
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA 50 year record for perfluoroalkyl acids in the high arctic: implications for global and local transport†
Daniel
Persaud
a,
Alison S.
Criscitiello
 b,
Christine
Spencer
c,
Igor
Lehnherr
d,
Derek C. G.
Muir
c,
Amila O.
De Silva
*c and
Cora J.
Young
*a
b,
Christine
Spencer
c,
Igor
Lehnherr
d,
Derek C. G.
Muir
c,
Amila O.
De Silva
*c and
Cora J.
Young
*a
aDepartment of Chemistry, York University, Toronto, ON, Canada. E-mail: youngcj@yorku.ca
bDepartment of Earth and Atmospheric Sciences, University of Alberta, Edmonton, Alberta, Canada
cAquatic Contaminants Research Division, Environmental and Climate Change Canada, Burlington, Ontario, Canada. E-mail: amila.desilva@ec.gc.ca
dDepartment of Geography, Geomatics and Environment, University of Toronto-Mississauga, Mississauga, Ontario, Canada
First published on 19th July 2024
Abstract
Perfluoroalkyl acids (PFAAs) are persistent compounds that are ubiquitous globally, though some uncertainties remain in the understanding of their long-range transport mechanisms. They are frequently detected in remote locations, where local sources may be unimportant. We collected a 16.5 metre ice core on northern Ellesmere Island, Nunavut, Canada to investigate PFAA deposition trends and transport mechanisms. The dated core represents fifty years of deposition (1967–2016), which accounts for the longest deposition record of perfluoroalkylcarboxylic acids (PFCAs) in the Arctic and the longest record of perfluoroalkylsulfonic acids (PFSAs) globally. PFCAs were detected frequently after the 1990s and have been increasing since. Homologue pair correlations, molar concentration ratios, and model comparisons suggest that PFCAs are primarily formed through oxidation of volatile precursors. PFSAs showed no discernible trend, with concentrations at least an order of magnitude lower than PFCAs. We observed episodic deposition of some PFAAs, notably perfluorooctane sulfonic acid (PFOS) and perfluorobutane sulfonic acid (PFBS) before the 1990s, which may be linked to Arctic military activities. Tracer analysis suggests that marine aerosols and mineral dust are relevant as transport vectors for selected PFAAs during specific time periods. These observations highlight the complex mechanisms responsible for the transport and deposition of PFAAs in the High Arctic.
Environmental significanceTo better understand the long-range transport of perfluoroalkyl acids (PFAAs), an ice core representing 50 years of deposition—from 1967 to 2016—was collected from the Canadian High Arctic. The deposition flux of perfluoroalkylcarboxylic acids increased from the early 1990s to 2016, while the perfluoroalkylsulfonic acids were deposited episodically. We found evidence for transport pathways that have not before been observed to contribute to remote terrestrial PFAA levels, including marine aerosols for some homologues. Understanding the deposition record of organic contaminants in remote pristine environments is an invaluable tool to help understand the global impacts of changing production and policy. |
Introduction
Per- and polyfluoroalkyl substances (PFAS, CnF2n+1–R) describe a diverse class of anthropogenic compounds that have been produced since the 1950s.1 A perfluoroalkyl moiety (CnF2n+1–) imparts unique properties when compared to its hydrocarbon counterparts including the ability to lower surface tension, form stable foams, as well as exceptional chemical and thermal stability.2,3 Thus, these compounds have numerous consumer and industrial applications including fluoropolymer-coated cookware, clothing, metal plating, and aqueous film-forming foams (AFFF).2 While many useful applications exist, they are highly persistent in the environment,4 bioaccumulative,5 toxic,6 and can undergo long-range transport.3 These are all hallmark characteristics of persistent organic pollutants (POPs), which prompted manufacturers and regulatory bodies to phase out and restrict the use of selected PFAS.7PFAS exists in several subgroups, the most widely known and studied is the perfluoroalkyl acids (PFAAs) which include both the perfluoroalkylcarboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs).1 Due to the ubiquitous nature of PFAAs, they are frequently detected in remote locations, including the Arctic.8–13 Two major pathways have been suggested to explain their long-range transport: direct transport and indirect formation in the atmosphere.3 With direct transport, PFAAs are released and transported in the stable carboxylic (PFCA) or sulfonic acid (PFSA) form after manufacture and use. This transport mechanism can occur in the gas phase, on particles (including marine aerosols) or by ocean currents.3,10 With indirect formation, PFAAs are formed through chemical transformation of volatile precursors in the atmosphere.14 Samples collected in remote locations can be used to investigate these different transport mechanisms. Ice caps and glaciers receive deposition primarily from the atmosphere due to their isolation from local sources and high elevation, in which a record of contamination can be preserved.9,10,15–17
Temporal variations over multiple decades can elucidate source and precursor contributions to long-range transport that can be used to understand the deposition mechanisms of PFAAs. Previously, ice core records from the Arctic have been measured, where PFAAs were detected in most samples with a few exceptions described in previous studies.8–10 To further improve our knowledge of PFAA deposition and transport mechanism in the High Arctic, we collected an ice core from Mt. Oxford icefield, located on Ellesmere Island, Nunavut, Canada. This ice core is geographically closer to possible emission sources highlighted in a previous study from Devon Ice Cap that recorded 38 years of deposition.10 Because Mt. Oxford icefield receives less precipitation annually (∼10 cm water equivalent per year) than the Devon Ice Cap (∼30 cm water equivalent per year),18 an ice core of similar length from Mt. Oxford icefield records over an additional decade of deposition compared to the Devon Ice Cap. This 50 year record represents the longest yet recorded for PFAS in the northern hemisphere and the longest for PFSAs globally. Here, we use the ice core record to examine: (1) PFAA depositional profiles; (2) temporal variations; (3) evidence of indirect and aerosol transport; and (4) causes of episodic pulses of high deposition.
Methods
Ice core collection and sectioning
Details of sampling procedures were described previously.10,19 Briefly, a 16.5 m ice core was collected from the summit of the Mt. Oxford icefield, Nunavut (82.19°N, 72.96°W, 1784 m above sea level) in May 2017 (Figure S1†). Ice core sections were retrieved using a 3-inch (7.7 cm) diameter aluminum and stainless-steel Kovacs ice drill. The ice core was drilled in one metre sections, packaged in polyethylene bags, and shipped frozen to the Canada Centre for Inland Waters (CCIW) in Burlington, ON, Canada.An age-depth scale was created using oxygen stable isotopes and ion chemistry measured in a co-located core that was drilled simultaneously within 5 m. Details of the dating methods have been previously described.20–22 The 16.5 m ice core represented 50 years of deposition (1967–2017) with a total dating error of ±1 year; further details can be found in the SI. The ice core was sectioned into discrete samples corresponding to individual years in a cold room (0 to −10 °C) using methanol (MeOH) rinsed stainless steel tools, with each section transferred to pre-cleaned high density polyethylene bottles. Samples were kept frozen (−30 °C) prior to sectioning, melting, extraction, and analysis. Data for the partial year of sample collection (2017) was not included in our analysis.
Sample preparation and extraction
Details of sample extraction have been described previously.10 We analyzed PFCAs (C5–C16), PFSAs (C4, C6, C8), perfluorooctanesulfonamide (FOSA), perfluoroethylcyclohexanesulfonate (PFECHS) and hexafluoropropylene oxide dimer acid (HFPO-DA), commonly known as GenX (See Table S1† for full analyte list). Measurement of PFCAs with four or fewer carbons in this ice core was reported previously.19 Samples were thawed prior to extraction and aliquoted to at least 500 mL melted volume which was required for solid phase extraction (SPE).9,10 Samples were shaken, sonicated for 10 minutes, and held for 30 minutes at room temperature.Samples were concentrated using OASIS® weak anion exchange SPE cartridges (6 cm3, 150 mg, 30 μm). Before extraction, each cartridge was conditioned with 5 mL 0.1% NH4OH/MeOH, followed by 5 mL MeOH and 5 mL SPE-cleaned HPLC grade water. Following sample concentration, cartridges were rinsed with 25 mM ammonium acetate buffer acidified to pH 4 with acetic acid and centrifuged at 4000 rpm for 2 minutes to remove any residual water. Elution was done in two fractions: the first fraction was eluted with 6 mL of MeOH for FOSA, while the second fraction was eluted with 8 mL of 0.1% NH4OH/MeOH for other PFAAs. Extracts were evaporated to dryness under a gentle stream of nitrogen and reconstituted in 0.5 mL 50/50 methanol–water containing the surrogate mixture (Table S2†), which was used to account for any matrix effects. Reconstituted samples were sonicated for 5 min, vortexed, and transferred to polypropylene vials for analysis.
Instrumental analysis
Extracts were analyzed using an Acquity UPLC I class liquid chromatography coupled to a XEVO® TQ-S tandem mass spectrometer in electrospray negative ionization mode (Waters Corporation, Massachusetts, USA). Analytes were separated using an Acquity UPLC BEH C18 column (Waters Acquity UPLC® BEH, 2.1 × 50 mm, 1.7 μm) with a water–methanol 2 mM ammonium acetate gradient method (Table S3 and S4†) and quantified based on relative response to isotopically labelled internal standards (Wellington Laboratories, Guelph, ON).Subsamples were also analyzed for major cations (Li+, Na+, K+, NH4+, Al3+, Ca2+, Mg2+) and anions (F−, Cl−, SO42−, PO42−, NO3−) by ion chromatography with conductivity detection. Separation was achieved using Dionex CS19 and AS11 columns for cations and anions, respectively (further details in the SI).
