Liu, C. W. Pao, Z. Hu, Q. Zhang, Y. Xu, H. Geng and X. Huang, J. Am. Chem. Soc., 2023, 145, 27757–27766 CrossRef CAS PubMed.
- P. Ren, T. Zhang, N. Jain, H. Y. V. Ching, A. Jaworski, G. Barcaro, S. Monti, J. Silvestre-Albero, V. Celorrio, L. Chouhan, A. Rokicinska, E. Debroye, P. Kustrowski, S. Van Doorslaer, S. Van Aert, S. Bals and S. Das, J. Am. Chem. Soc., 2023, 145, 16584–16596 CrossRef CAS PubMed.
- Q. Zhi, W. Liu, R. Jiang, X. Zhan, Y. Jin, X. Chen, X. Yang, K. Wang, W. Cao, D. Qi and J. Jiang, J. Am. Chem. Soc., 2022, 144, 21328–21336 CrossRef CAS PubMed.
- Z. Li, Y. Zhou, Y. Zhou, K. Wang, Y. Yun, S. Chen, W. Jiao, L. Chen, B. Zou and M. Zhu, Nat. Commun., 2023, 14, 5742 CrossRef CAS PubMed.
- Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202305355 CrossRef CAS PubMed.
- Y. Li, Z. Pei, D. Luan and X. Lou, J. Am. Chem. Soc., 2024, 146, 3343–3351 CrossRef CAS PubMed.
- E. Watanabe, H. Ushiyama and K. Yamashita, Catal. Sci. Technol., 2015, 5, 2769–2776 RSC.
- C. H. Choi, H. C. Kwon, S. Yook, H. Shin, H. Kim and M. Choi, J. Phys. Chem. C, 2014, 118, 30063–30070 CrossRef CAS.
- Z. Liu, X. Sheng, D. Wang and X. Feng, iScience, 2019, 17, 67–73 CrossRef CAS PubMed.
- X. Sheng, Z. Liu, R. Zeng, L. Chen, X. Feng and L. Jiang, J. Am. Chem. Soc., 2017, 139, 12402–12405 CrossRef CAS PubMed.
- H. Huang, R. Shi, Z. Li, J. Zhao, C. Su and T. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200802 CrossRef CAS PubMed.
- Y. Hou, P. Zhou, F. Liu, Y. Lu, H. Tan, Z. Li, M. Tong and J. Ni, Angew. Chem., Int. Ed., 2023, e202318562 Search PubMed.
- D. Kiper, M. Qui, G. Passard, C. Costentin and D. Nocera, Abstr. Pap. Am. Chem. Soc., 2019, 258, 8671–8679 Search PubMed.
- Z. Tian, C. Han, Y. Zhao, W. Dai, X. Lian, Y. Wang, Y. Zheng, Y. Shi, X. Pan, Z. Huang, H. Li and W. Chen, Nat. Commun., 2021, 12, 2039 CrossRef CAS PubMed.
- X. Liu, R. Qi, S. Li, W. Liu, Y. Yu, J. Wang, S. Wu, K. Ding and Y. Yu, J. Am. Chem. Soc., 2022, 144, 23396–23404 CrossRef CAS PubMed.
- J. Luo, X. Wei, Y. Qiao, C. Wu, L. Li, L. Chen and J. Shi, Adv. Mater., 2023, 35, e2210110 CrossRef PubMed.
- Q. Li, Y. Jiao, Y. Tang, J. Zhou, B. Wu, B. Jiang and H. Fu, J. Am. Chem. Soc., 2023, 145, 20837–20848 CrossRef CAS PubMed.
- J. N. Chang, J. W. Shi, Q. Li, S. Li, Y. R. Wang, Y. Chen, F. Yu, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202303606 CrossRef PubMed.
- J. Sun, H. Sekhar Jena, C. Krishnaraj, K. Singh Rawat, S. Abednatanzi, J. Chakraborty, A. Laemont, W. Liu, H. Chen, Y. Y. Liu, K. Leus, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Angew. Chem., Int. Ed., 2023, 62, e202216719 CrossRef CAS PubMed.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- P. Das, J. Roeser and A. Thomas, Angew. Chem., Int. Ed., 2023, 62, e202304349 CrossRef CAS PubMed.
- Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 5402–5406 CrossRef CAS PubMed.
- C. Zhao, J. Liu, B. Li, D. Ren, X. Chen, J. Yu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2003619 CrossRef CAS.
- J. H. Baek, T. M. Gill, H. Abroshan, S. Park, X. Shi, J. Norskoy, H. S. Jung, S. Siahrostami and X. Zheng, ACS Energy Lett., 2019, 4, 720–728 CrossRef CAS.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- Y. Xue, Y. Wang, Z. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469–10480 CrossRef CAS PubMed.
- Q. Wu, J. Cao, X. Wang, Y. Liu, Y. Zhao, H. Wang, Y. Liu, H. Huang, F. Liao, M. Shao and Z. Kang, Nat. Commun., 2021, 12, 483 CrossRef CAS PubMed.
- A. Gopakumar, P. Ren, J. Chen, B. V. Manzolli Rodrigues, H. Y. Vincent Ching, A. Jaworski, S. V. Doorslaer, A. Rokicinska, P. Kustrowski, G. Barcaro, S. Monti, A. Slabon and S. Das, J. Am. Chem. Soc., 2022, 144, 2603–2613 CrossRef CAS PubMed.
- W. Wang, Q. Song, Q. Luo, L. Li, X. Huo, S. Chen, J. Li, Y. Li, S. Shi, Y. Yuan, X. Du, K. Zhang and N. Wang, Nat. Commun., 2023, 14, 2493 CrossRef CAS PubMed.
- C. Zhang, F. Zhou, S. Zhan, Y. Song, F. Wang and J. Lai, Environ. Res., 2021, 197, 111129 CrossRef CAS PubMed.
- H. Zhang, S. Wang, J. Tian and X. Bai, J. Environ. Chem. Eng., 2024, 12, 112290 CrossRef CAS.
- W. Wang, Q. Luo, J. Li, Y. Li, L. Li, X. Huo, X. Du and N. Wang, Inorg. Chem. Front., 2023, 10, 1907–1918 RSC.
- J. Li, H. Shi, Z. Li, J. Wang, H. Si, F. Liao, H. Huang, Y. Liu and Z. Kang, Appl. Catal., B, 2024, 343, 123541 CrossRef CAS.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- J. Zhong, M. Kumar, J. S. Francisco and X. C. Zeng, Acc. Chem. Res., 2018, 51, 1229–1237 CrossRef CAS PubMed.
- A. Gaiduk, T. Pham, M. Govoni, F. Paesani and G. Galli, Nat. Commun., 2018, 9, 247 CrossRef PubMed.
- D. Ben-Amotz, J. Phys. Chem. Lett., 2011, 2, 1216–1222 CrossRef CAS PubMed.
- I. Dumitrescu, R. K. Anand, S. E. Fosdick and R. M. Crooks, J. Am. Chem. Soc., 2011, 133, 4687–4689 CrossRef CAS PubMed.
- J. K. Lee, S. Kim, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3898–3903 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Nat. Commun., 2018, 9, 1562 CrossRef PubMed.
- J. K. Lee, H. G. Nam and R. N. Zare, Q. Rev. Biophys., 2017, 50, 1–7 CrossRef CAS PubMed.
- K. Li, Q. Ge, Y. Liu, L. Wang, K. Gong, J. Liu, L. Xie, W. Wang, X. Ruan and L. Zhang, Energy Environ. Sci., 2023, 16, 1135–1145 RSC.
- C. Feng and L. Zhang, Mater. Horiz., 2024, 11, 1515–1527 RSC.
- K. Zhou, H. Su, J. Gao, H. Li, S. Liu, X. Yi, Z. Zhang and W. Wang, J. Am. Chem. Soc., 2024, 146, 2445–2451 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |