
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-dissolving Fe2N6 site-based carbon fiber membranes for binder-free Zn–air batteries with a 200-day lifespan†
Zian
Xu‡
ac,
Jian
Zhu‡
a,
Jingze
Shao‡
d,
Yu
Xia
g,
Pengfei
Liu
 f,
Guangshe
Li
d,
Rouxi
Chen
*h,
Shaoqing
Chen
e,
Jiacheng
Wang
i,
Shi
Chen
*c,
Fuqiang
Huang
*b and
Hsing-Lin
Wang
*a
f,
Guangshe
Li
d,
Rouxi
Chen
*h,
Shaoqing
Chen
e,
Jiacheng
Wang
i,
Shi
Chen
*c,
Fuqiang
Huang
*b and
Hsing-Lin
Wang
*a
aDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen, 518055, P. R. China. E-mail: wangxl3@sustech.edu.cn
bState Key Lab of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: huangfq@mail.sic.ac.cn
cJoint Key Laboratory of the Ministry of Education, Institute of Applied Physics and Materials Engineering, University of Macau, 999078, Macao, SAR, P. R. China. E-mail: shichen@um.edu.mo
dState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun, 130026, P. R. China
eCollege of Energy, Soochow Institute for Energy and Materials Innovations, Jiangsu Provincial Key Laboratory for Advanced Carbon Materials and Wearable Energy Technologies, Soochow University, Suzhou, 215006, P. R. China
fInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, P. R. China
gDepartment of Materials and Environmental Chemistry, Stockholm University, Stockholm, Sweden
hSchool of Innovation and Entrepreneurship, Southern University of Science and Technology, Shenzhen, 518055, P. R. China. E-mail: chenrx@sustech.edu.cn
iSchool of Materials Science and Engineering, Taizhou University, Taizhou, 318000, Zhejiang, P. R. China
First published on 4th October 2024
Abstract
Durable and highly efficient electrocatalysts for the oxygen reduction reaction (ORR) are central to rechargeable Zn–air batteries (ZABs). The current best-performing ORR electrocatalysts are FeN4-based powder materials among the non-noble metals, but they still suffer from peeling off and demetallation during long-term device operation. Herein, we constructed an anti-dissolving structure of dual-atomic Fe sites modified with carbon holes and pyridinic-N on carbon fiber membranes (Fe2N6-CMPCFs) as binder-free cathodes via two-step NH3-assisted carbonization. Experimental and theoretical studies implied that the energy barrier of Fe dissolution is significantly higher in Fe2N6-CMPCFs (1.41 eV) compared to that of conventional non-defective FeN4 (0.94 eV), which can significantly inhibit the demetallation of Fe sites during long-term electrocatalysis. Thus, the Fe2N6-CMPCFs-based cathode enabled ZABs to operate over 200 days (record-breaking 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cycles) with a remarkable peak power density of 261.4 mW cm−2. Furthermore, structure analysis uncovered the anti-dissolving origin of Fe sites in Fe2N6-CMPCFs, which can be attributed to the enhanced orbital interaction (Fe–N and Fe–Fe) and electrostatic force between the Fe and N atoms. This work provides a valuable route to design anti-dissolving atomic sites and binder-free cathodes for sustainable electronic devices.
500 cycles) with a remarkable peak power density of 261.4 mW cm−2. Furthermore, structure analysis uncovered the anti-dissolving origin of Fe sites in Fe2N6-CMPCFs, which can be attributed to the enhanced orbital interaction (Fe–N and Fe–Fe) and electrostatic force between the Fe and N atoms. This work provides a valuable route to design anti-dissolving atomic sites and binder-free cathodes for sustainable electronic devices.
Broader contextZn–air batteries (ZABs) with a high theoretical energy density, low cost, and good safety have attracted considerable attention as sustainable energy devices for promising large-scale commercialization. However, the major challenge associated with ZABs is the development of highly efficient and durable electrocatalysts for the oxygen reduction reaction (ORR). In this case, the current best-performing electrocatalysts, such as the typical Fe single atom-based powder materials, still suffer from peeling off and demetallation during long-term device operation. Herein, we report an anti-dissolving structure of dual-atomic Fe sites modified with carbon holes and pyridinic-N on a carbon fiber membrane (Fe2N6-CMPCFs) as a freestanding cathode via two-step NH3-assisted carbonization. Experimental and theoretical results disclose that the energy barrier of Fe dissolution is significantly higher in Fe2N6-CMPCFs (1.41 eV) compared to that in conventional non-defective FeN4 (0.94 eV), thus inhibiting the demetallation of Fe during long-term electrocatalysis. In addition, the aerophilic membrane-based cathodes can avoid the possible peeling off and cathodic flooding. As a result, it exhibits excellent ORR stability for 10000 cycles and a record-breaking lifespan of ZABs of over 200 days (14500 cycles) with negligible efficiency decay (∼2.5%). This work provides a valuable pathway to design anti-dissolving atomic sites and binder-free cathodes for renewable electronic devices.
|
Background
The electrocatalytic oxygen reduction reaction (ORR) plays a vital role in a range of energy conversion and storage technologies, such as fuel cells and rechargeable metal–air batteries.1–3 Zn–air batteries (ZABs) with a high theoretical energy density, low cost, and good safety have attracted considerable attention.4 However, a major challenge associated with the cathodes of ZABs is their sluggish kinetics and non-ideal stability of oxygen reduction, which involves a four proton-coupled electron transfer step.5,6 Thus, to date, platinum (Pt)-based catalysts have been mainly used to catalyze the ORR at the cathode, but their high cost, natural scarcity and poor durability severely impede the sustainable and commercial deployment of ZABs.7Single-atom catalysts (SACs) based on transition metals (e.g., Fe, Co, Ni, and Mn) have been widely used as ORR electrocatalysts owing to their maximum atomic utilization, tunable electronic structure, superior intrinsic activity, and low cost;8,9 thus, they are regarded as the most promising alternatives to noble-metal-based catalysts.10 Among them, Fe single atoms with four N coordinates in a carbon matrix (Fe–N4/C) are known to possess the highest intrinsic activity.11,12 Recently, to further enhance the ORR activity of Fe–N4/C, the introduction of heteroatoms (such as S and P) or metal atoms (such as Co and Mn) has been widely reported.13–16 Yang et al. reported the crucial role of S doping in regulating Fe–N4/C catalysts, which exhibited a positive half-wave potential of 0.89 V.17 Liu et al. proposed the preparation of contiguous dual single atom FeN4 and CoN4 sites embedded in N-doped graphitic carbon, demonstrating a remarkable half-wave potential of 0.877 V.18 However, despite recent advances, “big data” analysis revealed that the half-wave potential of most Fe–N4/C catalysts is below 0.90 V.19,20 Moreover, most reports on Fe–N4/C catalysts only focused on boosting their electrocatalytic activity, which cannot achieve long-term stability for practical applications such as ZABs.21 To date, studies on promoting the stability of atomic sites during catalytic reactions are still lacking.22 Therefore, it is highly desirable to enhance the inherent activity and durability of Fe–N4/C by further optimizing its electronic structure.
The rational design of carbon supports is another significant factor in improving the ORR activity and battery performance. To date, most SACs are fabricated in powder form, and then sprayed on carbon cloth or Ni foam as the cathode in ZABs. However, this method often causes the catalysts to peel off during the battery operation, which severely affects the long-term stability and lifespan of batteries.17 In addition, the use of extra binder (e.g., Nafion) for powder-like catalysts in the preparation of electrodes also leads to performance degradation due to their inactivity, insulation, and high resistance to oxygen transport.17,23 Therefore, designing binder-free cathodes with integrated catalysts is regarded as a promising strategy to address this issue.24,25 In this case, electrospinning carbon fiber membranes immobilized with single-atom moieties as cathodes have emerged as a revolutionary frontier in the field of sustainable ZABs. This innovative approach not only facilitates electron transport but also enhances electrolyte infiltration and gas diffusion to the active sites.26,27 Furthermore, with the increasing demand for wearable and portable electronic devices, the inherent bendability of membranes endows them with great potential to be used as flexible electrodes.28–30 However, despite the above-mentioned merits, the insufficient exposure of active sites is the main limitation of nanofibrous catalysts.31 Hence, increasing the porosity of carbon fibers is an effective solution to facilitate the exposure of the active sites.32–34 Currently, the main strategy is to adopt polyacrylonitrile (PAN) as the fiber precursor and mixing it with metal–organic frameworks (MOFs) to prepare porous carbon fibers but this will cause severe degradation of the flexibility and carbon yield.24 Therefore, it is imperative to investigate and explore alternative polymer precursors as potential sources for the preparation of porous carbon fibers.
Herein, we report a two-step carbonization strategy with the assistance of NH3 to construct anti-dissolving Fe2N6 sites on conjugated microporous polyimide (CMP)-derived carbon fibers (Fe2N6-CMPCFs) as freestanding cathodes for durable ZABs. This integrated cathode can avoid the possible peeling off from the substrate observed for powder-based catalysts and promote electron transfer to the active sites along the carbon fibers. In addition, our functionalized carbon fibers based on chemically modified polymers possess a large specific surface area of 1062 m2 g−1, high porosity, and rich pyridinic-N content. These characteristics play a pivotal role in exposing and promoting the Fe2N6 sites for electrocatalytic reaction. Thus, the as-synthesized Fe2N6-CMPCFs catalyst exhibited an exceptional positive half-wave potential of 0.91 V, outstanding accelerated kinetics (51.1 mV dec−1), and excellent ORR stability for 10000 cycles. When assembled as a freestanding cathode in liquid-state ZABs, it performed with an outstanding peak power density of 261.4 mW cm−2 and a record lifespan of over 200 days (14500 cycles) with an ultra-stable round-trip efficiency. In addition, its quasi-solid-state ZAB also afforded a remarkable peak power density (203.7 mW cm−2), demonstrating great potential for wearable energy devices. The theoretical simulation indicated that the dissolution energy barrier of the Fe sites in the dual-atomic models is significantly increased compared to that in the single-atomic structures, which can be ascribed to the enhanced orbital interaction (Fe–N and Fe–Fe bond) and electrostatic strength (Fe and N atoms). Besides, the modification of carbon holes and pyridinic-N also contributed to alleviating the demetallation of the Fe sites in Fe2N6-CMPCFs. This anti-dissolving structure and cathode design concept can promote the application of single atoms in the field of energy conversion and storage.
