
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined temperature and salinity effects on the passive sampling of PAHs with an assessment of impacts to petroleum toxicity†
Ibukun
Ola
 *a,
Carsten
Drebenstedt
a,
Robert M.
Burgess
b,
Lane
Tidwell
c,
Kim
Anderson
c,
Nils
Hoth
a and
Christoph
Külls
d
*a,
Carsten
Drebenstedt
a,
Robert M.
Burgess
b,
Lane
Tidwell
c,
Kim
Anderson
c,
Nils
Hoth
a and
Christoph
Külls
d
aInstitute of Mining and Special Civil Engineering, Technical University Mining Academy, Gustav-Zeuner Street 1A, Freiberg, 09599, Germany. E-mail: Ibukun.Ola@doktorand.tu-freiberg.de
bU.S. Environmental Protection Agency, Office of Research and Development, Center for Environmental Measurement and Modeling, Atlantic Coastal Environmental Sciences Division, 27 Tarzwell Drive, Narragansett, Rhode Island 02882, USA
cFood Safety & Environmental Stewardship Lab, Environmental & Molecular Toxicology, Oregon State University, USA
dLabor für Hydrologie und Internationale Wasserwirtschaft, Technische Hochschule Lübeck, Lübeck, 23562, Schleswig-Holstein, Germany
First published on 7th October 2024
Abstract
In equilibrium-based passive sampling applications, the accuracy of estimating freely dissolved concentration (Cfree) of hydrophobic organic compounds (HOCs) relies on the passive sampler–water partition coefficient (KPS–W) values applied. The vast majority of KPS–W are generated under standard conditions: 20 °C in deionized or freshwater. Few empirically derived values are available for non-standard conditions. In this study, polyethylene (PE)–water partitioning coefficients (KPE–W) were experimentally determined for 15 polycyclic aromatic hydrocarbons (PAHs, comprising 9 parent and 6 alkylated compounds) under three different temperature (10, 20, 30 °C) and salinity (0, 18 and 36‰) regimes, the KPE–W values were found to correlate strongly with a variety of molecular parameters (e.g., octanol–water partition coefficients (KOW), molecular weight (MW) and molecular volume (MVOL)). The effects of temperature and salinity on the magnitude of KPE–W were found to be substantial. For temperature, the values range between −0.005 and −0.023 log units per °C; these values indicate that every 10 °C rise in temperature would potentially decrease the KPE–W by a factor of between 0.4 to 1.6. For salinity, the values range from 0.0028 to 0.0057 log units per unit ‰, indicating that an 18‰ increase in salinity would likely increase the KPE–W by a factor of between 0.28 and 0.82. Moreover, temperature and salinity were shown to be independent of each other and non-interacting. Temperature effects were chemical-specific and moderately dependent on hydrophobicity (expressed as the KOW), whereas salinity effects were independent of hydrophobicity. We also assessed the combined impact of temperature and salinity, which showed increasing effects with the hydrophobicity of the PAHs studied. Based on the results, KPE–W values adjusted for site-specific temperature and salinity can be calculated. The impact of applying such site-specific values was demonstrated using a PE-based field monitoring dataset for PAHs from coastal waters of Grand Isle (LA, USA) collected during the 2010 Deepwater Horizon oil spill. When KPE–W values were adjusted to 10 °C and 30 °C, the final freely dissolved concentrations (Cfree) decreased or increased depending on the adjustment. Use of the results of this investigation allow for adjusting existing PE-based datasets to site-specific conditions resulting in more accurate Cfree values for estimating exposure and adverse ecological effects.
Environmental significanceThis study examines how temperature and salinity affect the estimation of freely dissolved concentrations (Cfree) of hydrophobic organic compounds (HOCs) using polyethylene (PE)-based passive samplers. By determining partition coefficient (KPE–W) values for polycyclic aromatic hydrocarbons (PAHs) under varying temperature and salinity conditions, we report significant temperature effects on KPE–W, which are chemical-specific and moderately dependent on hydrophobicity. Salinity effects, on the other hand, are independent of hydrophobicity. These results highlight the need for site-specific adjustments to improve the accuracy of Cfree estimations, crucial for assessing HOC exposure and ecological impacts in aquatic environments. Applying these adjustments to field monitoring datasets from areas affected by the 2010 Deepwater Horizon oil spill demonstrates the practical implications of our findings, aiding informed decision-making in environmental management and remediation efforts. |
Introduction
Several nonpolar organic compounds (NOCs) including polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs) preferentially sorb to sediments and particulate matter in the aquatic environment, making their freely dissolved concentrations (Cfree) very low and difficult to be reliably measured. Whole water sampling as currently recommended in the European Union Water Framework Directive (EU WFD) yields total concentration (i.e., total dissolved + colloidal bound + particulate) or when filtered yields total dissolved concentration (i.e., freely dissolved + colloid bound). This conventional approach does not allow the distinct measurement of Cfree, rather the measurement represents freely dissolved and absorbed concentrations which is a poor metric of a compound's chemical activity.1,2 This challenge has been recognised resulting in the development and use of an alternative approach involving the estimation of Cfree of nonpolar organic compounds from concentrations in other compartments (e.g., sediment, biota) through equilibrium partitioning theory3 and the relevant partitioning coefficients (e.g., organic carbon–water partition coefficient (KOC), bioaccumulation factor (BAF)). This approach is useful however it does have its own difficulties. For example, the natural variability in biota (e.g., biomagnification, metabolism) and in sediment (e.g., amount and quality of organic carbon content) may potentially influence KOC and BAF ultimately confounding Cfree estimates.2The ability of nonpolar passive sampling devices (PSDs) to effectively measure the Cfree of NOCs has been adequately demonstrated.4,5 Nonpolar PSDs mostly find use in measuring the Cfree of NOCs of environmental concern including PAHs, PCBs, hexachlorobenzene (HCB), and polybrominated diphenyl ethers (PBDEs)6–9 and other HOCs of emerging concern including triclosan,10,11 alkylphenols11 and organophosphate pesticides.12 Several types of nonpolar PSDs have been developed. The pioneer nonpolar PSD was biphasic (e.g., semipermeable membrane device (SPMD)) and filled with the synthetic lipid triolein. This type of sampler was used on a considerable scale in the early applications of passive sampling. Single phase polymeric materials (e.g., polyethylene (PE), polyoxymethylene (POM), polydimethylsiloxane (PDMS), silicon rubber (SR)) have been used more frequently recently. The initial group (i.e., the biphasic) are gradually being phased out by the latter (i.e., single phase polymers). This is in part, because, the single phase polymers allow simpler modelling of polymer–water kinetics and are less susceptible to damage in that they have no associated risk of lipid/triolein leakage during exposure and/or extraction.13,14
Single phase polymers when exposed to the aquatic environment act as a sorption phase by accumulating target compounds until system equilibrium is reached or polymer exposure terminated. The Cfree of the target compounds can be determined using the model (eqn (1)) from Huckins et al.1 and Smedes & Booij:15
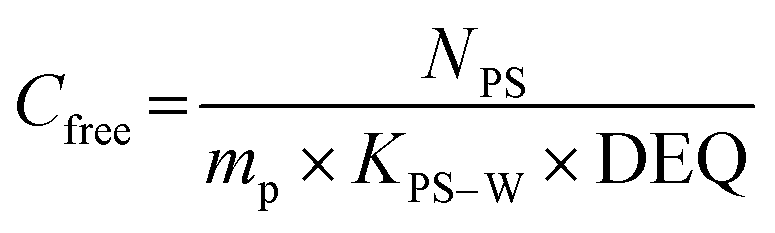 | (1) |
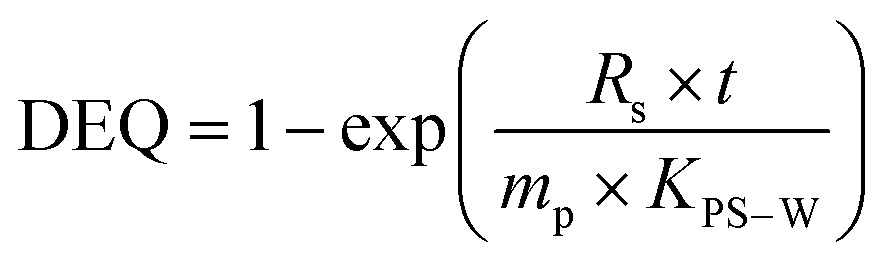 | (2) |
As seen in eqn (1) and 2, knowledge of KPS–W is crucial to the precise estimation of Cfree. For this reason, many investigations have been carried out to determine compound-specific KPS–W for several polymers including PE,6,14,20–25 POM,6,25–28 SR,21,29,30 and PDMS.25,31–36 The vast majority of KPS–W for all the types of passive samplers were experimentally determined or modelled under standard conditions (20 °C, deionized water).
Based on thermodynamic theory it is recognised that the partitioning of NOCs in a two-phase environmental system such as water and a solid phase is influenced by environmental parameters including temperature and salinity. At low temperatures and high salinity, the aqueous solubility of non-polar compounds decreases thereby increasing their hydrophobicity and preferential sorption to the solid phase such as a passive sampling polymer. This means, the use of KPS–W derived under standard conditions to estimate Cfree of compounds from laboratory experiments or field-deployments carried-out under non-standard conditions would likely yield inaccurate results.
Numerous passive sampling based studies focused on assessing Cfree of NOCs have been performed in non-standard environments including deep ocean waters37,38 and cold waterbodies.20,28,39,40 In these studies, the effects of changes in temperature and salinity on KPS–W were mostly corrected using the Van' t Hoff equation and the Setschenow constant, respectively. This correction approach is valid and useful for adjusting the individual effect of temperature and salinity; however, the method may not be appropriate for correcting for combined effects or assessing possible interacting effects of both temperature and salinity on KPS–W. Lohmann41 provided a comprehensive review of these correction methods, which forms a valuable basis for understanding how temperature and salinity individually affect KPS–W.
Alternative means using different single phase polymeric materials have been presented through laboratory studies. This new method based on empirical studies, depending on the experimental design, allows for the quantification and correction for both individual and combined effects of temperature and salinity on KPS–W. Furthermore, it also allows for the investigation of possible interacting effects between both parameters.
Currently, there are two issues with this alternative empirical approach. First, the results reported from several laboratory studies (using different polymers) are inconsistent. For example,16,30,32,42 using polymers including SR, PE and PDMS found substantial temperature effects on KPS–W values for low molecular weight (LMW) PAHs and PCBs. Interestingly, against theoretical principles, enhanced partitioning with increasing temperature was observed for methyl-triclosan, n-nonylphenol and some high molecular weight (HMW) PAHs and PCBs for PE.11 Conversely, no substantial temperature effects were observed in the sorption of some PAHs to PDMS, POM28 and PE,20 and the partitioning of triclosan and selected n- and t-octylphenols to PE.11 Similarly, substantial salinity effects have been reported on the partitioning of PAHs and PCBs to SR30 and PAHs sorption to PDMS28 and PE16 have been reported. In contrast, minor salinity effects on the partitioning of LMW PAHs, and sorption of methyl-triclosan and n-nonylphenol to PE11,22 have been observed. Overall, the mixed results are surprising given the effects of changes in temperature and salinity on KPS–W are supposedly driven by a compound's aqueous solubility and not by the differences in polymer properties.30 Variations in experimental design such as temperature ranges, salinity levels, equilibration times, and the presence of organic matter can significantly impact partitioning outcomes. Small differences in these experimental variables across studies can contribute to the observed inconsistencies.
A second concern with many of these laboratory studies except that of Jonker et al.30 is that they have largely focused on quantifying the individual effects of temperature and salinity leaving out the assessment of combined and possible interacting effects. Results from Jonker et al. show the effects of temperature and salinity on SR–water partition coefficients (KSR–W) of PAHs and PCBs are independent and non-interacting but their combined effects increase with chemical hydrophobicity.
Considering the current non-consensus in the literature on temperature and salinity effects on KPS–W, further studies focused on the quantification of not only individual effects, but also combined and possible interacting effects are needed for more commonly used nonpolar passive sampling polymers. Also, as noted earlier, the vast majority of KPS–W were determined under standard conditions (i.e., 20 °C in freshwater or deionized water). Consequently, an area of passive sampling application that needs further research is expansion of the experimental and modelling efforts for deriving these partition coefficients at temperatures and salinities beyond these narrow conditions. Access to KPS–Ws at higher salinities along a wider range of temperatures would expand the usefulness of passive sampling (e.g., in polar and tropical locations). To this end, the following exercises were carried out in this study: (1) using PE as the passive sampling polymer, we experimentally determined the PE–water partitioning coefficient (KPE–W) for 15 PAHs (10 parent, 5 alkylated) under three different temperature (10, 20, 30 °C) and salinity (0, 18 and 36‰) regimes while varying experimental temperatures and salinities on KPE–W, (2) assessed the joint effects of temperature and salinity on KPE–W values on the target compounds, and (3) using the relationships derived between temperature and salinity and KPE–W a field PE dataset collected during an oil spill was adjusted for site-specific conditions. Results of this investigation should allow for more accurate determinations of Cfree for estimating exposure and adverse ecological effects for a range of temperature and salinity conditions.
Materials and methods
Test materials
A standard solution of 15 target compounds 2-methylnapthalene, acenapthene, 2,6-dimethynapthalene, fluorene, phenanthrene, 1-methylflourene, fluoranthene, pyrene, 2-ethylanthracene, 1-methylpyrene, benzo(a)anthracene, benzo[e]pyrene, benzo(a)pyrene, 7,12-dimethylbenz [a] anthracene and benzo[ghi]perylene in acetonitrile was purchased from Neochema GmbH (all >98% purity; Bodenheim, Germany). Solvents used in this study were cyclohexane (99.5% purity; Th. Geyer GmbH & Co. KG, Renningen, Germany), methanol and acetone (GC grade; Sigma-Aldrich, Steinheim, Germany) and deionised water (Sigma-Aldrich, Steinheim, Germany). Internal standards (d10-acenaphthene, d12-chrysene, d8-napthalene, d12-perylene, d10-phenathrene and d10-pyrene) all with a purity of >99% were purchased from Dr Ehrenstorfer GmbH (Augsburg, Germany).Salts used in this study included sodium chloride (NaCl), sodium sulfate (Na2SO4), sodium tetraborate (Na2B4O7), calcium chloride (CaCl2·2H2O), strontium chloride (SrCl2·6H2O), sodium carbonate (NaHCO3), potassium chloride (KCl), potassium bromide (KBr), magnesium chloride (MgCl2·6H2O), and sodium azide (NaN3). All salts were of extra pure quality obtained from Merck KGaA (Darmstadt, Germany), Carl Roth Gmbh & Co. KG (Karlsruhe, Germany), and Sigma-Aldrich (Steinheim, Germany). Tables S1 and S2 in the ESI† contains additional information on the salt composition and the target compounds.
Polyethylene (PE) (70 μm thickness) was obtained from Brentwood Plastics (St. Louis, MO, USA) kindly supplied by K. Booij (PaSOC, The Netherlands). The PE was resized into several small square-shaped strips (0.05 g per strip) and the strips were cleaned by soaking twice in cyclohexane by shaking at 150 rpm. Each precleaning cycle lasted 24 hours.
Polyethylene–water partitioning coefficient (KPE–W)
Chemical-specific KPE–W were determined under three different temperature regimes (i.e., 10, 20 and 30 °C) and at three different salinity levels (i.e., 0, 18 and 36‰), representing freshwater, brackish and seawater, respectively.The KPE–W experiment was carried out using the desorption approach. This approach involved a first step that loaded the precleaned PE strips with fixed amount of target compounds (Table S2†) prior to the PE–water equilibration step. The loading process was carried out following the procedures detailed in the study by Booij et al.17 Briefly, PE strips were incubated in a target compound-spiked methanol/water (80/20) (V/V) mixture, the water content was increased in 10% steps (over 13 days mixed at 150 rpm) until a 50/50 methanol/water content was reached. Clean laboratory tissue paper was used to remove traces of methanol from the PE strips. Along with the methanol swelling the polymer to enhance HOC partitioning, the increasing aqueous fraction encourages the HOCs into the polymer rather than staying in the methanol.
Amber bottles (250 mL) were prepared by dishwashing twice with solvent and distilled water and oven dried for a 6 h period. Thereafter, 220 mL of aqueous solution was poured into each bottle followed by the addition of a piece of the loaded PE strips. The aqueous solution consisted of 50 mg L−1 sodium azide (to inhibit microbial growth) and sufficient salt contents for the treatments of 18‰ and 36‰ salinity levels. The filled amber bottles were swiftly closed by glass stoppers and transferred to corresponding rotating orbital shakers placed in temperature-controlled rooms. In total, there were three shakers, one each for the three different temperature regimes. Each treatment (i.e., combination of temperature and salinity level) was replicated three times. The overall shaking period for the partitioning experiment lasted 11 week.
Polyethylene and water – phase extraction
Upon the completion of the shaking period, accumulated PAHs in the PE and water phases was extracted from each bottle. The PE was extracted by submerging and soaking sequentially twice for 24 hours in cyclohexane and the extracts combined. The water phase was extracted with three sequential and composited aliquots (40 mL) of cyclohexane. Internal standards were added before the initial extraction of each phase. The extracts from both phases were reduced to appropriate volumes for gas chromatograph (GC) and mass spectrometry (MS) (GC/MS) analysis using a stream of nitrogen gas. Prior to analysis, the extracts were stored in 1.8 mL amber vials in the dark at 4 °C.Instrumental analysis
Chemical analysis of the target compounds was performed on an Agilent 6890 GC equipped with a 5973-mass selective detector (Agilent Technologies, Inc., Santa Clara, CA, USA) operated in select ion monitoring mode. Analytes and internal standards were separated using an Agilent DB – 5MS capillary column (30 m length, 250 μm diameter, 0.25 μm thickness, Agilent J&W Scientific, Santa Clara, CA, USA) and quantified with a 5- or 6-point calibration curve. The oven temperature was set at 40 °C for 3 min, then elevated to 280 °C at the rate of 10 °C min−1 (holding at each step for 6 min). The samples were injected in spitless mode. The temperatures of the injector, electron ionisation source, and analyzer were set to 250, 230 and 150 °C, respectively.Deepwater horizon dataset
Over a period of 13 months, the study by Allan et al..involved PE-based sampling conducted at four coastal locations: Grand Isle, Louisiana (LA); Gulfport, Mississippi (MS); Gulf Shores, Alabama (AL); and Gulf Breeze, Florida (FL).43 These sampling events encompassed the period before, during, and after the 2010 Deepwater Horizon oil spill. The study included a total of nine distinct sampling events, each assigned shorthand names such as ‘May 2010’ (May 10–13, 2010), ‘June 1 2010’ (June 8–11, 2010), ‘June 2 2010’ (June 11–July 7, 2010), ‘July 2010’ (July 7–August 5, 2010), ‘August 2010’ (August 5–September 8, 2010), ‘September 2010’ (September 8–October 13, 2010), ‘March 2011’ (February 9–March 15, 2011), ‘April 2011’ (March 15–April 29, 2011), and ‘May 2011’ (April 29–June 8, 2011).For the analysis in this study, only data from Grand Isle, Louisiana (LA) was investigated. For the specific data related to Grand Isle, Louisiana (LA), it should be noted that samplers were lost from Grand Isle in July 2010. The duration of sampling deployments at Grand Isle varied ranging from 3 to 41 days (Table S3†). The PAHs monitored at Grand Isle consisted mainly of 33 parent and alkylated PAHs (Table S4†). Throughout the sampling events at Grand Isle, the average temperature exhibited a range from 24.85 to 32.85 °C, with the salinity of the water consistently measured at 35 (‰). Detailed passive sampling conditioning, construction, cleanup, extraction and subsequent chemical analyses at Grand Isle are described in Tidwell et al.44 Briefly, approximately one meter long PSDs made from low-density PE (LDPE) tubing were fortified with deuterated PAHs (PRCs) for water sampling rate calculations. PRCs spanned a range similar to the target PAHs, with the most similar used for quantification. PSDs were transported in sealed polytetrafluoroethylene (PTFE) bags and stored at 4 °C until extraction within two weeks. All solvents were Optima grade or better, and standards had purities of ≥97%. PSDs were extracted as a composite sample for increased sensitivity, using n-hexane dialysis, and stored in amber glass vials at −20 °C until analysis.
Quality control
Laboratory blanks for the PE samplers and water phase (n = 3 per phase) were prepared, extracted, and analysed as described above. No PAHs were detected in the blanks above the limit of detection (LOD). Instrument LOD and quantitation (LOQ) were determined according to the German standard method DIN 32645.45 For this purpose, a calibration of 6–10 points with several points close to the expected limits was performed. For recovery determination, complete chemical recovery from PE was confirmed through repeated extraction. For the water phase, recovery of chemicals was verified by spiking a defined quantity of an independent PAH mixture (1000 ng) into clean water (n = 3). The mixture was then extracted, measured and evaluated analogously to the aqueous samples. The recovery rates were calculated by comparing analytically measured concentrations in the extract with the nominal concentrations of the spiked mixture.Data analysis
The concentrations of the target compounds in the PE samplers and the water were determined, and KPE–W values were calculated for each treatment by dividing concentrations in PE (CPE) extracts by concentrations in the aqueous phase extracts (Cw). A number of molecular parameters including molecular weight (MW), octanol–water partition coefficients (KOW), and molecular volume (MVOL) were used to correlate with the experimentally determined KPE–W values. Values for the molecular parameters used for the correlations are listed in Table S5.† Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KOW used were mostly sourced from Jonker35 a data review Ma et al.46 and from the U.S. EPA's EPI suite (Estimation Programs Interface Suite, v. 4.10). While MW and MVOL were sourced from the Molinspiration property calculator -MIPC- (https://molinspiration.com, accessed January 2022). Data processing, calculations and graphing were conducted using OriginPro 2021b (OriginLab Corporation USA, Northampton, MA, USA).
KOW used were mostly sourced from Jonker35 a data review Ma et al.46 and from the U.S. EPA's EPI suite (Estimation Programs Interface Suite, v. 4.10). While MW and MVOL were sourced from the Molinspiration property calculator -MIPC- (https://molinspiration.com, accessed January 2022). Data processing, calculations and graphing were conducted using OriginPro 2021b (OriginLab Corporation USA, Northampton, MA, USA).
Results and discussion
Polyethylene and water partitioning coefficient (KPE–W)
Experimentally-determined logKPE–W values for the PAHs investigated in this study are presented in Table 1. The standard deviation of the triplicate measurements are primarily within 0.1 log unit suggesting good reproducibility of the measurements. Plots of the logKPE–W values (determined under standard conditions: 20 °C in freshwater) versus logKow, molecular weight and molecular volume are shown in Fig. 1. The regression plots indicate good linear relationships with coefficients of determination (r2) greater than 0.931 suggesting these parameters are relatively good predictors of PAH logKPE–W values. Molecular weight values show a better correlation with logKPE–W than those of logKOW and molecular volume:| logKPE–W = 0.0264 MW − 0.3705 (r2 = 0.96, n = 15) | (3) |
KPE–W (average ± standard deviation) under different temperatures and salinities
| PAH | 10 °C | 20 °C | 30 °C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 0‰ | 18‰ | 36‰ | 0‰ | 18‰ | 36‰ | 0‰ | 18‰ | 36‰ | |
| 2-Methyl naphthalene | 3.40 ± 0.02 | 3.49 ± 0.07 | 3.56 ± 0.04 | 3.39 ± 0.09 | 3.44 ± 0.01 | 3.46 ± 0.01 | 3.28 ± 0.02 | 3.36 ± 0.04 | 3.46 ± 0.02 |
| Acenaphthene | 3.65 ± 0.01 | 3.75 ± 0.07 | 3.81 ± 0.02 | 3.63 ± 0.09 | 3.67 ± 0.02 | 3.69 ± 0.02 | 3.53 ± 0.05 | 3.59 ± 0.01 | 3.68 ± 0.00 |
| 2,6-Dimethyl naphthalene | 4.00 ± 0.01 | 4.12 ± 0.07 | 4.19 ± 0.04 | 3.99 ± 0.09 | 4.05 ± 0.01 | 4.08 ± 0.02 | 3.89 ± 0.05 | 3.96 ± 0.03 | 4.05 ± 0.02 |
| Fluorene | 3.81 ± 0.00 | 3.89 ± 0.07 | 3.95 ± 0.02 | 3.76 ± 0.09 | 3.8 ± 0.03 | 3.83 ± 0.03 | 3.65 ± 0.04 | 3.71 ± 0.03 | 3.8 ± 0.00 |
| Phenanthrene | 4.26 ± 0.02 | 4.36 ± 0.08 | 4.42 ± 0.02 | 4.21 ± 0.09 | 4.24 ± 0.01 | 4.28 ± 0.03 | 4.07 ± 0.05 | 4.12 ± 0.02 | 4.22 ± 0.03 |
| 1-Methylfluorene | 4.4 ± 0.00 | 4.49 ± 0.07 | 4.55 ± 0.01 | 4.34 ± 0.09 | 4.38 ± 0.01 | 4.43 ± 0.03 | 4.22 ± 0.04 | 4.27 ± 0.02 | 4.36 ± 0.00 |
| Fluoranthene | 4.95 ± 0.01 | 5.03 ± 0.09 | 5.08 ± 0.02 | 4.84 ± 0.08 | 4.86 ± 0.00 | 4.92 ± 0.05 | 4.7 ± 0.03 | 4.74 ± 0.04 | 4.81 ± 0.01 |
| Pyrene | 5.09 ± 0.01 | 5.16 ± 0.08 | 5.21 ± 0.02 | 4.97 ± 0.08 | 5.00 ± 0.00 | 5.05 ± 0.05 | 4.84 ± 0.03 | 4.86 ± 0.05 | 4.93 ± 0.01 |
| 2-Ethylanthracene | 5.52 ± 0.04 | 5.6 ± 0.07 | 5.65 ± 0.02 | 5.41 ± 0.09 | 5.42 ± 0.01 | 5.51 ± 0.04 | 5.32 ± 0.06 | 5.33 ± 0.04 | 5.46 ± 0.08 |
| 1-Methylpyrene | 5.6 ± 0.02 | 5.75 ± 0.07 | 5.8 ± 0.02 | 5.51 ± 0.12 | 5.52 ± 0.01 | 5.58 ± 0.03 | 5.32 ± 0.07 | 5.36 ± 0.01 | 5.48 ± 0.02 |
| Benzo(a)anthracene | 5.77 ± 0.01 | 5.85 ± 0.06 | 5.9 ± 0.03 | 5.64 ± 0.09 | 5.66 ± 0.00 | 5.71 ± 0.03 | 5.46 ± 0.07 | 5.5 ± 0.01 | 5.6 ± 0.02 |
| Benzo[e]pyrene | 6.31 ± 0.02 | 6.49 ± 0.12 | 6.52 ± 0.04 | 6.29 ± 0.06 | 6.36 ± 0.04 | 6.15 ± 0.09 | 6.1 ± 0.10 | 6.12 ± 0.01 | 6.22 ± 0.02 |
| Benzo(a)pyrene | 6.78 ± 0.16 | 6.80 ± 0.15 | 6.83 ± 0.08 | 6.52 ± 0.06 | 6.56 ± 0.09 | 6.61 ± 0.07 | 6.34 ± 0.11 | 6.38 ± 0.05 | 6.46 ± 0.03 |
| 7,12-Dimethylbenz[a]anthracene | 6.19 ± 0.09 | 6.31 ± 0.15 | 6.46 ± 0.03 | 5.69 ± 0.27 | 5.68 ± 0.11 | 5.76 ± 0.03 | 5.43 ± 0.05 | 5.29 ± 0.13 | 5.42 ± 0.24 |
| Benzo[ghi]perylene | 6.15 ± 0.22 | 7.17 ± 0.27 | 7.21 ± 0.13 | 7.05 ± 0.07 | 7.05 ± 0.03 | 6.28 ± 0.36 | 6.83 ± 0.12 | 6.9 ± 0.07 | 6.93 ± 0.13 |
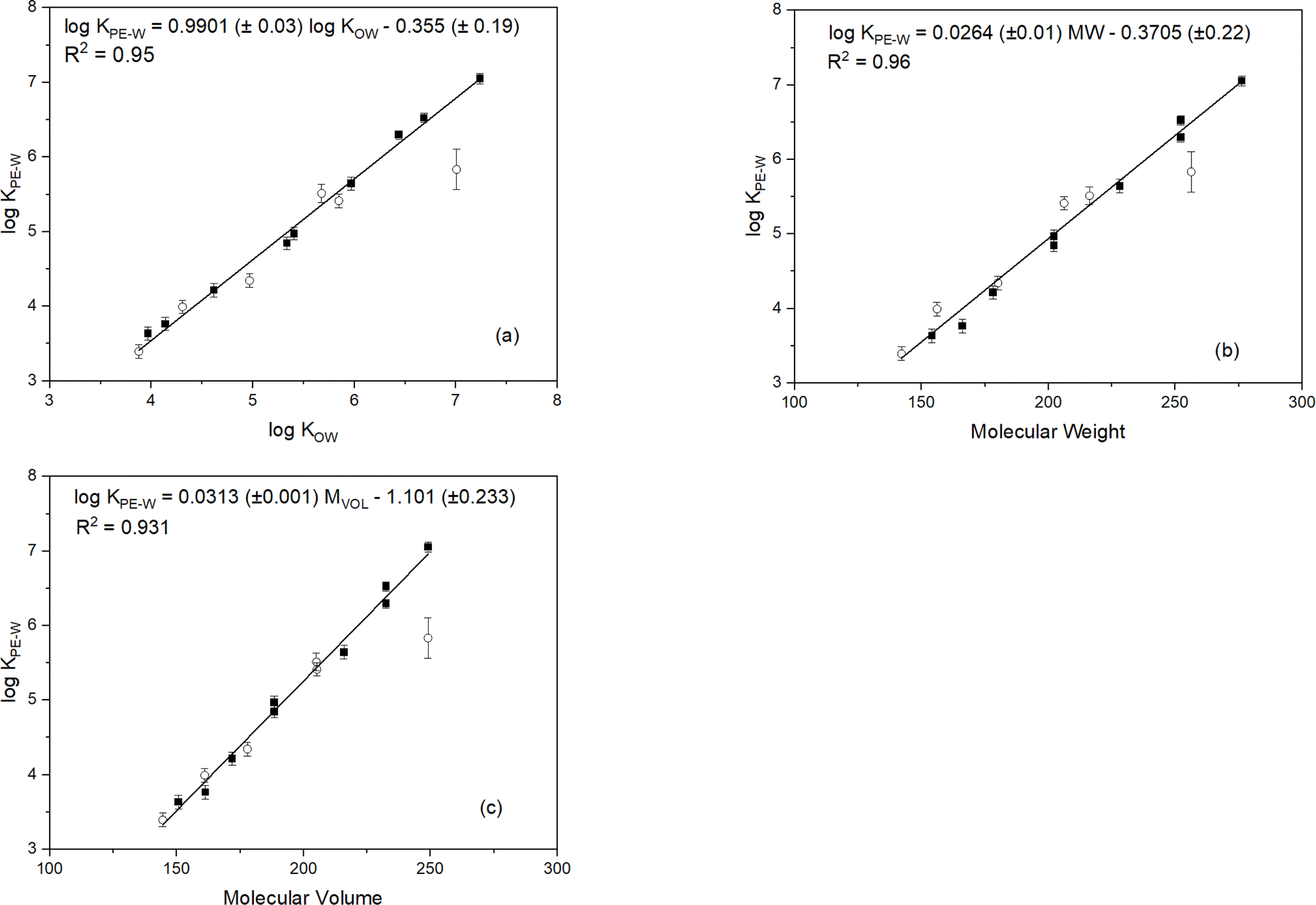 | ||
| Fig. 1 Relationships between logKPE–W and (a) molecular weight, (b) molecular volume, and (c) logKow for experimental system under standard conditions (20 °C, freshwater). Solid squares and open circles represent parent- and alkylated-PAHs, respectively, and the solid line represent the regression line. PAH consistently below the regression line (open circle) is 7,12 dimethyl benz[a]anthracene. | ||
As seen in Fig. 1, logKPE–W values increase with increasing values of the individual molecular parameters, except for 7,12-dimethylbenz[a]anthracene which showed an unusual interaction with the test polymer. Causes of the anomalous behaviour of this chemical are unclear but was noticed in all experimental treatments (Fig. S1†) and in the additional systems set-up for confirming partitioning equilibrium. The unusual behavior of 7,12-dimethylbenz[a]anthracene with the test polymer may be due to unique steric hindrance created by the methyl groups at the 7 and 12 positions, altering its conformation and interaction with the polymer in ways not seen with other methylated PAHs. Additionally, these specific positions uniquely alter the electron density, affecting interaction with the polymer differently. Unique non-covalent interactions, such as van der Waals forces or π–π stacking, may also occur between the polymer and the 7,12-dimethyl substituents, which are absent in other methylated PAHs.
For the PAHs in this study, addition of a methyl group to the ring structure consistently adds 0.4–0.5 log unit to the KPE–W values (Fig. S2†) but the expected ∼1.2 log unit increase for 7,12-dimethyl-benz(a)anthracene versus benz(a)anthracene is not observed, and, actually, becomes negative at higher temperatures (i.e., KPE–W values for 7,12- dimethyl-benz(a)anthracene are lower than those of benz(a)anthracene). This behaviour is inconsistent with theory, and it is also not supported by any of the other measurements (e.g., 2-ethylanthracene which is 1.2 log unit higher than phenanthrene has a similar KPE–W as anthracene (Figure S2†)). This observed inconsistency in the 7,12-dimethyl-benz(a)anthracene KPE–W values further supports the earlier mentioned unexpected behaviour of the compound with the test polymer.
Linear correlations between logKPE–W values and the molecular parameters (Fig. 1) indicates the reliability of the PAH KPE–W values determined in this investigation. To further validate the reliability of the results determined under standard conditions, the KPE–W values from this study and those reported in previous studies are listed in Table 1 for comparison. It is important to mention, for a fair comparison, only KPE–W values determined at standard conditions were considered. This means KPE–W values for PAHs determined from saline water exposure20,39,47 or laboratory experiments carried out at lower20,42 or higher temperatures16,42,48 were excluded. Also, the suggested best-fit KPE–W values for PAHs from the extensive literature review by Lohmann41 were excluded because the values were largely derived from pooled KPE–W data that included experimental conditions that were not standard.
As seen in Table 1, the logKPE–W values determined here are within the range of those reported in the literature. The logKPE–W values compare best with results from Jonker25 and Hale et al.49 varying by only ± 0.01 to ± 0.04 log units. Similar minor differences are observed when comparing with data from Smedes et al.21 (±0.01 to ± 0.47 log units), Fernandez et al.50 (±0.14 to ± 0.27 log units), Bao et al.24 (±0.09 to ± 0.62 log units), Reitsma et al.20 (±0.11 to ± 0.68 log units), and Belles et al.14 (±0.03 to ± 0.72 log units). Whereas the majority of our logKPE–W values (particularly the more hydrophobic PAHs) are higher than those from Perron et al.6 (0.03–1.24 log units) and Choi et al.22 (0.08–0.82 log units). In summary, it is evident the logKPE–W values determined under standard conditions in this study compare very well with those reported in the literature.
Temperature effects on KPE–W
Having discussed earlier the uncertainty in the derived KPE–W values of 7,12-dimethylbenz[a]anthracene, those data will not be presented relative to temperature and salinity effects. For the other KPE–W values for the remaining PAHs, it is more convenient to visualise the temperature dependency trends by plotting the partitioning data from Table 1versus temperature. These plots are presented in Fig. 2 for selected PAHs. | ||
| Fig. 2 LogKPE–Wversus temperature for selected PAHs as a function of salinity:(□) 0‰, (○)18‰, and (△) 36‰ based on this investigation. For comparison, the black diamonds represent adjustments based on corrections reported by Lohmann (2012). | ||
As expected, the logKPE–W values decrease with increasing temperature, this is consistent with theoretical principles and corroborates with observations from previous studies. As earlier discussed in the Introduction, Adams et al. and Booij et al. observed similar effects on the partitioning of low molecular weight (LMW) PAHs to PE polymer.16,42 Also, Jonker et al. reported comparable effects on the sorption of several LMW PAHs and PCBs to silicone rubber.30 The slopes of the plots (Fig. 2) differ an indication that temperature effects are chemical dependent.
Using the slope of the logKPE–W – temperature correlation plots, a numerical quantification of the change in logKPE–W per °C for each individual chemical was determined and is presented in Table S6.† The values range between −0.005 and −0.023 log units per °C (across the three salinities); these values indicate that every 10 °C rise in temperature would potentially decrease the KPE–Ws by a factor of between 0.4 to 1.6. The temperature effects (as quantified by the logKPE–W – temperature correlation plots) do not vary substantially across the different salinities (Table S6†), suggesting temperature influences on KPE–W are independent of the salinity of the experimental systems. Plotting each PAH's temperature effects (i.e., change in logKPE–W per °C derived at different salinities for each individual chemical (Table S6†)) against its respective logKOW, a moderately linear correlation with an r2 ranging from 0.54 to 0.69 is observed. The changes in logKPE–W per °C values becomes more negative with increasing PAH hydrophobicity (Fig. 3a).
 | ||
| Fig. 3 Temperature effects (determined by the change in logKPE–W per °C) versus logKOW (a) and the salinity effects (determined by the change in logKPE–W per unit salinity (‰)) versus logKOW (b). The regression equation for temperature effects in the absence of salt (i.e., 0‰) = −0.0041 logKOW + 0.010 (r2 = 0.69). | ||
To further understand the effect of temperature on KPE–W of the PAHs, we considered relationships with temperature expressed in van't Hoff form:
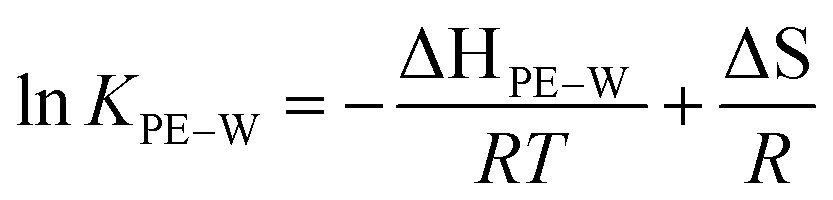 | (4) |
KPE–Wversus the reciprocal temperature (K−1), the contributions of enthalpy and entropy to logKPE–W were derived based on the slopes and intercepts of the plots, respectively (Fig. 4). Gibbs free energies were calculated using the fitted parameters. Resulting thermodynamic parameters are provided in Table S7.†
 | ||
| Fig. 4 Temperature effects for selected PAHs in van't Hoff equation form. Solid symbols represent the average value at a given temperature while open symbols are the results of experimental triplicates. | ||
For selected PAHs, the InKPE–W – temperature relationship demonstrated a linear correlation (Fig. 4). The positive slope of the individual chemicals and the negative ΔG values (i.e., ranging from −18.82 to −39.46, (Table S6†)) indicate an exothermic sorption process.51 Values for ΔHPE–W ranged from −9.5 to 34.7 kJ mol−1 (Table S6†) and showed a consistent trend with comparable values for PAHs with the same number of aromatic rings. The entropy term is typically greater than zero (Table S6†) for the individual chemicals. The ΔHPE–W values derived here are similar to those reported by Adams et al. (−20 to −30 kJ mol−1) and Booij et al. (−20 to −50 kJ mol−1) for PE.16,42 Muijs & Jonker reported PDMS–water partitioning enthalpy values between −15.7 and −34.7 kJ mol−1 for PAHs.32 Similarly Jonker et al. using silicone rubber (SR) observed SR–water partitioning enthalpy values of between −16.9 and −58.0 kJ mol−1 for PAHs.30 Polymers used in the latter two studies (i.e., PDMS and SR) though different from PE yielded comparable results, this suggests the sorption behaviour of PAHs to these polymers (i.e., PE, PDMS and SR) is likely similar thermodynamically.
Salinity effects on KPE–W
To quantify salinity effects, we plotted the logKPE–W – salinity correlation data at the three different experimental temperatures. As seen in Fig. 5, PAH partitioning to PE increased with salinity, this observation agrees with theoretical expectations explained by PAH aqueous solubility being reduced by the salting out effect41 and corresponds with previous results.6,16,30
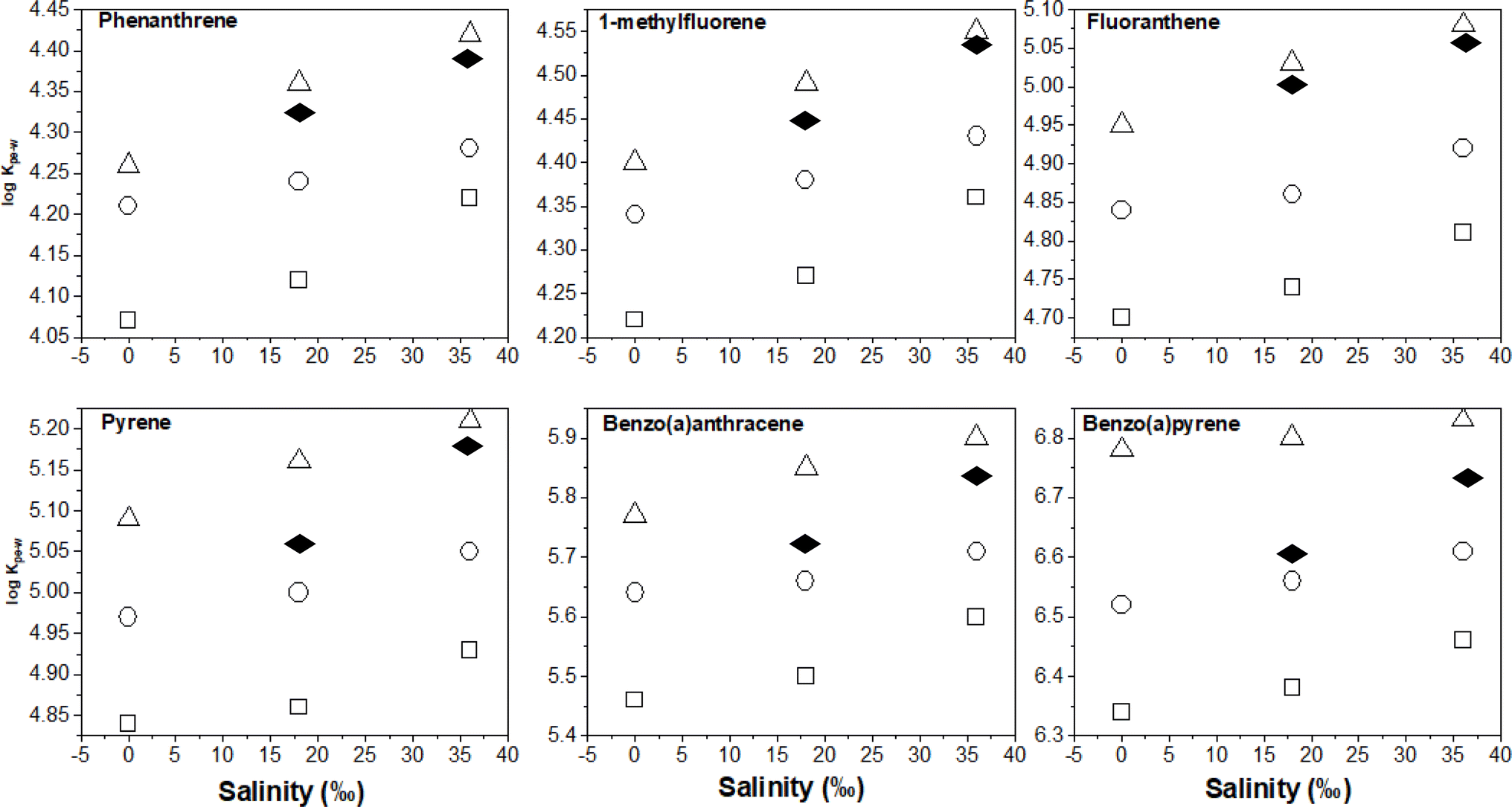 | ||
| Fig. 5 LogKPE–W – salinity correlation plots for selected PAHs as a function of temperature: (△) 10 °C, (○) 20 °C and (□) 30 °C based on this investigation. For comparison, the black diamonds represent adjustments based on corrections reported by Lohmann (2012). | ||
The changes in logKPE–W per unit salinity (‰) was quantified using the slope of plots in Fig. 5 and the results are presented in Table S8.† The values range from 0.002 to 0.0057 log units indicating that an 18‰ increase in salinity would likely increase KPE–W by a factor of between 0.28 and 0.82. Additionally, the data in Table S7† show salinity effects are similar across the three temperatures (observable but small), suggesting salinity effects on KPE–W is independent of temperature of the aqueous systems. A plot of the salinity effect as measured by the change in logKPE–W per unit salinity (‰) for individual PAHs determined at different temperature against their respective logKOW show appreciable scatter with no clear relationship between the two variables (Fig. 3b).
Combined effects of temperature and salinity on KPE–W
As discussed above, the effects of both temperature and salinity on KPE–W are independent and non-interrelated (i.e., temperature effects are similar at different salinities and salinity effects are similar at different temperatures), consequently, it is possible to assess the joint effects of both parameters and derive chemical-specific compensation equations involving the impacts of both environmental variables. A single equation of the following form, first developed by Jonker et al.30 may be used for this purpose:| logKPE–W(T,S) = logKPE–W(20,0) + β × (T − 20) + α × (S) | (5) |
KPE–W (20,0), β and α first need to be estimated based on the PAH's logKOW values using the relationships presented in Fig. 1a and 3a.
Results presented in Table S9† show the overall effect of temperature and salinity increasing with hydrophobicity and are largest for the more hydrophobic chemicals. For example, at freezing temperatures (i.e., 0 °C) and 36‰, a corrected logKPE–W value for benzo(a)pyrene would exceed its corresponding values determined at standard conditions by 0.63 log units (a factor of 5.4) while for a less hydrophobic chemical, like fluorene, the difference is smaller at 0.2 log units (a factor of 1.7). A visual representation of the combined effect of temperature and salinity on the logKPE–W of benzo(a)pyrene is presented in Fig. S3.† The plot shows that relative to logKPE–W values determined under standard conditions (benzo(a)pyrene: 6.52 ± 0.06), low temperatures combined with high salinities result in increased logKPE–W values ranging between 0.4 to 0.76 log units. However, high temperatures combined with the low salinities also reduces the logKPE–W by about 0.12 to 0.22 log units.
Estimating PAH Cfree under field conditions
In this section, we illustrate the practical implications of compensating for temperature and salinity effects on KPE–W using a case study. The case study draws from the PE-derived Deepwater Horizon spill monitoring data collected from the Grand Isle, Louisiana (USA) station in the Gulf of Mexico. As previously discussed, the original monitoring results, which encompassed the dissolved concentrations of several parent and alkylated PAHs, were presented in the study conducted by Tidwell et al.44 The raw data (i.e., amount of PAHs accumulated in the PE (NPE) and PRC release data (DEQ), and actual recorded temperatures of the water while sampling) underlying this study were used to recalculate Cfree under different theoretical temperature scenarios for Grand Isle, Louisiana station (i.e., 10 °C and 30 °C) over the duration of the monitoring. Details on the calculations can be found in the ESI section.†For one sampling event (April 2011) at the Grand Isle station, Fig. 6 evaluates the influence of adjustments in KPE–W due to variations in temperature (10 °C, 24.85 °C (actual field temperature), and 30 °C) and salinity (a constant salinity of 35 ppt is maintained to align with field conditions reported by Tidwell et al.44) on the estimated sum PAH of Cfree. As temperature increases from actual conditions to a hypothetical 30 °C, there is a consistent rise in sum PAH of Cfree. This indicates that the adjustments to KPE–W for higher temperatures lead to higher estimated PAH concentrations, highlighting the temperature–concentration relationship inherent in the adjusted partitioning behavior. Conversely, sum PAH of Cfree decreases relative to the actual conditions when adjusted for 10 °C (Fig. 6). PAHs with higher logKow values (>5.5) tend to exhibit a reduced sensitivity to the adjustments. Fig. 7 presents the sum PAH of Cfree for each of the eight sampling events applying the adjusted KPE–W values. Fig. 7b, indicates the relative change in sum PAH of Cfree compared to the actual field conditions. Overall, increasing the temperature to 30 °C resulted in an average 5.73 ± 2.52% increase in the Cfree while reducing the temperature to 10 °C affected a mean 29.9 ± 5.90% decrease in sum PAH of Cfree. It is important to note that the temperature correction does not directly affect the actual Cfree values but does influence the deviation in estimating Cfree. The direct inference from Fig. 7 is that the utilization of adjusted KPE–W values correlates with substantial increases or decreases in sum PAH of Cfree as temperatures adjusted up or down relative to actual conditions. This trend is consistently observed across the eight distinct sampling events. Even in cases such as June-1 and September sampling events, where field temperatures exceeded 30 °C, the use of adjusted KPE–W values led to an increase in Cfree.
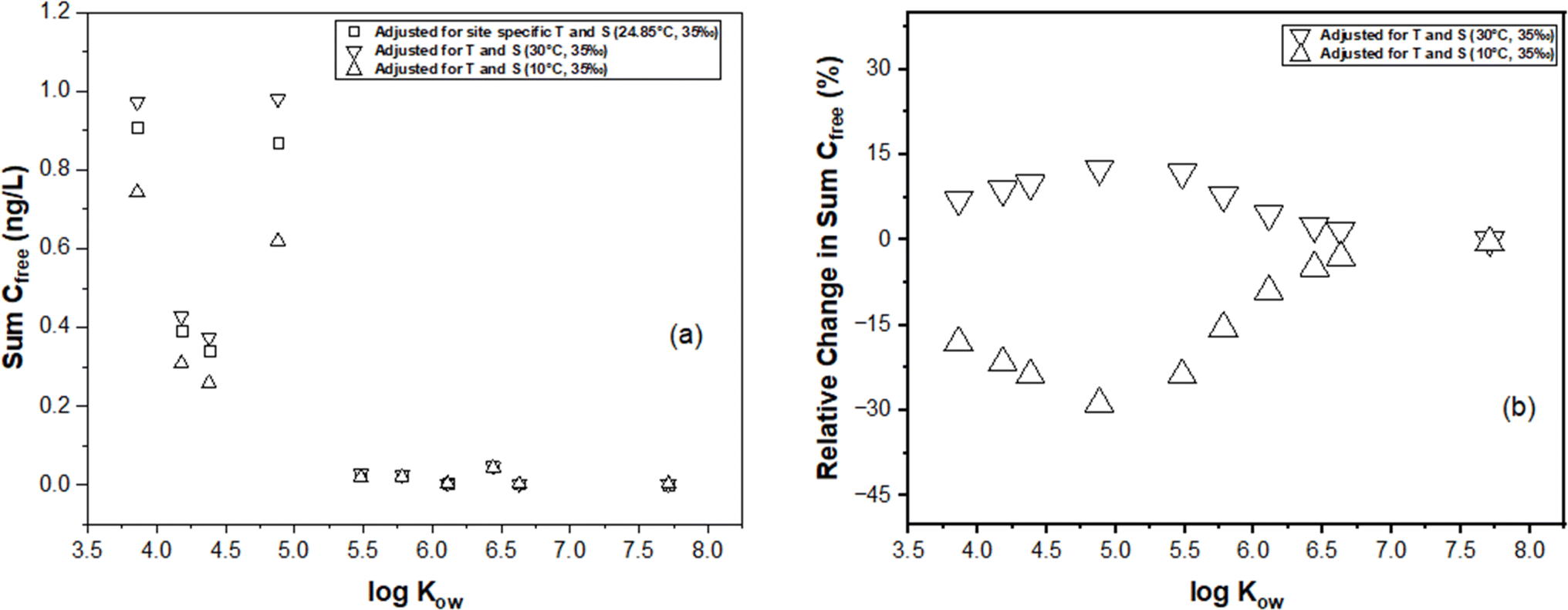 | ||
| Fig. 6 Passive sampling-based estimates of freely dissolved concentrations of selected PAHs at the Grand Isle (LA, USA) station monitored during the 2010 Deepwater Horizon oil spill. One 45 days deployment (March 15–April 29 2011) was selected for this example. (a) Treatments include the Cfree adjusted for site specific temperature salinity, the Cfree at an elevated temperature of 30 °C, and the Cfree at a reduced temperature of 10 °C. (b) The relative change in PAH Cfree at 30 °C and 10 °C compared to the actual station conditions. | ||
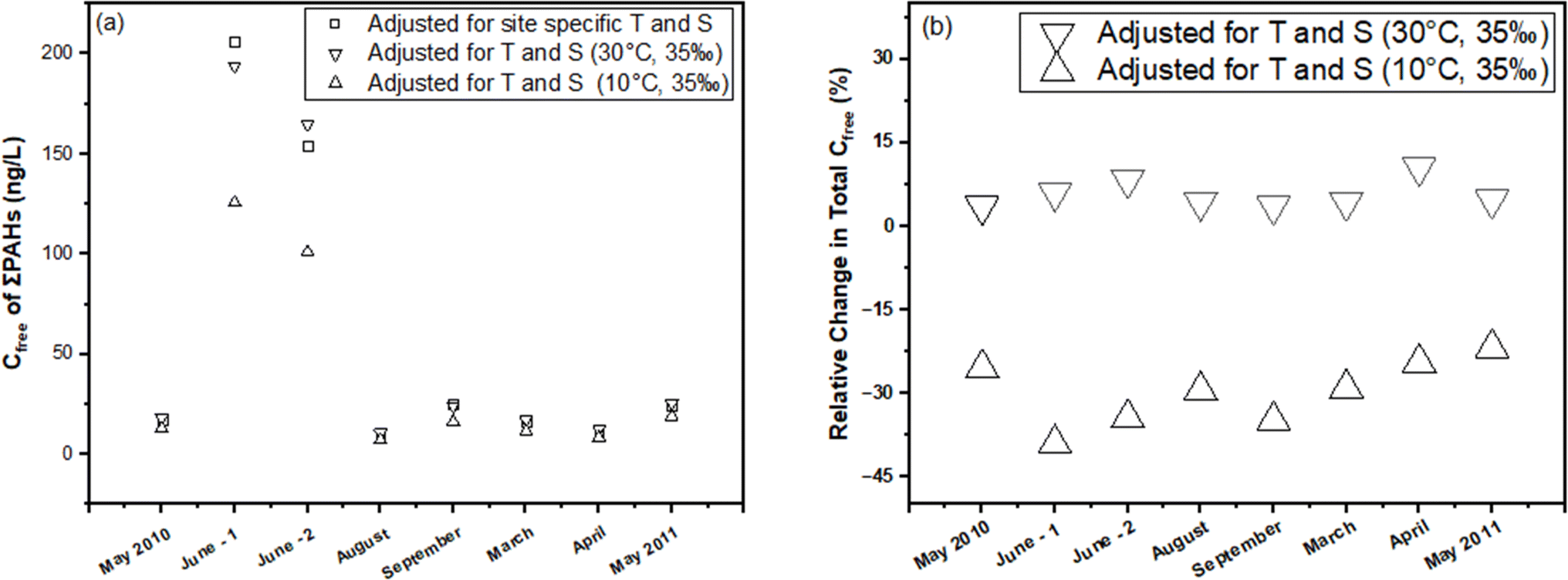 | ||
| Fig. 7 Sum PAH of Cfree for eight sampling events. (a) Treatments include the Cfree adjusted for site specific temperature and salinity, the Cfree at an elevated temperature of 30 °C, and the Cfree at a reduced temperature of 10 °C. (b) The relative change in PAH Cfree at 30 °C and 10 °C compared to the actual station conditions. | ||
To assess the impact of KPE–W adjustments on estimates of toxicity to aquatic life, an analysis was conducted across the eight distinct sampling events. Specifically, the sum PAH toxic units (TU) were estimated for each event (Fig. 8). The TU for an individual PAH is calculated by dividing its Cfree value (μg L−1) by the corresponding narcosis secondary chronic value (SCV) based on Di Toro et al.3 and Burgess et al.52 To obtain the sum PAH TU for a given event, the individual PAH TU values were summed. The estimated TU values for these events ranged from 0.001 to 0.045. A sum PAH TU exceeding 1 is typically indicative of potential sublethal toxicity to aquatic life.52 However, these findings indicate the TU values remain well below this threshold, implying a very low likelihood of toxicity resulting from the presence of PAHs from the Deepwater Horizon spill at these stations at the time of the passive sampler deployments.
 | ||
| Fig. 8 Estimated toxic units for total PAHs under various sampling events with different treatment conditions. | ||
Adjustments to the KPE–W values were found to have a minimal impact on the TU calculations. A comparison of TU values before and after the adjustments showed changes within a small range (0.001 to 0.003 units), indicating that while the adjustments refine the accuracy of the partitioning coefficients, they do not significantly alter the overall toxicity estimates. The TU calculations included 31 PAH compounds (Table S4†), selected for their prevalence and toxicological relevance in the context of the Deepwater Horizon spill. While it is acknowledged that petroleum spills contain thousands of compounds contributing to narcotic toxicity through additive effects, the selected PAHs serve as representative indicators. Therefore, despite the potential presence of additional unmeasured compounds, the low TU values observed for the analyzed PAHs indicate a relatively low risk of sublethal toxicity under the conditions and timeframe of this study.
This exercise demonstrates the power of having KPE–Ws that allow these temperature adjustments to be accurately performed. The continuing effects of climate change increasing oceanic water temperatures along with our on-going reliance on fossil fuels, and consequently, the occasional petroleum spill, provide a ‘real world’ application for this type of exercise. For example, modelling projected effects onsum PAH of Cfree under different future seawater temperatures and various spill volume magnitudes would be very useful for assessing petroleum exploration, transport and usage risks especially in sensitive habitats.
Conclusions
It is vital to understand the degree of potential bias using KPE–W values determined at standard conditions may introduce to Cfree values for passive samplers deployed under environmental conditions. Understanding this bias allows for a more accurate estimation and assessment of toxicity, bioaccumulation, and potential transfer of contaminants to the food web.Partition coefficients for PAHs were reliably determined at varying temperatures and salinities, the resulting data allowed for the direct quantification of individual and combined effects of both parameters on KPE–W values. Individual influences of both parameters on PAH KPE–W values were substantial: a factor of 0.4 to 1.6 decrease for a 10 °C increase in temperature and a factor between 0.28 to 0.82 increase per every 18‰. Observed temperature effects were chemical-specific, moderately dependent on hydrophobicity and independent of salinity, whereas salinity effects were independent of temperature and hydrophobicity. Furthermore, when combining both temperature and salinity effects, an increasing trend with respect to chemical hydrophobicity for KPE–W was observed. As shown in the current study, correcting PE-based monitoring data for site-specific environmental conditions (temperature and salinity) using the Deepwater Horizon oil spill dataset substantially influenced the final Cfrees. Notably, this correction led to a decrease in sum PAH of Cfree of about 30% when adjusted to 10 °C and an increase in Cfree of about 6% when adjusted to 30 °C.
This study benefits the application of passive sampling technology in a number of ways including: (1) a new set of reliable experimentally determined KPE–W for PAHs and (2) data on the direct quantification of temperature and salinity effects on PAH partitioning to PE demonstrates the greatest impacts particularly at low temperature and elevated salinities (i.e., 10 °C, 18‰ and 36‰). Normally, these trends and behavior are difficult to tease out due to non-equilibration issues with experimental systems. Finally, tools for determining the effects of temperature and salinity on PAH partitioning into PE are available herein and shown in Tables S5 and S7† while combined effects may be corrected for using the chemical specific equations given in Table S8.† Use of these chemical specific correction equations maybe extended to PAHs not specifically covered in this study if they are within the logKOW range used here (i.e., 3.88 to 7.24).
Disclaimer
Mention of trade names or commercial products does not constitute endorsement or recommendation for use.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ibukun Ola: conceptualization, data curation, project administration, writing – original draft, writing – review & editing. Carsten Drebenstedt: supervision, project administration. Robert M. Burgess: conceptualization, writing – original draft, writing – review & editing. Lane Tidwell: data curation, writing – review & editing. Kim Anderson: data curation, writing – review & editing. Nils Hoth: supervision, project administration. Christoph Külls: supervision, project administration.Conflicts of interest
The authors declare no conflicts of interest. The authors were not compensated for their contributions to this work.References
- J. N. Huckins, J. D. Petty and K. Booij, Monitors of Organic Chemicals in the Environment: Semipermeable Membrane Devices, Springer, 2006, DOI:10.1007/0-387-35414-X.
- R. Lohmann, K. Booij, F. Smedes and B. Vrana, Use of passive sampling devices for monitoring and compliance checking of POP concentrations in water, Environ. Sci. Pollut. Res., 2012, 19, 1885–1895, DOI:10.1007/s11356-012-0748-9.
- D. M. Di Toro, C. S. Zarba, D. J. Hansen, W. J. Berry, R. C. Swartz, C. E. Cowan, S. P. Pavlou, H. E. Allen, N. A. Thomas and P. R. Paquin, Technical basis for establishing sediment quality criteria for nonionic organic chemicals using equilibrium partitioning, Environ. Toxicol. Chem., 1991, 10, 1541–1583, DOI:10.1002/etc.5620101203.
- M. T. O. Jonker, R. M. Burgess, U. Ghosh, P. M. Gschwend, S. E. Hale, R. Lohmann, M. J. Lydy, K. A. Maruya, D. Reible and F. Smedes, Passive sampling protocol for ex situ determination of freely dissolved concentrations of hydrophobic organic chemicals in sediments and soils: Basis for interpreting toxicity and assessing bioavailability, risks, and remediation necessity and efficiency, Nat. Protoc., 2020, 15, 1800, DOI:10.1038/s41596-020-0311-y.
- R. M. Burgess, S. K. Driscoll, G. A. Burton, P. M. Gschwend, U. Ghosh, D. Reible, S. Ahn and T. Thompson, Laboratory, Field, and Analytical Procedures for Using Passive Sampling in the Evaluation of Contaminated Sediments: User's Manual, US Environmental Protection Agency, Alexandria, VA, 2017 Search PubMed.
- M. M. Perron, R. M. Burgess, E. M. Suuberg, M. G. Cantwell and K. G. Pennell, Performance of passive samplers for monitoring estuarine water column concentrations: 1. Contaminants of concern, Environ. Toxicol. Chem., 2013, 32, 2182–2189, DOI:10.1002/etc.2321.
- I. J. Allan, C. Harman, S. B. Ranneklev, K. V. Thomas and M. Grung, Passive sampling for target and nontarget analyses of moderately polar and nonpolar substances in water, Environ. Toxicol. Chem., 2013, 32, 1718–1726, DOI:10.1002/etc.2260.
- C. Harman, M. Grung, J. Djedjibegovic, A. Marjanovic, M. Sober, K. Sinanovic, E. Fjeld, S. Rognerud, S. B. Ranneklev and T. Larssen, Screening for Stockholm Convention persistent organic pollutants in the Bosna River (Bosnia and Herzogovina), Environ. Monit. Assess., 2013, 185, 1671–1683, DOI:10.1007/s10661-012-2659-0.
- V. P. Sacks and R. Lohmann, Freely dissolved PBDEs in water and porewater of an urban estuary, Environ. Pollut., 2012, 162, 287–293, DOI:10.1016/j.envpol.2011.11.028.
- M. M. Perron, R. M. Burgess, E. M. Suuberg, M. G. Cantwell and K. G. Pennell, Performance of passive samplers for monitoring estuarine water column concentrations: 2. Emerging contaminants, Environ. Toxicol. Chem., 2013, 32, 2190–2196, DOI:10.1002/etc.2248.
- V. P. Sacks and R. Lohmann, Development and use of polyethylene passive samplers to detect triclosans and alkylphenols in an urban estuary, Environ. Sci. Technol., 2011, 45, 2270–2277, DOI:10.1021/es1040865.
- I. J. Allan, C. Harman, S. B. Ranneklev, K. V. Thomas and M. Grung, Passive sampling for target and nontarget analyses of moderately polar and nonpolar substances in water, Environ. Toxicol. Chem., 2013, 32, 1718–1726, DOI:10.1002/etc.2260.
- F. Smedes, Dissertation, Vrije Universiteit, 2021.
- A. Belles, C. Alary and Y. Mamindy-Pajany, Thickness and material selection of polymeric passive samplers for polycyclic aromatic hydrocarbons in water: Which more strongly affects sampler properties?, Environ. Toxicol. Chem., 2016, 35, 1708–1717, DOI:10.1002/etc.3326.
- F. Smedes and K. Booij, Guidelines for passive sampling of hydrophobic contaminants in water using silicone rubber samplers, 2012, DOI:10.25607/OBP-236.
- R. G. Adams, R. Lohmann, L. A. Fernandez, J. K. MacFarlane and P. M. Gschwend, Polyethylene devices: Passive samplers for measuring dissolved hydrophobic organic compounds in aquatic environments, Environ. Sci. Technol., 2007, 41, 1317–1323, DOI:10.1021/es0621593.
- K. Booij, F. Smedes and E. M. van Weerlee, Spiking of performance reference compounds in low density polyethylene and silicone passive water samplers, Chemosphere, 2002, 46, 1157–1161, DOI:10.1016/S0045-6535(01)00200-4.
- I. J. Allan, K. Booij, A. Paschke, B. Vrana, G. A. Mills and R. Greenwood, Field performance of seven passive sampling devices for monitoring of hydrophobic substances, Environ. Sci. Technol., 2009, 43, 5383–5390, DOI:10.1021/es900608w.
- K. Booij, C. D. Robinson, R. M. Burgess, P. Mayer, C. A. Roberts, L. Ahrens, I. J. Allan, J. Brant, L. Jones and U. R. Kraus, Passive sampling in regulatory chemical monitoring of nonpolar organic compounds in the aquatic environment, Environ. Sci. Technol., 2016, 50, 3–17, DOI:10.1021/acs.est.5b04050.
- P. J. Reitsma, D. Adelman and R. Lohmann, Challenges of using polyethylene passive samplers to determine dissolved concentrations of parent and alkylated PAHs under cold and saline conditions, Environ. Sci. Technol., 2013, 47, 10429–10437, DOI:10.1021/es402528q.
- F. Smedes, R. W. Geertsma, T. van der Zande and K. Booij, Polymer− water partition coefficients of hydrophobic compounds for passive sampling: Application of cosolvent models for validation, Environ. Sci. Technol., 2009, 43, 7047–7054, DOI:10.1021/es9009376.
- Y. Choi, Y.-M. Cho and R. G. Luthy, Polyethylene–water partitioning coefficients for parent-and alkylated-polycyclic aromatic hydrocarbons and polychlorinated biphenyls, Environ. Sci. Technol., 2013, 47, 6943–6950, DOI:10.1021/es304566v.
- P. Lei, J. Zhu, K. Pan and H. Zhang, Sorption kinetics of parent and substituted PAHs for low-density polyethylene (LDPE): Determining their partition coefficients between LDPE and water (KLDPE) for passive sampling, J. Environ. Sci., 2020, 87, 349–360, DOI:10.1016/j.jes.2019.07.021.
- L.-J. Bao, S.-P. Xu, Y. Liang and E. Y. Zeng, Development of a low-density polyethylene-containing passive sampler for measuring dissolved hydrophobic organic compounds in open waters, Environ. Toxicol. Chem., 2012, 31, 1012–1018, DOI:10.1002/etc.1788.
- M. T. O. Jonker, Polyethylene–Water and Polydimethylsiloxane–Water Partition Coefficients for Polycyclic Aromatic Hydrocarbons and Polychlorinated Biphenyls: Influence of Polymer Source and Proposed Best Available Values, Environ. Toxicol. Chem., 2022, 41, 1370–1380, DOI:10.1002/etc.5333.
- S. B. Hawthorne, M. T. O. Jonker, S. A. van der Heijden, C. B. Grabanski, N. A. Azzolina and D. J. Miller, Measuring picogram per liter concentrations of freely dissolved parent and alkyl PAHs (PAH-34), using passive sampling with polyoxymethylene, Anal. Chem., 2011, 83, 6754–6761, DOI:10.1021/ac201411v.
- S. Josefsson, H. P. H. Arp, D. B. Kleja, A. Enell and S. Lundstedt, Determination of polyoxymethylene (POM)–water partition coefficients for oxy-PAHs and PAHs, Chemosphere, 2015, 119, 1268–1274, DOI:10.1016/j.chemosphere.2014.09.102.
- G. Cornelissen, A. Pettersen, D. Broman, P. Mayer and G. D. Breedveld, Field testing of equilibrium passive samplers to determine freely dissolved native polycyclic aromatic hydrocarbon concentrations, Environ. Toxicol. Chem., 2008, 27, 499–508, DOI:10.1897/07-253.1.
- K. Yates, I. Davies, L. Webster, P. Pollard, L. Lawton and C. Moffat, Passive sampling: partition coefficients for a silicone rubber reference phase, J. Environ. Monit., 2007, 9, 1116–1121, 10.1039/B706716J.
- M. T. O. Jonker, S. A. van der Heijden, M. Kotte and F. Smedes, Quantifying the effects of temperature and salinity on partitioning of hydrophobic organic chemicals to silicone rubber passive samplers, Environ. Sci. Technol., 2015, 49, 6791–6799, DOI:10.1021/acs.est.5b00286.
- E. L. DiFilippo and R. P. Eganhouse, Assessment of PDMS-water partition coefficients: implications for passive environmental sampling of hydrophobic organic compounds, Environ. Sci. Technol., 2010, 44, 6917–6925, DOI:10.1021/es101103x.
- B. Muijs and M. T. O. Jonker, Temperature-dependent bioaccumulation of polycyclic aromatic hydrocarbons, Environ. Sci. Technol., 2009, 43, 4517–4523, DOI:10.1021/es803462y.
- T. L. ter Laak, F. J. M. Busser and J. L. M. Hermens, Poly(dimethylsiloxane) as passive sampler material for hydrophobic chemicals: effect of chemical properties and sampler characteristics on partitioning and equilibration times, Anal. Chem., 2008, 80, 3859–3866, DOI:10.1021/ac800258j.
- G. Witt, G. A. Liehr, D. Borck and P. Mayer, Matrix solid-phase microextraction for measuring freely dissolved concentrations and chemical activities of PAHs in sediment cores from the western Baltic Sea, Chemosphere, 2009, 74, 522–529, DOI:10.1016/j.chemosphere.2008.09.073.
- M. T. O. Jonker, Determining octanol-water partition coefficients for extremely hydrophobic chemicals by combining “slow stirring” and solid-phase microextraction, Environ. Toxicol. Chem., 2016, 35, 1371–1377, DOI:10.1002/etc.3300.
- F. Smedes, SSP silicone–, lipid–and SPMD–water partition coefficients of seventy hydrophobic organic contaminants and evaluation of the water concentration calculator for SPMD, Chemosphere, 2019, 223, 748–757, DOI:10.1016/j.chemosphere.2019.01.164.
- K. Booij, R. van Bommel, H. M. van Aken, H. van Haren, G.-J. A. Brummer and H. Ridderinkhof, Passive sampling of nonpolar contaminants at three deep-ocean sites, Environ. Pollut., 2014, 195, 101–108, DOI:10.1016/j.envpol.2014.08.013.
- C. Sun, T. Soltwedel, E. Bauerfeind, D. A. Adelman and R. Lohmann, Depth profiles of persistent organic pollutants in the north and tropical Atlantic Ocean, Environ. Sci. Technol., 2016, 50, 6172–6179, DOI:10.1021/acs.est.5b05891.
- H. Lee, W. J. Shim and J.-H. Kwon, Sorption capacity of plastic debris for hydrophobic organic chemicals, Sci. Total Environ., 2014, 470, 1545–1552, DOI:10.1016/j.scitotenv.2013.08.023.
- E. J. Morgan and R. Lohmann, Detecting air–water and surface-deep water gradients of PCBs using polyethylene passive samplers, Environ. Sci. Technol., 2008, 42, 7248–7253, DOI:10.1021/es800518g.
- R. Lohmann, Critical review of low-density polyethylene's partitioning and diffusion coefficients for trace organic contaminants and implications for its use as a passive sampler, Environ. Sci. Technol., 2012, 46, 606–618, DOI:10.1021/es202702y.
- K. Booij, H. E. Hofmans, C. V. Fischer and E. M. van Weerlee, Temperature-dependent uptake rates of nonpolar organic compounds by semipermeable membrane devices and low-density polyethylene membranes, Environ. Sci. Technol., 2003, 37, 361–366, DOI:10.1021/es025739i.
- S. E. Allan, B. W. Smith and K. A. Anderson, Impact of the Deepwater Horizon oil spill on bioavailable polycyclic aromatic hydrocarbons in Gulf of Mexico coastal waters, Environ. Sci. Technol., 2012, 46, 2033–2039, DOI:10.1021/es202942q.
- L. G. Tidwell, S. E. Allan, S. G. O'Connell, K. A. Hobbie, B. W. Smith and K. A. Anderson, PAH and OPAH flux during the deepwater horizon incident, Environ. Sci. Technol., 2016, 50, 7489–7497, DOI:10.1021/acs.est.6b02784.
- K. Molt and U. Telgheder, Berechnung der Verfahrensstandardabweichung und Nachweis-, Erfassungs-und Bestimmungsgrenze aus einer Kalibrierung gemäβ DIN 32645, Online-Publikation der Universität Duisburg-Essen, 2010 Search PubMed.
- Y.-G. Ma, Y. D. Lei, H. Xiao, F. Wania and W.-H. Wang, Critical review and recommended values for the physical-chemical property data of 15 polycyclic aromatic hydrocarbons at 25 C, J. Chem. Eng. Data, 2010, 55, 819–825, DOI:10.1021/je900477x.
- M. M. Perron, R. M. Burgess, K. T. Ho, M. C. Pelletier, C. L. Friedman, M. G. Cantwell and J. P. Shine, Development and evaluation of reverse polyethylene samplers for marine phase II whole-sediment toxicity identification evaluations, Environ. Toxicol. Chem., 2009, 28, 749–758, DOI:10.1897/08-229.1.
- T. Zhu, C. T. Jafvert, D. Fu and Y. Hu, A novel method for measuring polymer-water partition coefficients, Chemosphere, 2015, 138, 973–979, DOI:10.1016/j.chemosphere.2014.12.040.
- S. E. Hale, T. J. Martin, K.-U. Goss, H. P. H. Arp and D. Werner, Partitioning of organochlorine pesticides from water to polyethylene passive samplers, Environ. Pollut., 2010, 158, 2511–2517, DOI:10.1016/j.envpol.2010.03.010.
- L. A. Fernandez, J. K. MacFarlane, A. P. Tcaciuc and P. M. Gschwend, Measurement of freely dissolved PAH concentrations in sediment beds using passive sampling with low-density polyethylene strips, Environ. Sci. Technol., 2009, 43, 1430–1436, DOI:10.1021/es802288w.
- K. Varani, S. Gessi, S. Merighi and P. A. Borea, Van't Hoff Based Thermodynamics, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, DOI:10.1002/9783527673025.ch2.
- R. M. Burgess, W. J. Berry, D. R. Mount and D. M. Di Toro, Mechanistic sediment quality guidelines based on contaminant bioavailability: Equilibrium partitioning sediment benchmarks, Environ. Toxicol. Chem., 2013, 32, 102–114, DOI:10.1002/etc.2025.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00133h |
| This journal is © The Royal Society of Chemistry 2024 |