
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivating iodine redox by enabling single-atom coordination to dormant nitrogen sites to realize durable zinc–iodine batteries†
Jisung
Lee‡
a,
Wooseok
Lee‡
b,
Seungho
Back‡
 a,
Seung Yeop
Yi
a,
Seonggyu
Lee
de,
Seongseop
Kim
f,
Joonhee
Moon
g,
Dong-Yeun
Koh
a,
Kyeounghak
Kim
c,
Seoin
Back
*b and
Jinwoo
Lee
*a
a,
Seung Yeop
Yi
a,
Seonggyu
Lee
de,
Seongseop
Kim
f,
Joonhee
Moon
g,
Dong-Yeun
Koh
a,
Kyeounghak
Kim
c,
Seoin
Back
*b and
Jinwoo
Lee
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daehak-ro 291, Daejeon 34141, Republic of Korea. E-mail: jwlee1@kaist.ac.kr
bDepartment of Chemical and Biomolecular Engineering, Institute of Emergent Materials, Sogang University, Seoul 04107, Republic of Korea. E-mail: sback@sogang.ac.kr
cDepartment of Chemical Engineering, Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul 04763, Republic of Korea
dDepartment of Chemical Engineering, Kumoh National Institute of Technology (KIT), Gumi 39177, Republic of Korea
eDepartment of Energy Engineering Convergence, Kumoh National Institute of Technology (KIT), Gumi 39177, Republic of Korea
fSchool of Chemical Engineering, Clean Energy Research Center, Jeonbuk National University, Jeonju 54896, Republic of Korea
gDivision of Materials Analysis, Korea Basic Science Institute (KBSI), Daejeon 34133, Republic of Korea
First published on 7th November 2023
Abstract
Aqueous rechargeable static zinc–iodine (Zn–I2) batteries are regarded as competitive candidates for next-generation energy storage devices owing to their safety and high energy density. However, their inherent limitations such as the shuttle effect, sluggish electrochemical kinetics, and the poor electrical conductivity of iodine have been challenging to mitigate when using methods that confer polarity to the surface of the carbon host through nitrogen doping. Moreover, the considerable prevalence of inactive pyridinic N sites significantly impedes the establishment of approaches to overcome issues associated with redox kinetics and iodine utilization. Herein, single Ni atoms were incorporated into an electrochemically inactive N-doped carbon matrix by carbonizing a zeolitic imidazolate framework and then thermally activating the Ni ions adsorbed onto the carbonized product. The single Ni atoms modulated the electronic structure of the surrounding N-doped carbon matrix, thereby improving its ability to adsorb polyiodides and exhibit bifunctional catalytic activity for iodine reduction and oxidation reactions. Consequently, the assembled Zn–I2 battery delivered an outstanding rate performance (193 mA h g−1 at a current density of 6 A g−1) and ultralong cyclability (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at a current density of 4 A g−1). Overall, this study illuminates the merits of using single-atom catalysts to revitalize inactive N pyridinic sites, thereby providing a promising direction for further advancement of Zn–I2 batteries.
000 cycles at a current density of 4 A g−1). Overall, this study illuminates the merits of using single-atom catalysts to revitalize inactive N pyridinic sites, thereby providing a promising direction for further advancement of Zn–I2 batteries.
Broader contextAqueous rechargeable static zinc-iodine (Zn-I2) batteries are gaining attention as potential replacements for conventional zinc-metal oxide batteries. This is due to their high theoretical capacity (211 mAh g−1), relatively high discharge potential plateau, and the abundance of iodine in seawater. However, the intrinsic insulating properties of iodine and soluble polyiodide intermediates can lead to sluggish redox kinetics and shuttling, thereby having a detrimental effect on power density and durability. Herein, we investigated the effects of modulating the electronic environment through the incorporation of atomically dispersed single Ni atoms into an N-doped carbon (NiNC) host material. We revealed that the coordination of the positively charged single Ni atoms to the previously inactive, negatively charged pyridinic N sites not only provided favorable adsorption sites for iodine species, significantly reducing the shuttling effect, but also acted as an electrocatalyst to promote iodine redox kinetics. This study highlights the crucial role of tailoring active sites for the iodine redox reaction, paving the way for the advancement of Zn-I2 batteries that outperform conventional zinc-metal oxide batteries. |
Introduction
Lithium-ion batteries (LIBs) have several advantageous features, such as high energy density, durability and scalability, and have achieved remarkable success as power sources in diverse settings, from portable electronic devices to grid-scale energy-storage systems.1 Notably, these beneficial attributes can be leveraged to advance the development of sustainable energy solutions for realizing a fossil-fuel-free economy.2–4 However, despite the numerous advantages of LIBs, their value is undermined by the fluctuating price of Li resources, which originates from the scarcity and geographically concentrated distribution of Li, and from the surging demand for LIBs.4–6 Furthermore, LIBs comprised of organic electrolytes pose significant safety concerns because of their high flammability, which causes fires that are relatively difficult to extinguish.4,7,8Rechargeable aqueous zinc metal batteries (ZMBs) are becoming increasingly attractive as viable options for powering utility-scale energy storage systems, owing to their use of environmentally benign and non-flammable aqueous electrolytes.9–11 Additionally, the abundant availability of Zn metal and its high theoretical volumetric capacity as an anode material (5855 mA h cm−3) make ZMBs a competitive alternative to conventional LIBs.11,12 However, progress in the development of ZMBs has been hindered by the challenge of finding suitable cathode materials that can effectively complement the Zn metal anode. The divalent Zn ions in Zn metal anodes have the tendency to induce strong electrostatic interactions with the crystal structures of cathode materials, leading to sluggish intercalation–deintercalation kinetics.13–16 Furthermore, the consecutive intercalation of Zn ions with protons can increase the pH in the vicinity of the cathode, resulting in the formation of insulating zinc hydroxide sulfate.17 Even worse, the dissolution of metal ions from the cathode material into the electrolyte considerably degrades the ZMB durability.14,16
Recently, aqueous rechargeable static zinc–iodine (Zn–I2) batteries have emerged as promising candidates to replace traditional zinc–metal oxide batteries owing to their high theoretical capacity (211 mA h giodine−1), relatively high discharge potential plateau (1.3 V vs. Zn/Zn2+), and the abundance of iodine in seawater.18–21 Additionally, the conversion reaction of iodine can facilitate a faster redox reaction than that of traditional metal-oxide-based cathodes and does not involve protons during the redox reaction, thereby maintaining the pH value in the vicinity of the cathode surface.22,23 Nevertheless, similar to lithium–sulfur batteries, the intrinsic insulating properties of iodine and the formation of soluble polyiodide intermediates can lead to sluggish redox kinetics and shuttling, thereby having a detrimental effect on power density and durability.24,25
Carbonaceous host materials for iodine have been comprehensively investigated for use in Zn–I2 batteries since Liu et al. reported a notable enhancement in the cell performance through the utilization of an active carbon fiber cloth.26 Essentially, the high surface area and micro pore volume of the carbon host material help in physically obstructing the vaporization of volatile iodine and the dissolution of water-soluble iodine intermediates.27–29 Furthermore, the superior electrical conductivity of the carbon host material can effectively complement the insulating properties of the iodine species, thereby facilitating electron transfer. However, the nonpolar characteristics of the carbon host material hamper attempts made to resolve the issues of iodine batteries.19,26,28 Therefore, heteroatom dopants have recently been introduced into nonpolar carbon surfaces to enhance the chemical interactions between the host materials and iodine species.29–32 For instance, upon examining the ability of different nitrogen species to adsorb polyiodides, Yasin et al. determined that graphitic N offered the most favorable sites for chemically adsorbing iodine.30 However, although nitrogen doping improves the adsorption ability and electrocatalytic performance of the carbon host, a high proportion of inactive pyridinic nitrogen is ineffective in this regard.29,33 Therefore, it is imperative to explore strategies that can effectively manipulate the electronic structure of inactive pyridinic N sites. Converting these sites into electrochemically active centers can potentially amplify iodine utilization and enhance the redox kinetics.
