
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrolyte engineering for thermally stable Li–S batteries operating from –20 °C to 100 °C†
Dong
Guo
 ab,
Simil
Thomas
a,
Jehad K.
El-Demellawi
c,
Zixiong
Shi
ab,
Zhiming
Zhao
ab,
Christian G.
Canlas
d,
Yongjiu
Lei
ab,
Jian
Yin
ab,
Yaping
Zhang
d,
Mohamed Nejib
Hedhili
d,
Muhammad
Arsalan
e,
Yunpei
Zhu
ab,
Osman M.
Bakr
ab,
Omar F.
Mohammed
ab and
Husam N.
Alshareef
*ab
ab,
Simil
Thomas
a,
Jehad K.
El-Demellawi
c,
Zixiong
Shi
ab,
Zhiming
Zhao
ab,
Christian G.
Canlas
d,
Yongjiu
Lei
ab,
Jian
Yin
ab,
Yaping
Zhang
d,
Mohamed Nejib
Hedhili
d,
Muhammad
Arsalan
e,
Yunpei
Zhu
ab,
Osman M.
Bakr
ab,
Omar F.
Mohammed
ab and
Husam N.
Alshareef
*ab
aMaterials Science and Engineering, Physical Science and Engineering (PSE) Division, King Abdullah University of Science and Technology (KAUST), Thuwal 239955-6900, Saudi Arabia. E-mail: husam.alshareef@kaust.edu.sa
bCenter for Renewable Energy and Storage Technologies (CREST), King Abdullah University of Science and Technology (KAUST), Thuwal 239955-6900, Saudi Arabia
cKAUST Upstream Research Center (KURC), EXPEC-ARC, Saudi Aramco, Thuwal, 23955-6900, Saudi Arabia
dCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
eEXPEC Advanced Research Center, Saudi Aramco, P.O. Box 5000, Dhahran 31311, Saudi Arabia
First published on 19th September 2024
Abstract
Lithium–sulfur (Li–S) batteries are deemed one of the most promising high-energy density battery technologies. However, their operation under thermal extremes, e.g., subzero and above 60 °C, remains largely underexplored. Especially, high temperatures (HT) accelerate sulfur dissolution and undesired side reactions, presenting significant challenges for electrolyte design. In this work, contrary to traditional understanding, we discovered that even (localized) high-concentration electrolytes (HCEs), which have shown promise within moderate temperature ranges (0–60 °C), fail at temperatures above 80 °C. Detailed investigations revealed that Li-anion aggregates in HCE trigger uncontrolled reductive decomposition at the Li anode side once the temperature exceeds a threshold of 80 °C. The resultant parasitic byproducts caused serious crosstalk and cathode oxidation in HT Li–S batteries. To counter this issue, we developed a localized medium-concentration electrolyte that features a well-mediated solvation structure and energy level, demonstrating excellent thermodynamic stability at high temperatures with superb kinetics at low temperatures. Consequently, high-performance and safely operating Li–S pouch cells are achieved over an unprecedented range of −20 to 100 °C. These findings link electrolyte microstructure, temperature, SEI structure, and degradation mechanism, offering a design protocol for the reliable function of batteries in extreme environments.
Broader contextThe demand for high-energy batteries that can function in extreme thermal conditions is rising in fields such as subsurface exploration, aerospace, and medical devices. These conditions often involve temperatures below 0 °C or exceeding 100 °C, posing significant challenges to existing lithium-ion batteries due to the limitations of electrolyte thermodynamics. Li–S batteries are poised to play a critical role in these harsh environments due to the moderate voltage plateau, which can secure a higher cathode safety and more feasible options for electrolyte design. Meanwhile, the extremely high specific capacity makes high-energy density possible. Nevertheless, the study of Li–S batteries under extreme temperatures remains largely unexplored. In this study, we illustrate the fundamental interplay between electrolytes and electrodes, which typically governs thermodynamics and kinetics. We found that Li–S batteries fail with most of the state-of-art electrolytes when temperature exceeded the threshold of 80 °C. The failure mechanism is well disclosed. Furthermore, we designed a new electrolyte to promote temperature tolerance. Such an electrolyte chemistry enables Li–S cells to operate safely from −20 °C to 100 °C. This work unlocks the potential of Li–S batteries and presents a design protocol for developing harsh-environment batteries. |
Introduction
Batteries in markets are generally considered to have a relatively fixed operation temperature range (0–60 °C). However, the ever-growing demands for high-energy batteries in specialized sectors, such as subsurface exploration, aerospace missions, and medical devices, call for battery functionality under extreme thermal conditions.1–4 Unfortunately, such environments, which can often dip below 0 °C or exceed above 100 °C, impose significant challenges for existing lithium-ion battery systems, especially regarding electrolyte design.5–7 In this context, exploring new electrolytes with wide-temperature and high redox tolerance is of great significance.For low-temperature (LT) battery designs, strategies focus on electrolytes with low viscosity and freezing points to enhance bulk ionic migration, alongside the construction of solid electrolyte interfaces (SEI) to facilitate interfacial desolvation.8–12 Yet, designing batteries for high-temperature operation poses greater challenges, especially for lithium metal batteries, which offer higher energy density but exhibit higher activity. Prior studies on “moderate temperature” (MT) batteries have typically capped their upper temperature limit at around 60 °C. Within this range, quality lithium deposition favored by inorganic SEI formation has been observed.13–15 However, the SEI properties above 80 °C are less investigated, and their failure mechanism is even barely reported. Importantly, anion-dominated solvation structures, achieved through weakly solvating electrolytes (WSE) or high concentration electrolytes (HCEs), have shown success in regulating durable SEI from low to moderate temperatures (Fig. 1).16–26 However, their effectiveness at extremely high temperatures (EHT), i.e., above 80 °C, remains unexplored.
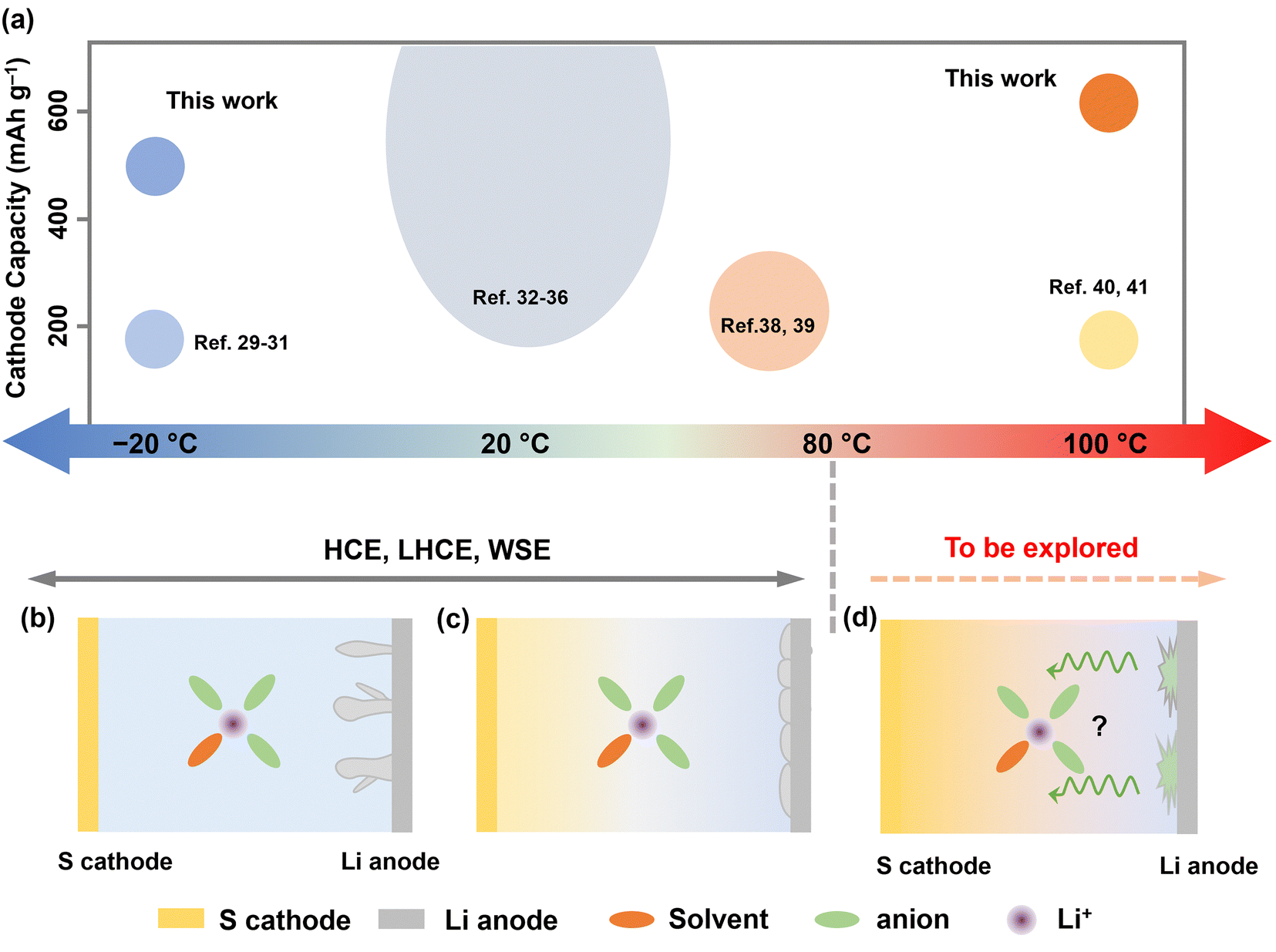 | ||
| Fig. 1 (a) A schematic illustration of the status of rechargeable batteries operating at various temperatures (top). The size of the circles represents the relative number of reports.8,10,17,21,24,27–39 (b) and (c) The conventional wisdom of electrolyte design for temperatures below 80 °C. HCE, LHCE, or WSE possess their advantages in regulating lithium metal anode from low to moderate temperature (<80 °C). However, these design strategies face challenges to Li–S cells operating at EHT (>80 °C) due to sulfur dissolution at the cathode and electrolyte failure at the anode (d). Therefore, their design protocol needs to be reconsidered. | ||
Additionally, high temperature causes positive shift of equilibrium potential according to the Nernst equation. Thermodynamically, this accelerates electrolyte degradation at high voltage, leading to catastrophic heat/gas formation. In this context, sulfur-based cathodes with moderate voltage (<3 V vs. Li+/Li) and high specific capacity present a more practical option for EHT operation. Nevertheless, lithium–sulfur (Li–S) batteries capable of functioning under thermal extremes have been barely reported due to several key challenges, including: (1) aggravated lithium polysulfide (LiPS) dissolution and shuttle effect at EHT; (2) increased parasite reactions on lithium metal anode at EHT; and (3) significantly reduced electrode kinetics for both cathode and anode at LT (Fig. 1b–d).11,40
For these reasons, we introduce an electrolyte design protocol for extreme-temperature Li–S batteries utilizing sulfurized polyacrylonitrile (SPAN) as the cathode (Fig. S1, ESI†). Although state-of-the-art (localized) HCEs showed significant success in improving the stability of both sulfur cathodes and lithium (Li) anodes from LT to MT, we observed their cycling failure at EHT. The primary cause of this failure is the crosstalk phenomenon, which has been gaining attention particularly in high-voltage lithium-ion batteries.41 Byproducts formed at the cathode generally pass through the separator and reach the anode, leading to anode degradation. In this work, we found that at high temperatures exceeding a threshold of 80 °C, (L)HCE electrolytes undergo continuous reductive decomposition on the anode side. The byproducts then diffuse to the cathode, causing endless oxidation on the cathode side and, ultimately, cell failure at EHT (Fig. 1d). To address this issue, we meticulously designed a localized medium concentration electrolyte (LMCE), which is illustrated in Fig. S2 (ESI†). In this system, the well-mediated solvation sheaths exhibit greater thermodynamic stability at EHT. Meanwhile, controlled anion reduction predominates at LT, creating a kinetically favorable interphase. This approach effectively balances the stability of the cathode and anode across a temperature range of −20 to 100 °C. Consequently, the electrolyte composed of 0.7 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) achieves 85% capacity retention following 150 cycles at 100 °C and 83% retention at −20 °C. Notably, it ensures a safe high-temperature storage shelf-life, evidenced by 90% of capacity retention after 60 days at 100 °C.
Results and discussions
Wide-temperature electrolyte design and screening
When operating a Li–S battery at 100 °C, the first concern is identifying suitable solvents since traditional ether-based electrolytes with low boiling points cannot safely operate.42 Tris(2,2,2-trifluoroethyl) phosphate (TFEP), with a high boiling point (186 °C), excellent thermal stability, and non-flammability (Fig. S3, ESI†), was screened as a promising solvent for high-safety battery at EHT. Moreover, the fluorinated moiety with its electron-withdrawing effect lessens ion-dipole (Li+–O) coordination, promoting the formation of a weak solvating environment, which is crucial for suppressing lithium polysulfide (LiPS) dissolution. In comparison, tetraethylene glycol dimethyl ether (TEGDME) and triethyl phosphate (TEP) with high boiling points were also tested. As shown in Fig. 2a and b, for the first time we demonstrated that an electrolyte of 1 M lithium LiTFSI in TFEP (1 M LiTFSI/TFEP) can enable Li-SPAN operate at 100 °C, with a remarkable capacity of 650 mA h g−1 in the initial cycles and 80% retention after 100 cycles at 0.5C. In contrast, 1 M LiTFSI/TEGDME and 1 M LiTFSI/TEP exhibited short lifetimes, retaining less than 30% and 10% capacity after merely 30 cycles, respectively (Fig. 2a). Their failure mechanisms are discussed in Fig. S4–S6 (ESI†).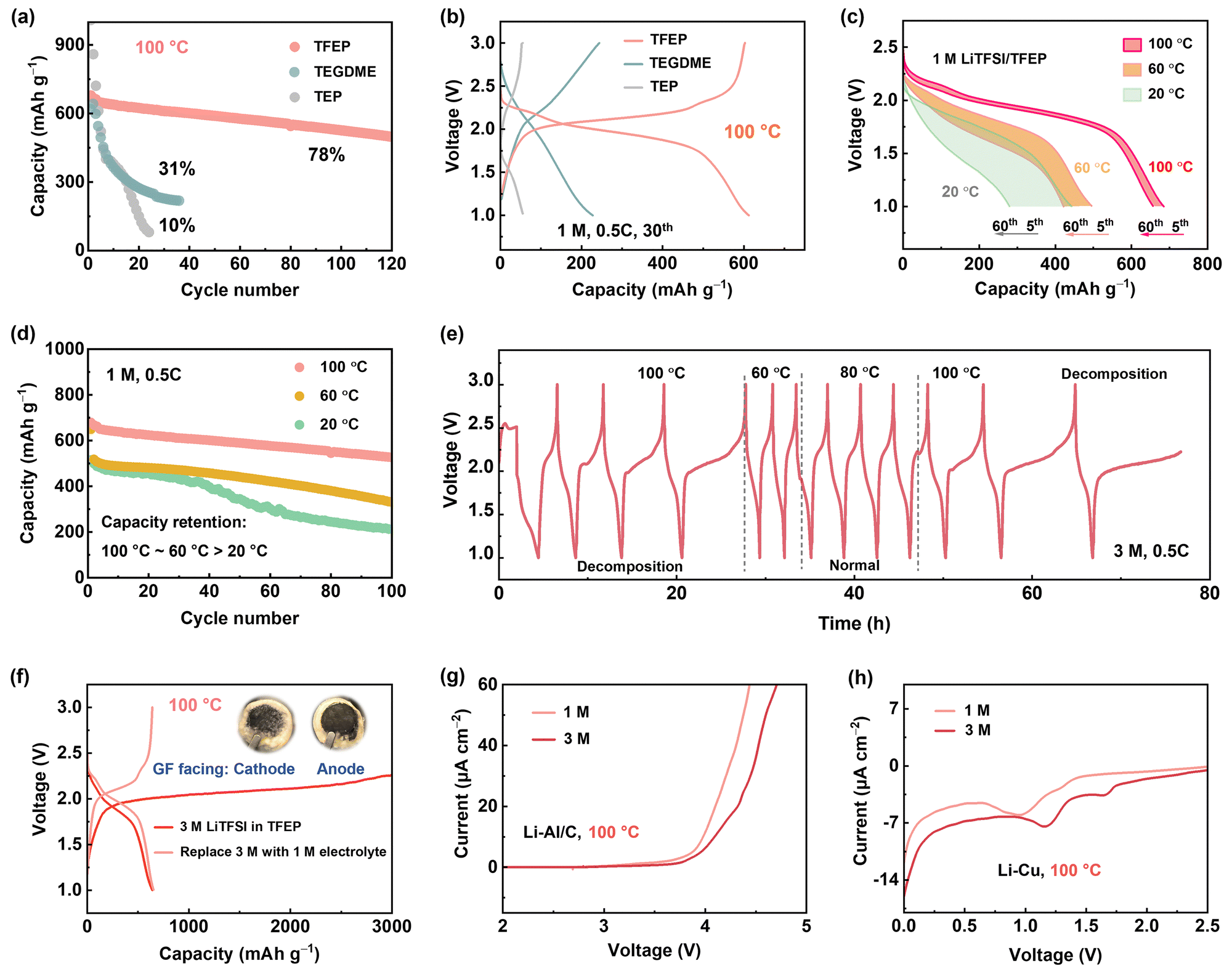 | ||
| Fig. 2 (a) Cycling performance of Li-SPAN cell at 0.5C at 100 °C with 1 M LiTFSI in different solvents, and (b) the corresponding voltage profiles at the 30th cycle. (c) Voltage profile of a cell with 1 M LiTFSI/TFEP electrolyte at 20 °C, 60 °C, and 100 °C, and (d) the corresponding cycling performance at 0.5C. (e) Voltage profile of a Li-SPAN cell with 3 M LiTFSI/TFEP electrolyte continuously cycled at different temperatures. (f) Voltage profile of the Li-SPAN cell at 100 °C. 3 M electrolyte-based cell was opened after 10 cycles and re-assembled with the 1 M electrolyte. Insert: Photos of the separator cycled in 3 M electrolyte at 100 °C. (g) Oxidation stability test. LSV curves for a Li|carbon coated Al cell at 100 °C. (h) Reduction stability test. LSV curves for Li|Cu cells at 100 °C. | ||
The stable cycling of 1 M LiTFSI/TFEP suggests the cathode dissolution is largely mitigated, and anode stability is maintained at 100 °C. Unfortunately, at lower temperatures (e.g., 20 °C and 60 °C), this electrolyte showed poor performance (Fig. 2c and d). For instance, the cell maintains only 40% capacity retention after 100 cycles at 20 °C. In principle, a lower temperature renders less sulfur dissolution, contributing to higher cathode stability, and ionic conductivity of 1 M electrolyte is not the limiting factor to the low stability (Fig. S7, ESI†). Therefore, the inferior performance at LT could only stem from its instability against the anode, which was also validated by the gradually increased overpotential in Li–Li symmetric cells at 20 °C (Fig. S4, ESI†), the boosted interfacial resistance (Fig. S8, ESI†), and the thick passivating layer on Li anode (Fig. S9, ESI†). By contrast, the 1 M electrolyte exhibited a stable overpotential at 100 °C (Fig. 2a).