Quality assurance and quality control
Our previous findings suggest that environmental exposure and shipping do not contribute to PFAS contamination.10 Cartridge blanks were analyzed to verify the integrity of the extraction procedure. A mixture of isotopically labelled standards was also used to determine analyte recoveries and matrix effects (Table S5†). The accuracy of the method was evaluated by spiking an Arctic freshwater composite (n = 4) with native analytes before or after extraction (Table S7†). Limits of detection (LOD) were based on signal to noise ratios of 3, while method detection limits were based on average plus 3 times the standard deviation of method blanks. All QA/QC data can be found in section 1.5 in the SI.Statistical analysis
Statistical analyses were performed using RStudio (Version 1.4.1106) with R version 4.3.0. Annual fluxes of PFAAs and ions were logarithm-transformed [log10 PFAA, ions] to approximate normality before Pearson's correlation (r) analysis using the lmodel2 package.23 Values below the detection limit were included as ½ detection limit for examining temporal variations, but were excluded from all statistical tests. Further details are provided in the SI.Results and discussion
PFAA concentrations and fluxes on the Mt oxford icefield
Annual concentrations were measured in the ice core samples dating from 1967 – 2016 for PFAAs and FOSA. Generally, PFCAs from PFPeA (C5) to PFDoDA (12) were detected on Mt. Oxford icefield. The PFCAs from PFOA to PFDA (C8–C10) were detected in 98% of samples, whereas PFPeA (C5), PFHxA (C6), PFHpA (C7), PFUnDA (C11) and PFDoDA (C12) were detected in 90, 68, 90, and 66 and 60% of samples, respectively. The concentrations of individual PFCAs ranged from <LOD to 2.33 ng L−1 (Table S9†), with PFOA (C8) and PFNA (C9) being the major contributors. The presence of C13–C16 and C18 PFCAs, as well as HFPO-DA, were sought but were <LOD for the entire 50 year record. HFPO-DA was introduced as a PFOA alternative and has been produced since 2009.24 Since its introduction, several studies have detected HFPO-DA within a few hundred kilometres of fluorochemical production facilities,25–27 as well as in precipitation samples in the United States.28 The lack of detection of HFPO-DA on Mt. Oxford icefield may be because (i) the compound is present at levels below our current detection limits; (ii) no known precursors exist to facilitate indirect transport; and/or (iii) not enough time has passed since its introduction for direct transport via ocean currents to the remote Canadian Arctic to occur.29 HFPO-DA was detected in surface water samples from Europe to the Arctic along two transects within the Fram Strait, with a concentration range of <MDL to 26 pg L−1.30 The concentration of HFPO-DA displayed a sequential decrease from Europe to the Fram Strait30 which aligns with our hypothesis that insufficient time has elapsed for a direct ocean transport mechanism to the High Arctic. Moreover, HFPO-DA was never registered for use in Canada.31 The PFSAs detected were PFBS (C4), PFOS (C8), and PFECHS (cyclic C8), as well as FOSA with individual concentrations ranging from <LOD to 4.16 ng L−1 (Table S10†). PFHxS (C6) and PFDS (C10) were <LOD in all samples. PFBS was the dominant homologue with a detection frequency of 98%, whereas PFOS, PFECHS, and FOSA were detected in 90, 94, and 84% of samples, respectively. The detection of PFECHS is not consistent among previous studies in the Canadian Arctic. The Devon Ice Cap summit was sampled in 2008 and 2015.9,10 In the 2008 study, PFECHS was detected up to 20 pg L−1 in snow pit samples whereas all PFECHS were <0.94 pg L−1 in the ice core record collected in 2015. The concentration of PFECHS on Mt Oxford icefield is greater (142 pg L−1) than the maximum observed in the 2008 Devon Ice Cap study.Concentrations (ng L−1) were converted to fluxes (ng m−2 a−1) to determine the annual deposition of PFAAs on Mt. Oxford icefield (Tables S11 and S12†). Fluxes of individual PFCAs were up to 316 ng m−2 a−1 and were consistently higher than other Arctic and sub-Arctic deposition measurements, but lower than lower-latitude precipitation samples8–10,32–35 (Table S13†). Fluxes of PFSAs were generally lower than PFCAs, averaging 13 ng m−2 a−1, but elevated levels of PFOS and PFBS up to 471 ng m−2 a−1 were observed in a few years before the 1990s, which will be discussed in the Episodic PFAA Transport Pathways section below.
PFAA temporal variations
Variations in PFAA deposition over time are useful to elucidate source and precursor contributions to the High Arctic. We do not expect the integrity of the temporal record to be affected by post depositional effects based on the detection of solstice peaks of major ions in the aforementioned co-located ice core (Fig. S2†). Nevertheless, to circumvent any possible errors associated with sectioning, dating, and post-depositional processes, we conservatively report fluxes as five-year moving averages for temporal trend analysis.In general, all fluxes of PFCAs were observed to be constant or increasing with time, with the increases being more prominent after 1985 (Fig. 1A and S3†). Before this period, fluxes were relatively constant except for episodes of elevated levels of C6-12 PFCAs in the 1970s, which will be discussed in the Episodic PFAA Transport Pathways section below. The C8 (PFOA) and C9 (PFNA) homologues were the dominant PFCAs observed at Mt. Oxford icefield, similar to previous observations.10 Contrary to the deposition trends at Devon Ice Cap, in which C8 (PFOA) and C9 (PFNA) plateaued after 1995, we observe a consistent increase for these two homologues. Additionally, PFOA fluxes were at least an order of magnitude higher at Mt. Oxford icefield compared to Devon Ice Cap after 1991 and the Lomonsovfonna Ice Cap in Svalbard16 (Fig. 2), suggesting potential differences in sources.
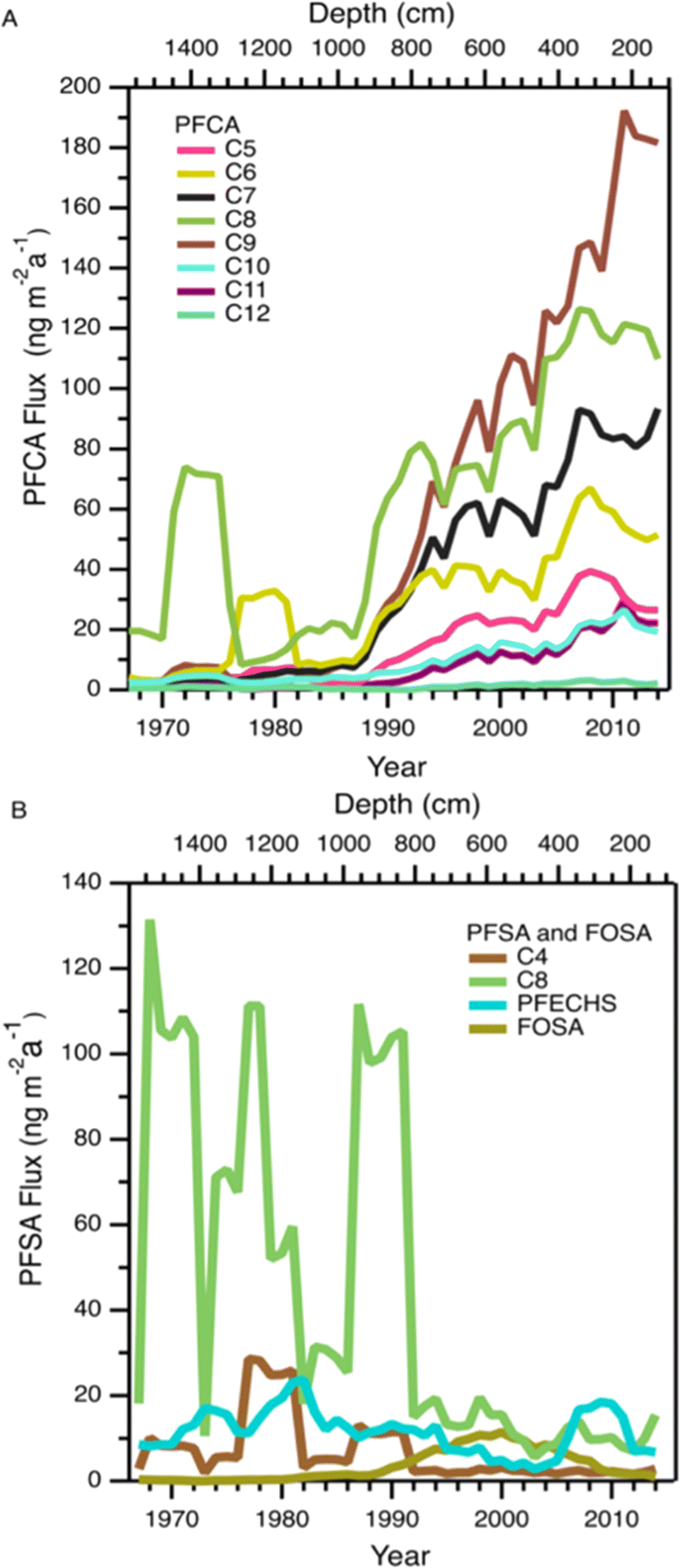 | ||
| Fig. 1 Five year moving averages of PFAA deposition flux on Mt. Oxford icefield: (A) PFCAs (C5–C12) and (B) PFSAs (C4, C8, PFECHS) and FOSA for the period 1967–2016. For values below detection limit, ½ detection limit was used to calculate flux. | ||
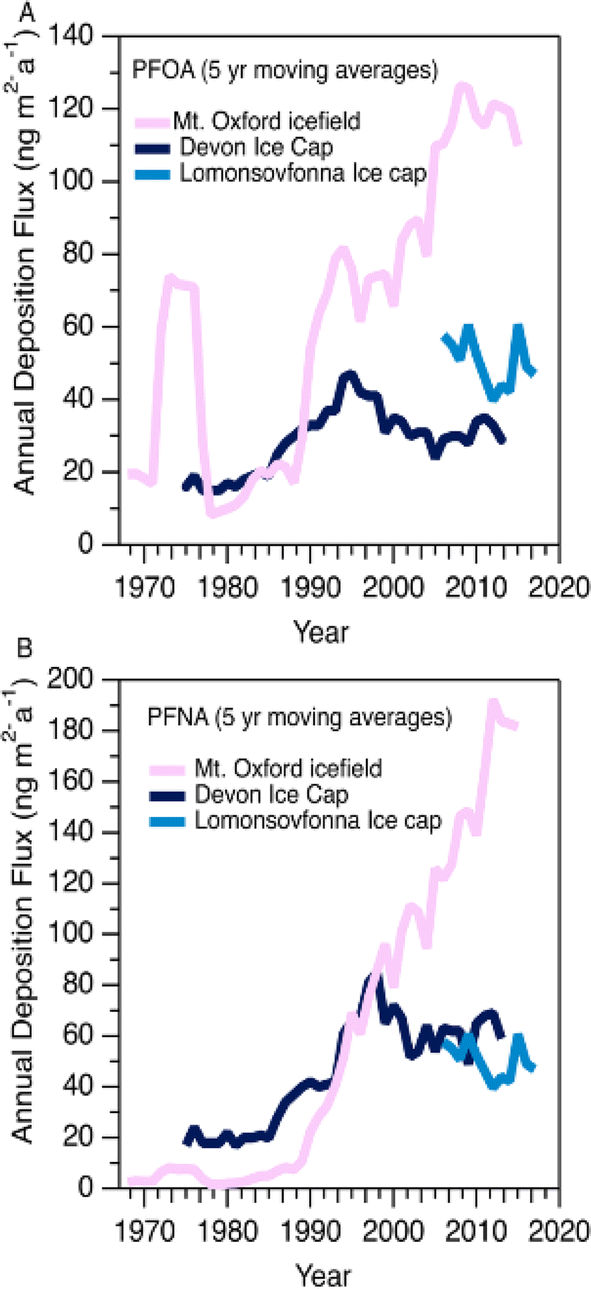 | ||
| Fig. 2 Five year moving averages of (A) PFOA and (B) PFNA deposition at Mt. Oxford icefield (1967–2016), Devon Ice Cap (1977–2015),10 and Lomonsovfonna Ice Cap (2006–2019).16 | ||
In 2006, the United States Environmental Protection Agency PFOA Stewardship program invited the eight major fluoropolymer and fluorotelomer manufacturers to voluntary eliminate emissions and production of PFOA, its precursor compounds, and the related longer chain homologues.26 Meanwhile in Canada, four participating companies agreed to eliminate residual PFOA, long chain PFCAs, and their precursors by 2015.27 Other manufacturers in Japan and Western Europe also implemented similar regulations. These regulations prompted a geographical shift in sources of PFCAs, as manufacturers that were not participants in the reduction programs (mainly in China and India) increased or began production of several classes of PFAS and its precursors.36 Ellesmere Island, where Mt. Oxford icefield is located, is influenced less by North American airmasses and more by Eurasian airmasses compared to more southern areas of the Canadian Arctic.37 Thus, we would expect that the more southern Devon Ice Cap might be more influenced by the reduction in emissions, while deposition to the more northern Mt. Oxford icefield would be more affected by continuing global production. This expected spatial pattern is generally consistent with our observations of PFCAs at Mt. Oxford icefield and the previous observations at Devon Ice Cap described above.
Fluxes of FOSA generally increased from 1980 to 2000 followed by a decrease over the next decade (Fig. 1B and S4†). This decrease is likely the effect of the voluntary phase out by 3 M of POSF-based chemistry (including PFOS and its precursors) by 2002.38 A similar temporal variation was observed at the Devon Ice Cap.10 However, at that location the decline happened over a faster timescale of ∼2 years. Following the decrease, FOSA fluxes at Mt. Oxford icefield remained higher than those at Devon Ice Cap. The slower decrease and higher levels of FOSA at the more northern Mt. Oxford icefield are again consistent with a larger influence of airmasses from Eurasia. No discernible trend with time was observed in the deposition of the other PFSAs. There were several years with deposition fluxes above 100 ng m−2 a−1 of the C8 PFSA (PFOS) and 10 ng m−2 a−1 of the C4 PFSA (PFBS) before 1995. Deposition events were isolated in time, suggesting episodic events that resulted in the deposition of these two homologues. This will be discussed further in the Episodic PFAA Transport Pathways section below.
Evidence for indirect transport of PFAAs
Indirect sources of PFAS may contribute to their presence and distribution to the Arctic. Laboratory chamber studies and chemical modelling demonstrated that volatile and semi-volatile precursor compounds including fluorotelomer alcohols (FTOHs), perfluoroalkane sulfonamides, and N-alkane perfluoroalkane sulfonamidoethanols can undergo OH-initiated oxidation in the gas phase to produce PFAAs.14,39,40 If a common indirect source exists, then PFAA homologues are expected to vary through time together. To explore this, we compared fluxes of PFAAs (Fig. 3 and Table S14†).9,10,28 Generally, all PFCA homologue pairs showed statistically significant moderate to very strong correlations (r: 0.5–0.96, p < 0.05). This is consistent with expected PFCA homologue production by gas phase atmospheric oxidation of fluorotelomer compounds.39 Weak but statistically significant correlations were also observed between FOSA and C5–C10 PFCAs (r: 0.34–0.46, p < 0.05). This has not been observed before in the Canadian Arctic9,10 but has been observed in Svalbard.16 Correlations between FOSA and PFCAs could be indicative of atmospheric formation, since sulfonamides have been identified as precursor of PFCAs with ≤8 carbons.40,41 These relationships suggest that most PFCAs originate from common sources and/or have experienced similar precursor oxidation and deposition processes. Our results are consistent with observations from other remote terrestrial locations where atmospheric deposition has been shown as a dominant source of PFCAs.9,10,16,17,32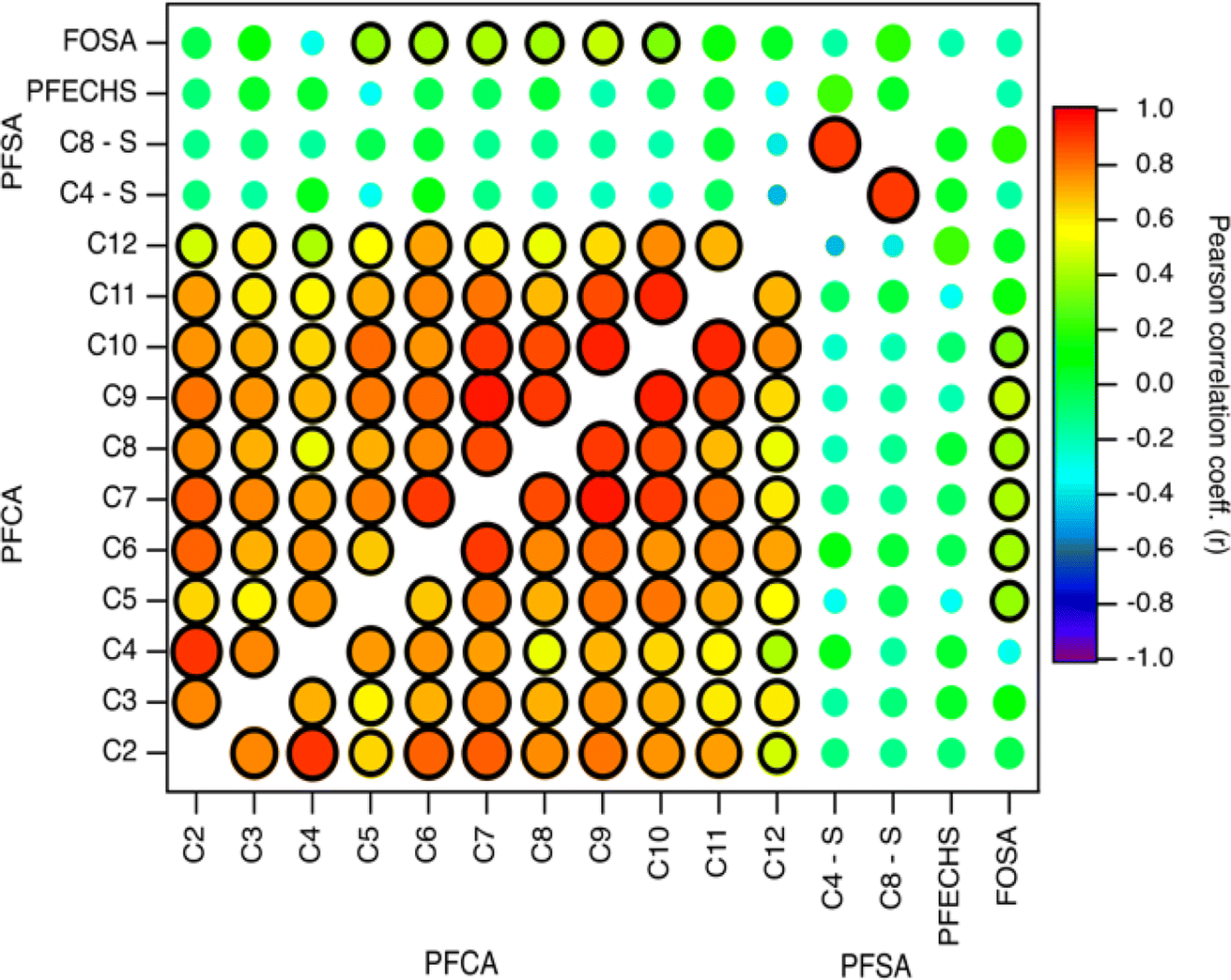 | ||
| Fig. 3 Pearson correlation map of log transformed PFCA, PFSA, and FOSA deposition fluxes on Mt. Oxford icefield for the entire sampling period (1967–2016). The strength of the correlation is illustrated by the colour and size of the circle, where larger circles denote a strong correlation based on the magnitude of r. The black outline represents a statistically significant correlation (p-value <0.05). Data for C2–C4 PFCAs from Pickard et al.19 | ||
Molar concentration ratios of even-odd homologue pairs are useful to discern the contribution of fluorotelomer atmospheric oxidation in PFCA deposition. Smog chamber studies suggest that the degradation of X![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 fluorotelomer compound yields PFCAs with X and X + 1 carbons.39 There will be some variability in the molar ratio of the even-odd PFCA pair depending on the atmospheric levels of NOx (NO + NO2) and peroxy radicals.14 If fluorotelomer oxidation is indeed a major pathway for PFCA formation, then sequential homologue pairs are expected to be present in roughly equal molar quantities (i.e., 1:1).10,42 Molar concentration ratios were calculated for three pairs of PFCAs: C6–C7, C8–C9, and C10–C11 (with the even carbon PFCA in the numerator and the odd carbon PFCA in the denominator), which represent the major PFCA oxidation products of 6:2, 8:2, and 10:2 fluorotelomers, respectively. Most flux ratio measurements were clustered around a 1:1 ratio within a factor of ± 2 after 1990 (i.e., between 2:1 and 1:2, Fig. S5†), comparable to previous measurements in the Arctic.10,16 The even homologues appear to be higher than the odd homologues (i.e., factor of 2 or more) prior to 1990. There is higher uncertainty in the data before 1990 because concentrations of odd PFCA homologues were close to detection limits, but may also suggest that PFCA sources pre-1990 were different.
:2 fluorotelomer compound yields PFCAs with X and X + 1 carbons.39 There will be some variability in the molar ratio of the even-odd PFCA pair depending on the atmospheric levels of NOx (NO + NO2) and peroxy radicals.14 If fluorotelomer oxidation is indeed a major pathway for PFCA formation, then sequential homologue pairs are expected to be present in roughly equal molar quantities (i.e., 1:1).10,42 Molar concentration ratios were calculated for three pairs of PFCAs: C6–C7, C8–C9, and C10–C11 (with the even carbon PFCA in the numerator and the odd carbon PFCA in the denominator), which represent the major PFCA oxidation products of 6:2, 8:2, and 10:2 fluorotelomers, respectively. Most flux ratio measurements were clustered around a 1:1 ratio within a factor of ± 2 after 1990 (i.e., between 2:1 and 1:2, Fig. S5†), comparable to previous measurements in the Arctic.10,16 The even homologues appear to be higher than the odd homologues (i.e., factor of 2 or more) prior to 1990. There is higher uncertainty in the data before 1990 because concentrations of odd PFCA homologues were close to detection limits, but may also suggest that PFCA sources pre-1990 were different.
The available evidence from homologue correlations and molar concentration ratios strongly suggests that most PFCAs are formed indirectly through the oxidation of volatile precursors. We can therefore use these data as an observational test of a recent GEOS-CHEM model (Fig. S6†)43 that includes oxidation chemistry of fluorotelomer compounds as well as deposition and transport of precursors and intermediates. Comparing the modelled and measured fluxes we found that modelled fluorotelomer oxidation can account for <50% of C9–C12 PFCAs and 3–46% of C5–C8 PFCAs, respectively, after the 1990s. Discrepancies between modelled and measured PFCA deposition highlight uncertainty in emission inventories used by the model44 and that gas-particle partitioning, which could contribute to the transport of PFCAs,43 has not yet been parameterized in model calculations. Thus, further work in chemical modeling of PFCA atmospheric formation and fate is needed.
In contrast to the PFCAs, pairs of PFSAs were not generally correlated with each other or with any PFCAs (Fig. 3 and Table S14†). This is consistent with the previous record from Devon Ice Cap,10 but inconsistent with a record from Svalbard,16 where correlation was observed between C8 PFSA (PFOS) and C2 PFCA (TFA). We observed a very strong, statistically significant correlation for the PFOS-PFBS pair (C8–C4, r: 0.90, p < 0.001). A similar observation was made at Devon Ice Cap (r2: 0.31, p < 0.05).10 These two compounds can be formed through the atmospheric oxidation of perfluoroalkane sulfonamido substances.40 Chamber experiments demonstrated that oxidation of N-methyl perfluorobutane sulfonamido ethanol (N-MeFBSE, C4 homologue) yields C2–C4 PFCAs and the C4 PFSA (PFBS). Similarly, the oxidation of N-methyl perfluorooctane sulfonamido ethanol (N-MeFOSE, C8 homologue) would be expected to yield the C8 PFSA (PFOS) and C2–C8 PFCAs. These precursors may co-occur in commercial products45 and may be emitted from similar sources.46 Both C4 and C8 PFSA precursors have been detected in Arctic environments.47,48 If this indirect formation were a significant source, we might expect to see correlations between C8 PFSA (PFOS) with one of its measured potential precursors, FOSA, and with C8 PFCA. We would also expect to see a correlation between C4 PFSA (PFBS) with the C4 PFCA. As discussed above, no correlations between any of these species were observed (Fig. 3). Thus, we have no substantive evidence that PFBS and PFOS found on Mt Oxford icefield were a result of indirect transport. However, another possibility for the correlation of PFBS and PFOS could be the use of these two homologues in AFFF formulations, where they could be released and subsequently transported to the Arctic. Potential impacts of local usage will be discussed in the next section.
Evidence for aerosol transport of PFAAs
Aerosol transport may also contribute to PFAA deposition on the Mt. Oxford icefield. Depending on the PFAA homologue and the aerosol source, transport on aerosols could be attributed to both direct and indirect sources. For example, aerosols could transport directly-emitted PFAAs or those formed from atmospheric oxidation. Most previous studies in remote regions have not observed clear evidence for marine aerosol-mediated long-range transport of PFAAs.9,16,17 While high volume atmospheric sampling has measured several PFAAs in aerosols (e.g., Wong et al., 2018, 2021),49,50 this sampling approach cannot accurately distinguish between gaseous and particulate PFAAs.51,52 The ice core record from Devon Ice Cap suggested a role for aerosols, in which there was some evidence for transport by mineral dust.10 Major ion analysis can be useful to determine the influence of sea spray, biomass burning, mineral dust, and anthropogenic aerosol contributions to the High Arctic.10,53To assess the contribution of marine aerosols to PFAA deposition, we compared the PFAA homologue profiles in the ice core to the surrounding ocean samples and conducted correlation analysis on individual PFAAs with a marine aerosol tracer (Na+). Consistent with other studies, the molar ratios are distinct to the typical ratios in oceans (Fig. S7†). The contribution of marine aerosols to PFAA deposition was also assessed by comparing the PFAA homologue profiles in the ice core and surrounding ocean samples (Fig. S8†).9,10 This transport mechanism is highly dependent on the concentration of the analyte in bulk seawater, which is expected to change with time, as well as the enrichment factor in marine aerosols.54,55 Enrichment is larger for PFSAs and increases with perfluorinated chain length.54,56 Thus, if a marine source is important, an icefield homologue profile relatively enriched in longer-chain homologues and PFSAs is expected.9 Consistent with previous observations from the Canadian Arctic10 and the Antarctic,17 we did not observe this (Fig. S8†). Instead, we observed a relative enrichment of short chain homologues and a lower abundance of PFSAs on the icefield when compared to the surrounding oceans.
Correlation with Na+ was also used to assess marine aerosol contributions to individual PFAA deposition. The ocean around Ellesmere Island is icebound for most of the year, with minimum sea ice in September.57 Despite this, evidence from measurements and simulations suggest that marine aerosols can be generated from salty blowing snow from sea ice surfaces and/or transport by frost flowers.58–62 In fact, model simulations estimate sea ice produces an order of magnitude more marine aerosols than the same unit area of open ocean under similar conditions.58,63 Polynyas also exist in the Canadian Arctic Archipelago year-round due to tidal currents and tidal mixing (e.g., North Water and Hell Gate polynya).64 Recently, the polynyas within Arctic Canada and the Canadian Basin of the Arctic Ocean were identified as a possible source of marine aerosols to Ellesmere Island.21 Correlation analysis revealed weak to moderate correlations (p < 0.05) between individual PFAA homologues and Na+ for the full time period (Fig. 4A, S10 and Table S18†). The C5–C10 and C12 PFCAs were all positively correlated with Na+ (r: 0.35–0.52, p < 0.05), while the C11 (PFUnDA) homologue showed no relationship with Na+ (r: 0.11, p = 0.54). Among the PFSAs, PFECHS was the only homologue that had a statistically significant correlation with Na+ (r = 0.52, p < 0.00001), whereas the other PFSAs (C4 and C8) and FOSA failed to show any correlations (r: −0.04–0.03, p: 0.83–0.84). This suggests transport by marine aerosols may be a contributing source of selected PFAAs to Mt. Oxford icefield.
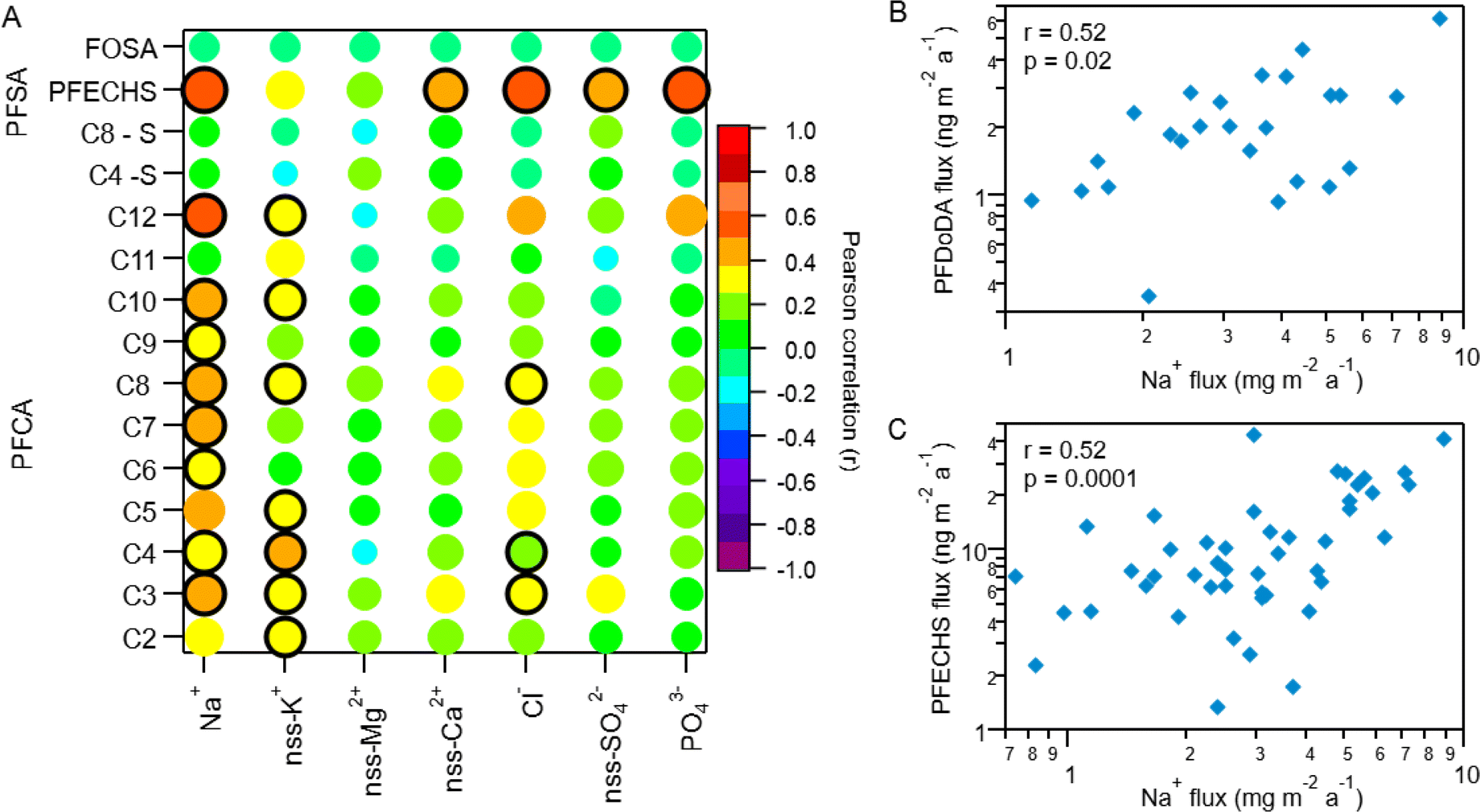 | ||
| Fig. 4 Correlations log transformed PFAA deposition fluxes and major ions on Mt. Oxford icefield for the entire sampling period (1967–2016). (A) Correlation matrix where the strength of the correlation is illustrated by the colour and size of the circle, where larger circles denote a strong correlation (larger Pearson correlation coefficient). The black outline represents a statistically significant correlation (p-value <0.05). (B) Correlation between Na+ and PFDoDA fluxes. (C) Correlation between Na+ and PFECHS fluxes (1967–2016). For (B) and (C), the Pearson correlation (r) and p-value for the line of best fit are also included. | ||
Although the ocean vertical distribution and composition profiles of PFAAs are variable, several studies have found enhanced PFAA concentrations near the air-sea interface.30,65 Recently, Garnett et al.66 investigated the distribution of PFAAs in seawater and sea ice compartments in the European High Arctic. They found elevated levels of PFAAs at the surface of sea ice and under-ice seawater, and, using salinity profiles and δ18OH2O composition, found a strong atmospheric source of PFAAs.66 It is possible that PFAAs are deposited to seawater or sea ice compartments through atmospheric transformation of precursors, followed by the subsequent transport to the terrestrial environment by marine aerosols.64 A similar association was made during a long-term air monitoring study in Norway, which found PFCAs and PFSAs significantly correlated with Na+ (r: 0.4–0.8, p < 0.05).56 However, that study collected particulate PFAA in low-lying sampling locations that were close to the ocean (∼1.3 and 20 km to open water)56 whereas Mt. Oxford icefield is located ∼100 km from the Arctic Ocean and at ∼1.8 km elevation. Interestingly, the results from our study are inconsistent with results from two other Arctic sites (Devon Ice Cap and Lomonsovfonna Ice Cap)10,16 where no correlation with Na+ was observed, although both sites can be influenced by marine aerosols. This may be due to competing sources other than marine aerosol transport (e.g., oxidation of volatile precursors). To the best of our knowledge, our observations represent the first evidence for marine aerosol transport contributing to PFAA loading in a remote polar terrestrial environment.
Among the statistically significant correlations between PFAAs and Na+, the C12 PFCA (PFDoDA) and PFECHS were most strongly correlated (r: 0.52 for both, p < 0.05, Fig. 4), which provides further evidence of marine aerosol transport. PFDoDA, with its long perfluorinated carbon chain, is one of the most surface-active PFCAs and has a higher enrichment factor in marine aerosols than other PFCAs.54,55 The transport of PFECHS must be driven by direct processes since no known precursors exist. Because PFECHS is permitted to be used as an abrasion inhibitor in aircraft hydraulic fluids,67 hydraulic fluid leakage from aircraft and airport activities may increase Arctic PFECHS concentrations. Multiple studies have linked PFECHS use in aviation to local environmental contamination (e.g., ref. 68 and 69). It is plausible that Arctic airport activity on and around Ellesmere Island (e.g., Canadian Forces Station Alert, located 162 km from Mt. Oxford icefield) could have led to elevated concentrations of PFECHS in the surrounding ocean. Though we cannot confirm our hypothesis since no measurements exist, this would be consistent with local contamination measurements elsewhere, and could explain this unusual observation.
Long range transport by mineral dust aerosols may also be a pathway for transport of PFAAs to this region. One previous study has suggested a role for PFAA transport mediated by mineral dust.10 Correlation with non-sea salt (nss) Ca2+ and nssMg2+ suggests either that a mechanistic relationship between PFAAs and mineral dust exists or that PFAAs and mineral dust are being transported within the same airmass. Other strong atmospheric acids are known to accumulate on dust and undergo long-range transport.70 Since PFAAs are strong acids, a similar mechanism could lead to their transport. To assess the potential role of this pathway, we examined correlations between PFAAs and typical indicators of mineral dust, nssCa2+ and nssMg2+.71 No correlations were observed when applied on a multidecadal scale, with the exception of PFECHS, which showed a moderate correlation with nssCa2+ (r = 0.49, p < 0.05). This is in contrast to the observations of Pickard et al.10 at the Devon Ice Cap where nssCa2+ and nssMg2+ were correlated with several PFAAs.
Episodic PFAA transport pathways
This ice core uniquely recorded several episodes of elevated deposition for PFSAs and PFCAs (Fig. 1); since the seasonal record is robust, we additionally reported the annual fluxes to highlight the episodic nature of deposition (Fig. 5). Episodic detection was based on at least two consecutive samples (e.g., 1973 and 1974), indicating the observations were not caused by contamination of a single sample. In previous ice core records, episodic deposition has not been observed, with the exception of one episode of elevated C8 PFSA (PFOS) on Devon Ice Cap in 2013.10 Because these events differed from the rest of the temporal record, they were examined separately to assess whether they could be attributed to different transport pathways.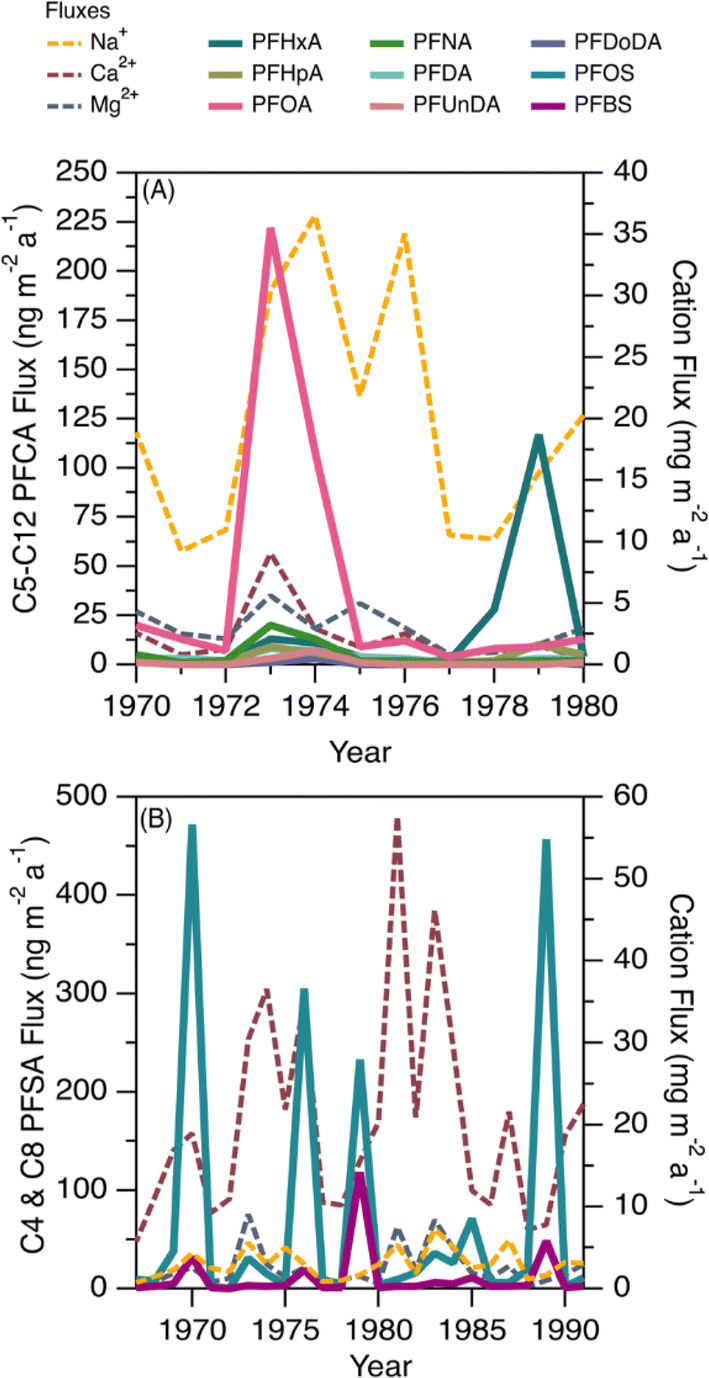 | ||
| Fig. 5 Annual deposition fluxes for (A) C6–C12 PFCAs (ng m−2 a−1, left) and Na+, nssCa2+, and nssMg2+ (mg m−2 a−1, right) for the period 1970–1980 and (B) C4 and C8 PFSAs (ng m−2 a−1, left) and Na+, nssCa2+ and nssMg2+ (mg m−2 a−1, right) for the period 1967–1991. The PFAAs are represented by solid lines while major ions are represented by broken lines and are presented on different scales. For values below detection limit, ½ detection limit was used to calculate flux. | ||
Prior to 1980, we observed two distinct periods of PFCA increases (Fig. 5A). One episode occurred during 1973–1974, where C6–C12 PFCA deposition fluxes showed more than a three-fold increase. Among these, C8 (PFOA) demonstrated the largest increase, rising by more than 34 times from the prior year. Another episode occurred in 1979, where C6 (PFHxA) deposition flux increased 100 times. The correlation analysis between PFCAs and aerosol tracers was repeated for the period 1967–1979 to determine if any relationship existed. We observed statistically significant correlations between C8–C10 PFCAs and Na+ (r: 0.55–0.70, p < 0.05) and moderate to strong correlations between C7–C10 PFCAs and nssCa2+ and nssMg2+ (r: 0.54–0.87, p < 0.05, Table S18†). These correlations were driven by the co-occurrence of elevated cation deposition during the 1973 episode, which may indicate a role for marine aerosols and/or mineral dust in the deposition events observed pre-1980. The long-range transport of mineral dust to the Arctic has been investigated to understand impacts on regional climate. Recently, Zhao et al.72 analyzed dust events in China during 2011–2015 and found two major transport routes to the Arctic: (i) mostly over land transport with a short duration (4–8 d), which was observed in Barrow, Alaska; and (ii) long-duration transport event (7–10 d) originating from the Gobi desert, passing through China, Korea, Japan, and the North Pacific ocean (where mixing with marine aerosols is possible) and finally deposited at Alert.72 Long-duration transport events could be consistent with our observation of elevated PFCAs along with Na+, nssCa2+, and nssMg2+ during the 1970s. The cation profile also shows high concentration of Ca2+ which is typical of Asian mineral dust,70 providing additional evidence for a synoptic scale event leading to enhanced transport from that region.73 Taken together, the evidence presented here suggests that direct transport during the period 1967–1980 may be responsible for the observed episodes of elevated PFCA deposition.
We also observed episodes of elevated PFSAs, with the C4 (PFBS) and C8 (PFOS) elevated in 1970, 1976, 1979, 1985, and 1989 (Fig. 5B). In all episodes, deposition fluxes of C8 were more than 74 times greater than the previous year, whereas the deposition fluxes of C4 increased 2 to 84 times. As discussed above, we have no clear evidence for C4 and C8 PFSA indirect formation or transport by marine aerosols or mineral dust. In the absence of other evidence, we hypothesize that episodic deposition may be related to direct release of AFFF from military Arctic activities. First developed in the 1960s, AFFF is primarily used to extinguish hydrocarbon fuelled fires at oil refineries, military bases, airports and firefighter training facilities.74 It is documented that AFFF was stored and used in military fire suppressant training at Thule Air Base, Qaanaaq, Greenland75 and in Alert, Canada.76,77 As of 2017, Thule Air Base was the only US Air Force base that still had AFFF stockpiles on site and had not transitioned to newer formulations that are typically characterized by shorter chain PFAS or the absence of fluorinated chemicals.75 The composition of commercially produced AFFF is proprietary and is known to vary between manufacturer and production time.78 While we cannot know for sure what formulations of AFFF were used at Alert and Thule, analysis of archived AFFF samples from military bases found that C8 (PFOS) was the dominant homologue in the formulation, with lesser contributions of other PFSAs, as well as PFCAs.79 The archived samples represented AFFF formulations from 1989 to 2001—well after our observed events—but represent the oldest AFFF characterizations available.72 Unfortunately, very little is known about emissions to air from AFFF, with the few studies that exist focusing on modern AFFF formulations.80,81 Given these uncertainties, our measurements are generally consistent with a possible legacy AFFF source.78,79 Air mass back trajectory analysis for two years (2011 and 2013) revealed that some of the air mass impacting Mt Oxford icefield originated from the Arctic Ocean and the western coast of Greenland (Fig. S11†), which is in close proximity to Thule Air Base. We caution that annual average air mass back trajectories may not capture source regions for sporadic long-range transport episodes that convey materials long distances to the Arctic; rather they are intended to identify possible source regions. It is also possible that anomalous events, such as those that have been shown to be responsible for the long-range transport of dust and pollen to the Arctic,73,82 could contribute to the transport of PFSAs.
Additionally, episodic pulses were observed for two organophosphate ester contaminants in Mt. Oxford icefield measured in a separate core sampled at the same time as this study.22 In that paper, pulses of tris(isobutyl)phosphate and tris-(1-chloro-2-propyl)phosphate were observed in several years between 1975 and 1992. These peaks were not coincident with the temporal profile at the Devon Ice Cap and were unique to Mt. Oxford icefield.22 Taken together, this may suggest a combination of local activity with climate episodes can result in depositional pulses.
Conclusions
Monitoring deposition of organic contaminants in remote pristine environments is an invaluable tool to help us understand the impacts of changing production, as well as different transport mechanisms. It is well established that PFAS can undergo long-range transport to remote locations;9,10,17,53 however, the mechanisms that facilitate this transport are not fully understood. This study emphasizes the power of ice cores to aid in understanding long-range transport of contaminants. To gain new insight into possible transport mechanisms, we presented the most extensive temporal record of PFCAs in the Northern Hemisphere, as well as the longest temporal record of PFSAs globally.Our results indicate that most PFAAs are present and there is evidence of increasing deposition of PFCAs over the past 50 years at Mt. Oxford icefield. Using homologue correlations, molar concentration ratios and model comparisons, we confirmed that PFCAs deposited at Mt. Oxford icefield are formed primarily through the long-range atmospheric transport and oxidation of volatile precursors in the atmosphere. The PFCA deposition was greater than observed deposition at Devon Ice Cap, which suggests a greater influence from Europe and Continental Asia. We also highlighted the importance of measuring supplementary analytes for source elucidation. Correlation analysis between marine and mineral dust tracers yielded two unique findings exclusive to this ice core: (i) marine aerosols have been identified as a possible transport mechanism for selected PFAAs to the terrestrial environment during specific time periods; and (ii) mineral dust was associated with an unusual deposition event of PFCAs observed during the 1970s.
The extended temporal record of this ice core allowed unique observations that were not possible in other previous studies. We observed episodic elevated deposition pulses for several PFAAs. Notably, C8 (PFOS) and C4 (PFBS) PFSAs were elevated in five (5) years prior to 1990. While one pulse of C8 PFSA was observed at the Devon Ice Cap in 2013 (70 ng m−2 a−1), the deposition fluxes observed in the current study (up to 471 ng m−2 a−1) are several times greater. We postulate that these events may be evidence of direct transport of AFFF used in nearby military Arctic activities. Further work is needed to better understand the causes of these historic pulses.
Ice caps represent a natural record of contaminant concentration, which is useful to discern trends and sources that may change over time. Ice cores provide particular utility for understanding PFAAs in remote locations, for which there are multiple potential long-range transport mechanisms. While ice cores can improve our understanding of long-range transport sources and mechanisms of PFAAs, as well as the efficacy of regulation, this valuable resource is disappearing as a result of climate change.83,84 Therefore, there is an urgent need to prioritize further research efforts to collect ice cores to discern temporal trends and possible sources.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Northern Contaminants Program and Natural Sciences and Engineering Research Council. Logistical support was provided by the Polar Continental Shelf Program. We thank Jocelyn Hirose for field sampling assistance, Shira Joudan for analytical assistance, and Colin Thackray for model outputs.References
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. De Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. J. van Leeuwen, Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins, Integr. Environ. Assess. Manage., 2011, 7(4), 513–541, DOI:10.1002/ieam.258.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. Dewitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, An Overview of the Uses of Per- And Polyfluoroalkyl Substances (PFAS), Environ. Sci.: Processes Impacts, 2020, 22(12), 2345–2373, 10.1039/d0em00291g.
- K. Prevedouros, I. T. Cousins, R. C. Buck and S. H. Korzeniowski, Sources, Fate and Transport of Perfluorocarboxylates, Environ. Sci. Technol., 2006, 40(1), 32–44, DOI:10.1021/es0512475.
- T. Frömel and T. P. Knepper, Biodegradation of Fluorinated Alkyl Substances, Rev. Environ. Contam. Toxicol., 2010, 208, 161–177, DOI:10.1007/978-1-4419-6880-7_3.
- J. M. Conder, R. A. Hoke, W. De Wolf, M. H. Russell and R. C. Buck, Are PFCAs Bioaccumulative? A Critical Review and Comparison with Regulatory Criteria and Persistent Lipophilic Compounds, Environ. Sci. Technol., 2008, 42(4), 995–1003, DOI:10.1021/es070895g.
- C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens and J. Seed, Perfluoroalkyl Acids: A Review of Monitoring and Toxicological Findings, Toxicol. Sci., 2007, 99(2), 366–394, DOI:10.1093/toxsci/kfm128.
- M. Land, C. A. De Wit, I. T. Cousins, D. Herzke, J. Johansson and J. W. Martin, What Is the Effect of Phasing out Long-Chain per- and Polyfluoroalkyl Substances on the Concentrations of Perfluoroalkyl Acids and Their Precursors in the Environment? A Systematic Review Protocol, Environ. Evid., 2015, 4(1), 1–13, DOI:10.1186/2047-2382-4-3.
- C. J. Young, V. I. Furdui, J. Franklin, R. M. Koerner, D. C. G. Muir and S. A. Mabury, Perfluorinated Acids in Arctic Snow: New Evidence for Atmospheric Formation, Environ. Sci. Technol., 2007, 41(10), 3455–3461, DOI:10.1021/es0626234.
- J. J. MacInnis, K. French, D. C. G. Muir, C. Spencer, A. Criscitiello, A. O. De Silva and C. J. Young, A 14-Year Depositional Ice Record of Perfluoroalkyl Acids in the High Arctic, Environ. Sci.: Processes Impacts, 2017, 19, 22–30, 10.1039/C6EM00593D.
- H. M. Pickard, A. S. Criscitiello, C. Spencer, M. J. Sharp, D. C. G. Muir, A. O. De Silva and C. J. Young, Continuous Non-Marine Inputs of per- and Polyfluoroalkyl Substances to the High Arctic: A Multi-Decadal Temporal Record, Atmos. Chem. Phys., 2018, 18(7), 5045–5058, DOI:10.5194/acp-18-5045-2018.
- D. Muir, R. Bossi, P. Carlsson, M. Evans, A. De Silva, C. Halsall, C. Rauert, D. Herzke, H. Hung, R. Letcher, F. Rigét and A. Roos, Levels and Trends of Poly- and Perfluoroalkyl Substances in the Arctic Environment – An Update, Emerging Contam., 2019, 5, 240–271, DOI:10.1016/j.emcon.2019.06.002.
- Y. Chen, J. Fu, T. Ye, X. Li, K. Gao, Q. Xue, J. Lv, A. Zhang and J. Fu, Occurrence, Profiles, and Ecotoxicity of Poly- and Perfluoroalkyl Substances and Their Alternatives in Global Apex Predators: A Critical Review, J. Environ. Sci., 2021, 109, 219–236, DOI:10.1016/j.jes.2021.03.036.
- Z. Xie and R. Kallenborn, Legacy and Emerging Per- and Polyfluoroalkyl Substances in Polar Regions, Curr. Opin. Green Sustainable Chem., 2023, 42, 100840, DOI:10.1016/j.cogsc.2023.100840.
- C. J. Young and S. A. Mabury, Atmospheric Perfluorinated Acid Precursors: Chemistry, Occurrence and Impacts, Reviews of Environment Contamination and Toxicology, P. De Voogt, Springer, New York, NY, 2010, vol. 208, DOI:10.1007/978-1-4419-6880-7_1.
- K. Y. Kwok, E. Yamazaki, N. Yamashita, S. Taniyasu, M. B. Murphy, Y. Horii, G. Petrick, R. Kallerborn, K. Kannan, K. Murano and P. K. S. Lam, Transport of Perfluoroalkyl Substances (PFAS) from an Arctic Glacier to Downstream Locations: Implications for Sources, Sci. Total Environ., 2013, 447, 46–55, DOI:10.1016/j.scitotenv.2012.10.091.
- W. F. Hartz, M. K. Björnsdotter, L. W. Y. Yeung, A. Hodson, E. R. Thomas, J. D. Humby, C. Day, I. E. Jogsten, A. Kärrman and R. Kallenborn, Levels and Distribution Profiles of Per- and Polyfluoroalkyl Substances (PFAS) in a High Arctic Svalbard Ice Core, Sci. Total Environ., 2023,(5), 161830, DOI:10.1016/j.scitotenv.2023.161830.
- J. Garnett, C. Halsall, H. Winton, H. Joerss, R. Mulvaney, R. Ebinghaus, M. Frey, A. Jones, A. Leeson and P. Wynn, Increasing Accumulation of Perfluorocarboxylate Contaminants Revealed in an Antarctic Firn Core (1958-2017), Environ. Sci. Technol., 2022, 56(16), 11246–11255, DOI:10.1021/acs.est.2c02592.
- J. B. Maxwell, Climatic Regions of the Canadian Arctic Islands, Arctic, 1981, 34(3), 225–240, DOI:10.14430/arctic2525.
- H. M. Pickard, A. S. Criscitiello, D. Persaud, C. Spencer, D. C. G. Muir, I. Lehnherr, M. J. Sharp, A. O. De Silva and C. J. Young, Ice Core Record of Persistent Short-Chain Fluorinated Alkyl Acids: Evidence of the Impact From Global Environmental Regulations, Geophys. Res. Lett., 2020, 47(10), e2020GL087535, DOI:10.1029/2020GL087535.
- M. M. Arienzo, J. R. McConnell, N. Chellman, A. S. Criscitiello, M. Curran, D. Fritzsche, S. Kipfstuhl, R. Mulvaney, M. Nolan, T. Opel, M. Sigl and J. P. Steffensen, A Method for Continuous 239Pu Determinations in Arctic and Antarctic Ice Cores, Environ. Sci. Technol., 2016, 50(13), 7066–7073, DOI:10.1021/acs.est.6b01108.
- A. S. Criscitiello, T. Geldsetzer, R. H. Rhodes, M. Arienzo, J. McConnell, N. Chellman, M. B. Osman, J. J. Yackel and S. Marshall, Marine Aerosol Records of Arctic Sea-Ice and Polynya Variability From New Ellesmere and Devon Island Firn Cores, Nunavut, Canada, J. Geophys. Res.: Oceans, 2021, 126(9), 1–20, DOI:10.1029/2021JC017205.
- A. O. De Silva, C. J. Young, C. Spencer, D. C. G. Muir, M. Sharp, I. Lehnherr and A. Criscitiello, Canadian High Arctic Ice Core Records of Organophosphate Flame Retardants and Plasticizers, Environ. Sci.: Processes Impacts, 2023, 2001–2014, 10.1039/d3em00215b.
- P. Legendre, lmodel2: Model II Regression. R Package Version 1.7-3, https://cran.r-project.org/web/packages/lmodel2/lmodel2.pdf Search PubMed.
- DuPont GenX Processing Aid for Making Fluoropolymer Resins, https://bladenonline.com/wp-content/uploads/2017/06/Chemours_GenX_Brochure_Final_07July2010.pdf, accessed 2022-05-10 Search PubMed.
- W. A. Gebbink, L. Van Asseldonk and S. P. J. Van Leeuwen, Presence of Emerging Per- and Polyfluoroalkyl Substances (PFASs) in River and Drinking Water near a Fluorochemical Production Plant in the Netherlands, Environ. Sci. Technol., 2017, 51(19), 11057–11065, DOI:10.1021/acs.est.7b02488.
- US EPA, PFOA Stewardship Program Baseline Year Summary Report, 2016, available at: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/epas-non-cbi-summary-tables-2015-company-progress, last access: 12 March 2020.
- Environment and Climate Change Canada, Environmental Performance Agreement, 2006, available at: http://ec.gc.ca/epe-epa/default.asp?lang=En&n=AE06B51E-1, last access: 13 March 2020.
- K. A. Pike, P. L. Edmiston, J. J. Morrison and J. A. Faust, Correlation Analysis of Perfluoroalkyl Substances in Regional U.S. Precipitation Events, Water Res., 2021, 190, 116685, DOI:10.1016/j.watres.2020.116685.
- R. W. Macdonald, L. A. Barrie, T. F. Bidleman, M. L. Diamond, D. J. Gregor, R. G. Semkin, W. M. J. Strachan, Y. F. Li, F. Wania, M. Alaee, L. B. Alexeeva, S. M. Backus, R. Bailey, J. M. Bewers, C. Gobeil, C. J. Halsall, T. Harner, J. T. Hoff, L. M. M. Jantunen, W. L. Lockhart, D. Mackay, D. C. G. Muir, J. Pudykiewicz, K. J. Reimer, J. N. Smith, G. A. Stern, W. H. Schroeder, R. Wagemann and M. B. Yunker, Contaminants in the Canadian Arctic: 5 Years of Progress in Understanding Sources, Occurrence and Pathways, Sci. Total Environ., 2000, 254(2–3), 93–234, DOI:10.1016/S0048-9697(00)00434-4.
- H. Joerss, Z. Xie, C. C. Wagner, W. J. Von Appen, E. M. Sunderland and R. Ebinghaus, Transport of Legacy Perfluoroalkyl Substances and the Replacement Compound HFPO-DA through the Atlantic Gateway to the Arctic Ocean - Is the Arctic a Sink or a Source?, Environ. Sci. Technol., 2020, 54(16), 9958–9967, DOI:10.1021/acs.est.0c00228.
- S. B. Gewurtz, A. S. Auyeung, A. O. De Silva, S. Teslic and S. A. Smyth, Per- and Polyfluoroalkyl Substances (PFAS) in Canadian Municipal Wastewater and Biosolids: Recent Patterns and Time Trends 2009 to 2021, Sci. Total Environ., 2024, 912, 168638, DOI:10.1016/j.scitotenv.2023.168638.
- X. Wang, C. Halsall, G. Codling, Z. Xie, B. Xu, Z. Zhao, Y. Xue, R. Ebinghaus and K. C. Jones, Accumulation of Perfluoroalkyl Compunds in Tibetan Mountain Snow: Temporal Patterns from 1980 to 2010, Environ. Sci. Technol., 2014, 48(1), 173–181, DOI:10.1021/es4044775.
- T. Kirchgeorg, A. Dreyer, J. Gabrieli, N. Kehrwald, M. Sigl, M. Schwikowski, C. Boutron, A. Gambaro, C. Barbante and R. Ebinghaus, Temporal Variations of Perfluoroalkyl Substances and Polybrominated Diphenyl Ethers in Alpine Snow, Environ. Pollut., 2013, 178(3), 367–374, DOI:10.1016/j.envpol.2013.03.043.
- K. Y. Kwok, S. Taniyasu, L. W. Y. Yeung, M. B. Murphy, P. K. S. Lam, Y. Horii, K. Kannan, G. Petrick, R. K. Sinha and N. Yamashita, Flux of Perfluorinated Chemicals through Wet Deposition in Japan, the United States, and Several Other Countries, Environ. Sci. Technol., 2010, 44(18), 7043–7049, DOI:10.1021/es101170c.
- B. F. Scott, C. Spencer, S. A. Mabury and D. C. G. Muir, Poly and Perfluorinated Carboxylates in North American Precipitation, Environ. Sci. Technol., 2006, 40(23), 7167–7174 CrossRef CAS PubMed.
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbuhler, Global Emission Inventories for C4-C14 Perfluoroalkyl Carboxylic Acid (PFCA) Homologues from 1951 to 2030, Part II: The Remianing Pieces of the Puzzle, Environ. Int., 2014, 69, 166–176, DOI:10.1016/j.envint.2014.04.006.
- K. Goto-Azuma and R. M. Koerner, Ice Core Studies of Anthropogenic Sulfate and Nitrate Trends in the Arctic, J. Geophys. Res.: Atmos., 2001, 106(D5), 4959–4969, DOI:10.1029/2000JD900635.
- W. Weppner, Letter by William Weppner, 3M, to Charles Auer, EPA OPPT, Regarding the Phase-Out Plan for POSF-Based Products; US EPA Public Docket EPA-HQ-OPPT-2002−0043−0009, 3M General Offices, St. Paul, MN, 2000 Search PubMed.
- D. A. Ellis, J. W. Martin, A. O. De Silva, S. A. Mabury, M. D. Hurley, M. P. Sulbaek Andersen and T. J. Wallington, Degradation of Fluorotelomer Alcohols: A Likely Atmospheric Source of Perfluorinated Carboxylic Acids, Environ. Sci. Technol., 2004, 38(12), 3316–3321, DOI:10.1021/es049860w.
- J. C. D'Eon, M. D. Hurley, T. J. Wallington and S. A. Mabury, Atmospheric Chemistry of N-Methyl Perfluorobutane Sulfonamidoethanol, C4F9SO2N(CH3)CH2CH2OH: Kinetics and Mechanism of Reaction with OH, Environ. Sci. Technol., 2016, 40, 1862–1868, DOI:10.1021/es0520767.
- J. W. Martin, D. A. Ellis, S. A. Mabury, M. D. Hurley and T. J. Wallington, Atmospheric Chemistry of Perfluoroalkanesulfonamides: Kinetic and Product Studies of the OH Radical and Cl Atom Initiated Oxidation of N-Ethyl Perfluorobutanesulfonamide, Environ. Sci. Technol., 2006, 40(3), 864–872, DOI:10.1021/es051362f.
- J. J. Macinnis, K. French, D. C. G. Muir, C. Spencer, A. Criscitiello, A. O. De Silva and C. J. Young, Emerging Investigator Series: A 14-Year Depositional Ice Record of Perfluoroalkyl Substances in the High Arctic, Environ. Sci.: Processes Impacts, 2017, 19(1), 22–30, 10.1039/c6em00593d.
- C. P. Thackray, N. E. Selin and C. J. Young, A Global Atmospheric Chemistry Model for the Fate and Transport of PFCAs and Their Precursors, Environ. Sci.: Processes Impacts, 2019, 22(2), 285–293, 10.1039/c9em00326f.
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbühler, Global Emission Inventories for C4-C14 Perfluoroalkyl Carboxylic Acid (PFCA) Homologues from 1951 to 2030, Part I: Production and Emissions from Quantifiable Sources, Environ. Int., 2014, 70, 62–75, DOI:10.1016/j.envint.2014.04.013.
- I. A. Titaley, J. Khattak, J. Dong, C. I. Olivares, B. Diguiseppi, C. C. Lutes and J. A. Field, Neutral Per- and Polyfluoroalkyl Substances, Butyl Carbitol, and Organic Corrosion Inhibitors in Aqueous Film-Forming Foams: Implications for Vapor Intrusion and the Environment, Environ. Sci. Technol., 2022, 56(15), 10785–10797, DOI:10.1021/acs.est.2c02349.
- H. Lin, J.-Y. Y. Lao, Q. Wang, Y. Ruan, Y. He, P. K. H. Lee, K. M. Y. Leung and P. K. S. Lam, Per- and Polyfluoroalkyl Substances in the Atmosphere of Waste Management Infrastructures: Uncovering Secondary Fluorotelomer Alcohols, Particle Size Distribution, and Human Inhalation Exposure, Environ. Int., 2022, 167, 107434, DOI:10.1016/j.envint.2022.107434.
- M. Shoeib, T. Harner and P. Vlahos, Perfluorinated Chemicals in the Arctic Atmosphere, Environ. Sci. Technol., 2006, 40(24), 7577–7583, DOI:10.1021/es0618999.
- N. L. Stock, V. I. Furdui, D. C. G. Muir and S. A. Mabury, Perfluoroalkyl Contaminants in the Canadian Arctic: Evidence of Atmospheric Transport and Local Contamination, Environ. Sci. Technol., 2007, 41(10), 3529–3536, DOI:10.1021/es062709x.
- F. Wong, M. Shoeib, A. Katsoyiannis, S. Eckhardt, A. Stohl, P. Bohlin-Nizzetto, H. Li, P. Fellin, Y. Su and H. Hung, Assessing Temporal Trends and Source Regions of Per- and Polyfluoroalkyl Substances (PFASs) in Air under the Arctic Monitoring and Assessment Programme (AMAP), Atmos. Environ., 2018, 172, 65–73, DOI:10.1016/j.atmosenv.2017.10.028.
- F. Wong, H. Hung, H. Dryfhout-Clark, W. Aas, P. Bohlin-Nizzetto, K. Breivik, M. N. Mastromonaco, E. B. Lundén, K. Ólafsdóttir, Á. Sigurðsson, K. Vorkamp, R. Bossi, H. Skov, H. Hakola, E. Barresi, E. Sverko, P. Fellin, H. Li, A. Vlasenko, M. Zapevalov, D. Samsonov and S. Wilson, Time Trends of Persistent Organic Pollutants (POPs) and Chemicals of Emerging Arctic Concern (CEAC) in Arctic Air from 25 Years of Monitoring, Sci. Total Environ., 2021, 775, 145109, DOI:10.1016/j.scitotenv.2021.145109.
- J. A. Faust, PFAS on Atmospheric Aerosol Particles: A Review, Environ. Sci.: Processes Impacts, 2022, 25(2), 133–150, 10.1039/d2em00002d.
- Y. Tao, T. C. VandenBoer, R. X. Ye and C. J. Young, Exploring Controls on Perfluorocarboxylic Acid (PFCA) Gas-Particle Partitioning Using a Model with Observational Constraints, Environ. Sci.: Processes Impacts, 2022, 264–276, 10.1039/d2em00261b.
- J. J. MacInnis, I. Lehnherr, D. C. G. Muir, K. A. St. Pierre, V. L. St. Louis, C. Spencer and A. O. De Silva, Fate and Transport of Perfluoroalkyl Substances from Snowpacks into a Lake in the High Arctic of Canada, Environ. Sci. Technol., 2019, 53(18), 10753–10762, DOI:10.1021/acs.est.9b03372.
- J. H. Johansson, M. E. Salter, J. C. Acosta Navarro, C. Leck, E. D. Nilsson and I. T. Cousins, Global Transport of Perfluoroalkyl Acids via Sea Spray Aerosol, Environ. Sci.: Processes Impacts, 2019, 21(4), 635–649, 10.1039/c8em00525g.
- M. Reth, U. Berger, D. Broman, I. T. Cousins, E. D. Nilsson and M. S. McLachlan, Water-to-Air Transfer of Perfluorinated Carboxylates and Sulfonates in a Sea Spray Simulator, Environ. Chem., 2011, 8(4), 381–388, DOI:10.1071/EN11007.
- B. Sha, J. H. Johansson, P. Tunved, P. Bohlin-Nizzetto, I. T. Cousins and M. E. Salter, Sea Spray Aerosol (SSA) as a Source of Perfluoroalkyl Acids (PFAAs) to the Atmosphere: Field Evidence from Long-Term Air Monitoring, Environ. Sci. Technol., 2022, 56(1), 228–238, DOI:10.1021/acs.est.1c04277.
- D. J. Cavalieri and C. L. Parkinson, Arctic Sea Ice Variability and Trends, 1979-2010, Cryosphere, 2012, 6(4), 881–889, DOI:10.5194/tc-6-881-2012.
- R. H. Rhodes, X. Yang and E. W. Wolff, Sea Ice Versus Storms: What Controls Sea Salt in Arctic Ice Cores?, Geophys. Res. Lett., 2018, 45(11), 5572–5580, DOI:10.1029/2018GL077403.
- R. H. Rhodes, X. Yang, E. W. Wolff, J. R. McConnell and M. M. Frey, Sea Ice as a Source of Sea Salt Aerosol to Greenland Ice Cores: A Model-Based Study, Atmos. Chem. Phys., 2017, 17(15), 9417–9433, DOI:10.5194/acp-17-9417-2017.
- B. Jourdain, S. Preunkert, O. Cerri, H. Castebrunet, R. Udisti and M. Legrand, Year-Round Record of Size-Segregated Aerosol Composition in Central Antarctica (Concordia Station): Implications for the Degree of Fractionation of Sea-Salt Particles, J. Geophys. Res.: Atmos., 2008, 113(14), 2007, DOI:10.1029/2007JD009584.
- D. Wagenback, F. Ducroz, R. Mulvaney, L. Keck, A. Minikin, M. Legrand, J. S. Hall and E. W. Wolff, Sea Salt Aerosol in Coastal Antarctic Regions, J. Geophys. Res., 1998, 103, 10961–10974 CrossRef.
- F. Domine, R. Sparapani, A. Ianniello and H. J. Beine, The Origin of Sea Salt in Snow on Arctic Sea Ice and in Coastal Regions, Atmos. Chem. Phys., 2004, 4(9–10), 2259–2271, DOI:10.5194/acp-4-2259-2004.
- X. Yang, J. A. Pyle and R. A. Cox, Sea Salt Aerosol Production and Bromine Release: Role of Snow on Sea Ice, Geophys. Res. Lett., 2008, 35(16), 1–5, DOI:10.1029/2008GL034536.
- C. G. Hannah, F. Dupont and M. D. Dunphy, Polynyas and Tidal Currents in the Canadian Arctic Archipelago, Arctic, 2009, 62(1), 83–95, DOI:10.14430/arctic115.
- M. Cai, H. Yang, Z. Xie, Z. Zhao, F. Wang, Z. Lu, R. Sturm and R. Ebinghaus, Per- and Polyfluoroalkyl Substances in Snow, Lake, Surface Runoff Water and Coastal Seawater in Fildes Peninsula, King George Island, Antarctica, J. Hazard. Mater., 2012, 209–210, 335–342, DOI:10.1016/j.jhazmat.2012.01.030.
- J. Garnett, C. Halsall, A. Vader, H. Joerss, R. Ebinghaus, A. Leeson and P. M. Wynn, High Concentrations of Perfluoroalkyl Acids in Arctic Seawater Driven by Early Thawing Sea Ice, Environ. Sci. Technol., 2021, 55(16), 11049–11059, DOI:10.1021/acs.est.1c01676.
- T. Stefanac, R. McCrindle, A. J. McAlees, N. Riddell, A. L. Brazeau and B. C. Chittim, Characterization of Nine Isomers in Commercial Samples of Perfluoroethylcyclohexanesulfonate and of Some Minor Components Including PFOS Isomers, Environ. Sci. Technol., 2018, 52(17), 9937–9945, DOI:10.1021/acs.est.8b02369.
- G. L. Lescord, K. A. Kidd, A. O. De Silva, M. Williamson, C. Spencer, X. Wang and D. C. G. Muir, Perfluorinated and Polyfluorinated Compounds in Lake Food Webs from the Canadian High Arctic, Environ. Sci. Technol., 2015, 49(5), 2694–2702, DOI:10.1021/es5048649.
- Y. Wang, R. Vestergren, Y. Shi, D. Cao, L. Xu, Y. Cai, X. Zhao and F. Wu, Identification, Tissue Distribution, and Bioaccumulation Potential of Cyclic Perfluorinated Sulfonic Acids Isomers in an Airport Impacted Ecosystem, Environ. Sci. Technol., 2016, 50(20), 10923–10932, DOI:10.1021/acs.est.6b01980.
- R. C. Sullivan, S. A. Guazzotti, D. A. Sodeman and K. A. Prather, Direct Observations of the Atmospheric Processing of Asian Mineral Dust, Atmos. Chem. Phys., 2007, 7(5), 1213–1236, DOI:10.5194/acp-7-1213-2007.
- T. Mochizuki, K. Kawamura, K. Aoki and N. Sugimoto, Long-Range Atmospheric Transport of Volatile Monocarboxylic Acids with Asian Dust over a High Mountain Snow Site, Central Japan, Atmos. Chem. Phys., 2016, 16(22), 14621–14633, DOI:10.5194/acp-16-14621-2016.
- X. Zhao, K. Huang, J. S. Fu and S. F. Abdullaev, Long-Range Transport of Asian Dust to the Arctic: Identification of Transport Pathways, Evolution of Aerosol Optical Properties, and Impact Assessment on Surface Albedo Changes, Atmos. Chem. Phys., 2022, 22(15), 10389–10407, DOI:10.5194/acp-22-10389-2022.
- Z. Huang, J. Huang, T. Hayasaka, S. Wang, T. Zhou and H. Jin, Short-Cut Transport Path for Asian Dust Directly to the Arctic: A Case Study, Environ. Res. Lett., 2015, 10(11), 114018, DOI:10.1088/1748-9326/10/11/114018.
- C. A. Moody and J. A. Field, Perfluorinated Surfactants and the Environmental Implications of Their Use in Fire-Fighting Foams, Environ. Sci. Technol., 2000, 34(18), 3864–3870, DOI:10.1021/es991359u.
- Department of Defence, Aqueous Film Forming Foam Report to Congress, https://denix.osd.mil/derp/home/documents/aqueous-film-forming-foam-report-to-congress/ Search PubMed.
- DRAFT State of Per- and Polyfluoroalkyl Substances (PFAS) Report Environment and Climate Change Canada Health Canada May 2023, https://www.canada.ca/en/environment-climate-change/services/evaluating-existing-substances/draft-state-per-polyfluoroalkyl-substances-report.html Search PubMed.
- PFAS Contamination sites in Airports and Military bases in Canada, https://cela.ca/wp-content/uploads/2023/03/PFAS-Contamination-Sites-Canada.pdf, accessed 2023-05-24 Search PubMed.
- B. J. Place and J. A. Field, Identification of Novel Fluorochemicals in Aqueous Film-Forming Foams Used by the US Military, Environ. Sci. Technol., 2012, 46(13), 7120–7127, DOI:10.1021/es301465n.
- W. J. Backe, T. C. Day and J. A. Field, Zwitterionic, Cationic, and Anionic Fluorinated Chemicals in Aqueous Film Forming Foam Formulations and Groundwater from U.S. Military Bases by Nonaqueous Large-Volume Injection HPLC-MS/MS, Environ. Sci. Technol., 2013, 47(10), 5226–5234, DOI:10.1021/es3034999.
- T. P. Riedel, J. R. Lang, M. J. Strynar, A. B. Lindstrom and J. H. Offenberg, Gas-Phase Detection of Fluorotelomer Alcohols and Other Oxygenated Per- and Polyfluoroalkyl Substances by Chemical Ionization Mass Spectrometry, Environ. Sci. Technol. Lett., 2019, 6(5), 289–293, DOI:10.1021/acs.estlett.9b00196.
- J. Roth, I. Abusallout, T. Hill, C. Holton, U. Thapa and D. Hanigan, Release of Volatile Per-and Polyfluoroalkyl Substances from Aqueous Film-Forming Foam, Environ. Sci. Technol. Lett., 2020, 7(3), 164–170, DOI:10.1021/acs.estlett.0c00052.
- J. C. Bourgeois, Seasonal and Annual Variation of Pollen Content in the Snow of a Canadian High Arctic Ice Cap, Boreas, 1990, 19(4), 313–322, DOI:10.1111/j.1502-3885.1990.tb00135.x.
- G. K. C. Clarke, A. H. Jarosch, F. S. Anslow, V. Radić and B. Menounos, Projected Deglaciation of Western Canada in the Twenty-First Century, Nat. Geosci., 2015, 8(5), 372–377, DOI:10.1038/ngeo2407.
- J. T. M. Lenaerts, J. H. Van Angelen, M. R. Van Den Broeke, A. S. Gardner, B. Wouters and E. Van Meijgaard, Irreversible Mass Loss of Canadian Arctic Archipelago Glaciers, Geophys. Res. Lett., 2013, 40(5), 870–874, DOI:10.1002/grl.50214.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00219a |
| This journal is © The Royal Society of Chemistry 2024 |