1. Results and discussion
The strategy for the synthesis of the Fe2N6-CMPCF membrane catalyst is presented in Fig. 1a. The precursor solution, containing two monomers, 1,3,5-tris(4-aminophenyl)benzene (TAPB) and naphthalene-1,4,5,8-tetracarboxylic acid (NTCA), generated the carboxylate ammonium salt after stirring. The template polymer, polyvinyl pyrrolidinone (PVP), was added to enhance the viscosity of the monomer solution for electrospinning. The specific preparation process included electrospinning, polycondensation and two-step carbonization. Fig. S1a (ESI†) illustrates the result of electrospinning the aforementioned solution, generating a yellow membrane with fibers with a diameter of around 950 nm. Fig. S1b (ESI†) shows that the heat treatment at 420 °C led to a cross-linked polycondensation reaction between NTCA and TAPT. This reaction produced the dark-brown CMP membranes with the decreased fiber diameter of 855 nm (Fig. S1b and S2, ESI†). Simultaneously, PVP underwent decomposition. Finally, the CMP membranes were subjected to carbonization at 900 °C, transforming into porous carbon fibers (CMPCFs), as shown in Fig. S1c (ESI†).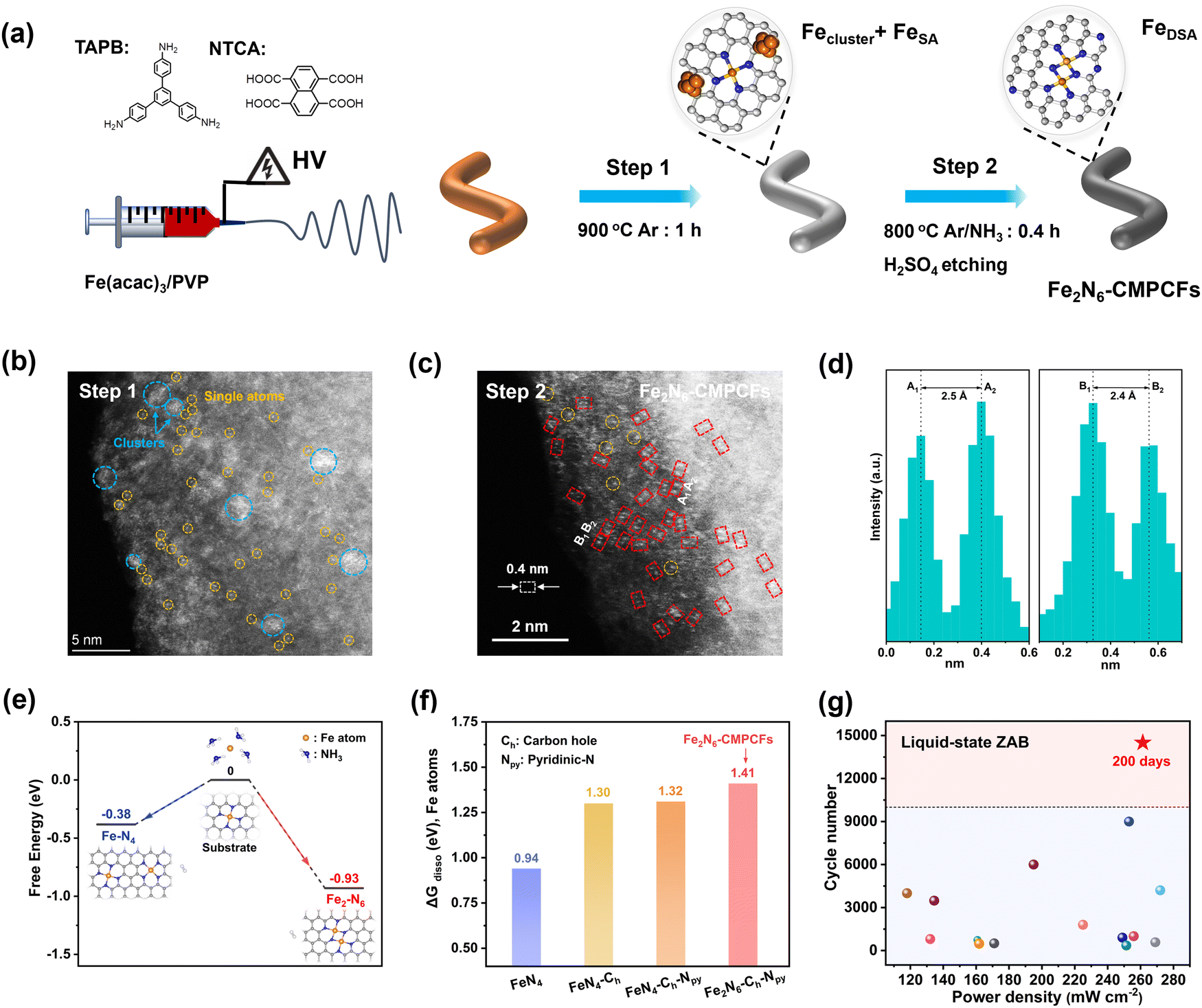 | ||
| Fig. 1 Synthesis strategies and design conception of constructing homonuclear dual Fe single atom-based carbon fibers (Fe2N6-CMPCFs) with enhanced anti-dissolving ability. (a) Schematic of the preparation of Fe-doped porous carbon fibers by electrospinning a mixture of TAPB, NTCA, Fe(acac)3, and PVP, followed by two-step carbonization. PVP was used to enhance the viscosity of the monomer solution for electrospinning. (b) Aberration-corrected high angle annular dark-field scanning TEM (AC-HAADF-STEM) image for the prepared catalysts after first-step carbonization. The blue and yellow circles highlight the coexistence of Fe clusters and single atoms in this step, respectively. (c) AC-HAADF-STEM image of the resultant Fe2N6-CMPCFs after second-step pyrolysis assisted by NH3. The dashed red rectangles show the homonuclear dual Fe single atoms. (d) Corresponding Z-contrast analysis of region A and region B in (c). (e) Theoretical simulation of Fe2N6 formation under an NH3 atmosphere. The blue, gray, white, and orange balls indicate N, C, H and Fe atoms, respectively. (f) Dissolution energy of one Fe atom (ΔGdisso) in different FeN4 and Fe2N6-based structures. (g) Performance comparison of liquid-state ZABs using Fe2N6-CMPCFs and other reported electrocatalysts, as listed in Table S12 (ESI†). | ||
The Fourier transform infrared spectra (FTIR) curves in Fig. S3 (ESI†) distinctly display peaks assigned to the amino group (3200 to 3500 cm−1) of TAPB and the carboxyl group of NTCA (broad peak at 3070 cm−1). Both peaks disappeared after the cross-linking reaction to form the CMP membranes. In addition, the three characteristic peaks of the imide unit are located at 1709, 1666, and 1343 cm−1 for CMP, among which the former two peaks are attributed to the asymmetric/symmetric vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in the six-membered imide rings and the latter peak corresponds to the stretching vibration of the formed C–N–C moiety. Simultaneously, the two characteristic peaks (2980 cm−1 for –CH2–, and 1661 cm−1 for CO) derived from PVP also disappeared due to its thermal decomposition.35 The chemical structure of CMP was also confirmed by solid-state 13C nuclear magnetic resonance spectroscopy (NMR), as shown in Fig. S4 (ESI†). The peak at 162 ppm originates from the carbonyl carbon of the imide ring and the two distinct peaks at 142 and 128 ppm are attributed to the naphthalene and phenyl carbon, originating from the incorporated building units of NTCA and TAPB, respectively. The thermogravimetric analysis (TGA) of TAPB/NTCA/PVP, as shown in Fig. S5 (ESI†), illustrates a high carbon yield of 46% at 900 °C. The resultant CMPCFs exhibited excellent flexibility, enduring over 1000 bending cycles (Fig. S6, ESI†), and possessed the good tensile strength of 3.2 MPa (Fig. S7, ESI†). It is significant to note that both CMP and CMPCFs exhibited a dominant pore size distribution of 1–2 nm, as shown in Fig. S8a (ESI†), and CMPCFs stand out with an impressive specific surface area of 1062 m2 g−1, as seen in Fig. S8b (ESI†). The micropore-hosted single atoms near the carbon edge are widely recognized as the primary active sites in catalytic systems.31 Additionally, the high porosity of carbon fibers can spatially suppress Fe aggregation and shorten the ion/electron transport pathways.
O groups in the six-membered imide rings and the latter peak corresponds to the stretching vibration of the formed C–N–C moiety. Simultaneously, the two characteristic peaks (2980 cm−1 for –CH2–, and 1661 cm−1 for CO) derived from PVP also disappeared due to its thermal decomposition.35 The chemical structure of CMP was also confirmed by solid-state 13C nuclear magnetic resonance spectroscopy (NMR), as shown in Fig. S4 (ESI†). The peak at 162 ppm originates from the carbonyl carbon of the imide ring and the two distinct peaks at 142 and 128 ppm are attributed to the naphthalene and phenyl carbon, originating from the incorporated building units of NTCA and TAPB, respectively. The thermogravimetric analysis (TGA) of TAPB/NTCA/PVP, as shown in Fig. S5 (ESI†), illustrates a high carbon yield of 46% at 900 °C. The resultant CMPCFs exhibited excellent flexibility, enduring over 1000 bending cycles (Fig. S6, ESI†), and possessed the good tensile strength of 3.2 MPa (Fig. S7, ESI†). It is significant to note that both CMP and CMPCFs exhibited a dominant pore size distribution of 1–2 nm, as shown in Fig. S8a (ESI†), and CMPCFs stand out with an impressive specific surface area of 1062 m2 g−1, as seen in Fig. S8b (ESI†). The micropore-hosted single atoms near the carbon edge are widely recognized as the primary active sites in catalytic systems.31 Additionally, the high porosity of carbon fibers can spatially suppress Fe aggregation and shorten the ion/electron transport pathways.