In the present study, porous N-doped carbon (NC) and single Ni atoms anchored onto NC (NiNC) were employed as iodine host materials, and their electrochemical and physical characteristics were systematically compared. The abundant microporous spaces in these materials enabled effective iodine confinement, whereas the active graphitic N sites served as catalytic sites and adsorption sites for both iodine and polyiodides. Comprehensive computational calculations revealed that the coordination of single Ni atoms to previously inactive pyridinic N sites intensified the chemical interactions of the sites with both iodine and polyiodides. With the benefits of a modulating electronic structure that favors adsorption, the I2@NiNC system exhibited a remarkable 40% higher reversible capacity (193 mA h g−1) than that of the I2@NC system at an extremely high current density (6 A g−1). Moreover, I2@NiNC demonstrated exceptional long-term cyclability with minimal capacity decay of 0.0018% per cycle and sustained a capacity of 181 mA h g−1 at a current density of 4 A g−1 even after 10000 cycles. Overall, this study provides valuable insight into the activation of dormant pyridinic N sites through single-atom coordination, paving the way for the development of high-performance Zn–I2 batteries.
Results and discussion
Zeolitic imidazolate frameworks (ZIF-8) are a class of nitrogen-doped carbon precursors, known for their orderly configuration comprising Zn nodes and organic linkers.34–37 Specifically, ZIF-8 and its carbonized materials have attracted significant attention in diverse fields owing to the abundant active nitrogen sites on their surfaces.38,39 We synthesized anchored single nickel atoms on N-doped carbon (NiNC) via a nickel ion adsorption–thermal activation method using ZIF-8 as the starting material (Fig. 1). Upon carbonization at 1100 °C under an Ar atmosphere, the organic linker used as the nitrogen source was converted into highly conductive and porous NC, presumably owing to the incomplete carbonization of the organic linker and Zn evaporation occurring simultaneously. Graphitic N, which exhibits noteworthy electrocatalytic activity toward iodine species, was deposited onto the carbon surface through a high carbonization temperature. The nitrogen species present in NC could then adsorb nickel ions from a solution and immobilize them within the carbon matrix, thereby preventing the formation of nickel clusters during the thermal activation.40,41 Following thermal activation, the resulting NiNC functional active sites had single Ni atoms that are strongly coordinated to the pyridinic nitrogen species.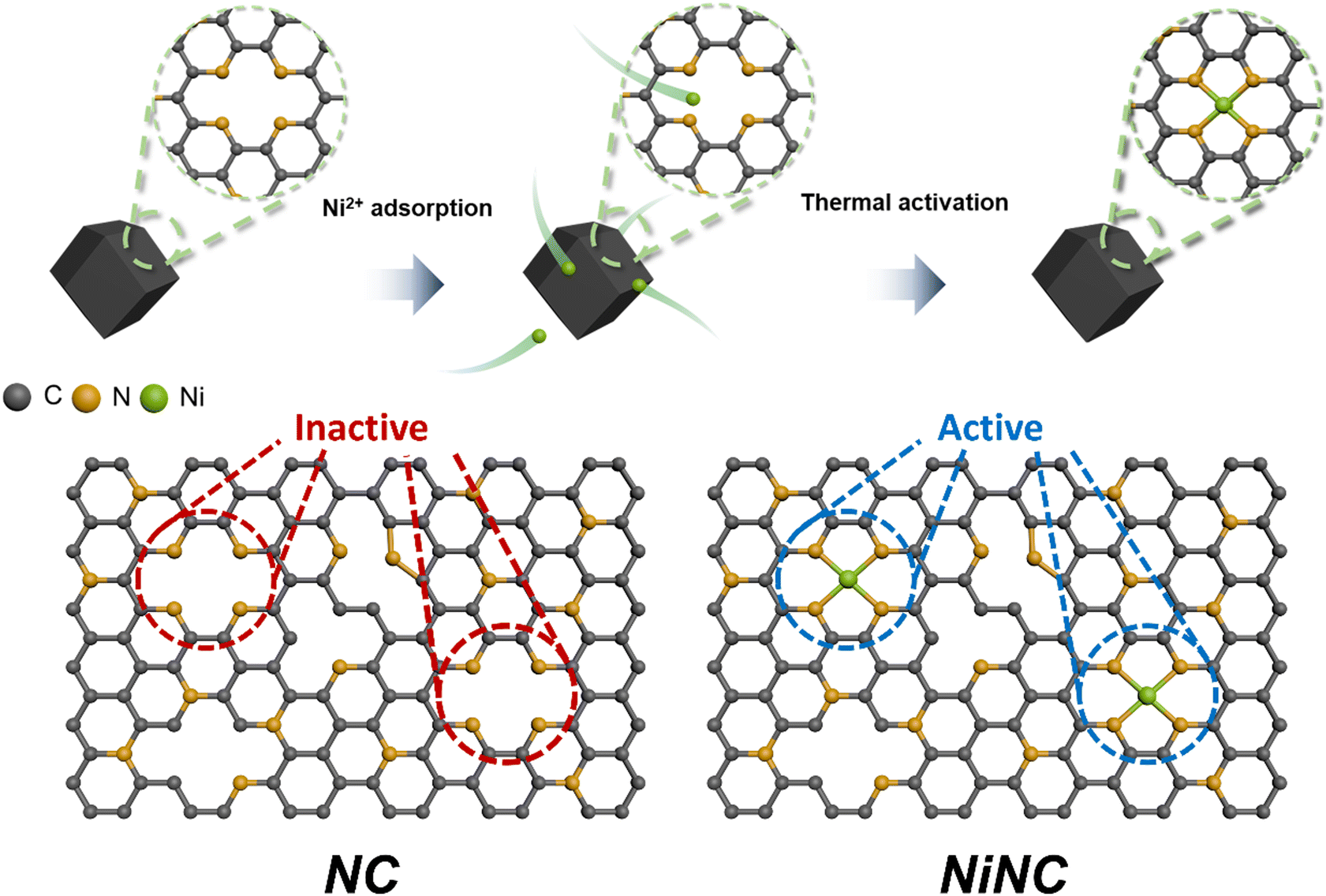 | ||
| Fig. 1 Schematic illustration of the synthesis of the N-doped carbon (NC) material and the single nickel atoms anchored on the N-doped carbon (NiNC). The inert pyridinic nitrogen sites convert into electrochemically active sites through the introduction of single Ni atoms. | ||
The morphology of the carbonized ZIF-8 was investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), which revealed that the NC has a size of 40 nm and exhibits some agglomeration. The agglomeration was attributed to the high surface energy of the ZIF-8 nanoparticles (Fig. S1, ESI†). Following the thermal activation, NiNC retained the original morphology and size of NC, which was devoid of Ni nanoparticles (Fig. 2a and b). This indicates that the thermal activation had no discernible impact on the carbon structure, a finding that is in agreement with results derived from Raman analysis. The Raman spectra of NC and NiNC displayed two characteristic peaks located at approximately 1330–1350 cm−1 (D band) and 1580–1600 cm−1 (G band) with low ID/IG ratios (1.01 and 1.04, respectively). These results signify negligible variations in the carbon properties and imply the existence of numerous defect sites that likely facilitate the adsorption of iodine species (Fig. S2, ESI†).27,29 Indeed, the X-ray diffraction (XRD) results confirmed the presence of amorphous carbon features, and the complete evaporation of the zinc species of ZIF-8. Furthermore, they highlighted the absence of a crystalline Ni phase, suggesting that the pyridinic N species effectively suppressed Ni nanoparticle growth by strongly bridging with the Ni ions (Fig. S3, ESI†). The distribution of single Ni atoms was confirmed from the high-angle annular dark-field scanning TEM image and its corresponding elemental mapping, indicating the atomic-level homogeneous dispersion of Ni. The atomically dispersed Ni could be a crucial factor in augmenting the electrochemical reactivity with respect to iodine (Fig. 2c and d). To ascertain the capacity of NC and NiNC to effectively accommodate volatile iodine species, N2 physisorption measurements were performed to provide further insight into their porous structures (Fig. S4 and Table S1, ESI†). The Brunauer–Emmett–Teller (BET) surface area and micropore volume of NC are 855 m2 g−1 and 0.25 cm3 g−1, respectively. With the BET surface area and micropore volume of NiNC measuring 1047 m2 g−1 and 0.34 cm3 g−1, respectively, it can be inferred that the thermal activation process has contributed to an increase in both the surface area and micropore volume. Therefore, the thermal-activation-induced enhancement in micropore volume evidently improved the ability of the carbon material to adsorb volatile iodine and polyiodide species.
 | ||
| Fig. 2 Characterization of anchored nickel single atom on N-doped carbon (NiNC). (a) SEM and (b) TEM images of NiNC. (c) HAADF-STEM and (d) its corresponding mapping images demonstrating homogeneous distribution of C (green), N (yellow), and Ni (red) elements. (e) Normalized Ni K-edge XANES spectra. (f) Fourier transformation (FT) of the EXAFS spectra in R-space for NiNC with reference samples (Ni foil, NiPc, and NiO). (g) Ni K-edge EXAFS (circles) and the curve-fit (line) for NiNC in k2-weight R-space. | ||
The surface elemental composition and electronic structure significantly influence the catalytic effects associated with the iodine redox reaction and the longevity of the I2 batteries. X-ray photoelectron spectroscopy (XPS) was utilized to probe these aspects. Elemental quantification analysis revealed that NiNC contained nitrogen species at a concentration of 5.53 at%. The N 1s spectrum was deconvoluted into three types of peaks at 398, 401, and 403 eV, which correspond to pyridinic N, graphitic N, and pyridine N oxide, respectively.42,43 Moreover, the additional peak at ∼399 eV was attributed to Ni–N coordination (Fig. S5 and Table S2, ESI†).40 The key components of nitrogen, pyridinic N and graphitic N, each exhibit unique electronic characteristics which induce distinct reactivity toward iodine. The higher electronegativity of nitrogen relative to carbon enables the pyridinic N to withdraw π-electrons from the π-conjugated system. This results in a negative charge on the nitrogen atom, assuming that it donates four electrons toward sp2 hybridization, including a lone pair.44 Conversely, graphitic N contributes only three electrons to its sp2 configuration, allowing the two residual electrons to interact with the stable π-conjugated system in graphite,45,46 thereby resulting in a positive charge on graphitic N. The positive charges on the nitrogen species and adjacent carbon atoms effectively prevent polyiodide shuttling by facilitating chemical adsorption. To gain further insight into the chemical states and coordination structures of the active functional NiNC sites, Ni K-edge X-ray absorption spectroscopy (XAS) was conducted. The near-edge spectra of NiNC were located between those of Ni foil and NiO, implying that the valence state of Ni was between 0 (metallic Ni) and +2 (NiO) (Fig. 2e). The presence of positively charged Ni in the NiNC matrix could potentially transform the electrochemically inactive NC matrix into an active state, consequently enhancing its electrochemical performance. The Fourier-transformed X-ray adsorption fine structure (EXAFS) spectra of NiNC exhibited a dominant peak at 1.21 Å, corresponding to Ni–N coordination. Importantly, the absence of peaks representing Ni–Ni and Ni–O oxide bonds at 2.20 and 1.62 Å, respectively, strongly suggested that atomically dispersed Ni was anchored onto the N-doped sites of the carbon matrix (Fig. 2f). According to the fitting results, a single Ni atom was coordinated to the four surrounding N atoms to form a Ni–N4 structure (Fig. 2f and Table S3, ESI†). Despite a relatively low Ni content of 1.1 wt%, as determined by inductively coupled plasma-mass spectroscopy (ICP-MS), the atomic-scale distribution of the Ni atoms evidently led to substantial performance enhancements (Table S4, ESI†).47
The encapsulation of iodine within the porous NC (I2@NC) and NiNC (I2@NiNC) materials was successfully achieved using a conventional vaporization–condensation technique.27–29 This method is feasible at low temperatures because of the inherently low vaporization temperature of iodine. Nonetheless, when considering this attribute from a different perspective, the low vaporization temperature of iodine renders it highly volatile. Consequently, there is a demand for host materials characterized by appropriate pore structures and robust capabilities to bond with iodine species. These attributes help in stably accommodating iodine, thereby effectively mitigating its inherent volatility. From this perspective, the high micropore volumes of NC and NiNC provided sufficient space to confine iodine to their micropores. To validate the successful encapsulation of vaporized iodine within the porous structures of NC and NiNC, SEM characterization was performed to detect any residual iodine on the surfaces of I2@NC and I2@NiNC after the iodine-loading step (Fig. S6, ESI†). The results verified the encapsulation of iodine within the porous structure, as there was no apparent formation of discrete iodine particles. Indeed, X-ray powder diffraction (XRD) demonstrated the absence of the crystalline iodine peak in the I2@NiNC pattern and that the intensity of the peaks of I2@NiNC is significantly decreased compared with NiNC, indicating that a large fraction of the pores in I2@NiNC are filled with I2 (Fig. S7, ESI†). Furthermore, the surface area and micropore volume of NiNC were significantly reduced after iodine loading to 263 m2 g−1 and 0.05 cm3 g−1, respectively, signifying the confinement of iodine within the micropores of NiNC (Fig. S8 and Table S1, ESI†). Subsequently, thermogravimetric analysis (TGA) was performed to verify the strong adsorption capability of the graphitic N and single Ni atom sites (Fig. S9a, ESI†). Focusing on the temperature at which 5% weight loss was achieved (T5%), the TGA yielded values of 103, 229, and 284 °C for pure iodine, I2@NC, and I2@NiNC, respectively. That the T5% value of I2@NC was higher than that of pure iodine was presumably due to the influence of the graphitic N, whereas the even higher T5% value of I2@NiNC was due to the single-atom effect. These results collectively signified an enhancement in thermal stability owing to the strong chemisorption of iodine (Fig. S9b, ESI†).48 In line with the T5% values obtained from the TGA study, no significant dissolution of iodine was observed after immersing I2@NiNC in a 2 M ZnSO4 electrolyte for one week, illustrating its superior stability relative to that of pure iodine (Fig. S10a, ESI†). Additionally, to effectively prevent the shuttle effect, not only the iodine adsorption capacity but also the superior ability to adsorb polyiodide species should be ensured.49 Both NC and NiNC were separately immersed in Lugol solution (polyiodide ion solution), and the resulting color changes were monitored (Fig. S10b, ESI†). Notably, the solution containing NiNC became transparent at the quickest rate, indicating its ability to quickly chemisorb polyiodide ions. The polyiodide ions left over in the Lugol solution after 24 h were analyzed by UV-vis spectroscopy (Fig. S10c, ESI†). Notably, no perceptible I3− peaks were detected in the NiNC spectrum, suggesting successful polyiodide-ion chemisorption had occurred, likely driven by the synergistic effect of the single Ni atoms and graphitic N.
Cyclic voltammetry (CV) tests were performed on I2@NC and I2@NiNC at a scan rate of 0.1 mV s−1 to reveal the role of nitrogen species and single Ni atoms as electrocatalysts in facilitating the kinetic iodine conversion reaction (Fig. 3a). Two distinct peaks appeared in the CV profile of I2@NiNC at 1.20 and 1.22 V, which corresponded to polyiodide reduction and iodine oxidation, respectively. In comparison to the cyclic voltammogram of I2@NC (1.22 and 1.25 V), the gap between the oxidation and reduction peaks in the I2@NiNC profile was smaller and the specific current was larger than that of I2@NC, indicating superior electrochemical kinetics and activation of iodine species. To meticulously probe the profound impact of the single Ni atom electrocatalytic sites on the redox reaction kinetics, comparative analyses of the Tafel slopes and b values were conducted for both the reduction and oxidation reactions.50,51 The cathodic reduction reaction of I2@NiNC exhibited a lower Tafel slope, derived from the cathodic CV, than I2@NC, pointing to iodine species with accelerated reduction kinetics (Fig. 3b). Notably, the oxidation reaction of I2@NC showed the steepest Tafel slope, suggesting that the oxidation reaction exhibited kinetics inferior to that of the reduction reaction. However, for I2@NiNC, the lowest Tafel slope was observed, implying that the single Ni atoms effectively served as an electrocatalyst for oxidizing the polyiodide species (Fig. 3c). The superior kinetic conversion of the iodine species was further analyzed by scrutinizing the charge-storage mechanism using CV curves scanning a range of sweep rates from 0.2 to 1 mV s−1. The redox peaks exhibited a marginal shift with increasing scan rate, demonstrating an effective iodine species conversion reaction (Fig. S11, ESI†). The relationship between current (i) and scan rate (v), as obtained from the CV curves, was used to elucidate the charge-storage mechanism at the I2@NiNC cathode using eqn (1):
| i = avb | (1) |
| i = k1v + k2v1/2 |
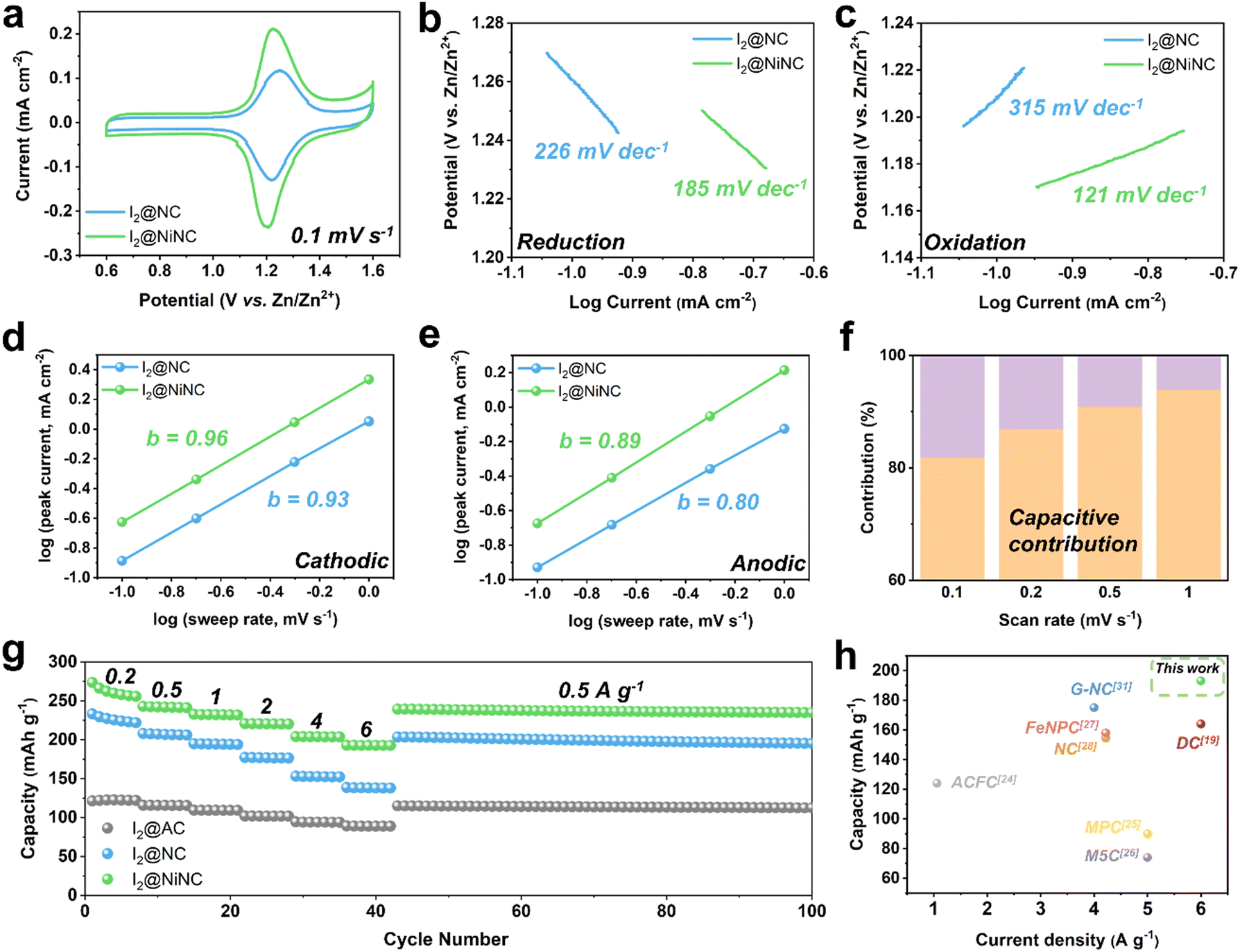 | ||
| Fig. 3 CV curves of I2@NC and I2@NiNC cathodes (a) at a scan rate of 0.1 mV s−1. Tafel plots for the conversion reactions of (b) iodine to polyiodide and (c) polyiodide to iodine. The fitted CV curves show the relationship between log(i) and log(v) used to obtain the b values for the (d) cathodic sweeps and (e) anodic sweeps. (f) The percentage of the capacitive contribution at scan rates of 0.1, 0.2, 0.5, and 1 mV s−1. (g) The rate performances of I2@AC, I2@NC, and I2@NiNC at current densities from 0.2 A g−1 to 6 A g−1. (h) Capacity of I2@NiNC at high current density in comparison with the reported results using carbonaceous host materials. | ||
The rate performance of the I2@activated carbon (AC), I2@NC, and I2@NiNC cathodes was evaluated at current densities ranging from 0.2 to 6 A g−1 (Fig. 3g). I2@NiNC exhibited reversible specific charge capacities of 255, 241, 232, 221, 204, and 193 mA h g−1 at 0.2, 0.5, 1, 2, 4, and 6 A g−1, respectively (Fig. S13, ESI†). Notably, even when subjected to an extremely high current rate of 6 A g−1, the specific charge capacity of I2@NiNC remained stable and then recovered to 239 mA h g−1 with negligible capacity decay when the current was reset to 0.5 A g−1. Furthermore, I2@NiNC achieved an energy efficiency superior to that of I2@AC and I2@NC, as indicated by their galvanostatic charge–discharge profiles, by exhibiting less voltage polarization at half-capacity (Fig. S14, ESI†). Although nitrogen doping endowed the carbon surface with polarity, leading to I2@NC exhibiting a higher rate performance than that of conventional porous carbon, the presence of inactive pyridinic N sites resulted in a relatively lower capacity retention of 62% at a current density of 6 A g−1 compared to I2@NiNC. In contrast, I2@NiNC not only transformed the electrochemically inactive pyridinic N sites into redox-conducive sites, but also utilized the activated graphitic N sites, resulting in a noteworthy capacity retention of 76%. Compared with the cathodes from previously reported iodine-loaded carbonaceous materials, I2@NiNC shows an extraordinary specific charge capacity at a high current density, demonstrating its superior kinetics and amenability to activity adjustment (Fig. 3h and Table S5, ESI†).
In addition to boosting the inherently sluggish kinetics of iodine, managing the volatility and dissolution of iodine and polyiodides is a vital but challenging endeavor for achieving enhanced cycling performance. The prevention of a shuttle effect induced by polyiodide dissolution was verified through cyclic performance testing at a current density of 1 A g−1 (Fig. 4a). After 10 activation cycles, the I2@NC cathode exhibited a specific charge capacity of 212 mA h g−1 and maintained a capacity of 168 mA h g−1 with a capacity decay ratio of 0.01% per cycle. Notably, the I2@NiNC cathode demonstrated exceptional performance, with a specific capacity of 241 mA h g−1 after the activation cycles, and still exhibited a reversible capacity of 207 mA h g−1, even after 2000 cycles with a capacity decay ratio of only 0.007% per cycle. This excellent cycle performance can be attributed to the ability of NiNC to effectively inhibit active material dissolution in the electrolyte, with coexisting graphitic N and pyridinic N sites coordinated to a single Ni atom, thereby fortifying the overall material stability. The robust adsorption behavior of NiNC also effectively impeded a shuttle effect of the charged state, as conclusively demonstrated by a self-discharge test. Specifically, the I2@NiNC cathode was charged to 1.6 V and was allowed to rest for 24 h before being discharged to 0.6 V, retaining 93% of its initial capacity, whereas the corresponding value for I2@NC is 90% (Fig. 4b and Fig. S15, ESI†). The remarkable efficacy of NiNC in preventing the detrimental effects of iodine species shuttling was also verified by analyzing its cycling performance, even at an extremely high current density of 4 A g−1 (Fig. 4c). Notably, I2@NiNC maintained a high capacity of 181 mA h g−1 with a retention ratio of 82% and capacity decay ratio of only 0.0018% per cycle without variations in the galvanostatic charge–discharge profile. This result is highly competitive with state-of-the-art reported values for long-term cycling (Fig. 4d and Fig. S16, Table S5, ESI†). As pyridinic N sites carry a negative charge within the carbon matrix, this feature typically hinders their ability to adsorb negatively charged polyiodide species. However, through the incorporation of single Ni atoms into the NC structure, the pyridinic N sites can be activated to effectively serve as strong adsorption sites for polyiodide species.
