
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect biomass valorisation to γ-valerolactone by Ru-PNP catalysed hydrogenation in acid†
Sakhitha
Koranchalil
and
Martin
Nielsen
 *
*
Department of Chemistry, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark. E-mail: marnie@kemi.dtu.dk
First published on 11th January 2024
Abstract
Converting carbohydrate-rich biomass waste directly to γ-valerolactone (GVL) is highly attractive but challenging owing to the inert nature and high complexity of biomass, necessitating a versatile and selective catalytic system. Herein, we describe the first direct conversion of monosaccharides (glucose, fructose, and xylose) and polysaccharides (cellulose and hemicellulose) in high yields under mild conditions. We also present the first direct conversion of raw lignocellulose, starch, and chitin biomass to GVL. Using the homogeneous catalyst Ru-MACHO-BH in H3PO4(aq) under 30 bar H2 at 125–140 °C for 24–120 hours provides GVL in excellent yields (26–48 mol%).
Broader contextDeveloping direct transformations of raw biomass to desired end-products is crucial for achieving viable and sustainable valorisation of biogenic substances. Performing such transformation under as mild and waste-less conditions as feasible and yet high-yielding is equally important. Gamma-valerolactone (GVL) is a highly attractive and biodegradable product, being valuable as, for example, a biofuel and industrial chemical or solvent. Hence, developing an environmentally benign synthesis route of GVL from raw biomass would potentially give access to a fuel and chemical that is both valuable and sustainable. However, to date there exists only a single example of this synthesis route carried out in a direct fashion, and it leads to a relatively low GVL yield (12 wt%) while employing heterogeneous catalysis, harsh conditions (800 W microwave heating to 180 °C) and using wasteful and expensive isopropanol as a H-donor. Our protocol is first-in-class to show the proof-of-concept of direct transformation of a range of carbohydrate-rich raw biomass substances to GVL in excellent yields under mild conditions. Thus, with 12 different raw biomass substances spanning three biogenic carbohydrate types, GVL yields up to 26 wt% are achieved using homogeneous catalysis and simple, cheap, and waste-free H2 as a H-donor under mild conditions (140 °C, no microwave). |
Introduction
As the only sustainable hydrocarbon resource available on Earth, biomass plays an essential role in the production of green fuels and chemicals.1 Indeed, biomass is the only perennial resource that might provide a negative carbon footprint, i.e., decrease the net atmospheric CO2 levels.2 Moreover, the replacement of petrochemicals by biogenic-derived substances is receiving increasing attention,3 and developing efficient and direct routes for biomass valorisation is of immense importance. However, biomass transformation is a complex chemical process, and it is highly challenging to develop selective and viable processes. For example, classical thermochemical methods, e.g., gasification, pyrolysis, and liquefaction,4 have severe drawbacks such as producing biofuels with complex and ill-defined compositions or generating high amounts of undesired side products, for example coke or CO2. Thus, an alternative conceptual approach of producing well-defined platform chemicals from biomass has emerged,5 which involves breaking down the biomass into one or a few molecules that can then be converted into biofuels or commodity chemicals.6One of the most promising sustainable and well-defined platform chemicals that can be obtained from biomass is γ-valerolactone (GVL),6a which is derived from biogenic carbohydrates in several steps. GVL has many applications, such as biofuel, fuel additive, green solvent, and in the syntheses of numerous end-products such as polymers or pharmaceuticals. Its production can be envisioned to follow two specific multistep cascade processes (Scheme 1).7 Using lignocellulose as model example of a biomass substrate, in the ‘glucose path’ acid mediates the hydrolysis of cellulose to glucose followed by the isomerization to fructose, dehydration to 5-hydroxymethyl furfural (HMF), rehydration to levulinic acid (LA), and finally hydrogenation followed by lactonization to GVL. In the ‘xylose path’, hemicellulose is first hydrolysed to xylose and dehydrated to form furfural, both steps mediated by acid. A subsequent hydrogenation leads to furfuryl alcohol, which is then rehydrated by acid to LA, and finally a second hydrogenation leads to GVL. Unfortunately, there are only very few examples of direct transformation of cellulose8 and hemicellulose9 to GVL, severely hampering the potential for using biogenic carbohydrates for obtaining GVL.
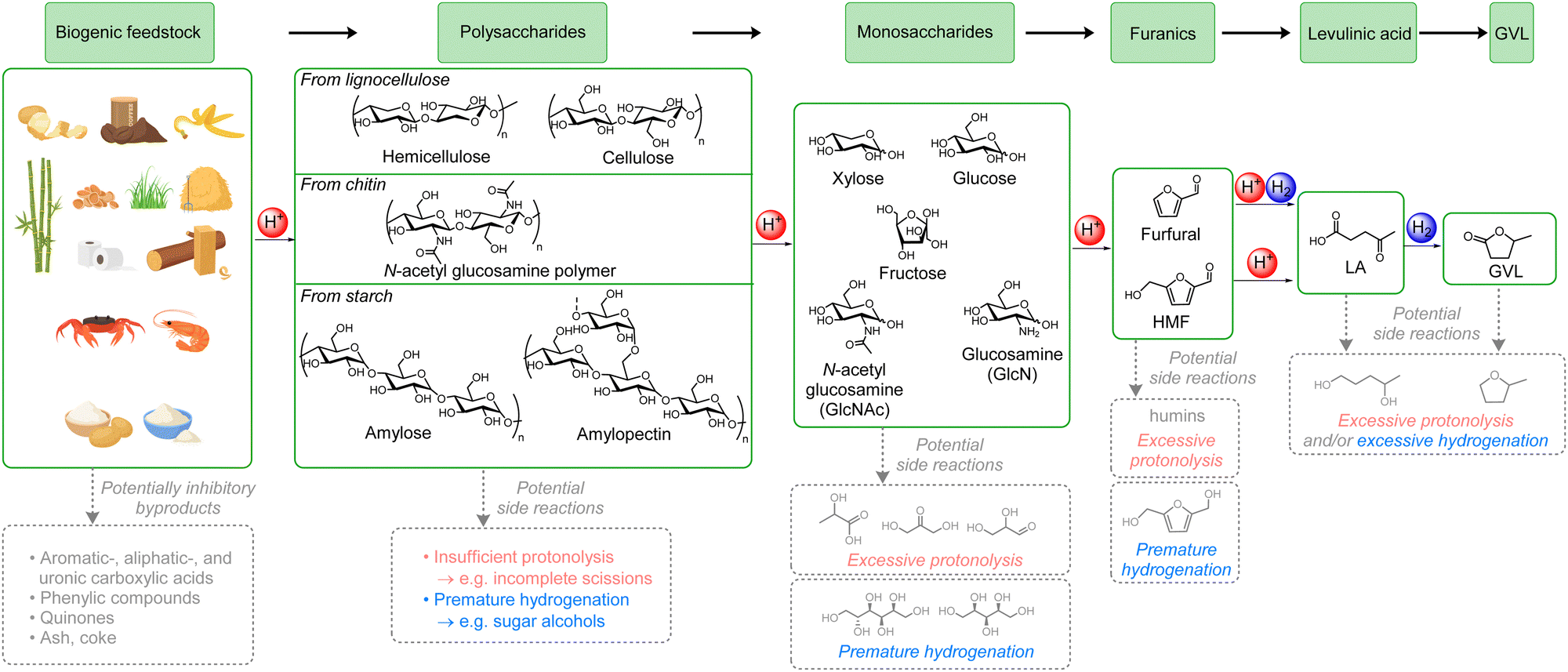 | ||
| Scheme 1 Generally accepted stepwise mechanism for the conversion of the three major types of carbohydrates-rich biomass, lignocellulose, chitin, and starch, into GVL (grey arrows and products indicate selected examples of the major pitfalls). | ||
Lignocellulosic residues, mainly originating from wood- and agricultural waste, exceed 180 Gtpa worldwide10 and therefore represent a very large non-edible resource. It is mainly composed of cellulose (40–50 wt%) and hemicellulose (16–33 wt%), with lignin constituting the remainder (15–30 wt%).11 It is, therefore, exceedingly important to develop procedures capable of transforming both cellulose and hemicellulose to GVL. Moreover, other major biogenic carbohydrate sources, such as chitin and starch, follow the ‘glucose path’ via either N-acetylglucosamine polymer12 or amylose/amylopectin,13 respectively.
The direct conversion of raw carbohydrate-rich biomass to GVL appears straightforward, involving a combination of several hydrogenations and acid mediated transformations. However, the lack of literature precedence underscores the substantial challenge that it in fact represents. Hence, as the only example of directly transforming real biomass to GVL, Huang converted poplar to GVL in merely 12 wt% yield by using a sacrificial H-donor (iPrOH) with mixed Al2(SO4)3 and Ru/ZrO2 catalyst, and 800 W microwave heating to 180 °C.8b In addition, most of the protocols developed for later-stage entries, such as poly- and monosaccharides, also employ very harsh conditions between 170–200 °C and either a sacrificial H-donor or high H2 pressures of up to approximately 100 bar.8,9 One exception is given by Li who reported the direct production of GVL by 20 wt% Ru/TiO2 catalyst in combination with 40 wt% of the strong Brønsted acid H3PW12O40 (HPA) in water/γ-butyrolactone mixture at 130–150 °C under 40 bar H2.8c GVL yields from fructose (130 °C), glucose (150 °C), starch (150 °C), and cellulose (150 °C) reached 68%, 55%, 48%, and 41%, respectively.
To the best of our knowledge, there are no examples of direct GVL production from raw biomass in high yields and, furthermore, there are no examples using mild conditions let alone using simple H2 as hydrogen source. All these factors are pivotal for developing viable biomass valorisation strategies. Interestingly, even though homogeneous catalysis is typically praised for its high selectivity and catalytic activity under mild conditions, the only examples of homogeneous catalysis for direct GVL production employ similarly harsh conditions as the heterogeneous ones8a only to reach yields below 40% from pure carbohydrates.
A major reason for the lack of literature examples can be attributed to the potential pitfalls associated with each of the reaction steps (see Scheme 1). Thus, there are numerous possible side products that might inhibit the catalyst(s), and there is an ominously high likelihood of carrying out insufficient, premature, or excessive protonolysis and/or hydrogenation leading to halting or overdoing the transformation process or to following other routes all-together. Thus, a catalytic system for a direct transformation of carbohydrate-rich biomass to GVL must operate with high conversion and selectivity for every distinct acid- and hydrogenation-mediated step and tolerate many different substrates simultaneously. As a note, even though there are numerous examples of direct formations of furanics9b,c,14 and LA,15 it is highly unattractive to isolate any of these intermediates in a two-step production of GVL. The furanics are prone to polymerise to humins and their conversion to GVL anyway requires a combination of acid and hydrogenation, and isolation of LA is exceedingly energetically demanding.16
Herein, we report a unique homogeneous catalytic protocol, using for the first time a well-defined catalyst for the direct production of GVL from several polysaccharides and monosaccharides with unparalleled high yields of GVL of up to 75% (Scheme 2). In addition, we demonstrate the first example of the direct conversion of raw biogenic waste materials to GVL in excellent yields of up to 48%, which corresponds to more than 90% yield in each reaction step. Furthermore, we achieve this with an unprecedented combination of weak acid and low H2 pressure. Finally, also as the first example, with identical reaction conditions we cover the three major types of carbohydrate-rich biogenic sources, i.e., lignocellulose, starch, and chitin.
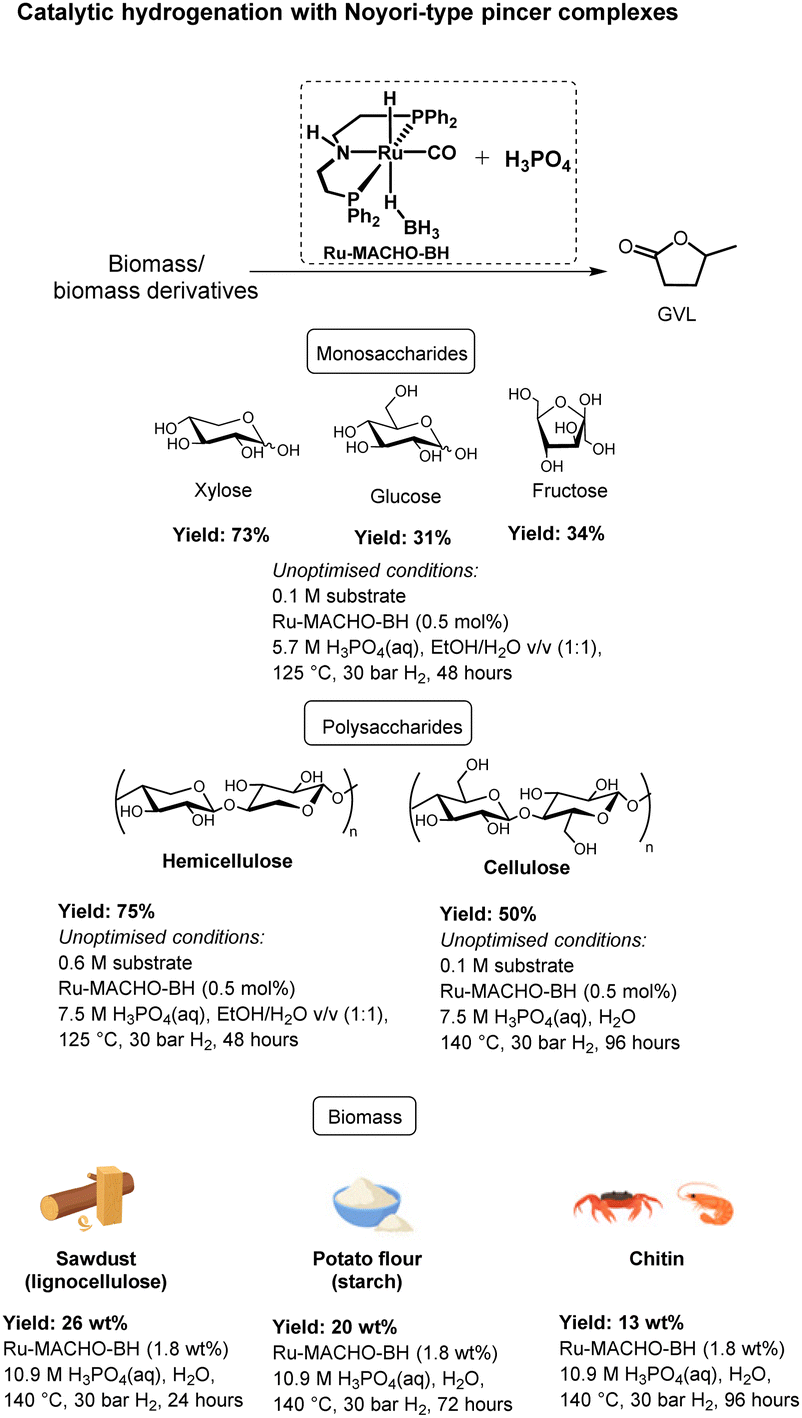 | ||
| Scheme 2 Catalytic hydrogenation of biomass, its derivatives, and this work. | ||
Our catalytic system consists of a novel combination of the PNP pincer complex Ru-MACHO-BH and the Brønsted acid H3PO4 under an atmosphere of H2 in water. Notably, even though Noyori-type PNP pincer complexes,17 such as the Ru-MACHO complex18 and analogues, have demonstrated excellent (de)hydrogenation activities in both basic,19 Lewis acidic,20 and neutral media,21 there exist, to the best of our knowledge, no examples of their use under Brønsted acidic conditions. In fact, compared to the plethora of examples in basic and neutral media, there are relatively few examples in general of hydrogenation reactions using homogeneous catalysis under Brønsted acidic conditions.22
Results and discussions
Direct conversion of monosaccharides to GVL
Initial efforts were focused on studying the monosaccharides such as xylose, glucose, and fructose. These three carbohydrates were chosen because they all represent important intermediates from either cellulose or hemicellulose to GVL, i.e., glucose and fructose are both derived from cellulose, and xylose arises from hemicellulose.Gratifyingly, with 0.5 mol% of Ru-MACHO-BH and 3.8 M H3PO4(aq) under 30 bar of H2 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v EtOH/H2O, the conversion towards GVL was 45%, 21%, and 25% from xylose, glucose, and fructose, respectively, after 48 hours at 125 °C (Table 1, entry 1). Employing 5.7 M H3PO4(aq) led to an encouraging 60% yield of GVL in case of xylose, whereas 16% and 29% were obtained from glucose and fructose, respectively, after 48 hours (entry 2). However, with higher concentration of H3PO4(aq) (7.5 M), the yield diminished to 50% for xylose, 13% for glucose, and 24% for fructose (entry 3). Noticeably, insoluble humins was observed in the reaction medium. Humins are usually produced from the polymerisation of furanics in acidic medium.23 Hence, we continued with 5.7 M H3PO4(aq). Moreover, glucose is first isomerized to fructose towards GVL,24 likely explaining why fructose consistently leads higher GVL yields than glucose. Next, the effect of concentration of the carbohydrate was investigated. The reaction afforded 73% GVL with 0.1 M xylose, dropped to 55% with 0.3 M xylose, and further lowered to 45% with 1.3 M xylose (entries 4–6). The similar trend was also observed with glucose and fructose, and 0.1 M glucose afforded 31% GVL and 0.1 M fructose led 34%. We speculate that humins are formed to a greater extent in the more concentrated samples.25
:1 v/v EtOH/H2O, the conversion towards GVL was 45%, 21%, and 25% from xylose, glucose, and fructose, respectively, after 48 hours at 125 °C (Table 1, entry 1). Employing 5.7 M H3PO4(aq) led to an encouraging 60% yield of GVL in case of xylose, whereas 16% and 29% were obtained from glucose and fructose, respectively, after 48 hours (entry 2). However, with higher concentration of H3PO4(aq) (7.5 M), the yield diminished to 50% for xylose, 13% for glucose, and 24% for fructose (entry 3). Noticeably, insoluble humins was observed in the reaction medium. Humins are usually produced from the polymerisation of furanics in acidic medium.23 Hence, we continued with 5.7 M H3PO4(aq). Moreover, glucose is first isomerized to fructose towards GVL,24 likely explaining why fructose consistently leads higher GVL yields than glucose. Next, the effect of concentration of the carbohydrate was investigated. The reaction afforded 73% GVL with 0.1 M xylose, dropped to 55% with 0.3 M xylose, and further lowered to 45% with 1.3 M xylose (entries 4–6). The similar trend was also observed with glucose and fructose, and 0.1 M glucose afforded 31% GVL and 0.1 M fructose led 34%. We speculate that humins are formed to a greater extent in the more concentrated samples.25
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate concentration [M] | H3PO4 [M] | GVL yielda [%] | ||
| Xylose | Glucose | Fructose | |||
| Standard reaction conditions: xylose (0.1–1.3 M), Ru-MACHO-BH (0.5 mol%), 85% w/w H3PO4, EtOH/H2O v/v (1:1), 125 °C, 30 bar H2 in 48 hours.a Determined by GC-FID using 1,4-dioxane as the internal standard. |
|||||
| 1 | 0.6 | 3.8 | 45 | 21 | 25 |
| 2 | 0.6 | 5.7 | 60 | 16 | 29 |
| 3 | 0.6 | 7.5 | 50 | 13 | 24 |
| 4 | 0.1 | 5.7 | 73 | 31 | 34 |
| 5 | 0.3 | 5.7 | 55 | 17 | 30 |
| 6 | 1.3 | 5.7 | 45 | 10 | 21 |

Hence, we here demonstrate the feasibility of achieving high yields of GVL from monosaccharides even under unoptimized conditions. As our goal is to directly transform real biomass to GVL, we decided not to pursue further optimisations with the monosaccharaides, and instead move a level up in complexity, i.e., to the polysaccharides.
Direct conversion of polysaccharides to GVL
Pure cellulose and hemicellulose were investigated as model substrates for lignocellulosic biogenic material. We commenced with cellulose with low concentration of acid in water as the sole solvent. Thus, to our delight, using 0.5 mol% Ru-MACHO-BH and 5.7 M H3PO4(aq) afforded 20% yield of GVL from 0.6 M cellulose after 48 hours at 125 °C (Table 2, entry 1), which increased to 22% after 96 hours (entry 2). Interestingly, increasing the acid concentration to 7.5 M H3PO4(aq) improved the yield to 28% yield (entry 3).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cellulose [M] | H3PO4 [M] | Solvent | P/T [bar]/[°C] | Time [h] | GVL yielda [%] |
| Reaction conditions: microcrystalline cellulose (0.6 and 0.1 mmol based on glucose monomers), 85% w/w H3PO4, Ru-MACHO-BH (0.5 mol%), H2O (1 mL), 30 bar H2, at 140 °C.a Determined by GC-FID using 1,4-dioxane as the internal standard. Catalyst loading and yield are relative to moles of glucose monomers contained in cellulose.b 1:1 v/v EtOH/H2O (1 mL).c Microcrystalline cellulose (7.8 mmol), H2O (15 mL). |
||||||
| 1 | 0.6 | 5.7 | H2O | 30/125 | 48 | 19 |
| 2 | 0.6 | 5.7 | H2O | 30/125 | 96 | 22 |
| 3 | 0.6 | 7.5 | H2O | 30/125 | 96 | 28 |
| 4 | 0.6 | 5.7 | H2O | 30/140 | 96 | 31 |
| 5 | 0.6 | 7.5 | H2O | 30/140 | 48 | 28 |
| 6 | 0.6 | 7.5 | H2O | 30/140 | 96 | 44 |
| 7 | 0.1 | 5.7 | H2O | 30/140 | 48 | 37 |
| 8 | 0.1 | 7.5 | H2O | 30/140 | 96 | 50 |
| 9c | 0.5 | 7.5 | H2O | 30/140 | 72 | 41 |
| 10 | 0.6 | 5.7 | EtOH/H2Ob | 30/125 | 48 | 5 |
| 11 | 0.6 | 7.5 | EtOH/H2Ob | 30/125 | 48 | 10 |
The effect of temperature was also studied as high temperature is known to be detrimental for the acid mediated hydrolysis of cellulose.26 Thus, at 140 °C with 5.7 M H3PO4(aq), 31% of GVL was obtained after 96 hours (entry 4). Using 7.5 M of H3PO4(aq) provided 28% of GVL after 48 hours (entry 5), which further yielded 45% after 96 hours (entry 6). Finally, lowering the substrate concentration to 0.1 M cellulose in 7.5 M H3PO4(aq) further increased the GVL yield to 50% (entry 8). Moreover, it is worth noting the excellent stability and high activity of Ru-MACHO-BH even in higher acid concentration, which is reflected in the full conversion of so-formed LA to GVL.
The reaction was then scaled up to 1.4 g of cellulose using 0.5 mol% of Ru-MACHO-BH at 140 °C and 30 bar H2 (entry 9). After 72 hours, a yield of 41% of GVL was obtained, demonstrating the reproducibility of the system at a larger scale. In order to investigate the influence of EtOH as a solvent on the yield of GVL, we performed the reaction with cellulose in a 1:1 v/v EtOH/H2O medium. Thus, employing 0.5 mol% Ru-MACHO-BH and 5.7 M of phosphoric acid under 30 bar H2 pressure in EtOH/H2O, afforded only 5% GVL after 48 hours at 125 °C (entry 10). Increasing the acid concentration to 7.5 M led to a significant increase in the yield to 10% (entry 11).
Next, we evaluated the performance of the catalytic system with hemicellulose. Corn core xylan and beechwood xylan were employed as two different xylan types from different biogenic sources. A reaction temperature of 125 °C was found sufficient to reach effective acid-mediated substrate turnover. However, a low yield of 8% of GVL was achieved from xylan from corn core using 0.5 mol% Ru-MACHO-BH and 7.5 MH3PO4(aq) in H2O after 48 hours (Table 3, entry 1). The yield from beechwood at 140 °C with 5.7 M H3PO4(aq) was 11% (entry 2). Increasing the acid concentration to 8.4 M H3PO4(aq) further diminished the yield to yield to 4% (entry 3). From these observations, we speculate that whether the diminished activity and low yield is as a result of humins formation, which is favoured under higher acid concentration and higher temperature when water is employed as the only solvent.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Xylan source (M) | H3PO4 [M] | Solvent | P/T [bar]/[°C] | GVL yielda [%] | |
| Corn core | Beechwood | |||||
| Reaction conditions: Ru-MACHO-BH (0.5 mol%), xylan from corn core (76.2% xylose monomers) or xylan from beech wood (95% xylose monomers), 85% w/w H3PO4, EtOH (1 mL), 30 bar H2, at 125 °C in 48 h. Catalyst loading and yield are relative to moles of xylose monomers contained in xylan.a Determined by GC-FID using 1,4-dioxane as the internal standard.b 1:1 v/v EtOH/H2O (1 mL). |
||||||
| 1 | 0.6 | 7.5 | H2O | 30/125 | 8 | |
| 2 | 0.6 | 5.7 | H2O | 30/140 | 11 | |
| 3 | 0.6 | 8.4 | H2O | 30/140 | 4 | |
| 4 | 0.6 | 5.7 | EtOH/H2Ob | 30/125 | 56 | |
| 5 | 0.6 | 5.7 | EtOH/H2Ob | 30/125 | 24 | |
| 6 | 0.6 | 7.5 | EtOH/H2Ob | 30/125 | 57 | |
| 7 | 0.6 | 7.5 | EtOH/H2Ob | 30/125 | 75 | |
| 8 | 0.6 | 5.7 | EtOH/H2Ob | 30/140 | 29 | |
| 9 | 0.6 | 8.4 | EtOH/H2Ob | 30/140 | 4 | |
| 10 | 0.6 | 5.7 | EtOH/H2Ob | 30/140 | 37 | |
We then carried out the reaction in 1:1 v/v EtOH/H2O mixture. Thus, using 5.7 M H3PO4(aq) in 1:1 v/v EtOH/H2O led to a significant increase in the yield to 56% at 125 °C (entry 4), but merely 29% at 140 °C (entry 8). With 7.5 M H3PO4(aq), 57% GVL is obtained at 125 °C (entry 6). To compare the results to that of corn core xylan, we performed the reaction with beechwood xylan under similar conditions. Thus, using 5.7 M H3PO4(aq) in 1:1 v/v EtOH/H2O at 125 °C yielded 24% of GVL after 48 hours (entry 5), whereas employing 7.5 M H3PO4(aq) yielded 75% of GVL at 125 °C (entry 7).
Having in our hands working conditions for both cellulose and hemicellulose that provide high yields, we decided to test various raw biomass materials.
Direct conversion of real biomass to GVL
We commenced our studies with beechwood sawdust, a lignocellulosic biomass. To our great pleasure, initial investigations revealed that the combination of Ru-MACHO-BH and H3PO4(aq) is catalytically active under a H2 pressure for the transformation to GVL. Hence, we first investigated the effect of H3PO4(aq) concentration while maintaining the catalyst loading, H2 pressure, and reaction temperature constant at 0.5 mol%, 30 bar, and 140 °C, respectively. Gratifyingly, employing 7.5 M H3PO4(aq) led to 15 wt% yield of GVL after 96 hours (ESI,† Table S1, entry 1), which was improved to 23 wt% by increasing the acid concentration to 9.3 M H3PO4(aq) (entry 3). Shortening or extending the reaction time to 24 or 168 hours, respectively, did not significantly change the yield (entries 2 and 4). Further increasing the H3PO4(aq) concentration to 10.1 M improved the yield to 25 wt%, which decreased again upon longer reaction times (entry 5 versus entries 6 and 7). With 10.9 M H3PO4(aq), the optimised yield of 26 wt% was obtained after 24 hours (entry 9). Considering that the theoretical yield of GVL from beechwood is limited to 54 wt% (assuming completely dry biomass and that only hemicellulose and cellulose are converted to GVL27), the yield is 48% (Table 4). Again, both shortening and extending the reaction time resulted in lower yields (entries 8 and 10). Moreover, after 18 hours, we still detected the intermediate LA, which was fully converted after 24 hours. It is noteworthy to mention that no hydrodeoxygenation products, such as hydrocarbons, were observed under these reaction conditions, neither in the liquid nor gas phase (ESI,† Fig. S36).28 Furthermore, a control experiment with GVL as substrate and otherwise identical conditions (Ru-MACHO-BH, 10.9 M H3PO4(aq), 30 bar H2, 140 °C, 72 hours) showed no decomposition products (ESI,† Fig. S37), underpinning the strong selectivity towards GVL production. When the catalyst loading was reduced to 0.9 wt%, the reaction afforded 15 wt% GVL under the optimized reaction conditions after 120 hours. However, LA was still detected in the reaction medium, suggesting the reaction had not finished. Finally, the reaction was also carried out in an EtOH/H2O mixture to study the solvent effect. With 1:1 v/v EtOH/H2O, a lower GVL yield of 14 wt% was observed under otherwise optimised conditions (entry 11).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Lignocellulosic source |
|
|
|
|
|
|
|
|
|
| Lignocellulosic content [wt%] | |||||||||
| Cellulose | 37 | 33 | 43 | 85–99 | 44 | 43 | 13 | 11 | 8 |
| Hemicellulose | 42 | 25 | 22 | 0 | 24 | 25 | 42 | 26 | 7 |
| Lignin | 19 | 17 | 27 | 0–15 | 17 | 16 | 25 | 10 | 33 |
| Starch | 23 | ||||||||
| GVL yield [wt% (mol%)] | |||||||||
| 24 h | 26(48) | 12(31) | 14(32) | 18(32–34) | 12(26) | 8(20) | 3(11) | 8(33) | |
| 48 h | 18(46) | 22(36–42) | 12(26) | 11(28) | 10(41) | ||||
| 72 h | 16(41) | ||||||||
| 96 h | 18(33) | 20(46) | 10(37) | ||||||
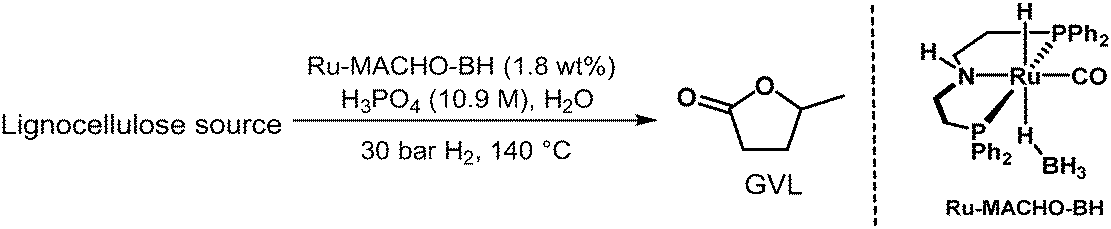









To demonstrate the power of our system and its potential to convert raw biowaste mixtures, the same reaction conditions were extended to other lignocellulosic biomass substrates (Table 4). Thus, 12 wt% yield of GVL is obtained from wheat straw after 24 hours, which is increased to 18 wt% after 48 hours. This yield accounts for 46% of the theoretical limit of 39 wt%.29 Further continuing the reaction to 72 hours results in a slight decrease of the yield. Moreover, these initial results validate the versatility of the catalytic system towards valorising different carbohydrate-rich biomass sources to GVL with approximately 90 mol% yield in each of the six-steps of the reaction pathway.
With bamboo stem, 14 wt% yield of GVL is obtained after 48 hours, with LA still detected in the reaction medium. Thus, extending the reaction hours to 96 hours completely consumed the LA and afforded 20 wt% of GVL, corresponding to a 46% yield of the theoretical limit of 43 wt%.30
Paper accounts for approximately 26% of total waste at landfills, contributing to air pollution and waste problems.31 Paper rich in cellulosic content32 can be utilized in this transformation and after 24 hours, 18 wt% GVL was obtained under the optimised reaction conditions. Extending the time to 48 hours increased the yield to 22 wt%.
Miscanthus, a perennial grass and an agricultural waste, contains high cellulosic content.33 Indeed, 12 wt% of GVL was obtained after 72 hours, by when the reaction seemed complete (no LA left).
Pistachio production and related dehulling processes generate large quantities of agricultural waste. Taking into consideration the generation volume (approximately 660000 tons) and lignocellulosic structure, pistachio residues can also be sustainably utilized to yield value-added compounds.34 Therefore, pistachio shells have been employed for this transformation as well, and a 12 wt% yield of GVL is obtained after 48 hours (26% yield35).
Biowaste such as used coffee grounds, banana peel, and potato peel were also tested. Despite having various valuable chemical components, used coffee grounds are in general considered as waste that ends up in landfills. Moreover, it is estimated that over 15 Mtpa of spent coffee grounds is produced.36 Food waste represents 60% of the total municipal biowaste in the EU and is estimated to contribute to global warming corresponding to 186 Mt of CO2 equivalent.37 Gratifyingly, employing the same reaction conditions on these biowaste feedstocks led to good GVL yields from all of them. Thus, 10 wt% was obtained from both banana peel and potato peel, whereas 11 wt% was achieved from used coffee grounds. These yields represent 28–41% of the theoretical maximum outcomes.38 Therefore, this approach demonstrates the feasibility and potential of utilizing day-to-day generated waste as feedstock towards fuels and chemicals.
We then moved our attention towards valorising the second-most abundant polysaccharide appearing in nature, the starch biomass.39 Rice grains and potato flour were tested, which contains 100% amylose and amylopectin.40 While these feedstocks are typically considered edible resources, there may be circumstances where it is necessary to classify them as non-food, especially if they are part of food waste. Thus, yields of 9 wt% and 8 wt% were obtained from rice grains and potato flour, respectively, after 24 hours (Table 5). Extending the reaction time to 72 hours improved the GVL yields to 16 wt% and 20 wt%, respectively. These results indicate that the catalytic system is very efficient in hydrolysing not only the α(1–4) glycosidic linkages, but also the α(1–6) linkages between glucose units of amylopectin, which is the major component of starch.
|
|
||
|---|---|---|
| Starch source |
|
|
| Reaction conditions: Ru-MACHO-BH (1.8 wt%) and 10.9 M H3PO4(aq) in H2O (1.7 mL) at 140 °C and 30 bar H2. Yields are calculated with respect to dry biomass and corresponding to the moles of cellulose and hemicellulose. | ||
| Starchy content [wt%] | ||
| Amylose | 18 | 35 |
| Amylopectin | 82 | 65 |
| GVL yield [wt% (mol%)] | ||
| 24 h | 9(15) | 8(13) |
| 72 h | 16(26) | 20(32) |
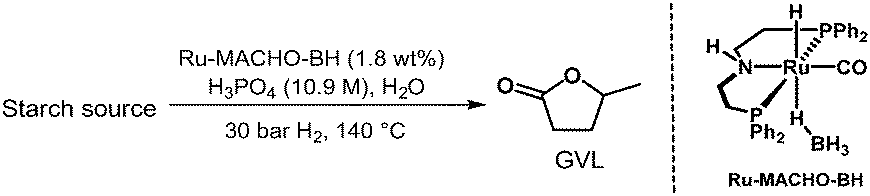


Next, chitin, which is usually obtained from e.g., shrimp or crab shells, fungi cell walls, or exoskeletons of arthropods,41 was tested. As a note, for safety reasons, we used industrially available chitin instead of the raw biomass sources in this category. To our delight, the reaction afforded 8 wt% of GVL after 72 hours under the optimized reaction conditions (Table 6). Extending the reaction conditions to 96 hours improved the yield to 13 wt%, corresponding to 29% yield or approximately 80% in every step of the seven-step cascade reaction. The comparatively lower yield from the chitin feedstock might be explained by the more challenging structure with a hard-to-hydrolyse amide unit contained in each carbohydrate unit41 or by the insoluble nature of chitin.42
|
|
|
|---|---|
| Chitin source |
|
| Reaction conditions: Ru-MACHO-BH (1.8 wt%) and 10.9 M H3PO4(aq) in H2O (1.7 mL) at 140 °C and 30 bar H2. Yields are calculated with respect to dry biomass and corresponding to the moles of N-acetyl-glucosamine. | |
| Chitin content [wt%] | |
| N-Acetyl-glucosamine | 100 |
| GVL yield [wt% (mol%)] | |
| 72 h | 8(18) |
| 96 h | 13(29) |
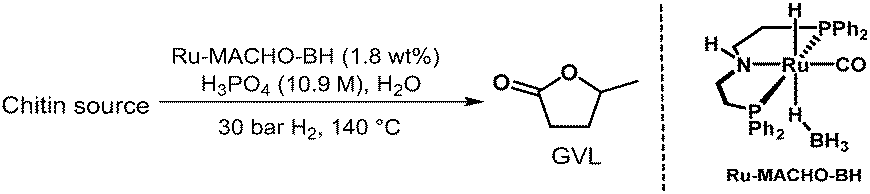

Furthermore, all the biomass materials were mixed in the same pot to evaluate the true versatility of the catalytic system (ESI,† Fig. S31). Thus, all the nine different types of lignocellulosic biomass tested here were mixed. Gratifyingly, 9 wt% of GVL was afforded after 96 hours. The mix of lignocellulosic biomass along with starch biomass (eleven substrates in one pot) gave 11 wt% yield after 96 hours and was improved to 17 wt% after 120 hours. Mixing all twelve substrates, i.e., including chitin, led to 15 wt% GVL after 120 hours. These results suggest the feasibility for the Ru-MACHO-BH/H3PO4(aq)/H2 combination to transform diverse mixtures of carbohydrate-rich biomass waste to GVL.
Finally, to investigate the fate of the catalyst, a test reaction was performed using Ru-MACHO-BH and beechwood sawdust under typical reaction conditions followed by NMR analysis. The persisting ligand phosphine shifts at approximately 63 ppm in 31P NMR suggests that the catalytic complex stays intact (ESI,† Fig. S39). The absence of a hydrido peak, including the disappearance of the hydrido triplet in 1H NMR at −12.3 ppm of Ru-MACHO-BH, might suggest that the resting species is a di-phosphato Ru-complex in presence of excess phosphoric acid and absence of a H2 pressure (ESI,† Fig. S40). Alternatively, the hydrido(s) might be interacting with the weak acid, thereby obscuring the NMR signal.43
Conclusions
In conclusion, we here demonstrate the direct conversion of monosaccharides (xylose, glucose, fructose), polysaccharides (cellulose, hemicellulose), and raw biomass (lignocellulose, starch, chitin) to GVL in good to excellent yields. The direct approach for the catalytic production of GVL from monosaccharides using the combination of Ru-MACHO-BH and H3PO4(aq) in EtOH/H2O with 30 bar of H2 at 125 °C produce GVL yields up to 73%. From the polysaccharides, 50% GVL was obtained from cellulose and 75% from hemicellulose. Finally, with raw biomass, using 1.8 wt% of the homogeneous catalyst Ru-MACHO-BH in 10.9 M H3PO4(aq) with 30 bar of H2 at 140 °C for 24–120 hours provides 10–26 wt% of GVL from twelve different biogenic sources, either as individual substrates or a combined pool. This corresponds to 26–48% yields, or an average of approximately 80–90 mol% yield in each reaction step. Finally, some studies on the catalyst suggest that it remains intact throughout the reaction.We are currently studying the recycling of the catalytic system for a more comprehensive understanding of the practical feasibility and sustainability of the proposed approach.
Author contributions
SK and MN conceived and designed the project. SK performed the experiments and analysis under the supervision of MN. Writing review and editing was done by SK and MN.Conflicts of interest
SK and MN are listed as inventors on the European patent application (application no. 22200625.6) by Technical University of Denmark (DTU) which describes this work. The authors declare no competing interests.Acknowledgements
The authors thank VILLUM FONDEN grant 19049 and COWI Foundation grant A149.10 for the generous funding. The authors acknowledge Professor Anders Riisager, Department of Chemistry, Technical University of Denmark, for providing the biomass feedstock and Leonhard Schill, Department of Chemistry, Technical University of Denmark, for the help with TGA as well as providing xylan samples. The authors also thank Torben Nielsen for providing the bamboo stem. The authors also thank Kasper Enemark-Rasmussen, Department of Chemistry, Technical University of Denmark, for carrying out a range of NMR measurements.References
- M. F. Demirbas, M. Balat and H. Balat, Energy Convers. Manag., 2009, 50, 1746–1760 CrossRef CAS.
- (a) D. Tilman, J. Hill and C. Lehman, Science, 2006, 314, 1598–6000 CrossRef CAS PubMed; (b) N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed; (c) C. Okkerse and H. V. Bekkum, Green Chem., 1999, 4, 107–114 RSC.
- (a) L. Petrus and M. A. Noordermeer, Green Chem., 2006, 8, 861–867 RSC; (b) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed; (c) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed; (d) T. Zhang, W. Li, S. An, F. Huang, X. Li and J. Liu, Bioresour. Technol., 2018, 264, 261–267 CrossRef CAS PubMed.
- (a) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC; (b) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- J. J. Bozell, Science, 2010, 329, 522–523 CrossRef CAS PubMed.
- (a) I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC; (b) F. Kerkel, M. Markiewicz, S. Stolte, E. Müller and W. Kunz, Green Chem., 2021, 23, 2962–2976 RSC; (c) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC; (d) K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292–304 CrossRef CAS.
- (a) C. Antonetti, D. Licursi, S. Fulignati, G. Valentini and A. M. R. Galletti, Catalysts, 2016, 6, 196 CrossRef; (b) L. Yea, Y. Hana, J. Fenga and X. Lu, Ind. Crops Prod., 2020, 144, 1120 Search PubMed; (c) P. Bajpai, Pretreatment of lignocellulosic biomass for biofuel production, Springer, Singapore, Structure of lignocellulosic biomass, 2016, pp. 7–12 CrossRef.
- (a) H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247–1255 RSC; (b) Y. Huang, T. Yang, Y. Luo, A. Liu, Y. Zhou, H. Pana and F. Wang, Catal. Sci. Technol., 2018, 8, 6252–6262 RSC; (c) J. Cui, J. Tan, T. Deng, X. Cui, H. Zheng, Y. Zhu and Y. Li, Green Chem., 2015, 17, 3084–3089 RSC; (d) N. Karanwal, R. G. Kurniawan, J. Park, D. Verma, S. Oh, S. M. Kim, S. K. Kwak and J. Kim, Appl. Catal., 2022, 314, 121466 CrossRef CAS.
- (a) T. Wang, J. He and Y. Zhang, Eur. J. Org. Chem., 2020, 1611–1619 CrossRef CAS; (b) S. Zhu, Y. Xue, J. Guo, Y. Cen, J. Wang and W. Fan, ACS Catal., 2016, 6, 2035–2042 CrossRef CAS; (c) B. Hernández, J. Iglesias, G. Morales, M. Paniagua, C. López-Aguado, J. L. García Fierro, P. Wolf, I. Hermanas and J. A. Melero, Green Chem., 2016, 18, 5777–5781 RSC.
- N. Dahmen, I. Lewandowski, S. Zibek and A. Weidtmann, Glob. Change Biol. Bioenergy, 2019, 11, 107–117 CrossRef.
- P. McKendry, Bioresour. Technol., 2002, 83, 37–46 CrossRef CAS PubMed.
- W. Hou, Q. Zhao and L. Liu, Green Chem., 2020, 22, 62–70 RSC.
- M. K. Marichelvam, M. Jawaid and M. Asim, Fibers, 2019, 7, 32 CrossRef CAS.
- (a) H. Winoto, B. Ahn and J. Jae, J. Ind. Eng. Chem., 2016, 40, 62–71 CrossRef CAS; (b) Y. Shao, Q. Li, X. Dong, J. Wang, K. Sun, L. Zhang, S. Zhang, L. Xu, X. Yuan and X. Hu, Fuel, 2021, 293, 120457 CrossRef CAS; (c) W.-P. Wu, Y.-J. Xu, S.-W. Chang, J. Deng and Y. Fu, ChemCatChem, 2016, 8, 3375–3380 CrossRef CAS; (d) F. Li, R. Yang, Z. Tian, Z. Du, J. Dai, X. Wang, N. Li and J. Zhang, Chem. – Eur. J., 2023, 29, e202300950 CrossRef CAS PubMed.
- (a) S. Wang, H. Huang, V. Dorcet, T. Roisnel, C. Bruneau and C. Fischmeister, Organometallics, 2017, 36, 3152–3162 CrossRef CAS; (b) A. D. Chowdhury, R. Jackstell and M. Beller, ChemCatChem, 2014, 6, 3360–3365 CrossRef; (c) J. M. Tukacs, M. Novak, G. Dibó and L. T. Mika, Catal. Sci. Technol., 2014, 4, 2908–2912 RSC; (d) C. Romain, S. Gaillard, M. K. Elmkaddem, L. Toupet, C. Fischmeister, C. M. Thomas and J.-L. Renaud, Organometallics, 2010, 29, 1992–1995 CrossRef CAS; (e) S. Wang, V. Dorcet, T. Roisnel, C. Bruneau and C. Fischmeister, Organometallics, 2017, 36, 708–713 CrossRef CAS; (f) J. Deng, Y. Wang, T. Pan, Q. Xu, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1163–1167 CrossRef CAS PubMed; (g) Y. Yi, H. Liu, L.-P. Xiao, B. Wang and G. Song, ChemSusChem, 2018, 11, 1474–1478 CrossRef CAS PubMed.
- (a) Q. Fang and M. A. Hanna, Bioresour. Technol., 2002, 81, 187–192 CrossRef CAS PubMed; (b) B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS; (c) A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC; (d) X. L. Du, Q. Y. Bi, Y. M. Liu, Y. Cao and K. N. Fan, ChemSusChem, 2011, 4, 1838–1843 CrossRef CAS PubMed.
- L. Piccirilli, D. L. J. Pinheiro and M. Nielsen, Catalysts, 2020, 10, 773 CrossRef CAS.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. I. Tanaka, K. Shida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166–171 CrossRef CAS.
- (a) S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed; (b) A. Kaithal, M. Hölscher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449–13453 CrossRef CAS PubMed; (c) M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed ; For examples from our lab, see: ; (d) L. Piccirilli, B. Rabell, R. Padilla, A. Riisager, S. Das and M. Nielsen, J. Am. Chem. Soc., 2023, 145, 5655–5663 CrossRef CAS PubMed; (e) Z. Ni, R. Padilla, L. dos Santos Mello and M. Nielsen, ACS Catal., 2023, 13, 5449–5455 CrossRef CAS; (f) D. L. J. Pinheiro and M. Nielsen, J. Org. Chem., 2022, 87, 5419–5423 CrossRef CAS PubMed; (g) R. Padilla, S. Koranchalil and M. Nielsen, Green Chem., 2020, 22, 6767–6772 RSC; (h) R. Padilla, M. S. B. Jørgensen, M. W. Paixão and M. Nielsen, Green Chem., 2019, 21, 5195–5200 RSC.
- (a) E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed; (b) J. E. Heimann, W. H. Bernskoetter and N. Hazari, J. Am. Chem. Soc., 2019, 141, 10520–10529 CrossRef CAS PubMed.
- (a) M. Nielsen, A. Kammer, D. Cozzula, H. Junge, S. Gladiali and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 9593–9597 CrossRef CAS PubMed; (b) R. A. Farrar-Tobar, B. Wozniak, A. Savini, S. Hinze, S. Tin and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 1129–1133 CrossRef CAS PubMed; (c) S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Nat. Catal., 2021, 4, 193–201 CrossRef CAS PubMed For examples from our lab, see: ; (d) R. Padilla, Z. Ni, D. Mihrin, R. W. Larsen and M. Nielsen, ChemCatChem, 2023, 15, e202200819 CrossRef CAS; (e) Z. Ni, R. Padilla, R. Pramanick, M. S. B. Jørgensen and M. Nielsen, Dalton Trans., 2023, 52, 8193–8197 RSC; (f) D. L. J. Pinheiro and M. Nielsen, Catalysts, 2021, 11, 558 CrossRef CAS.
- (a) Z. Xie and M. Schlaf, J. Mol. Catal. A: Chem., 2005, 229, 151–158 CrossRef CAS; (b) P. Ghosh, P. J. Fagan, W. J. Marshall, E. Hauptmann and R. M. Bullock, Inorg. Chem., 2009, 48, 6490–6500 CrossRef CAS PubMed; (c) F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed; (d) T. J. A. Foskey, D. M. Heinekey and K. I. Goldberg, ACS Catal., 2012, 2, 1285–1289 CrossRef; (e) A. Phanopoulos, A. J. P. White, N. J. Long and P. W. Miller, ACS Catal., 2015, 5, 2500–2512 CrossRef CAS; (f) F. K. Scharnagl, M. F. Hertrich, G. Neitzel, R. Jackstell and M. Beller, Adv. Synth. Catal., 2019, 361, 374–379 CrossRef CAS; (g) M. Trivedi, P. Sharma, I. K. Pandey, A. Kumar, S. Kumar and N. P. Rath, Chem. Commun., 2021, 57, 8941–8944 RSC.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- R. B. Stoffel, P. V. Neves and F. E. Felissia, Biomass Bioenergy, 2017, 107, 93–101 CrossRef CAS.
- S. Liu, Y. Zhu, Y. Liao, H. Wang, Q. Liu, L. Ma and C. Wang, Appl. Energy Combust. Sci., 2022, 10, 100062 Search PubMed.
- A. Orozco, M. Ahmad, D. Rooney and G. Walker, Process Saf. Environ. Prot., 2007, 85, 446–449 CrossRef CAS.
- E. M. Gucho, K. Shahzad, E. A. Bramer, N. A. Akhtar and G. Brem, Energies, 2015, 8, 3903–3923 CrossRef CAS.
- (a) D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schalf, Chem. – Eur. J., 2009, 15, 10132–10143 CrossRef CAS PubMed; (b) D. B. Lao, A. C. E. Owens, D. M. L. Heinekey and K. I. Goldberg, ACS Catal., 2013, 3, 2391–2396 CrossRef CAS; (c) Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 11162 CrossRef PubMed.
- F. A. Batzias, D. K. Sidiras, C. G. Siontorou, A. N. Bountri, D. V. Politi, O. N. Kopsidas, I. G. Konstantinou, G. N. Katsamas, I. S. Salapa and S. P. Zervopoulou, Int. J. Arts Sci., 2014, 7, 205–222 Search PubMed.
- J.-H. Chen, J. K. Xu, P.-L. Huang and R.-C. Sun, Cellulose, 2016, 23, 2727–2739 CrossRef CAS.
- Advancing Sustainable Materials Management: 2014 Fact Sheet (EPA530-R-17-01, EPA United States Environmental Protection Agency, 2014).
- R. L. Howard, E. Abotsi, E. L. Jansen van Rensburg and S. Howard, Afr. J. Biotechnol., 2003, 2, 602–619 CrossRef CAS.
- M. I. Jahirul, M. G. Rasul, A. A. Chowdhury and N. Ashwath, Energies, 2012, 5, 4952–5001 CrossRef CAS.
- J. Toghiani, N. Fallah, B. Nasernejad, A. Mahboubi, M. J. Taherzadeh and N. Afsham, Curr. Pollut. Rep., 2023, 9, 60–72 Search PubMed.
- X. Li, Y. Liu, J. Hao and W. Wang, Materials, 2018, 11, 1782 CrossRef PubMed.
- K. Johnson, Y. Liu and M. Lu, Front. Chem. Eng., 2022, 4, 31 Search PubMed.
- A. van der Linden and A. Reichel, Bio-waste in Europe-turning challenges into opportunities, European Environment Agency (EEA) Report 04, Denmark, 2020.
- (a) R. S. Orozco, P. B. Hernández, G. R. Morales, F. U. Núñez, J. O. Villafuerte, V. L. Lugo, N. F. Ramírez, C. E. B. Díaz and P. C. Vázquez, BioResources, 2014, 9, 1873–1885 Search PubMed; (b) M. Hijosa-Valsero, A. I. Paniagua-García and R. Díez-Antolínez, New Biotechnol., 2018, 46, 54–60 CrossRef CAS PubMed; (c) S. K. Karmee, Waste Manage., 2018, 72, 240–254 CrossRef CAS PubMed.
- M. M. Burrell, J. Exp. Bot., 2003, 54, 451–456 CrossRef CAS PubMed.
- M. K. Marichelvam, M. Jawaid and M. Asim, Fibers, 2019, 7, 32 CrossRef CAS.
- W. Hou, Q. Zhao and L. Liu, Green Chem., 2020, 22, 62–70 RSC.
- J. C. Roy, F. Salaün, S. Giraud, A. Ferri, G. Chen and J. Guan, Solubility of chitin: solvents, solution behaviors and their related mechanisms, Solubility Polysaccharides, IntechOpen, 2017, pp. 20–60 Search PubMed.
- N. Aebischer, U. Frey and A. E. Merbach, Chem. Commun., 1998, 2303–2304 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00247k |
| This journal is © The Royal Society of Chemistry 2024 |