To enhance the Li anode stability at LT, constructing anion-derived inorganic SEI has proven to be a popular strategy, which is achievable by increasing salt concentration to form anion-aggregated solvation sheaths, leading to a decreased lowest unoccupied molecular orbital (LUMO) for preferentially anion reduction. To this end, a 3 M LiTFSI/TFEP electrolyte was prepared, at its nearly saturated state. As expected, the 3 M electrolyte exhibited an improved Li stripping/deposition stability (Fig. 3b) and a higher Li-SPAN stability than the 1 M based cells at moderate temperatures of 60 °C and 80 °C (Fig. S10, ESI†). However, the 3 M electrolyte started to fail at 100 °C. It shows an abnormal and extremely high charge capacity (Fig. 2e and Fig. S11, ESI†), indicating uncontrolled side decomposition at 100 °C. Moreover, the decomposition is highly temperature-dependent with 80 °C the temperature threshold, as the voltage curve was back to normal once the temperature decreased to 60 °C or 80 °C (Fig. 2e). Overall, the 1 M electrolyte performed well at 100 °C but failed at 20 °C. By contrast, 3 M concentrated electrolyte can enhance battery stability below 80 °C, but failed at 100 °C. Interestingly, this failure behavior at EHT was found to be universal when salt concentration reaches a threshold. Taking the ether-based electrolyte as an example, 6 M LiTFSI/TEGDME works normally at 100 °C, but 7 M LiTFSI/TEGDME electrolyte showed the same failed charge process (Fig. S12, ESI†).
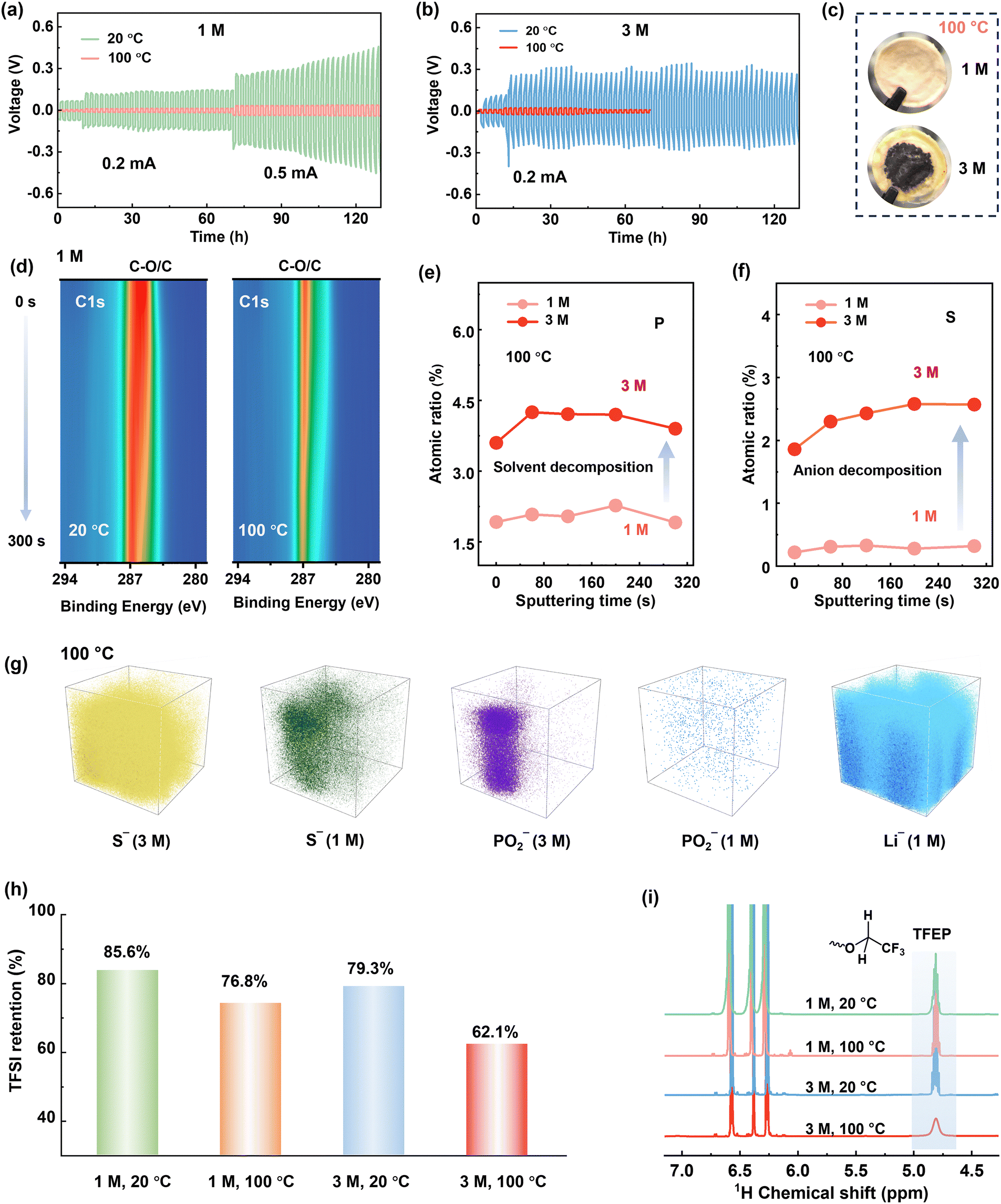 | ||
| Fig. 3 Voltage profiles of Li–Li symmetric cells at different temperatures for (a) 1 M LiTFSI/TFEP electrolyte, and (b) 3 M electrolyte. (c) Photographs of separators cycled at 100 °C. The parasite products penetrate the separators and potentially cause short circuit of the cell, as indicated by the reduced polarization after 20 cycles in Fig. 3b. (d) Contour plots of C1s XPS for cycled Li metal in 1 M LiTFSI/TFEP electrolyte. Depth profiling of P2p (e) and S2s (f) within SEI formed at 100 °C. (g) The 3D TOF-SIMS views of S− (anion fragment), PO2− (solvent fragment), and Li− in the SEI formed at 100 °C. (h) The retentions of TFSI− anions, determined by 19F NMR analyses. (i) 1H NMR of the electrolytes cycling at different cycling conditions after 30 cycles. The three 1H NMR peaks at 6.25–6.6 ppm belong to the internal reference (Fig. S23, ESI†). | ||
Failure mechanism of concentrated electrolytes at EHT
Disclosing the failure mechanism of concentrated electrolytes at EHT is critical. Since 3 M electrolyte has lower LiPS solubility and higher oxidative stability than the 1 M electrolyte at 100 °C (Fig. 2g and Fig. S13 and S14, ESI†), the failure of 3 M electrolyte at 100 °C is rooted in Li metal anode side instead of cathode degradation. Moreover, we reassembled the dyed Li-SPAN cell with the same cathode and anode, but refilled it with 1 M electrolyte, and found that the discharge capacity remained unchanged (Fig. 2f and Fig. S15, ESI†). That further confirmed the stability of cathode materials at EHT. To verify the observed failure on the Li metal anode side, we conducted a linear sweep voltammetry (LSV) test on Li–Cu cells at 100 °C with a negative scan to 0 V. Clearly, the 3 M electrolyte exhibited a much higher onset reduction potential (1.2 V) than the 1 M electrolyte (0.91 V), along with a significantly higher reduction current (Fig. 2h). This reveals aggressive reductive decompositions of the 3 M electrolyte. To elucidate this impact of potential-dependent electrolyte reduction on cell failure, a full cell composed of lithium titanium oxide (LTO) anode and lithium iron phosphate (LFP) cathode was tested. Since the Li+ insertion plateau of LTO anode (1.5 V, vs. Li+/Li) is higher than the reductive potential (1.2 V) of the 3 M electrolyte, electrolyte decomposition on LTO anode can be largely avoided at 100 °C. Consequently, the LTO-LFP cell achieved a normal discharge–charge process (Fig. S16a, ESI†) at 100 °C. But again, the 3 M electrolyte failed in Li–LTO cell (Fig. S16b, ESI†) because of the use of a Li metal anode. These results collectively reveal that when the concentrations reach high enough, disaster decompositions take place on those anodes having low reduction potentials under EHT conditions (Fig. S17, ESI†).It should be noted here that until now we only disclosed the failure of the 3 M electrolyte originating from the side reductions at the Li anode side. Cycling failure, specifically the abnormal charge capacity, appears more related to an oxidative decomposition process. The CV curves of Li-SPAN at 100 °C also confirmed this point (Fig. S18, ESI†). Given that cathode material and 3 M electrolyte are thermodynamically stable at cathode side as discussed above, this oxidation can only be induced by parasite products from anode, a crosstalk phenomenon observed in Li-ion batteries.41 This crosstalk can explain the fact that the abnormal charge process takes place after a couple of discharge–charge cycles rather than during the initial charge state (Fig. 2e). This is because only after the anode reduction species diffuse to cathode in the initial cycles, their oxidative decomposition could start.41 The abundant oxidative species including S–Ox and N–Ox at cathode were well detected by X-ray photoelectron spectroscopy (XPS) in Fig. S19 (ESI†). This crosstalk phenomenon at 100 °C was further visualized by the evolution of separators in the cycled Li-SPAN cell, which is detailed in Fig. S20 (ESI†). In summary, the 3 M electrolyte experienced uncontrolled reduction at lithium metal under EHT of 100 °C, and the crosstalk further leads to oxidation at cathode and cell failure.
Temperature-dependent electrolyte and interphase chemistry
To elucidate reductive decomposition, SEIs were resolved by depth-profiling XPS and time-of-flight secondary ion mass spectrometry (TOF-SIMS). The Li–Li symmetric cells were analyzed after 30 cycles (Fig. 3). For the 1 M electrolyte (Fig. 3d), the contour mappings of C1s XPS spectra showed that organic-rich SEI layer dominated at 20 °C, while an organic-less SEI layer formed at 100 °C. This difference corresponds to stable polarization curves at 100 °C and increased polarization at 20 °C (Fig. 3b). However, for 3 M electrolyte, stable polarization was observed at 20 °C (Fig. 3b), but vigorous decomposition occurred at 100 °C, evidenced by the substantial black side products accumulated on the anode (Fig. S21, ESI†) and the separator (Fig. 3c). More specifically, the SEI in 3 M electrolyte holds around 3 times of P atomic content (solvent decomposition), and 5–8 times of S atomic content (anion decomposition) than that in 1 M electrolyte at 100 °C (Fig. 3e and f). The above results are further visualized by TOF-SIMS. As illustrated in Fig. 3g, the 3D-rendering space of SEI exhibits thicker occupation of S− and PO2− fragments than 1 M electrolyte at 100 °C. This clearly reveals that Li metal anodes are highly corroded by the 3 M electrolyte at 100 °C.To further quantify the electrolyte decomposition, 1H and 19F nuclear magnetic resonance (NMR) spectra analyses were performed on electrolytes in the cycled Li-SPAN cells. As summarized in Fig. 3h and Fig. S22 (ESI†), the 3 M electrolyte demonstrated a much lower retention of TFSI− anion at (62.1%) at 100 °C than the 1 M electrolyte (76.8%), implying the faster degradation of anions. This severe anion decomposition is also accompanied by the fast consumption of solvent. As revealed in Fig. 3i, the peak at 4.8 ppm is assigned to the resonance of methylene from TFEP. Clearly, the 3 M electrolyte showed a much weaker signal of –O–CH2–CF3 at 100 °C, corresponding to a low solvent retention of 67.1%. The specific decomposition species are also explored by theoretical simulations and discussed later. Combined with the above electrochemical behavior, SEI analyses, and quantitative NMR results, we can conclude the decomposition of concentrated electrolytes, especially the anion-induced decomposition, is aggressive and catastrophic at 100 °C. Contrary to the widespread notion, the anion-derived SEI in (L)HCEs can protect electrolytes from further decomposition was only valid up to relatively moderate temperatures (<80 °C), yet this protection failed at EHTs of 100 °C. These investigations revisit and address the critical knowledge gap in understanding the electrolyte-SEI behavior across the temperature spectrum from moderate ranges to challenging EHT conditions.
Mediated solvation structures and temperature-tolerant electrolyte design
Given that electrolyte reduction is a process driven by Li-ion solvation, we deduced that increasing temperatures in concentrated electrolytes significantly impact solvation structures, leading to the failure of 3 M electrolyte at 100 °C. This temperature-dependent solvation is deciphered by NMR techniques. As shown in Fig. 4a, increasing salt concentration from 1 M to 3 M leads to a noticeable 19F downfield shift for TFSI− anion (deshielding effect) and an upshift shift for TFEP solvent at 20 °C, which suggests the strengthened Li+⋯anion pairing and less Li+⋯solvent coordination, respectively. This is typical for solvation behavior in HCEs.24 Notably, heating to 100 °C causes further deshielding of the TFSI− anion (Fig. 4b and Fig. S24, ESI†), revealing increased Li+⋯TFSI− binding with increasing temperatures.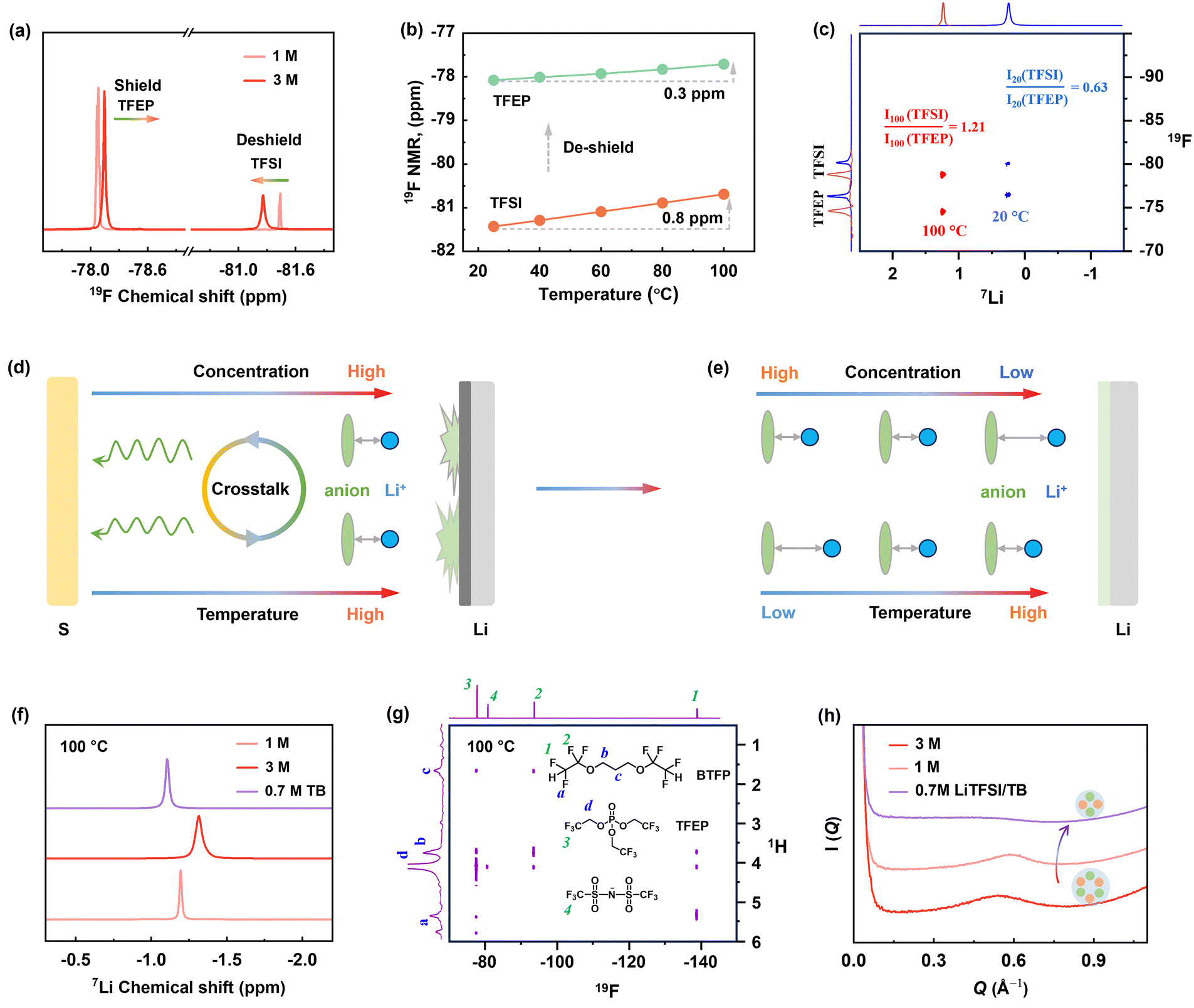 | ||
| Fig. 4 Characterization of electrolytes. (a) 19F NMR spectra of the 1 M and 3 M electrolyte. (b) Temperature-dependent 19F NMR chemical shift of 1 M electrolyte from 20 °C to 100 °C. (c) 7Li–19F HOESY spectra of 3 M electrolyte at 20 °C and 100 °C. (d) Schematic illustration of compact Li-anion spatial correlation leads to anode reduction and the resultant crosstalk to cathode. (e) Illustration of Li-anion spatial correlation impacted by temperature and concentration, along with the proposed relatively dilute concentration for high-temperature batteries. (f) 7Li NMR spectra of the electrolytes at 100 °C. (g) 1H–19F HOESY spectrum of 0.7 M LiTFSI/TB electrolyte at 100 °C. (h) SAXS profiles for the electrolytes. | ||
The Li-anion-solvent correlation at high temperatures was further quantified by hetero-nuclear Overhauser effect spectroscopy (HOESY), a powerful tool to detect spatial coupling in liquids.24 As shown in the 2D HOESY in Fig. 4c, the 3 M electrolyte presented two pronounced 7Li–19F cross peaks at both 20 and 100 °C, representing the spatial coupling of Li+ with TFSI− and TFEP, respectively. By integrating the 2D intensity (I) of Li–FTFSI and Li–FTFEP peaks, we found a ratio of I(TFSI)/I(TFEP) of 0.63 at 20 °C. This ratio distinctly increased to 1.21 at 100 °C, indicating a much shorter Li+⋯TFSI− distance at higher temperatures compared to Li+⋯TFEP. This behavior is also confirmed by 1 D 19F spectra as a function of temperature in Fig. 4b. The 19F NMR signal of the anion showed a distinctive upfield shift (0.8 ppm) compared to the slight shift (0.3 ppm) of the solvent signal upon heating to 100 °C, demonstrating that anions are more temperature sensitive to be involved in Li-ion solvation at HT. In summary, we disclose that high concentration promotes Li+⋯TFSI− coordination, and high temperature further strengthened this Li-anion pairing structure, as illustrated in Fig. 4d and e. Therefore, these closely bonded Li-anion species are more likely to be recruited into the Li/electrolyte interphase, which induces the decomposition and failure of concentrated electrolyte at the EHT of 100 °C.
From these investigations, we note that Li+⋯TFSI pairs should be regulated at relatively low levels to avoid the electrolyte failure under EHT. Although this can be achieved by lowering electrolyte concentration (such as 1 M), it compromises battery performance at room temperature due to either higher sulfur solubility or lower anode stability (Fig. 2d). To tackle this dilemma, we introduced a new fluorine-ether solvent, 1,3-bis(1,1,2,2-tetrafluoroethoxy) propane (BTFP), as a diluent. BTFP featuring a high boiling point (162 °C) and high safety are highly desirable for high-temperature Li–S operations. Importantly, a localized medium concentration electrolyte (LMCE) can be formulated with a 0.7 M LiTFSI in TFEP/BTFP (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, 0.7 M LiTFSI/TB), which shows negligible LiPS solubility (Fig. S25, ESI†). The tuned Li-anion spatial distance, as illustrated in Fig. 4e, effectively addressed the aforementioned issues across a wide temperature range.
:1, v/v, 0.7 M LiTFSI/TB), which shows negligible LiPS solubility (Fig. S25, ESI†). The tuned Li-anion spatial distance, as illustrated in Fig. 4e, effectively addressed the aforementioned issues across a wide temperature range.
The advantages of the solvation properties of LMCE are deciphered by NMR spectroscopy (Fig. 4f and g and Fig. S26 and S27, ESI†). The 1H–19F 2D HOESY spectrum of the 0.7 M LiTFSI/TB electrolyte was collected at 100 °C. As shown in Fig. 4g, the 1H peak at 4.1 ppm, associated with TFEP solvent, exhibited a prominent off-diagonal peak with 19F of TFSI at −81 ppm (d-4), and another cross peak with the 19F signal of BTFP at −138 ppm (d-1). By integrating the peak intensity, the normalized ratio of I(TFEP/TFSI):I(TFEP/BTFP) is 1:0.75, suggesting the closer distance between TFEP solvent and TFSI anion (<5 Å). Meanwhile, the 19F of TFSI at −81 ppm showed a cross peak with 1H of TFEP but no signals with protons of BTFP diluent. These results reveal that the solvation structure of LMCE is well maintained at 100 °C, with anions in inner sheath and relatively isolated from the outer diluent. More importantly, the 0.7 M LiTFSI/TB electrolyte shows a dispersity of TFSI anion around Li-ion in LMCE, unlike the over-strengthened ionic pairing in 3 M electrolyte that leads to drastic decomposition. This is also verified by the significantly upfield shift of 7Li peak compared to 3 M electrolyte at 100 °C in Fig. 4f. Moreover, as presented in 2D 7Li–19F NOESY (Fig. S28 and 29, ESI†) collected at 20 °C, the peak intensity ratio of I(Li–FTFSI):I(Li–FTFEP) is calculated to be 0.55:1 for the LMCE, which is higher than that of 1 M electrolyte (0.41:1) (Fig. S27, ESI†). This indicates that the average Li+⋯TFSI− distance in LMCE is shorter than in 1 M electrolyte at 20 °C, a favorable solvation structure to construct anion-derived SEI at low temperatures.20 Meanwhile, small-angle X-ray scattering (SAXS) results show that 0.7 M LiTFSI/TB electrolyte has an appreciably weaker scattering intensity, indicating the suppression of large cluster formation, which favors higher ion diffusivity according to the Stokes–Einstein equation.43 This is further verified by 7Li diffusion-ordered spectroscopy NMR (DOSY-NMR) (Fig. S30, ESI†), highlighting the significance of high ionic diffusivity at lower temperatures.
To gain further insight into the thermodynamic properties of the electrolytes, we performed density functional theory (DFT) and ab initial molecular dynamic (AIMD) simulations (Fig. S31, S32 and Table S1, ESI†). We examined the projected density of states (PDOS) of TFSI− at 100 °C and focused on conduction bands (i.e., unoccupied orbitals), which dominate the reduction reactions. As shown in Fig. 5a, the energy level of TFSI− in 0.7 M per TB electrolyte (2.95 eV) is distinctly higher than that in 3 M electrolyte (2.69 eV) at the lowest end of conduction bands, indicating the higher reductive stability of TFSI− in the diluted 0.7 M per TB electrolyte at 100 °C. In contrast, the lower energy level of TFSI− in 3 M electrolyte most likely leads to decomposition via the cleavage of S–C bond according to its lowest bond order of 0.71 (Fig. S33, ESI†). After defluorination, the associated byproducts of CF3SO2NSO2 and N(SO2)2 have HOMO of −7.72 eV and −7.07 eV, respectively, while TFSI− and TFEP solvent shows higher HOMO of −10.78 eV and −11.35 eV, respectively (Fig. 5b). Therefore, the byproducts would undergo further oxidative decomposition preferentially over the anions and solvent at the cathode side, causing the observed cell failure at high temperatures.
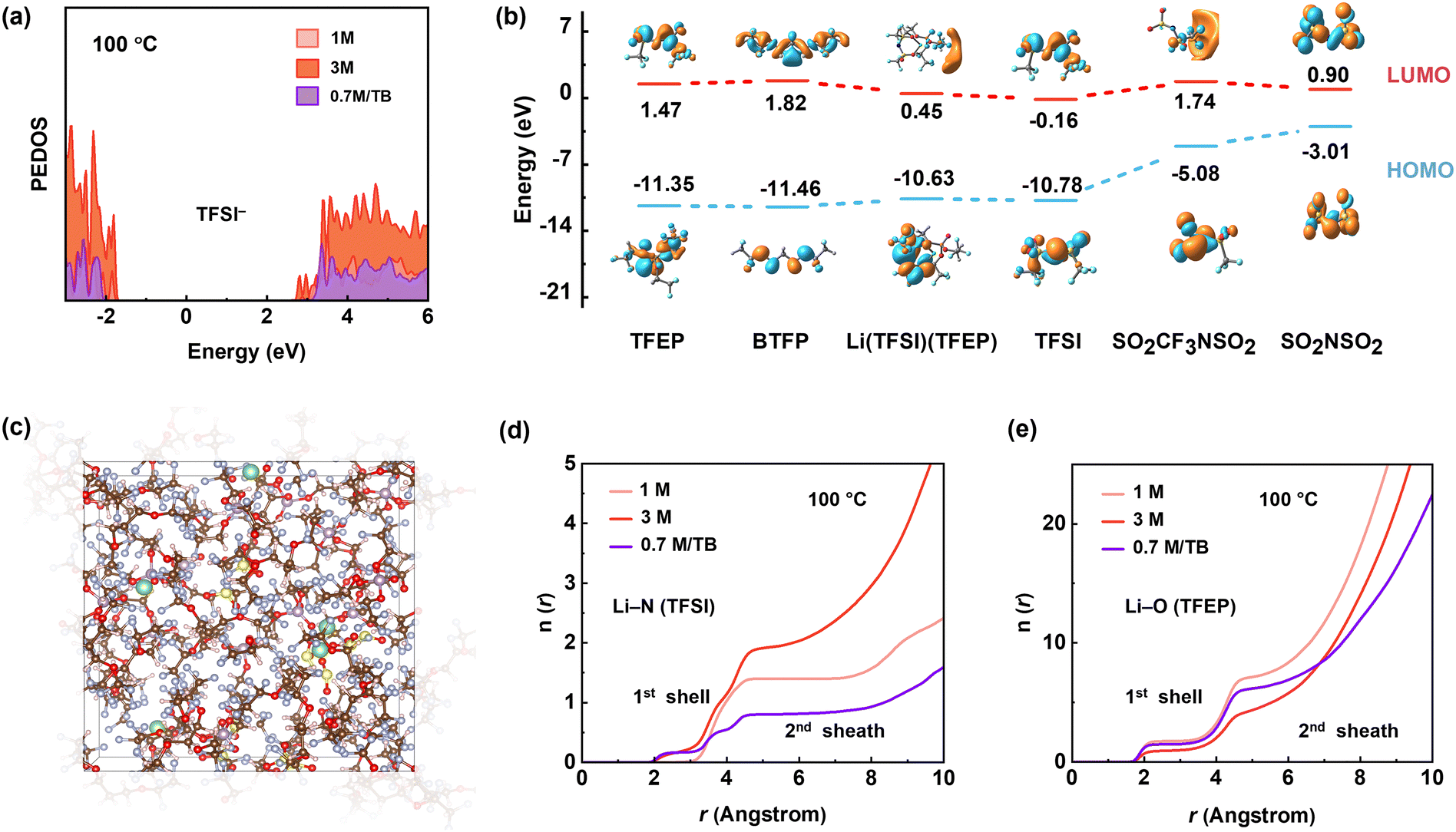 | ||
| Fig. 5 AIMD simulations at 100 °C: the projected density of states (PDOS) of the TFSI− anion in different electrolytes. (b) LUMO and HOMO energies of the compositions and byproducts. (c) Snapshoot of AIMD simulations of 0.7 M LiTFSI/TB. (d,f) The coordination number profiles of (e) Li–NTFSI and (f) Li–OTFEP for 1 M, 3 M LiTFSI/TFEP and 0.7 M LiTFSI/TB electrolytes. | ||
In view of kinetics, radial distribution functions (RDF) of Li–NTFSI suggest that the 0.7 M LiTFSI/TB electrolyte has a slightly lower Li–NTFSI coordination number in the first solvation shell and significantly decreased occupation in the second solvation shell compared to the 3 M electrolyte at 100 °C (Fig. 5d). This is beneficial to avoid severe side reactions at high temperatures. Meanwhile, RDF of Li–OTFEP illustrated that TFEP molecules in 0.7 M LiTFSI/TB electrolyte are less coordinated than in the 1 M electrolyte (Fig. 5e), contributing to a lower possibility of solvent decomposition at 100 °C. Opposite to the EHT scenario, the interfacial side reaction at low temperatures is thermodynamically limited. As a result, the 0.7 M LiTFSI/TB electrolyte, with its higher TFSI occupation in the first Li+ solvation sheath compared to the 1 M electrolyte (Fig. S34, ESI†), has formed a high-quality SEI at low temperatures (Fig. S35, ESI†).
Electrochemical performance of Li-SPAN from −20 °C to 100 °C
We further evaluated Li-SPAN full cells with the 0.7 M LiTFSI/TB electrolyte across an extremely wide temperature range from −20 °C to 100 °C. At 100 °C, the cells retained 85.3% of capacity (580 mA h g−1) after 150 cycles at 0.5C. At a 5C rate, they retained 75.2% capacity after 150 cycles (Fig. 6a). Even at a 10C rate, the cells delivered a decent capacity of 460 mA h g−1 with 70% retention after 100 cycles (Fig. S36, ESI†), the highest record for SPAN-based cells.32,33,36,44–48 The rate performance is summarized in Fig. 6d and Fig. S37 (ESI†). This high-rate stability demonstrates the reliability of the LMCE electrolyte under such harsh conditions. In contrast, an LHCE composed of the same salt and solvents exhibited a similar failure behavior to the concentrated 3 M electrolyte (Fig. S38, ESI†), further validating the unique capability of the LMCE electrolyte for EHT operation. Notably, the LMCE also performed well at lower temperatures, (Fig. S39, ESI†). Especially, at −20 °C, the Li-SPAN still maintained an 83% capacity retention (340 mA h g−1) over 110 cycles, outperforming the cells 1 M electrolyte (Fig. 6b) and traditional electrolytes.49,50 (Table S2, ESI†)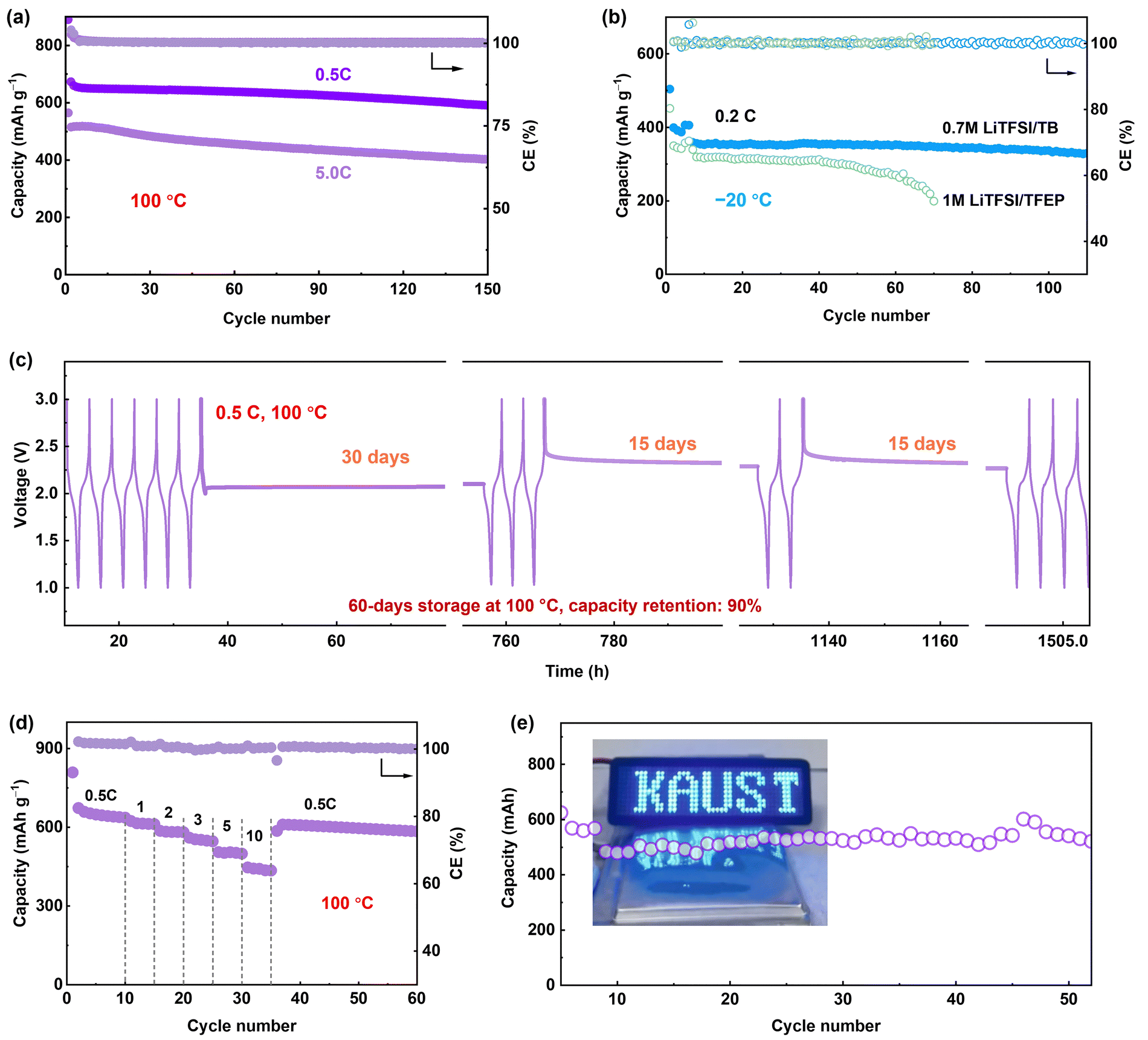 | ||
| Fig. 6 Electrochemical performance of the 0.7 M LiTFSI/TB electrolyte in a wide temperature range. Cycling performance at (a) 100 °C, and (b) at −20 °C. (c) Long-term storage performance of the Li-SPAN cell at 100 °C with different state of charge/discharge. (d) Rate performance of Li-SPAN cell cycled at 100 °C. (e) Cycling performance of the pouch cell with 0.7 M LiTFSI/TB electrolyte at 100 °C. Insert: Digital photos of the prepared Li-SPAN pouch cells. | ||
Another critical parameter for high-temperature batteries is calendar storage performance, as thermal-induced self-discharge is a huge challenge in practical applications. In this case, the Li-SPAN cell was first cycled at 0.5C at 100 °C, then rested at different states of charge/discharge for a specific time T. After each aging period T, cells were cycled at 0.5C for three cycles to track recoverable capacity. As recorded in Fig. 6c, after the first 30 days of storage at the discharge state of 2.1 V, no obvious voltage drop was observed, along with negligible capacity decay with 0.7 M LiTFSI/TB electrolyte (Fig. S40, ESI†). After another 30 days at a full-charge state, the cell retained 94% capacity retention with a CE of 99.8%, and cell voltage was maintained at the OCV state (∼2.3 V). Moreover, the storage performance at 100 °C shows negligible difference from that at 20 °C (Fig. S41, ESI†). Additionally, the cell maintained 75% capacity after 300 cycles at a shallow depth of discharge (Fig. S42, ESI†). These results clearly demonstrate that side reactions are well inhibited during EHT storage, and self-charge issues are mitigated by the LMCE electrolyte. Furthermore, the developed LMCE electrolyte was proven effective under lean electrolyte conditions. For instance, under a low electrolyte/SPAN (E/S) ratio of 8 μl mg−1, the Li–S batteries was able to maintain a capacity of 390 mA h g−1 at 20 °C, and an increased capacity of 550 mA h g−1 at 100 °C after 60 cycles (Fig. S43, ESI†). These results underline the significant potential of our developed approach for high-energy-density batteries operating under extreme thermal conditions.
Li-SPAN pouch cells were fabricated to evaluate feasibility under more practical conditions. The pouch cell with a total capacity of 0.5 A h was designed, as presented in Fig. 6e, and an average capacity of 0.4 A h was delivered. The 60-cycle lifespan of Li-SPAN pouch cell at 100 °C is reported for the first time, illustrating the extraordinary effectiveness of the electrolyte design in advancing extreme-temperature batteries. Given that commercially available polypropylene separators are not suitable to function at temperature above 100 °C, the next priority should be on developing lightweight separators that have high thermal and electrochemical stability, while also demonstrating techno-economic viability for large-scale implementation in other cell types, such as cylindrical cells.
Conclusions
In this work, we achieved a mechanistic understanding of Li-SPAN cell degradation at 100 °C in various electrolyte systems, including dilute and concentrated electrolytes. Contrary to widely followed protocols at room and moderate temperature, we discovered that increasing salt concentration does not enhance stability at 100 °C. Instead, coupling high temperature with high concentrations leads to catastrophic electrolyte reduction and crosstalk behavior that further causes cell failure. To address this, we introduced a new fluorine ether, BTFP, as an effective diluent to craft a localized medium-concentration electrolyte. This electrolyte reorganizes the solvation sheath with an appropriate anion population, Li-anion spatial distance, and electron energy level. Thermodynamically and kinetically, this reconfiguration balances durable interfacial stability across a wide temperature from −20 to 100 °C, thereby unlocking the potential of Li-SPAN cells. Our methodology of evaluating cell degradation and electrolyte design is applicable to many other metal-ion batteries operating in extreme temperatures. By tuning molecular structures and compositions of anions, electrolyte reduction can be modulated to extend high-temperature longevity, which is an urgent need for reliable batteries operating in harsh environments.Author contributions
D. G. and H. N. A. conceived the idea and conceptualized the work. D. G. conducted the materials synthesis, electrochemical tests, and characterizations. S. T. performed the simulations. J. K. D. revised the manuscript and provided valuable guidance. C. G. C. performed the NMR experiments. Y. Z. conducted the SAXS experiments. M. N. H. performed the XPS experiments. All authors contributed to the manuscript preparation and discussion.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by King Abdullah University of Science and Technology (KAUST) and by Saudi Aramco under grant number RGC/3/6003-01-01. This research used the resources of the Supercomputing Laboratory at KAUST in Thuwal, Saudi Arabia.References
- K. Kalaga, M. T. Rodrigues, H. Gullapalli, G. Babu, L. M. Arava and P. M. Ajayan, ACS Appl. Mater. Interfaces., 2015, 7, 25777–25783 CrossRef CAS PubMed.
- B. Liu, K. Fu, Y. Gong, C. Yang, Y. Yao, Y. Wang, C. Wang, Y. Kuang, G. Pastel, H. Xie, E. D. Wachsman and L. Hu, Nano Lett., 2017, 17, 4917–4923 CrossRef CAS PubMed.
- D. Luo, M. Li, Y. Zheng, Q. Ma, R. Gao, Z. Zhang, H. Dou, G. Wen, L. Shui, A. Yu, X. Wang and Z. Chen, Adv. Sci., 2021, 8, 2101051 CrossRef CAS PubMed.
- K. Tuul, S. M. Maher, C. Floras, W. Black, T. Taskovic, S. Chisholm, A. Clarke, E. Lust and J. R. Dahn, J. Electrochem. Soc., 2024, 171, 040510 CrossRef.
- A. Wang, Y. Song, Z. Zhao, X. Li, Z. Hu and J. Luo, Adv. Funct. Mater., 2023, 33, 2302503 CrossRef CAS.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X. Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- Y. Zhao, Z. Hu, Z. Zhao, X. Chen, S. Zhang, J. Gao and J. Luo, J. Am. Chem. Soc., 2023, 145, 22184–22193 CrossRef CAS PubMed.
- B. Nan, L. Chen, N. D. Rodrigo, O. Borodin, N. Piao, J. Xia, T. Pollard, S. Hou, J. Zhang, X. Ji, J. Xu, X. Zhang, L. Ma, X. He, S. Liu, H. Wan, E. Hu, W. Zhang, K. Xu, X. Q. Yang, B. Lucht and C. Wang, Angew. Chem., Int. Ed., 2022, 61, 202205967 CrossRef PubMed.