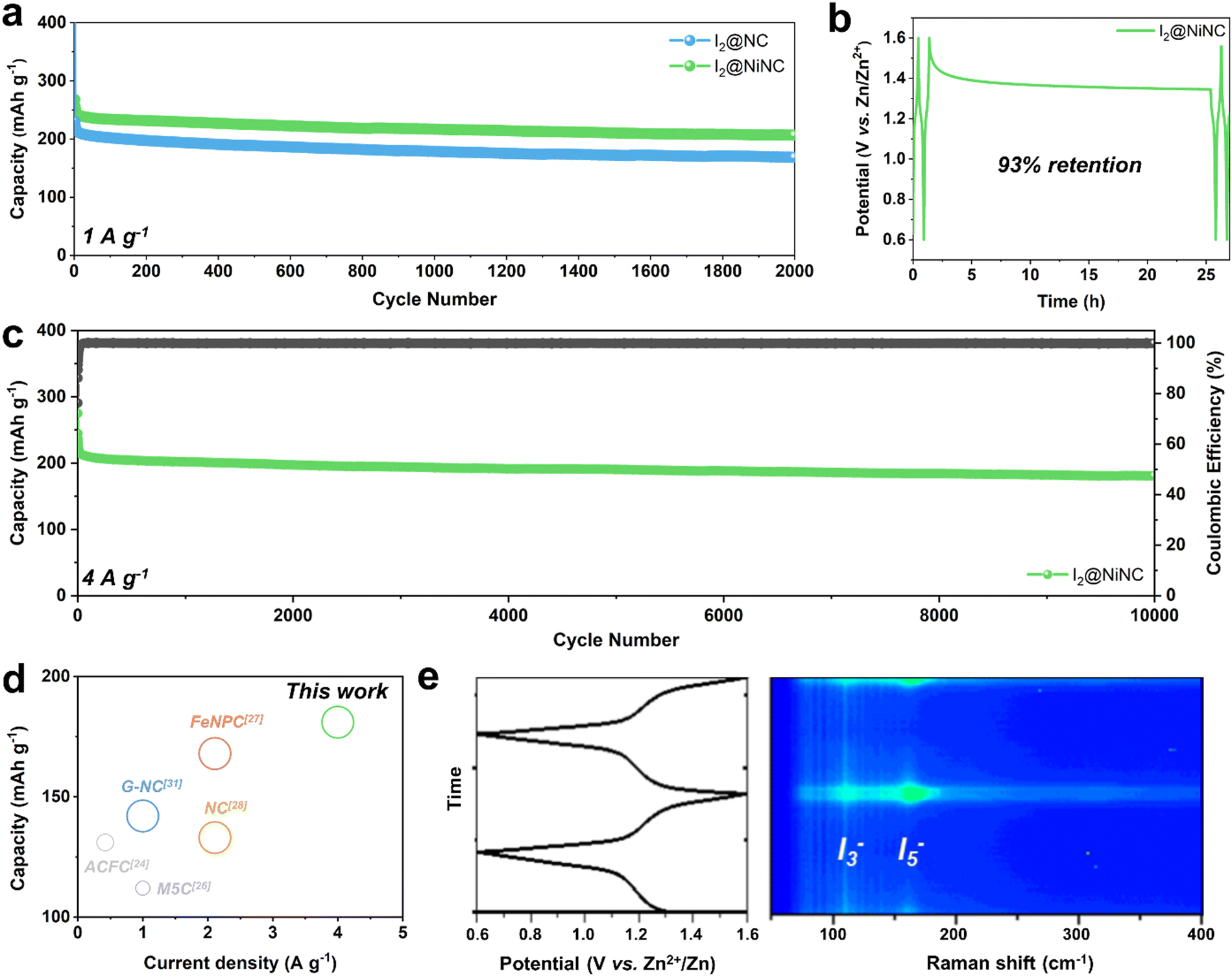 | ||
| Fig. 4 (a) The cycling performance of I2@NC and I2@NiNC at a low current density of 1 A g−1. (b) The self-discharge test of I2@NiNC resting for 24 hours after being fully charged. (c) The cycling performance of I2@NiNC at a high current density of 4 A g−1. (d) The cyclability of I2@NiNC in comparison with reported values using carbonaceous host materials (the diameter of the circle is directly proportional to the cycle number). (e) Operando Raman spectra of I2@NiNC at different discharge and charge states and the corresponding voltage profile. | ||
Additionally, the redox reversibility of the polyiodide species at the I2@NiNC cathode during charging–discharging was evaluated by performing operando Raman spectroscopy in a homemade cell (Fig. 4e). In the pristine state, characteristic Raman peaks appeared at 110 and 160 cm−1, which corresponded to the symmetric stretching bands of the intermediate species I3− and I5−, respectively.53 As the discharging process ensued, a continual decrease was observed in the peak intensities of I3− and I5−, indicative of the reduction of polyiodide. This finding signified the sequential transformation of these species into iodide, as follows: I5− → I3− → I−. Notably, the I3− and I5− peaks were no longer detected after the system was fully discharged, suggesting a complete transition to the I− species. Interestingly, the I3− and I5− peaks gradually re-emerged as the charging process advanced further. Overall, the high degree of reversibility observed in operando Raman spectroscopy helped extend the cycle life, thereby effectively minimizing any potential degradation in capacity.
The electrochemical performance was significantly enhanced through the modulation of the electronic environment, originating from the coordination of single Ni atoms with pyridinic N sites. To delve deeper into this observation, the electronic and geometric states and the adsorption energy were investigated by performing density-functional theory (DFT) calculations on three modelled structures (Fig. S17, ESI†). The adsorption energies and the corresponding charge density differences of the optimized arrangements of I2 molecules on the three different surfaces were estimated (Fig. 5a). Upon the incorporation of Ni, the adsorption configuration of I2 changed from a parallel position on the surfaces of C and NC to a vertical position on NiNC. The adsorption energy of I2 on NiNC was observed to be approximately twice as high, aligning well with the physical characterization results. When the binding energies were dissected into van der Waals and chemical interactions, the former were found to remain nearly identical across all samples, whereas the I2–NiNC chemical interactions were considerably stronger, indicating the occurrence of more potent chemical interactions on the surface (Fig. 5b). Additionally, to visualize and quantitatively demonstrate the charge transfer between iodine and the three modelled structures, the charge density difference and Bader charge were comprehensively analyzed. The adsorption of I2 onto C and NC involved a negligible amount of charge transfer, indicating a predominant reliance on van der Waals interactions for adsorption. In stark contrast, the adsorption onto NiNC involved substantial charge transfer, signifying a pronounced affinity resulting from chemical interactions. Furthermore, the changes in the electronic structure of the I–I chemical bonds in the I2 molecules on C, NC, and NiNC were investigated by conducting crystal orbital Hamilton population (COHP) analysis (Fig. 5c).54 It is worth noting that more negative values of the integrated COHP (ICOHP) indicate the existence of stronger I–I chemical bonds, implying weaker interactions with the surface. The integration of positively charged single Ni atoms into the negatively charged NC matrix led to robust surface binding with I2, thereby weakening the I–I bonding and consequently increasing the bonding distance. This phenomenon demonstrates the significant role of the unique chemical environment facilitated by Ni atoms, which drastically alters the interaction dynamics between the surface and I2 molecules (Fig. 5d). Thus, the facilitated cleavage of I–I bonds and the superior electron affinity contributed to the high utilization of I2, thereby promoting its high dynamic activity.
