 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSinglet oxygen formation in non-aqueous oxygen redox chemistry: direct spectroscopic evidence for formation pathways and reliability of chemical probes†
Soumyadip
Mondal
 ,
Rajesh B.
Jethwa
,
Bhargavi
Pant
,
Robert
Hauschild
and
Stefan A.
Freunberger
*
,
Rajesh B.
Jethwa
,
Bhargavi
Pant
,
Robert
Hauschild
and
Stefan A.
Freunberger
*
Institute of Science and Technology Austria (ISTA), Am Campus 1, 3400 Klosterneuburg, Austria. E-mail: stefan.freunberger@ist.ac.at
First published on 17th May 2023
Abstract
Singlet oxygen (1O2) formation is now recognised as a key aspect of non-aqueous oxygen redox chemistry. For identifying 1O2, chemical trapping via 9,10-dimethylanthracene (DMA) to form the endoperoxide (DMA–O2) has become the main method due to its sensitivity, selectivity, and ease of use. While DMA has been shown to be selective for 1O2, rather than forming DMA–O2 with a wide variety of potentially reactive O-containing species, false positives might hypothetically be obtained in the presence of previously overlooked species. Here, we first provide unequivocal direct spectroscopic proof via the 1O2-specific near-infrared (NIR) emission at 1270 nm for the previously proposed 1O2 formation pathways, which centre around superoxide disproportionation. We then show that peroxocarbonates, common intermediates in metal–O2 and metal carbonate electrochemistry, do not produce false-positive DMA–O2. Moreover, we identify a previously unreported 1O2-forming pathway through the reaction of CO2 with superoxide. Overall, we provide unequivocal proof for 1O2 formation in non-aqueous oxygen redox chemistry and show that chemical trapping with DMA is a reliable method to assess 1O2 formation.
Introduction
Exploiting oxygen redox chemistry in batteries holds enormous promise towards enabling a true step change in energy storage employing either transition-metal oxide (TMO) intercalation materials or metal–O2 cells. However, both families of cell chemistries suffer from irreversible reactions originating from oxygen redox chemistry. Parasitic chemistry has long been ascribed to reactions of the electrolyte or carbon electrodes with superoxides or peroxides, owing to their radical nature, nucleophilicity, or basicity. However, in recent years this view has changed, and parasitic chemistry is now recognized to predominantly originate from the highly reactive singlet oxygen (1O2 or 1Δg), the first excited state of ground-state triplet oxygen (3O2 or 3Σg−). The exceptional importance of 1O2 formation in battery chemistry stems from its high reactivity towards, and degradation of, vital organic cell components, ultimately leading to cell failure.1O2 was found to form at all stages of cycling in Li–O2 and Na–O2 cells at rates that correspond to the parasitic chemistries occurring in those cells.1,2 A central insight was that 1O2 forms in the electrochemical system through chemical reactions,1,3–5 with a key generation pathway being the disproportionation of superoxide to peroxide and oxygen (Fig. 1a) as shown experimentally,3,64 and rationalized theoretically.3,7,8 The 1O2 yield is further impacted by both Lewis6,9 and Brønsted acids,8,10–12 and electrochemical oxidation of Li2CO3 has also been shown to be a source of this highly reactive species.5,13 Understandably, these results have far-reaching implications for the long term cyclability of most currently studied battery cathodes,14 and justifies the fervent interest in the field towards understanding and mitigating 1O2 formation.
 | ||
| Fig. 1 (a) Superoxide disproportionation as the main source of 1O2 during charge and discharge of metal–air batteries. (b) Methods to detect singlet oxygen (1O2) in non-aqueous oxygen redox systems. | ||
The topic of 1O2 formation in non-aqueous electrochemistry goes back to 1973 when Mayeda and Bard observed signatures for 1O2 using a chemical trap whereby superoxide was itself oxidized by ferrocenium.15 For metal–O2 cells, possible 1O2 formation was first hypothesized at high charging voltages (>3.9 V) in 2010 by Armand and coworkers,16 a theory which remained unverified for some time due to the difficulties in detecting 1O2. Clarifying the involvement of 1O2 in battery chemistry necessitates the use of sensitive and reliable methods for its detection.
The most direct evidence for 1O2 formation are the characteristic light emissions at 1270 nm and 633 nm (Fig. 1b). Given the short lifetime of 1O2 and the low quantum yields, detecting these emissions is insensitive and the technique remains semi-quantitative, especially in heterogeneous systems. 1O2 detection based on chemical probes has therefore become more common.17 Highly sensitive fluorescent 1O2 probes developed for life science are not stable in the required potential range of ∼2 to 4 V for battery applications. The initially used chemical probes in non-aqueous electrochemistry, diphenylisobenzofuran15 and 2,2,6,6-tetramethyl-4-piperidone (4-oxo-TEMP)18 were found to be prone to false positives for 1O2.19,20 For example, 4-oxo-TEMP reacts to form 4-oxo-TEMPO not only with 1O2 but also with peroxodicarbonates21 and it is not electrochemically stable over the required potential range.
Current knowledge about 1O2 in metal–O2 cells stems primarily from experiments with chemical trapping with 9,10-dimethylanthracene (DMA) to form its endoperoxide (DMA–O2) as shown in Fig. 1b. Its conversion can be followed by fluorescence, UV-Vis absorbance, HPLC, and NMR. Introduced by Mahne and coworkers1 into battery chemistry, it has become the most widely used method1,3,4,6,12–14,22–33 because of its selectivity, sensitivity, simplicity, and compatibility with the cell environment. DMA fulfils the requirement to rapidly form its endoperoxide (DMA–O2), which makes it sensitive to sub-percent 1O2 yields, and both DMA and DMA–O2 are electrochemically stable between ∼2–4 V vs. Li/Li+. DMA does not form DMA–O2 with a wide variety of potentially reactive O-containing species: KO2, O2˙−, Li2O2, Li2CO3, CO2, O2, and H2O, as well as Li2O2 and CO2 in combination.1,5 Hence, DMA does not produce false positives in the presence of any of these species. Complementary to trapping with DMA, 1O2 formation can be further verified by adding physical quenchers and measuring the change in evolved O2 (Fig. 1b).5
Concerning the specificity of chemical probes, Cummins and coworkers,21 as mentioned above, have shown 4-oxo-TEMP to be non-specific with regards to 1O2, leading to false positives in the presence of peroxodicarbonates, which are commonly found species in environments where a peroxide or superoxide are present together with CO2. Therefore, the question arises whether DMA may equally produce false positives under such conditions. To prove the previously proposed 1O2 formation and underlying mechanisms independently of chemical probes, direct spectroscopic proof would be highly desirable.
Here, using the 1270 nm NIR emission, we give direct spectroscopic proof for 1O2 formation by chemical steps as previously proposed based on experiments with DMA. These pathways centre around superoxide disproportionation, influenced by the cations present. In accord with the validity of the previous findings, DMA does not produce false positive DMA–O2 with peroxocarbonates. Moreover, we identify a previously unreported 1O2-forming pathway through the reaction of CO2 with superoxide.
Experimental
Materials and syntheses
Ethylene glycol dimethyl ether (DME, >99.0%), lithium hydroxide (LiOH, reagent grade, >98%), 9,10-dimethyl anthracene (DMA, 99%), titanium(IV) oxysulfate solution (TiOSO4, ∼15 wt% in dilute sulfuric acid, 99.99% trace metals basis), potassium carbonate (K2CO3, >99.7%) and diethyl ether (anhydrous, >99.7%) were purchased from Sigma Aldrich. Hydrogen peroxide (30% v/v) and sulfuric acid (H2SO4) were purchased from Fisher Scientific. Potassium hydroxide (KOH pellets, >85%), potassium superoxide (KO2, 96%), and sodium carbonate were purchased from abcr. Sodium hydroxide (NaOH pellets, >98%) was bought from Honeywell. Ethanol was purchased from VWR. Lithium carbonate (Li2CO3, 99%) was purchased from Alfa Aesar. Deionised water, where used, was sourced from a Millipore purification unit. All non-aqueous solvents were distilled and dried over molecular sieves (4 Å) prior to use. DMA was also recrystallized from ethanol under an inert atmosphere with light exclusion before use. All other chemicals were used as received. The studied peroxocarbonates were synthesized according to a previously reported procedure.34Analytical methods
A UV-vis spectrophotometer (JASCO) was used to quantify the amount of peroxide in the peroxocarbonate samples. The product was dissolved in a Ti(IV)OSO4 solution (2 wt%) in sulphuric acid (1 M) according to a previously reported method.36 On mixing the sample with the TiOSO4 solution (0.5 mL), the colourless solution changed to a deep orange indicating the presence of peroxide functionalities. A small volume (100 μL) of this solution was then further diluted with water (Millipore, 3 mL) and analysed by UV-vis spectroscopy.High-performance liquid chromatography (HPLC) was used to determine the degree of the DMA-to-DMA–O2 conversion.1 The samples were analysed by a reversed-phase column (Poroshell 120 EC-C8, 3.0 mm × 100 mm, Ø 2.7 μm, Agilent Technology) using a gradient system of acetonitrile (solvent B) and water containing 0.01% formic acid (solvent A). A pre-column (UHPLC 3 PK, Poroshell 120 EC-C8 3.0 mm × 5 mm, Ø 2.7 μm, Agilent Technology) was connected before the reversed-phase column. The elution at a flow rate of 0.7 mL min−1 started with 50% solvent B and was then increased to 100% solvent B within 7 minutes. The column was held at 20 °C throughout the measurements. The eluent was monitored via an UV-vis detector at a wavelength of 210 nm.
The mass spectrometry setup was developed and built in-house and is similar to the one described previously.36 It consists of a commercial quadrupole mass spectrometer (Hiden Analytical) with a turbomolecular pump (Edwards), which was backed by a membrane pump and an in-house-made leak inlet, which samples from the purge gas stream. The setup was calibrated for different gases (Ar, O2, CO2, H2, N2 and H2O) using calibration mixtures in steps over the anticipated concentration ranges to capture nonlinearity and cross-sensitivity. All calibrations and quantifications were performed using in-house software written in MATLAB. The purge gas system consists of a digital mass flow controller (Bronkhorst) and stainless steel or PEEK tubing.
The sample setup was as described previously36 and consisted of a glass vial with a 5 mL volume equipped with a small stirring bar. A PEEK plug with glued-in PEEK tubes and an exchangeable septum was sealed against the glass vial using a flat silicone-rubber seal. The entire stack was then compressed using an aluminium clamp. During the measurement, the solutions were added through a septum using a gas-tight syringe and the gas flow was regulated using a four-way valve. The gas flow was fixed to 5 mL min−1. A few milligrams of compound was added to sulphuric acid (0.5 mL) and injected into the vial. Before the measurement, all solutions were degassed with nitrogen for at least 15 minutes to remove any carbon dioxide and oxygen that may have been dissolved in the solvent.
FTIR spectra were recorded on a diamond ATR crystal (Bruker Alpha). Thermogravimetric (TGA) analysis was performed on a Linseis TGA 1000 thermogravimetric analyser under an argon flow with a heating rate of 10 K min−1 with the temperature ranging from 298–598 K (0–300 °C).
Chemiluminescence from 1O2 at 1270 nm was recorded using a NIR-sensitive photomultiplier tube (Hamamatsu H10330C-45-C3), which is sensitive from 950–1400 nm. A band-pass filter with transmission between 1200–1300 nm, constructed by combining a short-pass and a long-pass filter (Edmund optics), was placed directly in front of the detector. The cell was constructed from a 10 mm fluorescence high-precision quartz cuvette (Hellma Analytics) with a purpose-made, gas-tight PEEK cap connected with PTFE tubing to a loop valve for gas purging and for injection of the reaction solutions. A focusing mirror was placed behind the cuvette to guide a larger fraction of the emitted light to the detector. The signal was recorded from the detector using an oscilloscope (Pico Technology). All recorded data was normalised to a gain of 600 V (control voltage).
Computational methods
Density functional theory (DFT) calculations were carried out using an in-house HPC linux cluster with the ORCA code.37–40 Molecular models were built using the current stable Microsoft Windows version of Avogadro 1.97 and then relaxed using the universal force field offered by the Microsoft Windows version of Avogadro 1.2.41 An initial estimate was made based on chemical intuition. The models were initially geometrically optimised at the B3LYP42/def2-SVP43–46 level of theory with empirical dispersion D4.47 The optimised geometry was then used as an initial estimate for further optimisation at the B3LYP42/def2-TZVPP43–46 level of theory, again with dispersion corrections. The optimised structure was then taken as an initial estimate for calculation for optimisation at the B3LYP42/def2-QZVPP43–46 level of theory. This relaxed structure was then optimised with implicit solvation Polarisable Continuum Model (PCM).48 Frequencies were checked to ensure that the structures had successfully reached the ground state. The Gibbs free energies were used to estimate the reaction energies herein.Results and discussion
Spectroscopic evidence for 1O2 from superoxide disproportionation
We start with revisiting the previously identified pathways for singlet oxygen (1O2) formation in non-aqueous media, which centre around superoxide disproportionation in the presence of Lewis acids.1,3,23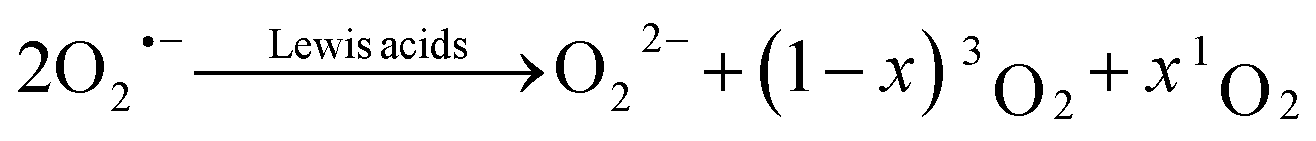 | (1) |
This reaction has been shown to be the dominant pathway to 1O2 generation during both charge and discharge of metal–O2 batteries. The mechanism of superoxide disproportionation (eqn (1)) and the impact of Lewis acids have previously been indirectly investigated via chemical trapping of 1O2 using 9,10-dimethylanthracene (DMA).1,3,4,23 Herein, these results will be independently verified by direct spectroscopic evidence.
Spectroscopic evidence of 1O2 formation can be obtained through two routes. The first is to observe the characteristic phosphorescence at 1270 nm (near-infrared, NIR) where a 1O2 molecule decays to a 3O2 molecule (monomol emission).49 The second is to observe the visible emission at 633 nm, which originates from the collision of two 1O2 molecules (dimol emission).18 Whilst it can be argued that visible light can be more sensitively detected than NIR radiation, the yield of the dimol emission scales with the square of the 1O2 concentration. This, when taken in combination with the short lifetime (in the order of μs) and correspondingly short diffusion lengths (sub-micron) of 1O2 results in low sensitivity at low concentrations.50,51 Moreover, most other luminescent processes in multicomponent systems occur at wavelengths below ∼1000 nm, which tends to preclude any unique assignment or attempt at identifying 1O2 formation through this. The NIR emission, however, is much preferred as the signal intensity is directly proportional to the 1O2 concentration, which makes it advantageous for quantification, especially at low 1O2 concentrations. The spectral separation of this process to other luminescent processes is also beneficial in this regard.
To sensitively probe the NIR emission from 1O2 generated in chemical reactions, we constructed a setup as detailed in the Experimental section. Briefly, it consisted of a sealed quartz cuvette, where solutions could be injected, a band-pass filter (1200–1300 nm) to sample the monomol emission at 1270 nm (Fig. S1, ESI†), and a commercial NIR photomultiplier tube (PMT), which has ∼2% quantum efficiency between 950 and 1350 nm. To assess the setup, we used the reaction of sodium hypochlorite (NaOCl) with hydrogen peroxide (H2O2), which produces 1O2 with a yield of 70%.52 Upon injecting H2O2 solution into the NaOCl solution, the output signal increased sharply before decaying within a couple of seconds (green trace in Fig. 2a). The integrated signal was directly proportional to the amount of 1O2 as shown in Fig. S3 (ESI†). Upon changing the solvent from water (H2O) to deuterium oxide (D2O), the integrated signal increased by a factor of ∼25 (purple trace in Fig. 2a and S4, ESI†). This increase is in accord with the ∼25-fold longer lifetime of 1O2 in D2O compared to H2O,49 and the generally recognised increase in lifetime afforded by deuterated solvents when compared to their standard analogues.53,54
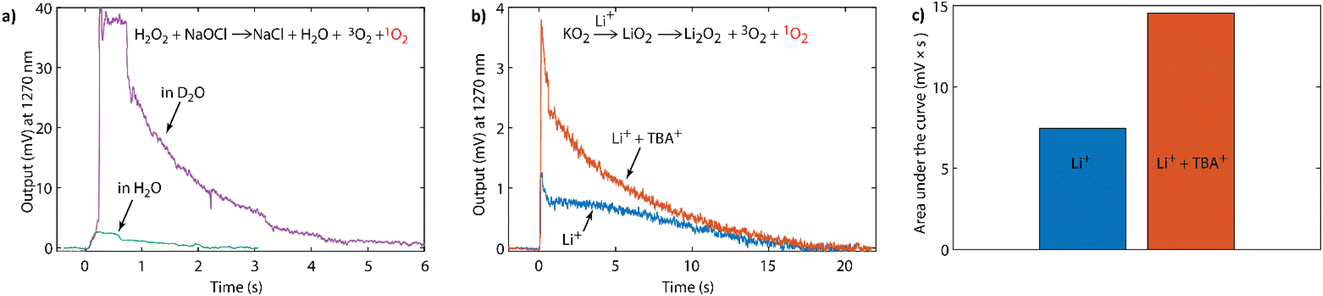 | ||
Fig. 2 (a) NIR-PMT signal upon injecting 0.5 mM NaOCl in water into 0.5 mM H2O2 with the solvent being either H2O or D2O. (b) NIR-PMT signal upon injecting 100 mM LiTFSI in CH3CN, or 100 mM Li+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TBA+ (1:9) TFSI− in CH3CN, into 1 mL suspension of KO2 mixed with 50 mM 18-crown-6 in CH3CN. (c) The integrated signals from (b). :TBA+ (1:9) TFSI− in CH3CN, into 1 mL suspension of KO2 mixed with 50 mM 18-crown-6 in CH3CN. (c) The integrated signals from (b). | ||
This NIR setup was used to detect the phosphorescence of 1O2 generated by superoxide disproportionation in non-aqueous media. Strong Lewis acids (such as Li+, Na+) drive the overall reaction in eqn (1) by stabilizing the peroxide relative to the superoxide.3 Weaker Lewis acids, in contrast, are unable to drive the disproportionation, but importantly reduce the energy barrier for 1O2 formation pathways and therefore strongly enhance the fraction of 1O2 produced. Weak Lewis acids for which this effect has been shown include organic cations found in ionic liquids, such as tetrabutylammonium3 and the oxidized forms of redox mediators.4 Potassium superoxide (KO2) is used as a convenient superoxide source due to its commercial availability.
Fig. 2b shows the intensity of the 1270 nm emission upon reacting KO2 with either pure Li+ electrolyte, pure TBA+ (tetra-nbutylammonium) electrolyte, or electrolyte containing both Li+ and TBA+ salts. KO2 powder was added with a crown-ether (50 mM, 18-crown-6) solution in acetonitrile (1 mL) into the sealed cuvette. The crown ether was used to increase the solubility of superoxide by increasing the ion-pair separation and hence the reaction rate. Upon addition of a solution of lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, 100 mM) in CH3CN the NIR emission signal rose sharply, before decaying within ∼20 s. In contrast, when KO2 was added to the lithium-free, TBA+-based solution no 1O2 emission was observed (Fig. S5, ESI†). However, reacting KO2 in a solution containing both Li+ and TBA+ enhanced the output signal compared to Li+ alone with a doubling of the integrated signal (Fig. 2b and c). This enhancement of 1O2 evolution in the presence of weak Lewis acids is in accord with our previous observations with organic cations3 and oxidized redox mediators.4
Additionally, if one compares the reaction traces between KO2 in the presence of LiTFSI (Fig. 2b), and H2O2 and NaOCl (Fig. 2a), it is also possible to semi-quantitatively estimate the 1O2 fraction. Whilst one expects there to be a difference in reaction rates, the longer reaction time of the former may also in part stem from the incomplete dissolution of the KO2 powder. Nonetheless, comparison of the integrated signal together with the consideration that 1O2 exhibits a similar lifetime in both D2O and CH3CN,49 suggests that the 1O2 yield from KO2/LiTFSI can be estimated to be a few percent of the total expected oxygen yield based on the amount of superoxide in the reaction. This is in accord with the previous quantification carried out by chemical trapping with DMA.1,3 However, as with H2O2 and NaOCl in Fig. 2a, it is possible to further enhance the signal. When changing the protic solvent to its deuterated analogue (CD3CN, see Fig. S6, ESI†), a ∼10-fold enhancement in the integrated signal of KO2/LiTFSI was observed, which is in agreement with the longer 1O2 lifetime.54 The fact that this isotope effect is observed can be seen as a further piece of evidence that the signal being observed at 1270 nm originates from 1O2.
When considering all the above results, the data strongly points towards 1O2 evolution during disproportionation of superoxide to peroxide and dioxygen. This is a key reaction step that occurs during both discharge and charge of Li– and Na–O2 cells (Fig. 1a). In situations where peroxide is the discharge product, superoxide disproportionation has been shown to be the dominant, or even exclusive, pathway in achieving full two-electron discharge3,23,55,56 and charge.4,57 Therefore, this reaction step, and its associated 1O2 generation mechanism, can be considered to be one of, if not the, major 1O2 forming pathways within metal–O2 batteries. The NIR results also further confirm the prominent influence of organic cations3 and oxidized redox mediators4 on this 1O2 formation pathway.
Reliability of DMA as a chemical 1O2 probe
While direct spectroscopic proof for 1O2 evolution as detailed above and before1 is most definitely desirable, there are certain limitations pertaining to its implementation. Firstly, the required NIR-PMT setups are typically expensive and may more commonly be found in specialist spectroscopic labs. Secondly, the technique remains semi-quantitative, especially in heterogeneous systems. Finally, the sensitivity is low, and the absence of a detectable signal cannot be considered definitive proof for the absence of sizable 1O2 formation, which may still be detected via chemical probes.Sodium and potassium peroxydicarbonate hydrates (Na2C2O6·H2O and K2C2O6·H2O), Fig. 3a, were synthesised using the method described by Jones et al.34 The compounds were characterized using multiple techniques, which are themselves described in the Experimental section. The FT-IR (Fig. S7, ESI†) results agree with the previous reports.34,35 FT-IR confirms the presence of OCO2 vibrations around 800 cm−1. These signals are lost following thermogravimetric analysis of the sample, as the metal carbonate is formed. The mass loss upon TGA (Fig. S8b, ESI†) is in agreement with the initial compounds being dominantly Na2C2O6·H2O and K2C2O6·H2O and smaller metal carbonate fractions. The peroxodicarbonate hydrates were further confirmed through MS analysis of the gases evolved upon heating of the compounds, where CO2, O2, and H2O (Fig. S8a, ESI†) were observed. Expelled CO2 was quantified by mass spectrometry and the peroxo-content by UV-vis spectroscopy of the Ti(IV)peroxo complex (Fig. S9, ESI†). Samples of Na2C2O6·H2O and K2C2O6·H2O, in a large excess, were exposed to a DMA (30 mM) solution in di-methoxy-ethane (DME) for 1 h and 2 days. The samples were analysed for the presence of DMA–O2 by HPLC (Fig. 3b and S7, ESI†), where analysis revealed that no DMA–O2 was generated, i.e., no false-positives for 1O2 were observed.
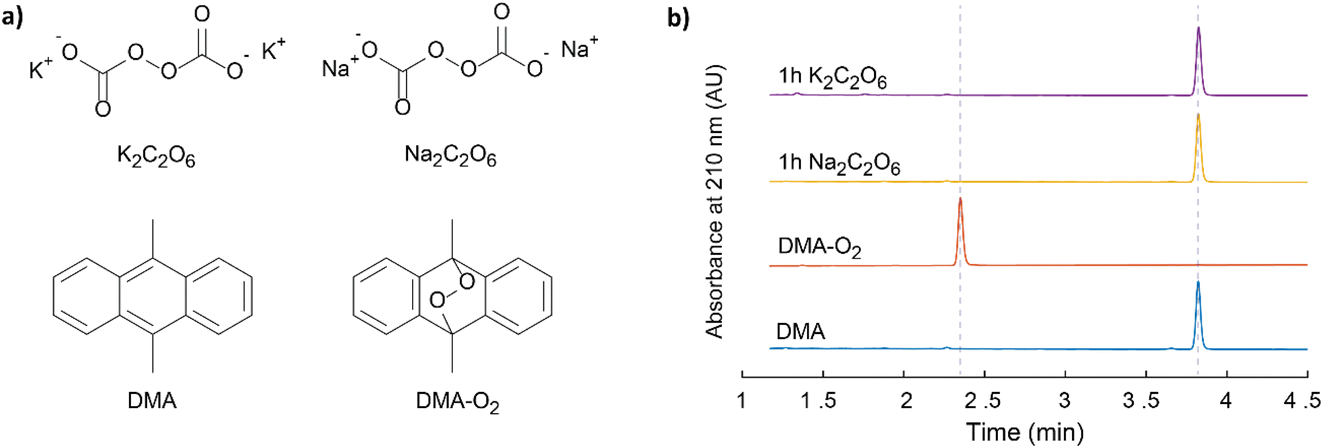 | ||
| Fig. 3 (a) The structures of the symmetric Na and K peroxodicarbonates, DMA and DMA–O2. (b) HPLC elugrams of these peroxodicarbonates in contact with 30 mM DMA in DME together with reference elugrams of DMA and DMA–O2. | ||
To fully explore the selectivity of DMA and its possible false-positives, the reaction of DMA˙+ with superoxide was also considered.58,59 This requires the simultaneous presence of two species, which have redox potentials of 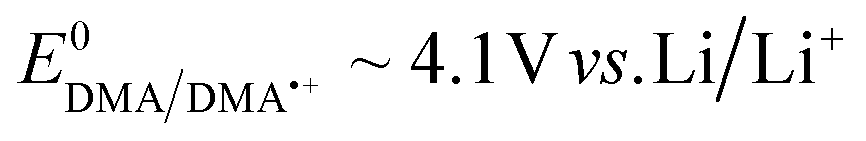 and
and 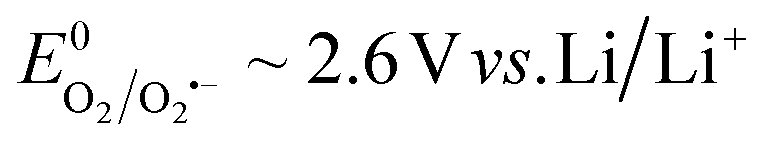 . The original work by Amatore achieved this using separated micro-band electrodes at both oxidizing and reducing potentials applied to a solution of dimethylanthracene in O2 saturated electrolyte, where DMA˙+ and superoxide could form in proximity.59 This is an atypical situation for a single electrode in metal–O2 cells. In the context of metal–O2 cells, simultaneous DMA˙+ and superoxide production could potentially arise in two ways. First, Li2O2 oxidation with an onset potential of ∼3 V has been shown to first produce LiO2, which then disproportionates to liberate 3O2 and 1O2.3,57 Beyond ∼4.1 V, DMA˙+ will form and could form DMA–O2. However, DMA–O2 formation has been observed from the onset of oxidation at ∼3 V and below 4.1 V, where DMA˙+ cannot form electrochemically.3 Second, DMA˙+ has been hypothesized to potentially form with peroxocarbonates as they were considered sufficiently oxidizing.58 However, this contradicts the previously shown oxidation potentials of peroxomonocarbonate (∼2.9 V vs. Li/Li+) and of peroxodicarbonate (∼3.3 V vs. Li/Li+).60 For either species, the initial intermediate on oxidation is the respective radical anion.61 Thermodynamically, metal peroxomono- or peroxodicarbonates and their radical anions cannot oxidise DMA because their oxidation potentials are well below the oxidation potential of DMA. To further confirm this, we exposed DMA to the synthesized peroxodicarbonates in DME solution and measured the UV-vis spectra of these solutions as well as the spectra of electrochemically formed DMA˙+. Figure S11 (ESI†) shows the complete absence of any DMA˙+ in the DMA solution that has been in contact with the peroxocarbonates.
. The original work by Amatore achieved this using separated micro-band electrodes at both oxidizing and reducing potentials applied to a solution of dimethylanthracene in O2 saturated electrolyte, where DMA˙+ and superoxide could form in proximity.59 This is an atypical situation for a single electrode in metal–O2 cells. In the context of metal–O2 cells, simultaneous DMA˙+ and superoxide production could potentially arise in two ways. First, Li2O2 oxidation with an onset potential of ∼3 V has been shown to first produce LiO2, which then disproportionates to liberate 3O2 and 1O2.3,57 Beyond ∼4.1 V, DMA˙+ will form and could form DMA–O2. However, DMA–O2 formation has been observed from the onset of oxidation at ∼3 V and below 4.1 V, where DMA˙+ cannot form electrochemically.3 Second, DMA˙+ has been hypothesized to potentially form with peroxocarbonates as they were considered sufficiently oxidizing.58 However, this contradicts the previously shown oxidation potentials of peroxomonocarbonate (∼2.9 V vs. Li/Li+) and of peroxodicarbonate (∼3.3 V vs. Li/Li+).60 For either species, the initial intermediate on oxidation is the respective radical anion.61 Thermodynamically, metal peroxomono- or peroxodicarbonates and their radical anions cannot oxidise DMA because their oxidation potentials are well below the oxidation potential of DMA. To further confirm this, we exposed DMA to the synthesized peroxodicarbonates in DME solution and measured the UV-vis spectra of these solutions as well as the spectra of electrochemically formed DMA˙+. Figure S11 (ESI†) shows the complete absence of any DMA˙+ in the DMA solution that has been in contact with the peroxocarbonates.
Overall, DMA–O2 formation via DMA˙+ and superoxide can therefore be excluded for charging potentials below 4.1 V vs. Li/Li+, as well as in the presence of peroxocarbonates.
Oxygenation of carbon dioxide by the superoxide ion
The superoxide anion and CO2 are ubiquitous in the context of metal–O2 batteries5,7,61,62 and O-redox transition metal oxides.14,63 Their reactions were first described by Sawyer64 and later detailed by Compton to be an ECE or disproportionation (DISP) mechanism.65Fig. 4a summarizes the previously proposed reaction steps to form species I, II, and IV. The other steps and the 1O2 formation are added based on our results below. First, nucleophilic addition of superoxide to CO2 to form the peroxocarbonate radical anion ˙OOC(O)O− or CO4˙− (I), which attacks another CO2 molecule to form the asymmetric peroxocarbonate radical anion ˙OOC(O)OC(O)O− or C2O6˙− (II). This radical is easily reduced to the dianion C2O62− (IV) either at an electrode or by another superoxide molecule, which evolves a dioxygen molecule. The latter steps can be regarded as DISP pathways given the superoxide state of the two involved O–O moieties, which then form peroxide and dioxygen. The peroxomonocarbonate radical anion CO4˙− (II) can also be reduced to its anionic form CO42− (III), which could carry out nucleophilic attack on another CO2 molecule to form the symmetric peroxodicarbonate anion C2O62− (V). Homolytic cleavage of this symmetric peroxodicarbonate anion, to form the carbonate radical, could result in the formation of metal carbonate (VI) and dioxygen via reaction with another equivalent of superoxide. The previously cited works used the poorly coordinating tetramethylammonium superoxide and therefore argued that the reduction of CO4˙− by a superoxide to the −OOC(O)O− was disfavoured by Coulombic repulsion. However, in the presence of chelating cations, such as lithium, this reaction pathway may become more viable. Nevertheless, the intermediates and steps were well verified in the previous works by NMR, IR, Raman spectroscopy, microelectrode studies, and chemical analysis.64,66 However, it remains unclear as to whether the asymmetric radical (C2O6˙−) or anion (C2O62−) could falsely produce DMA–O2 and what the multiplicity of the evolved dioxygen molecule might be. Therefore, we attempted to clarify these questions herein.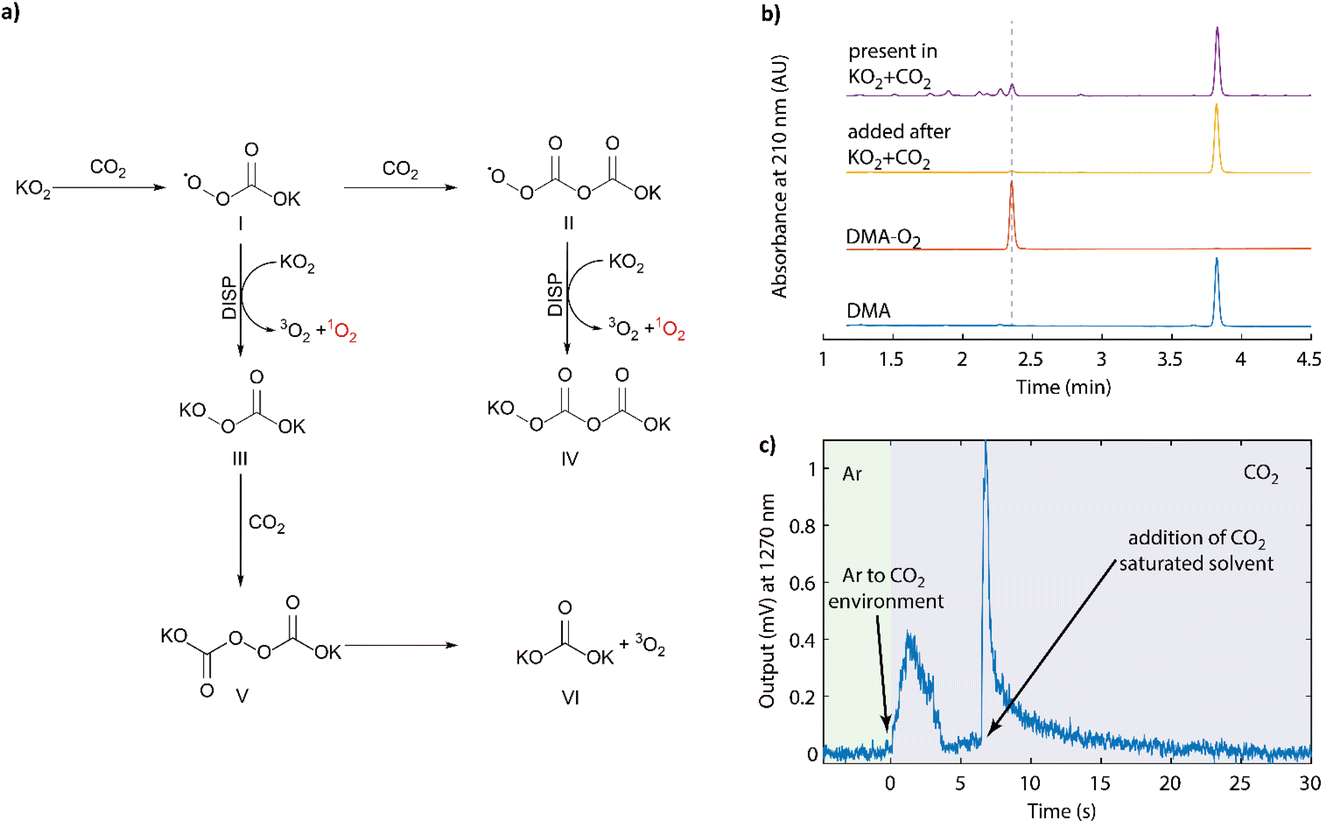 | ||
| Fig. 4 (a) Reaction sequence upon reacting superoxide with CO2 as initially described by Sawyer.64 The final DISP step forming 1O2 was identified in this work. (b) HPLC elugram of DMA exposed to asymmetric K2C2O6 (yellow trace) and DMA present during the reaction of KO2 and CO2 (purple trace). (c) NIR output signal around 1270 nm during the reaction of KO2 with CO2. The KO2 and 50 mM 18-crown-6 suspension in CH3CN was initially kept under Ar, then the headspace was purged by CO2, and then CO2-saturated CH3CN was added. | ||
First we produced the potassium salt of C2O62− by reacting KO2 and CO2 in DME as detailed in the Experimental section. The initially yellow suspension turned white within 20 h. The product was then isolated and analysed by FT-IR, which confirmed the formation of the asymmetric K2C2O6 along with some fraction of symmetric K2C2O6, K2CO3, and K2CO4 (Fig. S12, ESI†). This mixture of products can be easily explained when one considers the reactions depicted in Fig. 4a. The product was then stirred for 2 days in DMA (30 mM) solution in DME, and the filtered solution was then analysed by HPLC (Fig. 4b, yellow trace). Similar to the symmetric K2C2O6 discussed above, no DMA–O2 was observed indicating that the fully anionic peroxodicarbonate neither directly attacks DMA, nor forms DMA–O2via other reaction pathways.
To check the reactivity of the radical intermediates, we purged the headspace over a KO2 suspension in the presence of DMA with CO2. HPLC analysis showed that DMA–O2 formed in a large quantity (Fig. 4b, purple trace). The formation of a large quantity of DMA–O2 coupled with the absence of reactivity between K2C2O6 and DMA alone suggests that one of the intermediate species along the KO2/CO2 reaction pathway results in DMA–O2 formation. There are two possibilities to explain this behaviour: Firstly, one of the peroxocarbonate radical anions reacts with DMA to form DMA–O2, which would result in a false positive for 1O2 detection. Secondly, disproportionation of the peroxy-radicals I or II with KO2 to form the peroxy-carbonates III or IV could generate 1O2.
To probe whether 1O2 forms upon reacting KO2 with CO2, we detected the chemiluminescence around 1270 nm during the reaction in an analogous manner to Fig. 2a and b. The results are shown in Fig. 4c. The KO2/crown-ether (50 mM) suspension was prepared in acetonitrile as before and placed into the sealed cuvette under an argon atmosphere. After collecting a baseline output signal, the headspace gas in the cuvette was changed from argon to CO2, which resulted in a 1O2-specific NIR signal. Further CO2 was then added by adding CO2-saturated acetonitrile, which resulted in a sharp increase in the signal immediately after injection, followed by the expected decay. The result strongly suggests that the DISP step in the reaction sequence results in 1O2 formation. Tentatively, in combination with literature observations, we assign this to the reaction ˙OOC(O)OC(O)OK + KO2 → KOOC(O)OC(O)OK + 1O2, but it could also result from the initially formed KCO4˙.64
DFT calculations were undertaken to understand the energetics of the 1O2 pathway from oxygenation of CO2 by the superoxide ion. The relative Gibbs free energies ΔrG of the various reaction intermediates from Fig. 4a were considered for the lithiated, sodiated and potassiated superoxides and peroxomonocarbonates (see Fig. S13, ESI†) and the molecular species were used as proxies for the periodic structures. In all cases, formation of the mono-metallated peroxomonocarbonate radical resulted in an increase in the Gibbs free energy (Li: +68.71, Na: +42.36, K: +39.87 kJ mol−1) while formation of the di-metallated peroxomonocarbonate with a release of 3O2 resulted in a lowering of the Gibbs free energy (Li: −57.86, Na: −61.26, K: −29.80 kJ mol−1) relative to the energy of two superoxide molecules and a single molecule of carbon dioxide. This reduction in energy facilitates the overall disproportionation reaction of the two superoxide molecules resulting in oxygen release and peroxomonocarbonate formation. However, release of 1O2 by the analogous reaction path was found to result in an increase in free energy across all three alkali metal cations (Li: +103.72, Na: +100.32, K: +131.78 kJ mol−1). This may initially indicate a trend running in opposition to the experimental results presented here. However, when considering that these initial calculations do not incorporate the significant enthalpic driving force of peroxomonocarbonate crystallisation and the fact that the ΔrG of 1O2 release from LiO2 and LiCO4 disproportionation only has an energetic penalty of 35.01 kJ mol−1, it is not difficult to imagine that the true driving force for the disproportionation carries enough thermodynamic driving force to facilitate an observable fraction of 1O2 to be produced.
The discovery of 1O2 from the reaction of superoxide with CO2 has far-reaching implications as a 1O2-forming pathway in metal–O2/CO2 batteries and on transition metal oxide intercalation materials. It helps explain 1O2 formation upon carbonate oxidation5,7 and may be the key to the so far inconclusively explained degradation reactions on transition metal oxides14,67,68 and 1O2 generation mechanism.18,69
Conclusions
In conclusion, we have shown direct spectroscopic evidence of the previously identified pathways for singlet oxygen (1O2) formation in non-aqueous media, which centre around superoxide disproportionation in the presence of Lewis acids. The mechanism of superoxide disproportionation for 1O2 formation was previously studied by using DMA as a chemical trap owing to its selectivity, sensitivity, simplicity, and compatibility with the cell environment. The results described herein further support the previous studies and verify that the results were not due to the formation of false-positive endoperoxide (DMA–O2). Moreover, we rule out several potential pathways to false-positive DMA–O2, including exposure to the commonly observed peroxocarbonate anions. Through this, we further demonstrate the efficacy of DMA as a chemical trap for 1O2 detection. Finally, through our efforts here we identify through spectroscopy that the peroxocarbonate radical anion is capable of 1O2 formation and that this pathway likely proceeds via a disproportionation of the radical anion either with another molecule in this class, or with a molecule of superoxide.Author contributions
S. M. and B. P. carried out the synthesis and characterization. S. M. and R. H. developed the NIR setup and S. M. carried out the chemiluminescence study. R. B. J. carried out the DFT calculations. S. M. and S. A. F. wrote the initial version of the manuscript. S. A. F. conceptualized and supervised the work. S. M., R. B. J., and S. A. F. contributed to the interpretation of the results and manuscript revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. F. is indebted to ISTA for support. R. B. J. thanks the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101034413 for funding. We thank EL-Cell GmbH (Hamburg, Germany) for the OEMS test cell. Likewise, we thank E. Dutkiewicz for help with HPLC and P. Trosej for help with thermal analysis. This research was supported by the Scientific Service Units of ISTA through resources provided by the Imaging & Optics Facility, the Lab Support Facility, the Miba Machine Shop, and Scientific Computing.Notes and references
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 17036 CrossRef CAS.
- A. C. Luntz and B. D. McCloskey, Nat. Energy, 2017, 2, 17056 CrossRef.
- E. Mourad, Y. K. Petit, R. Spezia, A. Samojlov, F. F. Summa, C. Prehal, C. Leypold, N. Mahne, C. Slugovc, O. Fontaine, S. Brutti and S. A. Freunberger, Energy Environ. Sci., 2019, 12, 2559 RSC.
- Y. K. Petit, E. Mourad, C. Prehal, C. Leypold, A. Windischbacher, D. Mijailovic, C. Slugovc, S. M. Borisov, E. Zojer, S. Brutti, O. Fontaine and S. A. Freunberger, Nat. Chem., 2021, 13, 465 CrossRef CAS.
- N. Mahne, S. E. Renfrew, B. D. McCloskey and S. A. Freunberger, Angew. Chem., Int. Ed., 2018, 57, 5529 CrossRef CAS.
- S. Dong, S. Yang, Y. Chen, C. Kuss, G. Cui, L. R. Johnson, X. Gao and P. G. Bruce, Joule, 2022, 6, 185 CrossRef CAS.
- G. Houchins, V. Pande and V. Viswanathan, ACS Energy Lett., 2020, 5, 1893 CrossRef CAS.
- A. Pierini, S. Brutti and E. Bodo, npj Comput. Mater., 2021, 7, 126 CrossRef CAS.
- A. Pierini, S. Brutti and E. Bodo, ChemPhysChem, 2020, 21, 2060 CrossRef CAS.
- D. Córdoba, H. B. Rodríguez and E. J. Calvo, ChemistrySelect, 2019, 4, 12304 CrossRef.
- I. Lozano, D. Córdoba, H. B. Rodríguez, I. Landa-Medrano, N. Ortiz-Vitoriano, T. Rojo, I. R. de Larramendi and E. J. Calvo, J. Electroanal. Chem., 2020, 872, 114265 CrossRef CAS.
- D. Córdoba, H. B. Rodríguez and E. J. Calvo, J. Phys. Chem. C, 2023, 127(1), 78–84 CrossRef.
- D. Cao, C. Tan and Y. Chen, Nat. Commun., 2022, 13, 4908 CrossRef CAS PubMed.
- L. A. Kaufman and B. D. McCloskey, Chem. Mater., 2021, 33, 4170 CrossRef CAS.
- E. A. Mayeda and A. J. Bard, J. Am. Chem. Soc., 1973, 95, 6223 CrossRef CAS.
- J. Hassoun, F. Croce, M. Armand and B. Scrosati, Angew. Chem., Int. Ed., 2011, 50, 2999 CrossRef CAS PubMed.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181 RSC.
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825 CrossRef CAS.
- G. Nardi, I. Manet, S. Monti, M. A. Miranda and V. Lhiaubet-Vallet, Free Radical Biol. Med., 2014, 77, 64 CrossRef CAS.
- J. Moan and E. Wold, Nature, 1979, 279, 450 CrossRef CAS PubMed.
- S. Zhang, M. J. Nava, G. K. Chow, N. Lopez, G. Wu, D. R. Britt, D. G. Nocera and C. C. Cummins, Chem. Sci., 2017, 8, 6117 RSC.
- Y. K. Petit, C. Leypold, N. Mahne, E. Mourad, L. Schafzahl, C. Slugovc, S. M. Borisov and S. A. Freunberger, Angew. Chem., Int. Ed., 2019, 58, 6535 CrossRef CAS.
- L. Schafzahl, N. Mahne, B. Schafzahl, M. Wilkening, C. Slugovc, S. M. Borisov and S. A. Freunberger, Angew. Chem., Int. Ed., 2017, 56, 15728 CrossRef CAS.
- Z. Jiang, Y. Huang, Z. Zhu, S. Gao, Q. Lv and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202835119 CrossRef CAS PubMed.
- Z. Liang, Q. Zou, J. Xie and Y.-C. Lu, Energy Environ. Sci., 2020, 13, 2870 RSC.
- I. Ruiz de Larramendi and N. Ortiz-Vitoriano, Front. Chem., 2020, 8, 605 CrossRef.
- W.-J. Kwak, H. Kim, Y. K. Petit, C. Leypold, T. T. Nguyen, N. Mahne, P. Redfern, L. A. Curtiss, H.-G. Jung, S. M. Borisov, S. A. Freunberger and Y.-K. Sun, Nat. Commun., 2019, 10, 1380 CrossRef PubMed.
- K. Chaisiwamongkhol, C. Batchelor-McAuley, R. G. Palgrave and R. G. Compton, Angew. Chem., 2018, 130, 6378 CrossRef.
- A. T. S. Freiberg, M. K. Roos, J. Wandt, R. de Vivie-Riedle and H. A. Gasteiger, J. Phys. Chem. A, 2018, 122, 8828 CrossRef CAS.
- J. W. Mullinax, C. W. Bauschlicher and J. W. Lawson, J. Phys. Chem. A, 2021, 125, 2876 CrossRef CAS.
- M. Carboni, A. G. Marrani, R. Spezia and S. Brutti, J. Electrochem. Soc., 2018, 165, A118 CrossRef CAS.
- W.-J. Kwak, S. A. Freunberger, H. Kim, J. Park, T. T. Nguyen, H.-G. Jung, H. R. Byon and Y.-K. Sun, ACS Catal., 2019, 9, 9914 CrossRef CAS.
- A. Samojlov, D. Schuster, J. Kahr and S. A. Freunberger, Electrochim. Acta, 2020, 362, 137175 CrossRef CAS.
- D. P. Jones and W. P. Griffith, J. Chem. Soc., Dalton Trans., 1980, 2526, 10.1039/DT9800002526.
- P. A. Giguère and D. Lemaire, Can. J. Chem., 1972, 50, 1472 CrossRef.
- B. Schafzahl, E. Mourad, L. Schafzahl, Y. K. Petit, A. R. Raju, M. O. Thotiyl, M. Wilkening, C. Slugovc and S. A. Freunberger, ACS Energy Lett., 2018, 3, 170 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, e1327 CrossRef.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102(11), 1995–2001 CrossRef CAS.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685 CrossRef CAS.
- S. H. Whitlow and F. D. Findlay, Can. J. Chem., 1967, 45, 2087 CrossRef CAS.
- E. Furui, N. Akai, A. Ida, A. Kawai and K. Shibuya, Chem. Phys. Lett., 2009, 471, 45 CrossRef CAS.
- J. M. Aubry, J. Am. Chem. Soc., 1985, 107, 5844 CrossRef CAS.
- P. B. Merkel, R. Nilsson and D. R. Kearns, J. Am. Chem. Soc., 1972, 94, 1030 CrossRef CAS.
- P. R. Ogilby and C. S. Foote, J. Am. Chem. Soc., 1983, 105, 3423 CrossRef CAS.
- C. Prehal, S. Mondal, L. Lovicar and S. A. Freunberger, ACS Energy Lett., 2022, 7, 3112 CrossRef CAS.
- C. Prehal, A. Samojlov, M. Nachtnebel, L. Lovicar, M. Kriechbaum, H. Amenitsch and S. A. Freunberger, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021893118 CrossRef CAS.
- Y. Wang, N.-C. Lai, Y.-R. Lu, Y. Zhou, C.-L. Dong and Y.-C. Lu, Joule, 2018, 2, 2364 CrossRef CAS.
- A. Schürmann, B. Luerßen, D. Mollenhauer, J. Janek and D. Schröder, Chem. Rev., 2021, 121, 12445 CrossRef.
- C. Amatore and A. R. Brown, J. Am. Chem. Soc., 1996, 118, 1482 CrossRef CAS.
- Y. Qiao, J. Yi, S. Guo, Y. Sun, S. Wu, X. Liu, S. Yang, P. He and H. Zhou, Energy Environ. Sci., 2018, 11, 1211 RSC.
- H.-K. Lim, H.-D. Lim, K.-Y. Park, D.-H. Seo, H. Gwon, J. Hong, W. A. Goddard, H. Kim and K. Kang, J. Am. Chem. Soc., 2013, 135, 9733 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494 CrossRef CAS PubMed.
- R. A. House, J.-J. Marie, M. A. Pérez-Osorio, G. J. Rees, E. Boivin and P. G. Bruce, Nat. Energy, 2021, 6, 781 CrossRef CAS.
- J. L. Roberts Jr, T. S. Calderwood and D. T. Sawyer, J. Am. Chem. Soc., 1984, 106, 4667 CrossRef.
- J. D. Wadhawan, P. J. Welford, E. Maisonhaute, V. Climent, N. S. Lawrence, R. G. Compton, H. B. McPeak and C. E. W. Hahn, J. Phys. Chem. B, 2001, 105, 10659 CrossRef CAS.
- S. Zhang, M. J. Nava, G. K. Chow, N. Lopez, G. Wu, D. R. Britt, D. G. Nocera and C. C. Cummins, Chem. Sci., 2017, 8, 6117 RSC.
- S. E. Renfrew and B. D. McCloskey, J. Am. Chem. Soc., 2017, 139, 17853 CrossRef CAS.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684 CrossRef CAS.
- A. R. Genreith-Schriever, H. Banerjee, C. P. Grey and A. J. Morris, arXiv, 2022, preprint, DOI:10.48550/arXiv.2205.10462.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting methods, figures, and references. See DOI: https://doi.org/10.1039/d3fd00088e |
| This journal is © The Royal Society of Chemistry 2024 |