- D. Hubble, D. E. Brown, Y. Zhao, C. Fang, J. Lau, B. D. McCloskey and G. Liu, Energy Environ. Sci., 2022, 15, 550–578 RSC.
- H. Ji, Z. Wang, Y. Sun, Y. Zhou, S. Li, J. Zhou, T. Qian and C. Yan, Adv. Mater., 2023, 35, 2208590 CrossRef CAS PubMed.
- N. Zhang, T. Deng, S. Zhang, C. Wang, L. Chen, C. Wang and X. Fan, Adv. Mater., 2022, 34, 2107899 CrossRef CAS PubMed.
- J. Wang, W. Huang, A. Pei, Y. Li, F. Shi, X. Yu and Y. Cui, Nat. Energy, 2019, 4, 664–670 CrossRef CAS.
- Z. Zhao, B. Nian, Y. Lei, L. Zhao, M. N. Hedhili, D. Guo, Z. Shi, W. Zhao, J. K. El-Demellawi, Y. Wang, Y. Zhu, K. Xu and H. N. Alshareef, Adv. Mater., 2024, 2402626 CrossRef PubMed.
- D. Guo, D. B. Shinde, W. Shin, E. Abou-Hamad, A. H. Emwas, Z. Lai and A. Manthiram, Adv. Mater., 2022, 34, 2201410 CrossRef CAS PubMed.
- S. Wan, W. Ma, Y. Wang, Y. Xiao and S. Chen, Adv. Mater., 2024, 2311912 CrossRef CAS PubMed.
- Y. Mo, G. Liu, J. Chen, X. Zhu, Y. Peng, Y. Wang, C. Wang, X. Dong and Y. Xia, Energy Environ. Sci., 2024, 17, 227–237 RSC.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- Y. Chen, M. Li, Y. Liu, Y. Jie, W. Li, F. Huang, X. Li, Z. He, X. Ren, Y. Chen, X. Meng, T. Cheng, M. Gu, S. Jiao and R. Cao, Nat. Commun., 2023, 14, 2655 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- C. M. Efaw, Q. Wu, N. Gao, Y. Zhang, H. Zhu, K. Gering, M. F. Hurley, H. Xiong, E. Hu, X. Cao, W. Xu, J. G. Zhang, E. J. Dufek, J. Xiao, X. Q. Yang, J. Liu, Y. Qi and B. Li, Nat. Mater., 2023, 22, 1531–1539 CrossRef CAS PubMed.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- X. Liu, A. Mariani, H. Adenusi and S. Passerini, Angew. Chem., Int. Ed., 2023, 135, 202219318 CrossRef.
- D. Guo, J. Wang, T. Lai, G. Henkelman and A. Manthiram, Adv. Mater., 2023, 35, 2300841 CrossRef CAS PubMed.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- Z. Li, N. Yao, L. Yu, Y.-X. Yao, C.-B. Jin, Y. Yang, Y. Xiao, X.-Y. Yue, W.-L. Cai, L. Xu, P. Wu, C. Yan and Q. Zhang, Matter, 2023, 6, 2274–2292 CrossRef CAS.
- R. Xu, S. Zhang, X. Shen, N. Yao, J.-F. Ding, Y. Xiao, L. Xu, C. Yan and J.-Q. Huang, Small Struct., 2023, 4, 2200400, DOI:10.1002/sstr.202200400.
- Y. Lin, Z. Yang, X. Zhang, Y. Liu, G. Hu, S. Chen and Y. Zhang, Energy Storage Mater., 2023, 58, 184–194 CrossRef.
- Y. Liu, L. Xu, Y. Yu, M. He, H. Zhang, Y. Tang, F. Xiong, S. Gao, A. Li, J. Wang, S. Xu, D. Aurbach, R. Zou and Q. Pang, Joule, 2023, 7, 2074–2091, DOI:10.1016/j.joule.2023.07.013.
- Y. Liu, X. An, K. Yang, J. Ma, J. Mi, D. Zhang, X. Cheng, Y. Li, Y. Ma, M. Liu, F. Kang and Y.-B. He, Energy Environ. Sci., 2024, 17, 344–353 RSC.
- Y. Wu, W. Wang, J. Ming, M. Li, L. Xie, X. He, J. Wang, S. Liang and Y. Wu, Adv. Funct. Mater., 2018, 29, 1805978 CrossRef.
- S. Wang, B. Lu, D. Cheng, Z. Wu, S. Feng, M. Zhang, W. Li, Q. Miao, M. Patel, J. Feng, E. Hopkins, J. Zhou, S. Parab, B. Bhamwala, B. Liaw, Y. S. Meng and P. Liu, J. Am. Chem. Soc., 2023, 145, 9624–9633 CrossRef CAS PubMed.
- J. Lee, A. R. Jeon, H. J. Lee, U. Shin, Y. Yoo, H.-D. Lim, C. Han, H. Lee, Y. J. Kim, J. Baek, D.-H. Seo and M. Lee, Energy Environ. Sci., 2023, 16(7), 2924–2933, 10.1039/d3ee00157a.
- T. Meng, S. Yang, Y. Peng, X. Lan, P. Li, K. Hu and X. Hu, Energy Storage Mater., 2024, 71, 103598 CrossRef.
- H. Yang, C. Guo, J. Chen, A. Naveed, J. Yang, Y. Nuli and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 791–795 CrossRef CAS PubMed.
- A. L. Phan, C. Jayawardana, P. M. L. Le, J. Zhang, B. Nan, W. Zhang, B. L. Lucht, S. Hou and C. Wang, Adv. Funct. Mater., 2023, 33, 2301177 CrossRef CAS.
- M. Fang, X. Yue, Y. Dong, Y. Chen and Z. Liang, Joule, 2024, 8, 91–103 CrossRef CAS.
- T. Chen, Z. Jin, Y. Liu, X. Zhang, H. Wu, M. Li, W. Feng, Q. Zhang and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207645 CrossRef CAS PubMed.
- Z. Shi, Z. Tian, D. Guo, Y. Wang, Z. Bayhan, A. S. Alzahrani and H. N. Alshareef, ACS Energy Lett., 2023, 8, 3054–3080 CrossRef CAS.
- Y. Song, L. Wang, L. Sheng, D. Ren, H. Liang, Y. Li, A. Wang, H. Zhang, H. Xu and X. He, Energy Environ. Sci., 2023, 16, 1943–1963 RSC.
- Z. Li, H. Rao, R. Atwi, B. M. Sivakumar, B. Gwalani, S. Gray, K. S. Han, T. A. Everett, T. A. Ajantiwalay, V. Murugesan, N. N. Rajput and V. G. Pol, Nat. Commun., 2023, 14, 868 CrossRef CAS PubMed.
- S. C. Kim, J. Wang, R. Xu, P. Zhang, Y. Chen, Z. Huang, Y. Yang, Z. Yu, S. T. Oyakhire, W. Zhang, L. C. Greenburg, M. S. Kim, D. T. Boyle, P. Sayavong, Y. Ye, J. Qin, Z. Bao and Y. Cui, Nat. Energy, 2023, 8, 814–826 CrossRef CAS.
- T. Ma, Y. Ni, D. Li, Z. Zha, S. Jin, W. Zhang, L. Jia, Q. Sun, W. Xie, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2023, 62, 202310761 CrossRef PubMed.
- S. Dörfler, H. Althues, P. Härtel, T. Abendroth, B. Schumm and S. Kaskel, Joule, 2020, 4, 539–554 CrossRef.
- Y. Zhang, Z. Wang, Y. Pan, H. Yu, Z. Li, C. Li, S. Wang, Y. Ma, X. Shi, H. Zhang, D. Song and L. Zhang, Energy Environ. Sci., 2024, 17, 2576–2587 RSC.
- C. Li, Q. Zhang, J. Sheng, B. Chen, R. Gao, Z. Piao, X. Zhong, Z. Han, Y. Zhu, J. Wang, G. Zhou and H.-M. Cheng, Energy Environ. Sci., 2022, 15, 4289–4300 RSC.
- Z. Li, D. Guo, F. Li, G. Hou, X. Liu, C. Li, L. Cao, R. Wei, Z. Zhou and Z. Lai, Energy Storage Mater., 2022, 49, 575–582 CrossRef.
- D. Guo, M. Li, M. N. Hedhili, V. Tung, Y. Li and Z. Lai, Energy Storage Mater., 2019, 25, 33–40 CrossRef.
- D. Guo, X. Li, W. Wahyudi, C. Li, A.-H. Emwas, M. N. Hedhili, Y. Li and Z. Lai, ACS Nano, 2020, 14, 17163–17173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03191a |
| This journal is © The Royal Society of Chemistry 2024 |