
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing degradation mechanisms in 3D/2D perovskite solar cells under photothermal accelerated ageing†
Zijian
Peng
 *ai,
Andrej
Vincze
c,
Fabian
Streller
d,
Vincent M.
Le Corre
ab,
Kaicheng
Zhang
a,
Chaohui
Li
ai,
Jingjing
Tian
ai,
Chao
Liu
ab,
Junsheng
Luo
af,
Yicheng
Zhao
af,
Andreas
Späth
de,
Rainer
Fink
d,
Thomas
Heumüller
ab,
Andres
Osvet
a,
Ning
Li
abg,
Martin
Stolterfoht
h,
Larry
Lüer
*a and
Christoph J.
Brabec
*ab
*ai,
Andrej
Vincze
c,
Fabian
Streller
d,
Vincent M.
Le Corre
ab,
Kaicheng
Zhang
a,
Chaohui
Li
ai,
Jingjing
Tian
ai,
Chao
Liu
ab,
Junsheng
Luo
af,
Yicheng
Zhao
af,
Andreas
Späth
de,
Rainer
Fink
d,
Thomas
Heumüller
ab,
Andres
Osvet
a,
Ning
Li
abg,
Martin
Stolterfoht
h,
Larry
Lüer
*a and
Christoph J.
Brabec
*ab
aInstitute of Materials for Electronics and Energy Technology (i-MEET), Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany. E-mail: zijian.peng@fau.de; larry.lueer@fau.de; christoph.brabec@fau.de
bHelmholtz-Institute Erlangen-Nürnberg (HI ERN), 91058 Erlangen, Germany
cInternational Laser Centre SCSTI, 84104 Bratislava, Slovak Republic
dChair of Physical Chemistry II, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany
eCompetence Center Engineering of Advanced Materials, Friedrich-Alexander-Universität Erlangen-Nüünberg, 91058 Erlangen, Germany
fNational Key Laboratory of Electronic Thin Films and Integrated Devices, School of Integrated Circuit Science and Engineering, University of Electronic Science and Technology of China (UESTC), 611731 Chengdu, P. R. China
gState Key Laboratory of Luminescent Materials and Devices, Institute of Polymer Optoelectronic Materials and Devices, School of Materials Science and Engineering, South China University of Technology, 510640 Guangzhou, P. R. China
hElectronic Engineering Department, The Chinese University of Hong Kong, Hong Kong SAR, China
iGraduate School in Advanced Optical Technologies (SAOT), 91052 Erlangen, Germany
First published on 27th September 2024
Abstract
Three-dimensional/two-dimensional (3D/2D) heterojunctions in perovskite solar cells exhibit excellent optoelectronic properties and enhanced stability under mild ageing conditions. However, their performance degrades drastically under harsh ageing conditions. This study reveals the intrinsic instability of mono-ammonium based 2D perovskites (2D-mono) under photo-thermal ageing, which decompose into PbI2 and metallic lead (Pb0). The structural collapse promotes vacancy formation and facilitates iodide migration to the anode. As a result, it triggers a redox reaction that reduces the transport layer's mobility and doping concentration, leading to a significant increase in series resistance. Compared to mono-ammonium-2D structure, di-ammonium-2D (2D-di) based interfaces demonstrate superior structural stability and effectively block iodide migration into the transporting layer. However, blocking-induced uneven iodide distribution leads to interstitial defect formation in the 3D layer, exacerbating non-radiative recombination. To address it, we propose a strategical method by incorporating 2D-di in the 3D bulk instead of on the top surface, which effectively confines mobile ions within the grain and suppresses cation phase segregation. This optimization yields stable perovskite solar cells with an extrapolated operational T80 lifetime exceeding 560 hours under harsh conditions (85 °C and 2-sun illumination).
Broader context3D/2D perovskite heterojunctions have shown great potential in enhancing both efficiency and stability for perovskite solar cells (PSCs) under mild aging, though their performance suffers significantly under harsher stresses. This study sheds light on the underlying mechanisms responsible for the degradation of mono-ammonium based 2D/3D heterojunctions under photothermal aging. We unveil the intrinsic instability of mono-ammonium-2D/3D interfaces, leading to decomposition and detrimental interfacial reactions that seriously detoriate device performance. This knowledge paves the way for the development of more robust 2D/3D interfaces. We further explore di-ammonium cation spacer as an alternative for the 2D component. While di-ammonium-2D/3D interfaces exhibit superior structural stability, they introduce limitations due to uneven iodide distribution within the bulk and interstitial defect formation. Our work proposes a novel strategy to leverage the advantages of di-ammonium-2D perovskites by incorporating them within the 3D bulk rather than at the interface. This approach achieves exceptional operational lifetimes under harsh aging conditions (85 °C and light-soaking). Our work provides a theoretical basis for the development of PSCs accelerated aging standards. |
Introduction
Perovskite solar cells (PSCs) have impressive power conversion efficiencies (PCEs) exceeding 26%, rivalling silicon-based counterparts. This progress is largely attributed to innovative passivation strategies utilizing two-dimensional/three-dimensional perovskite (2D/3D) heterojunctions. Notably, 2D perovskites offer enhanced stability against humidity, temperature, and ionic migration compared to 3D perovskites thanks to their robust molecular structure and reduced volatilility.1,2 Despite the challenges posed by the large exciton binding energy and low charge carrier mobility in 2D perovskites, integrating them with 3D perovskites leverages the strengths of both,3,4 addressing these drawbacks while creating beneficial effects like reduced interfacial recombination, aligned energy levels for efficient carrier extraction, and improved long-term device stability.5–8Despite their advantages, 2D/3D heterojunctions exhibit limitations under harsh ageing conditions.9,10 Elevated temperatures or light exposure can promote cation diffusion between 2D and 3D layers, compromising the integrity and charge carrier dynamics of the 2D/3D interfaces.11–14 Mitigation strategies include cation-blocking cross-linking layers,15 but concerns remain regarding 2D ligands deprotonation affecting 3D structure10,16 and potentially causing the formation of metallic lead (Pb0) as deep traps.17–19 Consequently, some studies proposed avoiding 2D passivation to sidestep detrimental interactions with 3D perovskite under harsh ageing conditions.20–23 Although the reasons behind 2D/3D interfacial instability have been reported, the impact of 2D/3D deconstruction on device performance has not been quantified yet.
2D organic–inorganic perovskite, depending on the lateral displacement of the adjacent inorganic layers, can be categorized into Ruddlesden–Popper (RP) and Dion–Jacobson (DJ) structures,1,24 as shown in Fig. 1(a) and (b). The RP structure has staggered inorganic layers with ideal displacement of (0.5, 0.5) between successive layers, while the DJ structure exhibits perfect stacking and eclipsed inorganic layer with no displacement (0, 0). In many cases, 2D organic–inorganic perovskite can also be further classified according to the type of organic cation spacer into mono-ammonium spacer and di-ammonium spacer based 2D perovskites. The instability issues mentioned above primarily focus on 3D/2D-mono-ammonium interfaces. The mono-ammonium cation interacts with inorganic octahedra at one side and it leaves a van der Waals gap between adjacent octahedra layer, which facilitates the spacer diffusion and reconstruction of 2D phase.13 Considering the strong double side hydrogen-bonding and eliminated van der Waals gap in di-ammonium 2D phase, replacing mono-ammonium cation with di-ammonium cation has been proposed.25–30 However, the stability of 3D/2D-di-ammonnium interfaces under various accelerated ageing conditions remains poorly understood.31
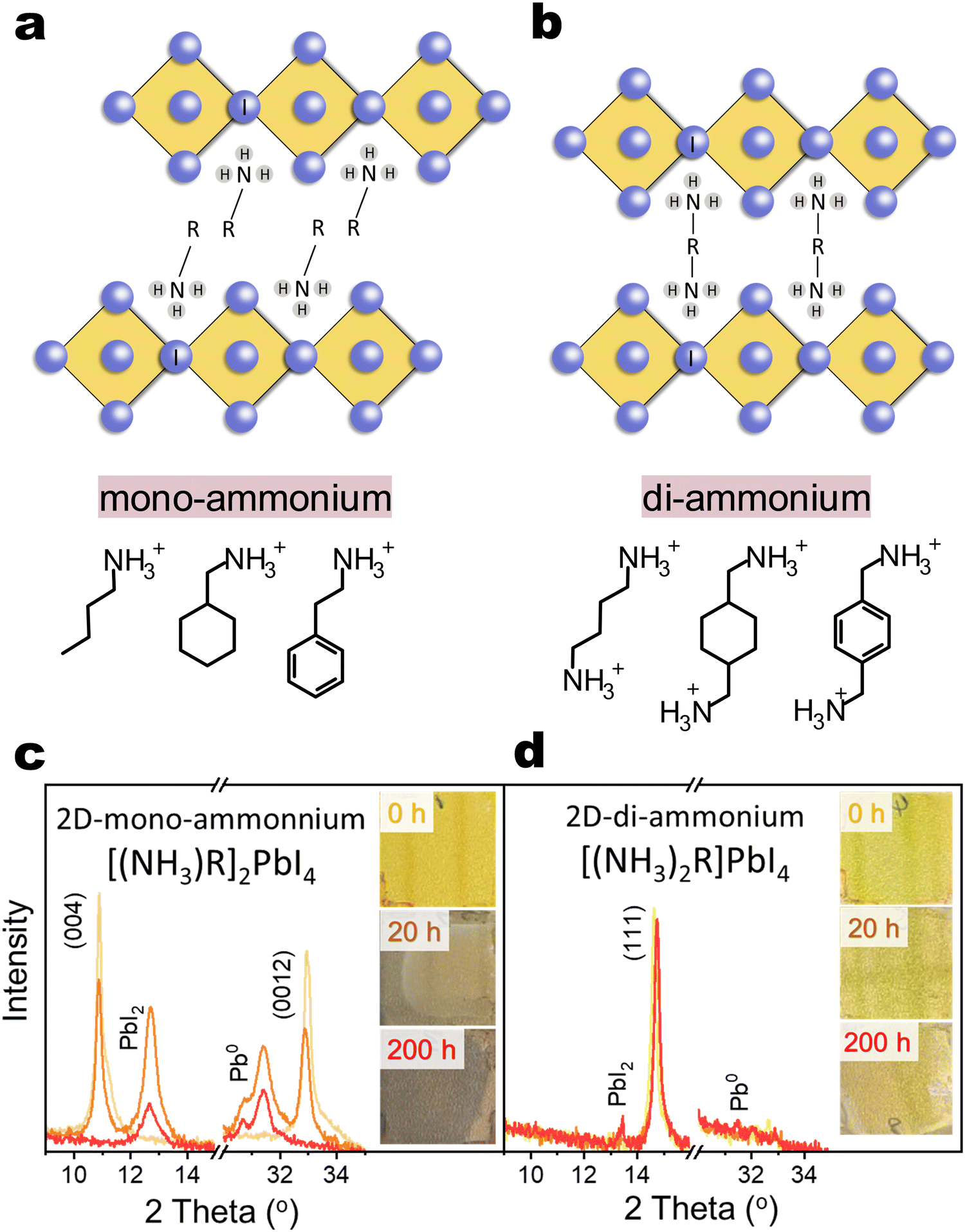 | ||