To impart ORR electrocatalytic activity, Fe precursors were introduced and converted to homonuclear dual Fe single atoms through the two-step NH3-assisted carbonization (Fig. 1b). The aberration-corrected high angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images in Fig. 1b and Fig. S9 (ESI†) confirm the coexistence of Fe single atoms and clusters in the sample after the first calcination step at 900 °C in an Ar atmosphere. However, after the second pyrolysis with NH3 treatment and acid etching, the Fe aggregates disappeared and all the elements (iron, nitrogen, and carbon) were homogeneously distributed throughout the fiber structure, as shown in Fig. S10 and S11 (ESI†). Notably, the AC-HAADF-STEM image in Fig. 1c demonstrates that numerous bright dots formed in pairs (highlighted by red rectangles) were detected and their average interatomic distance is in the range of 2.4 to 2.5 Å based on the Z contrast analysis (Fig. 1d and Fig. S12, ESI†). The thorough statistical analysis indicated that the dual-atomic sites occupy 82% among the identifiable bright spots in the resultant Fe2N6-CMPCFs (Fig. S13, ESI†). This formation of structure is mainly attributed to the introduction of the NH3 atmosphere, which could thermally transport the volatile Fe atoms from the Fe nanoparticles to bond with the isolated Fe single atoms, forming dual-atomic Fe sites on the carbon fibers.36,37 This formation mechanism was also verified by DFT simulation, as seen in Fig. 1e. The free energy diagram indicates that the mobile Fe atom under the NH3 atmosphere tended to form the Fe2N6 model more easily (ΔG = −0.93 eV) by coupling with the atomic Fe atom on the carbon substrate instead of forming new isolated Fe–N4 sites (ΔG = −0.38 eV). Furthermore, the content of metal loading also played a significant role in forming the dual single atoms. By adjusting the Fe contents in the precursor, two other types of Fe-doped CMPCFs could be prepared, i.e., FeN4-CMPCFs with Fe single atoms and Fe NPs-CNMCFs with Fe nanoparticles.38 The theoretical calculation indicated that the homonuclear Fe2N6 sites in Fe2N6-CMPCFs possess a highly superior anti-dissolving capacity to the conventional FeN4 sites, especially after decorating with carbon holes and pyridinic N (Fig. 1f and Fig. S14, Table S1, ESI†). This property can effectively prevent the demetallation of the metal sites from the carbon host. Consequently, when it was assembled as a freestanding air-electrode in practical liquid-state ZABs, it exhibited excellent cycling durability to a record-breaking 200-day lifespan (Fig. 1g).
Comprehensive characterization was further performed to verify the structure of Fe2N6-CMPCFs and other samples for comparison. The X-ray diffraction (XRD) patterns of FeN4-CMPCFs and Fe2N6-CMPCFs, as shown in Fig. S15 (ESI†), only display two broad peaks, which correspond to the graphitic carbon. No peaks ascribed to Fe particles were observed, indicating the high dispersion of Fe atoms. Alternatively, with a continuous increase in the Fe content, the emergence of a sharp graphitic peak at 26° and other very small peaks indicated the formation of Fe nanoparticles together with the graphitic carbon structure in Fe NPs-CMPCFs. Among them, the appearance of sharp graphitic peak can be attributed to the formation of ordered graphitic layers catalyzed by Fe nanoparticles.39 The intensity ratio (ID/IG) between the D and G bands in the Raman spectra further confirmed that the excess Fe dopant facilitated the formation of graphitic layers (Fig. S16, ESI†). The high-resolution TEM and HAADF-STEM images show that no metal aggregation could be detected in FeN4-CMPCFs, similar to Fe2N6-CMPCFs (Fig. S17, ESI†). However, the AC-HAADF-STEM result suggested that most of the bright dots (highlighted by orange circles) are isolated in FeN4-CMPCFs (Fig. S18, ESI†), which is different from that in Fe2N6-CMPCFs. In the case of Fe NPs-CMPCFs, we could distinctly observe the metal aggregation with the d-spacing of 0.206 nm, corresponding to the (110) facet of metallic Fe.40 This result confirmed the existence of Fe nanoparticles in Fe NPs-CMPCFs (Fig. S19, ESI†). The content of Fe loading in FeN4-CMPCFs, Fe2N6-CMPCFs, and Fe NPs-CMPCFs catalysts was determined to be 1.5, 2.6 and 3.9 wt%, respectively, by inductively coupled plasma-optical emission spectroscopy (ICP-OES), as shown in Table S2 (ESI†).
X-ray photoelectron spectroscopy (XPS) was used to investigate the element composition on the surface of the catalysts (Fig. S20–S22, ESI†). It was found that the Fe2N6-CMPCF catalyst exhibited a distinct intense peak at 399.4 eV, which is attributed to the contribution of Fe–N and pyrrolic N.41,42 This peak showed a significantly higher percentage compared to the other contrast samples (Fig. 2a), indicating the formation of an increased number of Fe–N active sites in Fe2N6-CMPCFs. Additionally, the high-resolution Fe 2p spectra of FeN4-CMPCFs and Fe2N6-CMPCFs could be well-fitted with Fe2+ and Fe3+ species. In contrast, the presence of metallic iron Fe0 could be observed at 707.3 eV for Fe NPs-CMPCFs.43,44 The content of different elements on the catalyst surface is also summarized in Table S3 (ESI†). X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were conducted to determine the chemical state and coordination environment of the Fe atoms in FeN4-CMPCFs and Fe2N6-CMPCFs. As shown in XANES (Fig. 2b), the absorption edge position of both FeN4-CMPCFs and Fe2N6-CMPCFs is located between the standard Fe foil and Fe2O3, which reveals that the valence of Fe is between 0 and +3.45,46 By comparing the absorption edge positions of FeN4-CMPCFs and FePc, a positive energy shift was observed in Fe2N6-CMPCFs. This shift suggests a higher proportion of high valence Fe in Fe2N6-CMPCFs, consistent with previous reports.47 Furthermore, the EXAFS spectra in Fig. 2c reveal that compared to the planar Fe–N4, the Fe–N peak in Fe2N6-CMPCFs became broader and exhibited a negative shift of 0.03 Å. The main reason for this is that unlike the typical D2h symmetry of Fe–N4, the asymmetric dual-atomic Fe2N6 sites result in electron redistribution, thus existing two different Fe–N bond lengths. In addition, the peak position of Fe–Fe in Fe2N6-CMPCFs (2.4 Å) showed a positive shift compared to that of Fe–Fe in the Fe foil (2.2 Å), demonstrating that the existence of Fe–Fe bonds in Fe2N6-CMPCFs did not originate from Fe clusters. The wavelet transform contour plots (Fig. 2d) confirmed the predominant Fe–N coordination in both FeN4-CMPCFs and Fe2N6-CMPCFs, with the maximum intensity at approximately 4.8 Å. To unveil the detailed coordination structure of Fe atoms, we fitted the Fe R-space EXAFS spectra of the FeN4-CMPCF and Fe2N6-CMPCF catalysts, as shown in Fig. 2e and f, respectively. The fitting result exhibited that the Fe–N bond length of FeN4-CMPCFs is 1.98 ± 0.01 Å with an N coordination number of 4.0 ± 0.2, which is consistent with the standard Fe–N4 model (Table S4, ESI†). Nevertheless, two different bond lengths of Fe–N (Fe–N1 and Fe–N2) existed in Fe2N6-CMPCFs. In this case, the bridging bond (Fe–N2) in the Fe2N6 structure possessed a shorter bond length of 1.89 Å. Furthermore, the fitting results also demonstrated a significantly lower coordination number (CN:1.0 ± 0.1) and a longer average bond length (R: 2.55 ± 0.01Å) of Fe–Fe in Fe2N6-CMPCFs, which is different with the coordination environment of Fe–Fe in Fe foil (CN: 8, R: 2.47 ± 0.01Å). The main reason for this is that the Fe atoms are separated by two N atoms in the Fe2N6 model, thus exhibiting a longer Fe–Fe distance than that in conventional Fe nanoparticles.38 These results not only verify the existence of the Fe2N6 structure but also exclude the presence of Fe clusters in Fe2N6-CMPCFs. Based on the aforementioned results, it can be reasonably concluded that the main coordination structure in FeN4-CMPCFs consists of isolated Fe–N4 single atoms, while Fe2N6-CMPCFs consist of homonuclear Fe2–N6 dual single atoms.
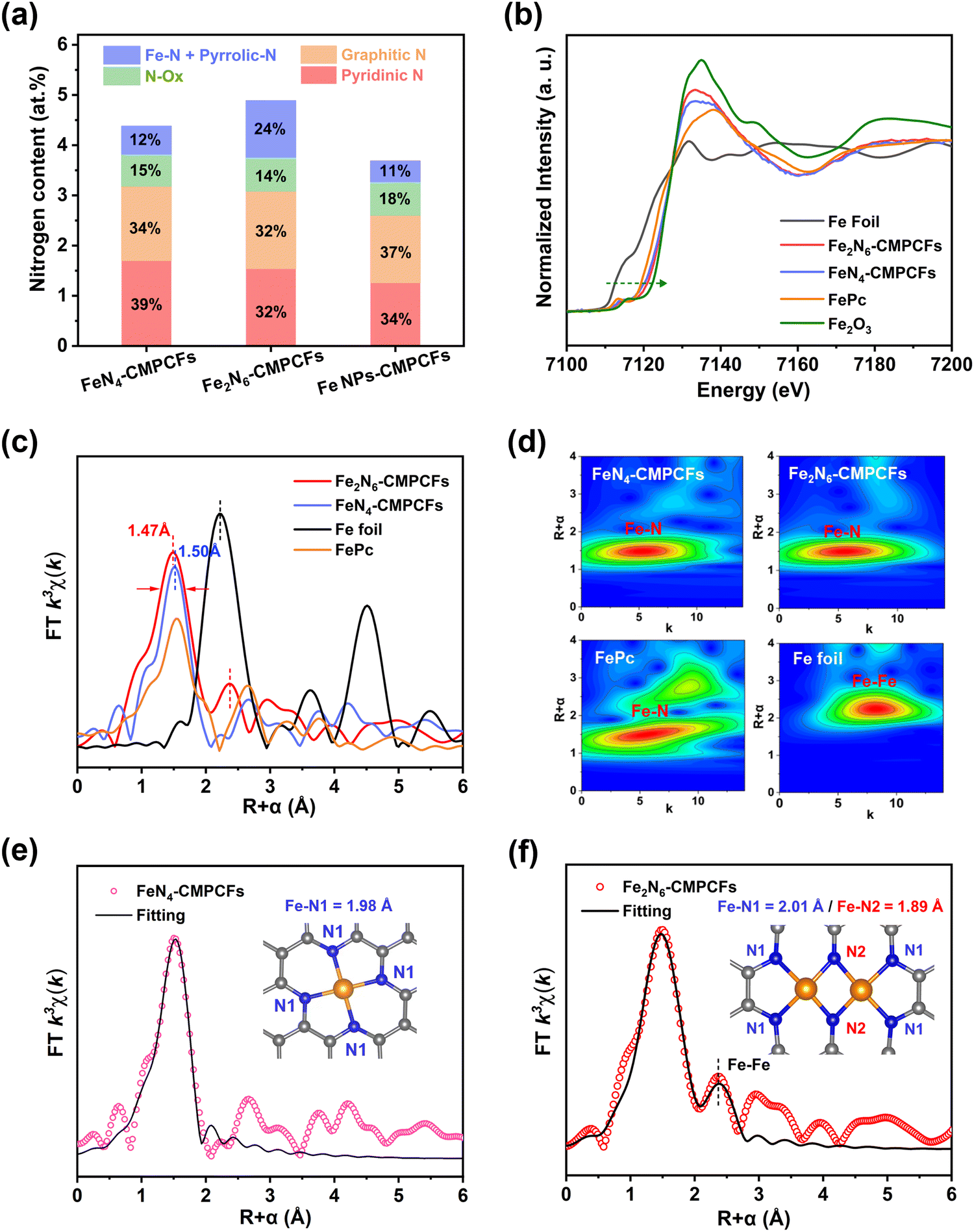 | ||
| Fig. 2 Structural analysis of Fe2N6-CMPCFs and the control samples. (a) Nitrogen contents and ratios of different nitrogen species in Fe2N6-CMPCFs, FeN4-CMPCFs and Fe NPs-CMPCFs. (b) X-ray absorption near-edge structure (XANES) spectra of the Fe K-edge of FeN4-CMPCFs, Fe2N6-CMPCFs, Fe foil, FePc, and Fe2O3. (c) Fourier transforms and (d) wavelet transforms of the extended X-ray absorption fine structure (EXAFS) spectra of the different samples. Corresponding fitting curve of the Fourier-transformed EXAFS spectra of (e) FeN4-CMPCFs and (f) Fe2N6-CMPCFs. The insets are the models of the deductive Fe–N4 and Fe2–N6 structure. Fe–N1 and Fe–N2 represent the existence of two different bond lengths of Fe–N in Fe2N6-CMPCFs. The blue, gray, and yellow balls indicate N, C, and Fe atoms, respectively. | ||
Considering the potential application of dual-single-atomic structures with homonuclear metallic center, the ORR performance of the catalyst was further assessed in 0.1 M KOH solution using a rotating disk electrode (Fig. S23, ESI†). The cyclic voltammetry (CV) curves of Fe2N6-CMPCFs and the control samples exhibit significant reduction peaks in the O2-saturated rather than N2-saturated electrolyte (Fig. S24, ESI†), indicating the advanced oxygen reduced activity. As shown in Fig. 3a and b, the Fe2N6-CMPCF catalyst exhibited an outstanding onset potential of 1.004 V and a positive half-wave potential of 0.91V vs. RHE (reversible hydrogen electrode), outperforming the commercial 20% Pt/C (0.975 V/0.863 V), FeN4-CMPCFs (0.955 V/0.882 V) and Fe NPs-CMPCFs (0.945 V/0.859 V), respectively. In addition, the catalyst without NH3 treatment in the second-step pyrolysis (Fe-CMPCFs-Ar) displayed inferior catalytic activity (Fig. S25, ESI†). The ORR kinetics of the different catalysts was further evaluated by the Tafel plot, as shown in Fig. S26 (ESI†). The Fe2N6-CMPCF catalyst displayed the smallest Tafel slope of 51.1 mV dec−1, demonstrating more favorable reaction kinetics. To estimate the four-electron ORR selectivity, the transferred electron number and produced H2O2 yield were studied by rotating ring disk electrode (RRDE) measurements. In the voltage range of 0.3–0.8 V, the Fe2N6-CMPCF catalyst consistently exhibited a low H2O2 yield of approximately 5%, as depicted in Fig. 3c. Furthermore, this yield is consistent with an average electron transfer number of 3.9, indicating the predominant four-electron transfer pathway. This result was further verified by the Koutecky–Levich (K–L) plots at various potentials, which can determine the electron transfer number according to the LSV curves at different rotating speeds from 400 to 2025 rpm (Fig. S27, ESI†). Based on the K–L equation, the electron transfer number of Fe2N6-CMPCFs was also calculated to be ≈ 4, which is consistent with the RRDE measurement. In addition, the kinetic current density (Jk) of the Fe2N6-CMPCF catalyst is about 65.1 mA cm−2 at 0.85 V, which is 8.3- and 15.5-times higher than that of FeN4-CMPCFs (7.8 mA cm−2) and Fe NPs-CMPCFs (4.2 mA cm−2), respectively, suggesting the enhanced reaction kinetics of Fe2N6-CMPCFs for ORR (Fig. S28, ESI†). Electrochemical impedance spectroscopy (EIS) was also performed to investigate the electrocatalytic kinetics and interface reactions in ORR. As illustrated in Fig. S29 (ESI†), the Fe2N6-CMPCF catalyst possessed the smallest semicircle, further demonstrating its superior reaction kinetics and charge transfer resistance of ORR.
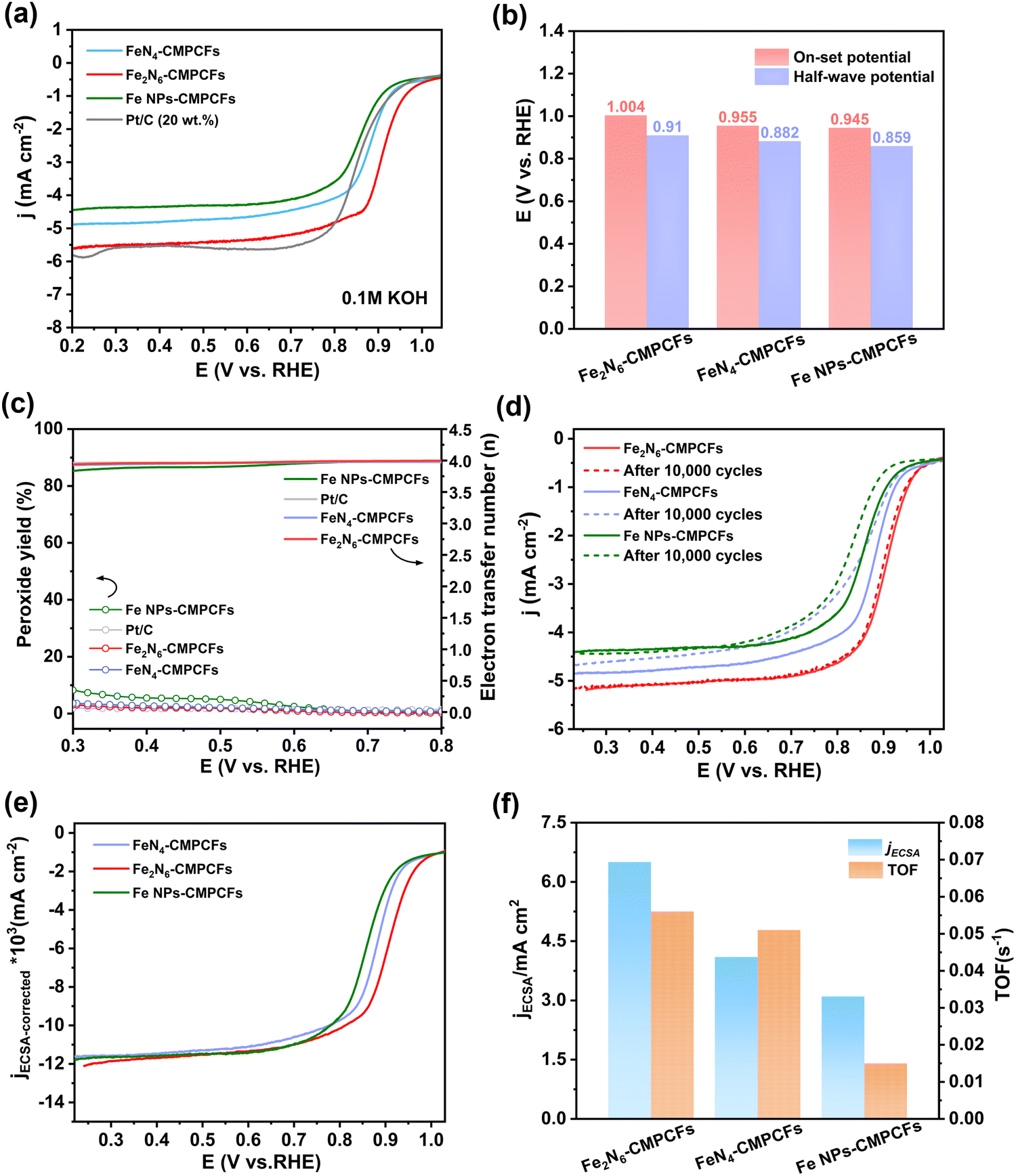 | ||
| Fig. 3 ORR activity evaluation Fe2N6-CMPCFs and the control samples. (a) Linear sweep voltammetry (LSV) curves at 1600 rpm for different catalysts in 0.1 M KOH. (b) Comparison of on-set and half-wave potential of the different samples. (c) By-product peroxide yield (H2O2%) and electron transfer number for Fe2N6-CMPCFs, FeN4-CMPCFs, Fe NPs-CMPCFs and Pt/C. (d) LSV curves after accelerated durability test (ADT) by 10000 cycles of measurements for Fe2N6-CMPCFs, FeN4-CMPCFs, and Fe NPs-CMPCFs. (e) ECSA-normalized LSV curves for Fe2N6-CMPCFs, FeN4-CMPCFs, and Fe NPs-CMPCFs. (f) Comparison of current density and turnover frequency (TOF) at 0.9 V vs. RHE for Fe2N6-CMPCFs, FeN4-CMPCFs, and Fe NPs-CMPCFs. | ||
Stability is another significant factor to evaluate the practical application of catalysts. After a fast accelerated durability test (ADT) of 10000 cycles for the Fe2N6-CMPCF catalyst, the decline in its ORR activity was negligible (Fig. 3d). Even after 30000 cycles, the decrease in its half-wave potential was still very slight (ΔE = 3 mV), as shown in Fig. S30 (ESI†), implying its outstanding stability. By contrast, nearly 20 mV decay of half-wave potential was seen for FeN4-CMPCFs and Fe NPs-CMPCFs after 10000 cycles. Additionally, a chronoamperometry test (i–t) was also conducted to assess the long-term stability of the catalysts. As shown in Fig. S31 (ESI†), Fe2N6-CMPCFs maintained 98% of their original current density after 15 h, which is superior to that of Pt/C (65%). After injecting methanol into the electrolyte, almost no disturbance in the i–t curve could be found for Fe2N6-CMPCFs compared to commercial Pt/C (Fig. S32, ESI†). These results confirm the excellent ORR stability and methanol tolerance of Fe2N6-CMPCFs.
To estimate the electrochemical active surface areas (ECSAs), we conducted a CV test to calculate the double layer capacitance values (Cdl), as shown in Fig. S33 (ESI†). The Cdl of the Fe2N6-CMPCF catalyst is 17.5 mF cm−2, which is higher than that of the FeN4-CMPCF (16.6 mF cm−2) and Fe NPs-CMPCF (14.8 mF cm−2) catalysts. As expected and shown in Fig. S34 (ESI†), Fe2N6-CMPCFs possessed the largest ECSA of 438 cm2, corresponding to more accessible active sites in Fe2N6-CMPCFs. The ECSA-corrected linear sweep voltammetry (LSV) curves in Fig. 3e confirm that the Fe2–N6 sites possess superior intrinsic catalytic activity compared to the other catalysts with isolated Fe–N4 and Fe nanoparticles. Moreover, the Fe2–N6 sites showed the highest turnover frequency (TOF) and ECSA-corrected current density at 0.9 V vs. RHE, which also revealed its excellent intrinsic ORR activity (Fig. 3f).
Given that the cathode of ZABs also needs OER catalytic ability, the OER performance of Fe2N6-CMPCFs and the contrast samples was measured in 1.0 M KOH, as shown in Fig. S35 (ESI†). The Fe2N6-CMPCF catalyst exhibited a remarkable overpotential of 346 mV, which is superior to that of FeN4-CMPCFs (385 mV), Fe NPs-CMPCFs (370 mV) and commercial RuO2 (350 mV). Moreover, the i–t curve in Fig. S31b (ESI†) demonstrates the outstanding OER stability of Fe2N6-CMPCFs at 10 mA cm−2.
Based on the above-mentioned characterization results, density functional theory (DFT) calculations were performed to shed light on the ORR activity and stability of the isolated single-atomic Fe (FeN4-CMPCFs) and homonuclear dual single-atomic Fe (Fe2N6-CMPCFs). In the case of the single-atom catalysts (SACs), as shown in Fig. 4a, the ORR reaction pathway involves the following steps: (I) O2 adsorption and hydrogenation into OOH*; (II) O–O bond cleavage of OOH* into O*; (III) protonation of O* into OH*; and (IV) OH* removal to form H2O on the structure of Fe–N4.11 Different from the traditional pathway (path I) on SACs, dual single-atom catalysts (DACs) provide two adjacent metal active sites for adsorption, resulting in a change in the O2 adsorption configurations on DAC (path II).48–50 Besides, by calculating the original Fe2–N6 site, as shown in Fig. S36 (ESI†), the thermodynamic reaction potential is relatively low; thus, the Fe2–N6 sites tend to coordinate by an extra OH ligand (Fe2N6OH) when they serve as the ORR active sites.51 Therefore, its reaction pathway includes (I) OH–O*, (II) OH–OH*, (III) OH*, and (IV) OH* removal,49 as presented in Fig. 4b. To prove the rationality of this two-site dissociation pathway (path II) on DACs, the Gibbs energy change of the Fe2N6OH–Ch–Npy sites undergoing the traditional path I and path II was calculated (Fig. S37, ESI†). As seen in Fig. S38 (ESI†), it is obvious that OH–O* and OH–OH* in path II are more stable reaction intermediates than OOH* and O*, respectively, in path I. Given that the thermodynamic process tends to energetically favorable intermediates, the Fe2N6OH–Ch–Npy site is more likely to follow the two-site dissociation pathway (path II). In addition, the calculated theoretical ORR activity on DACs by path II is more compatible with the experiment result from the free energy diagram of U = 1.23 V. Besides the metal coordination structure, the influence of carbon holes (Ch) and pyridinic-N (Npy) around the metal active sites was also considered and their specific structure diagrams are presented in Fig. S39 (ESI†).
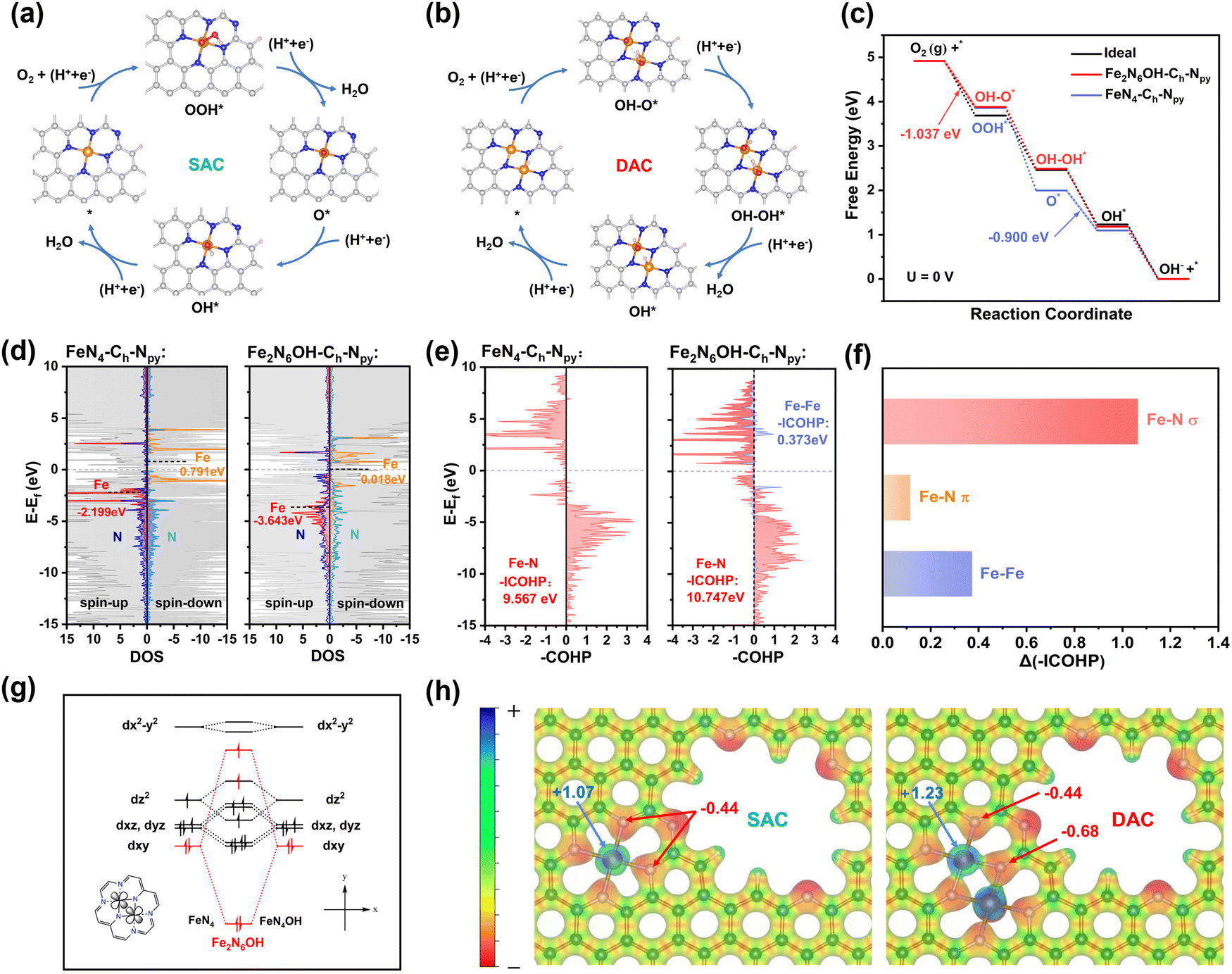 | ||
| Fig. 4 Mechanism studies by theoretical calculations. Reaction scheme of the ORR on (a) single-atom catalysts (SACs) and (b) dual single-atom catalysts (DACs). (c) Free energy diagrams of the ORR on FeN4–Ch–Npy and Fe2N6OH–Ch–Npy at U = 0 V. (d) Density of states (DOS) of FeN4–Ch–Npy and Fe2N6OH–Ch–Npy. (e) Crystal orbital Hamilton population (COHP) analysis with the corresponding ICOHP values of the Fe–N and Fe–Fe bond in FeN4–Ch–Npy and Fe2N6OH–Ch–Npy. (f) Enhanced orbit interaction of Fe–N and Fe–Fe in Fe2N6OH–Ch–Npy. (g) Qualitative molecular orbital of the d-orbitals in Fe2N6OH–Ch–Npy. (h) Electrostatic potential diagram and atomic charge between Fe and N atoms in SAC and DAC. | ||
The diverse ORR performances originate from the different binding strengths of the intermediate oxygen species on various catalysts. It is well-known that non-defective Fe–N4 has too strong adsorption energy for OH*. Therefore, fine-tuning the local coordination of Fe sites is imperative to optimize the binding strength of the oxygen intermediates.52 As shown in Table S5 (ESI†), the Fe2N6 sites modified with Ch and Npy (Fe2N6OH–Ch–Npy) exhibit weaker OH* adsorption energy than FeN4–Ch–Npy. Moreover, it is notable that the decoration of Ch and Npy on the Fe2N6 or FeN4 sites is also conducive to the removal of OH*, in contrast to the non-defective FeN4 and Fe2N6 system. Furthermore, the Gibbs free energy (ΔG) of each step along the 4e− ORR process was calculated on both SACs and DACs (Fig. 4d and Table S6, ESI†). It is obvious that all the electron-transfer steps are exothermic on the catalysts at U = 0 V, and thus the free energy pathway goes downhill. The limiting reaction barrier is a vital factor influencing the catalytic activity, which can be evaluated by the free energy of the potential-determining step (PDS). The PDS for Fe2N6OH–Ch–Npy was the formation of OH–O*, while that for FeN4–Ch–Npy was the formation of OH*. The ΔG value of PDS in the DAC system decreased, particularly for Fe2N6OH–Ch–Npy to only −1.037 eV, indicating that the synergistic interaction between Fe–Fe in DACs has a remarkable effect on the PDS and reaction barrier for ORR, which accelerated the catalytic process (Fig. 4d and Fig. S40, Table S7, ESI†). Moreover, according to the free energy diagrams of ORR at U = 1.23 and 0.85 V (Fig. S41, ESI†), it can be also seen that the ΔG of each step becomes more uniform and closer to the ideal ORR catalyst. The PDS step at the U = 1.23 V diagram directly reflects that the Fe2N6OH–Ch–Npy sites possess a lower thermodynamic overpotential (0.192 V) than that of FeN4–Ch–Npy (0.329 V).
To gain insight into the electronic structure of Fe SACs and DACs, the density of states (DOS) was further investigated for Fe2N6OH–Ch–Npy and FeN4–Ch–Npy (Fig. 4c and Table S8, ESI†). The negative shift in the Fe-3d band center in the structure of Fe2N6OH–Ch–Npy confirms that the optimized adsorption energy for ORR can be attributed to the weaker binding interaction between the Fe sites and OH* intermediate. Besides, the higher total density of states can be observed in Fe2N6OH–Ch–Npy near the Fermi level, demonstrating the better electron conductivity.
Additionally, studying the mechanism of ORR stability is equally crucial. It is commonly believed that the demetallation of metal sites from the carbon host is the main reason for the degradation in catalytic activity. Therefore, the dissolution energy barrier of the Fe atoms (ΔGdisso) in the FeN4 and Fe2N6 systems was calculated, as presented in Fig. 1f and Table S1 (ESI†). A higher ΔGdisso value was found for the Fe2N6 system, suggesting its better ORR stability. Moreover, the introduction of carbon holes and pyridinic-N also proved to be favorable for the anti-dissolving of Fe sites. To uncover the anti-dissolving origin of the Fe sites in DACs, an in-depth electronic structure analysis was further performed. The crystal orbital Hamilton population (COHP) analysis was utilized to quantitatively compare the orbit interaction of the chemical bonds, which is regarded as a descriptor of demetallation. As shown in Fig. 4e and Table S9 (ESI†), the negative area of integrated COHP (–ICOHP) for the Fe–N bond increased from 9.567 eV in the FeN4–Ch–Npy structure to 10.747 eV in Fe2N6OH–Ch–Npy, indicating the stronger Fe–N orbital interaction and bond strength in the latter case. Moreover, the unique Fe–Fe bond in Fe2N6OH–Ch–Npy can also have a positive contribution (0.373 eV) to the total orbit interaction. By further projecting to each orbital angular momentum, discovered that the enhanced σ interaction of Fe–N plays a dominant role in strengthening the orbital interaction, as seen in Fig. 4f and Table S10 (ESI†). Certainly, we cannot ignore the influence of the unique Fe–Fe bond in the Fe2N6OH–Ch–Npy structure. In this case, the Fe 4s–4s interaction creates partially filled bonding orbitals, which provide significant bonding stability, as shown in Table S11 (ESI†). In addition, the d-orbital splitting manner in Fig. 4g indicates that the Fe dxy–dxy interaction produces bonding and antibonding orbitals. The bonding orbitals filled with electrons contribute a large positive –COHP. In contrast, the strong antibonding orbital is lifted above the Fermi level, causing electron refluxing and fewer electrons filling the antibonding orbitals. Given that the antibonding orbitals only contribute negligible negative –COHP, the Fe dxy–dxy interaction also possesses remarkable contribution to the structure stability (Table S11, ESI†). Besides, the electrostatic potential with the atomic charge was also calculated to assess the electrostatic interaction between the Fe and N atoms (Fig. 4h and Table S12, ESI†). As displayed in Fig. 4h, the electrostatic potential is enhanced in Fe2N6OH–Ch–Npy mainly because the neighboring Fe3+ atom affects the atomic charge of the Fe2+ (more positive) and N atoms (more negative). In terms of the aforementioned theoretical support, we can conclude that the high stabilization of DACs can be primarily attributed to the enhanced orbital interaction (σ interaction of Fe–N, 4s–4s and dxy–dxy interaction of Fe–Fe) and electrostatic force between the Fe and N atoms.
Inspired by the excellent ORR performance of the Fe2N6-CMPCF catalyst, its practical application in ZABs was further investigated. In comparison to the conventional powder-like catalysts prepared through drop-casting, the as-synthesized Fe2N6-CMPCF membranes could directly function as freestanding air-electrodes for rechargeable ZABs. In the case of the liquid-state ZAB utilizing the Fe2N6-CMPCF membrane, it demonstrated a high open-circuit potential of 1.57 V (Fig. S42, ESI†), outperforming that of RuO2/Pt-based ZABs (1.45 V). Moreover, it exhibited a narrower charging/discharging voltage gap than RuO2/Pt-based ZABs under identical current densities (Fig. S43, ESI†), suggesting the enhanced electrocatalytic activity of Fe2N6-CMPCFs. Notably, the ZAB with the Fe2N6-CMPCF membranes achieved an outstanding peak power density of 261.4 mW cm−2, as shown in Fig. 5a, which is superior to that of commercial RuO2/Pt-based ZABs (102.9 mW cm−2). In addition, Fig. 5b shows the exceptional rate performance of the Fe2N6-CMPCF-based ZAB. It achieved a notable discharge voltage of 1.18 V at the current density of 50 mA cm−2, suppressing the benchmark set by RuO2/Pt-based ZABs (1.14 V). Remarkably, even after discharging at a high current density of 50 mA cm−2, the Fe2N6-CMPCF-based ZAB swiftly recovered to a steady discharge voltage, implying its outstanding rate performance. To further demonstrate its remarkable rate capability, its discharge–charge curve at various current densities ranging from 5 to 15 mA cm−2 was recorded, as presented in Fig. S44 (ESI†). It is obvious that the variation in the discharge–charge voltage plateaus was minimal with an increase in the current density. Notably, it could be easily recovered and maintained long-term stability as the current density returned to 10 mA cm−2. Moreover, the specific capacity of the Fe2N6-CMPCF-based ZABs was calculated to be 775 mA h gZn−1, which is superior to that of the RuO2/Pt-based ZABs (705 mA h gZn−1) at the current density of 10 mA cm−2, as shown in Fig. 5c. The cycling stability was evaluated by a continuous galvanostatic discharging–charging test. The freestanding Fe2N6-CMPCF membrane-based ZABs deliver an ultralong operation lifespan of over 4800 h at 5 mA cm−2, which is superior to that of the RuO2/Pt-based ZABs (500 h) (Fig. 5d and Fig. S45, ESI†). Notably, they could stably operate with a minor widening voltage gap, and after replacing the zinc plate and electrolyte, the voltage gap could be restored to its original state (0.73 V). This excellent performance in liquid-state ZABs has ascended to the top level among recent reports (Table S13, ESI†). To further assess the stability of the ZABs under different environmental conditions, cycling tests were conducted at different temperatures. As shown in Fig. S46 (ESI†), even under high-temperature (50 °C) and low-temperature (−10 °C) conditions, the ZABs using the Fe2N6-CMPCF membranes could still operate stably for a long period.
