
An energy-efficient tellurium electrode enabled by a Cs2TeI6 perovskite structure for durable aqueous Zn–Te batteries†
Jinye
Li
,
Chengjun
Lei
 ,
Pengjie
Jiang
,
Chen
Xu
,
Tingting
Liu
and
Xiao
Liang
*
,
Pengjie
Jiang
,
Chen
Xu
,
Tingting
Liu
and
Xiao
Liang
*
State Key Laboratory of Chem/Bio-Sensing and Chemometrics, Joint International Research Laboratory of Energy Electrochemistry, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China. E-mail: xliang@hnu.edu.cn
First published on 2nd October 2024
Abstract
Tellurium (Te) is a promising high-capacity electrode material for aqueous zinc-ion batteries, capable of multi-electron redox reactions. However, the inherent hydrolysis of oxidized Te4+ exhibits significant polarization during redox, rendering it highly coupled with water in the electrolyte. This study presents a comprehensive investigation into regulating the multi-electron transfer redox chemistry of Te by incorporating cesium iodide (0.3 M CsI) into a low-concentration aqueous electrolyte (2 M ZnSO4), facilitating the formation of a stable Cs2TeI6 double perovskite during oxidation. This phase formation effectively suppresses the hydrolysis and dissolution of Te4+ species and decouples the redox reactions from water participation, leading to significantly reduced polarization. The CsI regulated Zn–Te battery delivers a high energy efficiency of 92% for the 4-electron process (Te ⇌ Te4+) and high discharge capacity of 1248 mA h g−1 for the 6-electron process (Te2− ⇌ Te ⇌ Te4+). Furthermore, the 4-electron cell exhibits exceptional cycling stability, retaining 80% capacity after 1500 cycles. This study provides valuable insights into tailoring the redox chemistry of high-capacity electrode materials, paving the way for the development of high-performance aqueous battery systems.
Broader contextTellurium (Te), with lower electronegativity compared to other chalcogen analogues, enables a six-electron (6e) redox process in Zn–Te aqueous batteries through the conversion of ZnTe ⇌ Te ⇌ Te4+, providing a theoretical capacity up to 1260 mA h g−1. However, this glorious 6e redox exhibits significant polarization due to the formation of TeO2via hydrolysis. The polarization arises from the cleavage of the H–O bond in water during charging and the breakage of the Te–O bond during discharging. Moreover, the reversibility of this redox process is heavily dependent on the electrolyte volume, as water molecules are actively involved in the reaction. This study presents a comprehensive investigation into regulating the multi-electron transfer redox chemistry of Te by incorporating cesium iodide (0.3 M CsI) into a low-concentration aqueous electrolyte (2 M ZnSO4), facilitating the formation of a stable Cs2TeI6 double perovskite during oxidation. This phase formation effectively suppresses the hydrolysis of Te4+ species and decouples the redox reactions from water participation, leading to significantly reduced polarization. Additionally, the dissociation energy of the Te–I bond in the Cs2TeI6 perovskite structure is considerably lower than that of the Te–O bond in TeO2, sustaining fast redox kinetics in Zn–Te aqueous batteries. |
1. Introduction
Aqueous zinc-ion batteries have emerged as a promising energy storage technology due to their inherent safety, low cost, and environmental friendliness.1 However, the limited capacity of conventional electrode materials with intercalation chemistry restricts the practical energy density of aqueous zinc-ion batteries.2 Chalcogens with conversion chemistry have been widely pursued as a high energy density cathode choice, by pairing with Li, Na in nonaqueous batteries, or with zinc anode in aqueous batteries.3–6 In these batteries, the chalcogens are mostly electrochemically reduced through a two-electron transfer process. While chalcogens can also undergo oxidation due to the existence of multivalence electrons,7 the exploration of multi-electron transfer redox of chalcogens was undertaken recently to further improve the energy density. Tellurium (Te), with a lower electronegativity compared to other chalcogen analogues,8,9 potentially allows for the establishment of a multi-electron transfer redox process and has garnered significant interest as a high-capacity electrode material for aqueous batteries. A recently reported six-electron (6e) redox of Te electrode in Zn–Te aqueous batteries offers a theoretical capacity up to 1260 mA h g−1, accounting for a ZnTe ⇌ Te ⇌ Te4+ conversion.10 Moreover, compared to S and Se, Te exhibits higher conductivity and bulk density, which are crucial factors for high energy batteries.11,12 Nevertheless, the practical implementation of Te electrodes in aqueous Zn–Te batteries with this glorious 6e redox process faces several challenges.Early studies have revealed that Te can undergo a four-electron (4e) electrochemical oxidation process to form TeO2 in aqueous media (i.e. 2 M ZnSO4), according to the reaction: Te + 2H2O ⇌ TeO2 + 4H+ + 4e−.13,14 This multi-electron redox process of Te exhibits significant polarization, resulting in substantial energy loss during charge/discharge processes.13 The involvement of cleaving the H–O of water during charge to form the Te–O bond, and the breakage of Te–O bond during discharge, are attributed to the significant polarization.3,14 Moreover, such redox reactions are inherently coupled with the aqueous electrolyte, leading to the participation of water molecules, rendering the reversibility heavily dependent on the electrolyte volume.15 It is crucial to note that Te4+ cation is susceptible to hydrolysis in water, with the formation of TeO2 as the mostly stabilized phase in aqueous media.16,17 Alternatively, using nonaqueous electrolyte (ionic liquids) or highly concentrated electrolyte (30 m ZnCl2) for the redox of Te4+ was suggested to suppress the hydrolysis. Indeed, in both cases, Te4+ was stabilized as TeCl4 due to the strong nucleophilicity of chloride, consequently resulting in a much lower polarization.10,18,19 It is noteworthy that the nucleophilic stabilization of Te4+ with Cl− could only be effective in such a highly concentrated environment,19,20 attributed to the extensively suppressed water activity.21 However, these approaches are accomplished with the expense of limited conductivity of the viscous electrolyte and the dissolution of TeCl4,22 and potentially a compromised rate capability especially at subzero temperatures.23 Achieving stable Te4+/Te multi-electron redox in conventional dilute aqueous solutions remains an attractive, yet to be explored, avenue.
In this study, we present a systematic investigation into regulating the electrochemistry of Te through the incorporation of cesium iodide (CsI, 0.3 M) into a low-concentration (2 M ZnSO4) aqueous electrolyte. We elucidate the critical role of CsI in facilitating the formation of a stable Cs2TeI6 phase during the 4e oxidation process, effectively suppressing the hydrolysis of Te4+ species and de-coupling the redox process from water participation. Consequently, the Te4+/Te redox exhibits significantly reduced polarization and becomes independent of the electrolyte volume, enabling efficient and sustained multi-electron redox reactions. Through comprehensive characterizations, we reveal that iodide anions are enriched on the electrode interface, promoting favorable charge transfer kinetics for the formation of the Cs2TeI6 phase. Additionally, the large Cs+ cation could shield the Zn anode from dendrite formation. In combination with the successive redox of the Te ⇌ Te2− couple with an additional two-electron transfer process, the proposed aqueous Zn–Te batteries with 6e redox demonstrate high energy efficiency and a prolonged lifespan.
2. Results and discussion
Stabilizing Te4+ species in various dilute aqueous electrolyte
To evaluate the redox behavior of Te in diverse chemical environments, we first employ DFT to simulate the dissociation energy of the Te–X bond (X = Cl, Br, I) in the form of TeX4 compounds compared to the Te–O bond in TeO2. The dissociation energies of the Te–O and Te–X bonds exhibit significant discrepancies. DFT analysis shows that dissociation of the Te–O bond requires 3.792 eV, markedly higher than the 1.113, 0.895 and 0.691 eV required to break Te–Cl, Te–Br, and Te–I bonds, respectively (Fig. 1a). However, we observed that TeX4 was rapidly hydrolyzed in the aqueous solution (2 M ZnSO4), even within a weakly acidic aqueous electrolyte (2 M ZnSO4 + 1 M HOAc) (Fig. 1b),16,17 suggesting that the practical instability of TeX4 in aqueous solutions severely hinders its further application in aqueous Zn–Te batteries (Fig. S1, ESI†). This observation underscores the challenge of achieving reversible Te redox by simply incorporating halogen-containing salts into the aqueous electrolyte (Fig. 1c). The critical question remains how to stabilize the low dissociation energy characteristics of the Te–halogen bond in dilute aqueous electrolytes to achieve fast conversion kinetics. | ||
| Fig. 1 (a) Dissociation energies for different bonding modes. (b) Visualization of the stability of TeX4 in blank electrolyte. (c) Schematic illustration of the conversion paths of Te electrode in different aqueous media. | ||
The bond strength of Te4+ compounds and the hydrolysis of Te4+–halogens are correlated with the electrochemical signals of the aqueous Zn–Te batteries in various electrolytes. The Te electrode exhibits an oxidation signal at 1.26 V (vs. Zn2+/Zn) in blank electrolyte, corresponding to the oxidation of Te to TeO2 (Fig. 2a). TeO2 is reversible, however, it occurred with a high reduction polarization at 0.75 V, aligning with the high dissociation energy of the Te–O bond. X-ray diffraction patterns confirm the successive conversion path of the Te electrode in blank electrolyte, revealing the formation of ZnTe alloy at 0.05 V, metallic Te at 0.6 V and TeO2 at 1.6 V, respectively (Fig. 2b).
 | ||
| Fig. 2 (a) CV curve at a sweep rate of 0.2 mV s−1 of the Zn–Te cell in blank electrolyte. (b) Ex situ XRD patterns of the cathode in blank electrolyte. (c) Voltage profiles of the Zn–Te cell in blank electrolyte with different electrolyte volumes. (d) CV curve at a sweep rate of 0.2 mV s−1 of Zn–Te cell in CsI electrolyte. (e) Ex situ XRD patterns of the cathode in CsI electrolyte. (f) Voltage profiles of Zn–Te cells in CsI electrolyte with different electrolyte volumes. | ||
We further studied the conversion between Te and TeO2 (Fig. S2, ESI†), revealing that the 4e reaction can occur reversibly. The general multi-electron Te electrode reaction can be described as (Fig. 1c):
| Te + 2H2O − 4e− → TeO2 + 4H+ | (1) |
| Te + Zn2+ + 2e− → ZnTe | (2) |
While water participates in this conversion process, it is imperative to control the electrolyte volume within a reasonable range for the proper functioning of the battery.24 At an electrolyte volume/electrode mass ratio of 100 μL mg−1, this conversion reaction can sustain a reversible capacity of 500 mA h g−1 over the first 10 cycles with a low energy efficiency of 56% (Fig. 2c). However, reducing the electrolyte volume/electrode mass ratio to 10 μL mg−1, akin to other battery systems such as Zn–MnO2 alkaline batteries,25,26 leads to rapid decay of the Zn–Te battery (Fig. S3, ESI†).
The introduction of 0.3 M CsI into the 2 M ZnSO4 electrolyte significantly altered the electrochemical process of the Te electrode (Fig. 1c). A pair of reversible redox signals emerged at 1.1/1.2 V for the high-valence conversion (Fig. 2d), corresponding to a low electrochemical polarization of 0.1 V. XRD analysis conducted on the Te electrode at various states of charge in the CsI containing electrolyte revealed the formation of a Cs2TeI6 double perovskite during charging, indicative of a 4e process, which subsequently vanished during discharge (Fig. 2e). The electrode reaction can be interpreted as:
| Te + 6CsI − 4e− ⇌ Cs2TeI6 + 4Cs+ (4e charge) | (3) |
Note that the molar ratio of iodine ions to Te in the cell has to reach 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to accomplish the Cs2TeI6 formation (Fig. S4, ESI†). The Te electrode in the CsI electrolyte is highly reversible, sustains a high discharge capacity of 1248 mA h g−1 with 99.2% coulombic efficiency for the successive 6e redox process between Cs2TeI6 and ZnTe. Due to the significantly suppressed voltage polarization for the Te4+/Te redox couple, the aqueous Zn–Te cell demonstrates a high energy efficiency of 78.5%. Moreover, the electrode reaction excludes the participation of water from the aqueous electrolyte. A comparison of cells with different electrolyte volumes reveals that at an electrolyte volume/electrode mass ratio of 10 μL mg−1, the CsI electrolyte exhibits no significant decay (Fig. 2f and Fig. S4, ESI†), which is in sharp contrast to the electrolyte dependent redox in blank electrolyte. Note that CsI was blended into the electrode to accomplish the Cs2TeI6 phase formation for the lean electrolyte evaluation (see the methods section). Furthermore, the pH value of the blank and CsI electrolyte was in situ monitored using a pH meter, revealing a constant pH value for the CsI electrolyte during charge/discharge (Fig. S6, ESI†). Remarkably, CsI emerges as the sole optimal additive for the aqueous Zn–Te cell towards high conversion efficiency, a capability unattainable through the substitution of other anions (Br− and Cl−) and cations (Li+ and K+) (Fig. S7, ESI†).
:1 to accomplish the Cs2TeI6 formation (Fig. S4, ESI†). The Te electrode in the CsI electrolyte is highly reversible, sustains a high discharge capacity of 1248 mA h g−1 with 99.2% coulombic efficiency for the successive 6e redox process between Cs2TeI6 and ZnTe. Due to the significantly suppressed voltage polarization for the Te4+/Te redox couple, the aqueous Zn–Te cell demonstrates a high energy efficiency of 78.5%. Moreover, the electrode reaction excludes the participation of water from the aqueous electrolyte. A comparison of cells with different electrolyte volumes reveals that at an electrolyte volume/electrode mass ratio of 10 μL mg−1, the CsI electrolyte exhibits no significant decay (Fig. 2f and Fig. S4, ESI†), which is in sharp contrast to the electrolyte dependent redox in blank electrolyte. Note that CsI was blended into the electrode to accomplish the Cs2TeI6 phase formation for the lean electrolyte evaluation (see the methods section). Furthermore, the pH value of the blank and CsI electrolyte was in situ monitored using a pH meter, revealing a constant pH value for the CsI electrolyte during charge/discharge (Fig. S6, ESI†). Remarkably, CsI emerges as the sole optimal additive for the aqueous Zn–Te cell towards high conversion efficiency, a capability unattainable through the substitution of other anions (Br− and Cl−) and cations (Li+ and K+) (Fig. S7, ESI†).
The unique role of CsI for the stabilization of Te4+ in aqueous media
Previous studies have reported that certain double perovskite halides with the general formula A2TeX6 (where A represents the monovalent cation and X is the halogen ion) exhibit chemical stability in water.27 Our experimental findings indicate that the size of alkaline cations and halides significantly affects the stability of A2TeX6. Fig. S8 (ESI†) illustrates those three representative monovalent cations (Li+, K+, Cs+) were individually added to an acidic solution of Te4+–halogens. It was observed that upon increasing the size of the cation, a yellowish precipitate formed at the bottom of the solution with Cs+ cation, while the upper solution faded from its original yellow color. This phenomenon was similarly observed in solutions of bromides and iodides. Characterization of the solid precipitates by XRD (Fig. S9–S10, ESI†) and SEM (Fig. S11, ESI†) confirmed the formation of double perovskite type halides with Cs+ cation.28,29 The stability of the three double perovskites in the aqueous electrolyte has been tested. Cs2TeCl6 and Cs2TeBr6 cannot be stabilized in the weakly acidic ZnSO4 electrolyte, as shown by the faded color of the powder in the solution (Fig. 3a). UV-vis analysis of the supernatant of Cs2TeCl6 and Cs2TeBr6 confirms the existence of Cl− and Br− signals, implying significant hydrolysis or dissolution (Fig. 3b). The pH values of the Cs2TeCl6 and Cs2TeBr6 solutions reveal a significant decrease after hydrolysis (Fig. S12, ESI†). The diffraction pattern confirms the hydrolysis products are TeO2 for both Cs2TeCl6 and Cs2TeBr6 (Fig. S13, ESI†). However, Cs2TeI6 is stable in the aqueous electrolyte, as confirmed by negligible iodide ions in the solution, and no crystallinity reformation of the Cs2TeI6 phase (Fig. S14, ESI†). In the blank electrolyte, the pH value exhibited a significant periodic change with the deepening of the charge and discharge degree, whereas that of the CsI electrolyte remained relatively stable.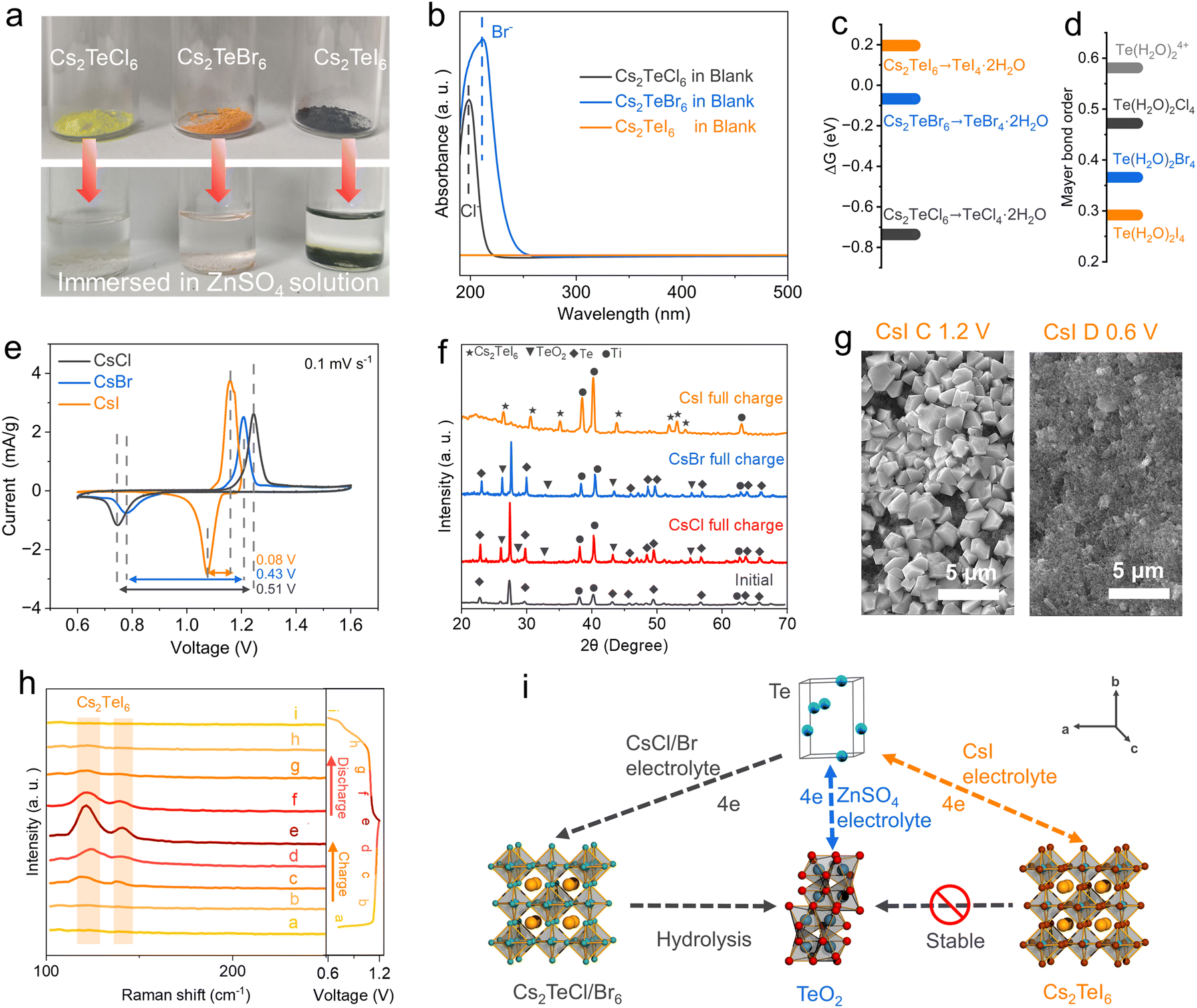 | ||
| Fig. 3 (a) Visualization on the stability of A2TeX6 in blank electrolyte. (b) UV spectrum of the supernatant of A2TeX6 powder in blank electrolyte. (c) The free energy changes of the hydrolysis of various A2TeX6. (d) Te–O Mayer bond order of various clusters. (e) CV curves at a sweep rate of 0.1 mV s−1 of the Te electrode in different electrolyte. (f) Ex situ XRD patterns of the fully charged Te electrode. (g) Ex situ SEM images of Te electrode in CsI electrolyte at selected potentials. (h) Ex situ Raman patterns of Te electrode in CsI electrolyte at different potentials. (i) Schematic demonstration of the electrode reaction with/without CsI. | ||
DFT was conducted to uncover the hydrolysis of the double perovskite Cs2TeX6 phase in water. The primary building block, the [TeX6]2− octahedral unit, was firstly subjected to water ligand substitution on halogen atoms to mimic the hydrolysis, with the formation of [Te(H2O)2X4] as the intermediates (Fig. S15–S17, ESI†). The free energy changes of this process exhibit a rational decrease from 0.196 eV for [TeI6]2−, −0.067 eV for [TeBr6]2−, and −0.736 eV for [TeCl6]2− (Fig. 3c). The positive free energy change of the [TeI6]2− octahedral unit means the water replacement on the iodine atom is not a thermodynamically favorable process, while chlorine is more susceptible to be substituted compared to bromine. These results are consistent with the stepwise enhanced stabilities of Cs2TeX6 reported in previous literature reports,30 which are attributed to the variations in the atomic radius of different halogen elements. Water ligands hooked in the distorted octahedral [Te(H2O)2X4] intermediates initiate the conversion to TeO2, and the conversion efficiency could be accessed by Mayer bond orders of Te–O in the octahedral structure.31,32 The Mayer bond order is typically defined as the degree of overlapping of two adjacent atomic orbitals to create a chemical bond.33 The Mayer bond orders of Te–O in [Te(H2O)2X4] are 0.472 (Cl), 0.366 (Br), and 0.292 (I), suggesting that [Te(H2O)I4] has the lowest probability for the formation of Te–O as an oxide (Fig. 3d). The Mayer bond order theory is also valid for the explanation of the spontaneous hydrolysis of TeX4. For the most predominant solvation structure of TeX4 in aqueous media, Te(H2O)64+ clusters, the Mayer bond order of Te–O is calculated to be 0.581. This explains the distinct stability of the Cs2TeX6 phase in the aqueous electrolyte, and the hydrolysis of TeX4 chemicals (Fig. 1b and 3a).
Indeed, the coordination anions could significantly affect the stability of double perovskite in aqueous solution.34,35 The hydrolysis ability of the [TeX6]2− octahedral is influenced by several factors including its number of ionic charges (M), and ionic potential (Z2/r, where Z is the number of electronic layers and r is the ionic radius). The empirical formula provides a general framework for understanding and predicting the hydrolysis constant:36,37
| pKm = 19.04r − 3.65Z + 3.56M − 0.74(Z2/r) + 1.16 | (4) |
Employing first principles calculations based on density functional theory (DFT) calculations, we conducted a comparative analysis of different coordination anions and the radius (r) of the Te4+ cation (Fig. S18, ESI†). For Cs2TeX6, the r values of Te4+ are 2.63, 2.81, and 3.05 Å in the Cs2TeCl6, Cs2TeBr6, and Cs2TeI6 structure, respectively. The corresponding hydrolysis constants calculated from eqn (4) indicate a rational increase, further support of the suppressed hydrolysis for Cs2TeI6.
The stability of the oxidized Te species is strongly correlated to the electrochemical performance. We focused on the electrochemical redox processes of Te0 ⇌ Te4+, since the introduction of CsX salts in the electrolyte does not significantly affect the Te/Te2− redox signal (Fig. S19, ESI†). It was observed that when CsCl and CsBr were used as additives, the redox signals of the aqueous Zn–Te cell are indistinguishable from those in the blank electrolyte (Fig. 3e and Fig. S1, ESI†), albeit with a slightly decreased polarization as shown by the CV curves. However, replacing the anion with I− resulted in distinct electrochemical signals, with a pair of redox peaks appearing at 1.1 V/1.2 V. Additional CV and GCD tests were conducted to exclude the contribution of I−/I2 redox for the CsI electrolyte by extending the voltage range (Fig. S20, ESI†).38 XRD patterns of the charged electrode indicate that TeO2 was the primary oxidation product for the CsCl and CsBr based aqueous electrolyte, whereas in the CsI electrolyte, the Cs2TeI6 phase was stabilized as the oxidation product (Fig. 3f). Moreover, a significant amount of Te was presented in the electrode after being fully charged in the CsBr and CsCl electrolyte, as uncovered by their strong diffraction peaks (Fig. 3f), indicative of incomplete conversion. This incomplete utilization of Te in the TeO2 conversion path has been widely reported in conventional aqueous Zn–Te batteries,15,20 which might be attributed to the passivation of the electrode by TeO2. The Te 3d XPS spectrum of the charged electrode in the CsI electrolyte shows the formation of Cs2TeI6, whereas TeO2 was formed in the CsBr electrolyte (Fig. S21, ESI†). We also conducted a self-discharge test on the battery. Following a 10 h period of standing, the battery exhibited a capacity retention rate of 99.0% (Fig. S22, ESI†).
The XPS spectra of Te electrodes at different states of charge were employed to elucidate the valence changes associated with the 6e process (Fig. S23, ESI†). Upon charging to 1.2 V and discharging to 0.05 V, the binding energy of Te 3d exhibited a shift towards higher and lower energies, respectively,39 indicative of the successive redox of ZnTe ⇌ Te ⇌ Cs2TeI6 in the CsI electrolyte. In accordance, the aqueous Zn–Te cell in the CsCl and CsBr electrolytes exhibit similar charge/discharge curves to that in the 2 M ZnSO4 electrolyte for the 4e redox process, demonstrating a rapid decay during cycling (Fig. S24, ESI†).
The electrode morphology after charge/discharge was further characterized by SEM. The initial SEM of the Te electrode is shown in Fig. S25 (ESI†). The fully charged Te electrode in CsI electrolyte exhibits numerous Cs2TeI6 crystals on the surface (Fig. 3g). EDS mapping reveals that the elemental ratios match those of Cs2TeI6 (Fig. S26, ESI†). These particles disappeared after discharge to 0.6 V, indicating reversible conversion. Conversely, no significant particle formation was observed on the electrode surface containing CsCl and CsBr after being fully charged, indicating the spontaneous conversion of the Cs2TeCl6 and Cs2TeBr6 phases to TeO2 surface passivation (Fig. S27, ESI†). Ex situ Raman spectroscopy was conducted to elucidate the Te conversion chemistry (Fig. 3h). The intensity of distinctive Raman bands corresponding to Cs2TeI6 reaches a maximum at the end of the charge. Thereafter, the pattern gradually disappears during the discharge process. Based on the above conclusions, a 6e conversion reaction mechanism with a sequential contribution from Zn2+ ion and CsI is proposed, as illustrated in Fig. 3i.
The improved Te electrode kinetics and Zn electrode stability
To experimentally investigate the Te electrode kinetics in both blank and CsI electrolyte, we conducted a series of electrochemical measurements including using electrochemical impedance spectroscopy (EIS) and galvanostatic intermittent titration technique (GITT). As shown in Fig. 4a, the cell with the CsI electrolyte exhibits a lower charge transfer resistance (Rct) of 7.48 Ω compared to that with blank electrolyte (17.36 Ω). Since Rct is a primary indicator of reaction kinetics, the lower Rct at an open-circuit voltage (OCV) suggests superior reaction kinetics for Te redox.10,40 Additionally, the cell with CsI electrolyte maintains a significantly lower average Rct than that in the blank electrolyte during charge and discharge (Fig. 4b and Fig. S28, ESI†). The voltage hysteresis of Te electrode investigated by GITT reveals a lower overpotential between the closed-circuit voltage (CCV, non-equilibrium) and the open-circuit voltage (OCV, quasi-equilibrium) for the cell with CsI electrolyte (Fig. 4c). The calculated diffusion coefficients for the Te/Cs2TeI6 conversion range from 10−6 to 10−8 cm2 s−1, whereas those for the TeO2/Te conversion range from 10−7 to 10−11 cm2 s−1 (Fig. S29, ESI†). We also collected the temperature dependence of Rct at the onset potential, using the Arrhenius equation to calculate the active energy barrier (Ea).41 The Ea was deduced from the fitted linear relationship between the logarithmic values of the reciprocal of the charge transfer resistance, which is closely related to the kinetic barrier of ion and electron transportation over active centers to participate in redox reactions. As shown in Fig. 4d, the Ea for the redox process in CsI electrolyte is 4.42 kJ mol−1, significantly lower than that in the blank electrolyte (6.19 kJ mol−1). Moreover, Cs2TeI6 exhibits a higher electrical conductivity of 4.21 × 10−8 S cm−1 compared to that of TeO2 (1.72 × 10−9 S cm−1) (Fig. S30, ESI†). | ||
| Fig. 4 (a) EIS measurement of the cells at OCV, and the equivalent circuit along with the fitted curves. (b) The charge transfer impedance at a different state of charge of the 4e process. (c) GITT profiles of the 4e process of Zn–Te cells with different electrolytes. (d) Arrhenius plots of Te electrode in different electrolytes. (e) The plating/stripping profile of Zn‖Zn symmetric cells at 1 mA cm−2 for 1 hour. (f) SEM images of a Zn anode after being plated at 1 mA h cm−2 in CsI electrolyte. | ||
The enhanced electrode kinetics can be attributed to the ion distribution at the electrode–electrolyte interface.42,43 A Zeta potential test shows that the surface potential of the KB/Te composite in the blank electrolyte was 7.51 mV, while it is −1.96 mV in the CsI electrolyte (Fig. S31, ESI†).44,45 This reduction suggests the enrichment of anions at the electrode interface. DFT modeling provides further insights into the surface ion distribution (Fig. S32, ESI†). The introduction of CsI in the electrolyte results in accumulation of I− anion on the Te (101) plane, as supported by its highest adsorption energy (Fig. S33, ESI†). This adsorption preference of I− anion results in an extended distance between Te and H2O (Fig. S34, ESI†), potentially mitigating the formation of oxides during Te oxidation.
We observed that CsI electrolyte also significantly influences the Zn anode electroplating process. In the blank electrolyte, the Zn anode exhibited an uneven deposition morphology (Fig. S35, ESI†), leading to pronounced dendrite formation and a reduced lifespan of Zn‖Zn symmetrical cell (Fig. 4e). In contrast, the cell with the CsI electrolyte demonstrated a dense plating of Zn with suppressed dendrite formation (Fig. 4f), sustaining a stable cycling performance for over 2000 h at 1 mA cm−2 and 1 mA h cm−2. Coulombic efficiency (CE) was evaluated in Zn‖Cu asymmetric cells. The Zn‖Cu cells with CsI electrolyte cycle stably for 100 cycles with an average CE of 99.2%, whereas it is only 97% for the blank electrolyte (Fig. S36, ESI†). While a higher current density usually results in faster failure of Zn metal anode, we collected the zinc anode morphology at high current (10 mA cm−2). The deposition of Zn in blank electrolyte results in a loose and disordered surface with abundant uneven Zn nanoflakes (Fig. S37a–c, ESI†), whereas a dense and smooth morphology was revealed in the CsI electrolyte (Fig. S37d–f, ESI†). The large Cs+ cation in the electrolyte is responsible for the enhanced Zn stability, which exerts a shielding effect through strong adsorption to promoting uniform deposition of Zn metal.46,47 Zeta potential tests of the blank, LiI, KI and CsI electrolytes further corroborate the strong adsorption of Cs+ cations (Fig. S38, ESI†). Moreover, the CsI electrolyte also contributed to a Zn corrosion, as evidenced by Tafel polarization curves in Zn‖Zn symmetric cells with different electrolytes (Fig. S39, ESI†).
The full cell performance
We tested the Zn–Te cell between 0.6–1.6 V by activating only the Te ⇌ Te4+ 4e conversion (Fig. 5a). The charging cutoff voltage of the cell with the CsI electrolyte was set to 1.2 V to avoid the iodine redox. The cell with CsI electrolyte delivers a higher discharge capacity of 818 mA h g−1 at a current density of 0.5 A g−1, while the cell with blank electrolyte only displays a reversible capacity of 600 mA h g−1. In addition, the polarization in the CsI electrolyte is only 50 mV, significantly lower than that in the blank electrolyte (400 mV). The 6e transfer processes were activated by decreasing the discharge cutoff voltage to 0.05 V (Fig. 5b). The successive redox of Cs2TeI6 ⇌ Te ⇌ ZnTe in CsI electrolyte results in a discharge capacity of 1248 mA h g−1, while it is only 864 mA h g−1 for the TeO2 ⇌ Te ⇌ ZnTe process in the blank electrolyte. Note that a poor cycling performance of the 6e process is possibly attributed to the sluggish reaction kinetics of the alloy process (Te ⇌ ZnTe),47 as also reflected by the fast decay of the cell being tested between 0.05–1 V (Fig. S40, ESI†).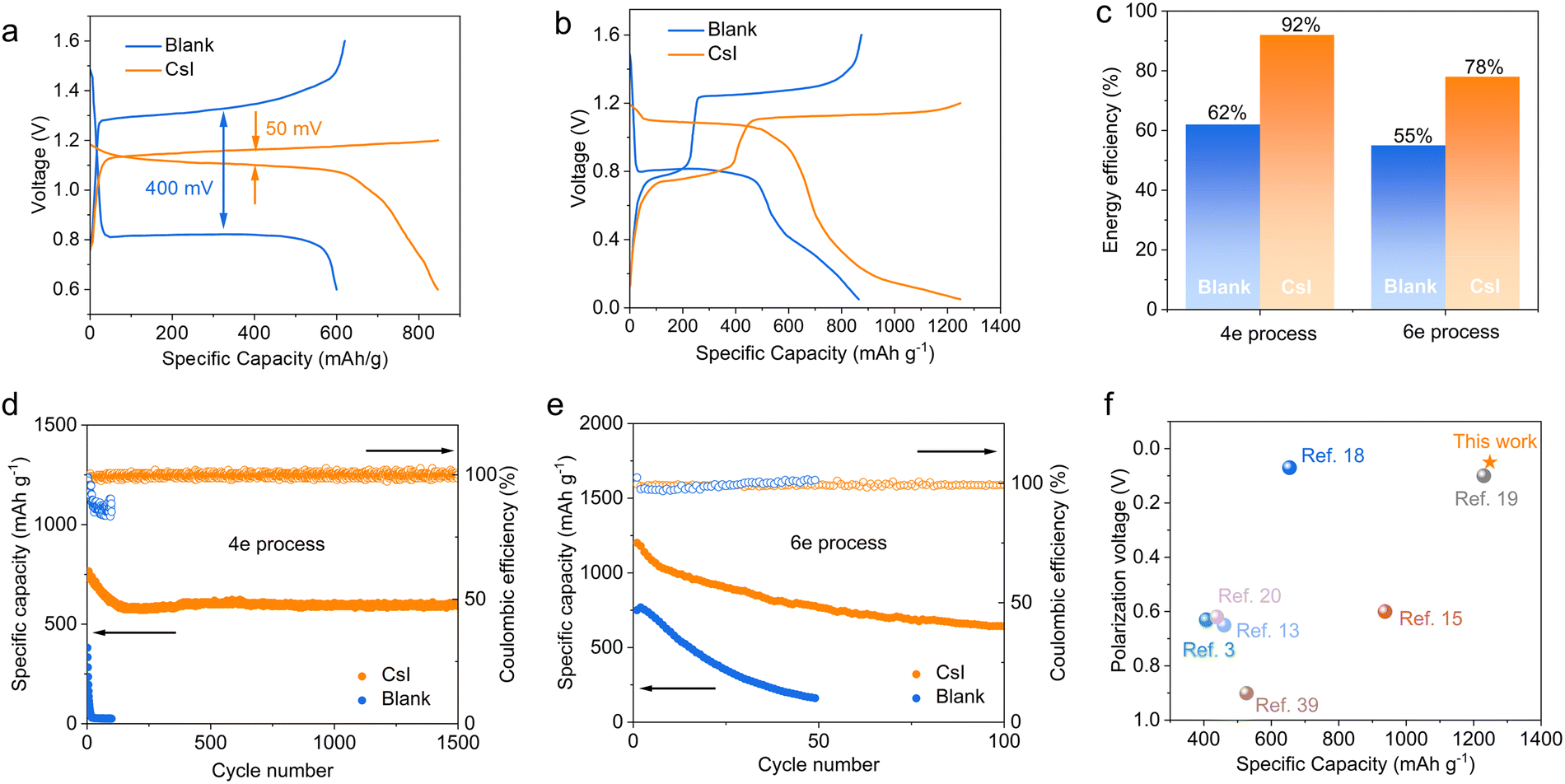 | ||