| Fig. 1 High-temperature photostability of 2D perovskite films (a) schematic structure of Ruddlesden–Popper perovskite using mono-ammonium as spacer. Representative linear (n-butylammonium), cyclic (cyclohexylmethylammonium) and aromatic (phenethylammonium) spacers are shown. (b) Schematic structure of Dion–Jacobson perovskite using di-ammonium as spacer. Representative linear (butyldiammonium), cyclic (1,4-cyclohexanedimethylammonium) and aromatic (1,4-phenylenedimethylammonium) spacers are shown. (c) and (d) X-ray diffraction (XRD) patterns of pure 2D-mono (PEA2Pb4) and 2D-di (BDAPbI4) films on glass substrates aged at different times under 85 °C and 2-sun illumination. The insets show images of the samples (2.5 cm × 2.5 cm) at different ageing time. | ||
The long-term stability of PSCs is heavily influenced by mobile ion migration. Iodide ions are particularly detrimental, exhibiting a possibility to: (a) migrate towards and undergo redox reactions with transport layers and electrodes, causing interfacial degradation;32–37 (b) move within the perovskite layer, leading to alterations in the internal electric field through mechanisms like screening;38–42 (c) induce halide phase segregation;43–45 (d) form vacancy defects as shallow traps and interstitial defects as deep traps.46–48 Similarly, cation migration and aggregation lead to undesired phase segregation within the 3D perovskite layer.49,50 While PSCs’ operational stability under conventional ageing conditions (e.g. ISOS consensus) has been widely reported (Table S1, ESI†),51–53 accelerated ageing tests under short-term and even more harsh conditions are crucial for evaluating the long-term stability of PSCs and predicting their commercial viability.
This work addresses these gaps. We first investigate the intrinsic stability of mono-ammonium based 2D (2D-mono) and di-ammonium based 2D (2D-di) structures, and find only the 2D structure with di-ammonium spacers can survive under photo-thermal ageing, while the 2D-mono decomposes rapidly. Then, a combination of optical experiments, element depth profiles, and drift-diffusion simulations suggests that reconfiguration or deconstruction of 3D/2D-mono interface creates substantial vacancies, subsequently attracting and enabling the diffusion of mobile iodide into the upper transport layer and electrode. This process reduces the doping concentration and mobility of the hole transport layer (HTL), as identified by the Kelvin probe measurement, drastically increasing the series resistance. While the 3D/2D-di structure effectively blocks iodide migration, this interaction unintentionally generates iodide interstitial defects that act as traps, hindering carrier extraction. Finally, instead of surface passivation forming a 3D/2D heterojunction, we incorporate the 2D-di perovskite as a growth seed to improve crystallization and minimize ion movement channels. This strategy yields highly stable devices (bandgap of 1.61 eV) with a T80 exceeding 560 hours under 85 °C and 2-sun illumination, surpassing 1100 hours when extrapolated to 85 °C and 1-sun condition.
Results
3D/2D PSCs stability under harsh ageing conditions
PSCs with a stack described as ITO/SnO2/Cs0.05MA0.1FA0.85PbI2.75Br0.25/passivation/PDCBT/PTAA doped with 10 wt% BCF/Au, utilizing various passivators, were fabricated and aged at 85 °C & N2 under 1-sun white light LEDs illumination (Fig. S1 and S2, ESI†). These passivation layers include (1) mono-ammonium spacers: phenethylammonium iodide (PEAI), 4-fluoro-phenethylammonium iodide (FPEAI), n-butylammonium iodide (BAI); (2) di-ammonium spacers: butane-1,4-diammonium iodide (BDAI2). The control (3D) devices retained 65% of their initial PCE after ageing for 200 hours. In contrast, devices with 2D-mono perovskite passivation exhibited a significantly faster PCE decline to ∼35% of their initial PCE, primarily due to the large loss in current density (JSC). The strong JSC loss with minimal fill factor (FF) loss suggests an increase in the concentration of mobile ions, likely due to the formation of numerous vacancies during 2D-mono reconstruction, which may screen the electric field.13,39,41 Conversely, devices with 2D-di passivation demonstrated a stability trend similar to the control devices, indicating enhanced structural stability compared to the 2D-mono structure. These results suggest that the commonly used passivation layers offer no significant benefit or even worsen device stability under harsh ageing conditions.Intrinsic stability of 2D perovskite films
To clarify the devices’ instability issue, we first studied the intrinsic stability of bare 2D iodide films. Representative linear (n-butylammonium, BA or BA2PbI4), cyclic (cyclohexylmethylammonium, CyMA or (CyMA)2PbI4) and aromatic (phenethylammonium, PEA or (PEA)2PbI4) mono-cation spacers based 2D structure (named as [(NH3)R]2PbI4), and representative linear (butyldiammonium, BDA or (BDA)PbI4), cyclic (1,4-cyclohexanedimethylammonium) (CyDMA or (CyDMA)PbI4) and aromatic (1,4-phenylenedimethylammonium (PhDMA or (PhDMA)PbI4) di-cation spacers based 2D structure (named as [(NH3)2R]PbI4)) were chosen for investigation. The as-deposited PEA2PbI4 film exhibited a bright yellow color and well-defined X-ray diffraction peaks along the (00l) planes (Fig. 1(c)), as well as the other two 2D-mono films of BA2PbI4 and (CyMA)2PbI4 (Fig. S3, ESI†). After 20 hours of photo-thermal ageing, these films showed visible darkening and their characteristic diffraction peaks diminished, being replaced by new peaks of PbI2 and metallic lead (Pb0). These 2D-mono structures completely decomposed into PbI2 and Pb0 after 200 hours. Pb0 formation is attributed to PbI2 decomposition, following the redox reaction PbI2 ⇌ Pb0 + I2, driven by light and high temperatures promoting I2 volatilization (shifting the reaction equilibrium towards Pb0). This decomposition can be accelerated through mono-ammonium deprotonation under light stress and reaction with PbI2,10,17,54 leading to the formation of Pb0 and the sublimation of volatile HI or I2. We tentatively propose the following reaction scheme under heat and light conditions:10,17| R-CH2–NH3+ ⇌ R-CH2–NH2 + H+ |
R-CH2–NH2 + PbI2 ⇌ R-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH + Pb0 + 2HI NH + Pb0 + 2HI |
In contrast, BDAPbI4 remained structurally stable after 200 hours (Fig. 1(d)), similar to other 2D-di films of (CyDMA)PbI4 and (PhDMA)PbI4 (Fig. S4, ESI†). It is interesting that although both mono-ammonium and di-ammonium cations have the similar acid dissociation constant (pKa), namely PEA+ pKa = 9.82 while BDA2+ pKa1 =9.90 pKa2 = 10.51, the di-ammonium spacer in 2D is much less reactive with PbI2 or PbI6 octahedra than mono-ammonium in 2D. To clarify the effect of cation charge number and the stacking mode of lead–iodide octahedra on the stability of 2D materials, we further studied the photo-thermal stability of compounds formed by 21 spacers (11 mono-cations and 10 di-cations) and PbI2 (Fig. S3, S4 and Table S1, ESI†). We found that only di-ammonium based 2D perovskites (DJ and near RP phase) are structurally stable after photothermal ageing. In contrast, if di-ammonium and PbI2 form a non-2D structure, fast degradation is observed. Mono-ammonium based compounds with near-DJ structures, RP structures, and non-2D structures degrade rapidly and form Pb0. The impressive photo-thermal stability of 2D-di structure can be due to the reason that in this 2D structure, the bidentate hydrogen bonding of the di-cation interacting with the PbI6 octahedron greatly enhances interaction between the adjacent inorganic layers and the rigidity of the structure,29,55 which suppresses the deprotonation process from ammonium to amine as well as the decomposition of PbI6 octahedron, thus inhibiting the sublimation of the amine and its reaction with PbI2. A similar increase in stability was observed when n values were increased to 2 and 3 (Fig. S5, ESI†). Thus, the di-ammonium based 2D perovskite demonstrate superior intrinsic stability under harsh conditions compared to mono-ammonium counterparts.
Impact of passivators on 3D films
To understand how the 2D-mono and 2D-di passivators affect the optical and structural properties of 3D perovskites during ageing, glass/perovskite/passivator films were subjected to controlled light and temperature conditions (Fig. 2(a)). PEAI is selected as the representative 2D-mono passivator, as it is the most widely used mono-ammonium passivator, while the BDAI2 is selected as the representative 2D-di passivator, as BDAI2 is also widely used and one of the shortest organic spacers that can form stable lead–iodide 2D-di structure (Fig. S4, ESI†), which favours layer to layer carrier transport.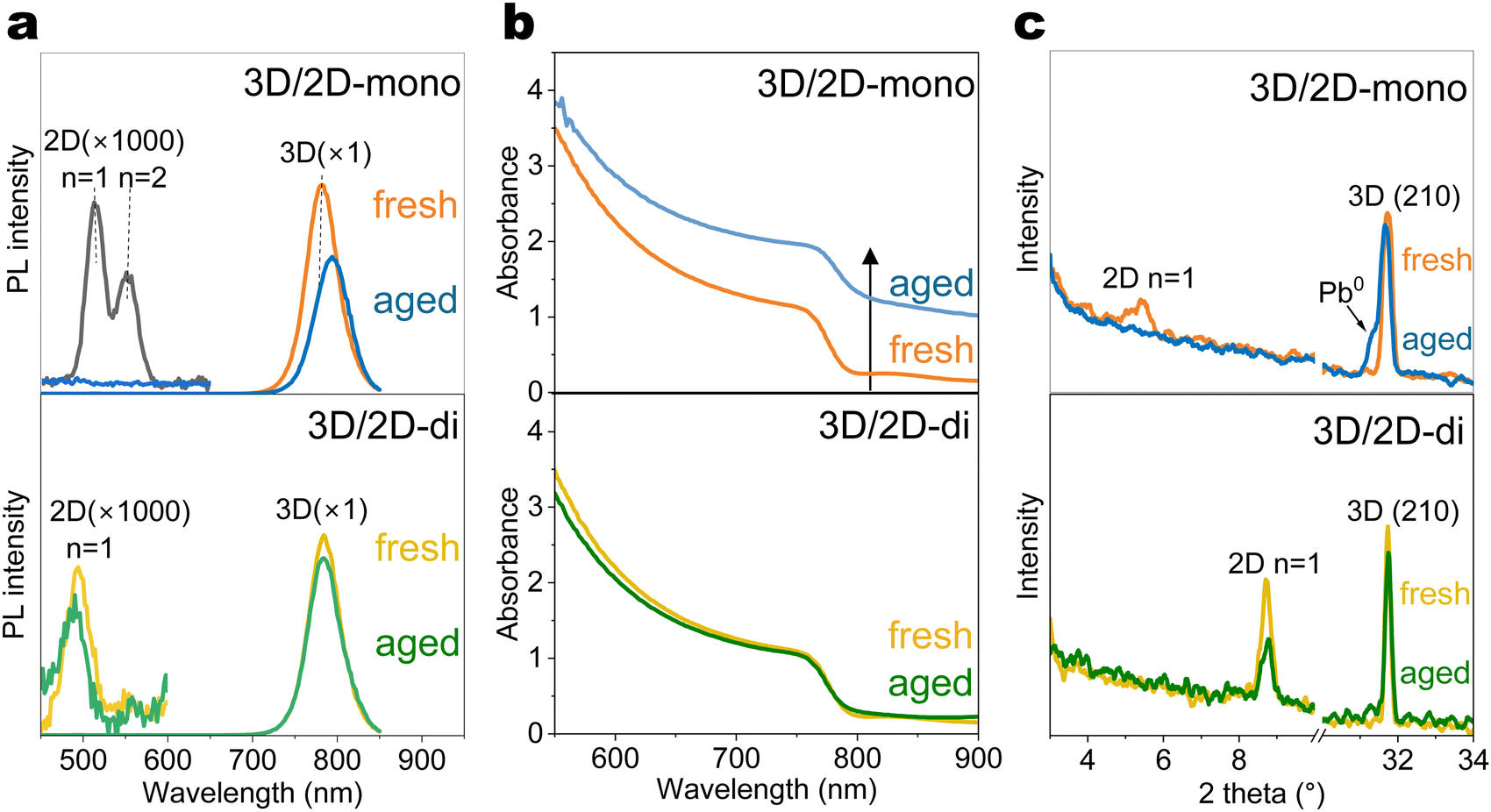 | ||
| Fig. 2 3D/2D perovskite films ageing. (a) Photoluminescence (PL) spectra, (b) UV-Vis spectra and (c) XRD patterns of the 3D perovskite films coated with mono-ammonium (PEAI) or di-ammonium (BDAI2) passivators before and after ageing under light-heat (85 °C and 1-sun illumination) for 300 hours. PEAI and BDAI2 passivators are respectively labelled as “3D/2D-mono” and “3D/2D-di”. | ||
Photoluminescence (PL) spectra (Fig. 2(a) and Fig. S6, ESI†) revealed a main peak at ∼780 nm corresponding to the 3D perovskite. The 3D/2D-mono (PEAI) sample initially showed strong n = 1 and n = 2 PL peaks at ∼520 and ∼560 nm, respectively. Under 1-sun light ageing, the n = 1 peak disappeared, while n ≥ 3 peaks emerged after heat (85 °C) ageing (Fig. S6, ESI†). This n-value change indicates the migration of PEA+ ligands and the reconstruction of the 2D layer. Furthermore, light exhibits a stronger influence in the 3D/2D hetero-junction than heat, likely due to reduced activation energy for ion migration under illumination.56,57 Notably, under the combination of light and heat, all 2D-mono PL peaks vanished and this phenomenon was further confirmed in XRD pattern (Fig. 2(c)). Similar trends were also observed in the perovskite films with other mono-ammonium passivators (Fig. S7, ESI†), highlighting their vulnerability under light-heat ageing conditions. Our results agree with previous literature that light has a strong impact on the 3D/2D-mono interface and heat accelerates this process.12 3D/2D-di (BDAI2) samples, however, exhibited improved stability, though the n = 1 PL peak at around 500 nm slightly weakened after light-heat ageing, as well as the (002) diffraction facet at 2θ = 8.8° (Fig. 2(a) and (c)).
Interesting trends were observed in the UV-Vis absorption spectra (Fig. 2(b) and Fig. S8, ESI†). 3D/2D-mono film exhibited upshifted absorbance after light-induced degradation, particularly under combined light and heat exposure. Typically, the decomposition of perovskites under elevated temperature or light reduces the density of states of perovskite,58 leading to decreased optical density (OD). An increase of OD in transmission mode (absorbance = log(1/transmittance)) may result from an enhanced reflectance or absorptance. The presence of Pb0 (Fig. 2(c)), suggests its shiny metallic nature could contribute to direct or indirect reflection due to film surface unevenness caused by Pb0 formation. Further analysis revealed significant bandgap changes upon combined light-heat degradation except 3D/2D-di (BDAI2) (Fig. S10, ESI†), likely due to MA or Br volatilization altering the active layer composition. This result demonstrates the outstanding structural stability of 3D/2D-di interface under harsh conditions.
Impact of 2D-mono or 2D-di passivation on device characteristics
To further explore the influence of mono- and di-ammonium based 2D passivators on device characteristics, PEAI (2D-mono) and BDAI2 (2D-di) were introduced into the top interface of the device. Device degradation was accelerated under harsher conditions (85 °C, 2-sun illumination) for quicker observation. Visual inspection revealed significant color changes in the top layers of 3D/2D-mono and 3D (without passivation) devices after ageing (Fig. 3(a)). This likely arises from iodide migration into upper layers, as XPS analysis detected higher I 3d core level signals in the HTL based on 3D/2D-mono device compared to that on 3D or 3D/2D-di sample (Fig. 3(b)). Besides, time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) further confirmed iodide migration from the perovskite to the upper layers, with the 3D/2D-mono device exhibiting several times higher I− signal in the HTL and Au electrode compared to the 3D sample (Fig. 3(d)). In contrast, the 3D/2D-di device displayed minimal I− signal at the surface, suggesting its effectiveness in blocking ion migration. Additionally, the increased AuI− signal in 3D/2D-mono devices (Fig. S12a, ESI†) suggests two species: Au0 with I−, or AuI with e−. Former implies serious iodide migration and latter can be caused through potential oxidation by accumulated holes and interaction with migrated I−,33,59 as triiodide complex is more reactive toward Au oxidation.32 Iodide migrating to HTL can de-dope the HTL by suppressing its oxidation.60–62 Kelvin probe measurements revealed a significant Fermi level shift in the HTL of 3D and 3D/2D-mono devices after ageing (−4.8 eV to −4.4 eV), reflecting a strong decrease of the doping level (Fig. 3(c)). The 3D/2D-di device exhibited a smaller shift (−4.8 eV to −4.56 eV), indicating a better conservation of HTL doping. The observed changes within the HTL and electrode are expected to contribute to increased series resistance, as discussed in following sections.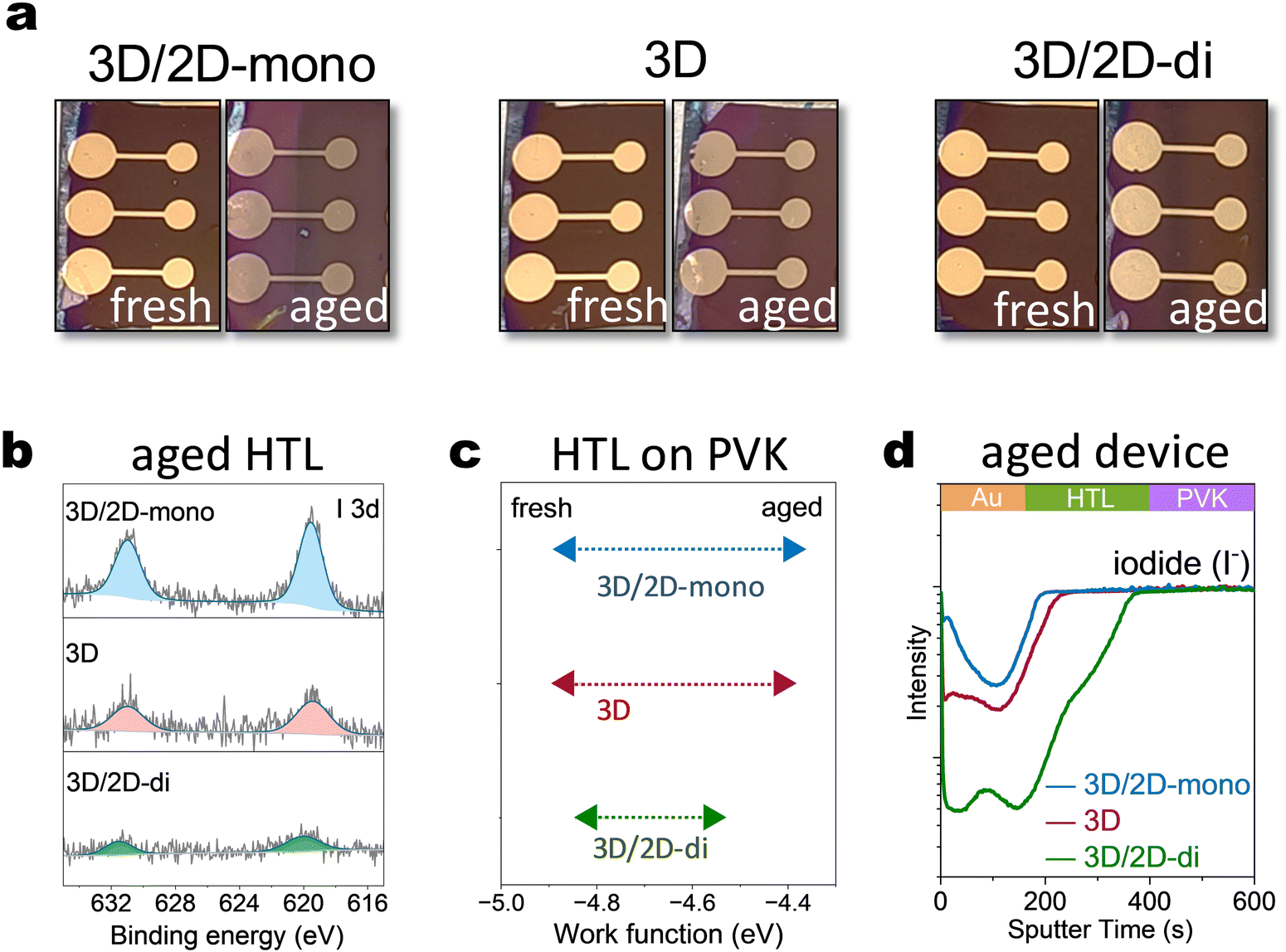 | ||
| Fig. 3 Devices characterization. (a) Image of the 3D/2D-mono, 3D and 3D/2D-di devices before and after 85 °C and 2-sun illumination for 300 hours. (b) XPS spectra of I 3d core level of the HTL on aged 3D/2D-mono, 3D and 3D/2D-di devices. (c) Work function change of the HTL of 3D/2D-mono, 3D and 3D/2D-di devices. (d) Iodide depth profiles of the aged 3D/2D-mono, 3D and 3D/2D-di devices measured by ToF-SIMS. | ||
Long-term operational stability
Fig. 4(a)–(c) and Fig. S13 (ESI†) illustrate the long-term performance of devices. Both 3D (without passivation), and 3D/2D-mono devices exhibit similar degradation trends, ultimately failing abruptly, suggesting similar degradation mechanisms. Although the 3D/2D-di devices initially experience a steeper PCE decline compared to the 3D, their decline rate slows down after 100 hours.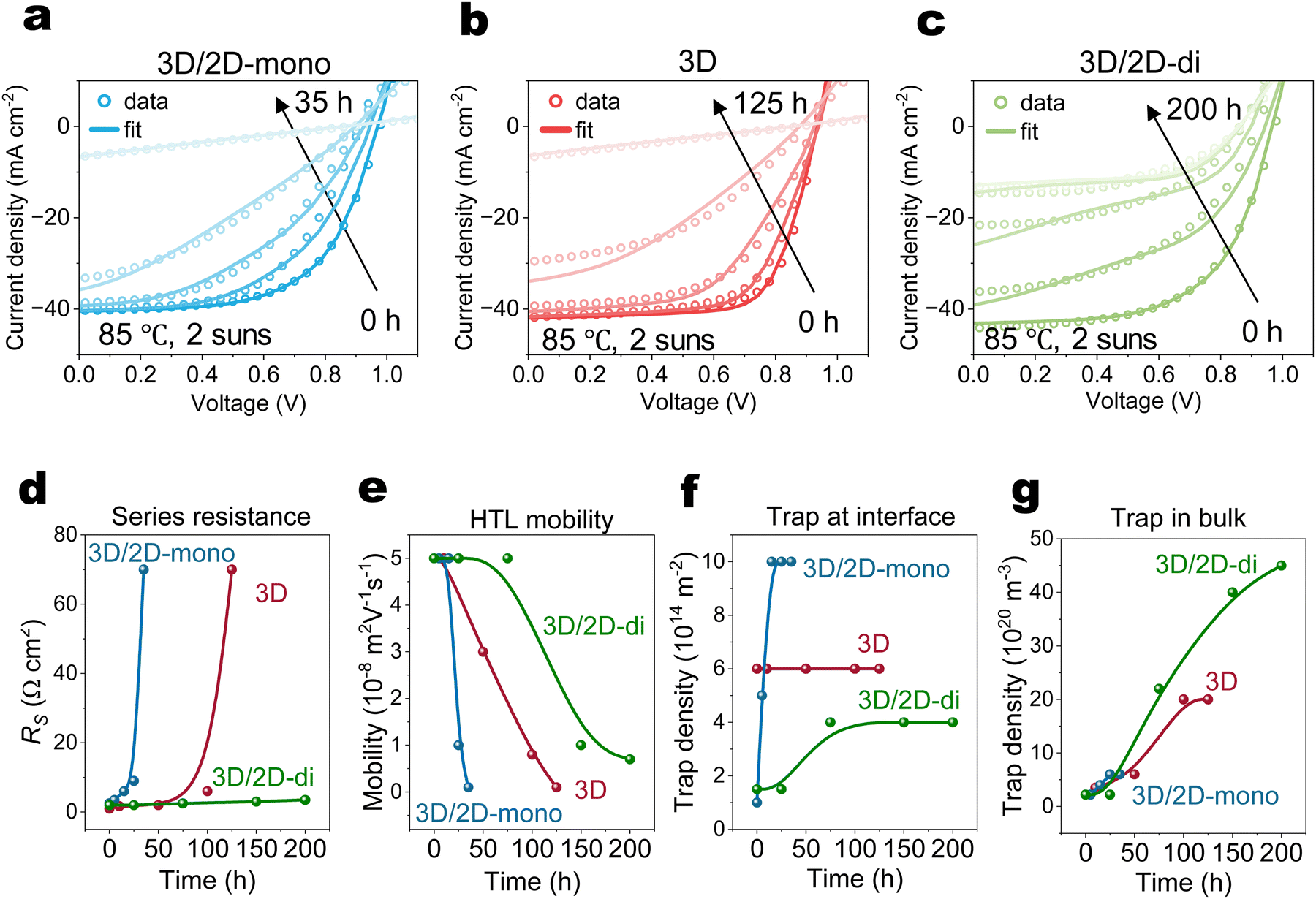 | ||
| Fig. 4 Drift diffusion simulation results. (a)–(c) The experimental JV (open circles) and simulation fitting (solid line) of operational stability of 3D/2D-mono, 3D and 3D/2D-di devices under 85 °C and 2-sun illumination. (d)–(g) Corresponding simulation parameters obtained at various ageing time. The points in (d)–(g) are the values of the different parameters obtained by the simulation, while the connecting lines only show the trend and do not represent the simulated values. | ||
Drift-diffusion simulations
To relate the evolution of electrical performance to degradation-induced changes of physical parameters, J–V curves at various ageing times were simulated using a drift-diffusion (DD) simulator (Fig. 4(a)–(c)).63 Simulated parameters in Fig. 4(d)–(g) and Fig. S14 (ESI†) track the evolution of series resistance (RS), HTL mobility, HTL doping concentration, interfacial traps (upper), bulk traps and mobile ions.In case of the 3D device, the simulations suggest that, over time, the concentrations of bulk traps increase steadily, while the interface trap concentration remains relatively constant. Rapidly rising RS coincides with significant reductions in HTL mobility and doping concentration, ultimately leading to device failure. 3D/2D-mono device exhibits a similar trend as the 3D but at a faster rate. Additionally, an increased concentration of interface traps suggests the deconstruction of the 2D-mono layer, in agreement with the observations from PL and XRD (Fig. 2(a) and (c)). The initial performance drop (0–25 h) in 3D/2D-di device likely arises from an increased excess density of mobile ions confined within the perovskite bulk, screening the electric field and hindering carrier extraction.39,40,64 The rising concentration of bulk traps after 75 h in 3D/2D-di samples can be attributed to the formation of interstitial iodide (Ii) defects in the bulk,47,65 as iodide migration is repelled and rebounded by the rigid structure of 2D-di perovskite. The subsequent performance decay might result from decreased HTL properties, causing slightly increased RS. The carrier density profile (Fig. S16, ESI†), as obtained from the DD simulations, reveals hole accumulation at the HTL/Au interface during degradation due to reduced HTL doping and mobility. In addition, impedance spectroscopy measurements show that aged 3D and 3D/2D-mono devices exhibit high resistance and lack of frequency-dependent capacitance (Fig. S17, ESI†). In contrast, the aged 3D/2D-di device displays higher low-frequency response and lower intermediate-frequency response, agreeing with our simulation results of increased recombination rate (due to increased traps density) and interfacial charge accumulation.66,67 Lastly, although the DD simulations are not definitive in decoupling the degradation losses and there are likely other parameter sets that can reproduce the trends, our finding in DD simulation agrees well with experimental findings, indicating reasonable result.
Degradation mechanisms
Fig. 5 summarizes the proposed degradation mechanisms for each device type. In the 3D samples, the early degradation originates from increased metallic lead Pb0 acting as a deep trap for charge carriers. I2 is generated simultaneously with Pb0 through PbI2 decomposition, increasing the vacancy concentration as well. Although iodide vacancy (VI) is a shallow trap in perovskite and is expected to have little influence on recombination properties,46 high vacancy concentrations can still affect the recombination (potentially dominating the recombination).68 They generate mobile ion species like I− and I3− (I2 + I−) that are known to migrate within the perovskite layer. In the case of a triphenylamine (TPA) based HTL (PTAA), iodine has been reported to react with the solitary pair of electrons of the TPA unit leading to a high iodine adsorption capacity.69 While iodine can dope specific hole conductors,70 iodide/polyiodide species was reported to suppress HTL oxidation60 and subsequently decrease the doping concentration and mobility of HTL. This leads to an overall reduction in the HTL conductivity which is observed by an increasing RS in the corresponding solar cell devices. As the binding energy of gold electrodes in aged devices shifts to higher energy (Fig. S18, ESI†), gold iodide species (AuIx) are likely produced at the HTL/Au interface (Fig. S12, ESI†) which as well will increase RS due to their lower conductivity. This scenario explains the sharp drop of JSC leading to device failure. The 3D/2D-mono devices exhibit a similar mechanism as the 3D device, but largely accelerated due to 2D-mono and PbI2 decomposition. Higher vacancy concentration and propensity for iodide migration result in a higher iodide/iodine concentration in HTL, causing much faster device degradation.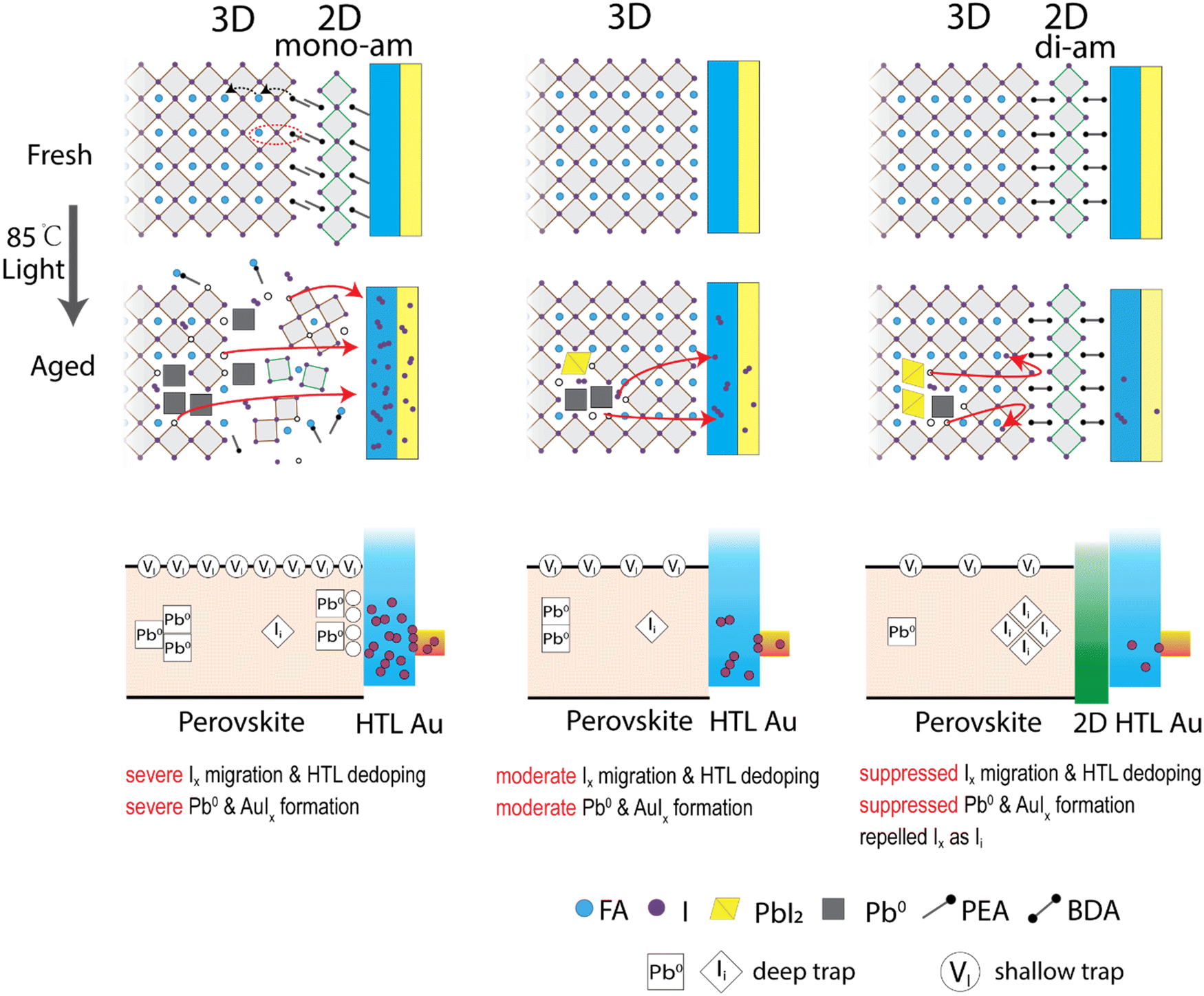 | ||
| Fig. 5 Proposed degradation mechanisms in 3D/2D-mono, 3D and 3D/2D-di perovskite devices under 85 °C and light-soaking. Mono-ammonium and di-ammonium are labelled as mono-am and di-am, respectively. | ||
In contrast, the 3D/2D-di devices show a different degradation mechanism. The robust 2D-di structure blocks mobile iodide from reaching the HTL during degradation and slows down the reaction equilibrium towards Pb0. Mobile ions remain confined within the perovskite bulk, leading to an initial performance drop caused by shielding the internal electrical field. A subsequent performance drop is caused by the formation of deep traps from interstitial iodide (Ii), arising from the enrichment of iodide at interface under external bias and rebounding of iodide by the 2D-di structure back into the lattice interstitial.65 Ii is known to have a deep trap state in perovskites and causes more serious non-radiative recombination, comparing with the iodide vacancy (VI).46,47,71 At the later stage, a small degree of Pb0 formation and limited iodine migration to HTL contributes to further gradual degradation.
Strategies for improved stability based on degradation mechanisms
Understanding the mechanism of electrical performance degradation for the various 2D cations provides valuable insights for designing stable perovskite cells. Two key areas for improvement are identified. First, developing transport layers and electrodes resistant to halide migration and iodide/polyiodide redox reactions is crucial. This is essential to provide unwanted reaction of iodide with either the hole extraction layers or the electrode. Second, minimizing the channels for mobile ions within the perovskite bulk reduces the potential probability for ion migration. The latter can be achieved by using perovskite compositions with less volatile A-site cations, such as Cs0.15FA0.85PbI2.75Br0.25 (CsFA) with a bandgap of 1.61 eV instead of Cs0.05MA0.1FA0.85PbI2.75Br0.25 (CsMAFA) with 1.57 eV bandgap. Fig. S19 (ESI†) demonstrates the improved stability of CsFA perovskite solar cells compared to CsMAFA devices. Slower Jsc and FF degradation suggest reduced Pb0 formation (Fig. S20, ESI†) and thus reduced recombination rate. To obtain the acceleration factor (AF), we define the degradation rate as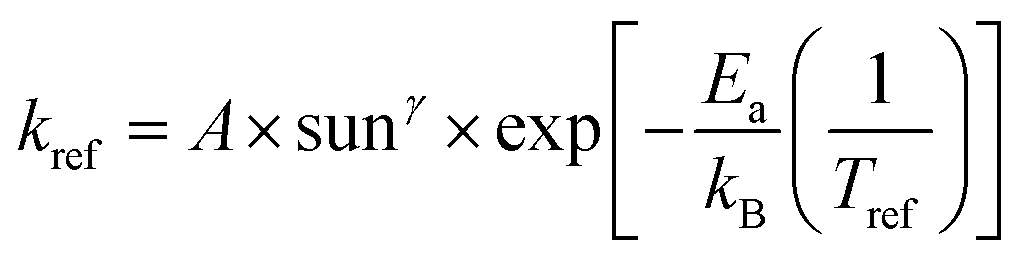 , where Ea is the activation energy, kB is the Boltzmann constant, Tref is the ageing temperature, sunref is the ageing light intensity, entering non-linearly into the equation via the exponent γ, and A is pre-exponential factor.72 Then AF is calculated according to:
, where Ea is the activation energy, kB is the Boltzmann constant, Tref is the ageing temperature, sunref is the ageing light intensity, entering non-linearly into the equation via the exponent γ, and A is pre-exponential factor.72 Then AF is calculated according to: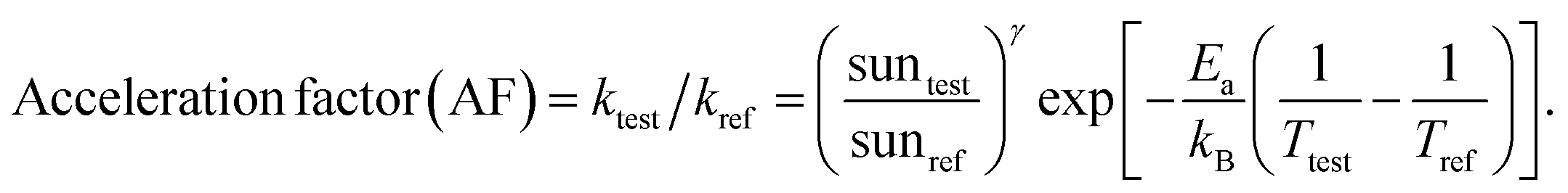
Since the ageing temperature is kept at 85 °C, γ can be obtained by fitting the equation kdecay = a × sunγ, where kdecay = 1/T80 or 1/T60, T80 and T60 is the time when the PCE drops to its 80% or 60% initial value. If γ is close to 1, it means the degradation rate increases linearly with light intensity. As shown in Fig. S21 (ESI†), for CsMAFA samples, γ changes from 1.07 (T80) to 1.86 (T60), indicating multiple degradation processes: a short-term, defect-generation and ion migration dominated phase, and a long-term dominated by halide migration to upper layers and subsequent redox reactions. In contrast, CsFA devices exhibit a linear light-dependence relationship at both T80 (γ = 1.01) and T60 (γ = 1.09), suggesting suppression of halide migration processes at long-term region due to reduction of vacancy resulted from volatile MA.
Having learned that di-ammonium cations or 2D-di based perovskites can effectively block iodide migration, and having understood the basic physical degradation processes related to iodide enrichment at an interface or the formation of iodide interstitials, it is natural to ask the question whether di-ammonium cations can suppress these effects throughout the whole perovskite layer. That concept is further fuelled by previous studies that have shown that 2D-di phase or di-ammonium halide molecules in or re-dissolved in the precursor can act as a seed for improved crystallization, whereas the di-ammonium molecules got located at the perovskite grain boundaries after annealing.73,74 To address this issue, we incorporated di-ammonium ligands into the perovskite by first depositing a layer of 2D-di on top of ETL at the n-type interface (target-2D-di) instead the top surface. When spin-coating the perovskite precursor on top of the 2D-di layer, most of the di-ammonium molecules will be redissolved and thereby introduced into the bulk perovskite. It is noted that no 2D signal is detected according to PL and XRD results, even when the thickness of the bottom 2D-di layer is increased ten times (Fig. S22, ESI†), suggesting the diffusion of di-ammonium ligand into bulk grain boundary and less probability of forming continuous and even 2D-di perovskite phase. This modification resulted a further remarkable improvement in stability: a T80 lifetime exceeding 560 h under 85 °C and 2 suns was observed (Fig. 6), which is about 5 times better than for the unpassivated devices (see Fig. 4 and Fig. S24, ESI†), and 10 times better than the 2D-mono passivated. Considering the light-related AF (γ = 1.01 under 85 °C), the extrapolated T80 lifetime under 85 °C and 1 sun is beyond 1100 hours, making it one of the most stable PSCs reported under harsh conditions (Table S2, ESI†). These grain-boundary-located di-ammonium ligands likely confine the mobile ions (I− or Cs+) within the grains, mitigating recombination, formation of Pb0, and phase segregation caused by A-site cation (Fig. S25 and S26, ESI†).
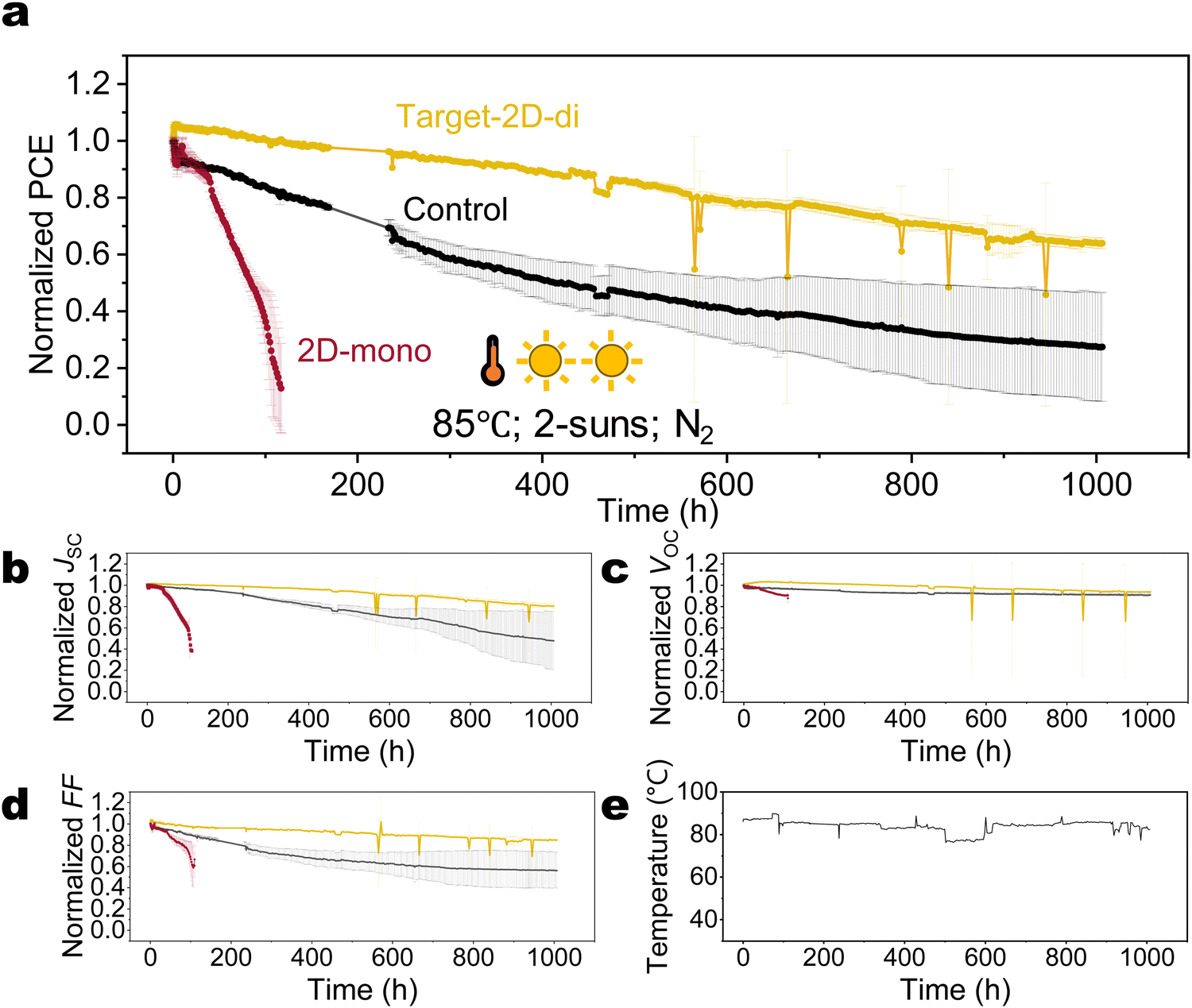 | ||
| Fig. 6 Operational stability performance of control and target devices under 85 °C and 2-suns illumination: (a)–(d) normalized PCE, JSC, VOC, and FF. Error bar represents the standard deviation of three individual devices. The data was normalized at 0.6 hour when the temperature increased to 85 °C. (e) The temperature profile during stability measurement. | ||
Conclusions
In this study, we have examined the impact of mono- and di-ammonium 2D perovskite passivation on device stability under photothermal accelerated ageing conditions. Both passivation types exhibited distinct degradation mechanisms. 3D/2D-mono interfaces undergo rapid reconstruction and decomposition, triggering Pb0, vacancy formation, and facilitated halide migration, leading to swift device degradation. Conversely, 3D/2D-di interfaces remain stable, effectively blocking halide migration but suffering from early performance losses due to mobile ion induced electric field shielding and interstitial defect formation within the bulk. By incorporating di-ammonium passivators into bulk and minimizing mobile ions channels in the CsFA based perovskite, we achieved highly-stable devices with a remarkable T80 lifetime exceeding 560 hours under 85 °C and 2-suns. We emphasize the need for holistic stability approaches beyond just passivator development, designing robust transport layers against iodide/polyiodide redox and intrinsic perovskite quality for the realization of highly stable PSCs. This work reveals the degradation mechanisms of PSCs under harsh ageing conditions, providing a theoretical basis for the development of PSCs accelerated ageing standards in the photovoltaic field.Author contributions
Z. P. conceived the idea and designed the experiment. L. L. and C. J. B. supervised the project. Z. P., J. L. and Y. Z. fabricated devices. A. V. performed the ToF-SIMS measurement. F. S., A. S. and R. F. performed the XPS measurement. Z. P. and V. L. C. performed the DD simulation. Z. P. and T. H. performed long-term stability test. Z. P. and A. O. conducted films’ optical measurement. S. Z., K. Z., Chaohui L., J. T., Chao L., L. N. and M. S. helped with the interpretation and analysis of the results.Data availability
Parameter sets used for simulation are listed in the ESI.†Conflicts of interest
M. S. is co-founders of FastChar UG.Acknowledgements
Z. P., J. T., K. Z., and C. H. L. acknowledge the financial support from China Scholarship Council. C. J. B. gratefully acknowledges the financial support through the Bavarian Initiative “Solar Technologies go Hybrid” (SolTech), the DFG-SFB953 (project no. 182849149), DFG-GRK2495 (ITRG2495), and DFG-INST 90/917-1 FUGG. Z. P. and J. T. gratefully acknowledge funding of the Erlangen Graduate School in Advanced Optical Technologies (SAOT) by the Bavarian State Ministry for Science and Art. M. S. acknowledges the Vice-Chancellor Early Career Professorship Scheme from CUHK for funding. J. L. is grateful to the National Natural Science Foundation of China (grant no. 62104031) and the Technical Field Funds of 173 Project (grant no. 24JJ210663A) for financial support. A. V. acknowledges funding from the Slovak Research and Development Agency (APVV 20-0310).References
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121(4), 2230–2291 CrossRef CAS.
- J. Gong, M. Hao, Y. Zhang, M. Liu and Y. Zhou, Angew. Chem., Int. Ed., 2022, 61(10), e202112022 CrossRef CAS PubMed.
- I. Metcalf, S. Sidhik, H. Zhang, A. Agrawal, J. Persaud, J. Hou, J. Even and A. D. Mohite, Chem. Rev., 2023, 123(15), 9565–9652 CrossRef CAS.
- G. Wu, R. Liang, M. Ge, G. Sun, Y. Zhang and G. Xing, Adv. Mater., 2022, 34(8), e2105635 CrossRef PubMed.
- S. Sidhik, Y. Wang, M. De Siena, R. Asadpour, A. J. Torma, T. Terlier, K. Ho, W. Li, A. B. Puthirath, X. Shuai, A. Agrawal, B. Traore, M. Jones, R. Giridharagopal, P. M. Ajayan, J. Strzalka, D. S. Ginger, C. Katan, M. A. Alam, J. Even, M. G. Kanatzidis and A. D. Mohite, Science, 2022, 377(6613), 1425–1430 CrossRef CAS PubMed.
- S. Tan, T. Huang, I. Yavuz, R. Wang, T. W. Yoon, M. Xu, Q. Xing, K. Park, D. K. Lee, C. H. Chen, R. Zheng, T. Yoon, Y. Zhao, H. C. Wang, D. Meng, J. Xue, Y. J. Song, X. Pan, N. G. Park, J. W. Lee and Y. Yang, Nature, 2022, 605(7909), 268–273 CrossRef CAS.
- C. Liu, Y. Yang, H. Chen, J. Xu, A. Liu, A. S. R. Bati, H. H. Zhu, L. Grater, S. S. Hadke, C. Y. Huang, V. K. Sangwan, T. Cai, D. Shin, L. X. Chen, M. C. Hersam, C. A. Mirkin, B. Chen, M. G. Kanatzidis and E. H. Sargent, Science, 2023, 382(6672), 810–815 CrossRef CAS.
- R. Chen, J. A. Wang, Z. H. Liu, F. M. Ren, S. W. Liu, J. Zhou, H. X. Wang, X. Meng, Z. Zhang, X. Y. Guan, W. X. Liang, P. A. Troshin, Y. B. Qi, L. Y. Han and W. Chen, Nat. Energy, 2023, 8(8), 839–849 CrossRef CAS.
- G. Szabó and P. V. Kamat, ACS Energy Lett., 2023, 9(1), 193–200 CrossRef.
- M. Wang, Z. Shi, C. Fei, Z. J. D. Deng, G. Yang, S. P. Dunfield, D. P. Fenning and J. Huang, Nat. Energy, 2023, 8(11), 1229–1239 CrossRef CAS.
- J. Chakkamalayath, N. Hiott and P. V. Kamat, ACS Energy Lett., 2022, 169–171 Search PubMed.
- F. Fiorentino, M. D. Albaqami, I. Poli and A. Petrozza, ACS Appl. Mater. Interfaces, 2022, 14(30), 34180–34188 CrossRef CAS.
- C. A. R. Perini, E. Rojas-Gatjens, M. Ravello, A. F. Castro-Mendez, J. Hidalgo, Y. An, S. Kim, B. Lai, R. Li, C. Silva-Acuna and J. P. Correa-Baena, Adv. Mater., 2022, e2204726 CrossRef PubMed.
- S. M. Park, M. Wei, J. Xu, H. R. Atapattu, F. T. Eickemeyer, K. Darabi, L. Grater, Y. Yang, C. Liu, S. Teale, B. Chen, H. Chen, T. Wang, L. Zeng, A. Maxwell, Z. Wang, K. R. Rao, Z. Cai, S. M. Zakeeruddin, J. T. Pham, C. M. Risko, A. Amassian, M. G. Kanatzidis, K. R. Graham, M. Gratzel and E. H. Sargent, Science, 2023, 381(6654), 209–215 CrossRef CAS.
- L. Luo, H. P. Zeng, Z. W. Wang, M. Li, S. You, B. Chen, A. Maxwell, Q. Y. An, L. M. Cui, D. Y. Luo, J. T. Hu, S. Z. Li, X. Q. Cai, W. X. Li, L. Li, R. Guo, R. Huang, W. X. Liang, Z. H. Lu, L. Q. Mai, Y. G. Rong, E. H. Sargent and X. Li, Nat. Energy, 2023, 8(3), 294–303 CAS.
- J. Liang, A. Sun, Z. Zhang, Y. Zheng, X. Wu, C. Tian, Z. Chen and C.-C. Chen, ACS Mater. Lett., 2023, 5(5), 1395–1400 CrossRef CAS.
- J. Hu, R. A. Kerner, I. Pelczer, B. P. Rand and J. Schwartz, ACS Energy Lett., 2021, 6(6), 2262–2267 CrossRef CAS.
- R. A. Kerner, T. H. Schloemer, P. Schulz, J. J. Berry, J. Schwartz, A. Sellinger and B. P. Rand, J. Mater. Chem. C, 2019, 7(18), 5251–5259 RSC.
- R. A. Kerner, T. H. Schloemer, P. Schulz, J. J. Berry, J. Schwartz, A. Sellinger and B. P. Rand, J. Mater. Chem. C, 2019, 7(18), 5244–5250 RSC.
- C. Liu, Y. Yang, K. Rakstys, A. Mahata, M. Franckevicius, E. Mosconi, R. Skackauskaite, B. Ding, K. G. Brooks, O. J. Usiobo, J. N. Audinot, H. Kanda, S. Driukas, G. Kavaliauskaite, V. Gulbinas, M. Dessimoz, V. Getautis, F. De Angelis, Y. Ding, S. Dai, P. J. Dyson and M. K. Nazeeruddin, Nat. Commun., 2021, 12(1), 6394 CrossRef CAS PubMed.
- D. Gao, R. Li, X. Chen, C. Chen, C. Wang, B. Zhang, M. Li, X. Shang, X. Yu, S. Gong, T. Pauporte, H. Yang, L. Ding, J. Tang and J. Chen, Adv. Mater., 2023, 35(23), e2301028 CrossRef.
- Q. Jiang, J. Tong, Y. Xian, R. A. Kerner, S. P. Dunfield, C. Xiao, R. A. Scheidt, D. Kuciauskas, X. Wang, M. P. Hautzinger, R. Tirawat, M. C. Beard, D. P. Fenning, J. J. Berry, B. W. Larson, Y. Yan and K. Zhu, Nature, 2022, 611(7935), 278–283 CrossRef CAS PubMed.
- H. Su, X. Lin, Y. Wang, X. Liu, Z. Qin, Q. Shi, Q. Han, Y. Zhang and L. Han, Sci. China: Chem., 2022, 65(7), 1321–1329 CrossRef CAS.
- M.-H. Tremblay, J. Bacsa, B. Zhao, F. Pulvirenti, S. Barlow and S. R. Marder, Chem. Mater., 2019, 31(16), 6145–6153 CrossRef CAS.
- X. Q. Jiang, J. F. Zhang, S. Ahmad, D. D. Tu, X. Liu, G. Q. Jia, X. Guo and C. Li, Nano Energy, 2020, 75, 104892 CrossRef CAS.
- Y. Zheng, X. Wu, R. Zhuang, C. Tian, A. Sun, C. Tang, Y. Liu, Y. Hua and C. C. Chen, Adv. Funct. Mater., 2023, 33(25), 2300576 CrossRef CAS.
- Y. Shang, Y. Liao, Q. Wei, Z. Wang, B. Xiang, Y. Ke, W. Liu and Z. Ning, Sci. Adv., 2019, 5(8), eaaw8072 CrossRef CAS PubMed.
- J. K. Wang, L. W. Zeng, D. Zhang, A. Maxwell, H. Chen, K. Datta, A. Caiazzo, W. H. M. Remmerswaal, N. R. M. Schipper, Z. H. Chen, K. V. Ho, A. Dasgupta, G. Kusch, R. Ollearo, L. Bellini, S. F. Hu, Z. W. Wang, C. W. Li, S. Teale, L. Grater, B. Chen, M. M. Wienk, R. A. Oliver, H. J. Snaith, R. A. J. Janssen and E. H. Sargent, Nat. Energy, 2023, 9, 70–80 CrossRef.
- S. Ahmad, P. Fu, S. Yu, Q. Yang, X. Liu, X. Wang, X. Wang, X. Guo and C. Li, Joule, 2019, 3(3), 794–806 CrossRef CAS.
- F. Zhang, S. Y. Park, C. Yao, H. Lu, S. P. Dunfield, C. Xiao, S. Ulicna, X. Zhao, L. Du Hill, X. Chen, X. Wang, L. E. Mundt, K. H. Stone, L. T. Schelhas, G. Teeter, S. Parkin, E. L. Ratcliff, Y. L. Loo, J. J. Berry, M. C. Beard, Y. Yan, B. W. Larson and K. Zhu, Science, 2022, 375(6576), 71–76 CrossRef CAS.
- T. L. Leung, I. Ahmad, A. A. Syed, A. M. C. Ng, J. Popovic and A. B. Djurisic, Commun. Mater., 2022, 3(1), 63 CrossRef.
- R. A. Kerner, L. Zhao, S. P. Harvey, J. J. Berry, J. Schwartz and B. P. Rand, ACS Energy Lett., 2020, 5(11), 3352–3356 CrossRef CAS.
- J. Pospisil, A. Guerrero, O. Zmeskal, M. Weiter, J. J. Gallardo, J. Navas and G. Garcia-Belmonte, Adv. Funct. Mater., 2019, 29(32), 1900881 CrossRef.
- X. Li, S. Fu, W. Zhang, S. Ke, W. Song and J. Fang, Sci. Adv., 2020, 6(51), eabd1580 CrossRef CAS.
- Y. M. Sha, E. N. Bi, Y. Zhang, P. B. Ru, W. Y. Kong, P. Zhang, X. D. Yang, H. Chen and L. Y. Han, Adv. Energy Mater., 2021, 11(5), 2003301 CrossRef CAS.
- S. Bitton and N. Tessler, Energy Environ. Sci., 2023, 16(6), 2621–2628 RSC.
- Z. Xu, D. D. Astridge, R. A. Kerner, X. Zhong, J. Hu, J. Hong, J. A. Wisch, K. Zhu, J. J. Berry, A. Kahn, A. Sellinger and B. P. Rand, J. Am. Chem. Soc., 2023, 145(21), 11846–11858 CrossRef CAS.
- C. Eames, J. M. Frost, P. R. Barnes, B. C. O'Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- J. Thiesbrummel, V. M. Le Corre, F. Pena-Camargo, L. Perdigon-Toro, F. Lang, F. J. Yang, M. Grischek, E. Gutierrez-Partida, J. Warby, M. D. Farrar, S. Mahesh, P. Caprioglio, S. Albrecht, D. Neher, H. J. Snaith and M. Stolterfoht, Adv. Energy Mater., 2021, 11(34), 2101447 CrossRef CAS.
- V. M. Le Corre, J. Diekmann, F. Peña-Camargo, J. Thiesbrummel, N. Tokmoldin, E. Gutierrez-Partida, K. P. Peters, L. Perdigón-Toro, M. H. Futscher, F. Lang, J. Warby, H. J. Snaith, D. Neher and M. Stolterfoht, Sol. RRL, 2022, 6(4), 2100772 CrossRef CAS.
- J. Thiesbrummel, S. H. Shah, E. Gutierrez-Partida, F. S. Zu, F. Pena-Camargo, S. Zeiske, J. Diekmann, F. Y. Ye, K. P. Peters, K. O. Brinkmann, P. Caprioglio, A. Dasgupta, S. Seo, F. A. Adeleye, J. Warby, Q. Jeangros, F. L. Lang, S. Zhang, S. Albrecht, T. Riedl, A. Armin, D. Neher, N. Koch, Y. Z. Wu, V. M. Le Corre, H. Snaith and M. Stolterfoht, Nat. Energy, 2024, 9(6), 664–676 CrossRef CAS.
- W. Tress, N. Marinova, T. Moehl, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Gratzel, Energy Environ. Sci., 2015, 8(3), 995–1004 RSC.
- M. C. Brennan, S. Draguta, P. V. Kamat and M. Kuno, ACS Energy Lett., 2017, 3(1), 204–213 CrossRef.
- T. Elmelund, B. Seger, M. Kuno and P. V. Kamat, ACS Energy Lett., 2019, 5(1), 56–63 CrossRef.
- Z. Xu, R. A. Kerner, S. P. Harvey, K. Zhu, J. J. Berry and B. P. Rand, ACS Energy Lett., 2022, 513–520 Search PubMed.
- D. Meggiolaro, S. G. Motti, E. Mosconi, A. J. Barker, J. Ball, C. A. R. Perini, F. Deschler, A. Petrozza and F. De Angelis, Energy Environ. Sci., 2018, 11(3), 702–713 RSC.
- J. L. Minns, P. Zajdel, D. Chernyshov, W. van Beek and M. A. Green, Nat. Commun., 2017, 8, 15152 CrossRef CAS.
- M. A. Uddin, P. J. S. Rana, Z. Ni, G. Yang, M. Li, M. Wang, H. Gu, H. Zhang, B. D. Dou and J. Huang, Nat. Commun., 2024, 15(1), 1355 CrossRef CAS.
- Y. Bai, Z. Huang, X. Zhang, J. Lu, X. Niu, Z. He, C. Zhu, M. Xiao, Q. Song, X. Wei, C. Wang, Z. Cui, J. Dou, Y. Chen, F. Pei, H. Zai, W. Wang, T. Song, P. An, J. Zhang, J. Dong, Y. Li, J. Shi, H. Jin, P. Chen, Y. Sun, Y. Li, H. Chen, Z. Wei, H. Zhou and Q. Chen, Science, 2022, 378(6621), 747–754 CrossRef CAS PubMed.
- Z. Liang, Y. Zhang, H. Xu, W. Chen, B. Liu, J. Zhang, H. Zhang, Z. Wang, D. H. Kang, J. Zeng, X. Gao, Q. Wang, H. Hu, H. Zhou, X. Cai, X. Tian, P. Reiss, B. Xu, T. Kirchartz, Z. Xiao, S. Dai, N. G. Park, J. Ye and X. Pan, Nature, 2023, 624(7992), 557–563 CrossRef CAS PubMed.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulovic, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y. B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Graetzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Koebler, M. S. Leite, S. Liu, Y. L. Loo, J. M. Luther, C. Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, C. Odabasi, A. Osherov, N. G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. B. Xie, R. Yildirim, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5(1), 35–49 CrossRef.
- Z. Li, X. Sun, X. Zheng, B. Li, D. Gao, S. Zhang, X. Wu, S. Li, J. Gong, J. M. Luther, Z. Li and Z. Zhu, Science, 2023, 382(6668), 284–289 CrossRef CAS.
- Q. Jiang, R. Tirawat, R. A. Kerner, E. A. Gaulding, Y. Xian, X. Wang, J. M. Newkirk, Y. Yan, J. J. Berry and K. Zhu, Nature, 2023, 623(7986), 313–318 CrossRef CAS PubMed.
- J. Liang, Z. Zhang, Y. Huang, Q. Xue, Y. Zheng, X. Wu, C. Tian, Y. Zhang, Y. Wang, Z. Chen and C. C. Chen, Adv. Funct. Mater., 2022, 2207177 CrossRef CAS.
- S. Min and J. Cho, Adv. Opt. Mater., 2024, 2302516 CrossRef CAS.
- W. Zhou, Y. Zhao, X. Zhou, R. Fu, Q. Li, Y. Zhao, K. Liu, D. Yu and Q. Zhao, J. Phys. Chem. Lett., 2017, 8(17), 4122–4128 CrossRef CAS.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365(6452), 473–478 CrossRef CAS.
- Y. Zhao, J. Zhang, Z. Xu, S. Sun, S. Langner, N. T. P. Hartono, T. Heumueller, Y. Hou, J. Elia, N. Li, G. J. Matt, X. Du, W. Meng, A. Osvet, K. Zhang, T. Stubhan, Y. Feng, J. Hauch, E. H. Sargent, T. Buonassisi and C. J. Brabec, Nat. Commun., 2021, 12(1), 2191 CrossRef CAS PubMed.
- J. C. Pérez-Martínez, M. Berruet, C. Gonzales, S. Salehpour, A. Bahari, B. Arredondo and A. Guerrero, Adv. Funct. Mater., 2023, 33(47), 2300576 CrossRef.
- J. Carrillo, A. Guerrero, S. Rahimnejad, O. Almora, I. Zarazua, E. Mas-Marza, J. Bisquert and G. Garcia-Belmonte, Adv. Energy Mater., 2016, 6(9), 1502246 CrossRef.
- S. Kim, S. Bae, S. W. Lee, K. Cho, K. D. Lee, H. Kim, S. Park, G. Kwon, S. W. Ahn, H. M. Lee, Y. Kang, H. S. Lee and D. Kim, Sci. Rep., 2017, 7(1), 1200 CrossRef PubMed.
- T. Wang, Y. Zhang, W. Kong, L. Qiao, B. Peng, Z. Shen, Q. Han, H. Chen, Z. Yuan, R. Zheng and X. Yang, Science, 2022, 377(6611), 1227–1232 CrossRef CAS PubMed.
- M. Koopmans, V. Corre and L. Koster, J. Open Source Softw., 2022, 7(70), 3727 CrossRef.
- M. Stolterfoht, A. Armin, B. Philippa and D. Neher, J. Phys. Chem. Lett., 2016, 7(22), 4716–4721 CrossRef CAS.
- C. Li, A. Guerrero, S. Huettner and J. Bisquert, Nat. Commun., 2018, 9(1), 5113 CrossRef.
- L. J. Bennett, A. J. Riquelme, J. A. Anta, N. E. Courtier and G. Richardson, Phys. Rev. Appl., 2023, 19(1), 014061 CrossRef CAS.
- W. Clarke, G. Richardson and P. Cameron, Adv. Energy Mater., 2024, 14, 2400955 CrossRef CAS.
- Y. Yuan, G. Yan, C. Dreessen, T. Rudolph, M. Hulsbeck, B. Klingebiel, J. Ye, U. Rau and T. Kirchartz, Nat. Mater., 2024, 23(3), 391–397 CrossRef CAS.
- T. Geng, C. Zhang, M. Liu, C. Hu and G. Chen, J. Mater. Chem. A, 2020, 8(5), 2820–2826 RSC.
- Y. Hou, X. Du, S. Scheiner, D. P. McMeekin, Z. Wang, N. Li, M. S. Killian, H. Chen, M. Richter, I. Levchuk, N. Schrenker, E. Spiecker, T. Stubhan, N. A. Luechinger, A. Hirsch, P. Schmuki, H. P. Steinruck, R. H. Fink, M. Halik, H. J. Snaith and C. J. Brabec, Science, 2017, 358(6367), 1192–1197 CrossRef CAS PubMed.
- W. Li, J. Liu, F.-Q. Bai, H.-X. Zhang and O. V. Prezhdo, ACS Energy Lett., 2017, 2(6), 1270–1278 CrossRef CAS.
- Y. Li, X. Huang, K. Ding, H. K. M. Sheriff, L. Ye, H. Liu, C. Z. Li, H. Ade and S. R. Forrest, Nat. Commun., 2021, 12(1), 5419 CrossRef CAS PubMed.
- C. Luo, G. H. Zheng, F. Gao, X. J. Wang, Y. Zhao, X. Y. Gao and Q. Zhao, Joule, 2022, 6(1), 240–257 CrossRef CAS.
- S. W. Liu, X. Y. Guan, W. S. Xiao, R. Chen, J. Zhou, F. M. Ren, J. A. Wang, W. T. Chen, S. B. Li, L. B. Qiu, Y. Zhao, Z. H. Liu and W. Chen, Adv. Funct. Mater., 2022, 32(38), 2205009 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03869j |
| This journal is © The Royal Society of Chemistry 2024 |