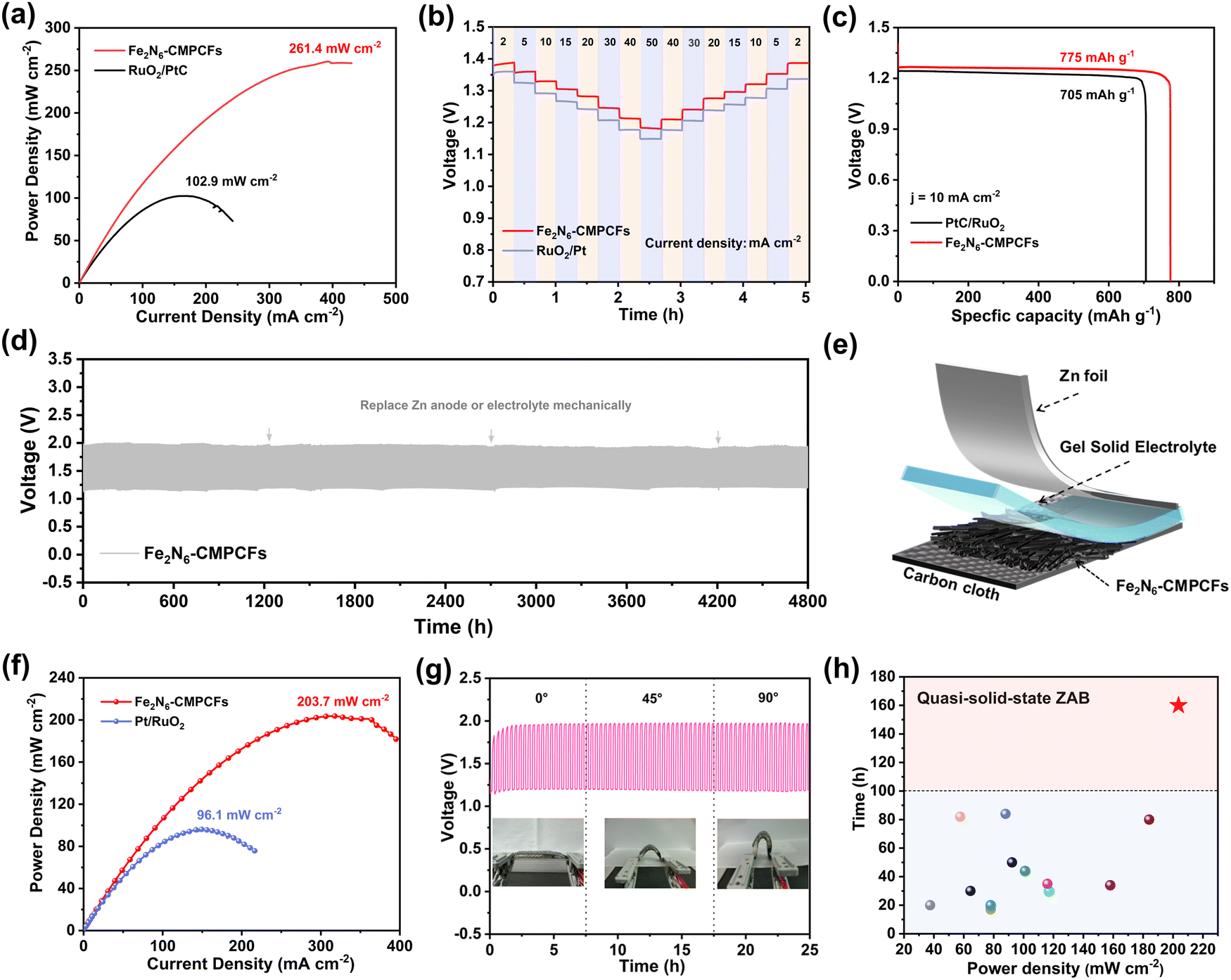 | ||
| Fig. 5 Assembly of ZABs and performance evaluation. (a) Power densities for liquid-state ZABs (electrolyte: 6 M KOH and 0.2 M Zn(CH3COO)2) using Fe2N6-CMPCFs or mixed Pt/RuO2 as the cathodes. (b) Comparison of the rate performance at different current densities for liquid-state ZABs. (c) Specific discharge capacity at 10 mA cm−2. (d) Galvanostatic discharge–charge test at 5 mA cm−2. (e) Schematic structure of freestanding flexible quasi-solid-state ZABs using Fe2N6-CMPCFs as the flexible cathodes and KOH/sodium polyacrylate (KOH/PANa) as the solid electrolyte. (f) Power densities of quasi-solid-state ZABs. (g) Galvanostatic discharge–charge test at different bending states for quasi-solid-state ZABs using Fe2N6-CMPCFs as the flexible cathodes. (h) Comparison of quasi-solid-state ZAB performance between Fe2N6-CMPCFs and recently reported electrocatalysts. | ||
The Fe2N6-CMPCF membrane after the long-term operation was further characterized. The SEM images in Fig. S47 (ESI†) show that the cross-linked carbon fibers were well-preserved after 4800 h of cycling. Some impurities, possibly KOH or KHCO3 from the electrolyte, were observed on the surface of the carbon fibers. The AC-HAADF-STEM images in Fig. S48 (ESI†) display that the fibrous structure was well-reserved with no noticeable Fe aggregation. Notably, a high ratio of dual-single-atomic sites could be detected, up to approximately 77%, demonstrating its remarkable anti-dissolving effect. Additionally, the Fe 2p XPS spectra displayed that the Fe element maintained its oxidation state (Fe2+ and Fe3+) in Fe2N6-CMPCFs. Only a tiny positive shift in the Fe 2p3/2 peak could be observed, indicating a slight decrease in the Fe2+ percentage. (Fig. S49, ESI†). Furthermore, the Fe2N6-CMPCF membrane-based cathode even remained flexible after 4800 h, as depicted in Fig. S50a (ESI†). In comparison, for the RuO2/Pt powder-based cathodes, a significant portion of the catalyst peeled off from the substrate and dissolved in the electrolyte (Fig. S50b, ESI†). Thus, we can reasonably conclude that powder-based catalysts are prone to peeling off from the substrate during long-term cycling, which also serves as the primary cause for their performance degradation. Besides, the O2 adhesion experiments demonstrated that the as-prepared Fe2N6-CMPCF membrane is highly aerophilic, which can promote the rapid diffusion of O2 bubbles across the interface (Fig. S51, ESI†). It is believed that this characteristic can alleviate cathodic flooding, and consequently improve the stability of ZABs. Therefore, in addition to the anti-dissolving active sites, the advantages of the membrane-based cathodes also play a significant role in achieving the durable ZABs with a record 200-day lifespan.
Due to its inherent flexible and binder-free nature, the Fe2N6-CMPCF membrane also holds great potential for application in wearable quasi-solid-state ZABs. The simplified schematic in Fig. 5e displays the utilization of KOH/sodium polyacrylate (KOH/PANa) gel as the solid electrolyte and Fe2N6-CMPCF membrane as the freestanding cathode in a quasi-solid-state ZAB. The as-assembled ZAB showed a smaller gap between the discharge and charge polarization curves compared to that of RuO2/Pt-based ZABs (Fig. S52, ESI†). Additionally, it demonstrated a distinguished peak power density of 203.7 mW cm−2, outperforming the Pt/RuO2 based ZAB (96.1 mW cm−2), as shown in Fig. 5f. Furthermore, compared to the Pt/RuO2-based ZAB, it demonstrated a higher voltage platform of 1.23 V at 30 mA cm−2, as shown in Fig. S53 (ESI†), indicating its excellent rate capability. Moreover, it could operate stably for 160 h at 2 mA cm−2 without replacing the hydrogel, as depicted in Fig. S54 (ESI†). Even at 5 mA cm−2, it still exhibited a steady discharge–charge cycling performance (Fig. S55, ESI†). Notably, after changing the hydrogel, the voltage gap of the ZAB could be restored significantly. In contrast, the Pt/RuO2-based ZABs experienced rapid decay within 40 h at 2 mA cm−2. Besides, it also exhibited consistent discharge–charge curves with minimal variation under different bending states, as shown in Fig. 5g. To showcase its potential application, it is noticeable that two sets of quasi-solid-state batteries using the Fe2N6-CMPCF membrane could easily power two LED screens (Fig. S56, ESI†). Undoubtedly, its remarkable performance also surpasses other reported quasi-solid-state ZABs in recent years, as shown in Fig. 5h and Table S14 (ESI†).
2. Conclusion
In summary, we successfully integrated dual-atomic Fe sites within flexible carbon nanofibers as freestanding cathodes for ZABs via a two-step NH3-assisted carbonization. The comprehensive characterization confirmed that Fe2N6 sites modified with carbon holes and pyridinic-N were homogeneously dispersed throughout the carbon fibers. Thus, the resultant Fe2N6-CMPCF catalyst demonstrated exceptional catalytic activity and stability for ORR. Moreover, the ZABs assembled with the freestanding Fe2N6-CMPCFs also exhibited an outstanding peak power density for both liquid-state and quasi-solid-state ZABs (261.4 and 203.7 mW cm−2, respectively). Notably, they delivered unprecedented durability for 4800 h without degradation in their performance. The theoretical simulation further demonstrated that the Fe2N6 structure possesses a remarkable anti-dissolving capacity, especially after decorating with carbon holes and pyridinic-N, which can alleviate the demetallation of the Fe atoms during long-term electrocatalysis. In addition, the aerophilic membrane-based cathodes can avoid the possible peeling off and cathodic flooding, which also promote the durability of ZABs. This study yielded valuable insights into the underlying mechanism for the stabilization of the dual single atoms and the design of freestanding air cathodes.Author contributions
Zian Xu: conceptualization, methodology, writing – original draft, writing – review & editing, visualization, formal analysis, investigation, resources, and data curation. Jian Zhu: conceptualization, methodology, visualization, formal analysis, investigation, resources, and data curation. Jingze Shao: methodology, formal analysis, investigation, resources, and data curation. Yu Xia: formal analysis and investigation. Pengfei Liu: investigation and resources. Guangshe Li: validation, investigation, and resources. Rouxi Chen: methodology, supervision, and project administration. Shaoqing Chen: investigation and resources. Jiacheng Wang: writing – review & editing. Shi Chen: writing – review & editing, supervision and project administration. Fuqiang Huang: supervision and project administration. Hsing-Lin Wang: conceptualization, supervision, and project administration.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Zian Xu, Jian Zhu, and Jingze Shao contribute equally to this work. The authors gratefully acknowledge the support of this work by Shenzhen Fundamental Research Scheme-General Program (JCYJ20220818100217037), Key-Area Research and Development Program of Guangdong Province (2019B010941001), National Natural Science Foundation of China (92163117, 22205153), Key University Laboratory of Highly Efficient Utilization of Solar Energy, Sustainable Development of Guangdong, Southern University of Science and Technology, Shenzhen 518055, China (Y01256331), Macau Science and Technology Development Fund FDCT 0092/2019/A2, 0013/2021/AMJ, and 0082/2022/A2, and Multi-Year research grant (MYRG2020-00283-IAPME and MYRG2022-00266-IAPME) from the Research & Development Office at University of Macau. This work is supported by the 1W1B beam line of the Beijing Synchrotron Radiation Facility (BSRF). This work is also supported by the Pico Center at SUSTech CRF which receives support from the Presidential Fund and Development and Reform Commission of Shenzhen Municipality.References
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- S. Chen, H.-M. Yan, J. Tseng, S. Ge, X. Li, L. Xie, Z. Xu, P. Liu, C. Liu, J. Zeng, Y.-G. Wang and H.-L. Wang, J. Am. Chem. Soc., 2024, 146, 13703–13708 CrossRef CAS PubMed.
- J. Liang, S. Li, X. Liu, Y. Wan, Y. Xia, H. Shi, S. Zhang, H.-L. Wang, G. Lu, G. Wu, Y. Huang and Q. Li, Nat. Catal., 2024, 7, 719–732 CrossRef CAS.
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- X. Lei, Q. Tang, Y. Zheng, P. Kidkhunthod, X. Zhou, B. Ji and Y. Tang, Nat. Sustainability, 2023, 816–826, DOI:10.1038/s41893-023-01101-z.
- J.-N. Liu, C.-X. Zhao, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Energy Environ. Sci., 2022, 15, 4542–4553 RSC.
- C.-X. Zhao, J.-N. Liu, J. Wang, C. Wang, X. Guo, X.-Y. Li, X. Chen, L. Song, B.-Q. Li and Q. Zhang, Sci. Adv., 2022, 8, eabn5091 CrossRef CAS PubMed.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- X. Li, X. I. Pereira-Hernandez, Y. Chen, J. Xu, J. Zhao, C. W. Pao, C. Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Nature, 2022, 611, 284–288 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- Y. Pan, X. Ma, M. Wang, X. Yang, S. Liu, H. C. Chen, Z. Zhuang, Y. Zhang, W. C. Cheong, C. Zhang, X. Cao, R. Shen, Q. Xu, W. Zhu, Y. Liu, X. Wang, X. Zhang, W. Yan, J. Li, H. M. Chen, C. Chen and Y. Li, Adv. Mater., 2022, 34, e2203621 CrossRef PubMed.
- C. Jiao, Z. Xu, J. Shao, Y. Xia, J. Tseng, G. Ren, N. Zhang, P. Liu, C. Liu, G. Li, S. Chen, S. Chen and H.-L. Wang, Adv. Funct. Mater., 2023, 33, 2213897 CrossRef CAS.
- T. Cui, Y. P. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CrossRef CAS PubMed.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B. A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J. N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS.
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Mullen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed.
- L. Yang, X. Zhang, L. Yu, J. Hou, Z. Zhou and R. Lv, Adv. Mater., 2022, 34, e2105410 CrossRef PubMed.
- M. Liu, N. Li, S. Cao, X. Wang, X. Lu, L. Kong, Y. Xu and X. H. Bu, Adv. Mater., 2022, 34, e2107421 CrossRef PubMed.
- Y. Li, Y. Ding, B. Zhang, Y. Huang, H. Qi, P. Das, L. Zhang, X. Wang, Z.-S. Wu and X. Bao, Energy Environ. Sci., 2023, 16, 2629–2636 RSC.
- M. Li, Q. Lv, W. Si, Z. Hou and C. Huang, Angew. Chem., Int. Ed., 2022, 61, e202208238 CrossRef CAS PubMed.
- H. Tian, A. Song, P. Zhang, K. Sun, J. Wang, B. Sun, Q. Fan, G. Shao, C. Chen, H. Liu, Y. Li and G. Wang, Adv. Mater., 2023, 35, e2210714 CrossRef PubMed.
- Z. Xu, J. Zhu, Z. Shu, Y. Xia, R. Chen, S. Chen, Y. Wang, L. Zeng, J. Wang, Y. Cai, S. Chen, F. Huang and H.-L. Wang, Joule, 2024, 8, 1790–1803 CrossRef CAS.
- T. Zhang, N. Wu, Y. Zhao, X. Zhang, J. Wu, J. Weng, S. Li, F. Huo and W. Huang, Adv. Sci., 2022, 9, 2103954 CrossRef CAS PubMed.
- Z. Xu, J. Zhu, J. Shao, Y. Xia, J. Tseng, C. Jiao, G. Ren, P. Liu, G. Li, R. Chen, S. Chen, F. Huang and H.-L. Wang, Energy Storage Mater., 2022, 47, 365–375 CrossRef.
- Y. Han, H. Duan, C. Zhou, H. Meng, Q. Jiang, B. Wang, W. Yan and R. Zhang, Nano Lett., 2022, 22, 2497–2505 CrossRef CAS PubMed.
- Q. Wei, F. Xiong, S. Tan, L. Huang, E. H. Lan, B. Dunn and L. Mai, Adv. Mater., 2017, 29, 1602300 CrossRef PubMed.
- B. Yang, Q. Han, L. Han, Y. Leng, T. O'Carroll, X. Yang, G. Wu and Z. Xiang, Adv. Mater., 2023, 35, 2208661 CrossRef CAS PubMed.
- Y. Li, J. Fu, C. Zhong, T. Wu, Z. Chen, W. Hu, K. Amine and J. Lu, Adv. Energy Mater., 2019, 9, 1802605 CrossRef.
- M. Jiao, L. Dai, H.-R. Ren, M. Zhang, X. Xiao, B. Wang, J. Yang, B. Liu, G. Zhou and H.-M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202301114 CrossRef CAS PubMed.
- T. Gu, D. Zhang, Y. Yang, C. Peng, D. Xue, C. Zhi, M. Zhu and J. Liu, Adv. Funct. Mater., 2023, 33, 2212299 CrossRef CAS.
- Y. He, H. Guo, S. Hwang, X. Yang, Z. He, J. Braaten, S. Karakalos, W. Shan, M. Wang, H. Zhou, Z. Feng, K. L. More, G. Wang, D. Su, D. A. Cullen, L. Fei, S. Litster and G. Wu, Adv. Mater., 2020, 32, e2003577 CrossRef PubMed.
- X. Wang, Y. Li, T. Jin, J. Meng, L. Jiao, M. Zhu and J. Chen, Nano Lett., 2017, 17, 7989–7994 CrossRef CAS PubMed.
- Q. Hu, Z. Wang, X. Huang, Y. Qin, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Energy Environ. Sci., 2020, 13, 5097–5103 RSC.
- J. Yan, K. Dong, Y. Zhang, X. Wang, A. A. Aboalhassan, J. Yu and B. Ding, Nat. Commun., 2019, 10, 5584 CrossRef CAS PubMed.
- G. Wang, N. Chandrasekhar, B. P. Biswal, D. Becker, S. Paasch, E. Brunner, M. Addicoat, M. Yu, R. Berger and X. Feng, Adv. Mater., 2019, 31, 1901478 CrossRef PubMed.
- X. Wang, L. Xu, C. Li, C. Zhang, H. Yao, R. Xu, P. Cui, X. Zheng, M. Gu, J. Lee, H. Jiang and M. Huang, Nat. Commun., 2023, 14, 7210 CrossRef CAS PubMed.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed.
- X. Cui, L. Gao, S. Lei, S. Liang, J. Zhang, C. D. Sewell, W. Xue, Q. Liu, Z. Lin and Y. Yang, Adv. Funct. Mater., 2021, 31, 2009197 CrossRef CAS.
- R. Chen, Y. Zhou and X. Li, Nano Lett., 2022, 22, 1217–1224 CrossRef CAS PubMed.
- A. Han, X. Wang, K. Tang, Z. Zhang, C. Ye, K. Kong, H. Hu, L. Zheng, P. Jiang, C. Zhao, Q. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19262–19271 CrossRef CAS PubMed.
- X. Xie, L. Shang, X. Xiong, R. Shi and T. Zhang, Adv. Energy Mater., 2022, 12, 2102688 CrossRef CAS.
- Q. Shi, Q. Liu, Y. Ma, Z. Fang, Z. Liang, G. Shao, B. Tang, W. Yang, L. Qin and X. Fang, Adv. Energy Mater., 2020, 10, 1903854 CrossRef CAS.
- G. Zhu, H. Yang, Y. Jiang, Z. Sun, X. Li, J. Yang, H. Wang, R. Zou, W. Jiang, P. Qiu and W. Luo, Adv. Sci., 2022, 9, 2200394 CrossRef CAS PubMed.
- S. Chen, N. Zhang, C. W. Narváez Villarrubia, X. Huang, L. Xie, X. Wang, X. Kong, H. Xu, G. Wu, J. Zeng and H.-L. Wang, Nano Energy, 2019, 66, 104164 CrossRef CAS.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed.
- N. Zhang, T. Zhou, J. Ge, Y. Lin, Z. Du, C. A. Zhong, W. Wang, Q. Jiao, R. Yuan, Y. Tian, W. Chu, C. Wu and Y. Xie, Matter, 2020, 3, 509–521 CrossRef.
- W. Zou, R. Lu, X. Liu, G. Xiao, X. Liao, Z. Wang and Y. Zhao, J. Mater. Chem. A, 2022, 10, 9150–9160 RSC.
- R. Cepitis, N. Kongi, J. Rossmeisl and V. Ivaništšev, ACS Energy Lett., 2023, 8, 1330–1335 CrossRef CAS PubMed.
- J. Liu, H. Xu, J. Zhu and D. Cheng, JACS Au, 2023, 3, 3031–3044 CrossRef CAS PubMed.
- T. Cui, Y.-P. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CrossRef CAS PubMed.
- L. Peng, J. Yang, Y. Yang, F. Qian, Q. Wang, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Adv. Mater., 2022, 34, 2202544 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03148b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |