
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-thermal plasma catalysis driven sustainable pyrolysis oil upgrading to jet fuel under near-ambient conditions†
Hoang M.
Nguyen
,
Ali
Omidkar
,
Wenping
Li
,
Zhaofei
Li
and
Hua
Song
 *
*
Green Catalysis Research Group, Department of Chemical and Petroleum Engineering, University of Calgary, 2500 University Dr NW, Calgary, AB T2N 1N4, Canada. E-mail: sonh@ucalgary.ca
First published on 2nd January 2024
Abstract
The global aviation industry is under increasing pressure to mitigate its environmental impact and achieve net-zero objectives. Reducing CO2 emissions alone is insufficient, as it overlooks crucial parts of the overall climate impacts. Addressing climate-neutral aviation fuel production from sustainable feedstock and effective technology is crucial. Here, we present a breakthrough in sustainable upgrading of pyrolysis oil with CH4 to jet fuel using non-thermal plasma triphase catalysis, characterized by an opposite gas–liquid flow and a fluidized catalyst under near-ambient conditions of atmospheric pressure and around 100 °C, resulting in an impressive liquid oil yield of almost 87%. This work delves into the strategic distribution of iridium (Ir) as minute nanoclusters on GaN, which serves to ensure robust catalytic stability and instigate synergistic interactions between plasma and catalysis. This unique configuration establishes an active surface conducive to the transformation of pyrolysis oil into essential jet fuel constituents, encompassing gasoline hydrocarbons (C5–C11), aromatics, and cycloalkanes. Both empirical experiments and computational analysis converge to confirm that methane activation can effectively occur within the plasma zone while surface reactions of cleavage of C–H, C–O, and C–N bonds with adsorbed H and CHx active species occurring on the Ir(100) surface play a crucial role in enhancing selectivity, resulting in upgraded oil meeting jet fuel commercial specifications. This novel approach holds substantial promise in the pursuit of sustainable and eco-friendly jet fuel production.
Broader contextIn the global pursuit of combating climate change and advancing sustainability, the transportation industry emerges as a substantial contributor to carbon emissions. As the urgency of addressing environmental concerns intensifies, the spotlight has increasingly turned towards sustainable fuel production from renewable sources with novel technologies, offering a pivotal avenue for mitigating environmental footprints. Our research marks a significant stride in the realm of fuel production powered by non-thermal plasma catalysis under ambient pressure and low-temperature conditions. Through the innovative integration of non-thermal plasma catalysis, featuring nanoclustered iridium supported on gallium nitride, we have unlocked a groundbreaking approach for converting pyrolysis oil into jet fuels with natural gas (CH4) in a triphase system characterized by CH4 gas, liquid oil, and fluidized catalyst particles. This pioneering method operates under mild conditions, yielding an 87% liquid oil output while rigorously meeting the demanding specifications of aviation fuels. The synergy of the nanoclustered iridium catalyst and energetic electrons injects a transformative element into our work. Both empirical data and computational modeling unequivocally affirm the pivotal role of plasma in effectively activating methane and generating active species, notably H* and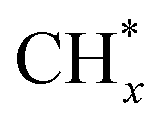 , which facilitate the cleavage of long-chain oil molecules on the highly active nanoclustered iridium surface. Beyond the fuel sector, our findings ripple into the broader landscape of sustainable energy or chemical production powered by plasma catalysis. , which facilitate the cleavage of long-chain oil molecules on the highly active nanoclustered iridium surface. Beyond the fuel sector, our findings ripple into the broader landscape of sustainable energy or chemical production powered by plasma catalysis.
|
1. Introduction
At the beginning of the 2020s, global concerns over energy insecurity, climate change, and mounting agricultural waste prompted the adoption of biofuels as renewable alternatives in countries worldwide. The depletion of fossil fuel reserves and the urgent need to address climate change have accelerated the pursuit of sustainable energy solutions in various sectors. Among these sectors, the aviation industry, a major contributor to greenhouse gas emissions, faces significant challenges in transitioning to greener alternatives. In 2018 alone, the aviation sector consumed approximately 94 billion gallons of jet fuels worldwide.1 The demand for jet fuels has seen substantial growth in recent years due to the expansion of air travel and the increasing number of commercial flights. The European Commission recognizes the importance of policies that address the complete climate impacts of aviation2 as indicated in their acknowledgement of the need for such measures. In the “Destination 2050” roadmap, aircraft manufacturers, airports, and airlines based in the European Union have set a target to reduce CO2 emissions by 92% by the year 2050.3 Many regions and countries have also followed a similar approach. The focus has long been on mitigation strategies involving enhancing air traffic management, developing larger and more fuel-efficient aircraft, promoting the use of sustainable aviation fuels, and implementing measures to offset any remaining effects. The aviation industry plays a significant role in global energy consumption and carbon emissions, necessitating a shift toward more sustainable fuel options. The development of renewable jet fuels that are compatible with existing aircraft engines is crucial for achieving a more sustainable aviation sector.There are pathways certified by the American Society for Testing and Materials (ASTM) for jet fuel production, including gas-to-jet (GTJ) such as Fischer–Tropsch synthesis (FTS), oil-to-jet (OTJ) such as hydrotreated renewable jet (HRJ), alcohol-to-jet (ATJ) such as ethanol-to-jet or butanol-to-jet, and sugar-to-jet (STG) such as catalytic upgrading of sugar to jet.4 However, of all the ASTM-certified options, only hydroprocessed esters and fatty acids (HEFA bio-jet), also known as HRJ, have reached a mature stage and are currently being commercially developed to produce drop-in biofuels.5 Bio-jet fuel, obtained from esters and fatty acids sourced from various materials, including oilseed crops, algae, and oils, fats, and greases, faces the challenge of a high feedstock cost, which constitutes about 80% of the total bio-jet production cost, even though triglyceride-based oils are easier to convert into liquid fuels compared to starch and cellulose.6 In contrast, the abundant renewable resource of lignocellulosic biomass, which yields approximately 150 to 170 × 109 tons annually,7 holds promise as a cost-effective feedstock for advanced biofuel production, offering potentially reduced or even neutral environmental impacts. Pyrolysis oil to jet fuel production uses renewable feedstocks like biomass, agricultural residues, or waste materials, which are plentiful and eco-friendly, reducing dependence on fossil fuels. Nonetheless, pyrolysis oil requires more extensive upgrading compared to oils from hydrothermal liquefaction given its high oxygen content, low heating value, poor thermal stability, acidity, chemical instability, corrosiveness, and immiscibility with petroleum.8
The debate revolves not so much only around the sustainability of the feedstocks but rather its level of technological advancement. It remains unclear from the available literature that which pathway offers superior overall economic feasibility. This suggests that the fuel must be produced using what is known as second-generation technology. Upgrading pyrolysis oil to hydrocarbon fuels under mild conditions (low temperature and low hydrogen pressure) by conventional catalytic thermal processes presents a significant challenge due to the high dissociation energy required to cleave perdurable bonds such as C–H (4.3 eV)9 and aromatic C–O bonds, which is approximately 4.8 eV.10 In general, the disruption of these stable bonds typically requires elevated temperatures exceeding 350 °C. However, this often gives rise to undesired outcomes like char or tar formation, as well as catalyst deactivation, imposing significant constraints on the reaction processes.11 Traditional hydrotreating processes for pyrolysis oil also require high and pure H2 pressures (>100 bars) in the presence of a catalyst,12 resulting in a product with a significant portion of hydrogen and approximately 20% by weight of liquid hydrocarbons, suitable for the direct use as gasoline and jet fuels.13 An alternative approach, fluid catalytic cracking, involves a single stage of hydrotreatment followed by fluid catalytic cracking using a zeolite as a catalyst,14 but this method is less commonly used for producing transportation fuels and primarily yields commodity chemicals.15 The utilization of pure H2 for chemical processes is considered economically and environmentally unsustainable.16 More importantly, the primary method for hydrogen production involves the energy-intensive reforming of natural gas.17 Leveraging CH4 directly as an active H2 source has the potential to substantially lower operational expenses and decrease greenhouse gas emissions.16 Non-thermal plasma (NTP) catalysis emerges as a viable technique for dissociating hydrogen from CH4 and breaking C–O and C–N bonds in pyrolysis oil under conditions closely approximating ambient parameters, including atmospheric pressure and low temperatures. This process is facilitated by the influence of high-energy electrons, reaching up to 10 eV.18 NTP offers high reactivity, straightforward on/off, versatile, and a tunable process.19 Furthermore, NTP relies on electrical energy and can seamlessly integrate into electrification systems fuelled by renewable sources like solar and wind power,20 with the aim of achieving carbon-neutral jet fuel production. The prospects of plasma catalytic technology for transforming pyrolysis oil into jet fuel are notably more promising in comparison to conventional thermal catalytic processes. However, a notable void exists in the literature regarding the application of NTP for directly upgrading liquid phase pyrolysis oil. The current hurdles within the plasma catalytic pathway encompass the need for a deeper understanding of the intricate interplay between the plasma and the catalyst, even more complicated with in-liquid plasma, as well as the efficient utilization of plasma energy.16 The successful integration of NTP into the jet fuel production value chain depends on overcoming technical and economic challenges and aligning with the evolving regulatory landscape. Continued research, innovation, and collaboration between academia, industry, and governments will play a crucial role in driving the development and deployment of this emerging technology, ultimately contributing to a more sustainable aviation sector.
In light of insights gleaned from the existing literature21,22 coupled with our prior experimental observations in plasma-driven methane activation,19,23,24 two pivotal challenges persist, preventing plasma catalytic processes from completely supplanting conventional thermal heating chemical methodologies. Often disregarded, these challenges encompass: (i) the in situ plasma-induced decomposition of intended products or excessive hydrogenation of oil, leading to gas production instead of the desired gasoline or aromatics and (ii) the dissipation of energy in the form of emitted photons and heat within the plasma reaction environment. Addressing the former hurdle may involve tailoring the design of catalyst porous structures and the strategic distribution of active centers. Pioneering research studies have indicated this possibility, citing the catalytic ability to shield products within the plasma regime.25 The second obstacle would be overcome by using a catalyst with photocatalytic properties to adsorb wasted photons to boost the catalytic performance further. Gallium nitride (GaN), a stable wurtzite-structure member of Group III nitrides and a well-known wide bandgap semiconductor with a fundamental bandgap energy of 3.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV, has gained significant attention as a revolutionary material due to its exceptional electronic and optical properties.26 In the realm of catalytic applications, GaN is increasingly being explored as a photocatalyst, thanks to its remarkable chemical and thermal stabilities.27 Recent research has shown that GaN exhibits catalytic abilities in activating C–H bonds, particularly in the non-oxidative aromatization of light alkanes such as methane, with evidence from a study suggesting a relatively low activation energy for methane coupling on GaN surfaces, where the rate-limiting step is the cleavage of C–H bonds in CHx species adsorbed on the Ga top site.28 In addition, Ir has been proven as an excellent active center for dehydrogenation from hydrocarbons and bio-oil.29,30 Despite significant advancements in the last decade, the development of an Ir catalyst with sufficient mass activity and stability remains a long-term objective, hindered by the challenge of identifying active sites and dealing with adverse reconstruction during operation.31 The highly effective catalysts would be those that feature smaller particle sizes of metallic Ir on GaN, serving as hybrid catalysts and supports. This configuration would result in enhanced electronic properties, significantly improved plasma interaction, and limited catalyst sintering. To the best of our knowledge, no prior research has reported the use of Ir-based catalysts for plasma-driven pyrolysis oil upgrading to jet fuel in the existing literature. Consequently, we aim to address and bridge this knowledge gap by offering valuable insights into catalyst development and exploring the synergistic effects of plasma and catalysis on the upgrading performance. This work seeks to contribute new perspectives and understanding to the field.
eV, has gained significant attention as a revolutionary material due to its exceptional electronic and optical properties.26 In the realm of catalytic applications, GaN is increasingly being explored as a photocatalyst, thanks to its remarkable chemical and thermal stabilities.27 Recent research has shown that GaN exhibits catalytic abilities in activating C–H bonds, particularly in the non-oxidative aromatization of light alkanes such as methane, with evidence from a study suggesting a relatively low activation energy for methane coupling on GaN surfaces, where the rate-limiting step is the cleavage of C–H bonds in CHx species adsorbed on the Ga top site.28 In addition, Ir has been proven as an excellent active center for dehydrogenation from hydrocarbons and bio-oil.29,30 Despite significant advancements in the last decade, the development of an Ir catalyst with sufficient mass activity and stability remains a long-term objective, hindered by the challenge of identifying active sites and dealing with adverse reconstruction during operation.31 The highly effective catalysts would be those that feature smaller particle sizes of metallic Ir on GaN, serving as hybrid catalysts and supports. This configuration would result in enhanced electronic properties, significantly improved plasma interaction, and limited catalyst sintering. To the best of our knowledge, no prior research has reported the use of Ir-based catalysts for plasma-driven pyrolysis oil upgrading to jet fuel in the existing literature. Consequently, we aim to address and bridge this knowledge gap by offering valuable insights into catalyst development and exploring the synergistic effects of plasma and catalysis on the upgrading performance. This work seeks to contribute new perspectives and understanding to the field.
Here, we present synergetic triphase phase catalysis plasma driven pyrolysis oil upgrading to jet fuel with CH4 as a H donor source under mild conditions of atmospheric pressure and low temperature of approximately 100 ± 5 °C. We use Ir supported on GaN as an active catalyst for CH4 activation to provide active species for the cleavage of C–H, C–N, and C–O bonds of oil molecules, and we propound that GaN with photocatalytic properties can use wasted photons induced in the plasma regime to promote the upgrading performance further. To initiate our investigation, we focused on devising a suitable support architecture essential for stabilizing the active Ir component. We infer that the agglomeration of large nanoparticles involves surface migration and coalescence; to prepare an Ir nanocluster catalyst, we therefore interfere these two steps by inducing a N-ligand to limit the fusion of Ir clusters to larger particle sizes. These ligands are considered essential for providing stability and influencing the electronic properties of mononuclear active sites through covalent bonding with the Ir metal centers and ligands. This understanding underscores the significance of designing a support material with electron-donating functional groups on its surface, mimicking the characteristics of a mononuclear Ir pincer complex. This design element stands as a pivotal factor in the advancement of an Ir-based nanocluster for plasma driven oil upgrading. Synergistic effects between clustered Ir and plasma lead to the formation of mobile electrons for CH4 activation and cleavage of heavy oil molecules to lighter components. The majority of the upgraded oil properties align with the ASTM standard for engine jet fuel with an excellent stability attributed to plasma-catalytic synergism. The observed outcomes lay the foundation for pioneering pathways in sustainable fuel production, promising novel routes for advancements within the field.
2. Experimental
2.1. Catalyst preparation
Ir/GaN catalysts were prepared by using a wet impregnation method with the assistance of 2-methylimidazole (99%, Sigma Aldrich) as a surface ligand to enable a cluster of Ir particles. In this study, three different catalyst loadings, 0.01%, 0.05%, and 0.1 wt% of Ir, were prepared. The use of such low Ir loadings serves the dual purpose of preventing potential agglomeration of Ir particles during synthesis, thereby inhibiting the formation of larger particles. Additionally, the approach ensures cost-effectiveness. This low catalyst loading strategy aligns with principles of efficiency and economy by optimizing the utilization of the catalyst, particularly one containing the precious metal Ir. It also minimizes the risk of saturation of active sites, enhancing selectivity while promoting the synergistic effects between the plasma and the catalyst for overall reaction efficiency. Furthermore, this practice adheres to green chemistry principles, reducing waste and considering the environmental impact. 2-Methylimidazole has been used as a genetic modulator to fine-tune ligands at the Co(II) center, inducing a self-adaptive coordination state for Co(II).32 In this work, 2 g of 2-methylimidazole was added into the deionized water and stirred at 500 rpm for 2 h at room temperature. Iridium(III) chloride (99.8%, Sigma-Aldrich) was weighted accurately and transferred to the mixing solution. The precursor solution was mixed at 500 rpm for 24 h at 60 °C. The pH of the solution diminished from 6.2 to 5.6, indicating the release of HCl from the reaction between IrCl3 and 2-methylimidazole. Gallium mononitride (GaN, 99.9%, Sigma Aldrich) was weighted accurately and added to the mixing solution. We expected that the unshared pair of electrons located on the nitrogen atom in 2-methylimidazole can readily rival the coordination effects of acids, impacting the iridium crystal's nucleation and growth. The presence of N-ligand enables the creation of numerous well-distributed iridium active sites with an iron atomic cluster (Fig. 1). This addresses the limitations of conventional plasma materials, which have fewer active sites and an uneven distribution. The lone pair of electrons on the nitrogen atom in 2-methylimidazole plays a pivotal role in stabilizing the cluster formation during its growth, contributing to the overall stability of the structure. However, it is important to note that during the subsequent calcination process, these ligands are expected to be removed. The calcination step is anticipated to eliminate the ligands, allowing the formation of the final cluster structure with the desired electronic properties. The dried catalyst precursor was calcined in a furnace by using a tube furnace at 500 °C for 4 h at a heating rate of 10 °C min−1 in air prior to further treatment in a tube furnace with a 5% H2/Ar mixture at 500 °C for 4 h at a heating rate of 3 °C min−1.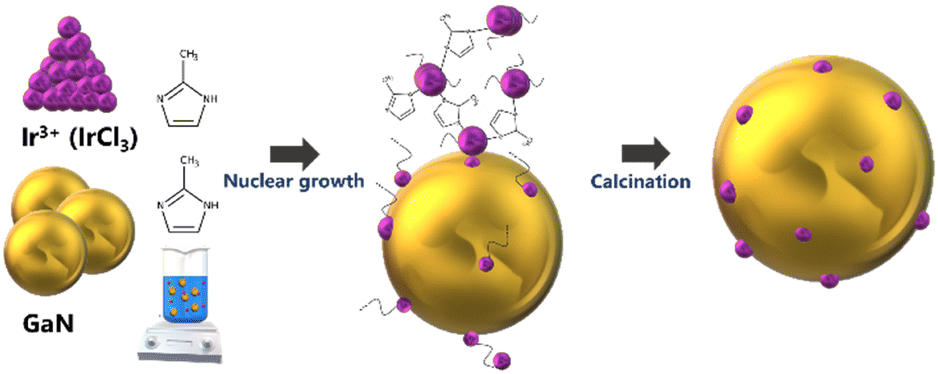 | ||
| Fig. 1 Schematic diagram of the synthesis process for the nanocluster Ir/GaN catalyst. | ||
2.2. Catalyst characterization
2.3. Evaluation of plasma catalytic performance
The experimental setup is depicted in Fig. S1 (ESI†). The oil upgrading reaction was conducted in a dielectric barrier discharge (DBD) reactor constructed from a quartz tube (an inner diameter of 8 mm, an outer diameter of 10 mm, and a thickness of 1 mm) with a coaxial stainless steel rod electrode (4 mm diameter) positioned at the center of the quartz tube and grounded with a silver mesh. For each trial, 0.1 g of catalysts (GaN and Ir/GaN) in a finely powdered form was packed at the reactor's core. The catalyst bed was secured using a quartz fritz with porosity #4, enabling gas passage. Prior to each experiment involving the catalyst, we commenced by introducing 0.1 cm3 of biomass-derived pyrolysis oil (supplied by FORGE Hydrocarbons Corp, Canada) to moisten the catalyst layer, followed by the introduction of CH4 gas to initiate the fluidized bed. Plasma was generated using a LEAP100© plasma generator with a maximum power of 700 W and an applied voltage of up to 80 kVpk–pk. The catalytic performance assessment was conducted at 12 W. To optimize mass transfer efficiency and product purity, a counter-current flow technique was employed, where CH4 gas and liquid oil were introduced in opposite directions. CH4 (ultra high purity, 99.99%, ALPHAGAZTM 1, Canada) was fed into the reactor from the bottom at a flow rate of 100 cm3 min−1, while pyrolysis oil was introduced from the top downward at a flow rate of 0.05 cm3 min−1 using a high-precision, high-pressure piston pump (Masterflex®). This configuration was used to enable the good mass transfer and prevent the liquid oil condensed in the reactor. This study included a control experiment in which the reaction was carried out using an external thermal heater set at 100 ± 5 °C to assess the impact of conventional thermal effects on the upgrading performance. However, no CH4 conversions or improvements in oil properties under these test conditions were observed, indicating that the influence of thermal factors on the reaction performance was effectively mitigated. For the plasma reaction test, the emitted light from the plasma reactor was analyzed using an optical system to provide in situ insights into the plasma reaction. Emission spectra from the glow region were captured at the reactor center. The measurements were recorded using a single-channel UV-VIS-NIR spectrophotometer in the scope mode (Avantes Inc., USB2000 Series) with a 400 μm fiber optic cable. The spectral range covered 200 to 800 nm, employing a line grating of 600 lines per mm and a 0.4 nm resolution. The temperature of the reactor was monitored using an infrared thermal camera (Flir©). Accurately measuring the temperature of the catalyst surface within the plasma reactor presents a challenging endeavor due to the intricate interactions involving high-energy electrons and plasma species. The measured temperature pertains to the external surface of the plasma reactor, serving as reference values for reproducible experiments and facilitating an assessment of the temperature effects on plasma catalytic reactions.2.4. Product analysis
2.5. Computational details
To determine optimized geometric configurations, the Dmol3 code integrated within the Material Studio package (Accelrys, San Diego, CA) was employed. The software utilizes the Perdew–Burke–Ernzerhof (PBE) functional method within the generalized gradient approximation (GGA) framework to effectively explore electron exchange and correlation interactions within crystal-based materials interacting with small gas molecules. The “DFT-D of Grimme” algorithm was employed to accurately account for van der Waals interactions, which significantly influence physisorption processes. By conducting Monte Carlo simulations across the substrate–adsorbate system's configurational space, the BIOVIA Materials Studio Adsorption Locator identifies potential adsorption arrangements as the temperature is gradually reduced by simulated annealing. This iterative process aids in identifying local energy minima. In most instances, adsorbates are molecular gases or liquids, whereas substrates typically encompass solid surfaces (crystalline or amorphous), porous materials, or substantial molecules (such as carbon nanotubes or nanocatalysts). The initial step in depicting the methane activation process on the Ir–GaN catalyst involves identifying the active site responsible for methane activation. This determination is made by evaluating the adsorption energy using eqn (1). The gallium nitride surface (GaN(110)) was chosen to represent the substrate, while the active metal nanocluster (Ir(100)) loaded onto this support was selected as the catalytic entity. Subsequently, the system's energy was recalibrated considering the dehydrogenation of methane within a plasma environment. Eqn (2) is employed to investigate the influence of plasma on the catalytic system and reevaluate its energy dynamics.| Eads = Esur+CH4 − Esur − ECH4 | (1) |
| Eads = Esur+CH+H − Esur − ECH4 − EH | (2) |
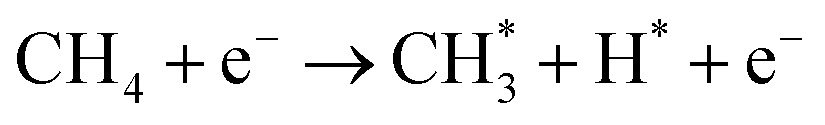 | (3) |
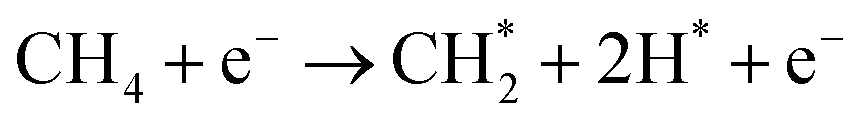 | (4) |
| CH4 + e− → CH* + 3H* + e− | (5) |
| CH4 + e− → C + 4H* + e− | (6) |
3. Results and discussion
3.1. Catalyst characterization
The screening test involving plasma-driven pyrolysis oil with different loadings of Ir on GaN revealed notable variations in performance. Specifically, 0.01 wt% Ir/GaN and 0.05 wt% Ir/GaN exhibited lower efficiency compared to 0.1 wt% Ir/GaN (Fig. S2 and S3, ESI†). The diminished performance at lower Ir loadings suggests an insufficient influence on upgrading capabilities, while higher loadings of noble metals may contradict principles of efficiency and economy. The chosen 0.1 wt% Ir/GaN loading strikes a balance, optimizing catalyst utilization and minimizing the risk of active site saturation. This deliberate approach enhances selectivity and facilitates synergistic effects between plasma and the catalyst, contributing to overall reaction efficiency. The subsequent thorough characterization of the optimal 0.1 wt% Ir/GaN catalyst underscores our commitment to methodical investigation and informed catalyst design in this work.33 from the fresh and spent Ir/GaN catalysts. Interestingly, Liu et al.34 reported observing diffraction peaks of Ir nanoclusters, ranging in size from 2 to 4 nm, supported on graphite at 2θ values of 40.7° and 47.3°. Nonetheless, neither the fresh nor the spent Ir/GaN samples displayed any detectable diffraction peaks related to Ir, indicating a smaller particle size distribution of Ir on the GaN support.
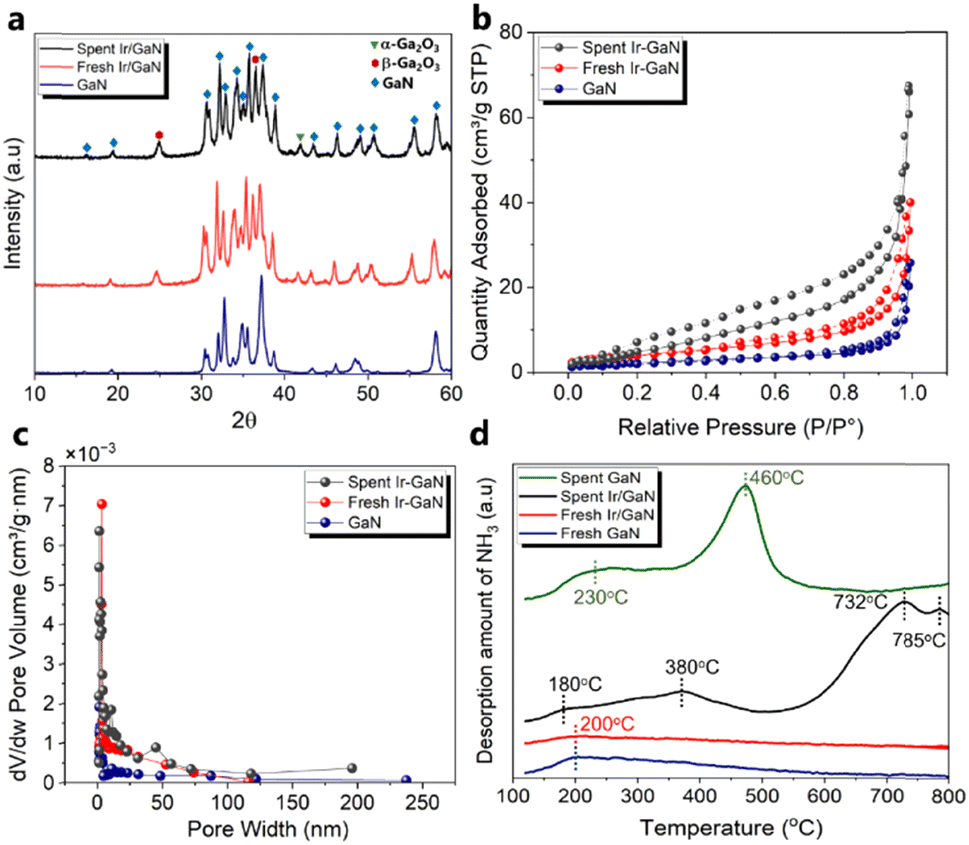 | ||
| Fig. 2 (a) XRD patterns of the GaN, fresh and spent Ir/GaN catalysts. (b) N2 adsorption isotherm profiles of the GaN, fresh and spent Ir/GaN catalysts. (c) Pore size distributions of the GaN, fresh and spent Ir/GaN samples. (d) NH3-TPD profiles of all fresh and spent samples. | ||
The results of the N2 adsorption and desorption experiment (Fig. 2b) provided confirmation of the existence of Ir with a small particle size. This was evident from the notable increase in the Brunauer–Emmett–Teller (BET) surface area of the fresh Ir/GaN sample compared to that of the GaN sample. The measured BET specific surface area of GaN was 8.0 m2 g−1 with an average corresponding pore size diameter derived from the Barrett–Joyner–Halenda (BJH) method of 19.0 nm and a pore volume of 0.031 cm3 g−1, as shown in Table S1 (ESI†). The large pore size provided shielding effects for partially dehydrogenated molecules diffused inside such pores25 to limit a deep dehydrogenation of oil molecule hydrocarbon gas. Incorporating 0.1 wt% of Ir into the GaN led to a rise in the BET surface area and pore volume to 14.6 m2 g−1 and 0.052 cm3 g−1, respectively, albeit with a slight decrease in the average pore size to 14.5 nm. These findings suggest that the Ir likely exists in the form of very small particles or nanoclusters, possessing a greater specific surface area than the GaN alone. Additionally, the presence of Ir nanoclusters partially obstructed the mesopores of the GaN, contributing to the observed reduction in pore diameters. Another highly possible reason is that the oxidation of GaN, likely facilitated by Ir, culminates in the production of Ga2O3, concurrently inducing the formation of defects and pores. This transformation is discernible through the outcomes of XRD and later TGA. Similar observation of the increment in the BET surface area of α-Fe2O3 with the addition of Pd nanocluster35 ascertains such data interpretation. The larger Type II hysteresis loop observed in the GaN sample confirms the presence of larger surface areas, allowing for the adsorption of molecular clusters around favorable sites on the GaN surface. The pore size distribution analysis of both GaN and fresh Ir/GaN samples reveals a varied pore size distribution, predominantly in the range of 5–20 nm (Fig. 2c). It is noteworthy that the surface area and pore volume of the Ir/GaN catalyst increased after the plasma reaction. Specifically, the BET surface area and pore volume of the spent Ir/GaN catalyst were increased to 23.1 m2 g−1 and 0.098 cm3 g−1, respectively. This finding aligns with our earlier assumption regarding the notable stability of the Ir/GaN catalyst, as indicated by the XRD patterns. Many reports on plasma catalysis have noted a decrease in the surface area and pore volume of catalysts after undergoing plasma reactions attributed to catalyst sintering. Our recent observations also indicate minimal sintering of Cu particles after plasma-driven gaseous phase reactions between N2 and CH4.19 The increment in the BET surface area of the spent Ir/GaN catalyst is possibly due to better dispersion of Ir atoms in the nanocluster form in the NTP environment, and in such smaller sizes, catalytic sintering is limited, and plasma discharge during the reaction possibly acted as a surface activation to increase the active surface area.36
The catalyst surface acidity plays a crucial role in facilitating dehydrogenation, deoxygenation reactions, and carbon formation. In this work, the surface acidity of the catalyst was assessed using the NH3-TPD technique, and the results are presented in Fig. 2d. The peak observed at 200 °C is attributed to NH3 absorption on the weak acid sites (<300 °C) of the GaN support, a consistency with the observations of other researchers.26 Significantly, the NH3-TPD profile of the fresh Ir/GaN catalyst exhibits minimal disparity, except for a slight reduction in the desorption peak area compared to that of the GaN. This reduction suggests a slightly lower amount of desorbed NH3, indicating marginally weaker acidity. This observation implies that Ir exists in the form of nanoclusters or single atoms, confined within the distinctive surface cavities of the GaN. This outcome further substantiates our previous hypothesis regarding the presence of Ir as either single atoms or nanoclusters, as corroborated by the absence of Ir-related XRD peaks in the dispersion of Ir as small particles. Catalysts with weak acidity tend to dissociate hydrogen protons, a trait favorable for dehydrogenation reactions, which leads to the creation of carbon cations serving as precursors for coke formation.37
Hence, catalysts displaying weak-to-moderate acidity, coupled with appropriate pore sizes facilitating the removal of coke precursors towards the reaction medium under plasma conditions, provide highly energetic electrons for enhancing the cleavage of C–H and C–O bonds. This combination represents a suitable equilibrium between upgrading performance and resistance to coke formation. Upon analyzing the spent Ir-GaN catalyst, four new peaks emerge at 180, 370, 732, and 785 °C, falling into three acidity regions: weak acidity (120–300 °C), moderate acidity (310–530 °C), and strong acidity (550–800 °C). These novel peaks suggest the formation of fresh acid sites on the catalyst's surface during the reaction.
These new acid sites appear to be more acidic than the weak sites on the fresh Ir/GaN and GaN catalysts but less acidic than the weak sites present on the surface of the initial catalyst. Motivated by this revelation, we conducted the NH3-TPD characterization of the spent GaN sample, uncovering a novel medium acid site arising from a peak at 460 °C. This newly formed acid site demonstrates greater acidity compared to both the weak and medium sites of the spent Ir/GaN and GaN catalysts. The efficient dispersion of Ir nanoclusters on GaN ensures the preservation of surface acidity without any modification. We firmly believe that alterations in the catalyst surface acidity are driven by surface restructuring, the redistribution of surface species, and intensified interaction with probe molecules. Plasma treatment has a well-established history as an effective technique for modifying catalyst surfaces. The synergistic interplay between the plasma and the catalyst can lead to the redistribution of surface species, resulting in an increased concentration of acidic sites. Additionally, alterations in surface electron density can influence the adsorption and desorption strength of NH3, thereby augmenting the observed acidity.
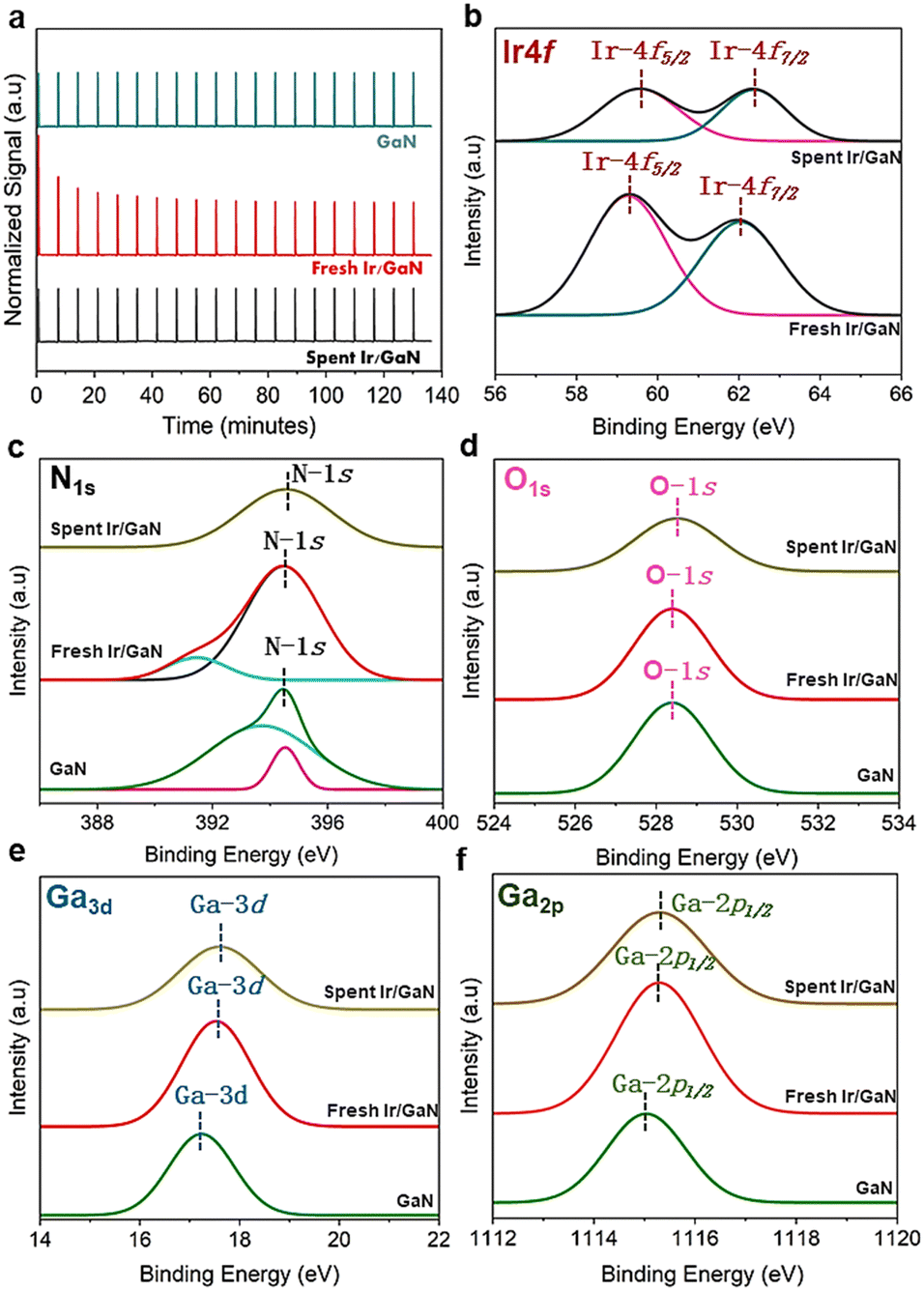 | ||
| Fig. 3 (a) CO pulse chemisorption profiles of the GaN, fresh and spent Ir/GaN catalysts. (b) High-resolution XPS spectra of the Ir4f and fresh and spent Ir/GaN catalysts. (c) XPS spectral surveys of N1s. (d) XPS spectral surveys of O1s. (e) XPS spectral surveys of Ga3d. (f) XPS spectral surveys of Ga2p. | ||
We are convinced that the reactions facilitated by plasma play a pivotal role in the additional decomposition or restructuring of GaN, leading to a significant increase in the surface area (as indicated by the BET surface area results). This process enhances the dispersion of Ir on the surface, and notably, Ir particles were not detectable. In addition, it has been indicated that particle size interpretations obtained through CO chemisorption typically tend to be larger compared to those acquired using XRD and TEM techniques.38 Even with their superior dispersion and small size, nanoclusters of this magnitude may escape the detection of Ir-related diffraction patterns by XRD. For a more comprehensive understanding of the surface electronic characteristics of the prepared catalyst, both before and after the plasma reaction, we conducted XPS analysis on all the catalyst samples. A detailed spectrum survey of the XPS data is shown in Fig. 3b. It is important to note that the XPS investigations were performed ex situ to evaluate potential surface oxidation. Specifically, we subjected the Ir-4f peak of the Ir/GaN to deconvolution using Gaussian components. As illustrated in Fig. 3b, the binding energy values of Ir-4f5/2 and Ir-4f7/2 for the fresh Ir/GaN catalyst (62.0 and 59.4 eV, respectively) exhibit negative shifts when compared to those of unsupported Ir nanoparticles, typically known to be at 63.8 and 61.8 eV.39 This shift indicates an electron transfer from GaN to Ir, which is a consequence of electron-mediated surface interactions involving Ir, Ga, and N. Although the electronegativity of Ir (2.20)40 is lower than that of N1s (3.04) but higher than Ga (1.81),41 the electron density in its proximity decreases. This reduced electron density induces an enhancement in binding energy, resulting in a negative shift in the peak position of the binding energy. This unequivocally illustrates the effective binding of Ir and GaN, establishing a robust interaction at the interfaces, which not only improves surface electrical properties but also facilitates the adsorption and catalytic upgrading of pyrolysis oil. A post-plasma reaction, a slight positive shift in comparison to the fresh Ir/GaN catalyst, was observed. This could stem from amplified electron donation, charge transfer, or alterations in the local electronic environment due to interactions between the plasma and the catalyst. Essentially, these collaborative interactions between the plasma and the catalyst are anticipated to enhance the performance of oil upgrading processes. The peak observed at 397.5 eV originates from the presence of N1s in GaN (Fig. 3c), and no other peaks corresponding to N–O interactions, such as the one at 400 eV, were identified. Furthermore, the electronic signal of O1s (Fig. 3d) detected in both the initial and used Ir/GaN catalysts confirms the presence of gallium oxides, which aligns with the XRD findings mentioned earlier. The incorporation of Ir into the GaN structure led to a positive shift in the binding energy of Ga3d (Fig. 3e) and Ga2p (Fig. 3f), thereby, reinforcing our earlier hypothesis regarding electron donation, specifically Ga, to Ir and N. Subsequent to the plasma oil upgrading reaction, we observed additional positive shifts in the binding energies of Ga2p, Ga3d, and O1s. These shifts could potentially arise from modifications in the local electronics given the plasma–catalyst interactions. It has been indicated that plasma can enhance the interaction between the catalyst and the support, resulting in better electron transfers.42
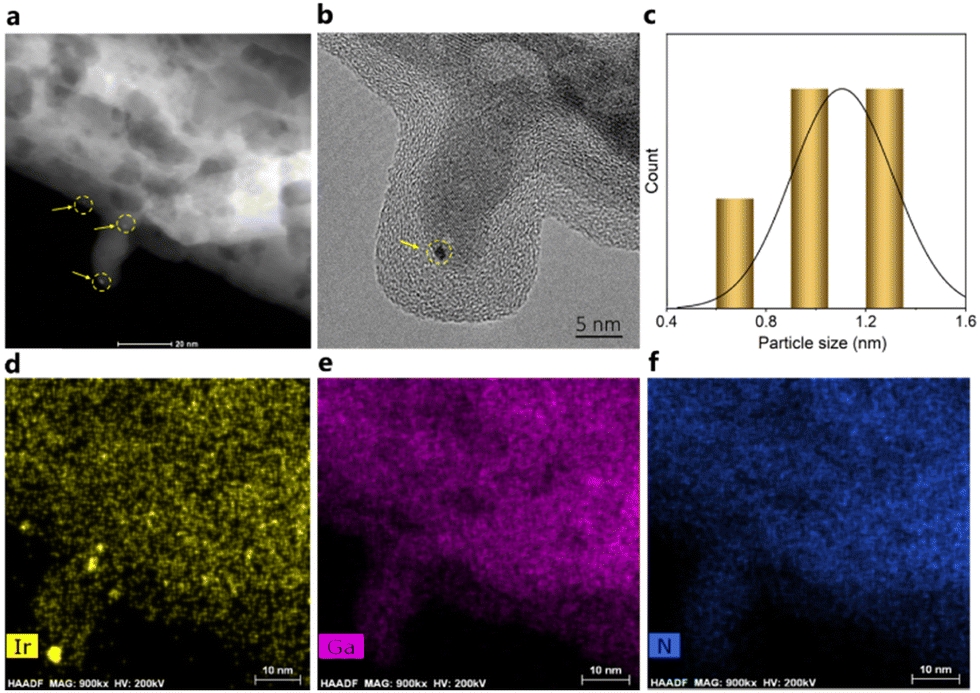 | ||
| Fig. 4 (a) STEM-HAADF image of the fresh Ir/GaN catalyst. (b) HR-TEM image of the fresh Ir/GaN catalyst. (c) Particle size distribution of the Ir clusters on the GaN surface. (d) EDX mapping of Ir. (e) EDX mapping of Ga. (f) EDX mapping of N. | ||
Furthermore, an investigation of the spent Ir/GaN catalyst by STEM-EDX (Fig. 5a) and HRTEM (Fig. 5b) unequivocally confirms the exceptional stability of the Ir/GaN catalyst. Notably, the clustered Ir atoms displays insignificant alteration during plasma reactions. These clusters persist, even exhibiting dimensions smaller than 1.5 nm in diameter, with certain regions measuring less than 1 nm (as indicated by the inset particle size distribution of Fig. 5a). Moreover, the Ir, Ga, and N elements maintain their good dispersion after the plasma reaction (Fig. 5c–f). At this juncture, these compelling observations lead us to hypothesize that Ir nanoclusters hold significant promise as catalysts for plasma-powered pyrolysis oil upgrading. The subsequent catalytic performance tests outlined in the forthcoming section are poised to provide a more comprehensive understanding of their activity, thereby substantiating this intriguing proposition.
 | ||
| Fig. 5 (a) STEM-HAADF image of the spent Ir/GaN catalyst with the inset particle size distribution of the Ir clusters on the GaN surface. (b) HR-TEM image of the spent Ir/GaN catalyst. (c) EDX mapping images of all element overlapping. (d) EDX mapping of Ir. (e) EDX mapping of Ga. (f) EDX mapping of N. | ||
3.2. Plasma catalysis driven pyrolysis oil upgrading performance
Inspired by the unique structural features of the Ir/GaN catalyst, we embark on an assessment of its intrinsic prowess in pyrolysis upgrading. This evaluation is conducted alongside the empty Tube, pristine GaN, and Ir nanoclusters loaded on GaN, all under a continuous liquid mode configuration. In this operational setup, the liquid feed oil is introduced into the reactor upper section, while CH4 is introduced into the plasma reaction zone from the reactor lower region. The liquid oil product is extracted from the upper-side portion of the reactor (Fig. S1, ESI†). Our primary objective in this context is to optimize mass transfer efficiency between the gaseous and liquid phases, thereby facilitating an enhanced upgrading performance. Control experiments were conducted to assess the influence of GaN and Ir/GaN catalysts on the pyrolysis upgrading process. The first set of control experiments involved plasma-powered CH4 conversion under two conditions: one without feed oil, referred to as “Empty 1,” and another with the presence of feed oil, referred to as “Empty 2” (Fig. 6a). The objective of these tests was to investigate the impact of feed oil on CH4 conversion and H2 formation. In the Empty 1 experiment, the CH4 conversion rate was approximately 9.2%, and the H2 yield reached around 77% at a flow rate of 100 cm3 min−1 and a power input of 12 W. Interestingly, the introduction of feed oil into the reaction mixture in the Empty 2 experiment maintained the CH4 conversion rate at around 9.1%. However, the H2 yield decreased to 3.1%, while the total C2–C5 gas yield slightly increased from 17.2% (CH4 only, Empty 1) to 2.1% (CH4 with feed oil, Empty 2). This suggests that the generated H2 was consumed in facilitating active H species for cleavage of pyrolysis oil molecules and stabilization of the formed cracking intermediates.43 In the absence of oil, the plasma-mediated conversion of CH4 resulted in a phenomenon known as methane liquefaction, leading to the formation of liquid products. However, the liquid yield from this reaction over the course of an hour remained relatively modest, measuring at 4.1%. Additionally, there was a coke yield of 1.7%. Notably, the predominant component in this process was gas, constituting 94.2% of the total gas products (H2 and C2–C5) generated. This finding aligns with a similar observation reported by our research group in the liquefaction of methane under plasma conditions.43 The presence of feed oil in the reaction with CH4 (Empty 2 experiment) diminished gas to 5.3% but increases the liquid yield to 91.2%. It is worth mentioning that the introduction of GaN or Ir/GaN had limited impact on CH4 conversion and upgraded the liquid oil yield, both remaining relatively stable at approximately 8.9% for CH4 conversion and 86.0 and 86.8% for oil yields, respectively. However, the H2 yield exhibited a decline to 4.8% for GaN and 4.5% for Ir/GaN. Additionally, a slight increase in the C2–C5 gas yield relative that of the Empty 2 test was observed from the upgrading reaction over the GaN and Ir/GaN catalyst, reaching approximately 3.4 and 3.2%, respectively. Therefore, it is reasonable to state that the liquid yield from pyrolysis oil upgrading in this study was not significantly affected by the liquid products of CH4 liquefaction.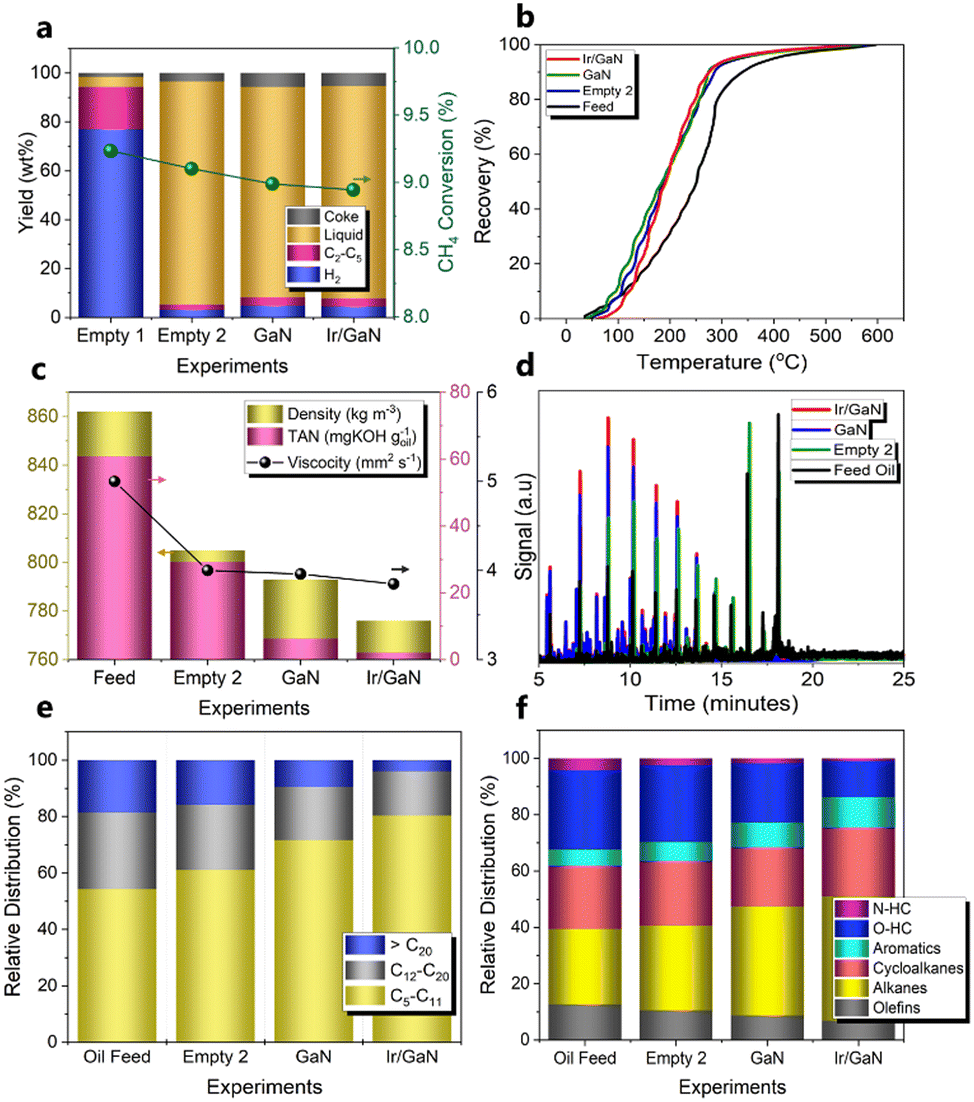 | ||
| Fig. 6 (a) CH4 conversion and product yields of experiments. (b) Boling curves of feed oil and upgraded oil produced from different plasma reactions. (c) Properties of feed oil and upgraded oil produced from different plasma reactions. (d) GC-MS spectra of the feed oil and upgraded oil produced from different plasma reactions. (e) Production distributions of feed oil and upgraded oil produced from different plasma reactions with gasoline components: C5–C11, diesel components: C12–C20, and heavy components: above C20. (f) Hydrocarbon distribution of feed oil and upgraded oil. [Reaction conditions: CH4 flowrate 100 cm3 min−1; feed oil flowrate: 0.05 cm3 min−1; catalyst weight: 0.1 g; plasma power: 12 W.] | ||
The significance of catalysts became evident during the characterization of the upgraded liquid oil. The primary objective of upgrading pyrolysis oil to jet fuels is two-fold: to facilitate refining through advanced refinery technology rivalling conventional processes and to enhance the yield of higher-value light distillates. From the boiling curves shown in Fig. 6b, a lightening effect is clearly visible. Approximately 10% of the distillates from both the feed and upgraded oil in all experiments underwent initial distillation at around 100–122 °C. Subsequently, a significant portion, about 90%, was distilled within the range of 205–300 °C for the upgraded oil with the empty tube test (305 °C), GaN (287 °C), and the Ir/GaN catalyst (275 °C), whereas the feed oil required a higher distillation temperature of 340 °C. This suggests that about 10% of the distillates could be attributed to water-soluble compounds such as oxygenated hydrocarbons due to their similar boiling points. In contrast, the remaining part of the distillates (distilled at 205–252 °C) may serve as potential fuels, as their distillation temperatures are comparable to those of jet fuels (205–300 °C) and diesel (<343 °C).44 The temperatures for 10% and 90% mass recovery of upgraded oil with the Ir/GaN in this work were 121 °C and 275 °C, respectively. While the maximum T10 temperature required for the aviation jet fuel is 205 °C, the real-time temperature for 90% mass recovery of the aviation fuel (T90) has not been reported.4 However, it is worth noting that the T90–T10 ≥ 40 °C requirement, as specified in ASTM D7566,45 has been met by the upgraded oil obtained in this research. Additionally, the feed oil had a high-boiling distribution with an average molecular weight (MW) of 210 g mol−1 (Table S2, ESI†). However, after upgrading, all the boiling curves shifted towards lower-boiling distributions, indicating that the upgraded oil contained lighter hydrocarbons with lower boiling points than the feed oil. The oil resulting from the plasma reaction with Ir/GaN had the lowest average molecular weight of 165.6 g mol−1, followed by GaN with an average molecular weight of 171.2 g mol−1, and the upgraded oil from the empty tube (Empty 2 experiment) with an average molecular weight of 181.2 g mol−1, in the increasing order. Such transformation of high-boiling compounds into low-boiling fractions caused a leftward shift in the boiling curve of the feed oil. This shift was brought about by the cracking of C–C bonds in the heavy components of feed oil.
In consistence with the reduction in the average molecular weight, other crucial properties including density, viscosity, and total acid number (TAN, derived from mg KOH consumed for titrating 1 g of oil) have also shown a reduction after treatment (Fig. 6c). For example, upgrading with the Empty Tube test reduced the density of the oil from 862 to 805 kg m−3 while with the presence of GaN and Ir/GaN, the densities were further diminished to 793 and 776 kg m−3, respectively. These results met the standard density requirement for jet A-1 fuel, which should be between 775 and 840 kg m−3 (measured by ASTM D4052 at 15 °C).46 The viscosity of the fuel significantly influences fuel injection, atomization, and combustion processes. Fig. 6c also presents the average viscosity values and their corresponding standard deviations for various distillates. Moreover, the viscosity also decreased from 5.00 mm2 s−1 (feed oil) to 4.00, 3.96, and 3.85 mm2 s−1 after upgrading with the empty tube (Empty 2 experiment), GaN, and Ir/GaN catalysts under plasma, respectively. While the viscosity of the oil upgraded through the plasma reaction over the Ir/GaN catalyst was still higher than the standard requirement for the jet fuel (ASTM D7042),47 this work demonstrated significant improvements compared to conventional processes, even when using only a single model compound (α-tetralone), which achieved a viscosity of 3.94 mm2 s−1 and a density of 927.8 kg m−3.48 This indicates the potential for further optimization for practical applications. Another critical parameter for the jet fuel is fuel acidity, with a maximum amount of 0.1 mg of KOH goil−1 (ASTM D3242).49 The pyrolysis oil, due to the presence of various oxygenated compounds, often exhibits significant acidity, which can lead to corrosion issues, necessitating the use of specialized materials in engines and fuel feed systems. However, after upgrading with the empty tube, GaN, and Ir/GaN catalysts under the plasma regime, the TAN of the feed oil significantly decreased from 60.8 mg of KOH goil−1 to 29.2, 6.3, and 2.1 mg of KOH goil−1, respectively. This nearly 30 times reduction in the acidity of the upgraded oil obtained over the plasma reaction with the Ir/GaN catalyst highlights its potential for further optimization. The insights gained from understanding the reaction mechanisms, discussed in the next section of this work, will be crucial for guiding future studies in this field.
Another crucial parameter of upgraded oils is their hydrocarbon product distribution characterized in detail to evaluate the upgrading efficiency and potential applications. First, Fig. S4 (ESI†) provides evidence for the alterations in the properties of the feed oil and the upgraded oil resulting from various experiments. The transition from the initial red-dark hue of the feed oil to a lighter yellow shade following the upgrading process with the empty tube, GaN, and Ir/GaN catalysts is clearly visible. Moreover, the GCMS signals of upgraded oils resulting from the plasma reaction using the empty tube, GaN, and Ir/GaN catalysts (Fig. 6d) exhibit a noticeable shift towards earlier retention times accompanied by a reduction in signal intensity under identical analysis conditions. This observation highlights substantial alterations in the composition of both the feed and upgraded oils. We thoughtfully integrated the GCMS results of the feed and upgraded oil, and we categorized the hydrocarbons present in both samples into three distinct groups: (1) gasoline components: C5–C11, (2) diesel components: C12–C20, and (3) heavy components: above C20. The C5 hydrocarbon listed here is cyclopentene as hexane (C5H12) evaporated strongly and quantified by micro-GC and listed in gas components, as shown in Fig. 6a. As presented in Fig. 6d, the feed oil contains 54.3% of C5–C11, 27.1% of C11–C20, and 18.6% of heavy component (>C20). In the absence of the catalyst (Empty 2 experiment), the plasma upgrading led to an increase in gasoline components up to 61.0%, and decreases in diesel and heavy components to 23.1% and 15.9%, respectively. Incorporating GaN and Ir/GaN in the plasma zone significantly increased the gasoline components to 71.7% and 80.4%, respectively, while reducing the diesel components to 18.8% and 15.7%, respectively. Notably, Ir/GaN exhibited the highest reduction in heavy components, with only 4% detected in the upgraded oil, compared to 9.5% in the plasma upgrading with GaN. This shift from heavier hydrocarbons to lighter components, such as gasoline, which falls within the high-density jet fuel range, is crucial in producing the aviation fuel with improved properties, including lower freezing points and higher energy density, thus meeting stringent aviation requirements. As shown in Fig. 6f, the most hydrocarbons of feed oil are oxygenated compounds (O-HC) with 28.2%, followed by alkanes with 26.8% and cycloalkanes with 22.5%. Olefins occupy 16.7% while aromatic and nitrogen containing hydrocarbons (N-HC) occupy 5.6 and 4.2%, respectively. After upgrading with plasma in the absence of catalysts (Empty 2 experiment), a visible reduction in olefins to 10.6%, oxygenated compounds to 27.3%, and nitrogen-containing hydrocarbons to 2.4% was observed. In contrast, plasma led to slight increases in alkanes to approximately 30%, cycloalkanes to 23%, and aromatic to 6.8%. Commercial aviation fuels, for example Jet A-1 and Jet A, are widely used for international civil airline operations and within the US, respectively, making them the most prevalent aviation turbine fuels.50 The composition of these jet fuels typically includes approximately 60% alkanes (comprising 20% alkane and 40% iso-alkane), 20% cycloalkanes, and 20% aromatics in terms of volume fraction.51 The relative proportions of these major components may vary between other commercial jet fuels; nonetheless, alkanes, cyclohexane, and aromatic plays an important role for jet engines.52 Aromatics have inferior combustion characteristics compared to alkanes, leading to elevated particulate emissions and lower specific energy. For jet engines, it is essential to have a certain number of aromatics to ensure appropriate swelling of elastomeric seals in the fuel systems, preventing leakage that could pose a fire safety hazard. Meanwhile, alkanes, particularly iso-alkanes, exhibit high specific energy, good thermal stability, and low freezing points.53 Cycloalkanes complement iso-alkanes by offering similar benefits to aromatics, enabling fuels to meet density requirements and potentially providing seal-swelling capacity similar to that of aromatics.54 Together, iso-alkanes and cycloalkanes have the potential to enhance fuels by providing high specific energy, increased energy density, and reduced emission characteristics. Due to their reactivity given by unsaturated bonds, olefins can lead to deposits in jet engines, making them unsuitable components for jet fuels. As a result, olefin content in jet fuel formulations is below 1%. The addition of GaN into the plasma regime boosts the content of alkanes, cycloalkanes, and aromatics to 38.6, 21.0, and 8.8%, respectively, while diminished olefins, oxygenated, and nitrogen-containing compounds to 8.8, 21.0 and 1.8%. Noticeably, plasma driven pyrolysis oil upgrading with the Ir/GaN lowers the nitrogen-containing, oxygenated, and olefin compounds to below 1.0, 12.7, and 6.8% while alkanes, cycloalkanes, and aromatics were increased to 44.2, 24.5, and 10.8%.
The distribution of these products closely resembles that of commercial jet fuels. It is important to highlight that conventional aviation fuels have only trace levels of oxygen contents;45 however, the utilization of oxygenated biofuels in aviation remains a subject of scientific debate. Certain oxygenated compounds, such as antioxidants and stabilizers, enhance the fuel's stability, preventing degradation and reducing the formation of gums and deposits during storage, while also increasing its energy content. Additionally, certain oxygenates, like alkyl ethers, can boost the octane rating of the fuel, benefiting aviation engines with higher compression ratios and improving engine performance, while also contributing to reduced emissions of pollutants and potentially modifying the fuel's viscosity for better flow properties and handling characteristics.55 This study also explored the impact of varying flowrates (0.05, 0.1, 0.15, and 0.2 cm3 min−1) on upgrading performance. Interestingly, an increase in the flow rate did not significantly alter the product yield, with the liquid yield improving from around 87% to 92%, coke yield reducing from 5.5% to 4.0%, and gas yield diminishing from 7.7% to 3.6% as the oil feed flow rate increased from 0.05 to 0.2% (Fig. S5, ESI†). Notably, the influence of the feed flow rate on upgrading performance became more apparent through the product distribution (Fig. S6, ESI†). Higher feed flow rates led to lower upgrading performance, evident, for instance, at 0.2 cm3 min−1, where N-HC reduced only from 4.2% (feed) to 3.0%, and O-HC declined from 28.2% (feed) to 26.2%. We attribute this trend to the suitability of the reactor size for lab-scale operations, suggesting that the utilization of high feed flow rates in this configuration is not optimal. While our primary focus is to provide novel insights into the activity of the Ir/GaN catalyst for plasma-driven biomass-derived pyrolysis oil to jet fuels, this research sets the stage for future work, including optimizing operational parameters, scaling-up studies, and conducting comprehensive techno-economic assessments.
3.3. Stability test
This work performed a 24 h stability test for plasma-driven pyrolysis oil upgrading using the Ir/GaN catalyst, with 8 h cycles of plasma on/off during each continuous reaction, mimicking the operational shifts of a practical industrial plant. Notably excelling in pyrolysis oil upgrading, the Ir/GaN catalyst demonstrates exceptional stability, maintaining nearly-consistent liquid product yields, CH4 conversion, and properties even after 24 h of continuous operation (Fig. 7a). The distribution of products within the upgraded oil, collected at the 1 h, 8 h, 16 h, and 24 h marks of the plasma reaction, exhibits slight changes, as shown in Fig. 7b. While other distributions exhibit insignificant alterations, the most notable change is approximately 7.5% reduction in the distribution of oxygenated hydrocarbons (O-HC) from 12.7% at 1 h to 20.2% at 24 h. Nevertheless, this highlights comparative stability compared to thermal catalytic processes for hydrocarbon upgrading, typically reported within durations below 12 h (Table S3, ESI†). These findings affirm the outstanding stability of the catalyst, indicating that catalyst regeneration is presently unnecessary, considering the tradeoff between the intricacy of regeneration steps and the incremental improvement in catalytic performance. However, future research endeavors will involve extended stability tests over months or years with a larger scale to delve deeper into the potential for piloting the scaling up of this biomass-derived oil upgrading to the jet fuel process, building on the novel insights gained in catalyst development and performance from this study. A slight increment is observed in olefins, oxygenates, and nitrogen-containing products, reaching 10.9%, 16.0%, and 1.1%, respectively. This shift is accompanied by a reduction in alkanes, cycloalkanes, and aromatics, which decrease to 42.0%, 22.0%, and 8.0%, respectively. The percentage of alkenes in our study still does not meet the current standards of commercial jet fuels. However, this pioneering work has unveiled novel avenues for future directions and offers a basis for further optimization efforts. | ||
| Fig. 7 (a) Stability test of the plasma driven pyrolysis oil with the nanocluster Ir/GaN catalyst over 24 h of a continuous reaction. (b) Hydrocarbon distribution of the upgraded oils at 1 h, 8 h, 16 h, and 24 h. [Reaction conditions: CH4 flow rate 100 cm3 min−1; feed oil flow rate: 0.05 cm3 min−1; catalyst weight: 0.1 g; plasma power: 12 W]. | ||
The TGA of the GaN and Ir/GaN catalysts spent over 8 h of the plasma reaction is shown in Fig. S7 (ESI†). As evident from the TGA profiles, the reduction in weight for the Ir/GaN catalyst is initiated within the temperature range of 300–350 °C and concludes at 550–600 °C with 13.5% followed by a gradual re-increase. Simultaneously, the TGA profile of the spent GaN sample exhibits a decline in mass starting around 100 °C and continuing until 500 °C with 14.2%, subsequently showing a lower degree of weight gain compared to that of the Ir/GaN catalyst. This reduction in weight within this specific temperature range could be attributed to the combustion of high molecular weight compounds originating from bio-oils, resulting from processes such as condensation and re-polymerization during the upgrading process, which become adsorbed on the catalyst structure and contribute to the formation of deposited coke. In the context of this study, the overall weight loss within this range is collectively referred to as deposited coke for the sake of simplicity. The subsequent increase in mass observed for the spent GaN and Ir/GaN catalysts could be attributed to the oxidation of Ga or Ir, leading to the formation of their respective oxides and resulting in a weight gain. In contrast to a thermal catalytic route for pyrolysis oil upgrading, which demonstrated a carbon deposition rate of 53.3% (using optimal CoMo supported on SBA-15 silica as an example),56 the utilization of Ir/GaN in the plasma upgrading process exhibits a significantly lower rate of carbon deposition. The unique conditions created by non-thermal plasma, such as high-energy electrons and reactive species under near-ambient conditions, facilitate more efficient cracking and conversion of feedstock molecules, reducing the tendency for coke formation. This phenomenon is attributed to the ability of plasma to activate and break down complex hydrocarbons, preventing their accumulation on the catalyst surface, particularly on the nanocluster surface.
3.4. Unravelling the reaction pathways of plasma-induced methane activation and oil molecule upgrading
Compared to other catalytic upgrading methods of pyrolysis oil to jet fuels using batch-mode thermal catalytic processes (with copper-based catalysts) at 300 °C, which yield 10% cycloalkanes and oxygenated compounds higher than 14.2%,57 the triphase plasma catalytic route employed in this study exhibits significantly better performance. Importantly, we have also observed promising results including better acidity, density, and viscosity properties, which warrant further investigation and exploration for large-scale applications. Despite this, the successful implementation of this technology for commercial purposes necessitates additional optimization and cannot be achieved without a comprehensive understanding of the underlying reaction mechanisms at a molecular level. We first characterized yielded plasma species fascinating insights into the plasma-chemistry of the investigated mixture. Fig. 8a displays the recorded in situ OES spectra of the upgrading mixture (CH4 + oil) and N2. In this work, the OES of N2 plasma was recorded to subtract strong N2 bands detected during the pyrolysis oil upgrading reaction from N-containing products. The in situ OES spectra of the upgrading reaction reveal prominent bands associated with the excited CH, C2, Hγ, Hβ, H2, and CN species, which differentiate from the N2 OES bands showing only nitrogen plasma species peaks. The CH band peaking at 430 nm can be attributed to A2Δ → X2Π. The peak at 422 nm of CN species was assigned to the B2Σ → X2Σ energy level transitions. The most intense peak of the OES profile at 434 nm belongs to the 5d2D → 2p2p° of Hγ while Hβ with 4d2D → 2p2p° energy level transitions was detected at 488 and 831 nm. The H2 bands at 441 and 456 nm attribute to the G1Σ+g → G1Σ+u. The C2 band captured at 473 nm belongs to d3Π8 → a3Πu. These energy level transitions are consistent with those in other research studies.58–60 Oxygen-related bands were not detected due to factors such as the weaker intensity of its emission lines relative to strong N2, H2, CH, and CN species in the oil and gas system and potential interferences from these compounds and radicals. The strong signal of H radicals during the plasma reactions initially took us falling into two scenarios: (i) H from CH4 dissociation and (ii) cleavage from oil molecules. Nonetheless, control experiments, as depicted in Fig. 6a above, show insignificant impact of oil on hydrogen formation and CH4 conversion. We hence conclude that the dominant presence of active H species in the plasma reactor originate from CH4 as a primary source and intermediates from dehydrogenation of pyrolysis oil molecules as a second source. In this manner, the counter flow of CH4 and oil facilitates the rapid transfer of H from gas–liquid interface to the fluidized GaN and Ir/GaN surface, contributing to catalytic oil upgrading reactions. This explains for the better upgrading performance from catalytic tests relative to the Empty experiments. In this work, the IR thermal camera indicated the temperature of the plasma reactor was around 100 ± 5 °C (Fig. 8b). As we mentioned earlier, the control experiment, conducted with an external thermal heater set at 100 ± 5 °C, revealed no CH4 conversions or improvements in oil properties, demonstrating the effective mitigation of conventional thermal factors' influence on the upgrading performance. The “thermal effects” of plasma, on the other hand, have long been overshadowed, given the characterization of plasma as non-thermal, with seemingly unchanged outlet gas and liquid temperatures. However, these thermal influences predominantly arise from interactions among the constituents of the gaseous, liquid, and catalyst medium within the reactor. The voltage–current profile depicting the gas (CH4)–liquid (oil)–solid (catalyst) system exhibits micro-pulsed plasma discharges, highlighting intricate interactions between the plasma and the reaction medium within the reactor (Fig. S8, ESI†). This results in a conversion of a fraction of the discharge power into thermal energy, thereby raising the catalyst and surrounding temperatures. These interactions involve inelastic collisions between high-energy electrons and substantial particles, leading to the release of energy by the formation of excited molecules and ionized atoms. Additionally, collisions between ions, neutral particles, and free electrons contribute further to the energy release. The presence of thermal effects cannot be disregarded, especially at the catalyst surface, where nanoclusters play a crucial role in promoting the upgrading reaction. We posit that while these thermal effects may not exhibit characteristics akin to traditional thermal catalytic processes due to their relatively moderate nature,61 they do play a significant role. Specifically, these effects contribute to promoting the desorption rate of adsorbed species on the catalyst surface. This, in turn, helps mitigate surface poisoning, ultimately leading to an enhanced upgrading performance. In addition to considering hydrogen (H), another crucial factor to take into account is the presence of active CHx species. These species play a significant role in lowering the activation energy required for the cleavage of C–H, C–N, and C–O bonds within the non-aqueous plasma environment. The sequential dehydrogenation reaction of CH4 initiates with the detachment of a single hydrogen atom from the CH4 molecule, progressing through transitional species to ultimately form C (CH4 → CH3 → CH2 → CH → C), with hydrogen being released at each step. In the final dehydrogenation stage, the most stable form of carbon (C) is produced, predominantly in the coke form, which does not significantly contribute to the cleavage of pyrolysis molecules. In contrast, our upgrading performance results, demonstrating a reduction in the molecular weight of upgraded oil compared to the feed oil, suggest that CH3 and CH2 are less likely to undergo oligomerization with other hydrocarbons in the oil, leading to the formation of heavier products. Instead, CHx species, along with H, are expected to play a dominant role in reacting with oil molecules. We propose the adsorption of CHx and H, originating from CH4 dissociation on the Ir(100) surface, as shown in Fig. 8c. Our DFT calculations (Fig. 8d and Table S4, ESI†) reveal that the adsorption energy of CH4 on GaN is −0.23 eV and that on Ir is −0.06 eV. Under plasma conditions, the adsorption energies of transitional species, including CH3, CH2, CH, and C, on the GaN(110) surface, with the proposed adsorption geometry depicted in Fig. S9 (ESI†), are −0.41, −0.5, −0.54, and 0.64 (Table S4, ESI†), respectively. These corresponding values obtained from the Ir(100) surface were −0.24, −0.3, −0.36, and −0.42 eV. Plasma augmentation enhances surface properties, promoting stronger molecule adsorption on the catalyst surface. Referring to the aforementioned XPS results, we observe a positive energy binding shift in the Ir/GaN comparison following the plasma reaction, indicating higher electron density in the surroundings under plasma conditions, leading to stronger molecule binding. When considering the GaN and Ir/GaN surfaces, the GaN surface exhibits stronger binding with CH4 and active species such as CH and H. Consequently, it displays higher dehydrogenation barriers, where site poisoning becomes the limiting factor rather than CH4 activation, resulting in lesser rate enhancements. Conversely, the Ir/GaN catalyst exhibits more substantial rate enhancements due to its weaker binding of CH4 and active species, enabling Ir to be the active surface that bind species weakly for catalytic reactions. This correlation between adsorption energy and reaction performance is consistent with findings in plasma-powered ammonia production.62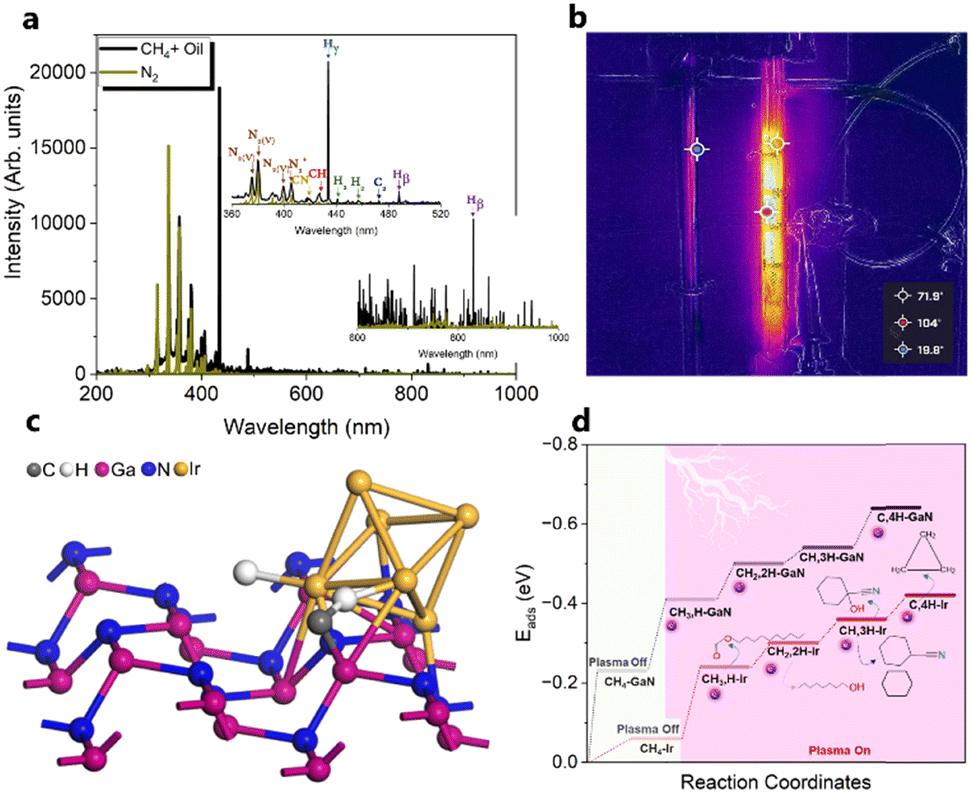 | ||
| Fig. 8 (a) In situ OES spectra acquired during the plasma driven pyrolysis oil upgrading reaction with CH4 over the Ir/GaN catalyst and N2 spectra for subtracting the detected strong N2 bands. (b) Temperature of the plasma reaction measured using an IR thermal camera. (c) Possible structure and adsorption of CH and H species on the Ir(110) nanoclusters supported on GaN. (d) Adsorption energy diagram and proposed reaction pathway of plasma driven pyrolysis oil upgrading with CH4 using the Ir/GaN catalyst. | ||
The exploration of the potential for H* and 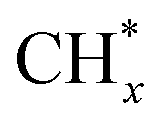 production from CH4 in the plasma was conducted to generate the necessary active species for the hydrodeoxygenation and cleavage of unsaturated hydrocarbons reactions. These reactions specifically target oxygen-containing compounds and olefins present in the feed chemical structure. Within the active volume of the plasma discharge reactor, a multitude of energetic species, including electrons, highly reactive species H* and CHx*, and metastable forms, engage in collisions with feed oil molecules. These collisions induce excitation or the generation of products through energy transfer. Consequently, compared to conventional thermal reactors, the mechanism of reactions in a plasma reactor is more intricate. Proposing a singular mechanism to explain the reaction pathways in the plasma reactor is challenging, even several studies63–65 used simpler model compounds to verify it. Nonetheless, integrating thermodynamic species databases with plasma discharge mechanisms allows us to suggest the primary reaction steps. Whereby, taking an oxygen contained-compound of CxHyOCH3 in the feed for an example, the excitation of this molecule to higher energy levels occurs due to collisions with high-energy electrons or temporarily excited collision partners such as H* and CHx*. The chemical bonds of CxHyOCH3 can be dissociated through collisions with high-energy electrons or temporarily excited collision partners such as H* and CHx*, and highly energetic electrons, resulting in decomposition of CxHyOCH3 to CxHyOH and then CxHy. This possible pathway coincides with other research studies.66
production from CH4 in the plasma was conducted to generate the necessary active species for the hydrodeoxygenation and cleavage of unsaturated hydrocarbons reactions. These reactions specifically target oxygen-containing compounds and olefins present in the feed chemical structure. Within the active volume of the plasma discharge reactor, a multitude of energetic species, including electrons, highly reactive species H* and CHx*, and metastable forms, engage in collisions with feed oil molecules. These collisions induce excitation or the generation of products through energy transfer. Consequently, compared to conventional thermal reactors, the mechanism of reactions in a plasma reactor is more intricate. Proposing a singular mechanism to explain the reaction pathways in the plasma reactor is challenging, even several studies63–65 used simpler model compounds to verify it. Nonetheless, integrating thermodynamic species databases with plasma discharge mechanisms allows us to suggest the primary reaction steps. Whereby, taking an oxygen contained-compound of CxHyOCH3 in the feed for an example, the excitation of this molecule to higher energy levels occurs due to collisions with high-energy electrons or temporarily excited collision partners such as H* and CHx*. The chemical bonds of CxHyOCH3 can be dissociated through collisions with high-energy electrons or temporarily excited collision partners such as H* and CHx*, and highly energetic electrons, resulting in decomposition of CxHyOCH3 to CxHyOH and then CxHy. This possible pathway coincides with other research studies.66
The comprehensive Techno-Economic Assessment (TEA) of NTP-driven pyrolysis oil to jet fuels falls outside the scope of the current work. Instead, our focus is on reporting the catalytic performance of nanoclustered Ir supported on GaN in a triphase plasma system. This marks the first instance of such an investigation, emphasizing the novelty of our research in this specific catalytic context. However, we performed a thoughtful comparison with other works in the literature, which upgraded the model compound of pyrolysis oil to jet fuels by thermal catalytic processes and also with other plasma process (Table S3, ESI†). The NTP process featured from the thermal catalytic one by its atmospheric pressure while thermal ones were operated at high pressures (8–120 bars) and much lower temperatures while thermal one run with higher than 350 °C. In our prior research,67 we conducted a thorough TEA that focused on evaluating the cost-effectiveness of renewable diesel production via CH4-assisted waste cooking oil. This was proposed as an alternative to the traditional pure H2 hydrotreating methods. Our findings indicate that utilizing CH4 in the hydrotreating process is significantly more cost-effective than using H2. Specifically, the production cost of the CH4-assisted catalytic process was determined to be $0.365 per kg of renewable diesel. In comparison, the cost of H2 was $0.574 per kg for renewable diesel and biodiesel. Building on these findings and considering similar observations in plasma-assisted heavy oil upgrading with CH4,23,68–70 we are confident in asserting that the economic benefits of plasma-driven CH4 treating pyrolysis oil are substantial for future jet fuel production. This confidence is based on the elimination of an additional step involving pure H2—a process often derived from energy-intensive and highly endothermic natural gas reforming.71 Consequently, we anticipate that plasma-driven pyrolysis oil with CH4 will emerge as a promising and sustainable approach for the production of premium fuels and chemicals even from heavy oils.
4. Conclusions
This work showcases a highly efficient H2-free process for pyrolysis oil upgrading using a triphase plasma catalytic system. Our experimental findings highlight impressive upgraded oil yields, approximately 87%, with the density and viscosity meeting the ASTM standards. Notably, the product distribution, including alkanes, cycloalkanes, and aromatics, aligns closely with requirements for commercial jet fuels. These remarkable results were achieved under mild operating conditions, with temperatures hovering around 100 °C and at ambient pressure. By harnessing the potent capabilities of cleavage reactions for long C–H bonds of plasma, we facilitated the effective transfer of active H and CHx species derived from CH4 to the oil liquid, primarily driven by highly energetic electrons. Our computational analysis supports the notion that nanoclustered Ir, with its weaker binding to CH4 and active species, coupled with the thermal effects on the Ir cluster surface, accelerates the desorption of adsorbed species. This dynamic interplay promotes rapid upgrading reactions, significantly lowering the barriers for cleaving C–H, C–N, and C–O bonds. As a result, pyrolysis oil can be efficiently upgraded to gasoline, all while operating under mild conditions. The implications of our findings lay the groundwork for scaling up and optimization, not only within central oil and gas plants but also in fostering regional economic growth. This work has the potential to stimulate the emergence of new jet fuel production technology, spanning from biomass feedstock cultivation and pyrolysis technology development to fuel production. The positive socio-economic effects are particularly promising for rural areas rich in biomass resources. By taking this leap forward, this work contributes to the more conscientious and energy-efficient future, signifying a significant stride toward the aviation industry's aspirations for a greener and more responsible path.Author contributions
Hoang M. Nguyen: methodology, investigation, formal analysis, writing – original draft, and data curation. Ali Omidkar: simulation. Wenping Li: visualization. Zhaofei Li: formal analysis and data curation. Hua Song: conceptualization, supervision, validation, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The TEM-EDX analysis in this work used the 4D LABS core facility at Simon Fraser University (SFU) supported by the Canada Foundation for Innovation (CFI), the British Columbia Knowledge Development Fund (BCKDF), and the Pacific Economic Development Canada (PacifiCan).References
-
L. Zhang, T. L. Butler and B. Yang, Recent trends, opportunities and challenges of sustainable aviation fuel, John Wiley & Sons, Inc., 2020 Search PubMed
.
- S. Arrowsmith, Directive Article, 2020, 30, 1–184 Search PubMed
- R. Sacchi, V. Becattini, P. Gabrielli, B. Cox, A. Dirnaichner, C. Bauer and M. Mazzotti, Nat. Commun., 2023, 14, 3989 CrossRef CAS PubMed
- P. Lahijani, M. Mohammadi, A. R. Mohamed, F. Ismail, K. T. Lee and G. Amini, Energy Convers. Manage., 2022, 268, 115956 CrossRef CAS
- R.-X. Chen and W.-C. Wang, Renewable Energy, 2019, 135, 819–835 CrossRef CAS
- W.-T. Chen, Y. Zhang, T. H. Lee, Z. Wu, B. Si, C.-F. F. Lee, A. Lin and B. K. Sharma, Nat. Sustainability, 2018, 1, 702–710 CrossRef
- X. Cui, X. Zhao and D. Liu, Green Chem., 2018, 20, 2018–2026 RSC
- C. J. Wrasman, A. N. Wilson, O. D. Mante, K. Iisa, A. Dutta, M. S. Talmadge, D. C. Dayton, S. Uppili, M. J. Watson, X. Xu, M. B. Griffin, C. Mukarakate, J. A. Schaidle and M. R. Nimlos, Nat. Catal., 2023, 6, 563–573 CrossRef
- W. Liu, W. You, W. Sun, W. Yang, A. Korde, Y. Gong and Y. Deng, Nat. Energy, 2020, 5, 759–767 CrossRef CAS
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS
- J. S. Wright and K. U. Ingold, J. Chem. Educ., 2000, 77, 1062 CrossRef
- A. H. Zacher, D. C. Elliott, M. V. Olarte, H. Wang, S. B. Jones and P. A. Meyer, Biomass Bioenergy, 2019, 125, 151–168 CrossRef CAS
- N. O. Burov, V. D. Savelenko, M. A. Ershov, A. O. Vikhritskaya, E. O. Tikhomirova, N. A. Klimov, V. M. Kapustin, E. A. Chernysheva, A. V. Sereda and T. M. Abdellatief, Renewable Energy, 2023, 215, 118898 CrossRef CAS
- A. Eschenbacher, T. Myrstad, N. Bech, H. D. Thi, M. Auersvald, K. M. Van Geem and A. D. Jensen, Fuel, 2021, 302, 121198 CrossRef CAS
- T. S. Ahamed, S. Anto, T. Mathimani, K. Brindhadevi and A. Pugazhendhi, Fuel, 2021, 287, 119329 CrossRef
- H. M. Nguyen, A. Omidkar and H. Song, ChemPlusChem, 2023, 88, e202300129 CrossRef PubMed
- H. M. Nguyen, C. M. Phan, G. H. Pham, Y. Asakuma, R. Vagnoni and S. Liu, J. Ind. Eng. Chem., 2021, 94, 173–179 CrossRef CAS
- H. M. Nguyen, F. Gorky, S. Guthrie and M. L. Carreon, Catal. Today, 2023, 418, 114141 CrossRef CAS
- H. M. Nguyen, A. Omidkar, W. Li, S. Meng, Z. Li and H. Song, Chem. Eng. J., 2023, 471, 144748 CrossRef CAS
- H. M. Nguyen and M. L. Carreon, ACS Sustainable Chem. Eng., 2022, 10, 9480–9491 CrossRef CAS
- D. Mei, X. Zhu, C. Wu, B. Ashford, P. T. Williams and X. Tu, Appl. Catal., B, 2016, 182, 525–532 CrossRef CAS
- Y. Zheng, Q. Ruan, J. Ren, X. Guo, Y. Zhou, B. Zhou, Q. Xu, Q. Fu, S. Wang and Y. Huang, Appl. Catal., B, 2023, 323, 122170 CrossRef CAS
- S. Meng, W. Li, H. Xu, Z. Li, Y. Li, J. Jarvis and H. Song, Appl. Catal., B, 2021, 297, 120459 CrossRef CAS
- S. Meng, A. Wang, P. He and H. Song, J. Phys. Chem. Lett., 2020, 11, 3877–3881 CrossRef CAS PubMed
- Y. Wang, W. Yang, S. Xu, S. Zhao, G. Chen, A. Weidenkaff, C. Hardacre, X. Fan, J. Huang and X. Tu, J. Am. Chem. Soc., 2022, 144, 12020–12031 CrossRef CAS PubMed
- C. Liu, J. Kang, Z.-Q. Huang, Y.-H. Song, Y.-S. Xiao, J. Song, J.-X. He, C.-R. Chang, H.-Q. Ge, Y. Wang, Z.-T. Liu and Z.-W. Liu, Nat. Commun., 2021, 12, 2305 CrossRef CAS PubMed
- M. G. Kibria, F. A. Chowdhury, S. Zhao, B. AlOtaibi, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 6797 CrossRef CAS PubMed
- K. Trangwachirachai, A. L. Huang, H. K. Chen, C. L. Chen, J. F. Lee, H. K. Tian and Y. C. Lin, Mater. Today Chem., 2023, 30, 101500 CrossRef CAS
- Z. Lu, V. Cherepakhin, T. Kapenstein and T. J. Williams, ACS Sustainable Chem. Eng., 2018, 6, 5749–5753 CrossRef CAS PubMed
- M. Tang, S. Mao, X. Li, C. Chen, M. Li and Y. Wang, Green Chem., 2017, 19, 1766–1774 RSC
- E.-J. Kim, J. Shin, J. Bak, S. J. Lee, K. H. Kim, D. Song, J. Roh, Y. Lee, H. Kim, K.-S. Lee and E. Cho, Appl. Catal., B, 2021, 280, 119433 CrossRef CAS
- Y.-X. Dai, W.-L. Xin, L.-H. Xu, J. Li, Y.-X. Li, J. Li, S. Cosnier, X.-J. Zhang, R. S. Marks and D. Shan, Appl. Surf. Sci., 2022, 591, 153066 CrossRef CAS
- T. Shao, P. Zhang, L. Jin and Z. Li, Appl. Catal., B, 2013, 142–143, 654–661 CrossRef CAS
- Q. Wang, C.-Q. Xu, W. Liu, S.-F. Hung, H. Bin Yang, J. Gao, W. Cai, H. M. Chen, J. Li and B. Liu, Nat. Commun., 2020, 11, 4246 CrossRef CAS PubMed
- M. Farrag, Microporous Mesoporous Mater., 2018, 257, 110–117 CrossRef CAS
- E. C. Neyts, K. Ostrikov, M. K. Sunkara and A. Bogaerts, Chem. Rev., 2015, 115, 13408–13446 CrossRef CAS PubMed
- H. Xu, Z. Li, Y. Li and H. Song, Commun. Chem., 2021, 4, 152 CrossRef CAS PubMed
- C. Dong, C. Lian, S. Hu, Z. Deng, J. Gong, M. Li, H. Liu, M. Xing and J. Zhang, Nat. Commun., 2018, 9, 1252 CrossRef PubMed
- V. Pfeifer, T. Jones, J. V. Vélez, C. Massué, M. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer and J. Allan, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC
- M. Kim, H. Kang, E. Hwang, Y. Park, W. Jeong, Y. J. Hwang and D.-H. Ha, Appl. Surf. Sci., 2023, 612, 155862 CrossRef CAS
- J. C. Boeyens, Z. Naturforsch., B: J. Chem. Sci., 2008, 63, 199–209 CrossRef CAS
- Z. Ye, L. Zhao, A. Nikiforov, J.-M. Giraudon, Y. Chen, J. Wang and X. Tu, Adv. Colloid Interface Sci., 2022, 308, 102755 CrossRef CAS PubMed
- S. Meng, W. Li, Z. Li and H. Song, Catal. Sci. Technol., 2023, 13, 4665–4672 RSC
-
A. ASTM, Annual Book of ASTM Standards, 2015 Search PubMed
-
A. Standard, ASTM International, West Conshohocken, PA, 2013 Search PubMed
-
A. Standard, 2011 ASTM Annual Book of Standards, 2018 Search PubMed
-
D. ASTM, ASTM International, West Conshohocken, PA, USA, 2016 Search PubMed
- G. Vijayakumar and A. Pandurangan, Energy, 2017, 140, 1158–1172 CrossRef CAS
- E. S. K. Why, H. C. Ong, H. V. Lee, W.-H. Chen, N. Asikin-Mijan and M. Varman, Energy Convers. Manage., 2021, 243, 114311 CrossRef CAS
- H. Zhang, Y. Fang, M. Wang, L. Appels and Y. Deng, J. Environ. Manage., 2020, 274, 111214 CrossRef CAS PubMed
- Z.-H. Jin, J.-T. Chen, S.-B. Song, D.-X. Tian, J.-Z. Yang and Z.-Y. Tian, Combust. Flame, 2021, 226, 190–199 CrossRef CAS
- Z. Yang, K. Qian, X. Zhang, H. Lei, C. Xin, Y. Zhang, M. Qian and E. Villota, Energy, 2018, 154, 289–297 CrossRef CAS
- R. C. Striebich, L. M. Shafer, R. K. Adams, Z. J. West, M. J. DeWitt and S. Zabarnick, Energy Fuels, 2014, 28, 5696–5706 CrossRef CAS
- P. Kallio, A. Pásztor, M. K. Akhtar and P. R. Jones, Curr. Opin. Biotechnol, 2014, 26, 50–55 CrossRef CAS PubMed
- M. D. Boot, M. Tian, E. J. Hensen and S. M. Sarathy, Prog. Energy Combust. Sci., 2017, 60, 1–25 CrossRef
- S. Ahmadi, Z. Yuan, S. Rohani and C. Xu, Catal. Today, 2016, 269, 182–194 CrossRef CAS
- X. Kong, C. Liu, Y. Fan, M. Li and R. Xiao, ACS Sustainable Chem. Eng., 2023, 11, 7454–7465 CrossRef CAS
- Z. Fan, H. Sun, S. Zhang, W. Han, C. Zhang, Q. Yang and T. Shao, Chem. Eng. J., 2022, 436, 135190 CrossRef CAS
- G. Wattieaux, M. Yousfi and N. Merbahi, Spectrochim. Acta, Part B, 2013, 89, 66–76 CrossRef CAS
- K. Clay, S. Speakman, G. Amaratunga and S. Silva, J. Appl. Phys., 1996, 79, 7227–7233 CrossRef CAS
- G. Giammaria, G. van Rooij and L. Lefferts, Catalysts, 2019, 9, 185 CrossRef
- P. Mehta, P. Barboun, F. A. Herrera, J. Kim, P. Rumbach, D. B. Go, J. C. Hicks and W. F. Schneider, Nat. Catal., 2018, 1, 269–275 CrossRef
- Y. Liu, L. Dou, R. Zhou, H. Sun, Z. Fan, C. Zhang, K. K. Ostrikov and T. Shao, Energy Convers. Manage., 2021, 250, 114896 CrossRef CAS
- L. Wang, Y. Yang, J. Sun, Y. Xin, X. Zhu and B. Sun, J. Anal. Appl. Pyrolysis, 2023, 170, 105931 CrossRef CAS
- H. Taghvaei, M. B. Hosseinzadeh, S. Rezazadeh, M. R. Rahimpour and A. Shariati, Chem. Eng. J., 2015, 281, 227–235 CrossRef CAS
- M. A. Mudassir, M. Batool, S. Kousar, M. A. Makarem, E. T. Razia, M. Meshksar, M. Murtaza, K. Tariq, M. A. Ud Din, M. A. Bodlah and M. R. Rahimpour, Fuel Process. Technol., 2023, 250, 107872 CrossRef CAS
- A. Omidkar, H. Xu, Z. Li, K. Haddadian and H. Song, J. Cleaner Prod., 2023, 414, 137512 CrossRef CAS
- H. Xu, Y. Song, Y. Zhang and H. Song, Fuel, 2022, 309, 122155 CrossRef CAS
- M. Gharibi, A. Khosravi, M. R. Khani, S. S. Shahabi, E. D. Guy and B. Shokri, J. Electrost., 2015, 76, 178–187 CrossRef CAS
- M. J. Gallagher, R. Geiger, A. Polevich, A. Rabinovich, A. Gutsol and A. Fridman, Fuel, 2010, 89, 1187–1192 CrossRef CAS
- A. Alagumalai and H. Song, Biofuel Res. J., 2023, 10, 1966–1973 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00309d |
| This journal is © The Royal Society of Chemistry 2024 |