| Fig. 5 (a) Voltage profiles of Zn–Te cells with a 4e transfer process at 0.5 A g−1. (b) Voltage profiles of Zn–Te cells with a 6e process at 0.5 A g−1. (c) Energy efficiencies of Zn–Te cells with different electrolytes. (d) and (e) Long-term cycling performance of Zn–Te cells. (f) The comparison of discharge capacity and polarization of various Zn–Te cells. | ||
The discharge capacity of the Zn–Te battery in CsI electrolyte is higher than that of state-of-art aqueous Zn batteries, such as Zn–MnO2, Zn–I2 batteries (refer to the radar plots, Fig. S41, ESI†).38,48 Due to the decreased polarization, the CsI electrolyte significantly enhances the average energy efficiency of the Zn–Te battery from 62% to 92% for the 4e process and from 55% to 78% for the 6e process, respectively (Fig. 5c).
The long-term cycling performance of the Zn–Te cells was investigated in the lean electrolyte state (10 μL mg−1). The cell with CsI electrolyte exhibits a discharge capacity of 599 mA h g−1 after 1500 cycles for the 4e transfer conversion, with a superior capacity retention of 80%, in comparison to the cell with blank electrolyte, which decays rapidly only after a few cycles (Fig. 5d). This drastic discrepancy is attributed to the decoupling of the redox process from the participation of water by CsI electrolyte. Moreover, we demonstrate the cycling stability of the 6e process at a 100 μL mg−1 electrolyte/electrode mass ratio. The 6e CsI electrolyte can also be cycled for more than 100 cycles at a current density of 1 A g−1 (Fig. 5e). The proposed CsI electrolyte affords a competitive specific capacity with significantly decreased polarization compared with the conventional TeO2 redox path (Fig. 5f), while the previously established TeCl4 conversions are highly dependent on the inhibition of hydrolysis by concentrated electrolyte.18,19
Our further study demonstrated the versatility of the perovskite strategy for the multi-electron conversion of chalcogens. Analogous to Te, elemental Se can also be oxidized to positive valence by the perovskite strategy to suppress Se4+ hydrolysis and dissolution (Fig. S42a, ESI†). Moreover, we also found that a quaternary ammonium cation could take the place of Cs+ to assemble Se4+ and Te4+ into perovskite solids (Fig. S42b, c, and S43, ESI†), sustaining improved reaction kinetics with low voltage polarization. Future studies on precise controlling of the electrolyte composition and electrode architecture are required to bring the multi-electron chalcogen based batteries to a more practical level.
3. Conclusion
In this study, we successfully regulated the redox chemistry of tellurium (Te) in aqueous zinc-ion batteries by incorporating cesium iodide (CsI) into a low-concentration ZnSO4 electrolyte. Through a combination of theoretical calculations and experimental investigations, we demonstrated that the introduction of CsI facilitates the formation of a stable Cs2TeI6 phase during the oxidation process. This phase formation effectively suppresses the hydrolysis of Te4+ species and decouples the redox reactions from water participation, leading to significantly reduced polarization. The CsI-regulated Zn–Te battery exhibited a remarkable electrochemical performance, with high reversible capacities of 818 mA h g−1 for the 4-electron process and 1248 mA h g−1 for the 6-electron process. These capacities were achieved with substantially enhanced energy efficiencies of 92% and 78%, respectively. The 4-electron process cell also demonstrated exceptional cycling stability, retaining 80% of its capacity after 1500 cycles, highlighting the long-term durability of the system. Our comprehensive characterizations elucidated the crucial role of iodide anions in stabilizing the Cs2TeI6 phase and enriching the electrode interface, which in turn promotes favorable charge transfer kinetics. The findings emphasize that the addition of CsI not only enhances the electrochemical performance but also provides a pathway to overcome the challenges associated with Te hydrolysis and redox process polarization. This work paves a new way for the development of energy-dense Te-based cathode materials in aqueous batteries, underscoring the potential of CsI integration to advance the performance and stability of aqueous zinc-ion batteries.Author contributions
J. Li and X. Liang proposed and designed the idea. J. Li designed the experiments, electrochemical tests and wrote the paper. C. Lei performed the theoretical calculations. C. Xu and J. Li performed Raman measurements, T. Liu and P. Jiang assisted with the data analysis and characterization. All the authors contributed to the discussion and revision of the manuscript.Data availability
The data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key Research and Development Program of China (2019YFA0210600), National Natural Science Foundation of China (22479043), the Major Program of the Natural Science Foundation of Hunan Province (2021JC0006), and the Postdoctoral Fellowship Program of China Postdoctoral Science Fund (GZC20230760).References
- Y. Liang and Y. Yao, Designing modern aqueous batteries, Nat. Rev. Mater., 2022, 8(2), 109–122 CrossRef.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Active Materials for Aqueous Zinc Ion Batteries: Synthesis, Crystal Structure, Morphology, and Electrochemistry, Chem. Rev., 2020, 120(15), 7795–7866 CrossRef CAS.
- Z. Chen, Q. Yang, F. Mo, N. Li, G. Liang, X. Li, Z. Huang, D. Wang, W. Huang, J. Fan and C. Zhi, Aqueous Zinc–Tellurium Batteries with Ultra flat Discharge Plateau and High Volumetric Capacity, Adv. Mater., 2020, 32(42), 2001469 CrossRef CAS.
- W. Li, Y. Ma, P. Li, X. Jing, K. Jiang and D. Wang, Electrochemically Activated Cu2–xTe as an Ultra flat Discharge Plateau, Low Reaction Potential, and Stable Anode Material for Aqueous Zn-Ion Half and Full Batteries, Adv. Energy Mater., 2021, 11(42), 2102607 CrossRef CAS.
- W. Li, K. Wang and K. Jiang, A Low Cost Aqueous Zn–S Battery Realizing Ultrahigh Energy Density, Adv. Sci., 2020, 7(23), 2000761 CrossRef CAS PubMed.
- Z. Yang, B. Wang, Y. Chen, W. Zhou, H. Li, R. Zhao, X. Li, T. Zhang, F. Bu, Z. Zhao, W. Li, D. Chao and D. Zhao, Activating sulfur oxidation reaction via six-electron redox mesocrystal NiS2 for sulfur-based aqueous batteries, Natl. Sci. Rev., 2023, 10(6), nwac268 CrossRef CAS.
- H. Li, R. Meng, Y. Guo, B. Chen, Y. Jiao, C. Ye, Y. Long, A. Tadich, Q.-H. Yang, M. Jaroniec and S.-Z. Qiao, Reversible electrochemical oxidation of sulfur in ionic liquid for high-voltage Al−S batteries, Nat. Commun., 2021, 12(1), 5714 CrossRef CAS.
- P. F. Lang and B. C. Smith, Ionic radii for Group 1 and Group 2 halide, hydride, fluoride, oxide, sulfide, selenide and telluride crystals, Dalton Trans., 2010, 39(33), 7786–7791 RSC.
- S. Nanda, A. Bhargav and A. Manthiram, Anode-free, Lean-Electrolyte Lithium-Sulfur Batteries Enabled by Tellurium-Stabilized Lithium Deposition, Joule, 2020, 4(5), 1121–1135 CrossRef CAS.
- Z. Yu, W. Wang, Y. Zhu, W.-L. Song, Z. Huang, Z. Wang and S. Jiao, Construction of double reaction zones for long-life quasi-solid aluminum-ion batteries by realizing maximum electron transfer, Nat. Commun., 2023, 14(1), 5596 CrossRef CAS PubMed.
- Y.-H. Wang, X.-T. Li, W.-P. Wang, H.-J. Yan, S. Xin and Y.-G. Guo, Chalcogen cathode and its conversion electrochemistry in rechargeable Li/Na batteries, Sci. China: Chem., 2020, 63(10), 1402–1415 CrossRef CAS.
- X. Wang, L. Liu, Z. Hu, C. Peng, C. Han and W. Li, High Energy Density Aqueous Zinc-Chalcogen (S, Se, Te) Batteries: Recent Progress, Challenges, and Perspective, Adv. Energy Mater., 2023, 13(44), 2302927 CrossRef CAS.
- J. Wang, J. Du, J. Zhao, Y. Wang, Y. Tang and G. Cui, Unraveling H+/Zn2+ Sequential Conversion Reactions in Tellurium Cathodes for Rechargeable Aqueous Zinc Batteries, J. Phys. Chem. Lett., 2021, 12(41), 10163–10168 CrossRef CAS.
- Z. Chen, C. Li, Q. Yang, D. Wang, X. Li, Z. Huang, G. Liang, A. Chen and C. Zhi, Conversion-Type Nonmetal Elemental Tellurium Anode with High Utilization for Mild/Alkaline Zinc Batteries, Adv. Mater., 2021, 33(51), 2105426 CrossRef CAS PubMed.
- Z. Yan, J. Li, H. Liu, H. Zhang, S. Xi and Z. Zhu, A Reversible Six-Electron Transfer Cathode for Advanced Aqueous Zinc Batteries, Angew. Chem., Int. Ed., 2023, e202312000 CAS.
- F. Arab, M. Mousavi-Kamazani and M. Salavati-Niasari, Synthesis, characterization, and optical properties of Te, Te/TeO2 and TeO2 nanostructures via a one-pot hydrothermal method, RSC Adv., 2016, 6(75), 71472–71480 RSC.
- H. Wang, Y. Diao, M. Rubin, L. M. Santino, Y. Lu and J. M. D’Arcy, Metal Oxide-Assisted PEDOT Nanostructures via Hydrolysis-Assisted Vapor-Phase Polymerization for Energy Storage, ACS Appl. Nano Mater., 2018, 1(3), 1219–1227 CrossRef CAS.
- Z. Chen, S. Wang, Z. Wei, Y. Wang, Z. Wu, Y. Hou, J. Zhu, Y. Wang, G. Liang, Z. Huang, A. Chen, D. Wang and C. Zhi, Tellurium with Reversible Six-Electron Transfer Chemistry for High-Performance Zinc Batteries, J. Am. Chem. Soc., 2023, 145(37), 20521–20529 CrossRef CAS.
- J. Du, Y. Zhao, X. Chu, G. Wang, C. Neumann, H. Xu, X. Li, M. Löffler, Q. Lu, J. Zhang, D. Li, J. Zou, D. Mikhailova, A. Turchanin, X. Feng and M. Yu, A High-Energy Tellurium Redox-Amphoteric Conversion Cathode Chemistry for Aqueous Zinc Batteries, Adv. Mater., 2024, 2313621 CrossRef CAS PubMed.
- J. Qi, Y. Tang, Z. Feng, J. Yan, G. Liu, M. Ye, W. Du, Q. Yang, Y. Wei, Y. Zhang, Z. Wen, X. Liu and C. C. Li, Offense-Defense-Balanced Strategy Escorting Tellurium Oxidation Conversion towards Energetic and Long-Life Zn Batteries, Adv. Energy Mater., 2023, 14(6), 2303616 CrossRef.
- Y. Zou, T. Liu, Q. Du, Y. Li, H. Yi, X. Zhou, Z. Li, L. Gao, L. Zhang and X. Liang, A four-electron Zn-I2 aqueous battery enabled by reversible I−/I2/I+ conversion, Nat. Commun., 2021, 12(1), 170 CrossRef CAS.
- C. Xu, C. Lei, J. Li, X. He, P. Jiang, H. Wang, T. Liu and X. Liang, Unravelling rechargeable zinc-copper batteries by a chloride shuttle in a biphasic electrolyte, Nat. Commun., 2023, 14(1), 2349 CrossRef CAS PubMed.
- J. Xu and C. Wang, Perspective-Electrolyte Design for Aqueous Batteries: From Ultra-High Concentration to Low Concentration?, J. Electrochem. Soc., 2022, 169(3), 030530 CrossRef CAS.
- Y. Hu, Z. Liu, L. Li, S. Guo, X. Xie, Z. Luo, G. Fang and S. Liang, Reconstructing interfacial manganese deposition for durable aqueous zinc–manganese batteries, Natl. Sci. Rev., 2023, 10(10), nwad220 CrossRef CAS PubMed.
- H. Yang, W. Zhou, D. Chen, J. Liu, Z. Yuan, M. Lu, L. Shen, V. Shulga, W. Han and D. Chao, The origin of capacity fluctuation and rescue of dead Mn-based Zn-ion batteries: a Mn-based competitive capacity evolution protocol, Energy Environ. Sci., 2022, 15(3), 1106–1118 RSC.
- S. Cai, X. Chu, C. Liu, H. Lai, H. Chen, Y. Jiang, F. Guo, Z. Xu, C. Wang and C. Gao, Water-Salt Oligomers Enable Supersoluble Electrolytes for High-Performance Aqueous Batteries, Adv. Mater., 2021, 33(13), 2007470 CrossRef CAS PubMed.
- P. J. Hendra and Z. Jovi, The Raman spectra of some complex anions of formula [MX6]2− in the solid phase and in solution, where M = Se and Te; X = Cl, Br, and I, J. Chem. Soc. A, 1968, 600–602 RSC.
- A. G. Christy, S. J. Mills and A. R. Kampf, A review of the structural architecture of tellurium oxycompounds, Mineral. Mag., 2018, 80(3), 415–545 CrossRef.
- Y. Jiang, M. R. Leyden, L. Qiu, S. Wang, L. K. Ono, Z. Wu, E. J. Juarez-Perez and Y. Qi, Combination of Hybrid CVD and Cation Exchange for Upscaling Cs-Substituted Mixed Cation Perovskite Solar Cells with High Efficiency and Stability, Adv. Funct. Mater., 2017, 28(1), 1703835 CrossRef.
- Q. Huang, C. Tian, J. Lai, F. Qi, N. Zhang, J. Liu, Z. Liang, D. Wu and X. Tang, Lead-Free Cs2TeX6 (X = Cl, Br, and I) Perovskite Microcrystals with High Stability for Efficient Photocatalytic CO Reduction, Inorg. Chem., 2022, 61(36), 14447–14454 CrossRef CAS PubMed.
- E. Y. Peresh, V. I. Sidei and O. V. Zubaka, Phase relations in the systems A2TeI6-Tl2TeI6 (A = K, Rb, Cs) and A2TeBr6-A2TeI6 (A = K, Rb, Cs, Tl(I)), Inorg. Mater., 2005, 41(3), 298–302 CrossRef CAS.
- M. C. Folgueras, J. Jin, M. Gao, L. N. Quan, J. A. Steele, S. Srivastava, M. B. Ross, R. Zhang, F. Seeler, K. Schierle-Arndt, M. Asta and P. Yang, Lattice Dynamics and Optoelectronic Properties of Vacancy-Ordered Double Perovskite Cs2TeX6 (X = Cl–, Br–, I–) Single Crystals. The, J. Phys. Chem. C, 2021, 125(45), 25126–25139 CrossRef CAS.
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, The Mayer bond order as a tool in inorganic chemistry, J. Chem. Soc., Dalton Trans., 2001, 14, 2095–2108 RSC.
- J. Sun, W. Zheng, P. Huang, M. Zhang, W. Zhang, Z. Deng, S. Yu, M. Jin and X. Chen, Efficient Near-Infrared Luminescence in Lanthanide-Doped Vacancy-Ordered Double Perovskite Cs2ZrCl6 Phosphors via Te4+ Sensitization, Angew. Chem., Int. Ed., 2022, 61(26), e202201993 CrossRef CAS PubMed.
- T. Chang, Q. Wei, R. Zeng, S. Cao, J. Zhao and B. Zou, Efficient Energy Transfer in Te4+-Doped Cs2ZrCl6 Vacancy-Ordered Perovskites and Ultrahigh Moisture Stability via A-Site Rb-Alloying Strategy, J. Phys. Chem. Lett., 2021, 12(7), 1829–1837 CrossRef CAS.
- K. H. Stern and E. S. Amis, Ionic Size, Chem. Rev., 2002, 59(1), 1–64 CrossRef.
- G. H. Cartledge, Studies on the Periodic System. I. The Ionic Potential as a Periodic Function, J. Am. Chem. Soc., 2002, 50(11), 2855–2863 CrossRef.
- H. Pan, B. Li, D. Mei, Z. Nie, Y. Shao, G. Li, X. S. Li, K. S. Han, K. T. Mueller, V. Sprenkle and J. Liu, Controlling Solid–Liquid Conversion Reactions for a Highly Reversible Aqueous Zinc–Iodine Battery, ACS Energy Lett., 2017, 2(12), 2674–2680 CrossRef CAS.
- J. Li, L. Zhang, W. Xin, M. Yang, H. Peng, Y. Geng, L. Yang, Z. Yan and Z. Zhu, Rationally Designed ZnTe@C Nanowires with Superior Zinc Storage Performance for Aqueous Zn Batteries, Small, 2023, 19(52), 2304916 CrossRef CAS.
- M. D. Levi and D. Aurbach, Simultaneous Measurements and Modeling of the Electrochemical Impedance and the Cyclic Voltammetric Characteristics of Graphite Electrodes Doped with Lithium, J. Phys. Chem. B, 1997, 101(23), 4630–4640 CrossRef CAS.
- L. Peng, Z. Wei, C. Wan, J. Li, Z. Chen, D. Zhu, D. Baumann, H. Liu, C. S. Allen, X. Xu, A. I. Kirkland, I. Shakir, Z. Almutairi, S. Tolbert, B. Dunn, Y. Huang, P. Sautet and X. Duan, A fundamental look at electrocatalytic sulfur reduction reaction, Nat. Catal., 2020, 3(9), 762–770 CrossRef CAS.
- X. Xie, H. Fu, Y. Fang, B. Lu, J. Zhou and S. Liang, Manipulating Ion Concentration to Boost Two-Electron Mn4+/Mn2+ Redox Kinetics through a Colloid Electrolyte for High-Capacity Zinc Batteries, Adv. Energy Mater., 2021, 12(5), 2102393 CrossRef.
- Z. Liu, L. Li, L. Qin, S. Guo, G. Fang, Z. Luo and S. Liang, Balanced Interfacial Ion Concentration and Migration Steric Hindrance Promoting High-Efficiency Deposition/Dissolution Battery Chemistry, Adv. Mater., 2022, 34(40), 2204681 CrossRef CAS.
- H. Bustamante and G. Woods, Interaction of dodecylamine and sodium dodecyl sulphate with a low-rank bituminous coal, Colloids Surf., 1984, 12, 381–399 CrossRef CAS.
- M. Kosmulski and J. B. Rosenholm, High ionic strength electrokinetics, Adv. Colloid Interface Sci., 2004, 112(1–3), 93–107 CrossRef CAS.
- J. Zhan, W. Zhu, Y. Liu, M. Li, J. Zhao, H. Su, J. Ding, J. Sun and Y. Xu, Low-Cost, Safe, and Ultra-Long Cycle Life Zn-K Hybrid Ion Batteries, Adv. Funct. Mater., 2023, 33(38), 2301935 CrossRef CAS.
- H. Xu, W. Yang, H. Liu, M. Li, S. Gong, F. Zhao, C. Li, J. Qi, H. Wang, W. Peng, X. Fan and J. Liu, Boosting kinetics of tellurium redox reaction for high-performance aqueous zinc-tellurium batteries, Chem. Eng. J., 2023, 465, 142896 CrossRef CAS.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J.-G. Zhang, Dendrite-Free Lithium Deposition via Self-Healing Electrostatic Shield Mechanism, J. Am. Chem. Soc., 2013, 135(11), 4450–4456 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02916j |
| This journal is © The Royal Society of Chemistry 2024 |