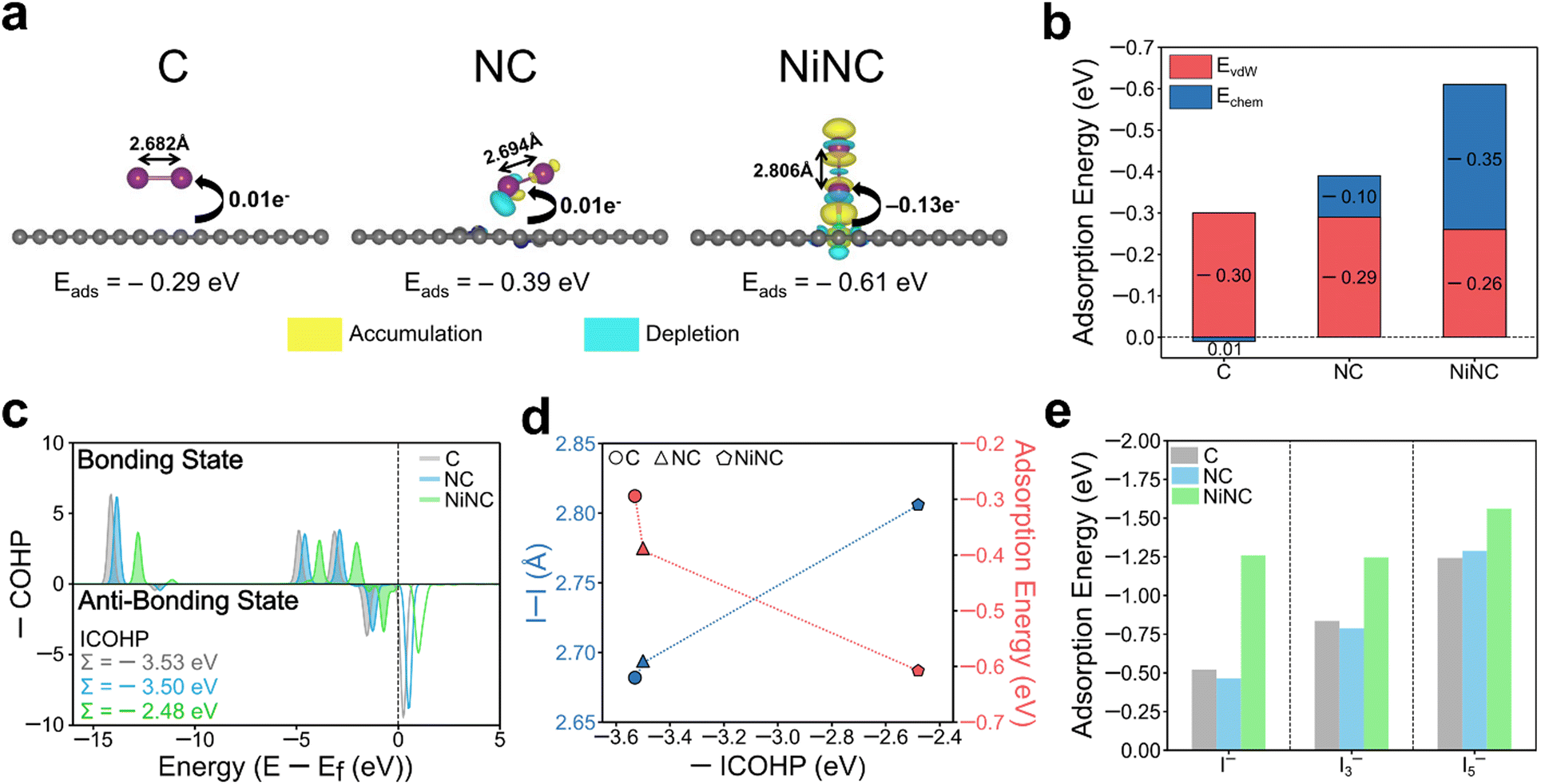 | ||
| Fig. 5 (a) Charge density difference plot during the adsorption of I2 on C, NC and NiNC. The amounts of electron transfer calculated from the Bader charge analysis and I–I intermolecular distance are also displayed. Color codes: purple (I), grey (C), blue (N), green (Ni). (b) Decomposition of binding energy into van der Waals (vdW) interactions (red) and chemical interactions (blue). (c) Crystal orbital Hamilton population (COHP) analysis and the corresponding integrated crystal orbital Hamilton population (ICOHP) for I–I bonding of I2 adsorbed on C, NC and NiNC. (d) I–I intermolecular distance and adsorption energy of I2 with respect to ICOHP for C, NC, and NiNC systems. (e) The adsorption energies of I−, I3− and I5−. | ||
The adsorption capability for other ionic species including iodide (I−) and polyiodides (I3− and I5−) also held substantial relevance to the electrochemical performance.55 The positively charged single Ni atoms served as favorable active sites that could proficiently adsorb negatively charged ionic species (Fig. 5e and Fig. S18, ESI†). This property emphasizes their effectiveness in suppressing the shuttling effect commonly induced by the negatively charged ionic species. Besides, graphitic N, which partially exists in NiNC, demonstrated superior adsorption ability, an observation that aligns with both the literature and our experimental findings (Fig. S19, ESI†). Theoretical simulations revealed the importance of modulating the electronic environment within the NC matrix and validated the effectiveness of introducing single Ni atoms to enhance the electrochemical performance.
Conclusions
In summary, we demonstrated the effect on the electrochemical performance of an N-doped carbon matrix that arises from modulating its electronic environment through the incorporation of atomically dispersed single Ni atoms. This material was then used to improve the performance of zinc–iodine (Zn–I2) batteries. As the positively charged single Ni atoms coordinated to the negatively charged pyridinic N sites, the resulting structure not only offered favorable adsorption sites for iodine species, greatly suppressing the shuttling effect, but also acted as an electrocatalyst to promote iodine redox kinetics. Furthermore, the microporous characteristics of the developed system provided ample space to effectively accommodate the iodine species. Consequently, the as-designed I2@NiNC cathode delivered a superior rate retention of 76% even at a considerably high current density of 6 A g−1. An ultra-stable cycling performance with a capacity retention of 82% after 10000 cycles, which corresponded to a low-capacity decay ratio of 0.0018% per cycle, was also observed. This study presents a pivotal way to tailor active sites for the iodine redox reaction, paving the way for the advancement of Zn–I2 batteries that outperform rechargeable aqueous zinc metal batteries using conventional metal oxide cathodes.
Author contributions
Jisung Lee and Jinwoo Lee conceived the idea and designed the experiments. Jinwoo Lee and Seoin Back supervised the project. Jisung Lee and Seungho Back conducted the experiments and data analysis. Wooseok Lee and Seoin Back contributed to the density functional theory study. Jisung Lee and Wooseok Lee organized and wrote the manuscript. Seung Yeop Yi and Seonggyu Lee helped with XAS analysis. Seongseop Kim performed N2 physisorption measurement and revised the manuscript. Joonhee Moon performed operando Raman measurement. Dong-Yeun Koh and Kyeounghak Kim assisted in writing the manuscript. All authors contributed to the discussion and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2023-00235596, 2019M3D1A1079306, and 2022R1A5A1033719), Institute of Information & Communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (RS-2023-00221723) and generous supercomputing time from KISTI.Notes and references
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- Y. Liang, C.-Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J.-Q. Huang, D. Yu, Y. Liu, M.-M. Titirici, Y.-L. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 1, 6–32 CrossRef CAS.
- H. Wang, B. D. Adams, H. Pan, L. Zhang, K. S. Han, L. Estevez, D. Lu, H. Jia, J. Feng, J. Guo, K. R. Zavadil, Y. Shao and J.-G. Zhang, Adv. Energy Mater., 2018, 8, 1800590 CrossRef.
- X. Ye, D. Han, G. Jiang, C. Cui, Y. Guo, Y. Wang, Z. Zhang, Z. Weng and Q.-H. Yang, Energy Environ. Sci., 2023, 16, 1016–1023 RSC.
- J.-N. Liu, C.-X. Zhao, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Energy Environ. Sci., 2022, 15, 4542–4553 RSC.
- Q. Li, Y. Zhang, Z. Chen, J. Zhang, Y. Tao and Q.-H. Yang, Adv. Energy Mater., 2022, 12, 2201574 CrossRef CAS.
- Y. Liang and Y. Yao, Nat. Rev. Mater., 2023, 8, 109–122 CrossRef.
- Z. Yu, S.-L. Shang, D. Wang, Y. C. Li, H. P. Yennawar, G. Li, H.-T. Huang, Y. Gao, T. E. Mallouk, Z.-K. Liu and D. Wang, Energy Storage Mater., 2019, 17, 70–77 CrossRef.
- L. Zhang and Y. Hou, Adv. Energy Mater., 2021, 11, 2003823 CrossRef CAS.
- N. Dong, F. Zhang and H. Pan, Chem. Sci., 2022, 13, 8243–8252 RSC.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- Q. Wang, M. Zhu, G. Chen, N. Dudko, Y. Li, H. Liu, L. Shi, G. Wu and D. Zhang, Adv. Mater., 2022, 34, 2109658 CrossRef CAS PubMed.
- J. Yu, B.-Q. Li, C.-X. Zhao, J.-N. Liu and Q. Zhang, Adv. Mater., 2020, 32, 1908488 CrossRef CAS PubMed.
- Y. Zeng, D. Luan and X. W. D. Lou, Chem, 2023 DOI:10.1016/j.chempr.2023.03.033.
- K. Zhu, S. Wei, H. Shou, F. Shen, S. Chen, P. Zhang, C. Wang, Y. Cao, X. Guo, M. Luo, H. Zhang, B. Ye, X. Wu, L. He and L. Song, Nat. Commun., 2021, 12, 6878 CrossRef CAS PubMed.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- Y. Kim, Y. Park, M. Kim, J. Lee, K. J. Kim and J. W. Choi, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
- H. Yu, Z. Wang, R. Zheng, L. Yan, L. Zhang and J. Shu, Angew. Chem., Int. Ed., 2023, e202308397 CAS.
- S. Niu, B. Zhao and D. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 25558–25566 CrossRef CAS PubMed.
- W. Li, H. Xu, H. Zhang, F. Wei, T. Zhang, Y. Wu, L. Huang, J. Fu, C. Jing and J. Cheng, Energy Environ. Sci., 2023, 16, 4502–4510 RSC.
- B. Yong, D. Ma, Y. Wang, H. Mi, C. He and P. Zhang, Adv. Energy Mater., 2020, 10, 2002354 CrossRef CAS.
- W.-G. Lim, X. Li and D. Reed, Small Methods, 2023, 2300965 CrossRef PubMed.
- J. Hao, L. Yuan, B. Johannessen, Y. Zhu, Y. Jiao, C. Ye, F. Xie and S. Z. Qiao, Angew. Chem., 2021, 133, 25318–25325 CrossRef.
- W.-G. Lim, C.-Y. Park, H. Jung, S. Kim, S. H. Kang, Y.-G. Lee, Y. C. Jeong, S. B. Yang, K. Sohn, J. W. Han and J. Lee, Adv. Mater., 2023, 35, 2208999 CrossRef CAS PubMed.
- W.-G. Lim, C. Jo, A. Cho, J. Hwang, S. Kim, J. W. Han and J. Lee, Adv. Mater., 2019, 31, 1806547 CrossRef PubMed.
- H. Pan, B. Li, D. Mei, Z. Nie, Y. Shao, G. Li, X. S. Li, K. S. Han, K. T. Mueller and V. Sprenkle, ACS Energy Lett., 2017, 2, 2674–2680 CrossRef CAS.
- Q. Guo, H. Wang, X. Sun, Y. N. Yang, N. Chen and L. Qu, ACS Mater. Lett., 2022, 4, 1872–1881 CrossRef CAS.
- Y. Hou, F. Kong, Z. Wang, M. Ren, C. Qiao, W. Liu, J. Yao, C. Zhang and H. Zhao, J. Colloid Interface Sci., 2023, 629, 279–287 CrossRef CAS PubMed.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- D. Yu, A. Kumar, T. A. Nguyen, M. T. Nazir and G. Yasin, ACS Sustainable Chem. Eng., 2020, 8, 13769–13776 CrossRef CAS.
- W. Liu, P. Liu, Y. Lyu, J. Wen, R. Hao, J. Zheng, K. Liu, Y.-J. Li and S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 8955–8962 CrossRef CAS PubMed.
- Y. Ji, J. Xu, Z. Wang, M. Ren, Y. Wu, W. Liu, J. Yao, C. Zhang and H. Zhao, J. Electroanal. Chem., 2023, 931, 117188 CrossRef CAS.
- T. Liu, H. Wang, C. Lei, Y. Mao, H. Wang, X. He and X. Liang, Energy Storage Mater., 2022, 53, 544–551 CrossRef.
- X.-C. Huang, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47, 2071–2073 RSC.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- W.-W. Zhan, Q. Kuang, J.-Z. Zhou, X.-J. Kong, Z.-X. Xie and L.-S. Zheng, J. Am. Chem. Soc., 2013, 135, 1926–1933 CrossRef CAS PubMed.
- M. Jana, R. Xu, X.-B. Cheng, J. S. Yeon, J. M. Park, J.-Q. Huang, Q. Zhang and H. S. Park, Energy Environ. Sci., 2020, 13, 1049–1075 RSC.
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- Y. He, Q. Shi, W. Shan, X. Li, A. J. Kropf, E. C. Wegener, J. Wright, S. Karakalos, D. Su, D. A. Cullen, G. Wang, D. J. Myers and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 9516–9526 CrossRef CAS PubMed.
- S. Kim, M. Ju, J. Lee, J. Hwang and J. Lee, J. Am. Chem. Soc., 2020, 142, 9250–9257 CrossRef CAS PubMed.
- U. Sim, T.-Y. Yang, J. Moon, J. An, J. Hwang, J.-H. Seo, J. Lee, K. Y. Kim, J. Lee and S. Han, Energy Environ. Sci., 2013, 6, 3658–3664 RSC.
- T. Kondo, S. Casolo, T. Suzuki, T. Shikano, M. Sakurai, Y. Harada, M. Saito, M. Oshima, M. I. Trioni and G. F. Tantardini, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035436 CrossRef.
- S. K. Singh, K. Takeyasu and J. Nakamura, Adv. Mater., 2019, 31, 1804297 CrossRef PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- Z. Zhao, S. Wang, R. Liang, Z. Li, Z. Shi and G. Chen, J. Mater. Chem. A, 2014, 2, 13509–13512 RSC.
- M. U. Patel and R. Dominko, ChemSusChem, 2014, 7, 2167–2175 CrossRef CAS PubMed.
- W.-G. Lim, Y. Mun, A. Cho, C. Jo, S. Lee, J. W. Han and J. Lee, ACS Nano, 2018, 12, 6013–6022 CrossRef CAS PubMed.
- J. Lee, S. Kim, J.-H. Park, C. Jo, J. Chun, Y.-E. Sung, E. Lim and J. Lee, J. Mater. Chem. A, 2020, 8, 3119–3127 RSC.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- J. Cambedouzou, J.-L. Sauvajol, A. Rahmani, E. Flahaut, A. Peigney and C. Laurent, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 235422 CrossRef.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- Z. Su, Z. Wei, C. Lai, H. Deng, Z. Liu and J. Ma, Energy Storage Mater., 2018, 14, 129–135 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00228d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |