
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRare-metal single atom catalysts for large scale hydrogen production under actual operating conditions
Jiaye
Li
 a,
Xu
Tian
a,
Changle
Yue
a,
Han
Guo
a,
Zhidong
Wang
a,
Mengdi
Guo
a,
Siying
Huang
a,
Yang
Song
b,
Wei
Lin
*b,
Yichuan
Li
a,
Bin
Liu
*a and
Yuan
Pan
*a
a,
Xu
Tian
a,
Changle
Yue
a,
Han
Guo
a,
Zhidong
Wang
a,
Mengdi
Guo
a,
Siying
Huang
a,
Yang
Song
b,
Wei
Lin
*b,
Yichuan
Li
a,
Bin
Liu
*a and
Yuan
Pan
*a
aState Key Laboratory of Heavily Oil Processing, China University of Petroleum (East China), Qingdao, 266580, China. E-mail: liubin@upc.edu.cn; panyuan@upc.edu.cn
bResearch Institute of Petroleum Processing, Sinopec, Beijing 100083, China. E-mail: linwei.ripp@sinopec.com
First published on 11th November 2024
Abstract
The electrocatalytic hydrogen evolution reaction (HER) is an efficient technology for hydrogen production and holds great significance for the development of renewable energy economies. Rare-metal-based catalysts are considered benchmark catalysts for the HER; however, their application in HER reactors is limited due to their high cost and poor stability. Rare-metal single atom catalysts (RMSACs) can be considered as promising candidates for the HER due to several advantages such as high activity, high stability, and high atom utilization. The rational design of RMSACs for HER reactors has become a research hotspot in this field. This paper reviews the research progress in the development of RMSACs for large scale hydrogen production under actual operating conditions, including high current density, seawater electrolysis, and long-term operation. Firstly, the mechanism, design and synthesis method of RMSACs for the HER are summarized. Then the atomic-level rational design strategy of RMSACs was proposed for enhancing the HER performance under actual operating conditions. Lastly, the opportunities and challenges for industrial applications of RMSACs are also discussed.
 Jiaye Li | Jiaye Li is currently pursuing his master's degree at the State Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China) under the supervision of Prof. Yuan Pan. His current research interest is focused on single atom catalysts for electrocatalysis. |
 Yuan Pan | Yuan Pan received his PhD degree from the China University of Petroleum (East China) in 2016. Then he worked as a postdoc at Tsinghua University from 2016 to 2018. In 2019, he joined the China University of Petroleum (East China). His research interest focuses on the rational design and controllable synthesis of single-atom catalysts for green energy chemical production processes. |
Broader contextRenewable hydrogen energy (H2) is currently seen as one of the most promising options capable of replacing fossil fuels. In the hydrogen production process, catalyst performance will directly determine the productivity of the plant. Currently, commonly used rare metal-based catalysts suffer from low activity, selectivity and stability. In recent years, rare metal single atom catalysts (RMSACs) have become a new frontier of research due to their highest atom utilization, uniform active sites and good stability. Although RMSACs have shown good activity in hydrogen production from electrolytic water, most of the current studies are still carried out in a laboratory setting without considering the real industrial environment. In this paper, the research progress in the development of RMSACs for large-scale hydrogen production under actual operating conditions is systematically discussed. Firstly, the dynamic mechanism of the HER process and the failure mechanism of seawater electrolysis are explored, and then the rational design strategy of RMSACs for hydrogen production from electrolytic water under real operating conditions is summarized. In addition, the current opportunities and challenges of RMSACs for hydrogen production by seawater electrolysis under practical operating conditions are analyzed. These insights contribute to the further development of RMSACs for hydrogen production on a large scale. |
1. Introduction
Since the advent of the first and second industrial revolutions, coal, oil, natural gas, and other fossil fuels have played a significant role as the primary energy sources for industrial production processes.1 However, it is important to recognize that these fossil fuels are non-renewable resources. Prolonged and excessive exploitation of these resources has resulted in a rapid decline in fossil energy reserves, thereby triggering a global energy crisis.2,3 Absolutely, the development of sustainable, renewable, green, and pollution-free energy sources has become an urgent task in our pursuit of a more sustainable future.4,5 Hydrogen (H2) is a substance known for its exceptional specific mass and energy density (142 MJ kg−1), making it the material with the highest value on earth. Consequently, it is commonly seen as one of the most promising options to replace fossil fuels. Hydrogen offers numerous advantages, including high combustion heat value, zero carbon emissions, and recyclability. These qualities have garnered significant attention worldwide, positioning hydrogen as a compelling solution for a cleaner energy future.6,7 Hydrogen energy holds the potential to replace various fossil fuels, including oil and coal, in a wide range of applications. However, the current industrial scale hydrogen production reactors face demanding operating conditions, such as high current density, utilization of seawater, and long-term operation. Fig. 1 illustrates the typical working conditions and the promising application prospects for hydrogen production through water electrolysis. Finding an environmentally friendly, cost-effective, safe, and efficient method of supplying hydrogen energy is a pressing challenge in the hydrogen energy industry. Under practical operating conditions, hydrogen evolution reaction (HER) catalysts often encounter challenges such as low activity, poor selectivity, and limited stability. These issues significantly hinder their widespread application in large scale hydrogen production.8,9 Therefore, the development of highly active HER catalysts is required.10–12 Rare-metal based catalysts (e.g., Pt, Ir, Ru and Pd) are currently commonly used as HER reference catalysts.13 However, the low abundance and difficulty in mining rare metals on earth, as well as their susceptibility to poisoning and poor stability during use, have significantly impeded the widespread utilization of rare metal-based catalysts in the HER.14,15 Therefore, the development of highly active and stable HER catalysts has become the focus of research in this field. | ||
| Fig. 1 The operating conditions for large scale hydrogen production and the application prospects of hydrogen energy. | ||
Single atom catalysts (SACs) refer to active metals dispersed on catalyst supports in the form of atoms, which are the minimum limit of the active site on the spatial scale, and theoretically enable the utilization of metal atoms to reach 100%.16–18 Compared to nanoparticles (NPs), SACs exhibit more excellent catalytic activity due to their more obvious size effects.19–24 Since Zhang and his co-workers first synthesized SACs in 2011,25 new regulatory mechanisms of active sites, coordination structures, and catalyst morphology have been continuously researched, greatly enriching the research on SACs.15,26–35 At the same time, due to their unique atomic structural characteristics and rich electronic structure, SACs have high activity and stability in the HER process. Fig. 2 illustrates the development of rare metal single atom catalysts. Consequently, it is imperative to engineer rare metals as the active sites of SACs.36 Recently, rare-metal SACs (RMSACs) have been synthesized and used in the HER process, showing excellent performance.37–40
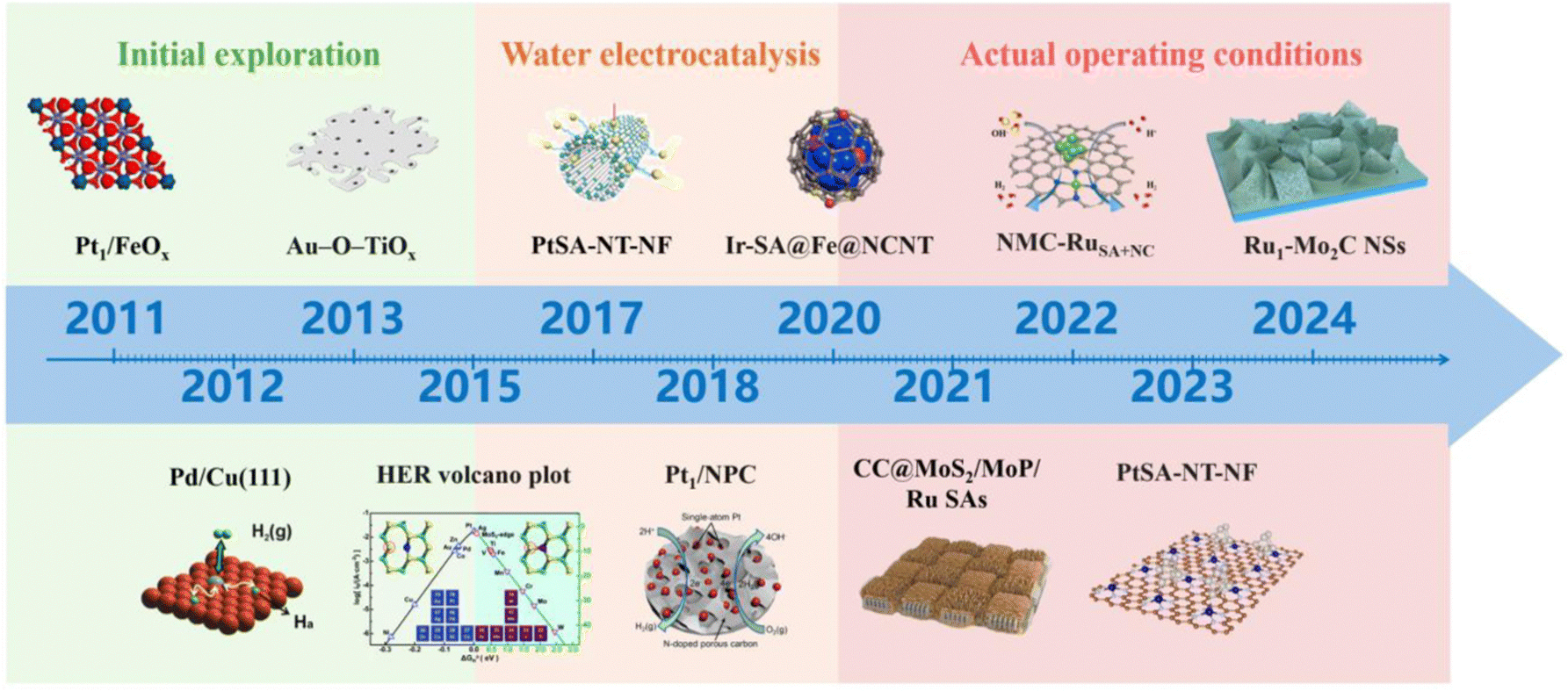 | ||
| Fig. 2 The progress in rare metal single atom catalyst development. Overall, the development of RMSACs has undergone three phases, i.e., initial exploration (2011–2015);25,41–43 in-depth research on HER activity and mechanism has expanded the application field of RMSACs, thus opening up the second phase: water electrolysis (2016–2020),43–46 and actual operation conditions (2021–2024).47–50 This figure has been adapted/reproduced from ref. 25 and 41–50 with permission from Springer Nature, The American Association for the Advancement of Science, American Chemical Society, RSC Publishing, John Wiley and Sons and Elsevier, copyright©2011, Springer Nature Limited. Copyright©2012, The American Association for the Advancement of Science. Copyright©2013, American Chemical Society. Copyright©Royal Society of Chemistry, 2015. Copyright©2017 The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA. Copyright©2018, American Chemical Society. Copyright©2020, American Chemical Society. Copyright©2021 Elsevier B.V. All rights reserved. Copyright©2022 Elsevier B.V. All rights reserved. Copyright©2023, American Chemical Society. Copyright©Royal Society of Chemistry, 2024. | ||
Unfortunately, most of the currently reported RMSACs cannot be applied to industrial reactors because of the damaged coordination structures or low selectivity under harsh industrial conditions (e.g. high current density, seawater, and electrolytic cells). Therefore, rational design and optimization of the atomic structure of RMSACs are of great significance for the large scale applications of RMSACs in the HER reactor.
This paper presents an extensive overview of the research advancements in large scale hydrogen production utilizing rare metal single atom catalysts under actual operating conditions. This review encompasses three key aspects: high current density conditions, seawater electrolysis, and long-term operation. Fig. 3 visually presents the key areas covered in this review. This paper first reviews the mechanism of the HER process. Furthermore, this review highlights the common synthesis methods employed for synthesizing RMSACs. These methods include high-temperature pyrolysis, atomic layer deposition (ALD), electrochemical methods and ball milling. Then the atomic-level rational design strategy of RMSACs was proposed for enhancing the HER performance under actual operating conditions. This review provides a comprehensive assessment of the progress made in these areas and offers prospects for the large scale applications of RMSACs. This work serves as a valuable reference for the design and synthesis of rare metal SACs in the context of large scale hydrogen production processes.
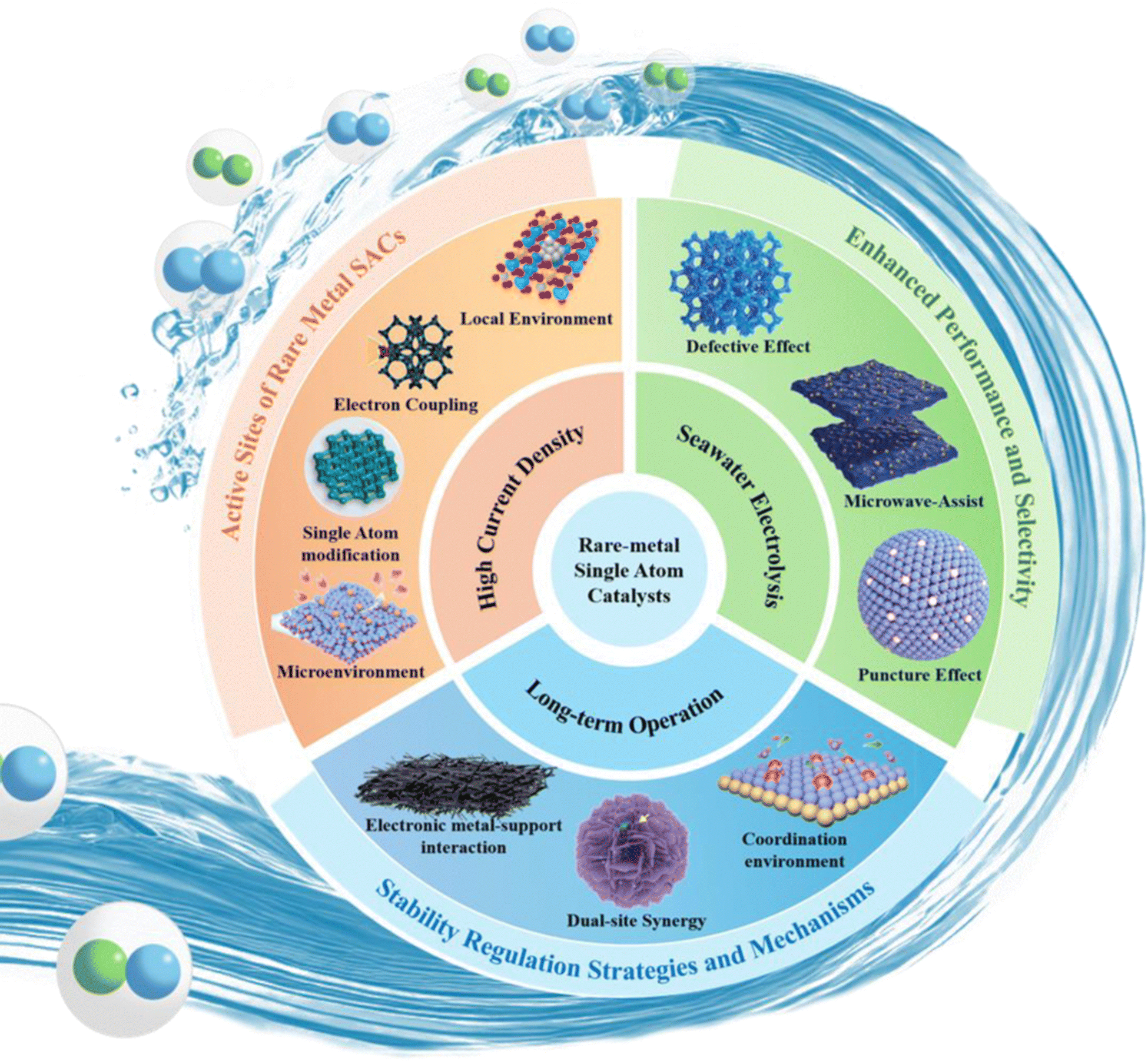 | ||
| Fig. 3 Rational design of RMSACs for large scale hydrogen production under actual operating conditions.51–60 This figure has been adapted/reproduced from ref. 51–60 with permission from Elsevier, John Wiley and Sons, RSC Publishing, Springer Nature and American Chemical Society, copyright©2024 Published by Elsevier Ltd on behalf of The editorial office of Journal of Materials Science & Technology. Copyright©2023 Wiley-VCH GmbH. Copyright©Royal Society of Chemistry, 2022. Copyright©2024, The Author(s). Copyright©2024, American Chemical Society. Copyright©2024 Wiley-VCH GmbH. Copyright©2024 Wiley-VCH GmbH. Copyright©2024 Elsevier Inc. All rights reserved. Copyright©2023 Wiley-VCH GmbH. Copyright©2024, American Chemical Society. | ||
2. Fundamental understanding of HER mechanisms
2.1 Overview
From an overall perspective, the HER involves the reduction of protons (H+) in acidic electrolytes or the reduction of water molecules (H2O) to produce hydrogen gas (H2) in alkaline and neutral electrolytes.61–63 The hydrogen evolution reaction (HER) mechanism can be generally categorized into two pathways: the Volmer–Heyrovsky pathway and the Volmer–Tafel pathway.64,65 The reactions shown in eqn (1)–(3) describe the HER process in acidic electrolytes.| H+ + e− + * → H* | (1) |
| H* + H+ + e− → H2 | (2) |
| H* + H* → H2 | (3) |
The mechanism of the hydrogen evolution reaction (HER) in alkaline electrolytes remains unclear due to the complexity of the process and the sluggish kinetics caused by high overpotential.66Eqn (4)–(6) describe the HER process in alkaline electrolytes.
| H2O + e− + * → H* + OH− | (4) |
| H* + H2O + e− → H2 + OH− | (5) |
| H* + H* → H2 | (6) |
In alkaline electrolytes, the prevailing perspective suggests that the initial step in the hydrogen evolution reaction (HER) process is still the Volmer process. However, unlike in acidic environments, the Volmer process in alkaline environments involves the dissociation of water molecules at the active site, resulting in the formation of protons and hydroxyl species. The proton subsequently attaches to the active site of the catalyst and engages in electronic reactions, resulting in the formation of hydrogen intermediates. In alkaline electrolytes, the second phase of the HER process exhibits two different pathways. The first pathway involves the hydrogen intermediate adsorbed on the active site reacting with water molecules and electrons simultaneously, ultimately generating hydrogen gas. This pathway is known as the Heyrovsky step. Alternatively, the hydrogen intermediates adsorbed on adjacent active sites can combine with each other, leading to the production of hydrogen gas. This pathway is referred to as the Tafel step. Therefore, the overall HER process in alkaline electrolytes includes the Volmer step for proton adsorption and hydrogen intermediate formation, followed by either the Heyrovsky step or the Tafel step for hydrogen gas generation.
In order to clearly understand the HER mechanism, researchers have proposed a variety of characterization methods, such as X-ray photoelectron spectroscopy (XPS),51,67–71 Raman spectroscopy,72–74 infrared (IR) spectroscopy,75–77 and X-ray absorption spectroscopy (XAS),78,79 as well as ab initio molecular dynamics (AIMD),80–84 density functional theory (DFT),85–97 and first principles.98–108 Nowadays, many researchers choose to use multiple characterization methods simultaneously, or use the characterization methods and theoretical calculations to verify each other, to understand the HER process occurring in the active site from multiple perspectives and to analyze and integrate the experimental results, so as to provide new ideas for the rational design of high-activity HER catalysts.55,109–117 For example, Dong et al.118 designed Ni single atom anchored on MoS2–FeS (Ni@amorphous MoS2–FeS) and investigated the effect of the relationship between the catalyst carrier structure and the coordination environment of Ni single atoms on the HER activity of the catalysts by first-principles calculations (Fig. 4(a)–(c)). Theoretical calculations showed that the amorphous MoS2–FeS has the lowest ΔGH* (0.312 eV), while the composite carrier can more stably ensure the effective loading of Ni single atoms, which can realize the HER process in a wider pH range. Cao et al.119 carried out ab initio calculations of molecular dynamics (AIMD) as well as constrained AIMD modeling using explicit solvation modeling and investigated the details of the kinetics of the acidic Volmer reaction over transition metal single atom catalysts (TM/N4C, TM = Mn, Cr, Fe, etc.) loaded onto N4C and re-optimized the constant potential free energy distribution of the Volmer reaction by using thermodynamic integration (TI) and charge extrapolation methods (Fig. 4(d)–(f)). It is shown that there is a strong charge–dipole interaction between the negatively charged intermediate H* produced by the Volmer process and the H2O molecules at the interface, resulting in the formation of a unique *H⋯HOH configuration. This unique effect is sufficient to alter the pathway of the acidic Volmer reaction and significantly increase its kinetic barrier, as evidenced by HER activity tests and Tafel slopes.
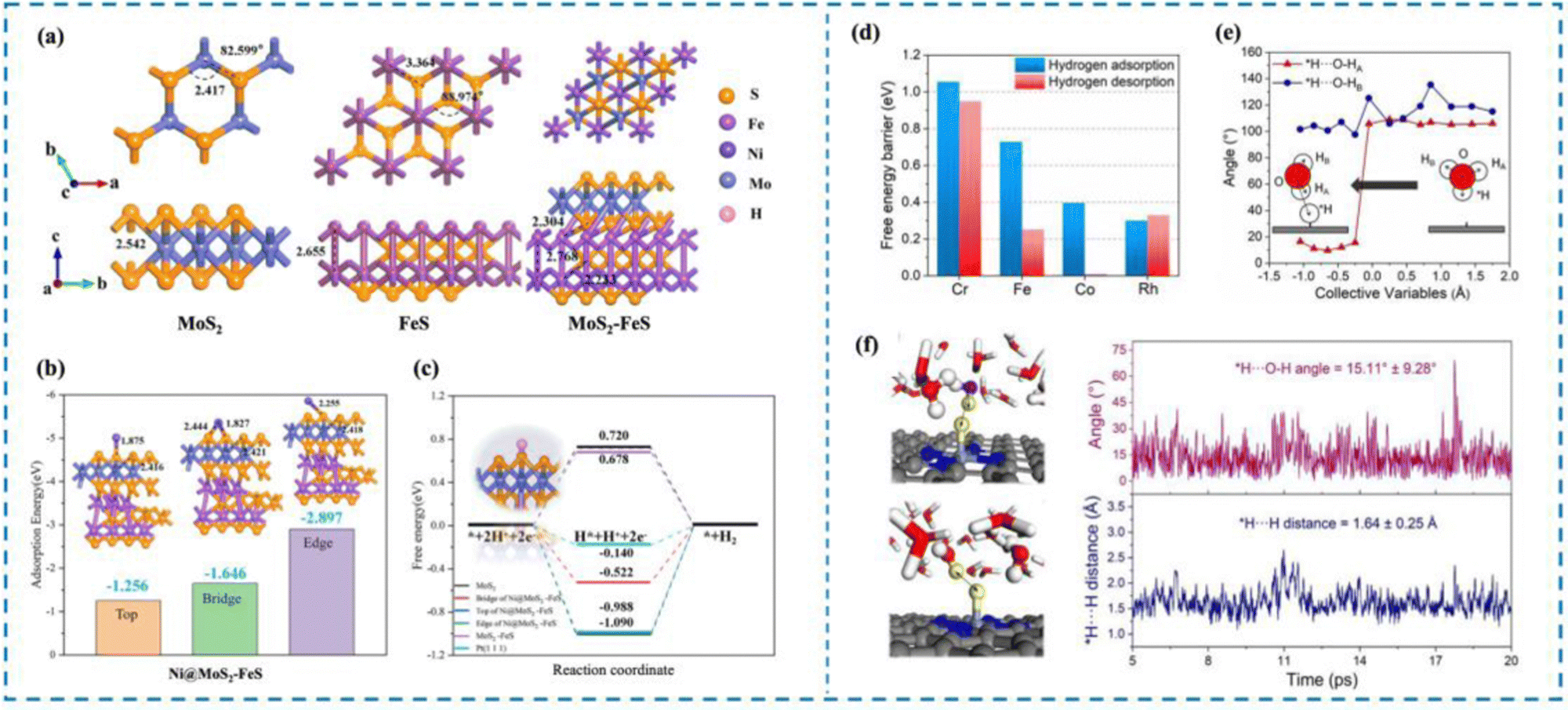 | ||
| Fig. 4 Examples of theoretical computational methods to study HER mechanisms. (a) MoS2, FeS and MoS2–FeS structure models. (b) Schematic diagram of adsorption energy of Ni@MoS2–FeS single-atom catalysts at different sites. (c) ΔG of electrocatalysts with different structures. (d) Hydrogen adsorption and hydrogen desorption on different catalysts. (e) Analysis of the angle between adsorbed hydrogen and the O–H bond of water. (f) AIMD simulation for illustration and analysis of the *H⋯O–H angle and *H⋯H distance.118,119 This figure has been adapted from ref. 118 and 119 with permission from IOP Publishing and American Chemical Society, copyright IOP Publishing. Copyright©2023, American Chemical Society. | ||
In summary, the ultimate goal of studying the HER process by combining theoretical calculations with experimental validation is to gain a deeper understanding of the kinetic process of HER, so as to find methods that can reduce the kinetic energy barriers and provide theoretical support for the rational design of high activity HER catalysts.
2.2 Dynamic mechanisms of the HER process
Theoretical calculations indicate that rare metal-based single-atom catalysts (RMSACs) with an atom utilization rate of up to 100% should have HER activities superior to those of the benchmark catalysts, such as Pt and Ru, which are commonly used today.46,120–128 However, the experimental results revealed that the HER activities of most of the RMSACs were still unsatisfactory. The puzzling discrepancy between the theoretical calculations and the experimental results suggests that the active sites and mechanisms of HER single atom catalysts are not yet comprehensively investigated, and new research tools are needed to further understand the dynamic processes occurring on SACs. Thanks to the development of in situ characterization and operational profiling techniques (e.g., in situ Raman, in situ/operational XAS, in situ ATR-SEIRAS, etc.), it is possible to follow the dynamic catalytic processes occurring on SACs, thus allowing researchers to gain insights into the fundamental mechanisms of the HER on SACs under operating conditions.129–137Wang et al.138 proposed an effective interfacial water molecule activation mechanism, which is a unique mechanism originating from the synergistic interaction between Co single atoms (Co–N3) and Co nanocrystals (Co NCs) to optimize the HER process, and thus this coupled catalyst (CoNC-SA/N*–C) exhibits excellent HER activity (Fig. 5(a)). In situ attenuated total reflectance-surface enhanced infrared absorption spectroscopy (in situ ATR-SEIRAS) demonstrated the Volmer–Heyrovsky process on the CoNC-SA/N*–C catalyst and that the H* intermediates originated from interfacial H2O molecules adsorbed on the catalyst surface. However, interfacial H2O molecules are able to form a relatively rigid interfacial water network, which hinders the mass transfer process of H2O molecules to the catalyst surface, whereas Co NCs are able to disrupt the rigid interfaces and produce more free H2O molecules, which enhances the adsorption of H2O molecules on the Co–N3 sites, which is specifically manifested in in situ ATR-SEIRAS by the appearance of Co–H peaks at a lower potential. Density-functional theory calculations revealed that the change in the free energy of dissociation of water molecules was significantly lower (0.16 eV) for the CoNC-SA/N*–C catalyst compared to the uncoupled CoSA/N–C and CoNC/N–C catalysts, indicating that H2O molecules were more readily dissociated on CoNC-SA/N*–C, which is consistent with the results of in situ ATR-SEIRAS. Meanwhile, the charge density difference results indicated that the electrons were transferred from Co–N3 to Co(111), while the Co-3d PDOS results found that the d-band center of Co in CoNC-SA/N*–C was −2.44 eV. The in situ ATR-SEIRAS and DFT results together indicate the synergistic effect between Co NCs and Co–N3, where Co NCs modulate the electronic structure of the Co–N3 site and optimize the HER process, thus enhancing the HER activity of CoNC-SA/N*–C, which is consistent with the experimental results.
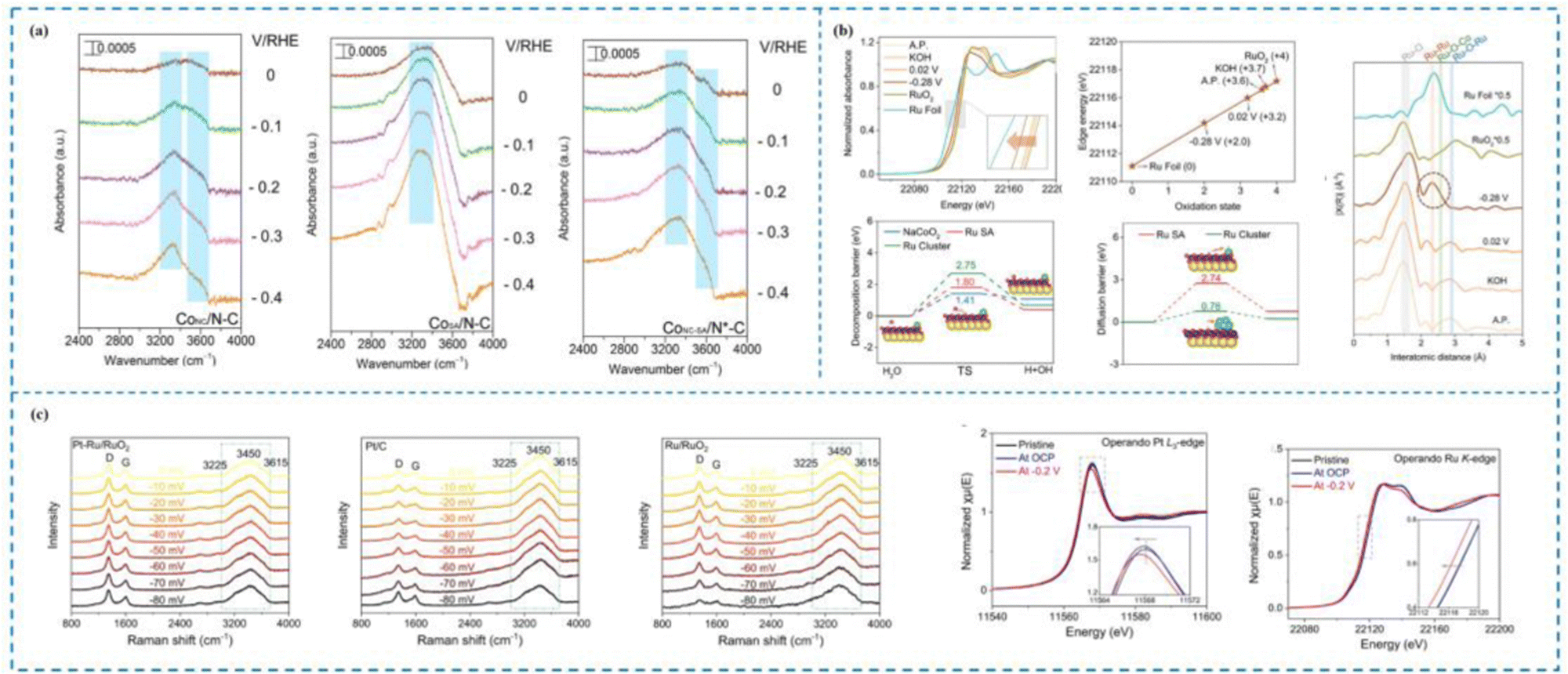 | ||
| Fig. 5 In situ characterization techniques used to investigate the dynamic mechanisms of the HER. (a) In situ ATR-SEIRAS of CoNC/N–C, CoSA/N–C and CoNC-SA/N*–C catalysts. (b) The operando Raman spectra of Pt–Ru/RuO2, Pt/C and Ru/RuO2; the Pt L3-edge XANES and the Ru K-edge XANES of Pt–Ru/RuO2. (c) In situ Ru K-edge XANES spectra, the oxidation state of Ru and the corresponding EXAFS R-space spectra of Ru/NC at various potentials and reference catalysts for the HER; kinetic energy barrier for water dissociation and energy barriers for hydrogen migration from the NC surface to the adjacent Ru SA and Ru cluster sites.138–140 This figure has been adapted/reproduced from ref. 138–140 with permission from American Chemical Society, Springer Nature and John Wiley and Sons, copyright©2022, American Chemical Society. Copyright©2024, The Author(s). Copyright©2023 Wiley-VCH GmbH. | ||
Zhu et al.139 synthesized an efficient and stable Pt single-atom catalyst (Pt–Ru/RuO2) and comprehensively investigated the different contributions of Pt SAs, Ru and RuO2 to the HER process by combining operando Raman and operando XAS spectroscopies (Fig. 5(b)). Specifically, operando Raman spectroscopy revealed that the peak ratios of both Pt–Ru/RuO2 and Ru/RuO2 showed a rapid decrease with increasing HER potential and the peak ratios decreased at almost the same rate, which indicated that both Pt–Ru/RuO2 and Ru/RuO2 had the ability to dissociate water efficiently, but Pt SAs did not contribute to the water dissociation step; operando XAS spectroscopy revealed that Pt SAs, Ru, and RuO2 contributed to the HER process; on the other hand, operando XAS and operando Ru K-edge XANES found that the valence states of Pt and Ru continued to decrease during the catalytic process, which suggests that both Pt and Ru are active sites for the HER process. Therefore, Zhu and his collaborators proposed that the HER process occurs on Pt–Ru/RuO2. It is noteworthy that the H2O molecule did not approach the Pt SAs directly and dissociate, but first dissociated into H* on the RuO2 carrier and then H* approached the single atom active site and finally generated H2. Consequently, throughout the process, RuO2 facilitated the slow dissociation process of the H2O molecule, whereas the Pt SAs together with Ru facilitated the H* adsorption and enhanced the H* adsorption.
Zhu et al.140 synthesized a Ru catalyst with a dual locally supported structure (Ru/NC) by co-dispersing amorphous ruthenium hydroxide clusters and adjacent Ru SAs on layered NaCoO2via a cation exchange reaction at room temperature. Experimental tests showed that Ru/NC possessed excellent HER activity with an onset overpotential close to 0 eV and an overpotential of 24 mV at 10 mA cm−2. Huang and colleagues utilized in situ XAS spectroscopy to reveal the dynamic structural evolution of Ru/NC during the HER process. Specifically, the Ru hydroxide clusters evolved toward Ru clusters during the HER process, while the oxidation state of Ru SAs remained almost unchanged, suggesting that Ru SAs with Ru clusters are the true HER active sites. The changes in Ru–O–Ru and Ru–Ru intensity peaks in the in situ EXAFS spectra proved this. Meanwhile, the combination of in situ Co K-edge XANES and EXAFS spectra revealed that the NaCoO2 substrate exhibits good structural stability, which suggests that NaCoO2 acts as a carrier in Ru/NC. The results of density functional theory calculations (DFT) further indicated that the excellent HER activity of Ru/NC originated from the action of the truly active substances, Ru SAs and Ru clusters (Fig. 5(c)).
In summary, tracking the dynamic processes occurring on SACs through advanced in situ characterization techniques, combined with theoretical simulation and experimental validation, is essentially a study of the dynamic transformation process of reactants, intermediates, and other species over catalysts. Studying the dynamic mechanism of the HER through in situ characterization enables researchers to gain a deep understanding of the mode of action between reactants, interfaces and active sites in the HER process, to probe deeply into the actual active sites and conformational relationships of SACs, and at the same time, to discover the root cause of the slow dynamics of the HER process and to propose a more reasonable design scheme for SACs based on it.
3. Synthesis strategy of rare metal SACs for the HER
3.1 High-temperature pyrolysis
The process of pyrolysis is frequently used to create SACs. This method offers advantages such as ease of operation and simplicity in terms of instrumentation.92 Typically, the current pyrolysis technique involves subjecting metal-containing precursors to pyrolysis under the protection of an inert gas. High temperatures are typically needed for pyrolysis, frequently between 800 and 900 °C. However, at such elevated temperatures, there is a risk of metal single atoms migrating and aggregating into larger particles. To mitigate this, a post-pyrolysis acid treatment is commonly employed after obtaining a single atom catalyst through high-temperature pyrolysis of metal precursors. Frequently, the high-temperature pyrolysis method is employed to synthesize SACs that are supported on N-doped carbon materials. For instance, Chen et al.141 demonstrated the synthesis of tungsten single atom catalysts (W-SACs) anchored on N-doped carbon supports via high-temperature pyrolysis of tungsten-containing precursors. Liu et al.142 developed a method to prepare a platinum single atom catalyst (Pt SA/NP-NC) with abundant Pt–N-4 sites using a high-temperature pyrolysis ammonium iodide etching strategy.3.2 Atomic layer deposition (ALD)
The ALD method involves depositing atoms onto a catalyst substrate in the form of a single atom layer. Precise manipulation of the morphology and structure of deposited atoms via this technique is crucial for investigating the catalytic mechanism and establishing the relationship between the structure and the activity of single atom catalysts.143 There are numerous advantages offered by ALD that cannot be found in other deposition methods. The first advantage of ALD is its ability to achieve precise control over the atomic film thickness. This level of control allows for the creation of films with exceptional uniformity and accuracy, making ALD an ideal choice for applications where precise film thickness is critical. Aside from achieving precise thickness control, films created using ALD also demonstrate outstanding shape retention. Moreover, ALD offers outstanding chemoselectivity, especially in the formation of single atom films.144 Jiao et al.145 utilized ALD to immobilize single Pt atoms on a MoS2 nanoarray, resulting in the fabrication of a highly efficient catalyst known as Pt/MoS2 NTA (the Pt single atom catalyst on the MoS2 nanoarray, Fig. 6(a) and (b)). When used in the hydrogen evolution process (HER), this catalyst performed exceptionally well. The precise control offered by ALD enabled the deposition of Pt atoms in a single-atom configuration, maximizing the catalytic activity of Pt. On a similar note, Thalluri et al.146 employed ALD to synthesize Ru single atom catalysts (Ru SACs) with remarkable activity in the HER. By carefully designing the ALD process, Thalluri was able to reduce the nucleation delay during Ru deposition. Due to this optimization strategy, Ru SACs are able to exhibit excellent HER performance.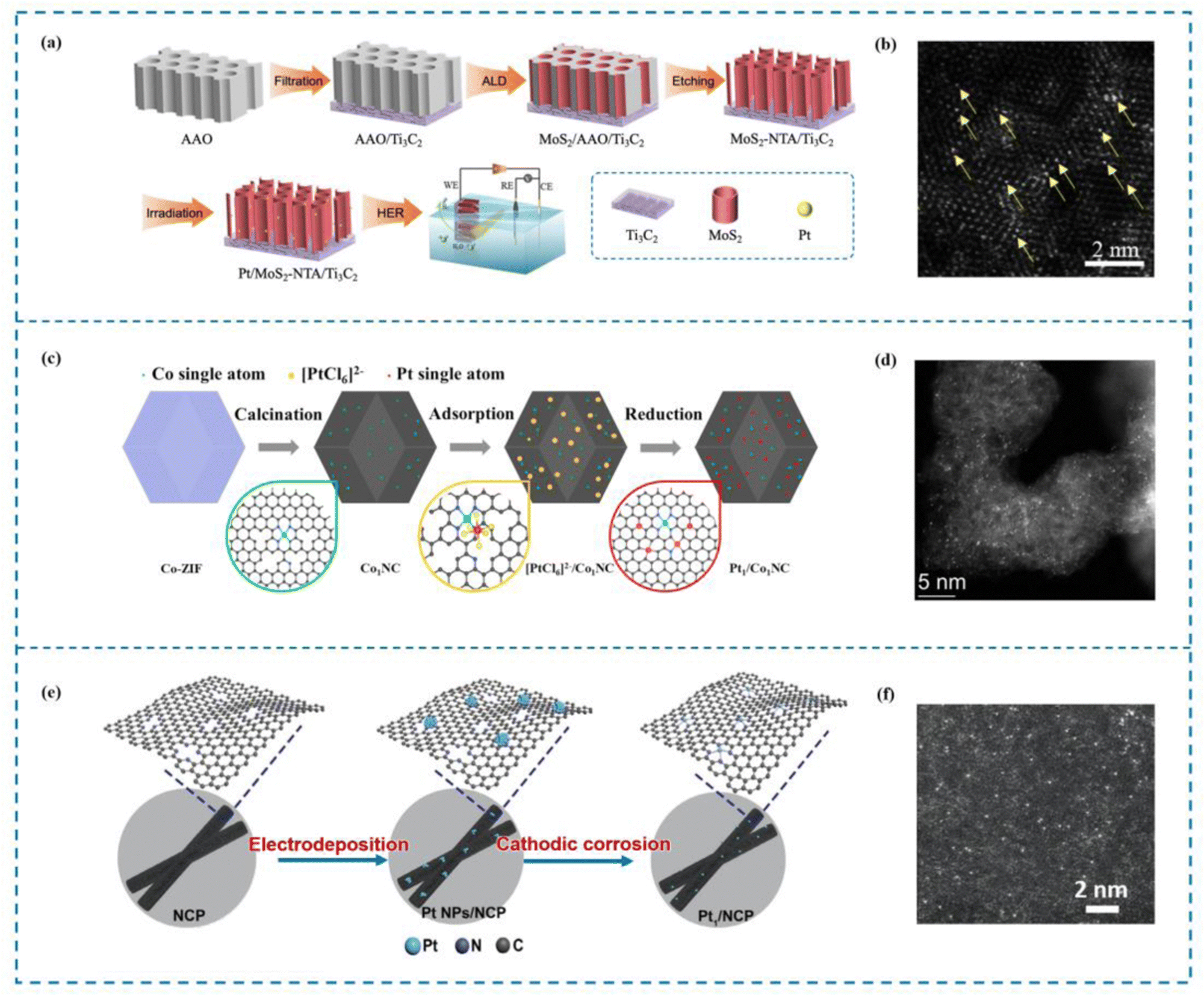 | ||
| Fig. 6 Examples of ALD and electrochemical methods. (a) Synthesis of Pt–MoS2 by ALD. (b) HAADF-STEM image of Pt–MoS2. (c) Synthesis of Pt1/Co1NC. (d) HAADF-STEM image of Pt1/Co1NC. (e) Synthesis of Pt1/NCP. (f) HAADF-STEM image of Pt1/NCP.145,147,148 This figure has been adapted/reproduced from ref. 145, 147, and 148 with permission from John Wiley and Sons, Elsevier and Springer Nature, copyright©2022 Wiley-VCH GmbH. Copyright©2021 Elsevier B.V. All rights reserved. Copyright©2021, Tsinghua University Press and Springer-Verlag GmbH Germany, part of Springer Nature. | ||
3.3 Electrochemical methods
Indeed, electrochemical methods offer a fast and simple approach to preparing SACs.149 Compared to pyrolysis methods that often require high temperatures, electrochemical methods can be conducted under relatively mild conditions. These mild conditions simplify the preparation process and help minimize energy loss during catalyst synthesis. Electrochemical techniques have garnered considerable interest in the domain of single-atom catalysts for the HER. Chen et al.148 demonstrated the synthesis of Pt1/Co1NC SACs with high HER activity using a mild electrochemical reduction method (Fig. 6(c) and (d)). This approach allows for the reduction of metal ions to single atoms on a support material, resulting in enhanced catalytic performance. Similarly, Li et al.147 developed a universal top-down route combined with a mild electrochemical reduction method to prepare uniformly dispersed Pt SACs (Pt1/NCP) (Fig. 6(e) and (f)). These catalysts exhibited excellent performance in the HER. The electrochemical reduction method enabled the precise control of Pt dispersion, ensuring the optimal utilization of Pt atoms for efficient catalysis.3.4 Ball milling
Ball milling is an intriguing technique for producing SACs. This technique involves the use of mechanical forces generated by the collision and grinding of balls in a milling jar.150 During ball milling, significant shear forces and localized high temperatures are generated, which can be beneficial for optimizing the chemical bonds and surface properties of catalyst materials. This process can also result in a more even distribution of metal atoms on the catalyst support.151 Ball milling has a number of noteworthy benefits, including simplicity and environmental friendliness. Unlike some other preparation methods that may involve complex procedures or require harsh conditions, ball milling is a relatively straightforward process. It typically requires minimal chemical additives and can be performed under ambient conditions, reducing the environmental impact associated with catalyst synthesis.152 Additionally, the ball milling technique allows for the efficient production of large quantities of SACs. This is particularly advantageous as it addresses the challenge of scaling up the production of SACs.153 Jin et al.154 employed a mechanical ball milling method to prepare nitrogen-doped carbon supported Pt SACs (represented as K2PtCl4@NC-M) with abundant defects. The ball milling process facilitated the incorporation of Pt atoms into the carbon matrix, resulting in the formation of SACs with nitrogen dopants. The resulting K2PtCl4@NC-M catalyst exhibited exceptional performance in the hydrogen evolution reaction (HER), showcasing its effectiveness as an electrocatalyst. The simplicity and speed of the preparation process further highlight the appeal of this ball milling method. This study provides new insights and potential avenues for the large scale synthesis of SACs. The mechanical ball milling method offers a straightforward and efficient approach to produce catalysts with excellent performance, opening up possibilities for the scalable production of SACs.4. Rational design of RMSACs under high current density conditions for hydrogen production
4.1 Pt SACs
The development of catalysts capable of high activity at high current density is an important requirement for the realization of industrial water electrolysis.155 However, unlike water electrolysis in the laboratory, the reaction rate of water electrolysis is gradually accelerated by the increase of current density, which leads to a large number of H2 bubbles accumulating on the surface of the electrodes, impeding the contact between the catalyst active sites and water molecules and thus reducing the efficiency of water electrolysis. In addition, high current density not only corrodes the carrier and destroys the local environment of the active site, but also damages the coordination structure of the active site, leading to the deactivation of the catalyst. Rare metal-based catalysts are currently the most widely used catalysts in high current density electrolysis of water for hydrogen production; however, rare metal-based catalysts still have great limitations.156 This is because, firstly, rare metal-based catalysts are mostly nanoscale catalysts, and their active sites often consist of rare metal clusters or particles such as Pt and Ru. During high current density electrolysis, these clusters or particles tend to agglomerate and form larger particles that eventually detach from the electrode, resulting in lower activity and stability of electrolyzed water. Second, the lower atomic utilization rate and unclear active sites are not conducive to the realization of high activity of rare metal-based catalysts and also lead to the need to use more rare metals in the preparation of rare metal-based catalysts, which raises the cost of preparation and use.156,157 RMSACs can effectively solve the limitations and deficiencies of rare metal-based catalysts. Firstly, RMSACs have a high atom utilization rate, which can effectively improve the catalytic activity for hydrogen production by electrolysis of water under high current density; secondly, RMSACs have a more clear and adjustable coordination structure of the active sites, which can effectively protect the active sites, and at the same time, the interaction between the active sites and the carriers can also slow down the corrosion of the carriers by high current, which makes the active sites of RMSACs able to preserve long-term effectiveness in the process of electrolysis of water at high current density. Therefore, it is very necessary to design Pt, Ru and other rare metals as the active sites of single-atom catalysts and optimize them using various modulation methods to achieve efficient hydrogen production by electrolysis of water at high current density.Pt/C catalysts have always been the preferred catalysts for the HER. Pt SACs are considered to be the best substitute for traditional rare metal catalysts.158 However, at high current density, Pt SACs are prone to metal atom aggregation, catalyst detachment, and incomplete mixing with electrolyte, limiting their application under high current density conditions. Pt single-atom catalysts are essential for their utilization in high current density reactors. Cao et al.159 reported the synthesis of Pt SACs (Pt/Co(OH)2) anchored on Co(OH)2 through a one-step method (Fig. 7(a)–(c)). Pt/Co(OH)2 exhibited outstanding HER performance, setting a new global benchmark with an overpotential of merely 4 mV and a current density of 10 mA cm−2. In this work, a current density of 1500 mA cm−2 was achieved by Pt/Co(OH)2, which is even more significant. X-ray fine extended structure spectroscopy (EXAFS) revealed that Pt single atoms existed in the form of Pt–Cl4, and no Pt–Pt bond was detected. Crystal orbital Hamilton population (COHP) and integrated crystal orbital Hamilton population (ICOHP) calculations demonstrated that incorporating Pt single atomic sites can effectively lower the occupancy of Pt–H antibonding orbitals, leading to enhanced hydrogen desorption efficiency. Meanwhile, the interaction between Pt single atom sites and Co(OH)2 optimized the local environment of Pt single atoms, which is the key to achieving high current density electrolytic water performance of Pt/Co(OH)2.
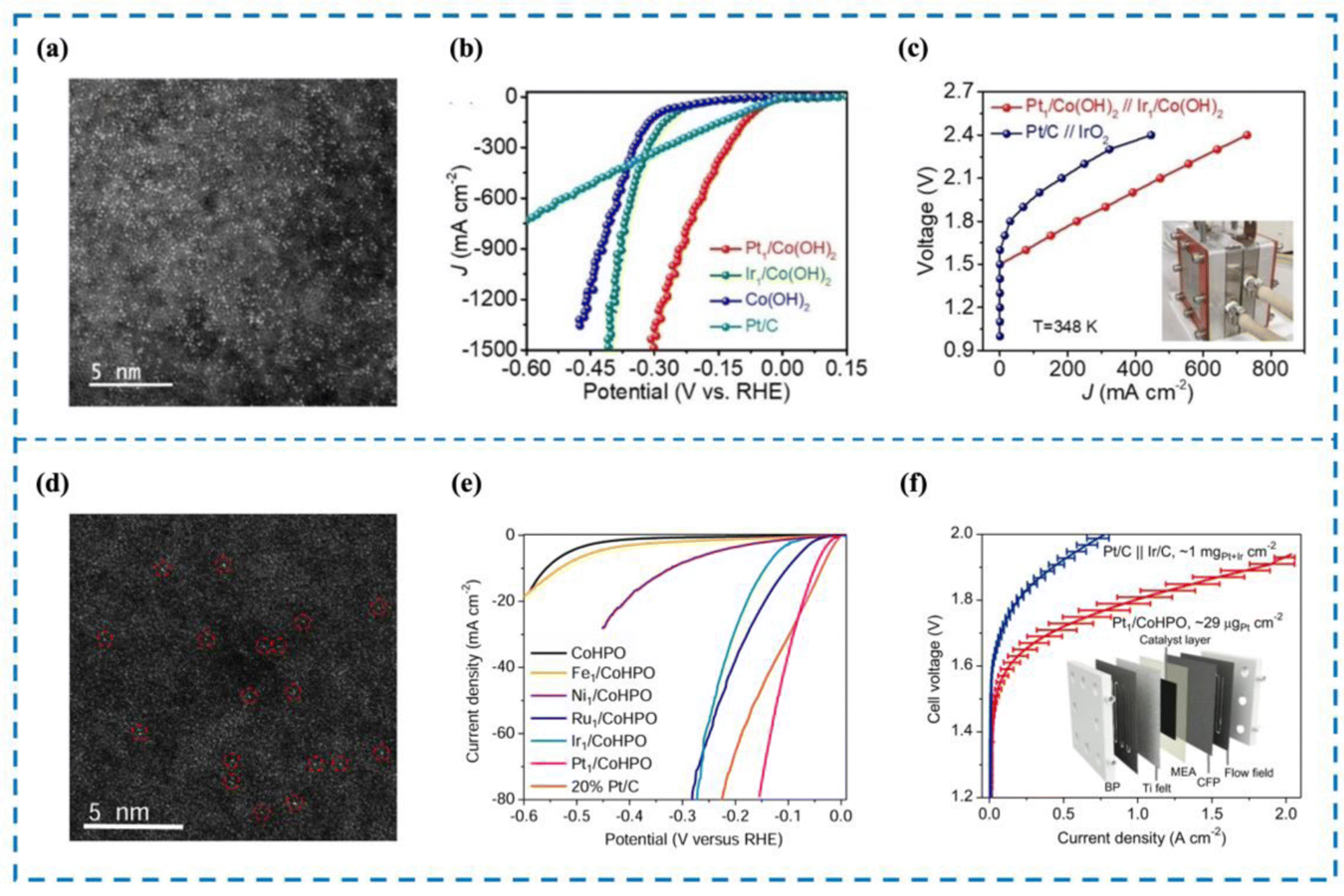 | ||
| Fig. 7 High current density performance of Pt/Co(OH)2 (a)–(c) and Pt1/CoHPO (d)–(f). (a) and (d) HAADF-SETM image of Pt/Co(OH)2 and Pt1/CoHPO. (b) and (e) The LSV curve of Pt/Co(OH)2 and Pt1/CoHPO. (c) and (f) The AEMWE curve of Pt/Co(OH)2 and Pt1/CoHPO.159,160 This figure has been adapted/reproduced from ref. 159 and 160 with permission from John Wiley and Sons and Springer Nature, copyright©2022 Wiley-VCH GmbH. Copyright©2022, The Author(s). | ||
Zeng et al.160 quickly frozen the solutions of cobalt hydrogen phosphate (CoHPO) and K2PtCl6 synthesized by liquid nitrogen to ensure the uniform dispersion of Pt precursors in the solution. Then xenon lamps with specific wavelengths of light were used to make the samples react and melt, and the products were collected by centrifugation to prepare Pt SACs dispersed on CoHPO (Pt1/CoHPO) (Fig. 7(d)–(f)). The effective production of Pt single atom species was demonstrated by X-ray diffraction (XRD) and aberration-corrected high angle annular dark field-scanning transmission electron microscopy (HAADF-STEM). Pt1/CoHPO showed excellent HER performance, with a Pt turnover frequency (TOF) of up to 12.8 s−1 at −100 mV, approximately 41 times that of commercial Pt/C. An anion exchange membrane water electrolyzer (AEMWE) was further assembled using this bi-functional catalyst as the anode and cathode with a mass load as low as 0.029 mgPt cm−2. The AEMWE achieves an industrial current density of 1 A cm−2 and durability of over 100 h at a low battery voltage of 1.8 volts. Combined with experimental studies and density functional theory (DFT) calculations, it is shown that Pt1/CoHPO has a unique Pt(OH)(O3)/Co(P) coordination. The primary reasons for the increased electrocatalytic activity and stability are the unique coordination structure of single-atom Pt sites and the robust electron coupling between isolated Pt atoms and nearby Co atoms.
DFT calculations indicate that the introduction of some axial ligands (such as –Cl and –CN) can enhance the HER performance of SACs. This is brought about by the axial ligands’ strong first electron affinity. When chelating metal single atom active sites, the metal active sites exhibit more excellent adsorption capacity for key intermediates *OH and *H, thereby promoting the speed limiting step (Volmer step) of HER kinetics. This allows the SACs to show more excellent HER activity and exhibit higher current density.161 Zhang et al.162 reported a series of Pt SACs (X-Pt/LDH, X = Cl, F, Br, I,OH) with axial coordination. Zhang and his co-workers introduced Pt SACs onto NiFe-layered-double-hydroxide by electrochemical deposition and then precisely adjusted the axial ligands on the Pt units through a simple radiation-impregnation procedure (Fig. 8(a)–(d)). The best HER performance was observed with Cl–Pt/LDH. When the overpotential in 1.0 M KOH was 10 mA cm−2, the mass activity reached 30.6 A mgPt−1, and when it was 100 mV, it reached 25.2 mV. It is 133 times more expensive than commercial 20% Pt/C and 5 times more expensive than HO–Pt/LDH. The membrane electrode assembly (MEA)-based water electrolytic cell Cl–Pt/LDH demonstrated exceptional performance, achieving a high current density of 1 A cm−2 and an energy conversion efficiency of 80%.
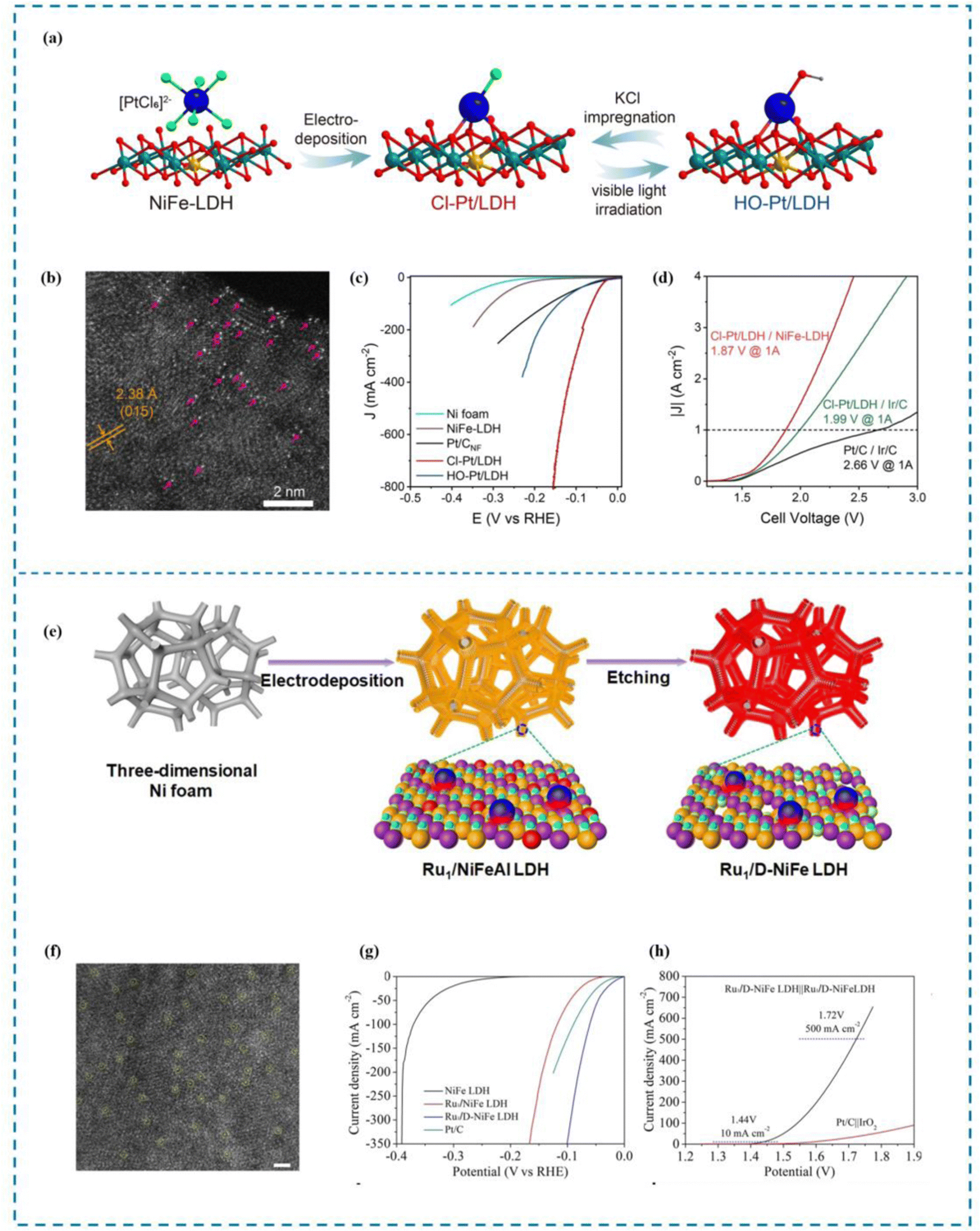 | ||
| Fig. 8 Synthesis, HAADF-STEM and HER performance of HO–Pt/LDH (a)–(d) and Ru1/D-NiFe LDH (e)–(h). (a) Synthesis of HO–Pt/LDH. (b) HAADF-STEM image. (c) High current density LSV curve. (d) AEMWE curve. (e) Synthesis of Ru1/D-NiFe LDH. (f) HAADF-STEM image. (g) High current density LSV curve. (h) AEMWE curve.162,163 This figure has been adapted/reproduced from ref. 162 and 163 with permission from Springer Nature, copyright©2022, The Author(s). Copyright©2021, The Author(s). | ||
Yue et al.164 proposed a “defect substitution”-induced strategy to construct a hierarchically stabilized system of Pt single atoms (PtSA@Mo2C@NC) by defect substitution, using a polyoxometalate (POM) (NH4)4[ZnMo6O24H6] (ZnMo6) as a template (Fig. 9(a)–(d)). The Pt SAs are designed to be embedded in the single atom defects on Mo2C and captured by axial N atoms. Due to this unique “three-dimensional modulation”, PtSA@Mo2C@NC exhibits excellent HER performance, showing overpotentials of only 25 and 66 mV under acidic and alkaline conditions, respectively, at current densities of up to 100 mA cm−2. The HER process can also be realized at a current density of 1000 mA cm−2. In addition, PtSA@Mo2C@NC exhibited excellent stability, with little or no decay in performance after 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. In situ electrochemical shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) together with theoretical calculations revealed that the excellent activity of PtSA@Mo2C@NC originated from the electron affinity of the ligand, in particular the strong electronic effect of axial N on the PtSA site, which facilitated the adsorption and dissociation of H2O, while decreasing the free energy of adsorption of H* and the detachment energy of H2, optimizing the active site and the HER kinetic process occurring at the active site, thus greatly improving the HER performance of PtSA@Mo2C@NC.
000 cycles. In situ electrochemical shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) together with theoretical calculations revealed that the excellent activity of PtSA@Mo2C@NC originated from the electron affinity of the ligand, in particular the strong electronic effect of axial N on the PtSA site, which facilitated the adsorption and dissociation of H2O, while decreasing the free energy of adsorption of H* and the detachment energy of H2, optimizing the active site and the HER kinetic process occurring at the active site, thus greatly improving the HER performance of PtSA@Mo2C@NC.
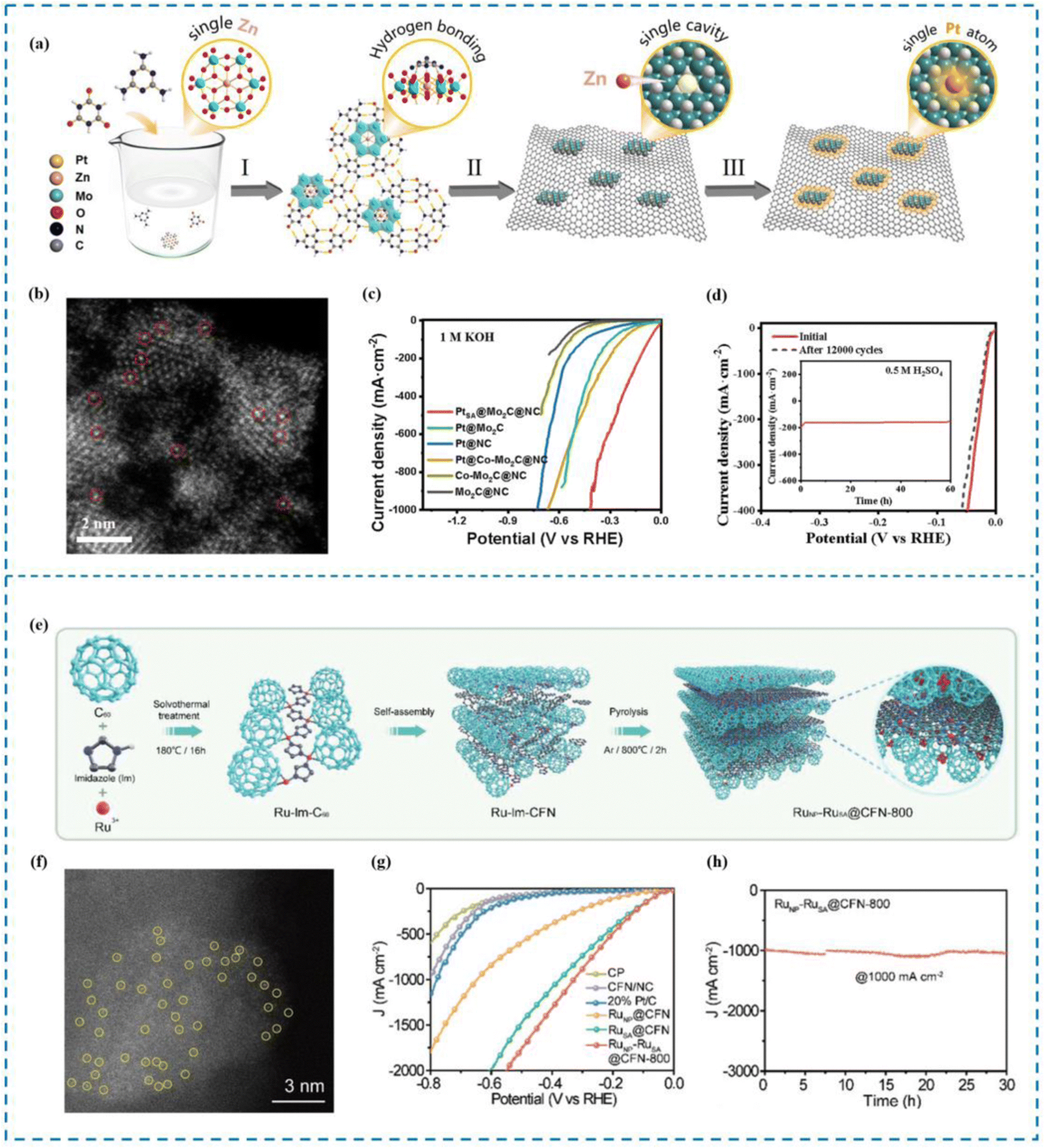 | ||
| Fig. 9 Synthesis, HAADF-STEM and HER performance of PtSA@Mo2C@NC (a)–(d) and RuNP–RuSA@CFN-800 (e)–(h). (a) Synthesis of PtSA@Mo2C@NC. (b) HAADF-STEM image. (c) High current density LSV curve. (d) Stability performance. (e) Synthesis of RuNP–RuSA@CFN-800. (f) AC-HAADF-STEM image. (g) High current density LSV curve. (h) Stability performance at high current density.164,165 This figure has been adapted/reproduced from ref. 164 and 165 with permission from The Royal Society of Chemistry and John Wiley and Sons, copyright©2023 Wiley-VCH GmbH. | ||
4.2 Ru SACs
The electronic and geometric structures of Ru are highly similar to Pt and Ru is cheaper than Pt, making it a viable alternative to Pt.120,166,167 Therefore, designing Ru as an active metal for SACs is important for realizing the application of SACs in high current density electrolysis of water. Zhai et al.163 synthesized a single atomic site Ru catalyst stabilized on defective NiFe LDH (Ru1/D-NiFe LDH) by a simple electrodeposition and subsequent etching procedure as a direct and practical method (Fig. 8(e)–(h)). The synthesized Ru1/D-NiFe LDH exhibits excellent HER properties, as demonstrated in performance tests that indicate a low overpotential of 18 mV at a current density of 10 mA cm−2 and a high turnover frequency of 7.66 s−1 at an overpotential of 100 mV. Importantly, the assembled two-electrode battery of Ru1/D-NiFe LDH achieved an industrial current density of 500 mA cm−2 at a low battery voltage of 1.72 V. According to the DFT calculations, the optimal regulation of H adsorption energy is achieved by Ru1/D-NiFe LDH. In addition, the abundant number of active sites accelerates water division kinetics, thereby enhancing intrinsic HER activity and achieving industrial current density.Research has demonstrated that the electrochemical properties of catalysts are highly sensitive to the atomic-level electronic environment due to the synergistic interaction between single atomic sites and metal nanoparticles.168–170 In a spherical core–shell structure, metal nanoparticles (NPs) can interact with multiple metal single atoms anchored on the core shell, resulting in an electron metal cage interaction (EMCI) that is similar to the endohedral metal fullerene (EMF).171 Based on this concept of electron metal cage interaction, rational design of RMSACs can be carried out. Enhanced catalytic performance, stability, and selectivity for various catalytic reactions can be achieved by customizing both the core–shell structure and the characteristics of the single metal atoms. By rational design, Feng et al.172 have created a Ru-based hybrid electrocatalyst, with Ru NPs serving as the core and single atom RuNx scattered over oxygen-like fullerene carbon cages serving as the shell. This catalyst has shown low overpotential in both acidic and alkaline media, excellent HER activity, and good stability (greater than 100 h). RuNP@RuNx-OFC/NC can deliver a high current output without high overvoltage under both acidic and alkaline conditions on an industrial scale. According to DFT calculations, efficient electronic communication between Ru NP and atomic Ru distribution on the carbon cage facilitates the transfer of charge from Ru NP to single atom Ru, altering the electronic density of the Ru site. This enhances the adsorption of hydrogen and the dissociation of water by reducing the Gibbs free energy, thereby improving the HER activity of the catalyst.
Luo et al.165 synthesized an Ru SAC (RuNP–RuSA@CFN-800) by embedding Ru single atoms (RuSA) and Ru nanoparticles (RuNP) into a 3D crystal fullerene network (CFN) through a new lattice limited domain strategy (Fig. 9(e)–(h)). Thanks to the synergistic effect between RuNP and RuSA, the synthesized RuNP–RuSA@CFN-800 showed significantly superior HER activity. In particular, in order to drive a current density of 10 mA cm−2, an overpotential of only 33 mV is required under alkaline conditions. Benefitting from the ultra-stable CFN substrate RuNP–RuSA@CFN-800 shows stable and long-term electrocatalytic stability in >1400 hours. Best of all, RuNP–RuSA@CFN-800 can deliver 1000 mA cm−2 current output at 251 mV overpotential at an industrial level, superior to commercial Pt/C. Theoretical calculations show that the electron synergistic effect between RuNP and RuSA can regulate the electronic structure of RuNP–RuSA@CFN-800 and reduce the Gibbs free energy of intermediate species during hydrolysis dissociation, thus accelerating the hydrogen precipitation process and achieving ultra-high current density.
In summary, the key factor for developing high current density RMSACs is to optimize the kinetic processes by applying different regulatory techniques to facilitate the production and release of crucial intermediates, decrease the free energy of the rate-determining step, improve the intrinsic HER activity of the catalyst, and consequently attain higher current density.
Rational design of RMSACs can improve their activity for electrolytic hydrogen production at high current density. The modulation strategies in essence modulate and improve Pt, Ru and other active sites, thus optimizing the electronic structure of the active sites, reducing the activation energy of the key decisive speed step in the HER process, which is conducive to the formation of intermediate products, and accelerating the adsorption and detachment process of the reactants on the catalysts. At the same time, reducing the destruction of the ligand structure of the active sites as well as the integrity of the carriers in a high current density environment allows RMSACs to exhibit more excellent performance in the high current density electrolysis process. However, due to the different electronic structures of different active sites, the means of modulation to enhance the performance of hydrogen production from water electrolysis at high current density are not the same. Meanwhile, the catalytic mechanisms occurring in the active sites of the catalysts are also different due to the different active metal elements, coordination structures and local environments. Therefore, it is even more necessary to rationally design the rare-metal single atom catalysts to show the optimal conformational relationship to adapt to the needs of hydrogen production by water electrolysis at high current density under different conditions.
5 Rational design of RMSACs under seawater electrolysis conditions for hydrogen production
Direct electrolysis of seawater has the potential to provide clean, low-carbon hydrogen while also purifying seawater, which is crucial for promoting and achieving sustainable hydrogen energy. In recent years, scientists have made significant efforts to advance the field of direct electrolysis of seawater for hydrogen production.173–177Although the electrode reaction that occurs at the cathode during the electrolysis of both freshwater and seawater is the HER, there are significant differences in the HER processes occurring in seawater electrolytes compared to those in freshwater. Firstly, unlike fresh water, seawater is compositionally complex. Complex impurities and ions in seawater (Na+, Mg2+, Ca2+, etc.) can lead to drastic changes in the pH of the electrode surface, which results in the formation of precipitates such as Ca(OH)2, Mg(OH)2, etc., and these precipitates can impede the active sites of the electrodes, resulting in poor stability.178–180 In addition, impurity ions may affect the adsorption/desorption behavior of reaction intermediates. High concentrations of chloride ions lead to chloride corrosion at the anode, which not only corrodes the electrodes, but also Cl− competes with the oxygen evolution reaction (OER) of the electrolyzed water, further inhibiting the electrolysis efficiency. Moreover, they can corrode most catalysts and electrodes containing metal elements.181–184 Secondly, the catalytic materials are not stable under high current density, strong alkali, and long-term operation conditions. Furthermore, compared to other hydrogen production technologies, seawater electrolysis is expensive, and it is currently difficult to achieve large scale electrolytic hydrogen production at current electricity prices. Therefore, finding solutions to the challenge of large scale green hydrogen production is key to transforming the future energy structure and building a clean and low-carbon energy system.185,186 Fortunately, the emergence of RMSACs has opened up the possibility of direct seawater electrolysis for hydrogen production.
5.1 Rational design of RMSACs to improve HER performance in seawater electrolysis
The most direct way to increase HER activity is to optimize the HER decisive step process on the active site. Therefore, it is essential to design RMSACs to accelerate the slow kinetics of HER and reduce the ultra-high overpotential of HER processes.187 Pan et al.188 developed an efficient catalytic material consisting of single-atom Ru–N4 sites and Ru nanoparticles anchored on nitrogen-doped carbon (Ru1+NPs/N–C) through the coordination pyrolysis strategy of formaldehyde melamine resin (Fig. 10). The HER performance of Ru1+NPs/N–C has been shown to be exceptional in performance tests. Ru1+NPs/N–C demonstrates an overpotential of 375 mV at a current density of 10 mA cm−2 and maintains stability in natural seawater (yellow sea seawater, pH = 7.8) for 24 hours. In artificially simulated seawater, it shows an overpotential of 58 mV and also remains stable for 24 hours. After 60 minutes of electrolysis, the Faraday efficiencies of Ru1+NPs/N–C in natural seawater and artificial simulated seawater were found to be 85.7% and 98.2%, respectively, displaying a noteworthy improvement over commercial 20% Pt/C. Meanwhile, the commercial 20% Pt/C failed to deliver comparable results. Subsequent experiments showed that the strong synergistic effect between the Ru–N4 site and Ru nanoparticles changed the electronic structure and accelerated the HER dynamics. Simultaneously, Ru nanoparticles can efficiently achieve the dissociation of H2O to produce adsorbed hydrogen and may also facilitate the process of combining adsorbed hydrogen atoms to form H2 followed by desorption from the single atom Ru–N4 site, thus improving the HER performance of the catalyst.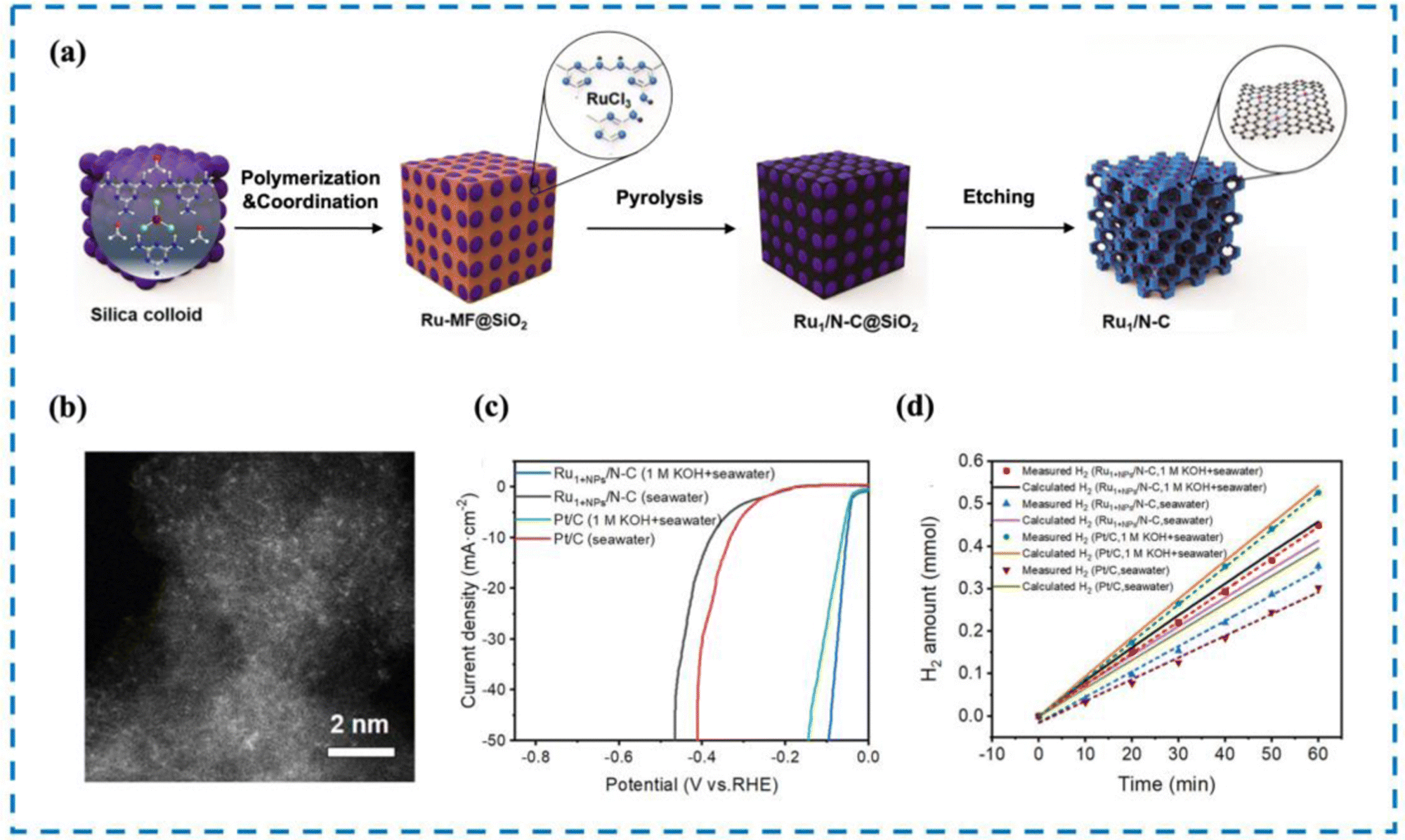 | ||
| Fig. 10 Synthesis, HAADF-STEM and HER performance of Ru1+NPs/N–C. (a) Synthesis of Ru1+NPs/N–C. (b) HAADF-STEM image. (c) LSV curve in seawater and 1 M KOH. (d) FE in seawater and 1 M KOH.188 This figure has been adapted/reproduced from ref. 188 with permission from American Chemical Society, copyright©2022, American Chemical Society. | ||
Through the use of DFT calculations, competitive coordination pairing (Ru–Ru bond) and single atoms (Ru–O bond) between Ru clusters, Qian and his colleagues189 were able to redistribute charge between Ru and O atoms in the ZnFe2O4 unit while balancing the formation of the new Ru–O bond and breaking of the ZnFe2O4 bond. This leads to the production and maintenance of individual Ru atoms, which boosts the binding of water molecules and optimizes the energy required for hydrogen adsorption (ΔGH*), resulting in significant HER activity at the Ru location. Meanwhile, Ru clusters contribute to the rapid evolution of electrons and protons into H2. These units share more free electrons to participate in the hydrogen desorption process, optimizing proton adsorption and hydrogen desorption. Based on this, the authors synthesized the Ru1,n–ZnFe2Ox–C catalyst. Thanks to the optimized coordination effect and electronic synergy between Ru clusters and single atoms, this catalyst exhibits excellent activity and stability in alkaline and seawater media with overpotentials as low as 10.1 and 15.9 mV, exceeding benchmark commercial Pt/C catalysts. In addition, it has significantly excellent mass activity, about 2 and 8 times higher than those of Pt catalysts in alkaline seawater (1 M KOH + seawater) and real seawater media, respectively.
It is worth mentioning that seawater electrolysis is a series reaction that includes the HER and OER, and the performance improvement of the HER can reduce the voltage loss of the whole electrolysis cell, thus reducing the energy consumption of the whole seawater electrolysis process. Even if the OER is the bottleneck step, optimization of the HER performance can improve the performance of the whole system to a certain extent, making the whole seawater electrolysis process more efficient. For example, by optimizing the cathode catalyst and design strategy, the electrical energy consumption can be effectively reduced, thus improving the efficiency of the whole electrolysis process. Efficient HER catalysts can accelerate hydrogen production, allowing more electrons to participate in the OER at the anode, thus indirectly improving the efficiency of the OER. Consequently, enhancing the HER activity is essential for the widespread application of RMSACs in seawater reactors. To design RMSACs for seawater reactors, it is crucial to consider the HER performance in seawater. However, solely focusing on HER activity is insufficient. To facilitate the large scale application of RMSACs in seawater reactors, it is also necessary to approach design from other perspectives.
5.2 Rational design of RMSACs to improve selectivity in seawater electrolysis
In seawater electrolysis, the elevated Cl− concentration in seawater causes it to move towards the anode due to the electric field, where it experiences an oxidation reaction.190 It has been shown that the key to the problem of catalyst selectivity in seawater electrolysis is the chlorine corrosion that occurs on the anode.191–194 Immobilization of reactive metals in the form of atoms on suitable carriers with optimized coordination environments and electronic structures is the key to solving the chlorine corrosion problems, such as unique coordination structure of the active sites, interactions between the active sites and carriers, etc. Therefore, rational design of RMSACs to optimize the coordination environments and electronic structures of rare metal atom centers and to increase the reaction energy barriers of ClOR and ClER while decreasing the OER activation energies of rare metal single-atom catalysts is an important idea to improve the selectivity of RMSACs in seawater electrolysis processes.Wu et al.195 synthesized a self-supported Pt–CoFe(II) layered double hydroxide (LDH) electrocatalyst using a self-assembly synthesis strategy (Fig. 11(a)–(c)). The Pt nanocatalysts were formed spontaneously by the oxidation of Fe2+ in the self-supporting CoFe(II) LDHs, without the need for external reducing agents or environments. With overpotentials of 214 mV and 15 mV at 10 mA cm−2, respectively, the catalyst demonstrated good performance in the OER and the HER in 1 M KOH. Even at high current densities of 100 and 500 mA cm−2, the catalyst remained stable for a minimum of 40 hours. In addition, when integrated into a real seawater electrolysis cell, the self-supported Pt–CoFe(II) LDHs required only 1.651 V and 1.858 V to achieve a current density of 500 mA cm−2 in 1 M KOH seawater electrolyte. The exceptional three-dimensional porous structure of self-supporting Pt–CoFe(II) LDH and the synergistic interaction between Pt clusters and CoFe hydroxide were the major factors influencing both its superior catalytic activity and improved chemical stability.
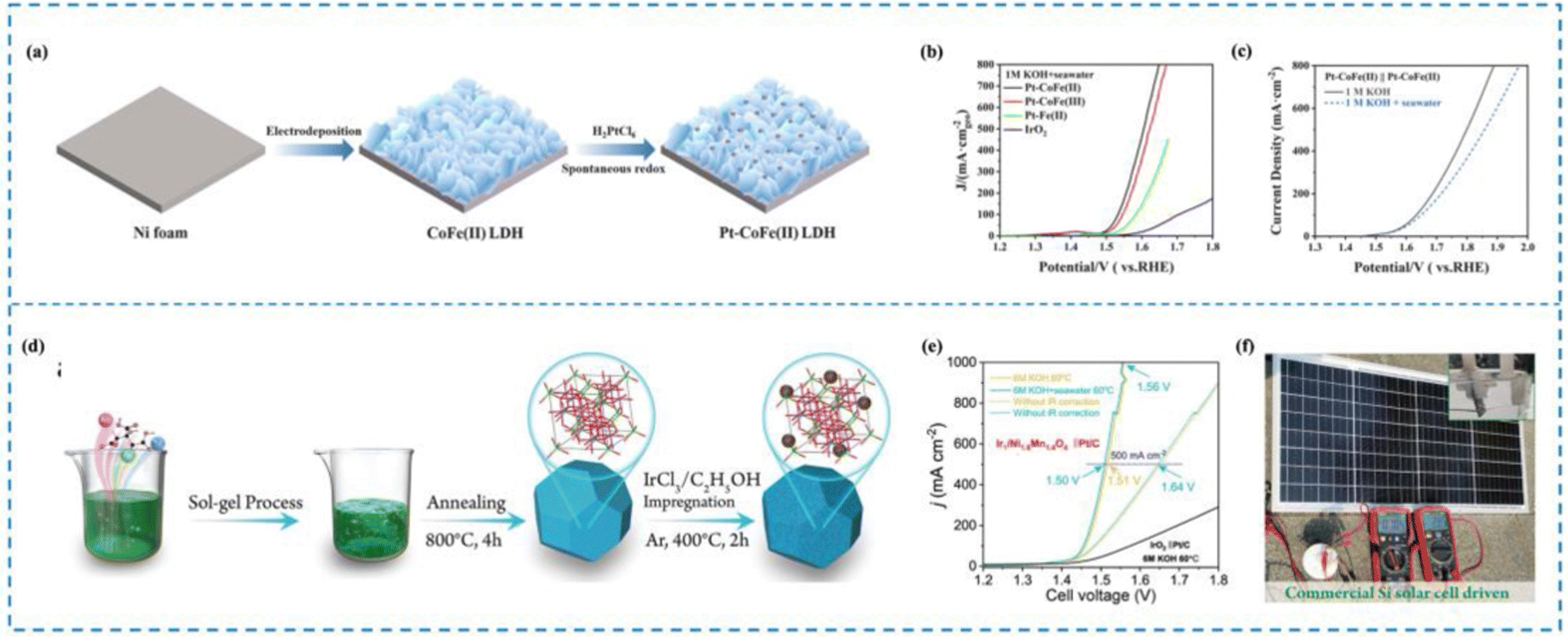 | ||
| Fig. 11 Synthesis and selectivity of Pt–CoFe(II) LDHs (a)–(c) and Ir1/Ni1.6Mn1.4O4 (d)–(f). (a) Synthesis of Pt–CoFe(II) LDHs. (b) AEMWE curve in seawater for Pt–CoFe(II) LDHs. (c) Overall water/seawater splitting performance of Pt–CoFe(II) LDHs in different electrolytes. (d) Synthesis of Ir1/Ni1.6Mn1.4O4. (e) AEMWE curve in seawater for Ir1/Ni1.6Mn1.4O4. (f) Optical image of Ir1/Ni1.6Mn1.4O4||Pt/C electrode pairs driven by silicon PV in 0.5 M KOH + seawater.195,196 This figure has been adapted/reproduced from ref. 195 and 196 with permission from Elsevier and John Wiley and Sons, copyright©2022 Elsevier B.V. All rights reserved. Copyright©2022 The Authors. Advanced Science published by Wiley-VCH GmbH. | ||
The study conducted by Wen et al.196 reported the preparation of Ir single atom (Ir SA)-immobilized spinel NixMn3−xO4 catalysts and screening of the best-performing catalyst, named Ir1/Ni1.6Mn1.4O4, for the OER in seawater (Fig. 11(d)–(f)). The catalyst exhibited low overpotentials of 330 and 350 mV for attaining 100 and 200 mA cm−2 current densities, respectively. Moreover, under industrial operating conditions, the assembled Ir1/Ni1.6Mn1.4O4 and Pt/C electrode pair achieved 500 mA cm−2 with only 1.50 V battery voltage. Subsequent experimental characterization and theoretical calculations have demonstrated that Mn-based oxides may exhibit excellent selectivity in alkaline electrolytes due to the surface polarization adsorption process of similar chloride ions. Moreover, the incorporation of Ir-SAs into the Mn-based oxide structure can further enhance the OER activity by facilitating the adsorption and activation of oxygen species on the catalyst surface. This is due to the unique electronic and structural properties of Ir-SAs, which provide highly active and stable active sites for the OER process. The authors attribute the excellent OER activity and selectivity of the catalyst in real seawater to the combined effect of the NixMn3−xO4 structure and the Ir SAs, which promote surface charge transfer kinetics and energy stable *OOH and unstable chloride ion adsorption.
Improving the selectivity of RMSACs for OER processes is an important means to enhance the hydrogen production rate of seawater electrolysis. Additionally, it can increase the lifespan of electrodes and decrease the formation of CER process products. In summary, the OER process is equally crucial for enhancing the rate of hydrogen production from seawater electrolysis as the HER process. It is unreasonable to solely focus on the HER activity of RMSACs while neglecting their selectivity for the OER process.
6 Rational design of RMSACs under long-term operation conditions for hydrogen production
The aggregation of metal single atoms during long cycle processes is the core issue that affects the stability of RMSACs. When the size of metal active particles decreases to the atomic level, their surface energy sharply increases, making single atoms more likely to aggregate.197 While N atoms can serve as anchor points for stabilizing individual metal atoms, the substrate of RMSACs can undergo corrosion over long periods of operation.198–203 This may disrupt the M–Nx structure, leading to aggregation of single atoms and ultimately reducing the stability of the catalyst. Significantly, poor long-term operation stability can increase the cost of catalyst synthesis and usage, which is not conducive to the large scale application of these catalysts in reactors. Therefore, designing RMSACs to improve their long-term stability is crucial for enabling the widespread adoption of RMSACs in long-term operation reactors on a large scale.6.1 Electronic metal–support interaction
The phenomenon of electron transfer between a metal and a support causing electronic disturbance is termed as electronic metal–support interaction (EMSI). Studies have indicated that EMSI has a distinctive function in managing the distribution and resilience of metallic particles, especially those metal atoms that have a high surface binding free energy.204,205 Therefore, the rational design of RMSACs and the incorporation of EMSI into them can effectively enhance the long-term cycle stability of RM SACs. Kuang et al.206 utilized a SiO2 template strategy to carbonize polydopamine (PDA) and prepare N-doped mesoporous hollow carbon spheres/Pt single-atom catalysts (Pt1/NMHCS) that possess a highly interconnected 3D porous structure and high specific surface area (Fig. 12(a) and (b)). In Pt1/NMHCS, isolated Pt atoms and adjacent C/N atoms are combined through covalent bonds to maximize the EMSI effect. Pt1/NMHCS exhibits excellent long-term stability in the reaction, as evidenced by only a 3 mV increase in overpotential after 3000 CV cycles at a current density of 10 mA cm−2. SEM, TEM, and HAADF-STEM characterization all demonstrate that Pt aggregation was not observed in Pt1/NMHCS before and after the reaction, unlike in 20 wt% Pt/C, which showed significant Pt particle aggregation after the reaction. Further experiments have shown that the stability of Pt1/NMHCS comes from its powerful EMSI effect. Compared to the feeble interaction between Pt and the carbon support observed in 20 wt% Pt/C, the robust interaction between Pt single atoms and NMHCS in Pt1/NMHCS induces substantial electron transfer. Notably, this interaction effectively impedes the movement and clustering of Pt single atoms during the reaction process. This is the reason why Pt1/NMHCS exhibits excellent long-term stability.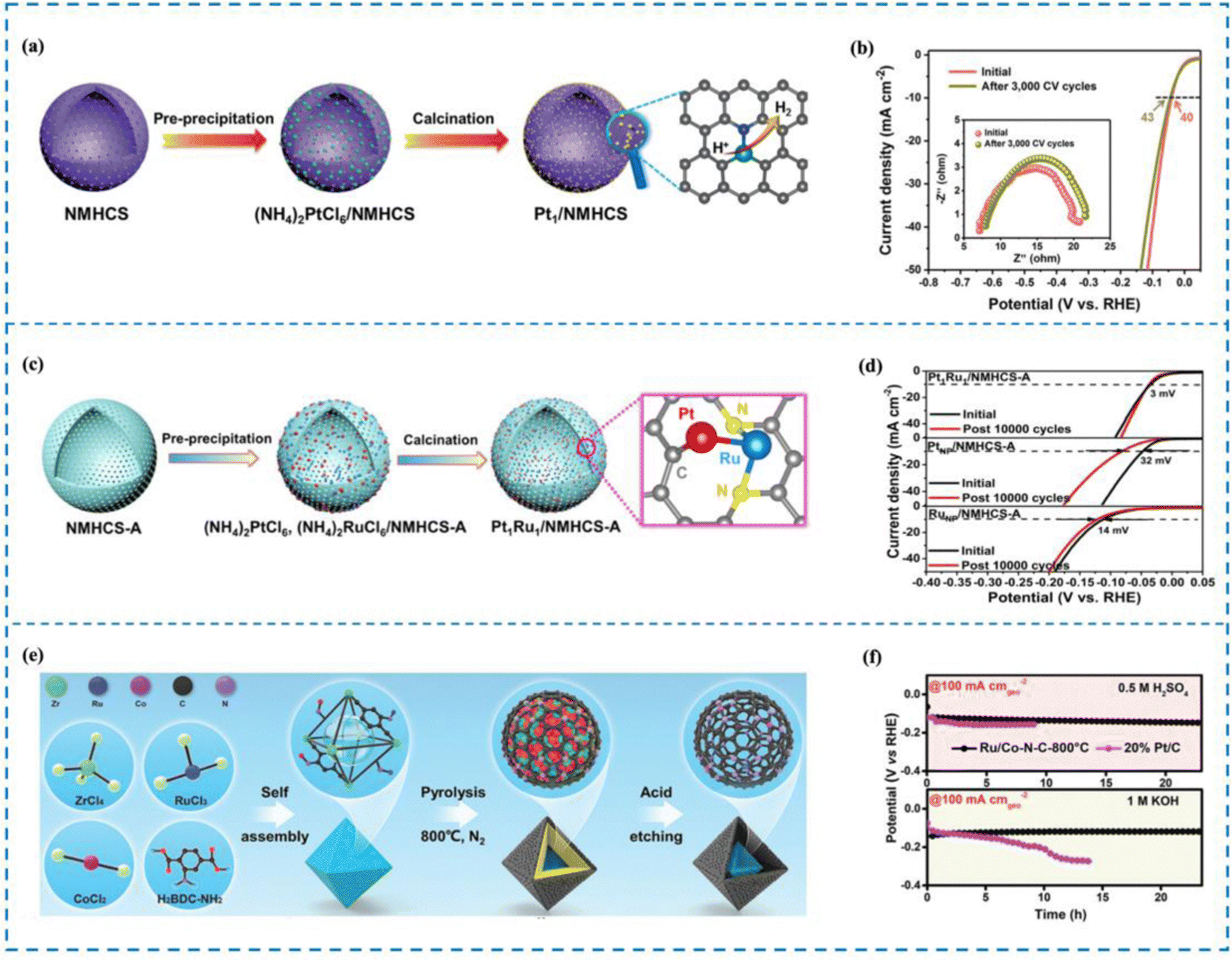 | ||
| Fig. 12 Synthesis and stability performance of Pt1/NMHCS (a) and (b), Pt1Ru1/NMHCS-ADAC (c), (d) and Ru/Co–N–C (e) and (f). (a) Synthesis of Pt1/NMHCS. (b) Stability performance of Pt1/NMHCS. (c) Synthesis of Pt1Ru1/NMHCS-ADAC. (d) Stability performance of Pt1Ru1/NMHCS-ADAC. (e) Synthesis of Ru/Co–N–C. (f) Stability performance of Ru/Co–N–C.206–208 This figure has been adapted/reproduced from ref. 206–208 with permission from John Wiley and Sons, American Chemical Society, copyright©2021, Wiley-VCH GmBh. Copyright©2022, American Chemical Society. Copyright©2022 The Authors. Advanced Materials published by Wiley-VCH GmbH. | ||
6.2 Dual-atom site regulation
When two metal atoms combine to form a dual-atom site within a SAC, their cooperation can have several beneficial effects.209–213 Firstly, this cooperation can optimize the electronic structure of the metal active site, thereby enhancing the activity of the catalyst for processes such as the HER. Additionally, the presence of the diatomic pair can induce the rearrangement of electrons around the metal active site, leading to further improvements in stability.214 Therefore, the stability of RMSACs in the HER process can be enhanced by introducing a second metal single atom. Zhao et al.207 carried out research in which Pt–Ru dual-site SACs (Pt1Ru1/NMHCS-ADAC) were synthesized using activated N-doped mesoporous hollow carbon spheres (NMHCS-A) as a support (Fig. 12(c) and (d)). They achieved remarkable performance in the HER by means of rational design. The Pt1Ru1/NMHCS-A DAC showed outstanding HER capabilities and had an overvoltage of just 22 mV when run at a current density of 10 mA cm−2. Importantly, the Pt1Ru1/NMHCS-A DAC demonstrated long-term stability. The overpotential increase (η10) was only 3 mV after 10000 cycles of CV. HAADF-STEM images indicated that Pt and Ru atoms maintained isolation and did not aggregate. In addition, the stability of the Pt1Ru1/NMHCS-A DAC in acidic environments was verified by chronoamperometric testing. Subsequent experiments demonstrated that the stable C1–Pt–Ru–N2 structure present in Pt1Ru1/NMHCS-A DAC facilitated strong interactions between Pt and Ru. Electronic rearrangement occurred around Pt atoms due to the presence of Ru atoms. This interaction and electronic arrangement contribute to the excellent HER stability observed for the Pt1Ru1/NMHCS-A DAC.
Rong and his co-workers208 developed Ru–Co diatomic catalysts immobilized on N-doped carbon through a pyrolysis etching strategy (Ru/Co–N–C) (Fig. 12(e) and (f)). The HER performance tests demonstrated that Ru/Co–N–C achieved a current density of 10 mA cm−2 in 1 M KOH with only 23 mV overpotential. Ru/Co–N–C demonstrated outstanding durability during the process of the HER, particularly. Chronoamperometric testing revealed that Ru/Co–N−C displayed exceptional durability for 20 hours at −50 mA cmgeo−2 and −100 mA cmgeo−2. In addition, the Ru/Co–N–C catalyst proved to be successful in functioning as a stable component of proton exchange membrane water electrolysis cells, sustaining operations at a high current density of 450 mA cm−2 for a total of 330 hours. Subsequent experiments elucidated that the introduction of the Co–N4 site triggered atomic separation structures and synergistic effects between the Ru–N4 site and Co–N4 site. This induced electron rearrangement around the Ru–N4 site, enhancing the corrosion resistance and long-term stability of Ru/Co–N–C during the HER process. These findings highlight the significance of the Co–N4 site in improving the performance and durability of the Ru/Co–N–C catalyst.
6.3 Coordination environment regulation
In the process of preparation of RMSACs, the stability of metal SACs for the HER can be affected by the bonding energy between the metal single atom and the ligand.215 If the bonding energy is low, the metal single atom should migrate on the support material and eventually agglomerate to form particles, leading to decreased stability of the SACs.216,217 A significant research finding is that the M–Nx structure can play a beneficial role in stabilizing metal single atoms (where M represents a metal).218–222 However, when considering an N-doped carbon support, the nitrogen species can vary, including pyridine-like nitrogen, pyrrole-like nitrogen, and quaternary nitrogen.223,224 Each nitrogen type possesses different abilities to stabilize metal single atoms. Generally, pyrrole-like nitrogen has been identified as achieving optimal stability and activity for metal single atoms. Therefore, it becomes crucial to optimize the coordination structure of the active site of RMSACs through rational design, aiming to achieve the pyrrole-like M–Nx structure. Zhang et al.225 conducted a comprehensive study investigating the influence of different types of nitrogen (N) on the stability of Pt SACs for the HER. The stability performance tests revealed that Pt single atoms anchored with pyrrole-like N exhibited the highest stability. In an accelerated degradation test (ADT) with 5000 cycles, the current density of the Pt SAC with pyrrole-like N was maintained at 88%. Subsequent investigations provided insights into how pyrrole-like N enhances the stability of Pt single atoms at multiple levels. Firstly, the introduction of pyrrole-like N induced more defects in the carbon support. These defects not only enhanced the HER activity of the catalyst but also stabilized individual Pt atoms, reducing their aggregation caused by migration within the support material. Secondly, the geometric effect of pyrrole-like N facilitated stronger binding between Pt single atoms and the pyrrole-like N groups. This enhanced binding helped capture and inhibit the aggregation of Pt single atoms during the catalyst synthesis process. Lastly, pyrrole-like N exhibited a stronger binding affinity for Pt single atoms, resulting in a more stable Pt–N bond. The overall improved stability of the catalyst can be attributed to the increased stability of the Pt single atom structure. The stability and long-term viability of Pt SACs for HER applications can be greatly improved by introducing pyrrole-like N and optimizing the coordination structure.6.4 Active site shielding/adsorption strategy
As previously described, impurities and ions in seawater can lead to poor stability of the electrolysis process. Cl− will be adsorbed on the active site of the catalyst and compete with the OER process, while the pH of the electrolyte will gradually change as the electrolysis process proceeds, generating OH−, which then combines with the metal ions to form a precipitate and prevents the electrolysis process from continuing. Therefore, in addition to rationally optimizing the active sites, coordination environments and local environments of the rare metal single-atom catalysts to maintain the structural stability, inhibiting the binding of Cl− to the active sites during the electrolysis process or stabilizing the pH of the electrolyte in the vicinity of the electrodes by altering the composition and structure of RMSACs is another important strategy to improve the performance of long-term electrolysis.Sun et al.57 synthesized a Pt single atom catalyst (Pt-SA) dispersed in ultrathin nickel-vanadium layered double hydroxide (NiV LDH) by a simple mixed-solvent preparation strategy. Tests showed that Pt-SA/NiV-LDH exhibited excellent activity and long-cycle electrolytic performance. In a simulated alkaline seawater environment, Pt-SA/NiV-LDH required 130 mV and 215 mV to achieve industrial-grade current densities of 1000 and 2000 mA cm−2, respectively, and could achieve a stability of 500 h at 500 mA cm−2, as well as exhibiting good chlorine resistance. Subsequent studies have shown that, thanks to the modification of the microscopic coordination environment of Pt-SA/NiV-LDH, the optimization of the NiV-LDH carrier, and the synergistic effect of Pt SA and NiV-LDH, Pt-SA/NiV-LDH is able to have a shielding effect on Cl− in the electrolyte while facilitating the adsorption and dissociation of the reaction intermediates of *H2O, *OH, and *H and can stabilize the pH of the electrolyte around the electrode and slow down the deposition of Ca(OH)2, Mg(OH)2 and other substances, thus enabling Pt-SA/NiV-LDH to exhibit excellent long-term operation stability.
Chen et al.226 synthesized a Ru–Mn diatomic catalyst (Ru/Mn–NiFe LDH) loaded onto NiFe LDH nanosheets. Tests showed that Ru/Mn–NiFe LDH exhibited excellent performance, requiring an overpotential of 44 mV and 210 mV to achieve 100 mA cmgeo−2 for HER and OER processes in alkaline seawater, respectively. What's more, Ru/Mn–NiFe LDH showed excellent activity and long-term operation stability in alkaline seawater, which was not only able to achieve a high current density of 2000 mA cmgeo−2, but also a stability of more than 252 h at industrial current density. In order to explain the source of the excellent activity and long-term operation stability of Ru/Mn–NiFe LDH, Chen et al. proposed a satellite shielding strategy. Specifically, the presence of Mn single atoms introduces a large number of Lewis acid sites, which are able to bind to OH− generated in situ at the cathode, slowing down the pH rise of the electrolyte and inhibiting the generation of metal hydroxide precipitates such as Ca(OH)2 and Mg(OH)2. At the same time, a large number of Lewis acid sites can rapidly replenish OH− at the anode to isolate Cl−, thus protecting the Ru single atom active sites, which improves the activity and long-term operation stability of Ru/Mn–NiFe LDH. At a high potential, the effectiveness of the Cl− shielding strategy may be greatly reduced. Therefore, Duan et al.227 improved the long-term stability of RMSACs in seawater electrolysis through a dynamic Cl− adsorption process that occurs at the active site. Specifically, Duan et al. synthesized an Ir single-atom catalyst loaded onto CoFe-LDH (Ir/CoFe-LDH). During the electrolysis of seawater, the interaction between the Ir SA and CoFe-LDH enables the Ir SA to adsorb Cl− from seawater, resulting in the formation of a specific Ir–OH/Cl coordination structure. This unique Cl adsorption state regulates the electronic structure of the Ir active center, which greatly improves the long-term operation stability of Ir/CoFe-LDH, achieving excellent stability of more than 1000 h at a current density of 400–800 mA cm−2.
7 Summary and outlook
7.1 RMSACs under high current operating conditions
Although RMSACs currently demonstrate excellent performance in the laboratory for the HER, there are significant challenges for their large scale applications in industrial HER processes.228 In order to achieve authentic industrial production, HER reactors must operate at industrial current densities exceeding 440 mA cm−2. However, at such high current densities, most RMSACs currently used face issues of poor HER performance under real operating conditions. Firstly, many RMSACs face limitations in achieving high current density due to factors such as the active site of the metal, coordination environment, and support. Secondly, under high current density, various side reactions can occur on the catalyst surface, hindering the HER process and leading to a decrease in catalyst performance. Continuous improvement and the pursuit of higher performance reactor catalysts are essential for addressing these challenges. It is worth noting that several reports have confirmed that optimizing metal–support interactions and interface effects, as well as using carbon materials as stable supports, can lead to the synthesis of RMSACs with high HER performance, even at high current densities.229,230 In conclusion, high current density electrolytic water reactors require high-efficiency catalysts with excellent performance and low price. Therefore, the development of RMSACs that can be adapted to high-current conditions and their scalable and controllable preparation technologies are of great significance for large scale hydrogen production.7.2 RMSACs for seawater electrolysis
The electrolysis of seawater for hydrogen production, which does not rely on limited freshwater resources, is anticipated to supplant current hydrogen production methods using water electrolysis and is viewed as a viable solution for large scale hydrogen generation. However, the complex composition of seawater poses challenges as various ions and impurities can affect the active sites of RMSACs, leading to catalyst poisoning and deactivation. Additionally, the high concentration of chloride ions (Cl−) in seawater can compete with the OER at the anode, hindering efficient hydrogen production through seawater electrolysis. Moreover, the precipitation of chlorine gas (Cl2) from the anode can cause electrode corrosion, damaging the electrode and reducing the efficiency of hydrogen production from electrolytic seawater.231 Currently, strategies such as electronic engineering, defect engineering, and interface engineering have been reported to precisely regulate the atomic orbital energy levels and electronic structures of RMSACs. These approaches strive to improve the inherent activity and self-sustainability of the catalyst. Nano-engineering techniques are employed to construct nano-porous structures, increase active sites, accelerate the mass transfer process, and improve the seawater electrolysis performance of RMSACs.232,233 With these advancements, it becomes possible to achieve seawater electrolysis even under industrial current densities. Therefore, the exploration and optimization of RMSAC structures are essential for improving the seawater HER performance and catalyst selectivity, ultimately driving the advancement of the seawater electrolysis hydrogen production sector.7.3 RMSACs under long-term operation conditions
A thorough investigation into the deactivation mechanism of RMSACs under actual working conditions can provide valuable insights for the design of more stable catalysts. The stability of RMSACs used for the HER is primarily evaluated in two aspects. Firstly, it involves their stability during storage, which can help reduce the costs associated with catalyst transportation. Secondly, the long-term stability of HER SACs under high current density is crucial for the industrialization. The primary cause of RMSAC deactivation during extended use is the aggregation that occurs as single atoms migrate. Research has revealed that when the particle size of active metals is reduced to the atomic level, the surface free energy dramatically increases, leading to the clear aggregation tendency of single atoms. Furthermore, under high current density, the electrode experiences more pronounced polarization, which affects the adsorption and mass transfer processes of intermediates on SACs. This interference severely impacts the activity and stability of the catalysts, limiting their industrial applications.234 Recently, numerous reports have successfully improved the stability of rare and precious metal-based single-atom catalysts under high current densities through rational design approaches. Investigating the long-term stability of RMSACs and enhancing the HER activity under prolonged operating conditions hold great significance in achieving large scale industrial applications of these catalysts.7.4 Application potential of non-rare-metal single atom catalysts
Some non-rare metal single atom catalysts (NRMSACs, Fe, Co, Ni, etc.) are also able to show better activity in the HER process. Therefore, through the continuous efforts of scientists, there are more reports on the application of NRMSACs for hydrogen production from electrolyzed water.235,236 Generally speaking, compared with rare metals, non-rare metals have higher elemental abundance and cheaper cost. At the same time, with the discovery of new strategies for regulating the active sites, coordination structures, electronic structures and local environments of non-rare metals, NRMSACs are sometimes able to exhibit the same catalytic activity as the RMSACs. Consequently, NRMSACs may be expected to replace RMSACs in the future, providing a new idea for low-cost and high-efficiency hydrogen production from electrolyzed water. However, there is still a long way to go before this goal can be realized. The applications of NRMSACs still have several key problems: (1) although the HER activities of some NRMSACs are very good, the activities of most of the NRMSACs still have a large gap compared with those of RMSACs, and at the same time there are deficiencies in the stability and selectivity and (2) currently, most of the studies on NRMSACs are still in the stage of laboratory exploration, and the researchers’ understanding of the active sites and catalytic mechanism is still unclear, which needs to be further explored. Overall, the development of low-cost, high-activity, selective and stable catalysts is an inevitable requirement for the realization of large scale electrolysis of water for hydrogen production. With continuous research on RMSACs and NRMSACs, a complete set of high-efficiency, cheap and green catalysts for water electrolysis will be designed in the future, which will provide new possibilities for the development of large scale hydrogen production by water electrolysis.Author contributions
Jiaye Li, Xu Tian, Changle Yue, Han Guo, Zhidong Wang, Mengdi Guo, Siying Huang and Song Yang: writing – original draft; Wei Lin, Yichuan Li, Bin Liu and Yuan Pan: writing – reviewing and editing; and Wei Lin and Yuan Pan: funding acquisition. All authors have given approval to the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the State Key Laboratory of Catalytic Materials and Reaction Engineering (RIPP, SINOPEC), National Natural Science Foundation of China (no. 22108306 and 22478432), Taishan Scholars Program of Shandong Province (no. tsqn201909065), and Shandong Provincial Natural Science Foundation (ZR2024JQ004 and ZR2021YQ15).Notes and references
- X. Tu, X. Liu, Y. Zhang, J. Zhu and H. Jiang, Green Carbon, 2024, 2, 131–148 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- X. Wang, W. Gao, Z. Zhao, L. Zhao, J. P. Claverie, X. Zhang, J. Wang, H. Liu and Y. Sang, Appl. Catal., B, 2019, 248, 388–393 CrossRef CAS.
- S. A. Lee, J. Kim, K. C. Kwon, S. H. Park and H. W. Jang, Carbon Neutralization, 2022, 1, 26–48 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- S. Yang, Z. H. Si, G. Z. Li, P. Zhan, C. Liu, L. Lu, B. X. Han, H. J. Xie and P. Y. Qin, Small, 2023, 19, 2207651 CrossRef CAS.
- F.-Y. Gao, P.-C. Yu and M.-R. Gao, Curr. Opin. Chem. Eng., 2022, 36, 100827 CrossRef.
- J. Liu, S. Duan, H. Shi, T. Wang, X. Yang, Y. Huang, G. Wu and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202210753 CrossRef CAS PubMed.
- T. Li, S. Y. Ren, C. Zhang, L. X. Qiao, J. Wu, P. He, J. Lin, Y. S. Liu, Z. G. Fu, Q. Z. Zhu, W. G. Pan, B. F. Wang and Z. W. Chen, Chem. Eng. J., 2023, 458, 141435 CrossRef CAS.
- J. N. Tiwari, N. K. Dang, S. Sultan, P. Thangavel, H. Y. Jeong and K. S. Kim, Nat. Sustain., 2020, 3, 556–563 CrossRef.
- S. A. Lee, S. E. Jun, S. H. Park, K. C. Kwon, J. H. Kang, M. S. Kwon and H. W. Jang, EES Catal., 2024, 2, 49–70 RSC.
- Z. X. Wei, Y. T. Zhu, J. Y. Liu, Z. C. Zhang, W. P. Hu, H. Xu, Y. Z. Feng and J. M. Ma, Rare Met., 2021, 40, 767–789 CrossRef CAS.
- Y. Q. Zhao, T. Ling, S. M. Chen, B. Jin, A. Vasileff, Y. Jiao, L. Song, J. Luo and S. Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 12252–12257 CrossRef CAS PubMed.
- P. Zhu, X. Xiong and D. Wang, Nano Res., 2022, 15, 5792–5815 CrossRef CAS.
- Y. Pan, S. J. Liu, K. A. Sun, X. Chen, B. Wang, K. L. Wu, X. Cao, W. C. Cheong, R. G. Shen, A. J. Han, Z. Chen, L. R. Zheng, J. Luo, Y. Lin, Y. Q. Liu, D. S. Wang, Q. Peng, Q. Zhang, C. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2018, 57, 8614–8618 CrossRef CAS.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
- S. Tian, M. Hu, Q. Xu, W. Gong, W. Chen, J. Yang, Y. Zhu, C. Chen, J. He, Q. Liu, H. Zhao, D. Wang and Y. Li, Sci. China Mater., 2021, 64, 642–650 CrossRef CAS.
- Y. Pan, C. Zhang, Z. Liu, C. Chen and Y. D. Li, Matter, 2020, 2, 78–110 CrossRef.
- X. Zheng, B. Li, Q. Wang, D. Wang and Y. Li, Nano Res., 2022, 15, 7806–7839 CrossRef CAS.
- R. Li and D. Wang, Nano Res., 2022, 15, 6888–6923 CrossRef CAS.
- Z. Zhang, M. Zhou, Y. Chen, S. Liu, H. Wang, J. Zhang, S. Ji, D. Wang and Y. Li, Sci. China Mater., 2021, 64, 1919–1929 CrossRef CAS.
- Z. Hou, L. Dai, J. Deng, G. Zhao, L. Jing, Y. Wang, X. Yu, R. Gao, X. Tian, H. Dai, D. Wang and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202201655 CrossRef CAS PubMed.
- Z. Liu, Y. Du, R. Yu, M. Zheng, R. Hu, J. Wu, Y. Xia, Z. Zhuang and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202212653 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- L. L. Cao, Q. Q. Luo, W. Liu, Y. K. Lin, X. K. Liu, Y. J. Cao, W. Zhang, Y. Wu, J. L. Yang, T. Yao and S. Q. Wei, Nat. Catal., 2019, 2, 134–141 CrossRef CAS.
- W. X. Chen, J. J. Pei, C. T. He, J. W. Wan, H. L. Ren, Y. Q. Zhu, Y. Wang, J. C. Dong, S. B. Tian, W. C. Cheong, S. Q. Lu, L. R. Zheng, X. S. Zheng, W. S. Yan, Z. B. Zhuang, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- J. Deng, H. B. Li, J. P. Xiao, Y. C. Tu, D. H. Deng, H. X. Yang, H. F. Tian, J. Q. Li, P. J. Ren and X. H. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- D. B. Li, X. Y. Li, S. M. Chen, H. Yang, C. D. Wang, C. Q. Wu, Y. A. Haleem, S. Duan, J. L. Lu, B. H. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef.
- K. Qi, X. Q. Cui, L. Gu, S. S. Yu, X. F. Fan, M. C. Luo, S. Xu, N. B. Li, L. R. Zheng, Q. H. Zhang, J. Y. Ma, Y. Gong, F. Lv, K. Wang, H. H. Huang, W. Zhang, S. J. Guo, W. T. Zheng and P. Liu, Nat. Commun., 2019, 10, 5231 CrossRef.
- H. J. Qiu, Y. Ito, W. T. Cong, Y. W. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. W. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS PubMed.
- X. P. Yin, H. J. Wang, S. F. Tang, X. L. Lu, M. Shu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 9382–9386 CrossRef CAS.
- X. J. Zeng, J. L. Shui, X. F. Liu, Q. T. Liu, Y. C. Li, J. X. Shang, L. R. Zheng and R. H. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef.
- L. H. Zhang, L. L. Han, H. X. Liu, X. J. Liu and J. Luo, Angew. Chem., Int. Ed., 2017, 56, 13694–13698 CrossRef CAS PubMed.
- A. W. Zhou, W. Ding-Sheng and L. Ya-Dong, Microstructures, 2022, 2, 2022005 CrossRef CAS.
- B. Wang, C. Cheng, M. Jin, J. He, H. Zhang, W. Ren, J. Li, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202207268 CrossRef CAS.
- D. W. Wang, Q. Li, C. Han, Z. C. Xing and X. R. Yang, Appl. Catal., B, 2019, 249, 91–97 CrossRef CAS.
- S. S. Niu, J. Yang, H. F. Qi, Y. Su, Z. Y. Wang, J. S. Qiu, A. Q. Wang and T. Zhang, J. Energy Chem., 2021, 57, 371–377 CrossRef CAS.
- G. M. Zhan, Y. C. Yao, F. J. Quan, H. Y. Gu, X. Liu and L. Z. Zhang, J. Energy Chem., 2022, 72, 203–209 CrossRef CAS.
- V. Ramalingam, P. Varadhan, H. C. Fu, H. Kim, D. L. Zhang, S. M. Chen, L. Song, D. Ma, Y. Wang, H. N. Alshareef and J. H. He, Adv. Mater., 2019, 31, 1903841 CrossRef CAS.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS.
- M. Yang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2013, 135, 3768–3771 CrossRef CAS PubMed.
- J. Deng, H. B. Li, J. P. Xiao, Y. C. Tu, D. H. Deng, H. X. Yang, H. F. Tian, J. Q. Li, P. J. Ren and X. H. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- L. Zhang, L. Han, H. Liu, X. Liu and J. Luo, Angew. Chem., Int. Ed., 2017, 56, 13694–13698 CrossRef CAS PubMed.
- T. Li, J. Liu, Y. Song and F. Wang, ACS Catal., 2018, 8, 8450–8458 CrossRef CAS.
- F. Luo, H. Hu, X. Zhao, Z. Yang, Q. Zhang, J. Xu, T. Kaneko, Y. Yoshida, C. Zhu and W. Cai, Nano Lett., 2020, 20, 2120–2128 CrossRef CAS PubMed.
- J. Li, C. Zhang, H. Ma, T. Wang, Z. Guo, Y. Yang, Y. Wang and H. Ma, Chem. Eng. J., 2021, 414, 128834 CrossRef CAS.
- H. Yao, X. Wang, K. Li, C. Li, C. Zhang, J. Zhou, Z. Cao, H. Wang, M. Gu, M. Huang and H. Jiang, Appl. Catal., B, 2022, 312, 121378 CrossRef CAS.
- L. Zeng, Z. A.-O. Zhao, Q. Huang, C. Zhou, W. A.-O. Chen, K. Wang, M. Li, F. Lin, H. Luo, Y. Gu, L. Li, S. Zhang, F. Lv, G. A.-O. Lu, M. Luo and S. A.-O. Guo, J. Am. Chem. Soc., 2023, 145, 21432–21441 CrossRef CAS.
- T. T. Chao, W. B. Xie, Y. M. Hu, G. Yu, T. H. Zhao, C. Chen, Z. D. Zhang, X. Hong, H. L. Jin, D. S. Wang, W. Chen, X. H. Li, P. Hu and Y. D. Li, Energy Environ. Sci., 2024, 17, 1397–1406 RSC.
- Y. Wu, M. Chen, D. Liu, H. Sun, T. Zhou, G. Na, G. Qiu, D. Li, Y. Chen, J. Zhao, Y. Zhang, J. Zhang, H. Pan, F. Liu, H. Cui and Q. Liu, J. Mater. Sci. Technol., 2025, 215, 111–120 CrossRef.
- L. Wang, M. Ma, C. Zhang, H.-H. Chang, Y. Zhang, L. Li, H.-Y. Chen and S. J. A. C. Peng, Angew. Chem., Int. Ed., 2023, 36, e202317220 Search PubMed.
- X. Q. Mu, X. Y. Gu, S. P. Dai, J. B. Chen, Y. J. Cui, Q. Chen, M. Yu, C. Y. Chen, S. L. Liu and S. C. Mu, Energy Environ. Sci., 2022, 15, 4048–4057 RSC.
- R. Yao, K. Sun, K. Zhang, Y. Wu, Y. Du, Q. Zhao, G. Liu, C. Chen, Y. Sun and J. Li, Nat. Commun., 2024, 15, 2218 CrossRef CAS.
- X. Lin, W. Hu, J. Xu, X. Liu, W. Jiang, X. Ma, D. He, Z. Wang, W. Li, L.-M. Yang, H. Zhou and Y. Wu, J. Am. Chem. Soc., 2024, 146, 4883–4891 CrossRef CAS PubMed.
- B. Zhou, J. Wang, L. Guo, H. Li, W. Xiao, G. Xu, D. Chen, C. Li, Y. Du, H. Ding, Y. Zhang, Z. Wu and L. J. A. E. M. Wang, Adv. Energy Mater., 2024, 2402372 CrossRef.
- H. Sun, H.-C. Chen, M. Humayun, Y. Qiu, J. Ju, Y. Zhang, M. Bououdina, X. Xue, Q. Liu, Y. Pang and C. Wang, Adv. Funct. Mater., 2024, 2408872 CrossRef CAS.
- Z. Xu, J. Zhu, Z. Shu, Y. Xia, R. Chen, S. Chen, Y. Wang, L. Zeng, J. Wang, Y. Cai, S. Chen, F. Huang and H.-L. Wang, Joule, 2024, 8, 1790–1803 CrossRef CAS.
- Z. Chen, X. Li, J. Zhao, S. Zhang, J. Wang, H. Zhang, J. Zhang, Q. Dong, W. Zhang, W. Hu and X. Han, Angew. Chem., Int. Ed., 2023, 62, e202308686 CrossRef CAS PubMed.
- Y.-C. Zhang, M. Zhao, J. Wu, Y. Wang, L. Zheng, F. Gu, J.-J. Zou, J. Gao and X.-D. Zhu, ACS Catal., 2024, 14, 7867–7876 CrossRef CAS.
- H. L. Duan, C. Wang, G. N. Li, H. Tan, W. Hu, L. Cai, W. Liu, N. Li, Q. Q. Ji, Y. Wang, Y. Lu, W. S. Yan, F. C. Hu, W. H. Zhang, Z. H. Sun, Z. M. Qi, L. Song and S. Q. Wei, Angew. Chem., Int. Ed., 2021, 60, 7251–7258 CrossRef CAS PubMed.
- J. J. Li, M. N. Banis, Z. H. Ren, K. R. Adair, K. Doyle-Davis, D. M. Meira, Y. Z. Finfrock, L. Zhang, F. P. Kong, T. K. Sham, R. Y. Li, J. Luo and X. L. Sun, Small, 2021, 17, 2007245 CrossRef CAS PubMed.
- L. Hou, H. Jang, X. Gu, X. Cui, J. Tang, J. Cho and X. Liu, EcoEnergy, 2023, 1, 16–44 CrossRef.
- S. Anantharaj, S. Noda, V. R. Jothi, S. Yi, M. Driess and P. W. Menezes, Angew. Chem., Int. Ed., 2021, 60, 18981–19006 CrossRef CAS PubMed.
- Q. Zhang and J. Guan, J. Power Sources, 2020, 471, 228446 CrossRef CAS.
- P. Aggarwal, D. Sarkar, K. Awasthi and P. W. Menezes, Coord. Chem. Rev., 2022, 452, 214289 CrossRef CAS.
- M. Kim, S.-h Kim, J. Park, S. Lee, I. Jang, S. Kim, C. Y. Lee, O. J. Kwon, H. C. Ham, J. T. Hupp, N. Jung, S. J. Yoo and D. Whang, Adv. Funct. Mater., 2023, 33, 2300673 CrossRef CAS.
- L. Zeng, Z. Zhao, Q. Huang, C. Zhou, W. Chen, K. Wang, M. Li, F. Lin, H. Luo, Y. Gu, L. Li, S. Zhang, F. Lv, G. Lu, M. Luo and S. Guo, J. Am. Chem. Soc., 2023, 145, 21432–21441 CrossRef CAS PubMed.
- T. Ma, H. Cao, S. Li, S. Cao, Z. Zhao, Z. Wu, R. Yan, C. Yang, Y. Wang, P. A. van Aken, L. Qiu, Y.-G. Wang and C. Cheng, Adv. Mater., 2022, 34, 2206368 CrossRef CAS.
- F. Yang, R. Yao, Z. Lang, F. Yu, H. Dong, Y. Wang, Y. Li and H. Tan, ACS Energy Lett., 2023, 8, 5175–5183 CrossRef CAS.
- Y. Da, Z. Tian, R. Jiang, G. Chen, Y. Liu, Y. Xiao, J. Zhang, S. Xi, W. Chen, X. Han and W. Hu, ACS Nano, 2023, 17, 18539–18547 CrossRef CAS PubMed.
- X. Chen, X.-T. Wang, J.-B. Le, S.-M. Li, X. Wang, Y.-J. Zhang, P. Radjenovic, Y. Zhao, Y.-H. Wang, X.-M. Lin, J.-C. Dong and J.-F. Li, Nat. Commun., 2023, 14, 5289 CrossRef CAS.
- L. Cao, X. Wang, C. Yang, J. Lu, X. Shi, H. Zhu and H.-P. Liang, ACS Sustainable Chem. Eng., 2021, 9, 189–196 CrossRef CAS.
- A. Han, X. Zhou, X. Wang, S. Liu, Q. Xiong, Q. Zhang, L. Gu, Z. Zhuang, W. Zhang, F. Li, D. Wang, L.-J. Li and Y. Li, Nat. Commun., 2021, 12, 709 CrossRef CAS.
- S. Zhu, X. Qin, Y. Yao and M. Shao, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef.
- Z. Lian, F. Wu, J. Zi, G. Li, W. Wang and H. Li, J. Am. Chem. Soc., 2023, 145, 15482–15487 CrossRef CAS PubMed.
- H. Tan, B. Tang, Y. Lu, Q. Ji, L. Lv, H. Duan, N. Li, Y. Wang, S. Feng, Z. Li, C. Wang, F. Hu, Z. Sun and W. Yan, Nat. Commun., 2022, 13, 2024 CrossRef CAS PubMed.
- R. Zhang, Y. Li, X. Zhou, A. Yu, Q. Huang, T. Xu, L. Zhu, P. Peng, S. Song, L. Echegoyen and F.-F. Li, Nat. Commun., 2023, 14, 2460 CrossRef CAS PubMed.
- B. B. Sarma, F. Maurer, D. E. Doronkin and J.-D. Grunwaldt, Chem. Rev., 2023, 123, 379–444 CrossRef CAS PubMed.
- Y. Cao, Y. Gao, H. Zhou, X. Chen, H. Hu, S. Deng, X. Zhong, G. Zhuang and J. Wang, Adv. Theory Simul., 2018, 1, 1800018 CrossRef.
- S. H. Talib, B. Bashir, M. A. Khan, B. Ali, S. Mohamed, A. Qurashi and J. Li, Energy Environ. Mater., 2024, 7, e12723 CrossRef CAS.
- F. Yaseen, M. A. Hashmi, Q. U. Ain, A. Lakhani and K. Ayub, Int. J. Hydrogen Energy, 2024, 57, 1389–1397 CrossRef CAS.
- P. Ge, Y. Liu, H. Liu, A. Sun, H. Yan, Y. Zhang, G. Ge, J. Yang and X. Yang, Mol. Catal., 2024, 553, 113775 CrossRef CAS.
- J. Ma, J. H. Cho, C. Lee, M. S. Kang, S. Choi, H. W. Jang, S. H. Ahn, S. Back and S. Y. Kim, Energy Environ. Mater., 2024, e12766 CrossRef CAS.
- F. Sun, Q. Tang and D.-E. Jiang, ACS Catal., 2022, 12, 8404–8433 CrossRef CAS.
- T. Li, S. Ren, C. Zhang, L. Qiao, J. Wu, P. He, J. Lin, Y. Liu, Z. Fu, Q. Zhu, W. Pan, B. Wang and Z. Chen, Chem. Eng. J., 2023, 458, 141435 CrossRef CAS.
- J. Kim, H.-E. Kim and H. Lee, ChemSusChem, 2018, 11, 104–113 CrossRef CAS.
- X. Liu, X. Song, G. Jiang, L. Tao, Z. Jin, F. Li, Y. He and F. Dong, Chem. Eng. J., 2024, 481, 148430 CrossRef CAS.
- H. Wang, C. Zhang, D. Zhang, L. Jiang, Y. Gao, T. Zhuang and Z. Lv, Small, 2024, 2403170 CrossRef CAS PubMed.
- J. W. Park, G. Park, M. Kim, M. Han, J. Jang, Y. Yamauchi, B. Yuliarto, P. Krüger, J. Kim, N. Park and H. Lim, Chem. Eng. J., 2023, 468, 143733 CrossRef CAS.
- V. Dao, G. Di Liberto, S. Yadav, P. Uthirakumar, K. Chen, G. Pacchioni and I.-H. Lee, Nano Lett., 2024, 24, 1261–1267 CrossRef CAS.
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W.-C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- Y. Huang, C. Zhu, J. Liao, X.-K. Gu and W.-X. Li, Chem. Eng. Sci., 2023, 270, 118551 CrossRef CAS.
- G. Di Liberto, L. A. Cipriano and G. Pacchioni, ACS Catal., 2022, 12, 5846–5856 CrossRef CAS.
- M. V. Jyothirmai, D. Roshini, B. M. Abraham and J. K. Singh, ACS Appl. Energy Mater., 2023, 6, 5598–5606 CrossRef CAS.
- D. Li, H. Li, Q. Wen, C. Gao, F. Song and J. Zhou, Adv. Funct. Mater., 2024, 34, 2313631 CrossRef CAS.
- W.-H. Lai, L.-F. Zhang, W.-B. Hua, S. Indris, Z.-C. Yan, Z. Hu, B. Zhang, Y. Liu, L. Wang, M. Liu, R. Liu, Y.-X. Wang, J.-Z. Wang, Z. Hu, H.-K. Liu, S.-L. Chou and S.-X. Dou, Angew. Chem., Int. Ed., 2019, 58, 11868–11873 CrossRef CAS PubMed.
- C. Liu, G. Pan, N. Liang, S. Hong, J. Ma and Y. Liu, Adv. Sci., 2022, 9, 2105392 CrossRef CAS.
- T. Bo, S. Cao, N. Mu, R. Xu, Y. Liu and W. Zhou, Appl. Surf. Sci., 2023, 612, 155916 CrossRef CAS.
- C. Yang, Z. Zhao and Q. Liu, Appl. Surf. Sci., 2022, 577, 151916 CrossRef CAS.
- Z. Shen, X. Fan, S. Ma, Y. An, D. Yang, N. Guo, Z. Luo and Y. Hu, Int. J. Hydrogen Energy, 2020, 45, 14396–14406 CrossRef CAS.
- D. Yi, F. Lu, F. Zhang, S. Liu, B. Zhou, D. Gao, X. Wang and J. Yao, Angew. Chem., Int. Ed., 2020, 59, 15855–15859 CrossRef CAS PubMed.
- S. Dong, Y. Li, H. Hu, R. Li, B. Yan, X. Zhang, Z. Wang, J. Zhang and L. Guo, J. Comput. Chem., 2023, 44, 15–26 CrossRef CAS PubMed.
- Y.-N. Zhu, X.-B. Li, Q. Zhang and F. Peng, ACS Appl. Mater., 2022, 14, 25592–25600 CrossRef CAS PubMed.
- L. Gao, D. Wu, S. Li, H. Li and D. Ma, J. Colloid Interface Sci., 2024, 676, 261–271 CrossRef CAS PubMed.
- Y. Li, S. Dong, X. Hu, W. Hou, J. Zhang, X. Zhang, H. Hu, W. Shang, C. Wu and H. Zhao, Int. J. Hydrogen Energy, 2024, 51, 957–969 CrossRef CAS.
- S. Dong, Y. Li, X. Zhang, H. Hu, B. Yan, J. Ren, L. Wan, H. Zhao and F. Sun, Mater. Des., 2023, 231, 112068 CrossRef CAS.
- S. H. Talib, Z. Lu, X. Yu, K. Ahmad, B. Bashir, Z. Yang and J. Li, ACS Catal., 2021, 11, 8929–8941 CrossRef CAS.
- Y. Wang, X. Zhang, Y. Yang, H. Wang, W.-M. Lau, C. Wang, Z. Fu, D. Pang, Q. Wang and J. Zheng, J. Colloid Interface Sci., 2025, 677, 491–501 CrossRef CAS PubMed.
- A. Munir, J. Abdul Nasir, T. U. Haq, J. Iqbal, I. Hussain and A. Qurashi, Coord. Chem. Rev., 2024, 521, 216112 CrossRef CAS.
- X. Gao, S. Dai, Y. Teng, Q. Wang, Z. Zhang, Z. Yang, M. Park, H. Wang, Z. Jia, Y. Wang and Y. Yang, Nano-Micro Lett., 2024, 16, 108 CrossRef CAS PubMed.
- R. Yao, Y. Wu, K. Zhang, S. Fan, Q. Zhao, J. Li and G. Liu, J. Energy Chem., 2024, 98, 503–511 CrossRef CAS.
- L. Feng, M. Zhou, D. He, H. Yin, Y. Huang, L. Cao, Y. Fang, D. Chu, Y. Liu, H. Chen, G. Li and J. Huang, Chem. Eng. J., 2024, 496, 154255 CrossRef CAS.
- X. Liu, Y. Zhou, J. Lin, X. Xiao, Z. Wang, L. Jia, M. Li, K. Yang, J. Fan, W. Yang and G. Li, Angew. Chem., Int. Ed., 2024, 63, e202406650 CrossRef CAS PubMed.
- J.-F. Huang, W.-J. Hsieh and J.-L. Chen, ACS Appl. Mater., 2024, 16, 27504–27510 CrossRef CAS PubMed.
- D. Yue, T. Feng, Z. Zhu, S. Lu and B. Yang, ACS Catal., 2024, 14, 3006–3017 CrossRef CAS.
- T. Zhang, L. Li, T. Huang, H. Wan, W.-Y. Chen, Z.-X. Yang, G.-F. Huang, W. Hu and W.-Q. Huang, Appl. Phys. Lett., 2024, 124, 063903 CrossRef CAS.
- S. Dong, Y. Li, X. Hu, S. Sun, B. Yan, H. Hu, X. Zhang, Z. Wang and L. Guo, Nanotechnology, 2022, 33, 275401 CrossRef CAS PubMed.
- H. Cao, Q. Wang, Z. Zhang, H.-M. Yan, H. Zhao, H. B. Yang, B. Liu, J. Li and Y.-G. Wang, J. Am. Chem. Soc., 2023, 145, 13038–13047 CrossRef CAS PubMed.
- D. Wang, Q. Li, C. Han, Z. Xing and X. Yang, Appl. Catal., B, 2019, 249, 91–97 CrossRef CAS.
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef.
- S. Niu, J. Yang, H. Qi, Y. Su, Z. Wang, J. Qiu, A. Wang and T. Zhang, J. Energy Chem., 2021, 57, 371–377 CrossRef CAS.
- J. Li, Y. Zhou, W. Tang, J. Zheng, X. Gao, N. Wang, X. Chen, M. Wei, X. Xiao and W. Chu, Appl. Catal., B, 2021, 285, 119861 CrossRef CAS.
- V. Ramalingam, P. Varadhan, H.-C. Fu, H. Kim, D. Zhang, S. Chen, L. Song, D. Ma, Y. Wang, H. N. Alshareef and J.-H. He, Adv. Mater., 2019, 31, 1903841 CrossRef CAS PubMed.
- Y. Han, H. Duan, W. Liu, C. Zhou, B. Wang, Q. Jiang, S. Feng, W. Yan, T. Tan and R. Zhang, Appl. Catal., B, 2023, 335, 122898 CrossRef CAS.
- L. Zhang, Q. Wang, L. Li, M. N. Banis, J. Li, K. Adair, Y. Sun, R. Li, Z.-J. Zhao, M. Gu and X. Sun, Nano Energy, 2022, 93, 106813 CrossRef CAS.
- T. Chu, G. Wang, X. Zhang, Y. Jia, S. Dai, X. Liu, L. Zhang, X. Yang, B. Zhang and F.-Z. Xuan, Nano Lett., 2024, 24, 9666–9674 CrossRef CAS.
- G. Liu, J. Li, C. Dong, L. Wu, D. Liang, H. Cao and P. Lu, Int. J. Hydrogen Energy, 2021, 46, 18294–18304 CrossRef CAS.
- J. Chen, G. Liu, Y.-Z. Zhu, M. Su, P. Yin, X.-J. Wu, Q. Lu, C. Tan, M. Zhao, Z. Liu, W. Yang, H. Li, G.-H. Nam, L. Zhang, Z. Chen, X. Huang, P. M. Radjenovic, W. Huang, Z.-Q. Tian, J.-F. Li and H. Zhang, J. Am. Chem. Soc., 2020, 142, 7161–7167 CrossRef CAS PubMed.
- Y. Zou, S. A. Kazemi, G. Shi, J. Liu, Y. Yang, N. M. Bedford, K. Fan, Y. Xu, H. Fu, M. Dong, M. Al-Mamun, Y. L. Zhong, H. Yin, Y. Wang, P. Liu and H. Zhao, EcoMat, 2023, 5, e12274 CrossRef CAS.
- C. Cai, K. Liu, L. Zhang, F. Li, Y. Tan, P. Li, Y. Wang, M. Wang, Z. Feng, D. Motta Meira, W. Qu, A. Stefancu, W. Li, H. Li, J. Fu, H. Wang, D. Zhang, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202300873 CrossRef CAS PubMed.
- S. A.-O. Lu, C. A.-O. Cheng, Y. A.-O. Shi, Y. A.-O. Wu, Z. A.-O. Zhang and B. A.-O. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2300549120 CrossRef CAS PubMed.
- K. Sun, X. Wu, Z. Zhuang, L. Liu, J. Fang, L. Zeng, J. Ma, S. Liu, J. Li, R. Dai, X. Tan, K. Yu, D. Liu, W.-C. Cheong, A. Huang, Y. Liu, Y. Pan, H. Xiao and C. Chen, Nat. Commun., 2022, 13, 6260 CrossRef CAS PubMed.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS.
- Y.-L. Sun, Y.-L. Deng, H.-N. Chen, X.-T. Yang, X.-M. Lin and J.-F. Li, Small Struct., 2023, 4, 2200201 CrossRef CAS.
- E. Zhu, W. Hu, J. Dong, J. Chen, Y. Li and L. Wang, Adv. Sustainable Syst., 2024, 2400224 CrossRef CAS.
- S. Xie, H. Dong, E. I. Iwuoha and X. Peng, Coord. Chem. Rev., 2024, 514, 215920 CrossRef CAS.
- M. Wang, K. Sun, W. Mi, C. Feng, Z. Guan, Y. Liu and Y. Pan, ACS Catal., 2022, 12, 10771–10780 CrossRef CAS.
- Y. Zhu, M. Klingenhof, C. Gao, T. Koketsu, G. Weiser, Y. Pi, S. Liu, L. Sui, J. Hou, J. Li, H. Jiang, L. Xu, W.-H. Huang, C.-W. Pao, M. Yang, Z. Hu, P. Strasser and J. Ma, Nat. Commun., 2024, 15, 1447 CrossRef CAS PubMed.
- Y. Zhu, K. Fan, C.-S. Hsu, G. Chen, C. Chen, T. Liu, Z. Lin, S. She, L. Li, H. Zhou, Y. Zhu, H. M. Chen and H. Huang, Adv. Mater., 2023, 35, 2301133 CrossRef CAS PubMed.
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W.-C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1800396 CrossRef.
- J. Liu, J. Liao, K. Huang, J. Dong, G. He, Z. Gong and H. Fei, Adv. Mater., 2023, 35, 2211398 CrossRef CAS.
- L. Lin, Z. Chen and W. Chen, Nano Res., 2021, 14, 4398–4416 CrossRef CAS.
- J. Fonseca and J. Lu, ACS Catal., 2021, 11, 7018–7059 CrossRef CAS.
- S. Jiao, M. Kong, Z. Hu, S. Zhou, X. Xu and L. Liu, Small, 2022, 18, 2105129 CrossRef CAS.
- S. M. Thalluri, J. Rodriguez-Pereira, R. Zazpe, B. Bawab, E. Kolíbalová, L. Jelinek and J. M. Macak, Small, 2023, 19, 2300974 CrossRef CAS PubMed.
- R. Li, J. Xu, Q. Zhao, W. Ren, R. Zeng, Q. Pan, X. Yan, J. Ba, T. Tang and W. Luo, Nano Res., 2022, 15, 1838–1844 CrossRef CAS.
- Y. Chen, R. Ding, J. Li and J. Liu, Appl. Catal., B, 2022, 301, 120830 CrossRef CAS.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed.
- Y. Hu, B. Li, C. Yu, H. Fang and Z. Li, Mater. Today, 2023, 63, 288–312 CrossRef CAS.
- T. Gan, Q. He, H. Zhang, H. Xiao, Y. Liu, Y. Zhang, X. He and H. Ji, Chem. Eng. J., 2020, 389, 124490 CrossRef CAS.
- X. Zhang, Y. Zhong, H. Chen, Y. Cheng, Q. Sun, H. Zhang, Q. He, Y. Zhang, G. Guo, X. He and H. Ji, Chem. Res. Chin. Univ., 2022, 38, 1258–1262 CrossRef CAS.
- T. Gan, Y. Liu, Q. He, H. Zhang, X. He and H. Ji, ACS Sustainable Chem. Eng., 2020, 8, 8692–8699 CrossRef CAS.
- H. Jin, S. Sultan, M. Ha, J. N. Tiwari, M. G. Kim and K. S. Kim, Adv. Funct. Mater., 2020, 30, 2000531 CrossRef CAS.
- G. Qian, J. Chen, T. Yu, L. Luo and S. Yin, Nano-Micro Lett., 2021, 13, 77 CrossRef PubMed.
- Y. Wan, L. Zhou and R. Lv, Mater Chem. Front., 2023, 7, 6035–6060 RSC.
- B. Zang, X. Liu, C. Gu, J. Chen, L. Wang and W. Zheng, Nanomaterials, 2024, 14, 1172 CrossRef CAS.
- Y. Zhu, X. Zhu, L. Bu, Q. Shao, Y. Li, Z. Hu, C.-T. Chen, C.-W. Pao, S. Yang and X. Huang, Adv. Funct. Mater., 2020, 30, 2004310 CrossRef CAS.
- D. Cao, Z. Zhang, Y. Cui, R. Zhang, L. Zhang, J. Zeng and D. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202214259 CrossRef CAS PubMed.
- L. Zeng, Z. Zhao, F. Lv, Z. Xia, S.-Y. Lu, J. Li, K. Sun, K. Wang, Y. Sun, Q. Huang, Y. Chen, Q. Zhang, L. Gu, G. Lu and S. Guo, Nat. Commun., 2022, 13, 3822 CrossRef CAS.
- M. Li, M. M. Wang, D. Y. Liu, Y. Pan, S. J. Liu, K. A. Sun, Y. J. Chen, H. Y. Zhu, W. Y. Guo, Y. P. Li, Z. M. Cui, B. Liu, Y. Q. Liu and C. G. Liu, J. Mater., 2022, 10, 6007–6015 CAS.
- T. Zhang, J. Jin, J. Chen, Y. Fang, X. Han, J. Chen, Y. Li, Y. Wang, J. Liu and L. Wang, Nat. Commun., 2022, 13, 6875 CrossRef CAS PubMed.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- C. L. Yue, C. Feng, G. X. Sun, N. Liu, H. Y. Hao, W. J. Bao, X. W. Zhang, F. Y. Sun, C. Zhang, J. H. Bi, Y. Zhou, H. C. Chen, Y. Pan, D. F. Sun and Y. K. Lu, Energy Environ. Sci., 2024, 17, 5227–5240 RSC.
- T. Luo, J. Huang, Y. Hu, C. Yuan, J. Chen, L. Cao, K. Kajiyoshi, Y. Liu, Y. Zhao, Z. Li and Y. Feng, Adv. Funct. Mater., 2023, 33, 2213058 CrossRef CAS.
- L. Zhu, H. Lin, Y. Li, F. Liao, Y. Lifshitz, M. Sheng, S.-T. Lee and M. Shao, Nat. Commun., 2016, 7, 12272 CrossRef CAS PubMed.
- S. E. Jun, S.-W. Myeong, B.-G. Cho, J. Kim, S. J. Park, C. Kim, T. H. Lee, S. Lee, J. Y. Kim, M. S. Kwon, J. H. Kang, K. C. Kwon, S. M. Choi, H. W. Jang and S. H. Park, Appl. Catal., B, 2024, 358, 124364 CrossRef CAS.
- Z. Liu, L. Zeng, J. Yu, L. Yang, J. Zhang, X. Zhang, F. Han, L. Zhao, X. Li, H. Liu and W. Zhou, Nano Energy, 2021, 85, 105940 CrossRef CAS.
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- M. Wang, M. Li, Y. Zhao, N. Shi, H. Zhang, Y. Zhao, Y. Zhang, H. Zhang, W. Wang, K. Sun, Y. Pan, S. Liu, H. Zhu, W. Guo, Y. Li, Y. Liu and C. Liu, J. Energy Chem., 2022, 67, 147–156 CrossRef CAS.
- Y. Wang, R. Morales-Martínez, X. Zhang, W. Yang, Y. Wang, A. Rodríguez-Fortea, J. M. Poblet, L. Feng, S. Wang and N. Chen, J. Am. Chem. Soc., 2017, 139, 5110–5116 CrossRef CAS.
- Y. Feng, W. Feng, J. Wan, J. Chen, H. Wang, S. Li, T. Luo, Y. Hu, C. Yuan, L. Cao, L. Feng, J. Li, R. Wen and J. Huang, Appl. Catal., B, 2022, 307, 121193 CrossRef CAS.
- C.-T. Dinh, A. Jain, F. P. G. de Arquer, P. De Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Zhang, M. Liu, D. Sinton, E. J. Crumlin and E. H. Sargent, Nat. Energy, 2019, 4, 107–114 CrossRef CAS.
- X. Y. Lu, J. Pan, E. Lovell, T. H. Tan, Y. H. Ng and R. Amal, Energy Environ. Sci., 2018, 11, 1898–1910 RSC.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef.
- H. Jin, X. Liu, A. Vasileff, Y. Jiao, Y. Zhao, Y. Zheng and S.-Z. Qiao, ACS Nano, 2018, 12, 12761–12769 CrossRef CAS.
- Y. Xu, H. Lv, H. Lu, Q. Quan, W. Li, X. Cui, G. Liu and L. Jiang, Nano Energy, 2022, 98, 107295 CrossRef CAS.
- H. Hu, Z. Zhang, Y. Zhang, T. Thomas, H. Du, K. Huang, J. P. Attfield and M. Yang, Energy Environ. Sci., 2023, 16, 4584–4592 RSC.
- L. Li, B. Wang, G. Zhang, G. Yang, T. Yang, S. Yang and S. Yang, Adv. Energy Mater., 2020, 10, 2001600 CrossRef CAS.
- N. Nie, D. Zhang, Z. Wang, W. Yu, S. Ge, J. Xiong, Y. Gu, B. Yang, J. Lai and L. Wang, Appl. Catal., B, 2022, 318, 121808 CrossRef CAS.
- Q. Lv, J. Han, X. Tan, W. Wang, L. Cao and B. Dong, ACS Appl. Energy Mater., 2019, 2, 3910–3917 CrossRef CAS.
- J.-P. Sun, Z. Zhao, J. Li, Z.-Z. Li and X.-C. Meng, Rare Met., 2023, 42, 751–768 CrossRef CAS.
- Y. Xu, Y. Fo, H. Lv, X. Cui, G. Liu, X. Zhou and L. Jiang, ACS Appl. Mater., 2022, 14, 10246–10256 CrossRef CAS PubMed.
- T. Yang, H. Lv, Q. Quan, X. Li, H. Lu, X. Cui, G. Liu and L. Jiang, Appl. Surf. Sci., 2023, 615, 156360 CrossRef CAS.
- H. Chen, Y. Zou, J. Li, K. Zhang, Y. Xia, B. Hui and D. Yang, Appl. Catal., B, 2021, 293, 120215 CrossRef CAS.
- G. Liu, M. Wang, Y. Xu, X. Wang, X. Li, J. Liu, X. Cui and L. Jiang, J. Power Sources, 2021, 486, 229351 CrossRef CAS.
- W. Zang, T. Sun, T. Yang, S. Xi, M. Waqar, Z. Kou, Z. Lyu, Y. P. Feng, J. Wang and S. J. Pennycook, Adv. Mater., 2021, 33, 2003846 CrossRef CAS.
- S. Wang, M. Wang, Z. Liu, S. Liu, Y. Chen, M. Li, H. Zhang, Q. Wu, J. Guo, X. Feng, Z. Chen and Y. Pan, ACS Appl. Mater., 2022, 14, 15250–15258 CrossRef CAS.
- C. Qian, W. Shao, X. Zhang, X. Mu, X. Gu, M. Yu, L. Ma, S. Liu and S. Mu, Small, 2022, 18, 2204155 CrossRef CAS PubMed.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- K. Izumiya, E. Akiyama, H. Habazaki, N. Kumagai, A. Kawashima and K. Hashimoto, Mater. Trans., JIM, 1997, 38, 899–905 CrossRef CAS.
- N. Menzel, E. Ortel, K. Mette, R. Kraehnert and P. Strasser, ACS Catal., 2013, 3, 1324–1333 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- L. Zhuang, J. Li, K. Wang, Z. Li, M. Zhu and Z. Xu, Adv. Funct. Mater., 2022, 32, 2201127 CrossRef CAS.
- J. Wu, Z. Nie, R. Xie, X. Hu, Y. Yu and N. Yang, J. Power Sources, 2022, 532, 231353 CrossRef CAS.
- N. Wen, Y. Xia, H. Wang, D. Zhang, H. Wang, X. Wang, X. Jiao and D. Chen, Adv. Sci., 2022, 9, 2200529 CrossRef CAS.
- J. Y. Li, P. Zhang and Y. Pan, Prog. Chem., 2023, 35, 643–654 CAS.
- H.-W. Liang, S. Brüller, R. Dong, J. Zhang, X. Feng and K. Müllen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- A. Han, W. Chen, S. Zhang, M. Zhang, Y. Han, J. Zhang, S. Ji, L. Zheng, Y. Wang, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1706508 CrossRef.
- C. Zhu, S. Fu, J. Song, Q. Shi, D. Su, M. H. Engelhard, X. Li, D. Xiao, D. Li, L. Estevez, D. Du and Y. Lin, Small, 2017, 13, 1603407 CrossRef PubMed.
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. Zhang, H.-L. Wang and L. Dai, Nano Energy, 2016, 29, 83–110 CrossRef CAS.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C.-W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 33, 2008599 CrossRef CAS PubMed.
- W. Zhao, C. Luo, Y. Lin, G.-B. Wang, H. M. Chen, P. Kuang and J. Yu, ACS Catal., 2022, 12, 5540–5548 CrossRef CAS.
- C. Rong, X. Shen, Y. Wang, L. Thomsen, T. Zhao, Y. Li, X. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS PubMed.
- X. Zheng, J. Yang, Z. Xu, Q. Wang, J. Wu, E. Zhang, S. Dou, W. Sun, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202205946 CrossRef CAS PubMed.
- W.-H. Li, J. Yang and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213318 CrossRef CAS PubMed.
- P. Zhang, K. Chen, J. Li, M. Wang, M. Li, Y. Liu and Y. Pan, Adv. Mater., 2023, 35, 2303243 CrossRef CAS.
- J. Zhang, Q.-a Huang, J. Wang, J. Wang, J. Zhang and Y. Zhao, Chin. J. Catal., 2020, 41, 783–798 CrossRef CAS.
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2021, 31, 2007423 CrossRef CAS.
- M. Li, H. Zhu, Q. Yuan, T. Li, M. Wang, P. Zhang, Y. Zhao, D. Qin, W. Guo, B. Liu, X. Yang, Y. Liu and Y. Pan, Adv. Funct. Mater., 2023, 33, 2210867 CrossRef CAS.
- X. Chen, W.-J. Ong, X. Zhao, P. Zhang and N. Li, J. Energy Chem., 2021, 58, 577–585 CrossRef CAS.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- G. M. Tomboc, T. Kim, S. Jung, H. J. Yoon and K. Lee, Small, 2022, 18, 2105680 CrossRef CAS.
- Y. Pan, M. Wang, M. Li, G. Sun, Y. Chen, Y. Liu, W. Zhu and B. Wang, J. Energy Chem., 2022, 68, 699–708 CrossRef CAS.
- Z.-F. Huang, J. Wang, Y. Peng, C.-Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef.
- T. Ling, D.-Y. Yan, H. Wang, Y. Jiao, Z. Hu, Y. Zheng, L. Zheng, J. Mao, H. Liu, X.-W. Du, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2017, 8, 1509 CrossRef PubMed.
- Y. Huang, L.-W. Jiang, B.-Y. Shi, K. M. Ryan and J.-J. Wang, Adv. Sci., 2021, 8, 2101775 CrossRef CAS PubMed.
- Z. Wang, H. Liu, R. Ge, X. Ren, J. Ren, D. Yang, L. Zhang and X. Sun, ACS Catal., 2018, 8, 2236–2241 CrossRef CAS.
- S. K. Kaiser, E. Fako, G. Manzocchi, F. Krumeich, R. Hauert, A. H. Clark, O. V. Safonova, N. López and J. Pérez-Ramírez, Nat. Catal., 2020, 3, 376–385 CrossRef CAS PubMed.
- D. Zhao, K. Sun, W.-C. Cheong, L. Zheng, C. Zhang, S. Liu, X. Cao, K. Wu, Y. Pan, Z. Zhuang, B. Hu, D. Wang, Q. Peng, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 8982–8990 CrossRef CAS.
- L. Zhang, Q. Wang, R. Si, Z. Song, X. Lin, M. N. Banis, K. Adair, J. Li, K. Doyle-Davis, R. Li, L.-M. Liu, M. Gu and X. Sun, Small, 2021, 17, 2004453 CrossRef CAS.
- H. Chen, Y.-Q. Wang, R. Ding, Z.-W. Zeng, B.-W. Liu, F.-R. Zeng, Y.-Z. Wang and H.-B. Zhao, Matter, 2024, 7, 3189–3204 CrossRef CAS.
- X. Duan, Q. Sha, P. Li, T. Li, G. Yang, W. Liu, E. Yu, D. Zhou, J. Fang, W. Chen, Y. Chen, L. Zheng, J. Liao, Z. Wang, Y. Li, H. Yang, G. Zhang, Z. Zhuang, S.-F. Hung, C. Jing, J. Luo, L. Bai, J. Dong, H. Xiao, W. Liu, Y. Kuang, B. Liu and X. Sun, Nat. Commun., 2024, 15, 1973 CrossRef CAS.
- S. Chen, T. Zhang, J. Han, H. Qi, S. Jiao, C. Hou and J. Guan, Nano Res. Energy, 2024, 3, e9120106 CrossRef.
- F. Lyu, S. Zeng, Z. Jia, F.-X. Ma, L. Sun, L. Cheng, J. Pan, Y. Bao, Z. Mao, Y. Bu, Y. Y. Li and J. Lu, Nat. Commun., 2022, 13, 6249 CrossRef CAS PubMed.
- E.-J. Lin, Y.-B. Huang, P.-K. Chen, J.-W. Chang, S.-Y. Chang, W.-T. Ou, C.-C. Lin, Y.-H. Wu, J.-L. Chen, C.-W. Pao, C.-J. Su, C.-H. Wang, U. S. Jeng and Y.-H. Lai, Appl. Surf. Sci., 2023, 615, 156372 CrossRef CAS.
- O. Fahad Aldosari, I. Hussain and Z. Malaibari, J. Energy Chem., 2023, 80, 658–688 CrossRef CAS.
- S. Pan, R. Li, J. Wang, Q. Zhang, M. Wang, B. Shi, P. Wang, Y. Zhao and X. Zhang, ACS Nano, 2023, 17, 4539–4550 CrossRef CAS PubMed.
- C. Feng, M. Chen, Z. Yang, Z. Xie, X. Li, S. Li, A. Abudula and G. Guan, J. Mater. Sci. Technol., 2023, 162, 203–226 CrossRef CAS.
- Y. Wang, X. Zheng and D. Wang, Nano Res., 2022, 15, 1730–1752 CrossRef CAS.
- Z. Li, X. Yan, D. He, W. Hu, S. Younan, Z. Ke, M. Patrick, X. Xiao, J. Huang, H. Wu, X. Pan and J. Gu, ACS Catal., 2022, 12, 7687–7695 CrossRef CAS.
- S. Eon Jun, S. Choi, J. Kim, K. C. Kwon, S. H. Park and H. W. Jang, Chin. J. Catal., 2023, 50, 195–214 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |